Investigations of the structural evolution of electrospun nanofibers using atomic force microscopy†
Peng
Xu
ab,
Wei
Li
*ac,
Huacong
Zhou
ab,
Feng
Pan
a,
Huifang
Xing
a and
Huizhou
Liu
*a
aState Key Laboratory of Biochemical Engineering, Key Laboratory of Green Process and Engineering, Institute of Process Engineering, Chinese Academy of Sciences, Beijing, China. E-mail: hzliu@home.ipe.ac.cn; Fax: +86-10-62554254; Tel: +86-10-62554264
bGraduate School of Chinese Academy of Science, Beijing, China. E-mail: qingkeer111@163.com; Fax: +86-10-62554254; Tel: +86-10-82544954
cDepartment of Chemistry, Capital Normal University, Beijing, China. E-mail: wli@cnu.ac.edu.cn; Fax: +86-10-68903086; Tel: +86-10-68903086
First published on 6th August 2012
Abstract
Stepwise structural evolution of nanofibers through electrospinning was captured by AFM imaging. Nanofiber evolution exhibited five characteristic structures in turn: columns, plates, micropores, bumps and smooth surface. Based on our results and previous studies, these five stages are discussed in detail and the mechanism of nanofiber formation is proposed. In addition, the drug incorporation process of blend-electrospun nanofibers was shown vividly and clearly by AFM. It suggested that drugs could be completely incorporated into nanofibers by blend-electrospinning if proper polymers and drugs were chosen. Furthermore, the effects of solution properties on nanofiber structural evolution were also studied systematically.
1 Introduction
Electrospinning is a straightforward and effective method to produce ultra-fine nanofibers, ranging from about 50 nm to several μm.1 During the spinning process, a polymer solution placed inside a syringe is driven out from a metal capillary connected to a high voltage power supply. Once the repulsive electrical forces overcome the surface tension, a charged jet (i.e., liquid filament) will be ejected from the apex of the cone. As solvent evaporates, jets become thinner and form nanofibers.2 Nanofibers fabricated by electrospinning have attracted researchers' attention due to their desired properties (e.g., large surface to volume ratio, high porosity, and easy surface functionalization and translation3). They have potential applications in many fields, especially in biomedical fields, such as tissue engineering and drug delivery systems.4–15Up to now, many studies have been reported on the mechanism of nanofiber formation. In these studies, observing the nanofiber path with a high-speed videographic camera (CCD)16–20 and building mathematical models18,19,21–23 were the two common methods. For example, Taylor used CCD to capture formation pictures of silicone and glycerin jets,17 which clarified jets’ stabilities and instabilities for the first time in 1969. In 2000, Reneker analyzed jet paths with a CCD camera and built a mathematical model to study the instability of jets in electrospinning.18 Though many models and theories had been proposed, almost all contributions concentrated on the macroscopic variations of the jet (e.g. the decrease of diameter) or the jet status (stability or instability), yet none of them had given a direct and clear description of the nanofiber structural evolution in electrospinning. The probable reason was that the nanofiber structural evolution was too fast to be observed.24 Besides, the structural variations are important and worthy of investigation, since they are the key elements to reveal the nanofiber status during electrospinning. Hence, it was urgent for us to find a way to investigate the nanofibers structural evolution in electrospinning.
