
On-demand activatable peroxidase-mimicking enzymatic polymer nanocomposite films†
Md. Lutful
Amin
a,
Ayad
Saeed
bc,
Le N. M.
Dinh
a,
Jiachen
Yan
b,
Haotian
Wen
b,
Shery L. Y.
Chang
 bd,
Yin
Yao
d,
Per B.
Zetterlund
a,
Tushar
Kumeria
*bce and
Vipul
Agarwal
*a
bd,
Yin
Yao
d,
Per B.
Zetterlund
a,
Tushar
Kumeria
*bce and
Vipul
Agarwal
*a
aCluster for Advanced Macromolecular Design (CAMD), School of Chemical Engineering, University of New South Wales, Sydney, NSW 2052, Australia. E-mail: agarwalvipul84@gmail.com
bSchool of Materials Science and Engineering, University of New South Wales, Sydney, NSW 2052, Australia. E-mail: t.kumeria@unsw.edu.au
cAustralian Centre for Nanomedicine, University of New South Wales, Sydney, NSW 2052, Australia
dElectron Microscopy Unit, Mark Wainwright Analytical Centre, University of New South Wales, Sydney, NSW 2052, Australia
eSchool of Pharmacy, University of Queensland, Brisbane, QLD 4102, Australia
First published on 4th July 2024
Abstract
Nanozymes continue to attract considerable attention to minimise the dependence on expensive enzymes in bioassays, particularly in medical diagnostics. While there has been considerable effort directed towards developing different nanozymes, there has been limited progress in fabricating composite materials based on such nanozymes. One of the biggest gaps in the field is the control, tuneability, and on-demand catalytic response. Herein, a nanocomposite nanozymatic film that enables precise tuning of catalytic activity through stretching is demonstrated. In a systematic study, we developed poly(styrene-stat-n-butyl acrylate)/iron oxide-embedded porous silica nanoparticle (FeSiNP) nanocomposite films with controlled, highly tuneable, and on-demand activatable peroxidase-like activity. The polymer/FeSiNP nanocomposite was designed to undergo film formation at ambient temperature yielding a highly flexible and stretchable film, responsible for enabling precise control over the peroxidase-like activity. The fabricated nanocomposite films exhibited a prolonged FeSiNP dose-dependent catalytic response. Interestingly, the optimised composite films with 10 wt% FeSiNP exhibited a drastic change in the enzymatic activity upon stretching, which provides the nanocomposite films with an on-demand performance activation characteristic. This is the first report showing control over the nanozyme activity using a nanocomposite film, which is expected to pave the way for further research in the field leading to the development of system-embedded activatable sensors for diagnostic, food spoilage, and environmental applications.
 Vipul Agarwal | Dr Vipul Agarwal is a Senior Lecturer at the University of New South Wales (UNSW). He joined UNSW in 2018 as a prestigious Australian National Health and Medical Research Council (NHMRC) Research Fellow in the School of Chemical Engineering. Prior to this, he was awarded the SERB-DST National Postdoctoral Fellowship to undertake postdoctoral training at the Indian Institute of Science, India. Dr Agarwal graduated with a PhD degree in Chemistry from The University of Western Australia, Australia in 2015. Dr Agarwal's current research interest is in materials chemistry focusing on developing synthesis and fabrication strategies towards two- and three-dimensional polymer nanocomposites for a range of applications including neural tissue engineering. |
Introduction
Natural enzymes are commonly used as catalysts in a variety of fields, such as environmental monitoring, agriculture, and the food industry.1–3 Peroxidase is a natural enzyme that catalyses the transfer of electrons from an electron donor to peroxides, which are electron acceptors.4–6 Convenient detection of hydrogen peroxide is of practical importance as it plays important roles in several fields including food, pharmaceutical and plastic industries, environmental chemistry, biological analysis, and clinical diagnostics.7–10 The use of peroxidase enzymes in diagnostics is most prominent and forms the basis for majority of ELISA-based assays, where hydrogen peroxide acts as an electron acceptor in the oxidation of chromogenic substrates into coloured compounds.1,11–13 However, the inherent disadvantages of natural enzymes, such as high cost, high pH