
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ion-conductive vs. non-ion-conductive ceramic fillers in silane-linked polyethylene oxide-based composite polymer electrolytes with high room-temperature ionic conductivity†
Eun Ju
Jeon
 abc,
Sharif
Haidar
abc,
Laura
Helmers
ab,
Arno
Kwade
abc and
Georg
Garnweitner
*abc
abc,
Sharif
Haidar
abc,
Laura
Helmers
ab,
Arno
Kwade
abc and
Georg
Garnweitner
*abc
aTechnische Universität Braunschweig, Institute for Particle Technology, Volkmaroder Str. 5, 38104 Braunschweig, Germany. E-mail: g.garnweitner@tu-braunschweig.de
bTechnische Universität Braunschweig, Battery LabFactory Braunschweig (BLB), Langer Kamp 19, 38106 Braunschweig, Germany
cCluster of Excellence SE2A – Sustainable and Energy Efficient Aviation, Braunschweig, Germany
First published on 28th August 2024
Abstract
Polyethylene oxide (PEO)-based polymer electrolytes, despite their cost-effectiveness and ease of processing, suffer from low ionic conductivity at lower temperatures due to the semi-crystalline nature of PEO. Incorporating ceramic filler particles into the polymer matrix offers a potential solution by disrupting its rigid crystalline structure, thereby improving the flexibility of the polymer chains. However, the Li ion conduction pathway within these composite polymer electrolytes (CPEs) remains predominantly within the polymer matrix if the filler particles are only physically mixed. The surface modification of filler particles can improve the interfacial compatibility and ionic conductivity. In this work, two types of filler particles, passive ZrO2 and active Li7La3Zr2O12 (LLZO), are compared and incorporated into PEO–polyethylene glycol (PEG)–lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) CPEs. The surface of the filler particles is functionalized with a silane ligand ((3-glycidyloxypropyl)trimethoxysilane (GPTMS)) prior to their integration into the PEO matrix. This modifies the interfacial properties between the polymer and the filler particles, hence influencing the ionic conductivity. The functionalized ZrO2 fillers enhance the ionic conductivity of the CPEs by reducing the crystallinity of PEO. The PEO–PEG–LiTFSI CPE with 15 vol% of GPTMS–ZrO2 achieved an ionic conductivity of 6.66 × 10−4 S cm−1 at 20 °C, which is significantly higher than that of the standard PEO–LiTFSI (9.26 × 10−6 S cm−1). Additionally, coupling GPTMS to PEO chains without the introduction of filler particles also improved the ionic conductivity, while the incorporation of functionalized LLZO fillers does not, which is attributed to a LiCO3 passivation layer. The results suggest a viable strategy to overcome the inherent limitations of PEO electrolyte, thus offering valuable insights into the design and optimization of CPEs for practical applications.
Introduction
All-solid-state batteries (ASSBs) are regarded as highly promising for energy storage in a multitude of applications due to their high energy density and improved safety resulting from the utilization of solid electrolytes (SEs).1–4 Their advantages include (1) the absence of volatile and flammable organic liquid electrolytes, (2) a high potential to suppress Li dendrites, and (3) the possibility of using high-voltage cathodes.5,6 One promising class are solid polymer electrolytes (SPEs), which are favorable due to their broad availability, low cost, and good processability. However, several critical challenges limit the practical use of SPEs, such as (1) low ionic conductivity at room temperature (RT) compared to liquid electrolytes, (2) significant interfacial resistance at the electrolyte/active material interface, and (3) poor electrochemical compatibility with certain active materials such as Li metal anodes and high-voltage cathodes.7 Among SPEs, polyethylene oxide-lithium bis(trifluoromethanesulfonyl)imide (PEO–LiTFSI)-based solid electrolytes have been extensively investigated since the 1970s.8–10 They have a favorable Li salt dissociation ability and electrochemical stability towards Li metal anodes.11–15 In addition, PEO is inexpensive and easy to process due to its high flexibility, and its strong adhesion property allows good interfacial contact with the electrodes. Despite these advantages, PEO electrolytes suffer from low ionic conductivity at RT (about 10−7 S cm−1) due to its high intrinsic crystallinity at low temperatures.16–18 To mitigate this problem, several strategies such as the incorporation of filler particles19–22 and polymer structure engineering, including cross-linking19,23,24 and copolymerization,25,26 have been suggested.Thereby, the firstly mentioned strategy has been widely studied since the 1980s. The resulting composite polymer electrolytes (CPEs) containing different types of filler particles were shown to possess improved ionic conductivity and mechanical strength while maintaining good flexibility.27–31 The incorporated fillers change the structure of the polymer phase, and thus reduce the crystallinity of the polymer, increasing the ionic conductivity. They promote Lewis acid–base interactions between fillers and the polymer matrix, helping to separate immobilized Li ions from a large Li cluster bound to the ethylene oxide group of the polymer.32–37 The fillers can be distinguished into two types, namely passive and active fillers, depending on the presence of Li ions in their composition. The applied passive fillers such as Al2O3, SiO2, MgO, and ZrO2 particles improve the ionic conductivity by affecting the crystallinity of PEO and increasing the salt dissociation. Active fillers from oxide ceramics such as garnet-type Li7La3Zr2O12 (LLZO), NaSiCON Li1+xAlxTi2−x(PO4)3 (LATP), Li1+xAlxGe2−x(PO4)3 (LAGP), and sulfides have been incorporated into the polymer matrix of PEO to obtain CPEs.38–41 In particular, LLZO in its cubic phase is considered as one of the most promising SEs due to its high ionic conductivity (up to 1 mS cm−1 at RT), good chemical stability, and a wide electrochemical stability window of 0–6 V vs. Li+/Li.42,43 A practical application of ceramic LLZO SEs in batteries is challenging due to their intrinsic nature of brittleness, leading to difficulty in processing.4,44 Also, Li dendrite penetration may occur following the defects and grain boundaries of LLZO, leading to a localized current with battery failure.45,46 When LLZO particles are employed as active fillers in the polymer matrix, better interfacial contact to the electrodes and strongly improved processability are reached compared to the purely ceramic SE. Fu et al. showed that 20 wt% of LLZO nanofibers in PEO-based CPEs led to high ionic conductivity of 2.5 × 10−4 S cm−1 at RT, which was enabled by Li ion conducting channels formed by the LLZO fibers in the PEO matrix.47 The CPEs showed electrochemical stability up to 6 V vs. Li+/Li. Li et al. introduced a CPE with 16 vol% Ga-doped LLZO in PEO exhibiting an ionic conductivity of 7.2 × 10−5 S cm−1, a Li transference number of 0.39, being electrochemically stable up to 4.6 V vs. Li+/Li.48 These examples suggest that positive results can be achieved by combining the two solid electrolyte classes. However, the addition of LLZO does not always guarantee an increase in ionic conductivity. Some studies described that the LLZO fillers did not significantly improve the ion transport properties.49,50 Most of the reported ionic conductivity values are in the limited range of 10−6 to 10−4 S cm−1 at RT.50 Nonetheless, active fillers are expected to have a stronger impact on the ionic conductivity of CPEs compared to passive fillers due to the presence of Li ions in their structures.51
The ion transport in CPEs consisting of a polymer matrix filled with inorganic particles is a complex process, and needs to be further understood for its rational improvement.48,52 The transport mechanism varies based on the properties, dimensions, and content of the polymer and fillers, as well as their interfacial properties.28 The potential Li-ion pathways in passive-filler CPEs have been postulated as two-fold: (1) through the polar segmental movement of the PEO chains within the amorphous region, and (2) via the interphase between the PEO and passive filler, known as the space charge region (SCR).48 In the alternative case of CPEs with active fillers, Li ions can migrate via three pathways: (1) through the PEO, (2) through the polymer-ceramic interphase (SCR), and (3) directly through the ceramic filler particles.38,53 The third mechanism can only occur if a percolation network of the filler particles exists. However, achieving such a network is challenging, and is only possible for high filler contents. As a result, this pathway is rarely observed.54 Instead, lithium ion transport is predominantly effected through the polymer matrix and the polymer-ceramic interphase. Fig. 1 shows these transport pathways, including the fast Li transport path along the SCR of the filler particles.