In our work, the key to the successful observation of nanofibers structural evolution was the selection of chitosan and poly(ethylene oxide) (PEO) as the polymers, because the velocity of chitosan–PEO nanofiber evolution is relatively slow and the critical length25 of jet is long (the critical length of most polymers is very short and fibers thinned too fast in the unstable region2,20,25). Acetic acid was chosen as the solvent for its low-toxicity and environmental-friendliness. In addition, AFM (atomic force microscope) was employed to visualize the nanofiber structural evolution during electrospinning due to its unique advantages over other microscopy techniques (e.g. SEM and TEM). First, AFM needs no difficult sample preparation and can be operated in any atmosphere, even in liquids. What's more, it is biologically friendly. The sample can avoid high radiation and keep the surface morphology unchanged, which is critical since protein will be decomposed under high radiation.26–28
Controlled drug release is one of the most important potential applications of electrospun nanofibers and plenty of work has focused on it.1,14,15 Blend-electrospinning and coaxial electrospinning are the two main ways to fabricate drug delivery systems.15 Compared to coaxial electrospinning, blend-electrospinning is much easier to operate, whereas it may lead to burst drug release due to the incomplete incorporation of drugs.1,14 In this work, BSA (Bovine Serum Albumin) as a model drug3,29,30 was employed to show the drug position and status in the blend-electrospun nanofibers. BSA was found to be entirely incorporated into nanofibers, which indicated that drugs could be completely packed inside by blend-electrospinning if proper polymers were selected. It might be helpful to deal with the issue of burst drug release.
In the present work, the whole process of nanofiber structural evolution is given graphically and clearly with AFM images. It was found that electrospinning was not only a simple fiber thinning process, but also involved a series of structural changes: columns, plates, micropores, bumps and smoothness. Routes toward these typical structures are discussed and a possible mechanism of nanofiber formation is proposed based on structural changes. What's more, the drug incorporation process and the effects of solution properties (chitosan molecular weight (Mw) and weight ratios of chitosan to PEO) on nanofiber formation were also studied. Our study not only gives more information for us to have a better understanding of the mechanism of nanofiber formation, but also gives guidance to obtain nanofibers with desired structures (e.g. porous nanofibers31 and rough nanofibers32).
2 Experimental section
2.1 Materials
Chitosan from crab shells with Mw = 560 kDa (93.6% deacetylation), Mw = 200 kDa (90% deacetylation) and Mw = 100 kDa (91.3% deacetylation) were purchased from Qingdao Hecreat Bio-tech Company Ltd. PEO (Mw = 600 kDa) was obtained from Shanghai Liansheng Chemical Co., Ltd. Acetic acid and other reagents were purchased from Sinopharm Chemical Reagent Beijing Co., Ltd. BSA was obtained from Beijing XinJingKe Biotechnology Co., Ltd. Double-distilled water was used throughout the experiments.2.2 Preparation of the polymer solution for electrospinning
Chitosan and PEO were separately dissolved in 2 v% acetic acid at a concentration of 3 wt%, and then the two solutions were mixed with different weight ratios varying from 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :67 to 80:20. The mixture was dissolved in 10 mL 90 v% acetic acid and stirred for 8 h.33 Next, the solution was centrifuged to remove the air bubbles, and the viscosity and conductivity were measured. For solution with BSA, BSA aqueous solution was added to the polymer blended solution to reach a concentration of 0.14 wt% and stirred gently for 3 h.
:67 to 80:20. The mixture was dissolved in 10 mL 90 v% acetic acid and stirred for 8 h.33 Next, the solution was centrifuged to remove the air bubbles, and the viscosity and conductivity were measured. For solution with BSA, BSA aqueous solution was added to the polymer blended solution to reach a concentration of 0.14 wt% and stirred gently for 3 h.
2.3 Electrospinning and AFM imaging
The electrospinning process was carried out at room temperature under the following conditions: the applied voltage was 25 kV, the distance between tip and collector was 25 cm and the feed rate was 1 mL h−1. A typical experimental setup is illustrated in Fig. 1(a), including a high voltage power supply (a DC high-voltage generator, purchased from Hengbo Co., Ltd.), a highly accurate syringe pump with a digital controller (TJ-3A W−10109-1B, purchased from Baoding Longer Precision Pump Co., Ltd.), a spinneret system and a nanofibers collector. The solution was loaded into a 5 mL syringe with a stainless needle (the inner diameter was about 0.8 mm) and fed by the syringe pump controller at a constant rate. | ||
| Fig. 1 A schematic representation of the electrospinning unit (a) and the three positions where the samples were fetched (b). Region A was close to the nozzle, region B was in the middle between region A and the beginning of instable stage, and region C corresponded to the instable region. | ||
Samples were fetched at different regions between the collector and nozzle, just as shown in Fig. 1(b). Each sample was fetched 3 times by sliced silicon and observed by AFM (AC Mode) after being totally dried, using n+-silicon probes (PPP-NCHR, Nanosensors, 10–130 N m−1) with a nominal spring constant of 40 N m−1 and resonance frequencies of about 270 kHz. The morphologies of jets were observed by Agilent Technologies 5500 Scanning Probe Microscope. Conductivity and viscosity of solution were measured by a Conductivity Meter (DDS-307, Shanghai Precision & Scientific Instrument Co., Ltd.) and Viscosity Meter (NDJ-8S, Shanghai Jingtian Electronic Instrument Co., Ltd.), respectively. Fourier Transform Infrared Spectrometer (Vector 22) was purchased from Bruker AXS GmbH. SEM images were collected on a JSM-6700F SEM operating at 5 kV, 10 μA.