sensitivity, and poor thermal stability challenge their practical applications.14–18 Hence, artificial peroxidase-mimicking materials as an alternative to natural enzymes have attracted considerable attention in recent years.14,19–22 To this end, various nanoparticles have emerged as natural enzyme-mimics with the ability to catalyse reactions in the absence of a natural enzyme. These nanoparticles, referred to as nanozymes, offer better stability, ease of synthesis and modification, comparable catalytic activity, and lower cost overcoming the limitations of the natural enzymes.Various nanomaterials, such as metallic nanoparticles, metallic oxides and salts, conducting polymer nanoparticles, and quantum dots, have been studied for peroxidase-mimicking activity in applications such as immunoassays and the detection of biochemicals including glutathione, ascorbic acid, glucose, and cholesterol.1,19,23–25 Iron oxide nanoparticles have attracted a great deal of attention as artificial non-proteinaceous nanozymes that mimic the catalytic characteristics of peroxidase enzymes.26–28 These nanoparticles can oxidise the substrate 3,3′,5,5′-tetramethyl benzidine (TMB) in the presence of hydrogen peroxide, resulting in a colourimetric change from colourless to blue, which can be observed by the naked eye and analysed using ultraviolet-visible (UV-vis) spectrophotometry.13,29 Unlike natural peroxidase enzyme, iron oxide nanoparticles are not susceptible to proteolytic degradation or affected by other environmental factors, such as temperature, pH, ionic strength, and heavy metals.30
Despite iron oxide nanoparticles being commonly studied as nanozymes, they undergo aggregation in particular when used in isolation in an aqueous environment, decreasing their total surface area, and thus significantly compromising their catalytic activity.30 To this end, iron oxide nanoparticles have been immobilised on solid substrates including nanoparticles and polymer matrices. For example, iron oxide nanozyme particles were attached to cotton-based textiles for pollutant removal, exhibiting elimination of dye with nanozyme activity.31 Geleto et al. fabricated nanocellulose-based iron oxide–silver nanozymes for enhanced antibacterial and wound healing applications.32 Also, electrospun composites with iron oxide nanoparticles have been fabricated, showing high catalytic activity.33 Another report by Satvekar et al. demonstrated the fabrication of a silica/chitosan organic–inorganic hybrid material, assimilated with iron oxide magnetic nanoparticles, for hydrogen peroxide biosensing, exhibiting high selectivity and sensitivity.34 The focus of these approaches has been to stabilise iron oxide nanoparticles to overcome nanoparticle aggregation-induced activity loss. However, these substrate-based approaches do not provide any tuneability or on-demand catalytic response.
Herein, we report the development of polymer nanocomposite films with on-demand activatable catalytic nanozymatic response. We used in situ miniemulsion polymerisation to prepare colloidally stable poly(styrene-stat-n-butyl acrylate)/iron oxide-embedded silica nanoparticle (P(St-stat-nBA)/FeSiNP) nanocomposites with an innate ability to undergo film formation at ambient temperature virtually on any kind of substrate. The porous FeSiNPs were used as peroxide mimicking nanozyme sensing fillers. It is important to note that nanocomposite films with tuneable and activatable nanozymatic activity have to the best of our knowledge never been explored previously.
The developed P(St-stat-nBA)/FeSiNP nanocomposite films have several advantages: (i) conjugation in silica nanoparticles limits the extent of iron oxide nanoparticle agglomeration; (ii) the hydrophilic nature of the silica shell enables their potential use in miniemulsion polymerisation as a surfactant stabilising polymer particle; (iii) the nanocomposite films can be stretched to different lengths exposing fresh FeSiNPs to tailor and enhance catalytic response on-demand. The present nanocomposite film strategy makes this approach highly versatile, thus challenging the status quo in functional composites for sensing and industrial applications, providing the channel to develop long-lasting sensors with tuneable responses.