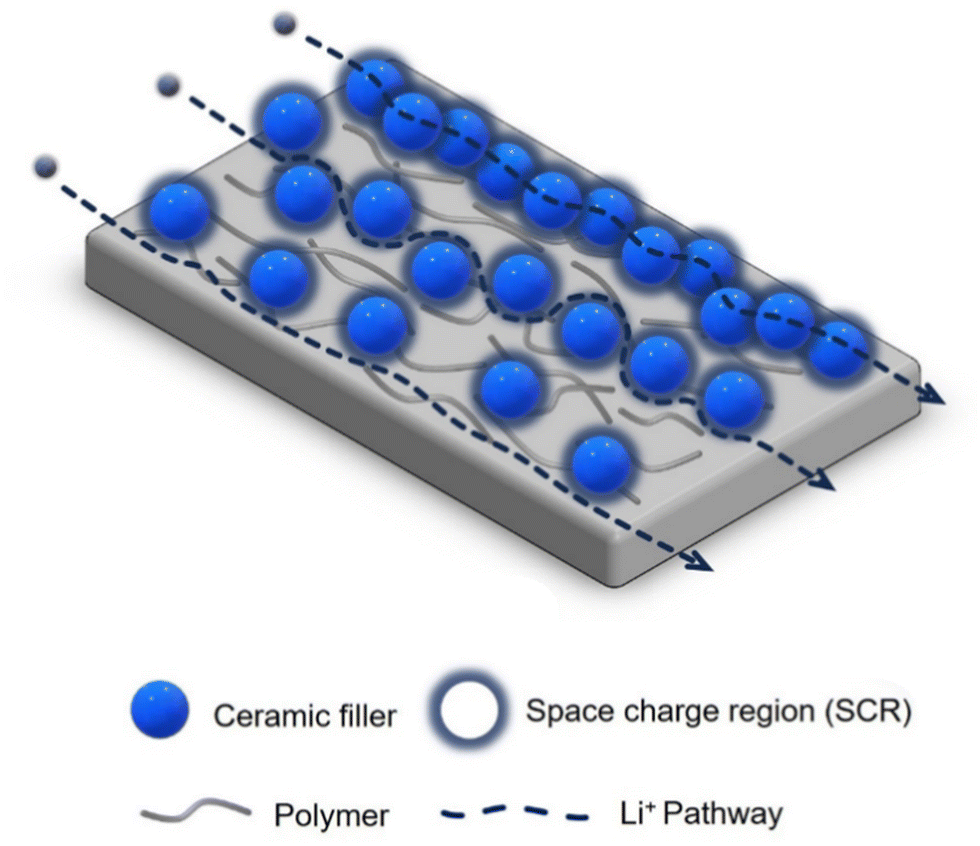 | ||
| Fig. 1 Illustration of the three proposed main mechanisms of Li ion migration through a composite polymer electrolyte (CPE) composed of a polymer matrix and ceramic filler particles. | ||
Hence, in CPEs with both active and passive fillers, the interphase between the filler particles and the polymer plays a significant role regarding the electrochemical performance. The two phases should be chemically and physically compatible and have a low interfacial resistance. The surface of ceramic particles can be modified by chemical reactions with functional ligands to enable covalent coupling to the polymer, improving their interfacial compatibility.55–57 This surface modification stabilizes the ceramic particles against agglomeration and thus enhances a homogeneous distribution. The functional ligands can also act as a buffer to prevent direct contact of the LLZO particles with the Li metal anode. The direct LLZO/Li contact leads to voltage instabilities during battery cycling. Table 1 and Table S1 (ESI†) list a number of previous studies on PEO-based CPEs with silane functional ligands attached to the surface of filler particles/fibers, including their respective electrochemical performance as reported. For example, a decrease in interfacial resistance of PEO CPEs from 5 MΩ cm2 to 500 Ω cm2 was achieved by the functionalization of LLZTO particles with (3-glycidyloxypropyl)trimethoxysilane (GPTMS).55 Hence, the functionalization of ceramic particles in the PEO matrix can effectively improve the ionic conductivity and overall performance of CPEs.
| Polymer–salt | Filler and filler content in the CPE | Silane agent and its content on functionalized LLZO | Ionic conductivity | E a | Cycling performance | Ref. |
|---|---|---|---|---|---|---|
| PEO–LiTFSI | 30 wt% of LLZTO | (3-Glycidyloxypropyl)-trimethoxysilane | 9.0 × 10−3 S cm−1 at 20 °C | — | — | 55 |
PEO–LiTFSI (EO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Li 12:1) :Li 12:1) |
7.5 wt% of Al–LLZO (porous scaffold) | Ca-coordinated Dynasylan IMEO (triethoxy-3-(2-imidazolin-1-yl)-propylsilane) | 6.38 × 10−4 S cm−1 at 25 °C | 0.029 eV | Initial discharge capacity: 130.2 mA h g−1 at 0.1 C at 25 °C NCM622/CPE/Li metal | 58 |
| PEO–LiTFSI (EO:Li 16:1) |
10 wt% of LLZO nanofibers | Dynasylan IMEO (triethoxy-3-(2-imidazolin-1-yl)-propylsilane) and Ca2+ | 5.44 × 10−5 S cm−1 at 30 °C | 0.17 eV | Initial discharge capacity: 144.3 mA h g−1 at 60 and 0.5 C LFP/CPE/Li metal | 59 |
| PEO–LiTFSI (EO:Li 12:1) |
20 wt% of Ta–LLZO nanoparticles (500 nm) | 2 wt% of 3-aminopropyl-triethoxysilane | 7.29 × 10−5 S cm−1 at 25 °C | — | Initial discharge capacity: 155 mA h g−1 at 60 °C and 0.1 C LFP/CPE/Li metal | 60 |
| PEO–LiTFSI + PEGMA–LiTFSI (in situ polymerized on the PEO–LLZO scaffold) | 8 wt% of LLZO particles | 8 wt% of 3-(trimethoxysilyl)-propyl methacrylate | 1.78 × 10−4 S cm−1 at 60 °C | — | Initial discharge capacity: 169 mA h g−1 at 0.1 C and 60 °C LFP/CPE/Li metal | 61 |
| PEO–succinonitrile–LiTFSI (EO:Li 32:1 & SN:Li 4:1) |
10 wt% of LLZTO nanoparticles (3D PAN-APTS-LLZTO nanofiber framework) | (3-Aminopropyl)-triethoxysilane | 1.58 × 10−4 S cm−1 at 25 °C | — | Initial discharge capacity: 173.63 mA h g−1 at 0.1 C and 60 °C NCM811/CPE/Li metal | 62 |
| PEO–LiTFSI (EO:Li 14:1) |
10 vol% of LATP particles | Chlorotris-(trimethylsilyl)silane | 4.8 × 10−5 S cm−1 at 20 °C | — | — | 22 |
In this study, we examine the impact of different filler types (active and passive fillers) on the ionic conductivity of PEO–PEG–LiTFSI-based CPEs. Thereby, high-molecular weight PEO is combined with short-chain PEG (when the molecular weight of PEO is less than 20000 g mol−1, it is commonly referred to as PEG) to provide more –OH groups, offering faster Li transport than only long-chain PEO.63,64 LLZO and ZrO2 particles, which both possess Zr–O surface groups and comparable physical properties, notably with similar particle diameters (D50,LLZO = 1.2 μm and D50,ZrO2 = 1 μm), were selected for the comparison. To enhance their compatibility with the polymer and to reduce interfacial resistance, the surface of these filler particles was modified with (3-glycidyloxypropyl)trimethoxysilane (GPTMS) ligands. Additionally, we explored CPEs without fillers, where the GPTMS ligand is directly bound to the PEO–PEG chains. By adjusting the concentrations of fillers and GPTMS ligands in the CPEs, the aim of this work is to develop electrolytes with superior ionic conductivity that can overcome the challenges of PEO-based electrolytes. What makes this study special is the careful comparison between PEO–PEG–LiTFSI CPEs with functionalized active fillers, functionalized passive fillers, and those that are filler-free but contain silane which is bound to the polymer chain, to further investigate the mechanisms of ionic transport in CPEs.
Materials and methods
Synthesis of CPEs and processing of separator films
In this study, four different CPEs are compared: PEO–PEG–LiTFSI (in short, referred to as PEO SPE in the following), PEO–PEG–LiTFSI–GPTMS (GPTMS CPE), PEO–PEG–LiTFSI–GPTMS–ZrO2 (GPTMS–ZrO2 CPE), and PEO–PEG–LiTFSI–GPTMS–LLZO (GPTMS–LLZO CPE). A three-step synthesis process of the CPEs was carried out: (1) silanization of the filler particles, (2) dispersion of the particles into a PEO–PEG–LiTFSI polymer mixture solution in acetonitrile by a dissolver, and (3) tape-casting of this dispersion using a film applicator (Fig. 2). The filler particles, ZrO2 (D50: 1 μm, specific surface area 4.5–7.5 m2 g−1, Saint-Gobain) and LLZO (D50: 1.2 μm, specific surface area 2.5 m2 g−1, Schott AG) were functionalized with a commercially available (3-glycidyloxypropyl)trimethoxysilane (GPTMS, Gelest) silane ligand in isopropanol (IPA, Thermo Fisher Scientific) via simple magnetic stirring for 24 h at room temperature. Different volume fractions of 6, 10, 15, and 20% of filler particles were used, as at least 6 vol% was required to obtain a processable material, and filler contents of up to 20% had been identified as suitable for enhancing ionic conductivity without significantly affecting the mechanical properties of CPEs.22 Volume fractions rather than weight fractions were used for the fillers to maintain a consistent volume of fillers across the CPEs, facilitating direct comparison of the effects of each type of filler which have different bulk density (LLZO: 4.8 g cm−3 and ZrO2: 5.6 g cm−3). The volume ratio between the ceramic particles and the GPTMS was 1:2, and of the ceramic particles to the IPA solvent 20:80. The functionalized ZrO2 and LLZO are referred to as GPTMS–ZrO2 and GPTMS–LLZO, respectively. Following the silanization, these particles were dispersed into a solution of PEO–PEG–LiTFSI in acetonitrile. The polymer mixture consists of two polymers, 80 wt% of PEO (MW = 900000 g mol−1, Dow) and 20 wt% of PEG (MW: 2000 g mol−1, Merck). 30 wt% LiTFSI (Solvionic) was added to the dissolved PEO–PEG polymer mixture ([EO]:[Li] = 14:1) in 93 vol% of acetonitrile (Thermo Fisher Scientific). The respective ceramic particle dispersion and the polymer mixture were combined and dispersed using a dissolver (DISPERMAT LC, VMA-Getzmann GmbH) at 2000 rpm for 30 min. In case of the GPTMS CPE without ceramic particles (PEO–PEG–LiTFSI–GPTMS), 6, 10, 15, and 20 vol% of GPTMS were added to the PEO–PEG solution and the GPTMS–PEO–PEG mixture was dissolved in ACN with 7 vol%. Immediately after mixing, the obtained coating suspension was tape-casted with three steps using a film applicator (Zehntner Testing Instruments, ZAA 2300). The casting was done at 30 °C with a coating speed of 5 mm s−1. Then the plate of the film application was heated to 80 °C for 10 min. After 10 min, the resulting electrolytes were detached from the non-stick foil which was placed on the film applicator.