3 Results and discussions
SEM images of chitosan–PEO nanofibers (75:25) are shown in Fig. 2. Homogeneous nanofibrous mats were fabricated and no significant differences were observed between nanofibers without (Fig. 2a) and with BSA (Fig. 2b). The average diameter of nanofibers without BSA was ∼520 nm, which was larger than that of nanofibers with BSA (∼480 nm). The possible reason was that the solution with BSA had lower viscosity (23025 mPa·s) and higher conductivity (2550 μs cm−1) compared with solution without BSA (viscosity = 23312 mPa·s, conductivity = 2400 μs cm−1). Viscosity was considered to have a positive impact on the diameter (i.e., increasing the diameter), while conductivity was a negative factor.34–36 In Fig. 2c, the surface of nanofibers is smooth and BSA was not observed on the surface, which was further confirmed by the corresponding phase image (there is no significant colour difference in Fig. 2d, which meant only polymer was detected).
 | ||
| Fig. 2 SEM images of chitosan–PEO (75:25) nanofibers without (a) and with BSA (b). AFM topography of nanofibers with BSA (c) and the corresponding phase image (d). | ||
In order to investigate the structural variations and the drug incorporation process through electrospinning, nanofibers without/with BSA obtained at different regions (regions A, B, C, Fig. 1(b)) were observed by AFM.
3.1 Structural evolution of nanofibers without BSA during electrospinning
Fig. 3 describes the structural evolution of electrospun nanofibers without BSA. Fig. 3a–g show the magnified topographies of different stages and the inset images at the lower right corner are the full morphologies of the jets. The corresponding 3D reconstruction images confirmed the real surface structure of each stage. From Fig. 3, it was evident that the microstructures of the jets’ surface changed dramatically with five characteristic features in turn: columns (Fig. 3a), plates (Fig. 3b), micropores (Fig. 3c and d), bumps (Fig. 3e and f) and smooth nanofibers (Fig. 3g). | ||
| Fig. 3 Structural evolution of chitosan–PEO (75:25) jets through electrospinning. AFM topographies (a–g) are the larger magnification morphologies of the surface (the full morphologies of jets are inserted in the lower right corner; the corresponding 3D images are shown below the topographic images). The characteristic structures are highlighted by yellow curves or circles: columnar (a), plate-like (b), microporous (c–e), bumpy (f) and smooth (g). | ||
To further illustrate the structural evolution, the diameters and surface roughness of jets corresponding to Fig. 3 are listed in Table 1 (calculated by “PicoImage” software, professional software provided by Agilent Technology Co. Ltd.). The diameters of jets decreased from 81.6 μm to 2.18 μm, whereas surface roughness changed randomly. So the jet surface profile (horizontal cross-curve) of each stage is also provided in Fig. 4. To get a complete view of the surface, Fig. 4a and b were sketched from the full morphologies of Fig. 3a and b (the inset images). Fig. 4c–g are from the magnified topographies in Fig. 3c–g, respectively. The blue lines in Fig. 4 (determined according to Fig. 3) represent the surface outlines of jets, which helped us to judge whether there was a bump or a micropore, i.e., bumps should above the blue line and micropores should be beneath it.