Materials and methods
Materials
Iron(III) chloride hexahydrate (FeCl3·6H2O, 97%), iron(II) chloride tetrahydrate (FeCl2·4H2O, 98%), and anhydrous sodium hydroxide pellets (NaOH, 98%) were used in the synthesis of iron oxide nanoparticles and were purchased from Sigma-Aldrich (Australia). Hexadecyltrimethylammonium bromide (CTAB, 98%, powder), cetyltrimethylammonium chloride (CTAC, 25 wt% in H2O), cyclohexane, triethanolamine (TEA), tetraethyl orthosilicate (TEOS), magnesium powder (99%), and hydrochloric acid (HCl, 37%) were purchased from Sigma-Aldrich (Australia) and were used in the synthesis of iron oxide-embedded silica nanoparticles. Acetic acid, sodium acetate (NaCH3COO, ≥99%), hydrogen peroxide (H2O2, 30% w/w in H2O), 3,3′,5,5′-tetramethylbenzidine (TMB, ≥98%), and L-ascorbic acid were purchased from Sigma-Aldrich (Australia). Styrene (St, Sigma-Aldrich, ≥99%) and n-butyl acrylate (nBA, Sigma-Aldrich, ≥99%) were purified through a filtration column filled with activated basic aluminium oxide powder (Sigma-Aldrich, Brockmann I) to remove the inhibitor. Azobisisobutyronitrile (AIBN, Sigma-Aldrich) was recrystallised in water from acetone. Hexadecane (HD, Sigma-Aldrich, 99%) and sodium dodecyl sulfate (SDS, Sigma-Aldrich, ≥99%) were used as received. Dimethyl sulfoxide (DMSO) and glycine were purchased from Chem-Supply (Australia). Sodium acetate buffer (0.2 M) was prepared by dissolving sodium acetate in Milli-Q water and adjusting the pH with acetic acid and NaOH. The water used in all experiments was Milli-Q (18.2 MΩ.cm at 25 °C) water.Synthesis of FeSiNPs
Iron oxide (γ-Fe2O3) nanoparticles were synthesised via co-precipitation according to the method reported previously.35 Iron(III) chloride hexahydrate (2.703 g) and iron(II) chloride tetrahydrate (0.994 g) were combined in a 150 mL beaker, maintaining a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ferric to ferrous ratio. NaOH (1.6 g) dissolved in 40 mL of deionised water was introduced into the iron mixture under stirring at 400 rpm for 1 min. The solution was heated to 50 °C and stirred for 30 min. Upon observing an inadequate pH level, an additional 0.4 g of NaOH was added in 10 mL of water, and the solution was stirred at the same speed for another 30 min at the same temperature. The nanoparticles formed were magnetically separated and washed thrice with Milli-Q water, followed by centrifugation (8000 rpm). The resultant paste containing the nanoparticles was oven-dried at 100 °C for 12 h, yielding 1.211 g of dry product.
:1 ferric to ferrous ratio. NaOH (1.6 g) dissolved in 40 mL of deionised water was introduced into the iron mixture under stirring at 400 rpm for 1 min. The solution was heated to 50 °C and stirred for 30 min. Upon observing an inadequate pH level, an additional 0.4 g of NaOH was added in 10 mL of water, and the solution was stirred at the same speed for another 30 min at the same temperature. The nanoparticles formed were magnetically separated and washed thrice with Milli-Q water, followed by centrifugation (8000 rpm). The resultant paste containing the nanoparticles was oven-dried at 100 °C for 12 h, yielding 1.211 g of dry product.
The synthesis of FeSiNPs was conducted based on the method described previously (Fig. S1, ESI†).36 The procedure commenced with the ultrasonication of iron oxide nanoparticles in 3 mL of deionised water until achieving a uniform dispersion. The dispersed nanoparticles were then transferred to a 150 mL conical flask containing 8 mL of CTAC, 360 mg of TEA, and 72 mL of deionised water. The mixture was stirred at 400 rpm for 1 h, maintaining a temperature of 60 °C. Subsequently, 32 mL of cyclohexane and 8 mL of TEOS were added, and the solution was aged for 14 h under the same conditions. The sample was then washed with acetone and centrifuged at 10000 rpm until the supernatant became clear. The particles were air-dried in a fume cupboard for 72 h. Magnetic-responsive particles were separated using a strong magnet bar for further analysis.