 | ||
| Fig. 2 Process route of PEO–PEG–LiTFSI-based CPEs. | ||
Thermal properties
Differential scanning calorimetry (DSC, DSC822e, Mettler Toledo) was used to evaluate the phase change behavior of the CPEs. The measurements were performed in 3 stages; in the first stage, the samples were heated from −80 °C to 120 °C, in the second stage the samples were cooled down from 120 °C to −80 °C, and in the third stage they were heated again from −80 °C to 120 °C with a heating rate of 10 °C min−1. The crystallinities (χc) of the CPEs are calculated using eqn (1), where ΔHm is the melting enthalpy of the electrolyte, ΔHPEO is the melting enthalpy of fully crystalline PEO (196.4 J g−1),65 and fPEO is the mass percentage of PEO in the CPEs. | (1) |
Materials characterization
The homogeneity of the CPEs was characterized using scanning electron microscopy (SEM, Helios G4 CX, FEI) coupled with energy-dispersive X-ray spectroscopy (EDX) (Octane Elite-70, EDAX). The chemical structure of the CPEs was analyzed using Fourier-transform infrared spectroscopy (FT-IR, VERTEX 70, Bruker). X-Ray photoelectron spectroscopy (XPS, Kratos Analytical Ltd) was performed to further reveal the successful functionalization of the silane ligand on the surface of the LLZO particles. A monochromatic Al Kα X-ray source (1486.6 eV) with an emission current at 20 mA was used; the chamber pressure was 1.7 × 10−9 torr. The collimation mode was set to slot, the lens mode to hybrid, and the resolution was fixed at 20. Regions including C 1s, Si 2p, Li 1s, and Zr 3d were analyzed with a step size of 0.1 eV over 5–10 sweeps. For the analysis, data were processed using Casa XPS software. The obtained XPS spectra were charge-corrected using the reference value by shifting them to the difference between the adventitious C 1s peak and the value for adventitious carbon at 284.8 eV. To confirm the crystalline phase of the functionalized and non-functionalized ceramic particles as well as CPEs, X-ray diffraction (XRD) was performed with Cu Kα radiation (Empyrean Cu LEF HR goniometer, Malvern PANalytical) on a Si sample holder at 40 kV and 30 mA over the 2θ range of 10°–90° (Empyrean Series 2, PIXcel-3D detector, Malvern PANalytical).Electrochemical properties
Electrochemical impedance spectroscopy (EIS) was conducted using a potentiostat (BioLogic VSP-300). For the measurement, symmetrical coin cells with a copper foil |CPE| copper foil setup were assembled (Fig. S1, ESI†). The coin cells were measured in the frequency range from 7 MHz to 100 mHz at an amplitude of 10 mV. The investigations were performed at 20, 40, 60 and 80 °C in a climate chamber (Binder GmbH). The Nyquist plots are fitted as shown in Fig. S2 (ESI†), dependent on the obtained plots (Fig. S3, ESI†) to determine the resistance of the CPEs.The ionic conductivity (σ) was determined by taking into account the thickness (d) and area (A) of electrolyte and the resistance (Rion) with the following eqn (2).
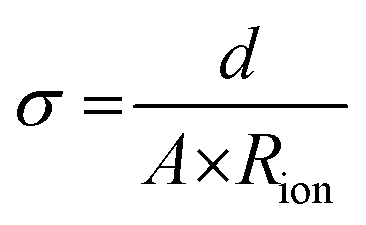 | (2) |
The following eqn (3) was used to determine the activation energy (Ea) of the CPEs by fitting the temperature-dependent ionic conductivity with the Vogel–Tamman–Fulcher (VTF) model, whereby A is pre-exponential factor, T is temperature, and k is the rate constant.
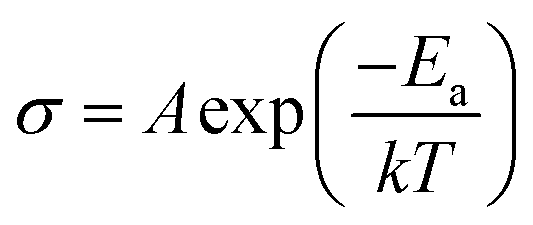 | (3) |
Results and discussion
Incorporation of fillers in CPEs
In this work, the effects of the type of filler incorporated into the PEO–PEG matrix on the ionic conductivity of CPE films are investigated by the comparison of LLZO and ZrO2 filler particles. A (3-glycidyloxypropyl)-trimethoxysilane (GPTMS) ligand was previously attached to the surface of the particles, as depicted in Fig. 3. This surface treatment might lead to the formation of covalently bound GPTMS layers on the fillers, which, in turn, is capable of creating a chemical bond with the PEO matrix via the GPTMS oxirane groups. Such surface functionalization is expected to reduce the free volume between the ceramic particles and the PEO matrix, potentially diminishing the interfacial resistance between the ceramic and polymer components.22,66,67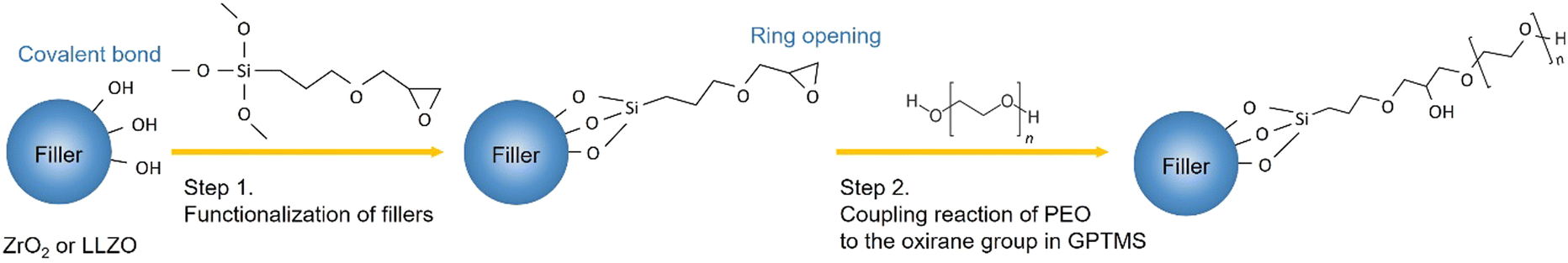 | ||
| Fig. 3 Illustration of the possible chemical reactions occurring during the synthesis of CPEs from the functionalization of fillers to the linking of PEO to the oxirane group in GPTMS. | ||
DSC measurement of the obtained electrolytes was performed to evaluate the influence of GPTMS and the fillers on the phase transition and the crystallinity. The DSC curves reveal that the PEO SPE has a glass transition temperature (Tg) of −50.7 °C, alongside an exothermic peak centered at −0.2 °C, indicative of polymer crystallization (Fig. 4(a)). The melting temperature (Tm) of PEO is determined as 47.9 °C. The GPTMS CPE exhibits a Tg at −63.0 °C and a single endothermic peak representing Tm at 40.5 °C. Both Tg and Tm are decreased compared to the PEO SPE, indicating that the presumed linkage of GPTMS to the PEO chain effectively increases the amorphous region of the PEO matrix. The CPEs with GPTMS–ZrO2 show similar results to the GPTMS CPE with the Tg and Tm reduced to −61.3 °C and 39.1 °C, respectively. Interestingly, the GPTMS–LLZO CPE does not possess a significantly reduced Tg and Tm compared to the GPTMS CPE. The higher Tm reveals that more energy is required to melt the physically restricted polymer chains between the densely packed LLZO particles.68
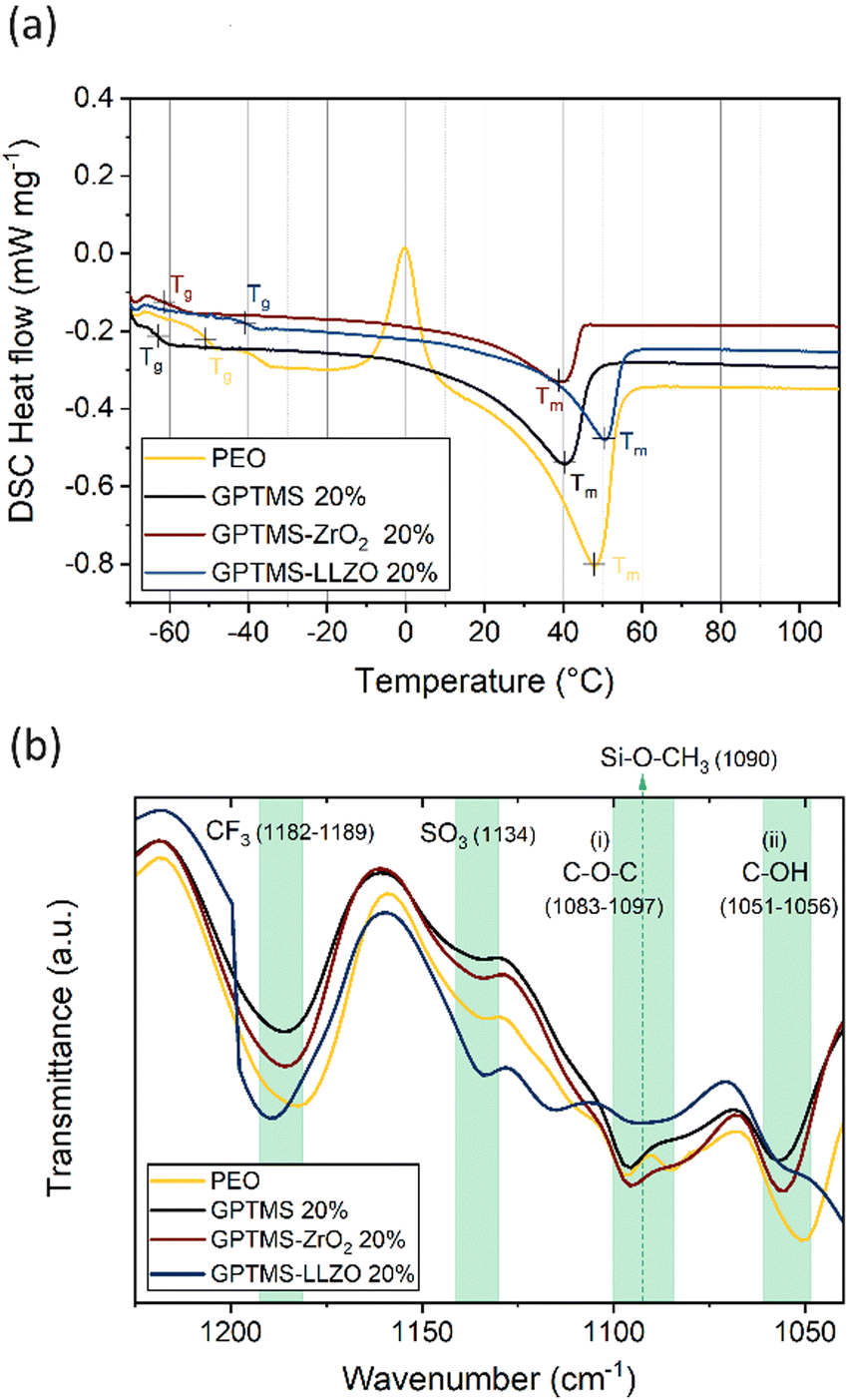 | ||
| Fig. 4 (a) The second heating curves of the DSC measurements and (b) FT-IR spectra of the PEO–LiTFSI SE, filler-free GPTMS 20% CPE, GPTMS–ZrO2 20% CPE, and GPTMS–LLZO 20% CPE. | ||
In Table 2, the determined thermal properties are summarized. The CPEs exhibit lower Tg, Tm and χc than PEO–LiTFSI SPE, implying that the incorporation of fillers and GPTMS effectively decreases the crystallinity of the polymer. However, the crystallinity of the GPTMS–LLZO CPE is higher than that of the filler-free GPTMS CPE. This result indicated that the effect of GPTMS–LLZO on the ion mobility of the segmental motion of polymer might be relatively weak.69