 | ||
| Fig. 4 The corresponding horizontal cross-curves of images in Fig. 3, which showed the surface profiles of jets. Fig. 4a and b were obtained from the full morphologies of Fig. 3a and b; Fig. 4c–g are from Fig. 3c–g, respectively. The blue lines represent the outlines of the jets’ surface; structures beneath the blue lines are micropores (c–e), and embellishments marked by blue circles in (a), (b) and (f) are columns, plates and bumps, respectively. (g) shows a single jet with a smooth surface. | ||
In Fig. 4a, the surface roughness was extremely high (63.5 nm, Table 1) and many columns caused by the electrical field37 (marked by dark blue circles) were evenly aligned on the surface. Then columns were stretched into plates (Fig. 4b, highlighted by dark blue circles) by the longitudinal electric stretch and radial electrostatic repulsion19,23 and, consequently, the surface roughness decreased to 7.28 nm (Table 1). However, the surface roughness didn't continue to decrease—the formation of micropores made the surface rough again (surface roughness increased to 23.3 nm). As electrospinning went on, micropores became much denser and smaller (Fig. 4c–e), which is in good agreement with Table 1 (the surface roughness decreased from 23.3 nm to 3.51 nm). The size decrease of micropores indicated that micropores faded during the process, which was further demonstrated by the 3D topographies of Fig. 3c–e. In addition, it was interesting that the depth of micropores became shallower (decreased from 12 nm to 4 nm, from Fig. 4c–e) as the process proceeded. After the stage of micropores, lots of bumps came out, as shown in Fig. 3f and Table 1 (surface roughness increased to 10.4 nm). Subsequently, the jet was thinned to 2.18 μm and the surface became smooth (see the jet profile in Fig. 4g, the surface roughness of the jet was only 1.67 nm). Eventually, nanofibers (∼520 nm) with a smooth surface were fabricated, as shown in Fig. 2a.
3.2 BSA incorporation process in blend electrospinning
It was obvious that no BSA was detected on the nanofiber surface, as can be seen from Fig. 2c and d. However, the FT-IR spectra (see Supporting Information†) proved that BSA had been successfully loaded into nanofibers. So we inferred that BSA had been entirely packed inside the nanofibers. The results were different from the classical model of drug-loaded nanofibers produced by blend-electrospinning38 and might be helpful to deal with the issue of burst drug release associated with partial incorporation of drugs. We then studied the incorporation process of BSA with AFM.Topographies of chitosan–PEO (75:25) jets with BSA are given in Fig. 5. The structural variations of jets are similar to those of nanofibers without BSA: columns (Fig. 5a), plates (Fig. 5b), micropores (Fig. 5c and d), bumps (Fig. 5e) and smoothness (Fig. 5f). Differently, lots of elliptic particles were detected on the surface initially (Fig. 5a and b). To figure out the material identity of these particles, AFM phase images were applied because they could tell the physical property differences of samples that were not readily discernible in topographic images.39Fig. 6a2–f are the phase images corresponding to Fig. 5a–f; Fig. 6a1 is the phase image of Fig. 3a (it shows the phase image of a jet without BSA). Compared to nanofibers without BSA (Fig. 6a1), nanofibers with BSA had lots of white dots on the surface (Fig. 6a2), which meant another kind of substance was detected. Given that the average diameter was about 10 nm, we confirmed that these white dots were BSA. These white dots disappeared from Fig. 6a2–f, indicating that BSA was gradually incorporated into nanofibers during blend-electrospinning.
 | ||
| Fig. 5 AFM topographies of jets electrospun from chitosan–PEO (75:25) with BSA; images (a–f) show the larger magnification of the jets’ surface, the inset images are the corresponding full morphologies of jets. | ||
 | ||
| Fig. 6 AFM phase images: (a2–f) are the phase images corresponding to Fig. 5a–f; (a1) is the phase image of Fig. 3a (it shows the phase image of a jet without BSA). | ||
Based on our results, the incorporation process of BSA in blend-electrospinning is shown in Fig. 7. At the initial stage, BSA and polymers were well blended, so BSA was detected on the surface. As the process proceeded, BSA was gradually packed inside. The reason might be that both chitosan and BSA carried positive charges, which made BSA and chitosan attracted to the surface.2 Since chitosan had a much higher charge density and weight ratio than that of BSA (BSA was only 0.14 wt% in the solution), chitosan migrated outside faster than BSA and formed a polymer skin. The results inferred that drugs could also be completely incorporated into nanofibers by blend-electrospinning if polymers and drugs were selected properly.
