
Photoredox sheds new light on nickel catalysis: from carbon–carbon to carbon–heteroatom bond formation
Yong-Yuan
Gui
,
Liang
Sun
,
Zhi-Peng
Lu
and
Da-Gang
Yu
*
Key Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, China. E-mail: dgyu@scu.edu.cn
First published on 3rd February 2016
Abstract
This highlight describes the recent emergence of photoredox/nickel dual catalysis to generate carbon–carbon and carbon–heteroatom bonds. To address the challenges in traditional nickel-catalyzed couplings, the application of photoredox catalysis facilitates transmetallation via single-electron transfer and generation of highly reactive nickel(III) intermediates to promote reductive elimination, thus providing new opportunities for nickel-catalysis.
 Yong-Yuan Gui, Da-Gang Yu, Liang Sun, Zhi-Peng Lu (from left to right) | Dr Yong-Yuan Gui (first from the left) was born in Henan, China in 1987. He received his Ph.D. degree in chemistry at the Chengdu Institute of Organic Chemistry, Chinese Academy of Sciences in 2015 (with Prof. Li-Xin Wang). He then joined the group of Prof. Da-Gang Yu as a Postdoctoral Fellow. His research interests focus on photoredox catalysis. Liang Sun (second from the right) was born in Hebei, China in 1988. He received his B.Sc. degree in chemistry from Sichuan University in 2011. He worked as a pharmaceutical research scientist for about 4 years in CSPC Pharmaceutical Group Limited. In 2015 he joined the research group of Prof. Da-Gang Yu. His current research interests focus on photochemistry, especially photoredox catalysis. Zhi-Peng Lu (first from the right) was born in Wenzhou, China, 1996. He is a junior undergraduate in Sichuan University and mainly focuses on photoredox/transition metal dual catalysis. Prof. Dr Da-Gang Yu (second from the left) was born in Jiangxi province, China, 1986. He received his Ph.D. with Prof. Dr Zhang-Jie Shi from Peking University, 2012. Then he carried out postdoctoral research with Humboldt fellowship in the group of Prof. Dr Frank Glorius, Muenster University. Since 2015, he has been working independently in Sichuan University with support from “The Thousand Young Talents Plan”. His research interests mainly focus on novel transition metal-catalysis, and green and sustainable chemistry. |
Introduction
Over the past few decades, transition metal-catalyzed cross-coupling reactions have evolved into one of the most robust and widely used methods to construct carbon–carbon and carbon–heteroatom bonds.1 Nickel, owing to its abundance, relatively low price and unique catalytic properties, has attracted much attention in catalysis and played an important role in facilitating many intriguing, valuable and challenging transformations.2 For example, nickel is less electronegative than palladium, allowing for the relatively facile oxidative addition into otherwise “inert” chemicals such as phenolic derivatives,3 aromatic nitriles4 or fluorides.5 There are also a number of readily available oxidation states of nickel that are primed for distinct reaction modes. Although there exists a well-known array of nickel-catalyzed transformations based on the two-electron Ni(0)/Ni(II) cycle, the easily accessible oxidation states of Ni(I) and Ni(III) also allow for the incorporation of radical species.6 Despite these advantages mentioned, some important challenges have not been fully addressed in this field. For example, two-electron transmetallation with alkylboranes is a big problem in nickel-catalysis, requiring high temperature, excess base or transition metal salts as promoters.7 Moreover, bond formation involving sp3 carbons and carbon–heteroatom bond construction have been less explored because of the sluggish reductive elimination.1Recently, visible-light photoredox catalysis has emerged as an enabling paradigm to accomplish novel chemical transformations via unique single-electron-transfer (SET) pathways.8 A huge effort in this field has been devoted to employ sunlight as a safe, inexpensive, abundant, and renewable energy source. Using transition metal polypyridyl complexes or organic dyes as photocatalysts, a variety of reactive radicals can be generated under mild conditions to engage in redox processes which previously have been elusive in traditional transition metal-catalysis. Moreover, the direct interaction between photocatalysis and transition metal-catalysis provides new redox processes. Therefore, introduction of the photoredox manifold might realize novel reactions with distinct mechanisms as well as provide new opportunities for transition metal-catalysis.9 Furthermore, functional group tolerance can be significantly improved and side reactions will be limited under the relatively mild reaction conditions. Indeed, many groups, including Sanford, Glorius, Toste and others, have successfully realized the dual catalysis of photoredox with Pd- or Au-catalysis through the reductive activation of electrophiles.10 This highlight will detail the recent advances in photoredox/nickel dual catalysis through the oxidative activation of alkyl and other nucleophiles ordered by the types of bonds formed.