| Solid electrolyte | T g (°C) | T m (°C) | ΔHm (J g−1) | χ c (%) |
|---|---|---|---|---|
| PEO–LiTFSI SPE | −50.7 | 47.9 | 65.9 | 55.8 |
| GPTMS 20% CPE | −63.0 | 40.5 | 31.3 | 43.7 |
| GPTMS–ZrO2 20% CPE | −61.3 | 39.1 | 15.8 | 37.1 |
| GPTMS–LLZO 20% CPE | −40.7 | 50.6 | 21.7 | 47.9 |
To further analyze the CPEs, FT-IR spectroscopy was used to identify their chemical structure and main functional groups (Fig. 4(b)). For the analysis, the PEO SPE and the GPTMS CPE containing 20 vol% GPTMS, as well as the CPEs with 20 vol% of GPTMS–ZrO2 and GPTMS–LLZO were chosen for comparison. The peaks at 1182–1189 cm−1 and 1134 cm−1 are attributable to CF3 stretching and SO3 stretching, respectively; both are characteristic peaks of LiTFSI.70 The C–O–C ether group at 1083–1097 cm−1 and the C–OH terminal group at 1055 cm−1 in the PEO chains were identified in all the CPEs.71,72 To reveal the presence of GPTMS within the CPE, a peak around 1090 cm−1, corresponding to the Si–O–CH3 stretching vibration of the methoxy group bound to Si, should be detected.73,74 If GPTMS is effectively bound to the particles, the methoxy group on the terminal group in the silane should not be detectable.75 The presence of the methoxy group was observed as shoulders in the spectra of the GPTMS CPE and the GPTMS–ZrO2 CPE, being slightly more prominent in GPTMS CPE. In contrast, the methoxy group band was barely visible in the spectrum of GPTMS–LLZO CPE, and was observed as a very weak shoulder in the PEO CPE spectrum. This peak, however, overlaps with the C–O–C band of PEO, and the small amounts of GPTMS compared to other components in the CPEs make it difficult to clearly distinguish the methoxy peak.76 Previously, the Zr–O–Si band at 1058 cm−1 has been taken as proof of covalent binding of GPTMS to LLZTO particles,55 and the shifted C–OH peak might indicate the presence of this band for the GPTMS–ZrO2 and GPTMS–LLZO samples, however again due to the small amount of GPTMS a clear distinction is not possible. The spectrum of the GPTMS–ZrO2 CPE was similar to that of the GPTMS CPE, suggesting that the polymer did not undergo significant changes upon addition of the ZrO2 filler. This similarity indicates the absence of any undesirable side reactions. A slight change in peak intensity and a shift in the GPTMS–ZrO2 CPE spectrum at (i) and (ii) in Fig. 4(b) might result from the increase in the amorphous phase of PEO upon incorporation of GPTMS–ZrO2. In contrast, the GPTMS–LLZO CPE spectrum significantly differed from the others, which can be explained by the accelerated hydrolysis reaction of GPTMS by the LiOH generated from the Li+/H+ exchange of LLZO, as an alcohol containing –OH groups was used as medium for LLZO particle functionalization.74 LiOH was reported to be deposited on the surface of LLZO and might even form a Li2CO3 insulating layer, which deteriorates the electrochemical performance.22,49
The presence of Li2CO3 on the surface of pristine LLZO particles was confirmed by XPS and TGA analyses, as proven by the Li2CO3 peak at 55 eV in the XPS Li 1s spectra and the less profound ZrO2 peak in the Zr 3d spectra due to coverage by Li2CO3, as visible in Fig. 5. Additionally, the TGA thermograms demonstrated that pristine LLZO exhibited higher weight loss compared to with acetic acid-washed particles and GPTMS–LLZO particles, suggesting the presence of a decomposable impurity such as Li2CO3 on the pristine particles, as shown in Fig. S4 (ESI†). Furthermore, the difference in weight loss trends between washed LLZO and GPTMS–LLZO, particularly noted at 250 °C where washed LLZO maintained 99.5% of its weight and GPTMS–LLZO 98.8%, indicates the successful grafting of GPTMS silane ligands onto the LLZO surface. The presence of grafted GPTMS on PEO chain and fillers in CPEs was further confirmed by TGA measurements (Fig. S5, ESI†) to support the FT-IR results.
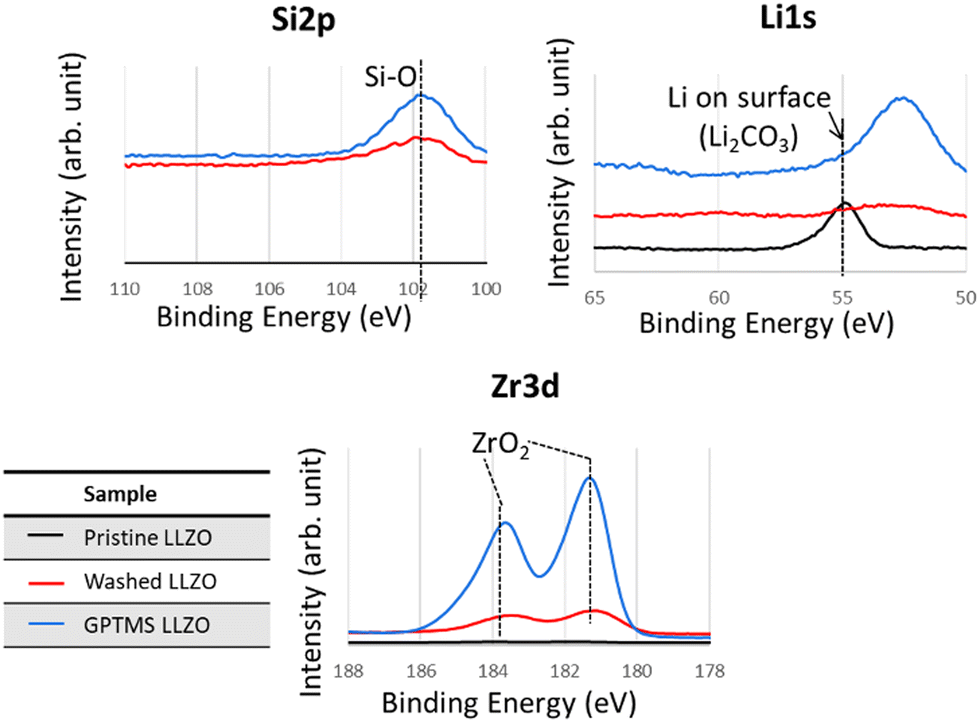 | ||
| Fig. 5 XPS Si 2p, Li 1s, and Zr 3d spectra of pristine (before functionalization), washed (acid-treated), and GPTMS–LLZO particles. The peak at around 53 eV in Li 1s corresponds to Zr 4s. | ||
Impacts of active fillers, passive fillers, and silanes on the ionic conductivity of SPEs
High ionic conductivity (>10−4 S cm−1) over a wide temperature range is an essential prerequisite for the practical application of SEs.77 First, the effects of GPTMS or filler concentration (6, 10, 15, and 20 vol%) on the ionic conductivity of the CPEs were investigated at four different temperatures and compared with the PEO SPE (Fig. 6). The PEO reference had an ionic conductivity of 9.26 × 10−6 S cm−1 at 20 °C, 1.66 × 10−4 S cm−1 at 40 °C, 6.57 × 10−4 S cm−1 at 60 °C, and 1.44 × 10−3 S cm−1 at 80 °C.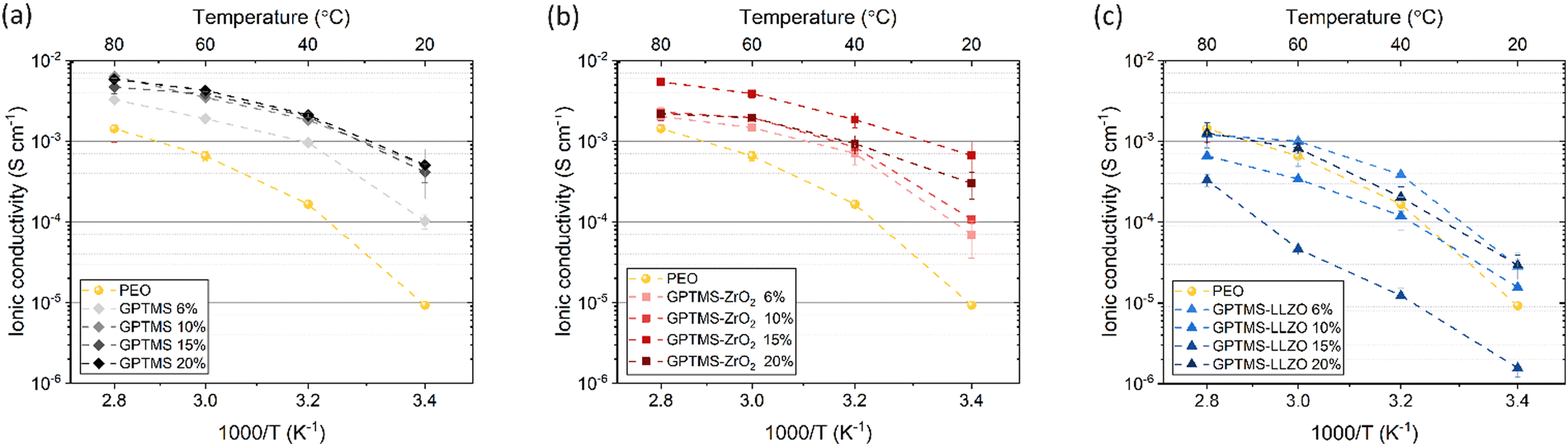 | ||
| Fig. 6 Ionic conductivity of 6, 10, 15, 20 vol% of (a) GPTMS CPE, (b) GPTMS–ZrO2 CPE and (c) GPTMS–LLZO CPE measured at 20, 40, 60, and 80 °C in comparison of PEO SE. | ||
The GPTMS CPEs exhibited higher ionic conductivity than the standard PEO SPE at all measured temperatures (Fig. 6(a)). Among the GPTMS CPE variants, the GPTMS 6% CPE had the lowest ionic conductivity of 1.02 × 10−4 S cm−1. The GPTMS 10%, 15%, and 20% CPEs achieved similar ionic conductivities ranging from 4.15 × 10−4 to 5.1 × 10−4 S cm−1 at 20 °C. Again at 80 °C, the GPTMS 6% CPE achieved a relatively low ionic conductivity of 3.27 × 10−3 S cm−1 and the GPTMS CPEs with higher silane content showed similar ionic conductivities, reaching about 5 × 10−3 S cm−1 at 80 °C. This reveals that the addition of GPTMS can effectively improve the ionic conductivity, which is unexpected, and could be advantageous of not having to deal with particles.