 | ||
| Fig. 7 The incorporation process of BSA in electrospinning (red dots represent BSA and the green part is polymer). | ||
3.3 Effects of solution properties on the structural evolution of nanofibers
The effects of solution properties (weight ratios of chitosan:PEO and Mw of chitosan) on the structural evolution of nanofibers were also investigated in our work (see Supporting Information for details†). The results showed that different weight ratios of polymers greatly influenced the process by varying solution viscosity, conductivity and glass transition temperature (Tg). A solution with a low chitosan content (low viscosity) not only made the jet transformation fast, but also restricted the formation of a porous structure due to the Tg of solution being too low.26,27 What's more, Mw of chitosan mainly affected the viscosity of the solution. So a solution with a higher Mw of chitosan was more difficult to form noticeable structures in electrospinning than a solution with a lower Mw. Although different Mw of chitosan and weight ratios of chitosan:PEO made the nanofibers formation process a little different, the tendency and sequence of the five stages were invariable.3.4 Possible mechanism of nanofiber formation based on the structural variations in electrospinning
From the above discussions, the mechanism of chitosan–PEO nanofiber formation is illustrated in Fig. 8. It typically underwent five stages before nanofibers were produced: columns, plates, micropores, bumps and smoothness. Taking previous reports and our results together, the explanation of electrospinning is as follows. At the beginning, a fluid jet was elongated and accelerated in the electrical field.2,40 Because the jet had a relatively large diameter (81.6 μm, Table 1) initially and behaved like a liquid layer, lots of columns were formed due to the fluctuations enhanced by electrical field.37 However, these columns could not exist for a long time; they were stretched into plates along its axis by electrostatic forces.23 Then rapid solvent evaporation and phase separation made the surface porous.26,27,41 As the process proceeded, these micropores became denser and smaller, and the depth of micropores became shallower as well. The possible reason was that the jet became more concentrated and viscous when flying to the collector.27 Subsequently, whipping instability played the leading role21 and jets were bent into a complex path and thinned by very large ratios. Micropores faded due to the excessive bending and massive stretch.21 Then lots of bumps were extruded out of the surface by charge repulsion,18 which was due to the significant increase in the surface charge density caused by the fast decrease of jet diameter.40 After this stage, jets were all stretched and bent into smooth jets. The diameter of jets (Fig. 3g, diameter = 2.16 μm) continued to decrease and finally nanofibers (Fig. 2a, diameter = 520 nm) were generated.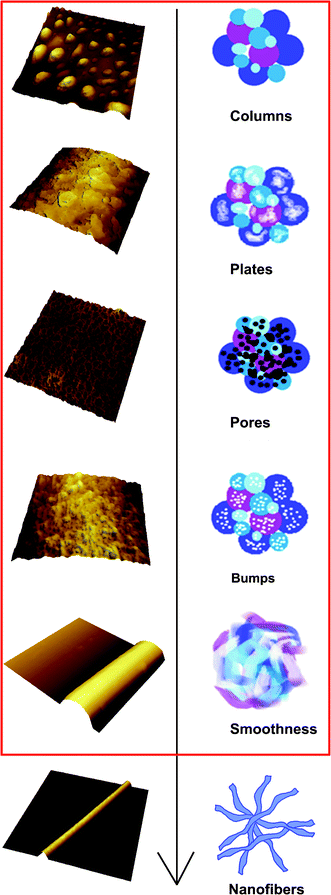 | ||
| Fig. 8 A schematic illustration of chitosan–PEO nanofibers formation in electrospinning. | ||
4 Conclusions
Herein, the structural evolution and drug behavior in electrospinning were investigated with AFM. We found that electrospinning not only thinned the nanofibers, but also varied their surface structures. During electrospinning, jets of chitosan–PEO went through five distinct stages, that is, columns, plates, micropores, bumps and smooth surface. Nanofibers with BSA had an evident drug incorporation process in electrospinning, which inferred that drugs could be entirely incorporated into nanofibers by blend-electrospinning if proper polymers were chosen. The tendency and sequence of the five stages were found to be unchanged under various conditions. Based on our results and previous reports, a possible mechanism of nanofiber formation was proposed. This work not only provides useful information for researchers to further understand the nanofiber formation process from the microscopic point of view, but also could be used to produce nanofibers with desired structures.Acknowledgements
The authors thank the National Natural Science Foundation of China (No. 21136009) and Innovative Research Group Science Found (No. 20221603).References
- S. Maretschek, A. Greiner and T. Kissel, J. Controlled Release, 2008, 127, 180–187 CrossRef CAS.