Carbon–carbon bond formation
In 2014, Molander,11a MacMillan and Doyle11b simultaneously disclosed two elegant examples of photoredox/nickel catalyzed cross coupling of alkyl nucleophiles with aryl halides. In each of their research disclosures (Fig. 1), first, the Ir-catalyst A is excited to generate B (Ered1/2 [*IrIII/IrII] = +1.21 V versus saturated calomel electrode (SCE) in CH3CN), which undergoes single-electron oxidation of the potassium trifluoroborates 1a (benzyltrifluoroborate, Eox1/2 = −0.67 V versus [FeCp2]/[FeCp2]+ couple in CH3CN)12 or carboxylic acid 1b (tert-butyl carbamoyl (Boc)-Pro-OCs, Ered1/2 = +0.95 V versus SCE in CH3CN)11b to give the Ir-catalyst C and carbon-centered radical D in the presence of a base. Meanwhile, oxidative addition of organohalides 2 to Ni(0) complex E forms the Ni(II) complex F. Subsequent capture of D by F yields the highly reactive Ni(III) species G, which readily undergoes reductive elimination to provide the desired product 3 and Ni(I) complex H. Lastly, both A and E are regenerated to complete the dual catalytic cycle through reduction of H (Ered1/2 [NiII/Ni0] = −1.2 V versus SCE in N,N′-dimethylformamide (DMF)) by the reduced form of the Ir-catalyst C (Ered1/2 [IrIII/IrII] = +1.37 V versus SCE in CH3CN).11b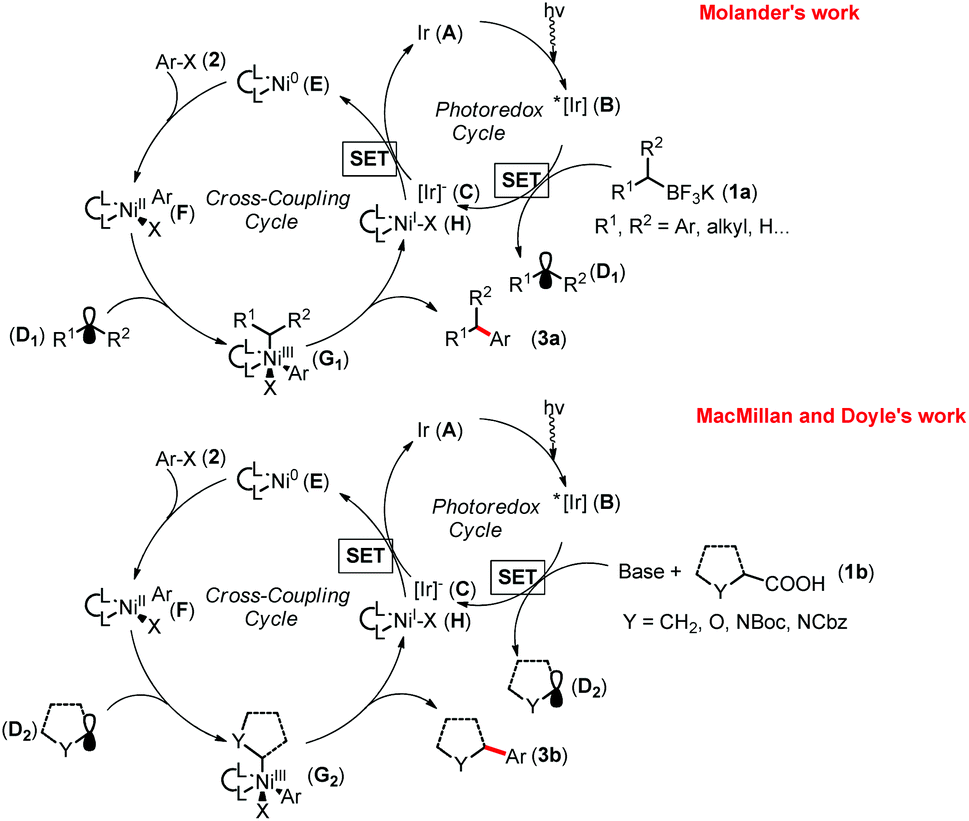 | ||
| Fig. 1 Catalytic cycles for nickel/photoredox dual catalysis. Boc = tert-butyloxycarbonyl, Cbz = carbobenzyloxy. | ||
Previously, Akita demonstrated the photo-catalyzed generation of radicals from organoborates and laid the foundation.12 In Molander's research, the merger with photoredox significantly improved the cross coupling by replacing the challenging two-electron transmetallation with a facile single-electron event (Scheme 1-a).11a A wide range of benzyl trifluoroborates 1a and aryl bromides 2 with electronically and sterically diverse substituents gave diarylmethanes 3a in moderate to excellent yields. Importantly, the coupling reaction of a sec-benzyl borate 1aa generated 3aa with moderate enantioselectivity in the presence of a chiral bisoxazoline ligand L1, demonstrating the radical D1 as a key intermediate and providing great potential for enantioselective coupling reactions (Scheme 1-b). A successive joint work with Kozlowski investigating the mechanistic details13 suggested that the alkylnickel(I) complex generated by addition of an alkyl radical D1 to Ni(0) complex E is also likely to operate in these reactions.