The GPTMS–ZrO2 CPE series also exhibited substantially higher ionic conductivity than the PEO SPE (Fig. 6(b)). The GPTMS–ZrO2-6% CPE with the lowest content of GPTMS–ZrO2 particles showed an ionic conductivity of 6.94 × 10−5 S cm−1 and 2.02 × 10−3 S cm−1 at 20 °C and 80 °C, respectively. The ionic conductivity values of GPTMS–ZrO2-10% CPE are similar/slightly higher to that of ZrO2-6% CPE. The GPTMS–ZrO2-15% CPE had the highest ionic conductivity of 6.66 × 10−4 S cm−1 at 20 °C and 5.43 × 10−3 S cm−1 at 80 °C. For the highest GPTMS–ZrO2 content of 20 vol%, the ionic conductivity decreased again to 3 × 10−4 S cm−1 at 20 °C and 2.19 × 10−3 S cm−1 at 80 °C. The decrease in ionic conductivity can be attributed to a reduction in the content of the conductive PEO–PEG polymer matrix with increasing content of non-conductive ZrO2 particles, as the more non-conductive material, the lower the conductivity of the composite according to the volume rule. Furthermore, increasing the content beyond a certain threshold leads to particle agglomeration, which can disrupt the conductive pathways through the polymer matrix and thus reduce the ionic conductivity. However, the ionic conductivity of these CPEs is still higher than that of the PEO SPE, suggesting that the particles may reduce the crystallinity of PEO.13
On the other hand, the GPTMS–LLZO-based CPEs exhibited unexpectedly low ionic conductivity which is similar or even lower than the reference PEO SPE (Fig. 6(c)). Whilst due to the lower specific surface area of the LLZO particles, less enhancement would be expected for smaller volume contents, a non-consistent trend is observed. The values of GPTMS–LLZO-6% CPE (2.83 × 10−5 at 20 °C and 1.2 × 10−3 S cm−1 at 80 °C) were higher than the GPTMS–LLZO-10% and 15% CPEs. At 15%, the GPTMS–LLZO CPE had the lowest ionic conductivity of 1.56 × 10−6 S cm−1 and 3.32 × 10−4 S cm−1 at 20 °C and 80 °C, respectively. GPTMS–LLZO-20% showed the highest ionic conductivity (2.93 × 10−5 S cm−1 at 20 °C and 1.27 × 10−3 S cm−1 at 80 °C). The possible reason is that from 20 vol% of filler particles, a percolation network of particles starts to exist which can slightly promote the ion transport through the particles. It is however also possible that the particles are highly agglomerated and the separation of PEO and LLZO is more pronounced (as indicated in SEM images, Fig. S6, ESI;† see also below), so that the pathways along the PEO chains are not blocked by LLZO as much as at other concentrations.
Overall, as shown in the direct comparison of the ionic conductivities at 20 °C where the PEO has a mostly crystalline structure and at 80 °C where the melted PEO is present (Fig. 7 and Fig. S7, ESI†), the GPTMS–LLZO CPEs had the lowest ionic conductivity. This can be explained by the intrinsic reactivity of LLZO78 and the presence of Li2CO3 on the surface of the pristine LLZO particles as demonstrated above. This increases the resistance of CPEs79 and possibly also hinders the coordination of GPTMS to the particles. The incorporation of LLZO filler particles into the electrolyte was conducted without additional surface cleaning treatments such as acetic acid washing to remove Li2CO3. While this study focuses on the commonly used LLZO particles with inherent surface Li2CO3, we acknowledge that removing Li2CO3 could potentially improve lithium ion transport through the electrolyte. Previous studies have shown that the removal of surface impurities can significantly enhance ionic conductivity and improve other related properties.80,81 Therefore, an inert processing atmosphere, solvent-free processing, and pre-treatment to remove surface impurities of the particles using tetrafluoromethane (CF4) plasma treatment or acidic solvents is required when handling LLZO materials.14,49 The residual Li2CO3 impurities on the pristine LLZO particles could potentially persist even after silanization. Additionally, the poor ionic conductivity can be ascribed to the uneven distribution of high-density LLZO particles within the polymer matrix. Such distribution not only impedes the migration pathways of Li ions but may also hinder the segmental dynamics of PEO chains, a process depicted schematically in Fig. 8.28 If the LLZO particles were carefully dispersed in the polymer matrix and the filler agglomeration were negligible, the inherent an influence of LLZO particles on the ionic conductivity could be evaluated more accurately. However, achieving a homogeneous distribution of LLZO particles in ceramic-rich CPEs (>4 vol% (20 wt%) LLZO content) is challenging due to the sedimentation tendencies of these high-density micron-sized particles.82 Some literature reports an improved ionic conductivity of LLZO-containing CPEs compared to pure SPEs,14,83 whereas other reports do not find such an enhancement, and even contradicting results were presented.49,84 J. Zagórski et al. have detected Li-ion exchange between the PEO phase and LLZO by 2D EXSY NMR investigation, which reveals a potential of LLZO for CPEs with high ionic conductivity.68 However, they found that this Li-ion exchange occurs only locally at the interphase of electrolyte/electrode with slow kinetics, and thus the absolute ionic conductivity of LLZO-containing PEO-based CPEs was lower than the filler-free PEO SPE, as the Li-ion pathway was still dominated by the polymer phase. In contrast, in our work, the GPTMS–ZrO2 CPEs exhibited higher conductivity than the PEO SPE and the GPTMS–LLZO CPEs, which is attributed to the relatively high stability of the ZrO2 particles against moisture and their covalent linkage to the polymer network. But the ionic conductivity of GPTMS–ZrO2 CPEs was usually lower than that of the GPTMS CPEs. Only GPTMS–ZrO2-15% CPE achieved similar and slightly higher ionic conductivity than the corresponding GPTMS CPE. This indicates that the addition of the GPTMS to the PEO chain can effectively deliver improved ionic conductivity, which also is more cost-efficient than adding ceramic fillers, as shown by a material cost assessment (Fig. 7). However, it should be noted that an increase in ionic conductivity does not always equate to improved battery performance, but it is necessary to evaluate the electrochemical performance of electrolytes more extensively.
