Recyclable Ru/C catalyzed oxidative cyanation of tertiary amines with TBHP†
K. Harsha Vardhan
Reddy
,
G.
Satish
,
V. Prakash
Reddy
,
B. S. P. Anil
Kumar
and
Y. V. D.
Nageswar
*
Organic Chemistry Division-I, Indian Institute of Chemical Technology, Hyderabad 500607, India. E-mail: dryvdnageswar@gmail.com; Fax: +91-40-27160512
First published on 10th October 2012
Abstract
The recyclable Ru/C catalyzed oxidative α-cyanation of tertiary amines with ethyl cyanoformate by using TBHP as an oxidant under ambient conditions has been developed. Utilizing this protocol, α-aminonitrile derivatives were synthesized in good to excellent yields with high selectivity. The cyanide source (ethyl cyanoformate) employed herein was relatively cheap and less toxic, which would be beneficial. The catalyst was also inexpensive and commercially available as well as recyclable up to four cycles, without significant loss of its catalytic activity.
Introduction
The development of novel methods for C–C bond formation through C–H activation has attracted the attention of researchers across the world.1 Transition metal catalyzed activation of C–H bond formation, especially sp3 C–H bonds and subsequent C–C bond formation, is a valuable and straightforward synthetic strategy,2 as it does not need a prefunctionalized starting material.The oxidative cyanation of tertiary amines via direct functionalization of the C(sp3)–H bond provides access to α-aminonitriles.3 α-Aminonitriles are highly useful and versatile synthetic intermediates (for the preparation of α-amino carbonyl compounds, α-amino alcohols and vicinal diamines), which have widely been used in the construction of a variety of synthetically, as well as biologically important compounds, such as alkaloids and functional materials.4 Therefore, the synthesis of these α-aminonitriles is significantly important both in synthetic as well as medicinal chemistry.
The synthesis of α-amino nitriles, by the reaction of carbonyl compound, an amine, and a cyanide, (Strecker reaction) is well reported in the literature.5 However, the new synthetic strategy for the preparation of these compounds involves the oxidation of a tertiary amine into an iminium ion followed by the nucleophilic attack of the cyanide ion. In this regard several metal catalysts such as Ru,6 V,7 Au,8 Mo,9 Fe,10 Ir11 and Ti,12 and metal free conditions,13 have been reported for the oxidative cyanation of tertiary amines. However, most of these protocols involve expensive and moisture sensitive catalytic systems as well as toxic cyanide reagents such as NaCN or TMSCN. Therefore, the development of a less toxic cyanide source, and a catalyst for the cyanation of tertiary amines, is highly desirable. Ethyl cyanoformate has been employed in various applications as a less toxic, cheap and readily available cyanide source. Even though few heterogeneous systems have been developed recently,10b,14 preparation of all these heterogeneous catalytic systems involve tedious reaction procedures with expensive starting materials. Due to advances in sustainable chemistry, exploration of an inexpensive and commercially available catalytic system is desirable for this transformation. Ru/C15 has emerged as a versatile heterogeneous catalyst for the oxidation of various functional groups in synthetic organic chemistry. To the best our knowledge there is no report on the oxidative cyanation of tertiary amines over a heterogeneous Ru/C catalyst. In continuation of our efforts to design and develop novel methodologies under environmentally benign conditions,16 we report here a recyclable Ru/C catalyzed oxidative cyanation of several tertiary amines to the corresponding α-aminonitriles, using ethyl cyanoformate as a cyanide source and TBHP as an oxidant at 60 °C (Scheme 1).
 | ||
| Scheme 1 Ru/C catalyzed oxidative α-cyanation of tertiary amines. | ||
Results and discussion
Initially, N,N-dimethylaniline, ethyl cyanoformate and Ru/C were used as model substrates to optimize the reaction conditions (Table 1). The reaction was screened to assess the effect of different oxidants such as TBHP in decane, H2O2 and O2. Among these oxidants TBHP was found to be the most suitable to give the desired product (Table 1, entries 1, 2 and 5). The effects of different concentrations of TBHP and ethyl cyanoformate on the course of the reaction were examined and 2.5 equivalents of TBHP (Table 1, entries 3–5) and 2.0 equivalents of ethyl cyanoformate (Table 1, entries 5–7) were observed to be optimal for this reaction. Different solvents such as MeOH, EtOH, PhCH3, CH3CN, EtOAc, CHCl3 and H2O were screened (Table 1, entries 5, 8–12), and MeOH was found to be efficient and produced the desired product in good yield (Table 1, entry 5). Water was observed to be ineffective and did not yield the desired product (Table 1 entry 13). The reaction is very sluggish at room temperature and required a longer time to get a reasonable yield (Table 1, entry 14). Nevertheless, a high yield was obtained at 60 °C within a short reaction time (Table 1, entry 5). The optimum reaction conditions were found to be N,N-dimethylaniline (1.0 mmol), ethyl cyanoformate (2.0 mmol), 5 wt% Ru/C (20 mg), TBHP (in decane) (2.5 equiv) in MeOH (3 mL) at 60 °C for 6 h.| Entry | Solvent | Oxidant (equiv.) | T (°C) | Yield (%)b |
|---|---|---|---|---|
| a Reactions were carried out with N,N-dimethylaniline (1.0 mmol), ethyl cyanoformate (2.0 mmol), ruthenium 5 wt% on carbon (20 mg) and oxidant in solvent (3 ml) at 60 °C for 6 h. b Isolated yield of the pure product. c Ethyl cyanoformate (1.5 mmol). d Ethyl cyanoformate (1.0 mmol). | ||||
| 1 | MeOH | H2O2 | 60 | 50 |
| 2 | MeOH | O2 | 60 | 40 |
| 3 | MeOH | TBHP in decane (1.0) | 60 | 40 |
| 4 | MeOH | TBHP in decane (2.0) | 60 | 74 |
| 5 | MeOH | TBHP in decane (2.5) | 60 | 95 |
| 6 | MeOH | TBHP in decane (2.5) | 60 | 84c |
| 7 | MeOH | TBHP in decane (2.5) | 60 | 71d |
| 8 | EtOH | TBHP in decane (2.5) | 60 | 61 |
| 9 | PhCH3 | TBHP in decane (2.5) | 60 | 25 |
| 10 | CH3CN | TBHP in decane (2.5) | 60 | 46 |
| 11 | EtOAc | TBHP in decane (2.5) | 60 | 51 |
| 12 | CHCl3 | TBHP in decane (2.5) | 60 | 30 |
| 13 | Water | TBHP in decane (2.5) | 60 | 0 |
| 14 | MeOH | TBHP in decane (2.5) | rt | 60 |
Under the optimized reaction conditions, the effect of different cyanide reagents was observed for the cyanation of tertiary amines. Ethyl cyanoformate was found to be a better cyanide source than other reagents (Table 2, entries 1–6). No product formation was observed with zinc cyanide (Table 2, entry 6).
| Entry | Cyanide source | Yield (%)b |
|---|---|---|
| a Reactions were carried out with N,N-dimethylaniline (1.0 mmol), cyanide source (2.0 mmol), ruthenium 5 wt% on carbon (20 mg), TBHP (in decane, 2.5 equiv) in MeOH (3 ml) at 60 °C for 6 h. b Isolated yield of the pure product. c 0.5 equivalents. | ||
| 1 | CNCOOEt | 95 |
| 2 | CNCOOC3H7 | 32 |
| 3 |