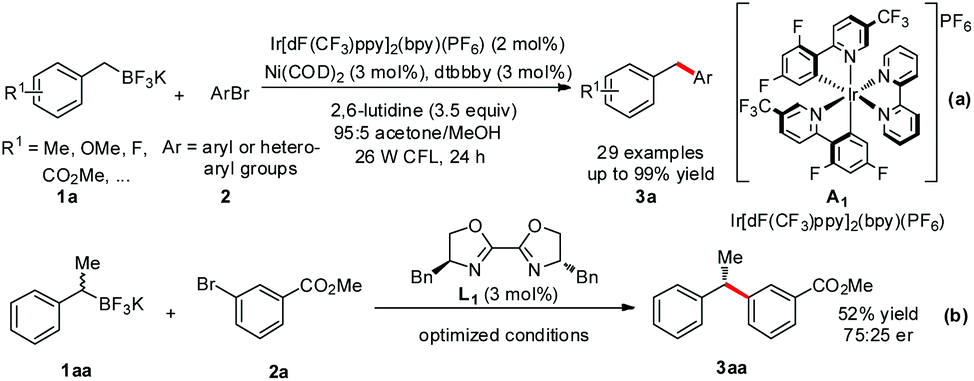 | ||
| Scheme 1 Molander's dual catalysis cross coupling. dF(CF3)ppy = 2-(2,4-difluorophenyl)-5-(trifluoromethyl)-pyridine, bpy = bipyridine, dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine, COD = cyclooctadiene, CFL = compact fluorescent light. | ||
Under MacMillan and Doyle's catalytic system,11b various kinds of carboxylic acids 1b were transformed in situ to the corresponding cesium carboxylates, which easily undergo single-electron oxidation with the excited photocatalyst B to generate the reactive radical species D2 (Fig. 1 and Scheme 2-a). (Hetero)aryl halides 2 bearing both electron-donating and electron-withdrawing groups could afford products 3b in 64–90% yields. The activated aryl chlorides are also efficient coupling partners. Of particular note is that this catalytic method can also be applied toward the direct cross couplings of the methyl C(sp3)–H in dimethylaniline 4via the key radical intermediate D3, delivering the arylated amines 5 in good to excellent yields (Scheme 2-b).