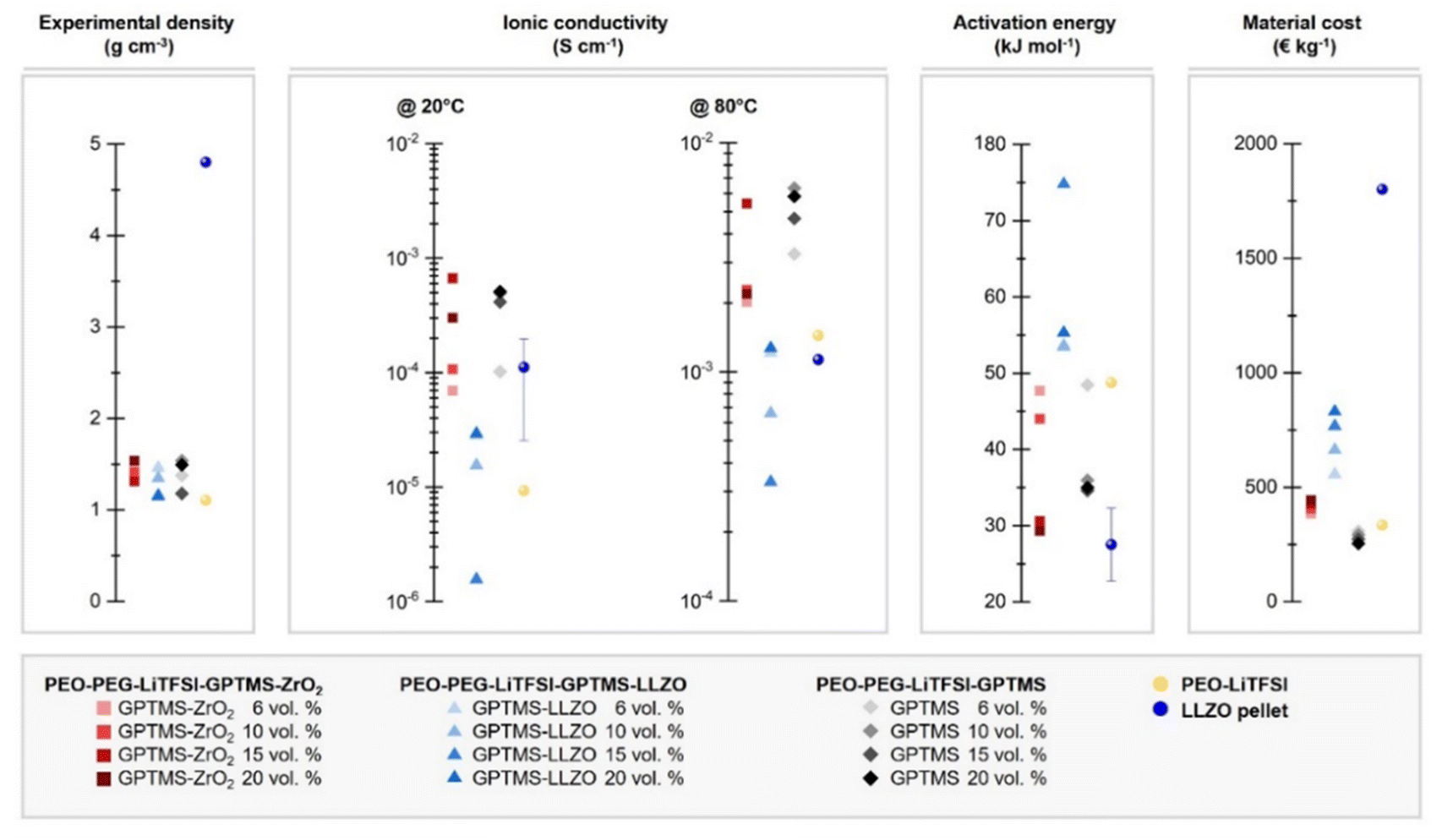 | ||
| Fig. 7 Characteristics of the fabricated PEO-based CPEs, and comparison to a LLZO pellet SE*, with respect to their experimental density, ionic conductivity at 20 °C and 80 °C, and activation energy. The material costs of the CPEs are estimated based on the literature from M. Balaish et al.85 Some properties of LLZO pellet SE were acquired from literature; the density: 4.8 g cm−3; ionic conductivity: 1.11 × 10−4 S cm−1@20 °C86 and 1.13 × 10−3 S cm−1@80 °C;87 activation energy: 24–35 kJ mol−1.86 | ||
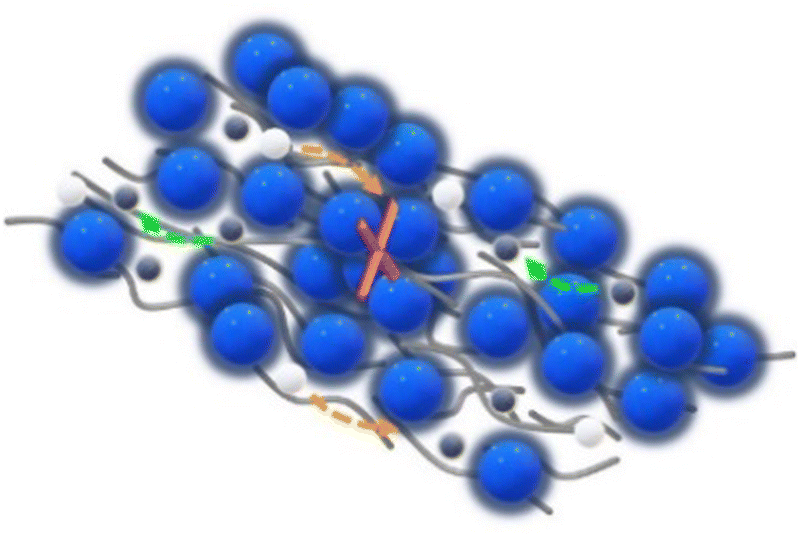 | ||
| Fig. 8 Illustration of a limited ion pathway due to the agglomeration of ceramic particles. | ||
The activation energy (Ea) is another critical parameter in analyzing ion mobility; a lower Ea is preferable because it indicates less energy is required to initiate the ion movement, contributing to higher overall battery efficiency and potentially faster charging. Especially for evaluating PEO-based CPEs, whose ionic conductivity is typically temperature-dependent, Ea can be useful to estimate whether the Li transport is dominated by the segmental motion of PEO or by other mechanisms. The GPTMS–ZrO2-15% and 20% CPEs featured the lowest Ea, suggesting that high loading of ZrO2 particles of over 15 vol% can reduce the crystallinity of PEO. The LLZO-containing CPEs exhibited high Ea, indicating that ion mobility is restricted. We postulate that the PEO chains are physically trapped between LLZO particles, resulting in denser PEO networks with confined chain mobility, thus limiting ion mobility.88 GPTMS-20% CPE showed low Ea (29.31 kJ mol−1), suggesting that this CPE requires low energy to start the ion movement. This implies again that linking GPTMS to the PEO/PEG chain without any fillers enhances the mobility of Li ions.
In addition, the Li ion transference number (TLi) of the CPEs was calculated (Fig. S9, ESI†). High TLi values indicate a lower Li concentration gradient near the surface of the Li anode. The typical TLi of an SPE is low due to the presence of free anions from the Li salt. The calculated TLi values of GPTMS-20%, GPTMS–ZrO2-20%, and GPTMS–LLZO-20% CPEs are 0.27, 0.37 and 0.26, respectively, all of which are higher than for the standard PEO–LiTFSI SPE (TLi of 0.14). The addition of GPTMS and GPTMS-modified fillers results in an increase in TLi, which can be attributed to the immobilization of the anions. The similar TLi values for the various CPEs further confirm that the fillers do not actively participate in ion transport but instead facilitate the Li transport. Moreover, linear sweep voltammetry (LSV) measurements were conducted to investigate the electrochemical stability window of the CPEs at 40 °C (Fig. S10, ESI†). Compared to the GPTMS CPE, the addition of fillers has no distinct effect on the electrochemical window of the CPEs. All the CPEs showed an anodic limit above 4.2 V (vs. Li/Li+), indicating that SPEs containing functionalized fillers can be applied for high-voltage electrode materials. Considering that all the CPEs showed a similar potential stability window, it can be concluded that the stability is mainly determined by the host polymer matrix.
Fig. 9 shows the surface morphology of the CPEs with 15 vol% of GPTMS–ZrO2 and GPTMS–LLZO, the GPTMS-15% CPE, and the PEO SPE. The PEO SPE shows a smooth, homogeneous surface free of particles. The addition of fillers or GPTMS significantly changed the surface morphology of the CPEs. The filler-free GPTMS CPE exhibited a fibrous and uneven structure, contrasting the smooth surface of the PEO SPE. Together with its sticky nature, this might offer enhanced interfacial adhesion to the electrodes or current collectors, which would increase the ionic conductivity compared to the PEO-based SPE – a result consistent with the ionic conductivity results. The SEM image of the GPTMS–ZrO2 CPE showed a homogeneous surface devoid of fibrous structure, and a uniform distribution of particles on the surface, thereby enabling efficient Li-ion migration along the space charge region of the GPTMS–ZrO2 particles, which explains the improved ionic conductivity of this solid electrolyte. In contrast, the GPTMS–LLZO CPE exhibited poor homogeneity on the surface with large particle agglomerates being visible, which would disrupt the Li ion transport pathway along and between the polymer chains. In addition, the space charge region is present to a much lower amount here due to the strongly reduced interface area between the particles and PEO – an explanation for the lowest ionic conductivity observed for the GPTMS–LLZO CPE. Moreover, the surface of the GPTMS–LLZO CPE does not exhibit the malleability observed in other CPEs; instead, it exhibits an appearance reminiscent of ceramic materials. This suggests potential difficulties in achieving optimal electrode–electrolyte adhesion, a critical aspect in subsequent cell assembly and performance.
 | ||
| Fig. 9 SEM images and digital photos (top right) of PEO SPE with smooth surface, GPTMS 15% CPE with fibrous and uneven structure, GPTMS–ZrO2 15% CPE with homogeneous surface and a uniform distribution of particles, and GPTMS–LLZO 15% CPE with heterogeneous surface and large particle agglomerates. | ||
Another comparison of the surface morphology was conducted for all CPEs with an additive volume fraction of 20% (Fig. S6, ESI,† with a higher magnification). The surface of the GPTMS–ZrO2 CPE appeared to be heterogeneous, displaying particle agglomeration and a fibrous structure, though less pronounced than observed for the GPTMS CPE. In contrast, the GPTMS–LLZO CPE surface was smoother around the filler particles with no fibrous structure. Nevertheless, SEM images of both GPTMS–ZrO2 and GPTMS–LLZO CPEs reveal the presence of particle agglomerates, indicating a poor dispersion of the particles, which could account for the reduced ionic conductivity in both CPEs compared to the filler-free GPTMS CPEs.