- G. C. Rutledge and S. V. Fridrikh, Adv. Drug Delivery Rev., 2007, 59, 1384–1391 CrossRef CAS.
- S. Tungprapa, I. Jangchud and P. Supaphol, Polymer, 2007, 48, 5030–5041 CrossRef CAS.
- S. Agarwal, J. H. Wendorff and A. Greiner, Polymer, 2008, 49, 5603–5621 CrossRef CAS.
- X. Y. Geng, O. H. Kwon and J. H. Jang, Biomaterials, 2005, 26, 5427–5432 CrossRef CAS.
- W. J. Li, C. T. Laurencin, E. J. Caterson, R. S. Tuan and F. K. Ko, J. Biomed. Mater. Res., 2002, 60, 613–621 CrossRef CAS.
- L. A. Smith and P. X. Ma, Colloids Surf., B, 2004, 39, 125–131 CrossRef CAS.
- K. S. Rho, L. Jeong, G. Lee, B. M. Seo, Y. J. Park, S. D. Hong, S. Roh, J. J. Cho, W. H. Park and B. M. Min, Biomaterials, 2006, 27, 1452–1461 CrossRef CAS.
- S. P. Zhong, W. E. Teo, X. Zhu, R. Beuertnan, S. Ramakrishna and L. Y. L. Yung, Mater. Sci. Eng., C, 2007, 27, 262–266 CrossRef CAS.
- M. I. Santos, K. Tuzlakoglu, S. Fuchs, M. E. Gomes, K. Peters, R. E. Unger, E. Piskin, R. L. Reis and C. J. Kirkpatrick, Biomaterials, 2008, 29, 4306–4313 CrossRef CAS.
- J. E. Gautrot, B. Trappmann, F. Oceguera-Yanez, J. Connelly, X. M. He, F. M. Watt and W. T. S. Huck, Biomaterials, 2010, 31, 5030–5041 CrossRef CAS.
- S. C. Neves, L. S. M. Teixeira, L. Moroni, R. L. Reis, C. A. Van Blitterswijk, N. M. Alves, M. Karperien and J. F. Mano, Biomaterials, 2011, 32, 1068–1079 CrossRef CAS.
- M. Y. Li, M. J. Mondrinos, M. R. Gandhi, F. K. Ko, A. S. Weiss and P. I. Lelkes, Biomaterials, 2005, 26, 5999–6008 CrossRef CAS.
- S. Chakraborty, I.-C. Liao, A. Adler and K. W. Leong, Adv. Drug Delivery Rev., 2009, 61, 1043–1054 CrossRef CAS.
- T. J. Sill and H. A. von Recum, Biomaterials, 2008, 29, 1989–2006 CrossRef CAS.
- M. Lee, S. B. Kang and J. H. Park, J Mech Sci Technol, 2006, 20, 409–417 CrossRef.
- G. Taylor, Proc. R. Soc. London, Ser. A, 1969, 313, 453–475 CrossRef.