|
60 |
| 4 | K3Fe(CN)6c | 10 |
| 5 | CH2(CN)2 | 19 |
| 6 | Zn(CN)2 | 0 |
While expanding the substrate scope of the reaction, a series of tertiary amines were employed to investigate the generality of the reaction, and the results are depicted in Table 3. We examined the oxidative cyanation with various N,N-dimethylanilines and cyclic amines. Substituted N,N -dimethylanilines bearing both electron-donating and electron-withdrawing substituents afforded the corresponding cyanated products in good to excellent yields (Table 3, entries 2–5). It was noteworthy that in the case of unsymmetrical tertiary amines the nucleophile CN− attacked chemoselectively (Table 3, entries 7–9). This protocol was also effective for cyclic amines. Piperidine, pyrrolidine, and tetrahydroisoquinoline afforded almost quantitative yields in all cases (Table 3, entries 10–17). Various substituted piperidine derivatives were prepared following our previously reported procedure17 to further react with ethyl cyanoformate, affording corresponding α-amino nitriles in high yields (Table 3, entries 11–16). In the case of the aliphatic tertiary amine, the desired product was not obtained (Table 3, entry 18).
| Entry | Substrate | Product | Yield (%)b |
|---|---|---|---|
| a Reactions were carried out with tertiary amine (1.0 mmol), ethyl cyanoformate (2.0 mmol), ruthenium 5 wt% on carbon (20 mg), TBHP (in decane, 2.5 equiv) in MeOH (3 ml) at 60 °C for 6 h. b Isolated yield of the pure product. | |||
| 1 |