Preparation of P(St-stat-nBA)/FeSiNP nanocomposite latexes using miniemulsion polymerisation
An aqueous dispersion of FeSiNPs was prepared by mixing FeSiNPs (concentrations of 5, 10, and 20 wt% relative to monomers) with 15 mL of water for 15 min, followed by ultrasonication (Branson digital sonifier) at 20% amplitude on ice for 5 min. The organic phase comprised St and nBA as monomers with a weight ratio of 1:1 (total of 7 wt% relative to water), HD as a co-stabiliser (5 wt% relative to monomers), AIBN as an initiator (0.25 M relative to HD and monomers), and SDS as a surfactant (1 wt% relative to the organic phase – monomer, HD and AIBN). The FeSiNP dispersion was subsequently mixed with the organic phase in a 20 mL glass bottle for 15 min on ice, followed by 10 min ultrasonication at 20% amplitude on ice to obtain monomer droplets decorated with FeSiNP. After that, the acquired miniemulsion underwent degassing for 20 min on ice. Finally, polymerisation was conducted at 70 °C for 24 h (Scheme 1).
 | ||
| Scheme 1 Schematic diagram showing the nanocomposite film preparation strategy, starting with polymer nanocomposite latex synthesis by miniemulsion polymerisation, followed by drop casting to prepare a film. | ||
Nanocomposite film preparation
To fabricate P(St-stat-nBA)/FeSiNP nanocomposite films, the as-synthesised nanocomposite latex was initially degassed in a vacuum chamber for 30 min to remove microbubbles. Subsequently, 10 mL of latex was drop-casted into a 25 × 25 mm silicone mould and then left to undergo film formation under ambient conditions (∼20 °C and 1 atm) for approximately 2 weeks to obtain robust standalone films (Scheme 1).Characterisation
:2 mL, followed by filtration using a syringe filter (13 mm-Ø, PTFE membrane, 0.45 μm, Adelab Scientific) to remove the FeSiNP.
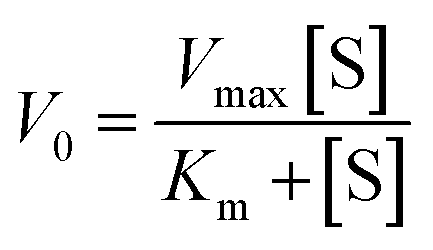 | (1) |
Results and discussion
Characterisation of FeSiNP
Fig. 1a shows the morphology of FeSiNPs as imaged using annular dark field scanning transmission electron microscopy (ADF-STEM). We observed a dendritic porous silica nanoparticle structure embedded with iron oxide nanoparticles. The iron oxide nanoparticles used for the FeSiNPs formulation were prepared using a precipitation process, which resulted in iron oxide nanoparticles of approximately 