In conclusion, we found that the performance of the composite polymer electrolytes is highly dependent on the properties of the polymer. The incorporation of fillers can improve the ionic conductivity, but the impact on the overall performance can be limited due to other considerations such as the mechanical properties of CPEs. The selection of a suitable polymer and careful attention to the surface functionalization and processing of the fillers are therefore critical to ensure that the surface properties of the fillers are improved and that the particles can be effectively dispersed in the polymer matrix preventing agglomeration.
Conclusion
In this study, the effects of functionalized active and passive fillers on the ionic conductivity of PEO–PEG–LiTFSI solid polymer electrolytes (SPEs) were investigated. Our results show that the integration of passive fillers, in particular 15 vol% GPTMS-grafted ZrO2 fillers, considerably increased the ionic conductivity of the PEO-based composite polymer electrolyte (CPE) at 20 °C to 6.66 × 10−4 S cm−1 at 20 °C, compared to the base PEO–SPE conductivity of 9.26 × 10−6 S cm−1 at 20 °C. Conversely, CPEs with active GPTMS–LLZO fillers showed lower conductivity, ranging from 1.56 × 10−6 S cm−1 to 2.93 × 10−5 at 20 °C. An interesting observation was the significant increase in ionic conductivity (to about 5 × 10−4 S cm−1 at 20 °C) achieved by anchoring the PEO–PEG chain to the GPTMS ligands in the absence of fillers, which can be attributed to the resulting decrease in PEO crystallinity. In particular, the study highlights the necessity of controlling surface impurities, such as Li2CO3, on LLZO fillers, which have been shown to form resistive layers that are detrimental to ionic conductivity.Furthermore, the study reveals that the obstruction to the Li-ion movement by the excessive and non-uniform distribution of high-density LLZO fillers strongly diminished the ionic conductivity, which underscores the importance of both an optimal filler content and its homogeneous dispersion within the matrix to achieve the desired performance. Additionally, the strategic incorporation of reactive silane compounds not only improves the ionic conductivity of PEO–PEG–LiTFSI electrolytes, but also represents a cost-effective method compared to the incorporation of filler particles. These findings provide a facile way for the development of efficient polymer electrolytes with improved performance and offer promising prospects for their application in solid-state lithium batteries.
Author contributions
Conceptualization: E. J. J., L. H., A. K., G. G.; methodology: E. J. J., L. H., G. G.; investigation: E. J. J., S. H., L. H.; visualization: E. J. J., S. H.; funding acquisition: A. K., G. G.; project administration: E. J. J., L. H.; supervision: A. K., G. G.; writing – original draft: E. J. J., S. H.; writing – review & editing: L. H., A. K., G. G.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Daniela Scholz for FT-IR, TGA and XPS measurements. We acknowledge funding by the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG) under Germany's Excellence Strategy EXC 2163/1 “Sustainable and Energy Efficient Aviation”, Project ID 390881007.References
- J. Janek and W. G. Zeier, Nat. Energy, 2023, 8, 230–240 CrossRef
.
- H. Huo and J. Janek, Natl. Sci. Rev., 2023, 10, nwad098 CrossRef PubMed
- T. Ye, L. Li and Y. Zhang, Adv. Funct. Mater., 2020, 30, 2000077 CrossRef CAS
- K. J. Kim, M. Balaish, M. Wadaguchi, L. Kong and J. L. M. Rupp, Adv. Energy Mater., 2021, 11, 2002689 CrossRef CAS
- Y. Pang, J. Pan, J. Yang, S. Zheng and C. Wang, Electrochem. Energy Rev., 2021, 4, 169–193 CrossRef CAS
- Z. Shen, W. Zhang, G. Zhu, Y. Huang, Q. Feng and Y. Lu, Small Methods, 2020, 4, 1900592 CrossRef CAS
- S. Xia, X. Wu, Z. Zhang, Y. Cui and W. Liu, Chem, 2019, 5, 753–785 CAS
- D. E. Fenton, J. M. Parker and P. V. Wright, Polymer, 1973, 14, 589 CrossRef CAS
- M. Armand, Solid State Ionics, 1983, 9–10, 745–754 CrossRef CAS
- W. H. Meyer, Adv. Mater., 1998, 10, 439–448 CrossRef CAS PubMed
- J. M. G. Cowie and S. H. Cree, Annu. Rev. Phys. Chem., 1989, 40, 85–113 CrossRef CAS
- G. Homann, L. Stolz, J. Nair, I. C. Laskovic, M. Winter and J. Kasnatscheew, Sci. Rep., 2020, 10, 4390 CrossRef CAS PubMed
- L. Froboese, J. F. van der Sichel, T. Loellhoeffel, L. Helmers and A. Kwade, J. Electrochem. Soc., 2019, 166, A318–A328 CrossRef
- L. Helmers, L. Froböse, K. Friedrich, M. Steffens, D. Kern, P. Michalowski and A. Kwade, Energy Technol., 2021, 9, 2000923 CrossRef CAS
- L. Froboese, L. Groffmann, F. Monsees, L. Helmers, T. Loellhoeffel and A. Kwade, J. Electrochem. Soc., 2020, 167, 20558 CrossRef CAS
- A. Manthiram, X. Yu and S. Wang, Nat. Rev. Mater., 2017, 2, 16103 CrossRef CAS
- J. Mindemark, M. J. Lacey, T. Bowden and D. Brandell, Prog. Polym. Sci., 2018, 81, 114–143 CrossRef CAS
- Y. Jiang, X. Yan, Z. Ma, P. Mei, W. Xiao, Q. You and Y. Zhang, Polymers, 2018, 10, 1237 CrossRef PubMed
- E. J. Jeon, A. Jean-Fulcrand, A. Kwade and G. Garnweitner, Nano Energy, 2022, 104, 107912 CrossRef CAS
- F. Croce, G. B. Appetecchi, L. Persi and B. Scrosati, Nature, 1998, 394, 456–458 CrossRef CAS
- X. Yang, J. Liu, N. Pei, Z. Chen, R. Li, L. Fu, P. Zhang and J. Zhao, Nano-Micro Lett., 2023, 15, 74 CrossRef CAS PubMed
- L. Helmers, F. Frankenberg, J. Brokmann, C. Burmeister, A. Buchheit, A. Kwade and P. Michalowski, ChemElectroChem, 2023, 10, e202300310 CrossRef CAS
- Y. Zhu, J. Cao, H. Chen, Q. Yu and B. Li, J. Mater. Chem. A, 2019, 7, 6832–6839 RSC
- X.-X. Zeng, Y.-X. Yin, N.-W. Li, W.-C. Du, Y.-G. Guo and L.-J. Wan, J. Am. Chem. Soc., 2016, 138, 15825–15828 CrossRef CAS PubMed
- V. St-Onge, M. Cui, S. Rochon, J.-C. Daigle and J. P. Claverie, Commun. Mater., 2021, 2, 83 CrossRef CAS
- J. Rolland, J. Brassinne, J.-P. Bourgeois, E. Poggi, A. Vlad and J.-F. Gohy, J. Mater. Chem. A, 2014, 2, 11839–11846 RSC
- Z. Lv, Q. Zhou, S. Zhang, S. Dong, Q. Wang, L. Huang, K. Chen and G. Cui, Energy Storage Mater., 2021, 37, 215–223 CrossRef
- Z. Li, J. Fu, X. Zhou, S. Gui, L. Wei, H. Yang, H. Li and X. Guo, Adv. Sci., 2023, 10, e2201718 CrossRef PubMed
- J. Fu, Z. Li, X. Zhou and X. Guo, Mater. Adv., 2022, 3, 3809–3819 RSC
- J. E. Weston and B. C. H. Steele, Solid State Ionics, 1982, 7, 75–79 CrossRef CAS
- M. C. Borghini, M. Mastragostino, S. Passerini and B. Scrosati, J. Electrochem. Soc., 1995, 142, 2118 CrossRef CAS
- F. Croce, L. Persi, B. Scrosati, F. Serraino-Fiory, E. Plichta and M. A. Hendrickson, Electrochim. Acta, 2001, 46, 2457–2461 CrossRef CAS
- L. C. Merrill, X. C. Chen, Y. Zhang, H. O. Ford, K. Lou, Y. Zhang, G. Yang, Y. Wang, Y. Wang, J. L. Schaefer and N. J. Dudney, ACS Appl. Energy Mater., 2020, 3, 8871–8881 CrossRef CAS
- W. Wieczorek, A. Zalewska, D. Raducha, Z. Florjańczyk and J. R. Stevens, J. Phys. Chem. B, 1998, 102, 352–360 CrossRef CAS