|

|
95 |
| 2 |

|

|
96 |
| 3 |

|
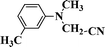
|
92 |
| 4 |
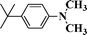
|

|
89 |
| 5 |

|

|
86 |
| 6 |

|

|
60 |
| 7 |

|

|
83 |
| 8 |

|

|
74 |
| 9 |
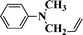
|

|
72 |
| 10 |
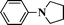
|

|
76 |
| 11 |

|

|
89 |
| 12 |

|
|
91 |
| 13 |

|

|
85 |
| 14 |
|

|
82 |
| 15 |

|

|
84 |
| 16 |
|

|
82 |
| 17 |

|

|
78 |
| 18 | (C2H5)3N | — | 0 |
Based on experimental observation, a plausible reaction mechanism was proposed for Ru/C catalyzed oxidative α-cyanation of N,N-dimethylaniline with TBHP in the presence of ethyl cyanoformate (Scheme 2). The Ru/C 1 undergoes reaction with TBHP to give the Ru–oxo species 2.18 This Ru–oxo species 2 reacts with tertiary amine 3, to produce an iminium ion intermediate 4 by electron transfer and subsequent hydrogen transfer. The iminium ion intermediate subsequently reacts with CN− (in situ generated from CNCOOEt) to afford α-aminonitrile 5, regenerating the Ru/C 1 species.
 | ||
| Scheme 2 Proposed reaction mechanism for the Ru/C catalyzed oxidative cyanation of tertiary amines with ethyl cyanoformate. | ||
The reusability of the Ru/C catalyst was examined and the results are summarized in Fig. 1. After the reaction, Ru/C was separated by centrifugation, washed with ethyl acetate followed by acetone and dried in a hot air oven. This dried Ru/C was reused directly for the next batch without any treatment. No significant loss of catalytic activity was observed up to four cycles (Fig. 1). It was observed from TEM analysis that the morphology, shape and size of the catalyst after reuse (Fig. 2b), did not differ much from the native catalyst (Fig. 2a).
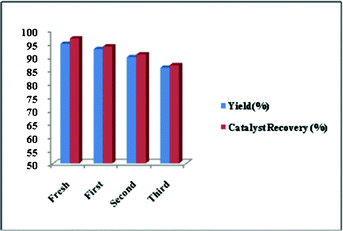 | ||
| Fig. 1 Proposed heterogeneous Ru/C recyclability data. | ||
 | ||
| Fig. 2 Proposed TEM images of (a) native Ru/C (b) Ru/C after four cycles. | ||
Conclusions
In conclusion, we have developed a simple and more convenient route for the oxidative cyanation of tertiary amines, using a recyclable ruthenium catalytic system with ethyl cyanoformate. The recyclability of the catalyst, inexpensive cyanide source, short reaction time, wide substrate scope and excellent product yields make this protocol very practical.Experimental section
General experimental procedure for oxidative cyanation of tertiary amines