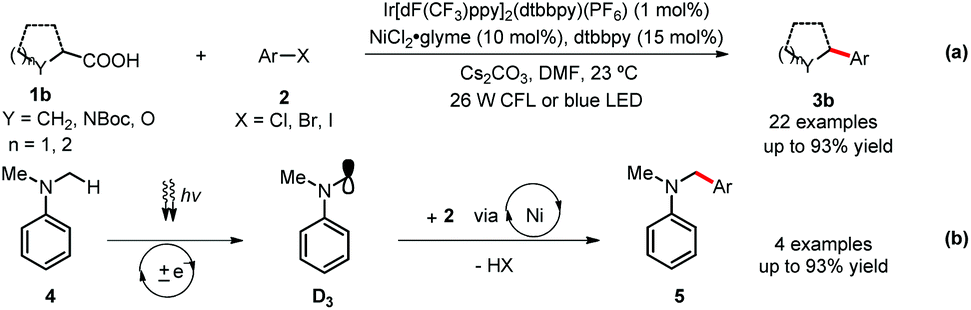 | ||
| Scheme 2 MacMillan and Doyle's dual catalysis cross coupling. glyme = dimethoxyethane. LED = light-emitting diode. | ||
After the successful realization of the cross couplings of benzylic trifluoroborates with aryl bromides, the Molander group applied this strategy to other trifluoroborates, including the sterically-hindered secondary alkyl-, α-alkoxymethyl- and α-aminomethyl trifluoroborates 1a (Scheme 3a–c).14 Under similarly mild conditions, most of these cross couplings gave the products 3 in moderate to excellent yields. Based on the significantly different reactivity of aryl versus alkyl trifluoroborates in the SET process, the same group recently applied borylated aryl bromides 2b as electrophiles in such a dual catalysis in which the aromatic carbon–borane bond was untouched. This advance provides a great opportunity for further functionalization of the products 3abvia Suzuki–Miyaura coupling, 1,4-addition or oxidation (Scheme 3-d).15
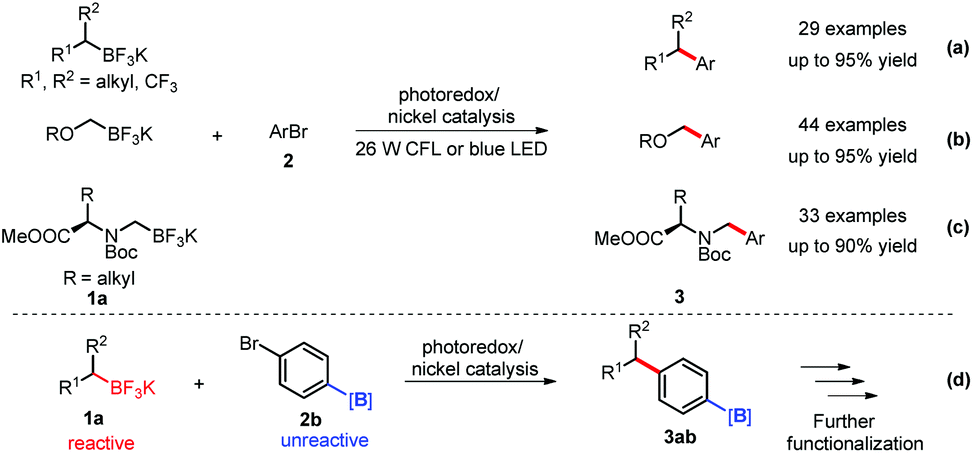 | ||
| Scheme 3 Other examples of dual catalysis of organoborates. | ||
Similarly, the MacMillan group further contributed to this field by significantly expanding the substrate scope with their strategy. Firstly, vinyl iodides 2, which have similar reactivity to aryl halides, have been investigated to generate important functionalized alkenes 3 using this dual catalytic system (Scheme 4-a).16 Vinyl iodides possessing various functional groups, such as amines, ethers, and C–Cl bond, delivered the corresponding products in good yields. A broad range of carboxylic acids, including α-amino acids, α-oxy acids, cyclic and acyclic alkyl carboxylic acids, also readily underwent this reaction. Secondly, simple and abundant α-keto acids 6 also have been applied to couple with various kinds of organohalides (Scheme 4-b).17 Importantly, one alkyl halide, bromocyclopentane, was employed to generate a dialkyl ketone 7 in high yield, which highlights the power of the photoredox/nickel dual catalysis. Thirdly, an elegant example of decarboxylation of anhydrides was reported by the same group. Intramolecular CO2 extrusion–recombination of mixed anhydrides 8 (formed in situ from carboxylic acids and acyl chlorides) provided ketones 7 in moderate to high yields (Scheme 4-c).18
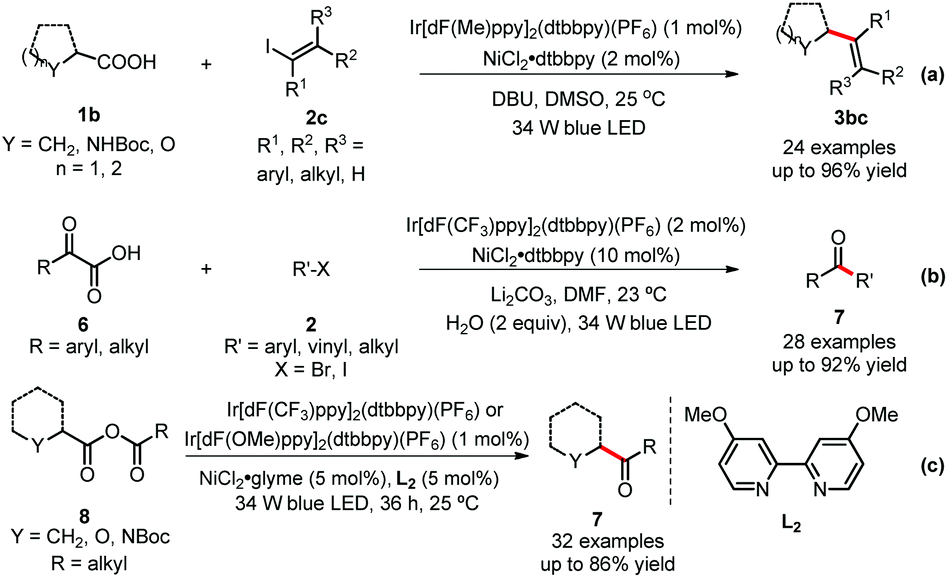 | ||
| Scheme 4 Other examples of decarboxylative dual catalysis. | ||