100 nm size as evident in the DLS and STEM data (Table S1 and Fig. S2, ESI†). The average size of the FeSiNPs was approximately 100 nm in diameter. The oxidation states of iron and silicon as well as the distribution of iron oxide particles in FeSiNP nanoparticle were further confirmed using EELS analysis (Fig. 1b) and STEM-EELS mapping (Fig. 1c). EELS spectra of Si L2,3 and Fe L2,3 edges of FeSiNP revealed characteristic spectral features corresponding to silica and iron oxide. The presence and morphology of iron oxide particles embedded in the silica of FeSiNPs were further confirmed by STEM-EELS maps.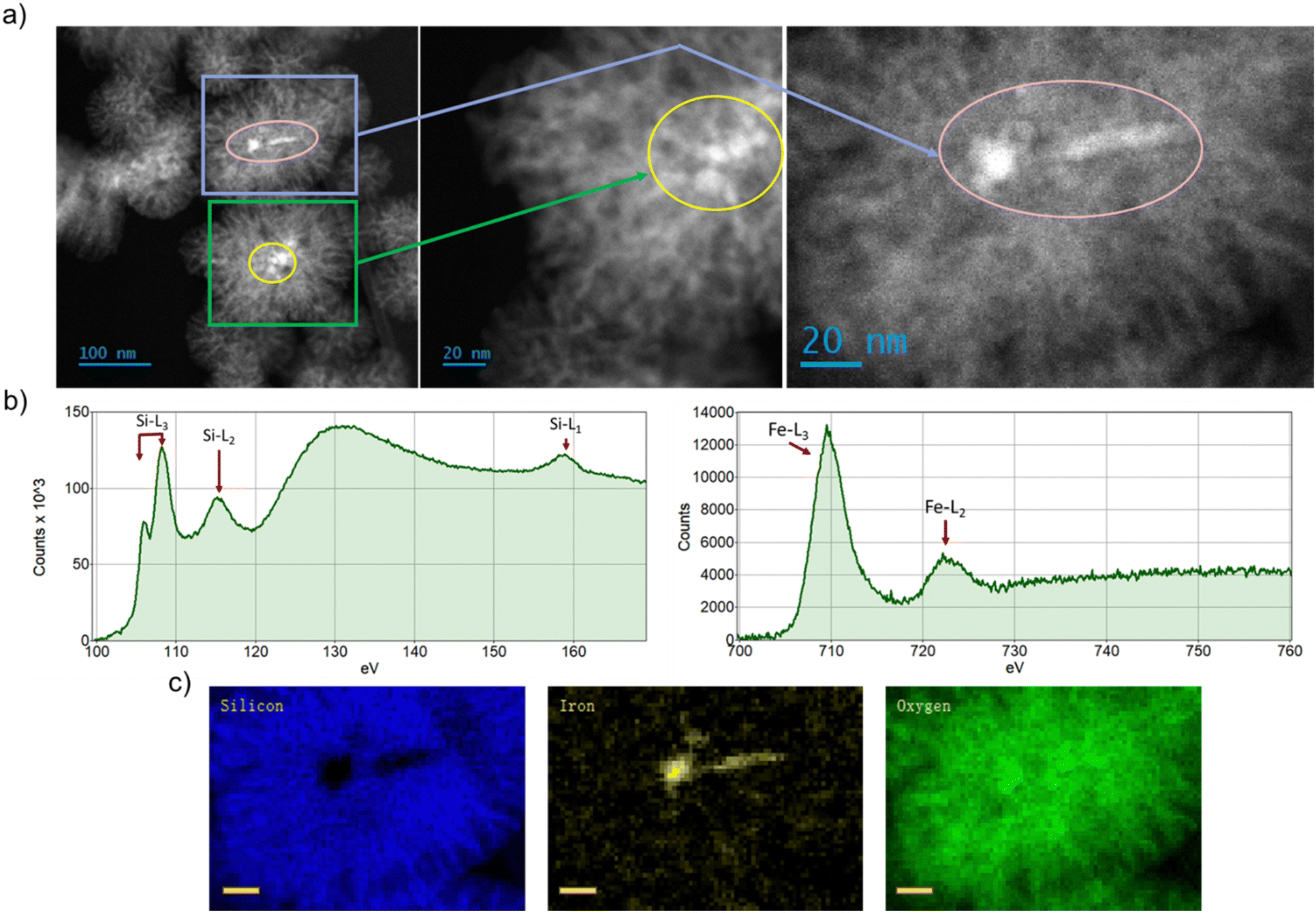 | ||
| Fig. 1 FeSiNP characterisation – (a) ADF-STEM images showing iron oxide in the central region and porous silicon dendritic structure outside, (b) Si L- and Fe L-edges EELS spectra confirming the oxidation states of Si and Fe as silica and iron oxide, and (c) STEM-EELS maps of silicon, iron and oxygen showing the localisation of different elements in the core and shell of synthesised FeSiNPs (scale bar = 20 nm). | ||
Characterisation of polymer latex
Colloidally stable poly(styrene-stat-n-butyl acrylate) (P(St-stat-nBA))/FeSiNP nanocomposite latexes were synthesised using miniemulsion polymerisation. Miniemulsion polymerisation is a one-pot synthesis strategy to prepare polymer/filler nanocomposite colloids. In miniemulsion, polymerisation proceeds by droplet nucleation within submicron-sized monomer droplets, produced using high energy mixing, ideally resulting in one-to-one conversion of monomer droplets into the polymer particles.37 Miniemulsion polymerisation typically forms filler-decorated polymer particles potentially at the interface in the case of nanocomposites when using fillers such as graphene oxide.38–43In this work, we employed a statistical copolymer of styrene (St) and n-butyl acrylate (nBA) due to the ability of the resulting polymer to undergo film formation at ambient temperature as previously reported by us and others.44–49 The weight ratio of St and nBA was maintained at 1:1 to obtain a theoretical glass transition temperature of ∼3 °C (based on the Fox equation).50 The polymerisation was conducted at 70 °C for 24 h using AIBN as an initiator, FeSiNP (5, 10, and 20 wt%; relative to monomers) and 