The reaction was carried in a 25 mL round bottomed flask equipped with magnetic stir bar, charged with N,N-dimethylaniline (1 mmol), ethyl cyanoformate (2 mmol), 5–6 M TBHP solution in decane (2.5 mmol), methanol (3 mL) and Ru/C (20 mg) catalyst. The resulting reaction mixture was stirred at 60 °C for 6 h. The reaction progress was monitored by TLC. After the reaction, Ru/C was separated by centrifugation and the solvent was evaporated and the crude product purified by column chromatography. The identity and purity of the product was confirmed by 1H NMR, 13C NMR and ESI-MS.2-(Methyl(phenyl)amino)acetonitrile (Table 3, entry 1)6d
1H NMR (300 MHz, CDCl3): δ 7.30–7.21 (m, 2H), 6.93–6.80 (m, 3H), 4.13 (s, 2H), 3.01 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 147.7, 129.4, 120.3, 114.9, 96.1, 42.2, 39.2. ESI-MS: m/z 147 [M+1].2-(Methyl(p-tolyl)amino)acetonitrile (Table 3, entry 2)6d
1H NMR (−300 MHz, CDCl3): δ 7.08–7.05 (m, 2H), 6.76–6.74 (m, 2H), 4.08 (s, 2H), 2.96 (s, 3H), 2.28 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 145.6, 129.8, 129.7, 115.4, 115.3, 42.6, 39.3, 20.2. ESI-MS: m/z 161 [M+1].2-(Methyl(m-tolyl)amino)acetonitrile(Table 3, entry 3)6d
1H NMR (300 MHz, CDCl3): δ 7.16–7.10 (m, 1H), 6.72–6.59 (m, 3H), 4.10 (s, 2H), 2.98 (s, 3H), 2.33 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 147.7, 139.1, 129.1, 120.9, 115.5, 111.9, 42.1, 39.1, 21.6. ESI-MS: m/z 161 [M+1].2-((4-(tert-Butyl)phenyl)(methyl)amino)acetonitrile (Table 3, entry 4)7
1H NMR (300 MHz, CDCl3): δ 7.29–7.24 (m, 2H), 6.79–6.76 (m, 2H), 4.09 (s, 2H), 2.97 (s, 3H), 1.29 (s, 9H). 13C NMR (75 MHz, CDCl3): δ 145.5, 143.0, 126.2, 115.0, 42.5, 39.3, 34.0, 31.5. ESI-MS: m/z 203 [M+1].2-((4-Bromophenyl)(methyl)amino)acetonitrile (Table 3, entry 5)6d
1H NMR (300 MHz, CDCl3): δ 7.40–7.34 (m, 2H), 6.74–6.68 (m, 2H), 4.10 (s, 2H), 2.99 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 146.7, 132.2, 116.3, 115.0, 112.5, 42.1, 39.3. ESI-MS: m/z 225 [M+1].2-(Methyl(naphthalen-1-yl)amino)acetonitrile (Table 3, entry 6)
1H NMR (300 MHz, CDCl3): δ 8.07–8.04 (m, 1H), 7.85–7.76 (m, 1H), 7.66–7.55 (m, 1H), 7.50–7.21 (m, 4H), 4.05 (s, 2H), 3.04 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 146.2, 134.8, 128.7, 128.7, 126.1, 125.4, 122.7, 117.1, 115.0, 46.1. 41.3. ESI-MS: m/z 197 [M+1].2-(Ethyl(phenyl)amino)acetonitrile (Table 3, entry 7)8
1H NMR (300 MHz, CDCl3): δ 7.32–7.17 (m, 2H), 6.91–6.75 (m, 3H), 4.09 (s, 2H), 3.41 (q, J = 7.1 Hz, 2H), 1.25 (J = 7.1 Hz, 3H). 13C NMR (75 MHz, CDCl3): δ 146.8, 129.5, 120.0, 116.0, 115.1, 114.0, 46.2, 39.5, 12.2. ESI-MS: m/z 161 [M+1].2-(Benzyl(phenyl)amino)acetonitrile (Table 3, entry 8)
1H NMR (300 MHz, CDCl3): δ 7.44–7.21 (m, 7H), 6.96–6.84 (m, 3H), 4.49 (s, 1H), 4.02 (s, 1H). 13C NMR (75 MHz, CDCl3): δ 147.9, 136.7, 129.5, 128.9, 127.8, 127.6, 120.7, 115.7, 114.1, 55.7, 39.4. ESI-MS: m/z 223 [M+1].2-(Allyl(phenyl)amino)acetonitrile (Table 3, entry 9)