Hypercoordinate silicates, due to their high solubility, low oxidation potentials and benign byproducts generated upon oxidation, were introduced as new sources of alkyl radicals19 in dual catalysis by the groups of Fensterbank and Goddard,20a and Molander,20b respectively (Scheme 5). Both systems worked highly efficiently under mild and base-free conditions. In Fensterbank and Goddard's work (Scheme 5-a), catechol-based silicates 1c were tested as alkyl radical precursors in a number of homolytic transformations, including photoredox/nickel dual catalysis with 4-bromobenzonitrile 2d. Although tert-butyl silicates gave no desired products, the cross couplings worked well with allyl, benzyl, secondary alkyl silicates and α-amino alkyl silicates. Meanwhile, Molander and colleagues extensively investigated the cross coupling of (hetero)aryl bromides 2 with primary and secondary ammonium alkylsilicates 1c (Scheme 5-b). Both 1° and 2° alkylsilicates afforded coupled products with a wide variety of aryl and heteroaryl bromides 2 in good to excellent yields. Importantly, various kinds of functional groups were well tolerated, including unprotected primary and secondary alkylamines, pyrazoles and indoles.
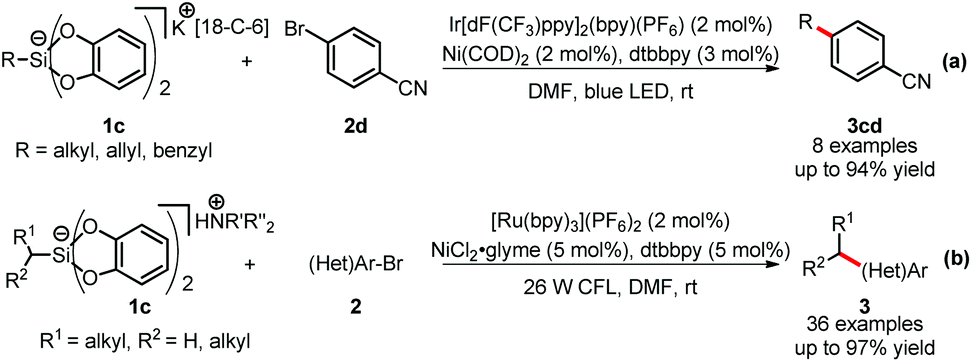 | ||
| Scheme 5 Photoredox/Ni dual catalysis of alkylsilicates. 18-C-6 = 18-crown-6. | ||
Carbon–heteroatom bond formation
Although it is highly important to generate valuable ethers, amines and other compounds containing heteroatoms, the reductive elimination to form carbon–heteroatom bonds (C–O, C–N, C–P and other bonds) is also very difficult in Ni(0)/Ni(II) catalysis. However, introduction of photoredox provides easy access to highly reactive Ni(III) complexes, which are prone to undergo reductive elimination, unlocking new pathways to generate carbon–heteroatom bonds. Therefore, much effort has been devoted in this direction.At the beginning of 2015, the Xiao and Lu group reported the first example of carbon–heteroatom bond formation using such a dual catalysis strategy (Scheme 6).21 The couplings of aryl iodides 2 with diarylphosphine oxides 9 generated C–P bonds efficiently under mild conditions. These reactions showed good substrate scope and excellent functional group tolerance. Phosphine oxide 9 is proposed to undergo SET oxidation and subsequent base-promoted deprotonation to give the P-centered radical D3. Then, D3 is captured by the Ar–Ni(II)–I complex F to form Ni(III) intermediates G3, which is highly reactive to give the desired product 10via facile reductive elimination.