- D. H. Reneker, A. L. Yarin, H. Fong and S. Koombhongse, J. Appl. Phys., 2000, 87, 4531–4547 CrossRef CAS.
- D. H. Reneker, T. Han and A. L. Yarin, Polymer, 2008, 49, 1651–1658 CrossRef.
- D. H. Reneker and A. L. Yarin, Polymer, 2008, 49, 2387–2425 CrossRef CAS.
- Y. M. Shin, M. M. Hohman, M. P. Brenner and G. C. Rutledge, Polymer, 2001, 42, 9955–9967 CrossRef CAS.
- M. P. Brenner, M. M. Hohman, M. Shin and G. Rutledge, Phys. Fluids, 2001, 13, 2201–2220 CrossRef.
- J. J. Feng, Phys. Fluids, 2002, 14, 3912–3926 CrossRef CAS.
- P. Dayal, J. Liu, S. Kumar and T. Kyu, Macromolecules, 2007, 40, 7689–7694 CrossRef CAS.
- J. H. He, Y. Wu and W. W. Zuo, Polymer, 2005, 46, 12637–12640 CrossRef CAS.
- S. Megelski, J. S. Stephens, D. B. Chase and J. F. Rabolt, Macromolecules, 2002, 35, 8456–8466 CrossRef CAS.
- C. L. Casper, J. S. Stephens, N. G. Tassi, D. B. Chase and J. F. Rabolt, Macromolecules, 2004, 37, 573–578 CrossRef CAS.
- K. T. Shalumon, K. H. Anulekha, C. M. Girish, R. Prasanth, S. V. Nair and R. Jayakumar, Carbohydr. Polym., 2010, 80, 413–419 CrossRef CAS.
- X. J. Loh, P. Peh, S. Liao, C. Sng and J. Li, J. Controlled Release, 2010, 143, 175–182 CrossRef CAS.
- Z. Ma, M. Kotaki and S. Ramakrishna, J. Membr. Sci., 2006, 272, 179–187 CrossRef CAS.
- M. Bognitzki, W. Czado, T. Frese, A. Schaper, M. Hellwig, M. Steinhart, A. Greiner and J. H. Wendorff, Adv. Mater., 2001, 13, 70–72 CrossRef CAS.
- M. M. Demir, I. Yilgor, E. Yilgor and B. Erman, Polymer, 2002, 43, 3303–3309 CrossRef CAS.
- B. Duan, C. H. Dong, X. Y. Yuan and K. D. Yao, J. Biomater. Sci., Polym. Ed., 2004, 15, 797–811 CrossRef CAS.
- H. Homayoni, S. A. H. Ravandi and M. Valizadeh, Carbohydr. Polym., 2009, 77, 656–661 CrossRef CAS.
- S. H. Tan, R. Inai, M. Kotaki and S. Ramakrishna, Polymer, 2005, 46, 6128–6134 CrossRef CAS.
- S. A. Theron, E. Zussman and A. L. Yarin, Polymer, 2004, 45, 2017–2030 CrossRef CAS.
- Z. Q. Lin, T. Kerle, T. P. Russell, E. Schaffer and U. Steiner, Macromolecules, 2002, 35, 3971–3976 CrossRef CAS.
- Y. Z. Zhang, X. Wang, Y. Feng, J. Li, C. T. Lim and S. Ramakrishna, Biomacromolecules, 2006, 7, 1049–1057 CrossRef CAS.
- C. González-García, S. R. Sousa, D. Moratal, P. Rico and M. Salmerón-Sánchez, Colloids Surf., B, 2010, 77, 181–190 CrossRef.
- J. Doshi and D. H. Reneker, J. Electrost., 1995, 35, 151–160 CrossRef CAS.
- M. Srinivasarao, D. Collings, A. Philips and S. Patel, Science, 2001, 292, 79–83 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Effects of solution properties (weight ratios of chitosan to PEO and molecular weight of chitosan on the formation process of nanofibers) and the FT-IR analysis. See DOI: 10.1039/c2ra20146a/ |
| This journal is © The Royal Society of Chemistry 2012 |