1H NMR (300 MHz, CDCl3): δ 7.28–7.21 (m, 2H), 6.93–6.80 (m, 3H), 5.96–5.79 (m, 1H), 5.37–5.26 (m, 2H), 4.09 (s, 2H), 3.93 (d, J = 5.2 Hz, 2H). 13C NMR (75 MHz, CDCl3): δ 147.4, 132.9, 129.5, 120.4, 115.3, 96.2, 54.5, 39.3. ESI-MS: m/z 173 [M+1].1-Phenylpyrrolidine-2-carbonitrile (Table 3, entry 10)8
1H NMR (300 MHz, CDCl3): δ 7.37–7.21 (m, 2H), 6.98–6.81 (m, 1H), 6.74–6.62 (m, 2H), 4.43 (d, J = 6.9 Hz, 1H), 3.50–3.33 (m, 2H), 2.49–2.10 (m, 4H). ESI-MS: m/z 173 [M+1].1-Phenylpiperidine-2-carbonitrile (Table 3, entry 11)8
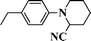
1H NMR (300 MHz, CDCl3): 7.29–7.24 (m, 2H), 6.97–6.94 (m, 3H), 4.55 (s, 1H), 3.39 (d, J = 12.2 Hz, 1H), 3.05 (t, J = 11.7 Hz, 1H), 2.03–1.55 (m, 6H). ESI-MS: m/z 187 [M+1].1-(4-Ethylphenyl)piperidine-2-carbonitrile (Table 3, entry 12)
1H NMR (300 MHz, CDCl3): δ 7.12 (d, J = 8.5 Hz, 2H), 6.91 (d, J = 8.5 Hz, 2H), 4.55 (s, 1H), 3.39–3.29 (m, 1H), 3.07–2.93 (m, 1H), 2.57 (q, J = 7.5 Hz, 2H), 2.01–1.59 (m, 6H), 1.20 (t, J = 7.5 Hz, 3H). 13C NMR (75 MHz, CDCl3): δ 147.6, 138.0, 128.5, 118.4, 117.1, 52.4, 46.7, 29.1, 27.9, 25.0, 20.1, 15.5. ESI-MS: m/z 215 [M+1].1-(4-Cyanophenyl)piperidine-2-carbonitrile (Table 3, entry 13)
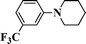
1H NMR (300 MHz, CDCl3): δ 7.56 (d, J = 8.8 Hz, 2H), 6.9 (d, J = 8.8 Hz, 2H), 4.76 (s, 1H), 3.64 (d, J = 12.1 Hz, 1H), 3.05 (t, J = 12.1 Hz, 1H), 2.16–1.56 (m, 6H). 13C NMR (75 MHz, CDCl3): δ 152.3, 133.5, 119.1, 116.6, 103.5, 49.3, 45.6, 28.8, 24.6, 19.8. ESI-MS: m/z 212 [M+1].1-(3-(Trifluoromethyl)phenyl)piperidine-2-carbonitrile (Table 3, entry 14)
1H NMR (300 MHz, CDCl3) δ 7.43–7.39 (m, 1H), 7.24–7.14 (m, 3H), 4.65 (s, 1H), 3.50–3.47 (m, 1H), 3.09–3.02 (m, 1H), 2.12–1.95 (m, 2H), 1.90–1.83 (m, 2H), 1.79–1.63 (m, 2H). 13C NMR (75 MHz, CDCl3): δ 150.0, 129.9, 120.8, 118.5, 116.7, 114.9, 51.5, 46.4, 29.6, 24.9, 19.9. ESI-MS: m/z 255 [M+1].1-(Naphthalen-1-yl)piperidine-2-carbonitrile (Table 3, entry 15)
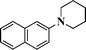
1H NMR (300 MHz, CDCl3) δ 8.06–7.96 (m, 1H), 7.88–7.79 (m, 1H), 7.68–7.60 (m, 1H), 7.53–7.40 (m, 3H), 7.38–7.22 (m, 1H), 4.40 (s, 1H), 3.45–3.31 (m, 1H), 3.17–3.13 (m, 1H), 2.33–2.09 (m, 1H), 2.08–1.69 (m, 5H). 13C NMR (75 MHz, CDCl3): δ 146.8, 134.6, 128.6, 125.8, 125.1, 122.2, 117.4, 54.5, 48.2, 29.3, 25.5, 20.3. EI-MS: m/z 237 [M+1].1-(Naphthalen-2-yl)piperidine-2-carbonitrile (Table 3, entry 16)
1H NMR (300 MHz, CDCl3): δ 7.77–7.73 (m, 3H), 7.46–7.15 (m, 4H), 4.75 (s, 1H), 3.59 (d, J = 11.3 Hz, 1H), 3.16–3.04 (m, 1H), 2.15–1.55 (m, 6H). 13C NMR (75 MHz, CDCl3): δ 147.2, 134.1, 129.0, 127.3, 127.1, 126.4, 124.3, 120.0, 113.3, 51.9, 46.6, 29.1, 25.0, 20.1. ESI-MS: m/z 237 [M+1].2-Phenyl-1,2,3,4-tetrahydroisoquinoline-1-carbonitrile (Table 3, entry 17)6d