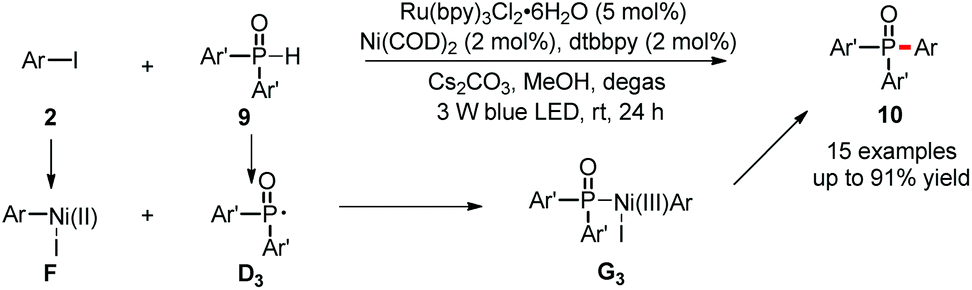 | ||
| Scheme 6 C–P bond formation via dual catalysis. | ||
Besides the generation of free carbon- or phosphine-based radicals to react with the Ni(II) complex, the photoredox cycle could also come into contact with nickel catalysis via direct oxidation and reduction of nickel complexes. Recently, MacMillan's group has enabled the challenging C(sp2)–O bond formation by using photoredox/nickel dual catalysis. The highly efficient reaction between aryl bromides and alcohols or water 11 took place under mild reaction conditions (Scheme 7).22 The aryl–Ni(II)-OR species F1, generated from oxidative addition and ligand exchange, is proposed to undergo SET oxidation with the excited photocatalyst to form the transient Ni(III) intermediate G4. This highly reactive species readily undergoes reductive elimination to afford the desired products 12. Importantly, this hypothesis has been supported by mechanistic studies, including testing stoichiometric reactions of F1 under photoredox conditions, cyclic voltammetry and Stern–Volmer fluorescence quenching experiments.
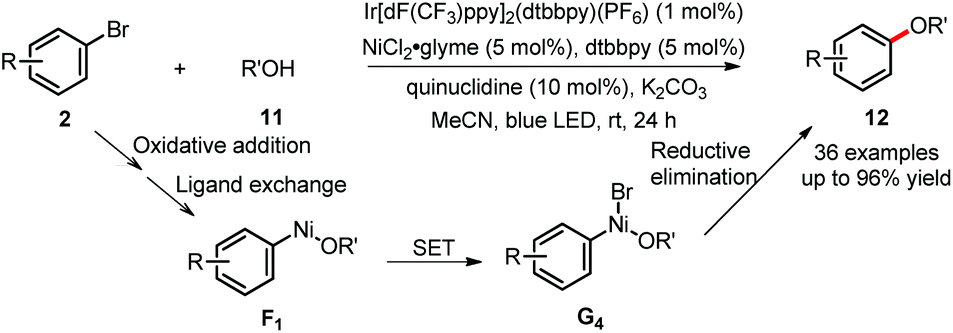 | ||
| Scheme 7 C–O bond formation via dual catalysis. | ||
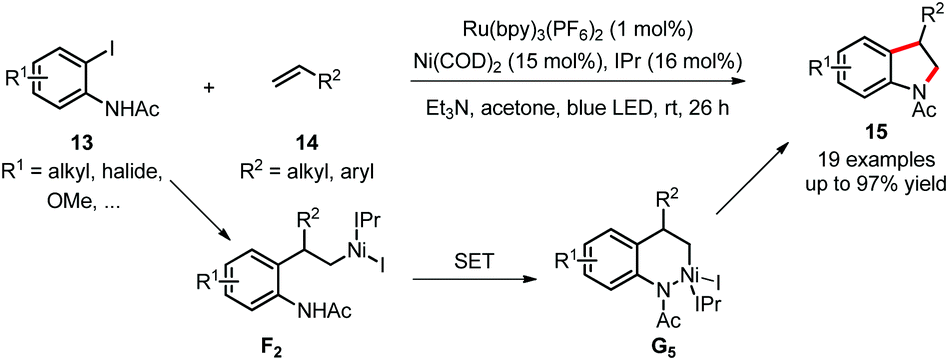
Simultaneously, the Jamison group developed a highly regioselective synthesis of indolines 15 and addressed the challenges of C(sp3)–heteroatom bond formation with dual catalysis (Scheme 8).23 The Ni(II) complex F2, formed via selective insertion of aliphatic or styrenyl alkenes 14 into the aryl–nickel bond, is oxidized by the excited photocatalyst (or other common oxidants including air) to generate the Ni(III) complex G5, which promotes C(sp3)–N bond formation by reductive elimination. The Heck-type products, generated from undesired β-hydride elimination, are significantly inhibited and show no reactivity under such dual catalysis. Mechanistic studies similar to those from MacMillan's C–O bond formation also provided deep insight into this reaction.