- H. Gao and K. Lian, ACS Appl. Mater. Interfaces, 2014, 6, 464–472 CrossRef CAS PubMed
- D. Lin, W. Liu, Y. Liu, H. R. Lee, P.-C. Hsu, K. Liu and Y. Cui, Nano Lett., 2016, 16, 459–465 CrossRef CAS PubMed
- N. L. Grotkopp, M. Horst, M. Batzer, G. Garnweitner and A. Jean-Fulcrand, Adv. Energy Sustainability Res., 2023, 4, 2200146 CrossRef CAS
- L. Chen, Y. Li, S.-P. Li, L.-Z. Fan, C.-W. Nan and J. B. Goodenough, Nano Energy, 2018, 46, 176–184 CrossRef CAS
- G. Piana, F. Bella, F. Geobaldo, G. Meligrana and C. Gerbaldi, J. Energy Storage, 2019, 26, 100947 CrossRef
- J. Lee, T. Howell, M. Rottmayer, J. Boeckl and H. Huang, J. Electrochem. Soc., 2019, 166, A416–A422 CrossRef CAS
- S. Chen, J. Wang, Z. Zhang, L. Wu, L. Yao, Z. Wei, Y. Deng, D. Xie, X. Yao and X. Xu, J. Power Sources, 2018, 387, 72–80 CrossRef CAS
- R. Murugan, V. Thangadurai and W. Weppner, Angew. Chem., Int. Ed., 2007, 46, 7778–7781 CrossRef CAS PubMed
- V. Thangadurai, S. Narayanan and D. Pinzaru, Chem. Soc. Rev., 2014, 43, 4714–4727 RSC
- Y. Jin and P. J. McGinn, J. Power Sources, 2013, 239, 326–331 CrossRef CAS
- D. Cao, X. Sun, Q. Li, A. Natan, P. Xiang and H. Zhu, Matter, 2020, 3, 57–94 CrossRef
- C. Zhu, T. Fuchs, S. A. L. Weber, F. H. Richter, G. Glasser, F. Weber, H.-J. Butt, J. Janek and R. Berger, Nat. Commun., 2023, 14, 1300 CrossRef CAS PubMed
- K. K. Fu, Y. Gong, J. Dai, A. Gong, X. Han, Y. Yao, C. Wang, Y. Wang, Y. Chen, C. Yan, Y. Li, E. D. Wachsman and L. Hu, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7094–7099 CrossRef CAS PubMed
- Z. Li, H.-M. Huang, J.-K. Zhu, J.-F. Wu, H. Yang, L. Wei and X. Guo, ACS Appl. Mater. Interfaces, 2019, 11, 784–791 CrossRef CAS PubMed
- M. Keller, G. B. Appetecchi, G.-T. Kim, V. Sharova, M. Schneider, J. Schuhmacher, A. Roters and S. Passerini, J. Power Sources, 2017, 353, 287–297 CrossRef CAS
- H. Yang and N. Wu, Energy Sci. Eng., 2022, 10, 1643–1671 CrossRef CAS
- Y. Zheng, Y. Yao, J. Ou, M. Li, D. Luo, H. Dou, Z. Li, K. Amine, A. Yu and Z. Chen, Chem. Soc. Rev., 2020, 49, 8790–8839 RSC
- R. Usiskin and J. Maier, Adv. Energy Mater., 2021, 11, 2001455 CrossRef CAS
- M. B. Dixit, W. Zaman, Y. Bootwala, Y. Zheng, M. C. Hatzell and K. B. Hatzell, ACS Appl. Mater. Interfaces, 2019, 11, 45087–45097 CrossRef CAS PubMed
- B. Zhang, R. Tan, L. Yang, J. Zheng, K. Zhang, S. Mo, Z. Lin and F. Pan, Energy Storage Mater., 2018, 10, 139–159 CrossRef
- E. Kuhnert, L. Ladenstein, A. Jodlbauer, C. Slugovc, G. Trimmel, H. M. R. Wilkening and D. Rettenwander, Cell Rep. Phys. Sci., 2020, 1, 100214 CrossRef CAS
- C. Yan, P. Zhu, H. Jia, Z. Du, J. Zhu, R. Orenstein, H. Cheng, N. Wu, M. Dirican and X. Zhang, Energy Storage Mater., 2020, 26, 448–456 CrossRef
- Y.-Y. Sun, Q. Zhang, L. Fan, D.-D. Han, L. Li, L. Yan and P.-Y. Hou, J. Colloid Interface Sci., 2022, 628, 877–885 CrossRef CAS PubMed
- Y. Xie, L. Huang and Y. Chen, J. Membr. Sci., 2023, 683, 121784 CrossRef CAS
- X. Zheng, J. Wei, W. Lin, K. Ji, C. Wang and M. Chen, ACS Appl. Mater. Interfaces, 2022, 14, 5346–5354 CrossRef CAS PubMed
- J. Cheng, H. Zhang, D. Li, Y. Li, Z. Zeng, F. Ji, Y. Wei, X. Xu, Q. Sun, S. Wang, J. Lu and L. Ci, Batteries, 2022, 8, 141 CrossRef CAS
- M. Wu, J. Song, X. Zhu, H. Zhan, T. Tian, R. Wang, J. Lei and H. Tang, Sci. China Mater., 2023, 66, 522–530 CrossRef CAS
- T. Wang, X. Liu, L. Xie, Y. He, H. Ji, L. Wang, X. Niu and J. Gao, J. Alloys Compd., 2023, 945, 168877 CrossRef CAS
- K. Pan, L. Zhang, W. Qian, X. Wu, K. Dong, H. Zhang and S. Zhang, Adv. Mater., 2020, 32, e2000399 CrossRef PubMed
- Y. Ito, K. Kanehori, K. Miyauchi and T. Kudo, J. Mater. Sci., 1987, 22, 1845–1849 CrossRef CAS
- N. A. Stolwijk, C. Heddier, M. Reschke, M. Wiencierz, J. Bokeloh and G. Wilde, Macromolecules, 2013, 46, 8580–8588 CrossRef CAS
- D. Bresser, S. Lyonnard, C. Iojoiu, L. Picard and S. Passerini, Mol. Syst. Des. Eng., 2019, 4, 779–792 RSC
- M. Echeverri, N. Kim and T. Kyu, Macromolecules, 2012, 45, 6068–6077 CrossRef CAS
- J. Zagórski, J. M. Del López Amo, M. J. Cordill, F. Aguesse, L. Buannic and A. Llordés, ACS Appl. Energy Mater., 2019, 2, 1734–1746 CrossRef
- M. Kato, K. Hiraoka and S. Seki, J. Electrochem. Soc., 2020, 167, 70559 CrossRef
- N. F. Yusof, A. A. Raffi, N. Z. S. Yahaya, K. H. Abas, M. H. D. Othman, J. Jaafar and M. A. Rahman, Membranes, 2023, 13, 253 CrossRef CAS PubMed
- N. S. Vrandečić, M. Erceg, M. Jakić and I. Klarić, Thermochim. Acta, 2010, 498, 71–80 CrossRef
- H. Wang, X. Cui, C. Zhang, H. Gao, W. Du and Y. Chen, Polymers, 2020, 12, 1889 CrossRef CAS PubMed
- R. Jain, Waterborne inorganic–organic hybrid coatings on magnesium by sol–gel route, 2015.
- J. Chai, B. Chen, F. Xian, P. Wang, H. Du, J. Zhang, Z. Liu, H. Zhang, S. Dong, X. Zhou and G. Cui, Small, 2018, 14, e1802244 CrossRef PubMed
- A. Zarinwall, T. Waniek, R. Saadat, U. Braun, H. Sturm and G. Garnweitner, Langmuir, 2021, 37, 171–179 CrossRef CAS PubMed
- E. Kuhnert, L. Ladenstein, A. Jodlbauer, C. Slugovc, G. Trimmel, H. M. R. Wilkening and D. Rettenwander, Cell Rep. Phys. Sci., 2020, 1, 100214 CrossRef CAS
- C. Zuo, M. Yang, Z. Wang, K. Jiang, S. Li, W. Luo, D. He, C. Liu, X. Xie and Z. Xue, J. Mater. Chem. A, 2019, 7, 18871–18879 RSC
- L. Li, Y. Deng and G. Chen, J. Energy Chem., 2020, 50, 154–177 CrossRef
- Y. Li, X. Chen, A. Dolocan, Z. Cui, S. Xin, L. Xue, H. Xu, K. Park and J. B. Goodenough, J. Am. Chem. Soc., 2018, 140, 6448–6455 CrossRef CAS PubMed
- K. He, S. H.-S. Cheng, J. Hu, Y. Zhang, H. Yang, Y. Liu, W. Liao, D. Chen, C. Liao, X. Cheng, Z. Lu, J. He, J. Tang, R. K. Y. Li and C. Liu, Angew. Chem., Int. Ed., 2021, 60, 12116–12123 CrossRef CAS PubMed
- T.-T. Wu, S. Guo, B. Li, C.-Y. Shen, X.-H. Liu and A.-M. Cao, Rare Met., 2023, 42, 3177–3200 CrossRef CAS
- W. Liu, N. Liu, J. Sun, P.-C. Hsu, Y. Li, H.-W. Lee and Y. Cui, Nano Lett., 2015, 15, 2740–2745 CrossRef CAS PubMed
- R. F. Samsinger, S. O. Schopf, J. Schuhmacher, P. Treis, M. Schneider, A. Roters and A. Kwade, J. Electrochem. Soc., 2020, 167, 120538 CrossRef CAS
- F. Langer, I. Bardenhagen, J. Glenneberg and R. Kun, Solid State Ionics, 2016, 291, 8–13 CrossRef CAS
- M. Balaish, J. C. Gonzalez-Rosillo, K. J. Kim, Y. Zhu, Z. D. Hood and J. L. M. Rupp, Nat. Energy, 2021, 6, 227–239 CrossRef CAS
- C. Shao, H. Liu, Z. Yu, Z. Zheng, N. Sun and C. Diao, Solid State Ionics, 2016, 287, 13–16 CrossRef CAS
- K.-w Kim, S.-H. Yang, M. Y. Kim, M. S. Lee, J. Lim, D. R. Chang and H.-S. Kim, J. Ind. Eng. Chem., 2016, 36, 279–283 CrossRef CAS
- G. J. Schneider, Curr. Opin. Chem. Eng., 2017, 16, 65–77 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: Physicochemical properties of different filler materials; schematics on cell setup; details on EIS analysis; TGA thermograms; chronoamperometry and LSV data. See DOI: https://doi.org/10.1039/d4ya00231h |
| This journal is © The Royal Society of Chemistry 2024 |