1H NMR (300 MHz, CDCl3): δ 7.34–7.17 (m, 6H), 7.05–6.94 (m, 3H), 5.43 (s, 1H), 3.77–3.67 (m, 1H), 3.52–3.38 (m, 1H), 3.19–3.05 (m, 1H), 2.96–2.91 (m, 1H). 13C NMR (75 MHz, CDCl3): δ 148.4, 134.5, 129.5, 128.7, 128.7, 127.1, 122.0, 117.7, 53.2, 44.2, 28.6. ESI-MS: m/z 235 [M+1].Acknowledgements
The authors thank the Council of Scientific and Industrial Research (CSIR), New Delhi for financial assistance.References
- (a) G. Dyker, Angew. Chem., Int. Ed., 1999, 38, 1698 CrossRef; (b) V. Ritleng, C. Sirlin and M. Pfeffer, Chem. Rev., 2002, 102, 1731 CrossRef CAS; (c) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS; (d) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS; (e) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 CrossRef CAS; (f) X. Chen, K. M. Engle, D. H. Wang and J. Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS; (g) L. Ackermann, Chem. Commun., 2010, 4866 RSC; (h) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS.
- (a) C. J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS; (b) J. J. Li, T. S. Mei and J. Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 6452 CrossRef CAS; (c) D. H. Wang, M. Was, R. Giri and J. Q. Yu, J. Am. Chem. Soc., 2008, 130, 7190 CrossRef CAS; (d) O. Basle and C. J. Li, Org. Lett., 2008, 10, 3661 CrossRef CAS.
- (a) Yu. M. Shafran, V. A. Bakulev and V. S. Mokrushin, Russ. Chem. Rev., 1989, 58, 148 CrossRef (Usp. Khim., 1989, 58, 250); (b) D. Enders and J. P. Shilvock, Chem. Soc. Rev., 2000, 29, 359 RSC; (c) M. North, Angew. Chem., Int. Ed., 2004, 43, 4126 CrossRef CAS; (d) A. Schmidt, in Science of Synthesis, ed. S.-I. Murahashi, Thieme, Stuttgart, 2004, 19, ch. 19.5.3, p. 133 Search PubMed.
- (a) L. M. Weinstoc, P. Davis, B. Handelsm and R. Tull, J. Org. Chem., 1967, 32, 2823 CrossRef; (b) W. L. Matier, D. A. Owens, W. T. Comer, D. Deitchma, H. C. Ferguson, R. J. Seideham and J. R. Young, J. Med. Chem., 1973, 16, 901 CrossRef CAS; (c) R. O. Duthaler, Tetrahedron, 1994, 50, 1539 CrossRef CAS; (d) G. Dyker, Angew. Chem., Int. Ed. Engl., 1997, 36, 1700 CrossRef CAS.
- (a) H. Ishitani, S. Komiyama and S. Kobiyashi, Angew. Chem., Int. Ed., 1998, 37, 3186 CrossRef CAS; (b) K. Surendra, N. S. Krishnavani, N. Mahesh and K. R. Rao, J. Org. Chem., 2006, 71, 2532 CrossRef CAS; (c) F. Cruz- Acosta, A. Santos-Exposito and P. Tellado, Chem. Commun., 2009, 6839 RSC; (d) J. Wang, Y. Masui and M. Onaka, Eur. J. Org. Chem., 2010, 1763 CrossRef CAS; (e) P. Galleti, M. Pori and D. Giacomini, Eur. J. Org. Chem., 2011, 3896 CrossRef; (f) G. K. S. Prakash, T. Mathew, C. Panja, S. Alconcel, H. Vaghoo, C. Do and G. A. Olah, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3703 CrossRef CAS.