 | ||
| Scheme 8 C–N bond formation via dual catalysis. IPr = 1,3-bis(2,6-diisopropylphenyl)imidazolylidene. | ||
Conclusions
The photoredox/nickel dual catalysis has come to the fore as a powerful tool for the discovery of novel reactivity. As demonstrated by the examples summarized in this highlight, the challenging C(sp3)–C and carbon–heteroatom bond formation have been successfully realized by merging photoredox with Ni-catalysis under extremely mild, environmentally friendly and sustainable conditions. Transmetallation via single-electron transfer and generation of highly reactive nickel(III) intermediates to promote reductive elimination are two key points in such successful dual catalysis.Although significant progress has been made in this field, many challenges remain to be addressed. For example, the scope of both nucleophiles and electrophiles need to be significantly expanded in the dual catalysis.24 In addition, much more attention should be paid to the mechanisms of the dual catalytic cycle, which will undoubtedly accelerate the development of novel transformations and the design of more effective catalytic systems. Last but not least, the asymmetric dual catalysis24d should be further developed based on the nice pioneering work.11a Considering the relative youth of this emerging field, we envision that the photoredox/nickel dual catalysis will find more applications in developing new and valuable transformations, thus allowing for the efficient construction of complex molecules in the future.
Acknowledgements
We thank the “1000-Youth Talents Plan”, a Start-up Grant from Sichuan University, and the National Natural Science Foundation of China (21502124) for financial support. We are also grateful to Prof. Jason Chruma for kind help in revision.Notes and references
- Metal-Catalyzed Cross-Coupling Reactions and More, ed. A. de Meijere, S. Bräse and M. Oestreich, Wiley-VCH, Weinheim, Germany, 2014 Search PubMed.
- (a) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299 CrossRef CAS PubMed; (b) V. P. Ananikov, ACS Catal., 2015, 5, 1964 CrossRef CAS.
- (a) B.-J. Li, D.-G. Yu, C.-L. Sun and Z.-J. Shi, Chem. – Eur. J., 2011, 17, 1728 CrossRef CAS PubMed; (b) B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A.-M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2011, 111, 1346 CrossRef CAS PubMed; (c) J. Yamaguchi, K. Muto and K. Itami, Eur. J. Org. Chem., 2013, 19 CrossRef CAS; (d) J. Cornella, C. Zarate and R. Martin, Chem. Soc. Rev., 2014, 43, 8081 RSC; (e) M. Tobisu and N. Chatani, Acc. Chem. Res., 2015, 48, 1717 CrossRef CAS PubMed.
- S. M. Bonesi and M. Fagnoni, Chem. – Eur. J., 2010, 16, 13572 CrossRef CAS PubMed.
- H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119 CrossRef CAS PubMed.
- (a) V. B. Phapale and D. J. Cárdenas, Chem. Soc. Rev., 2009, 38, 1598 RSC; (b) J. Breitenfeld and X. Hu, Chimia, 2014, 68, 235 CrossRef CAS PubMed.
- (a) H. Doucet, Eur. J. Org. Chem., 2008, 2013 CrossRef CAS; (b) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417 CrossRef CAS PubMed.
- For selected reviews on visible-light-induced photoredox catalysis, see: (a) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527 CrossRef CAS PubMed; (b) F. Teply, Collect. Czech. Chem. Commun., 2011, 76, 859 CrossRef CAS; (c) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC; (d) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828 CrossRef CAS PubMed; (e) L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687 RSC; (f) J. Xuan, L.-Q. Lu, J.-R. Chen and W.-J. Xiao, Eur. J. Org. Chem., 2013, 6755 CrossRef CAS; (g) Y. Xi, H. Yi and A. Lei, Org. Biomol. Chem., 2013, 11, 2387 RSC; (h) D. P. Hari and B. König, Angew. Chem., Int. Ed., 2013, 52, 4734 CrossRef CAS PubMed; (i) X.-J. Dai, X.-L. Xu and X.-N. Li, Chin. J. Org. Chem., 2013, 33, 2046 CrossRef CAS; (j) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (k) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 6174 CrossRef PubMed.