1 wt% SDS (relative to organic phase) for 24 h. We obtained highly colloidally stable latexes at all filler loadings with monomer conversions of >85% (Fig. 2a and Table S2, ESI†). The polymer molecular weights were in the typical range for such nanocomposites.51,52 GPC analysis revealed a unimodal distribution with the Mn of ∼100 to 110 kg mol−1 (Fig. 2b, c and Table S3, ESI†). Polymer particle size determined using DLS was also in the typical range (80–150 nm) reported for polymer nanocomposites prepared using miniemulsion polymerisation (Table S2, ESI†).47,53
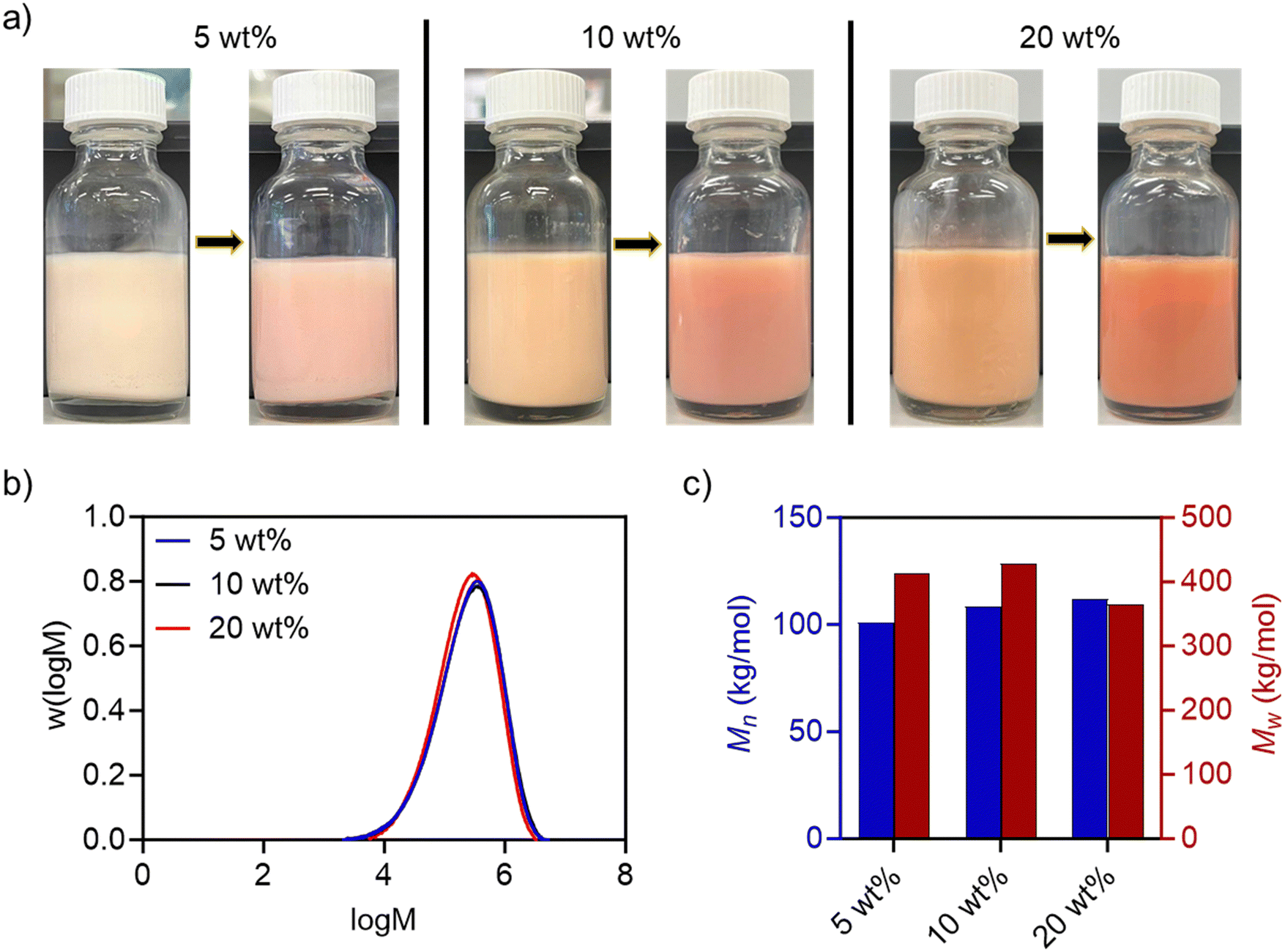 | ||
| Fig. 2 Characterisation of P(St-stat-nBA)/FeSiNP polymer latex- (a) conversion from monomers (left bottles) to polymers (right bottles) with three different FeSiNP concentrations. The size of the glass bottles is 20 mL. A clear increase in the colour for both monomers and resulting polymer dispersions was observed with increasing FeSiNP loading. (b) Molecular weight distribution of P(St-stat-nBA) at different FeSiNP loadings, and (c) comparison of number average (Mn) and weight average (Mw) molecular weights of P(St-stat-nBA). | ||
Next, the obtained P(St-stat-nBA)/FeSiNP nanocomposite latexes comprising different concentrations of FeSiNPs were drop-cast in silicone moulds to prepare nanocomposite films at ambient temperature (Scheme 1). SEM imaging was conducted to characterise the surface profile of the obtained films under both unstretched and stretched conditions to visualise any change to the surface profile as a result of stretching. In the case of unstretched films, SEM images revealed rough surfaces in all films regardless of FeSiNP loading (Fig. 3). In all films, we observed features akin to crevices on the film surface exposing embedded FeSiNPs. Higher magnification imaging revealed the presence of more aggregated spherical nanoparticles within these crevices, indicating potential interference during film formation and leading to their incomplete coverage, which could explain the emergence of crevices. These crevices do not result from film cracking, instead we hypothesise that during the film formation, decorated polymer particles start to coalesce, enhancing inter-FeSiNP interactions and resulting in FeSiNP aggregation. Such FeSiNP aggregation will in