- (a) S.-I. Murahashi, N. Komiya, H. Terai and T. Nakae, J. Am. Chem. Soc., 2003, 125, 15312 CrossRef CAS; (b) S.-I. Murahashi, N. Komiya and H. Terai, Angew. Chem., Int. Ed., 2005, 44, 6931 CrossRef CAS; (c) S. Varma, S. J. Jain and B. Sain, ChemCatChem, 2011, 3, 1329 CrossRef; (d) S. I. Murahashi, T. Nakae, H. Terai and N. Komiya, J. Am. Chem. Soc., 2008, 130, 11005 CrossRef CAS.
- S. Singhal, S. J. Jain and B. Sain, Chem. Commun., 2009, 2371 RSC.
- Y. Zhang, Y. Peng, M. Zhang and Y. Cheng, Chem. Commun., 2011, 47, 2354 RSC.
- K. Alagiri and K. R. Prabhu, Org. Biomol. Chem., 2012, 10, 835 CAS.
- (a) W. Han and A. R. Ofial, Chem. Commun., 2009, 5024 RSC; (b) S. Singhal, S. J. Jain and B. Sain, Adv. Synth. Catal., 2010, 352, 1338 CrossRef CAS.
- M. Rueping, S. Zhu and R. M. Koenigs, Chem. Commun., 2011, 47, 12709 RSC.
- M. Rueping, J. Zoller, D. C. Fabry, K. Poscharny, R. M. Koenigs, T. E. Weirich and J. Mayer, Chem.–Eur. J., 2012, 3478 CrossRef CAS.
- (a) Y. Pan, S. Wang, C. W. Kee, E. Dubuisson, Y. Yang, K. P. Loh and C.-H. Tan, Green Chem., 2011, 13, 3341.D Search PubMed; (b) P. Hari and B. König, Org. Lett., 2011, 13, 3852 CrossRef.
- K. T. V. Rao, B. Haribabu, P. S. S. Prasad and N. Lingaiah, ChemCatChem, 2012, 4, 1173 CrossRef CAS.
- (a) A. V. Kumar, V. P. Reddy, R. Sridhar, B. Srinivas and K. R. Rao, Synlett, 2009, 5, 739 Search PubMed; (b) A. V. Kumar, V. P. Reddy and K. R. Rao, Synlett, 2010, 17, 2571 Search PubMed.
- (a) K. Swapna, S. N. Murthy, M. T. Jyothi and Y. V. D. Nageswar, Org. Bio. Chem. Org. Bio. Chem., 2011, 9, 5978 CAS; (b) K. Swapna, S. N. Murthy, M. T. Jyothi and Y. V. D. Nageswar, Org. Biomol. Chem., 2011, 9, 5989 RSC; (c) K. H. V. Reddy, V. P. Reddy, J. Shankar, B. Madhav, B. S. P. A. Kumar and Y. V. D. Nageswar, Tetrahedron Lett., 2011, 52, 2679 CrossRef CAS; (d) K. H. V. Reddy, V. P. Reddy, J. Shankar and Y. V. D. Nageswar, Synlett., 2011, 9, 1268 Search PubMed; (e) K. H. V. Reddy, A. A. Kumar, G. Kranthi and Y. V. D. Nageswar, Beilstein J. Org. Chem., 2011, 7, 886 CrossRef CAS; (f) G. Satish, K. H. V. Reddy, K. Ramesh and K. Karnakar, Tetrahedron Lett., 2012, 53, 2518 CrossRef CAS; (g) K. H. V. Reddy, G. Satish, K. Ramesh, K. Karnakar and Y. V. D. Nageswar, Chem. Lett., 2012, 41, 585 CrossRef CAS; (h) B. S. P. A. Kumar, K. H. V. Reddy, B. Madhav, K. Ramesh and Y. V. D. Nageswar, Tetrahedron Lett., 2012, 53, 4595 CrossRef.
- K. H. V. Reddy, G. Satish, K. Ramesh, K. Karnakar and Y. V. D. Nageswar, Tetrahedron Lett., 2012, 53, 3061 CrossRef CAS.
- S.-I. Murahashi, Angew. Chem., Int. Ed. Engl., 1995, 34, 2443 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra21630b |
| This journal is © The Royal Society of Chemistry 2012 |