- For elegant reviews on dual catalysis involving visible-light photocatalysis, see: (a) M. N. Hopkinson, B. Sahoo, J. Li and F. Glorius, Chem. – Eur. J., 2014, 20, 3874 CrossRef CAS PubMed; (b) E. Jahn and U. Jahn, Angew. Chem., Int. Ed., 2014, 53, 13326 CrossRef CAS PubMed; (c) C. Vila, ChemCatChem, 2015, 7, 1790 CrossRef CAS.
- Selected examples, see: (a) D. Kalyani, K. B. McMurtrey, S. R. Neufeldt and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18566 CrossRef CAS PubMed; (b) B. Sahoo, M. N. Hopkinson and F. Glorius, J. Am. Chem. Soc., 2013, 135, 5505 CrossRef CAS PubMed; (c) X. Shu, M. Zhang, Y. He, H. Frei and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 5844 CrossRef CAS PubMed.
- (a) J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433 CrossRef CAS PubMed; (b) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437 CrossRef CAS PubMed.
- Reviews, see: (a) T. Koike and M. Akita, Synlett, 2013, 2492 CrossRef CAS; (b) M. Akita, T. Koike and A. Inagaki, J. Synth. Org. Chem., Jpn., 2014, 72, 538 CrossRef CAS.
- O. Gutierrez, J. C. Tellis, D. N. Primer, G. A. Molander and M. C. Kozlowski, J. Am. Chem. Soc., 2015, 137, 4896 CrossRef CAS PubMed.
- (a) D. N. Primer, I. Karakaya, J. C. Tellis and G. A. Molander, J. Am. Chem. Soc., 2015, 137, 2195 CrossRef CAS PubMed; (b) D. Ryu, D. N. Primer, J. C. Tellis and G. A. Molander, Chem. – Eur. J., 2016, 22, 120 CrossRef CAS PubMed; (c) I. Karakaya, D. N. Primer and G. A. Molander, Org. Lett., 2015, 17, 3294 CrossRef CAS PubMed; (d) M. El Khatib, R. A. M. Serafim and G. A. Molander, Angew. Chem., Int. Ed., 2016, 55, 254 CrossRef CAS PubMed.
- Y. Yamashita, J. C. Tellis and G. A. Molander, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12026 CrossRef CAS PubMed.
- A. Noble, S. J. McCarver and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 624 CrossRef CAS PubMed.
- L. Chu, J. M. Lipshultz and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 7929 CrossRef CAS PubMed.
- C. C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 11938 CrossRef CAS PubMed.
- (a) Y. Nishigaichi, A. Suzuki and A. Takuwa, Tetrahedron Lett., 2007, 48, 211 CrossRef CAS; (b) D. Matsuoka and Y. Nishigaishi, Chem. Lett., 2014, 43, 559 CrossRef CAS.
- (a) V. Corcé, L. M. Chamoreau, E. Derat, J. P. Goddard, C. Ollivier and L. Fensterbank, Angew. Chem., Int. Ed., 2015, 54, 11414 CrossRef PubMed; (b) M. Jouffroy, D. N. Primer and G. A. Molander, J. Am. Chem. Soc., 2016, 138, 475 CrossRef CAS PubMed.
- J. Xuan, T.-T. Zeng, J.-R. Chen, L.-Q. Lu and W.-J. Xiao, Chem. – Eur. J., 2015, 21, 4962 CrossRef CAS PubMed.
- J. A. Terrett, J. D. Cuthbertson, V. W. Shurtleff and D. W. C. MacMillan, Nature, 2015, 524, 330 CrossRef CAS PubMed.
- S. Z. Tasker and T. F. Jamison, J. Am. Chem. Soc., 2015, 137, 9531 CrossRef CAS PubMed.
- For the most recent progress reported during the proofing of this highlight, see: (a) J. Amani, E. Sodagar and G. A. Molander, Org. Lett., 2016 DOI:10.1021/acs.orglett.5b03705; (b) N. R. Patel, C. B. Kelly, M. Jouffroy and G. A. Molander, Org. Lett., 2016 DOI:10.1021/acs.orglett.6b00024; (c) M. S. Oderinde, M. Frenette, D. W. Robbins, B. Aquila and J. W. Johannes, J. Am. Chem. Soc., 2016 DOI:10.1021/jacs.5b11244; (d) Z. Zuo, H. Cong, W. Li, J. Choi, G. C. Fu and D. W. C. MacMillan, J. Am. Chem. Soc., 2016 DOI:10.1021/jacs.5b13211.
| This journal is © the Partner Organisations 2016 |