turn interfere with polymer particle coalescence to some extent, resulting in incomplete coverage on the film surface. These crevice-like features increased in films with increasing FeSiNP loading as anticipated (Fig. S3, ESI†). When the films were stretched, crevices became more evident leading to the exposure of FeSiNPs embedded within the films under all conditions. As shown in Fig. 3, FeSiNPs became clearly visible on all films, with an increasing number of FeSiNPs becoming exposed with increasing filler loading. SEM-EDS analysis of unstretched and stretched P(St-stat-nBA)/FeSiNP films confirmed the presence of silicon and iron from the FeSiNPs on the film surface (Fig. S4 and S5, ESI†). EDS mapping indicated potentially higher concentrations of silicon inside these crevices (Fig. S6, ESI†).
 | ||
| Fig. 3 Characterisation of P(St-stat-nBA)/FeSiNP films – SEM images in the unstretched condition showing overall surface features with the presence of FeSiNPs and a view of FeSiNPs through the surface openings of the films in the stretched condition. | ||
The effect of FeSiNP on mechanical properties in standalone nanocomposite films was studied using uniaxial tensile testing. The tensile strength increased with increasing FeSiNP concentration from 5 wt% (0.78 ± 0.08 MPa) to 10 wt% (1.71 ± 0.21 MPa) (Table S4, ESI†). However, a further increase to 20 wt% caused no further increase in tensile strength (1.15 ± 0.09 MPa). On the contrary, elongation at break decreased with increasing FeSiNP concentration from 5 wt% (∼1448%) to 10 wt% (847 ± 27%). However, no further change in tensile strength was observed when FeSiNP concentration was increased to 20 wt% (860 ± 46%) (Table S4, ESI†). We postulate that increasing the filler concentration from 10 wt% to 20 wt% may have induced a greater extent of FeSiNPs agglomeration during the formation of the film. The evidence of a greater extent of FeSiNP agglomeration at 20 wt% loading compared to 10 wt% is supported by the SEM data, where the presence of nanoparticle agglomeration became more clearly visible within the crevices under stretched film conditions. Such agglomeration compromises the ability of nanocomposite films to dissipate stress with increasing strain, thus compromising both tensile strength and elongation at break.
Catalytic nanozyme activity
The peroxidase-mimicking catalytic nanozyme activity of the nanocomposite films was subsequently investigated using TMB as a substrate. TMB is commonly used in ELISA assays to determine the catalytic activity of natural enzymes. Typically, natural peroxidase enzymes in the presence of H2O2 generate hydroxyl radicals (˙OH), which react with TMB to produce a blue coloured oxidised product (TMBox) that absorbs light at 652 nm and can be utilised for both qualitative and quantitative analysis (Fig. 4a).54 Before testing the catalytic activity, we optimised reaction conditions for the as-synthesised FeSiNPs. First, the effects of buffer pH and reaction temperature were investigated by changing the pH from 2.5 to 7.5 and the reaction temperature from room temperature (25 °C) to 45 °C. The catalytic activity increased initially, reaching the maximum activity at pH 3.5 (Fig. 4b). However, further increase in pH caused a continuous and significant reduction in the catalytic activity. The catalytic activity decreased systematically with an increase in the reaction temperature, exhibiting the best performance at room temperature (25 °C) and the lowest performance at 45 °C (Fig. 4c). These conditions were selected and maintained in all subsequent experiments unless stated otherwise. Next, the peroxidase-mimicking activity of the as-synthesised neat FeSiNPs was investigated. As anticipated, a linear correlation between FeSiNPs concentration and catalytic performance was observed with a regression coefficient (R2) of 0.973 (Fig. 4d). To determine the mechanism of catalytic activity of FeSiNPs, ascorbic acid was used as an oxygen free radical (˙OH and O2˙−) scavenger.55 The catalytic reactions were conducted similarly as described above with the incorporation of ascorbic acid, and the resulting absorbance of the product was quantified to determine the change in the catalytic activity. The absorbance decreased significantly with the incorporation of ascorbic acid (Fig. 4e). These results indicate that ascorbic acid reacted with ˙OH, inhibiting the oxidation of TMB, and thus causing a reduction in the formation of a blue colour product, confirming that the mechanism of action is indeed mediated by the ˙OH radicals. Based on the results, the catalytic mechanism is proposed to follow a previously published report as described in Fig. S7 and eqn (S1)–(S4) (ESI†).56 In summary, iron (Fe2+) in FeSiNP catalyses the reduction of H2O2 to generate hydroxyl radicals (˙OH) and oxidised Fe3+ in the first step. This reaction is the rate limiting step. Next, the generated ˙OH radicals react with TMB to form a blue coloured oxidised product (TMBox). In the third step, ˙OH radicals react with H2O2 to form HO2˙ radicals, which then reduce Fe3+ to generate the catalyst Fe2+ and a molecule of oxygen. | ||
| Fig. 4 Optimisation of reaction conditions using neat FeSiNPs – (a) schematic diagram showing the FeSiNP structure and nanozymatic catalytic reaction, (b) effects of buffer pH, (c) effects of temperature, (d) standard curve of FeSiNP, and (e) mechanism of action using ascorbic acid (AA) as a radical scavenger, (f) rate of reaction with changing H2O2 concentration, (g) rate of reaction with changing TMB concentration, (h) Lineweaver–Burk plot obtained by altering the concentration of H2O2 (100–1000 mM) at a fixed amount of TMB (1000 μM), and (i) Lineweaver–Burk plot obtained by varying the concentration of TMB (100–1000 μM) at a fixed amount of H2O2 (700 mM). | ||
A steady-state kinetics study was subsequently carried out to investigate the peroxidase-like catalytic activity of neat FeSiNPs by varying either the concentration of H2O2 or TMB while keeping the other component constant. The kinetic parameters (Km and Vmax) were calculated using the molar absorption coefficient (ε) of 39000 M−1 cm−1 (at 652 nm) for TMBox.57 Since natural peroxidase enzyme activity usually follows the Michaelis–Menten eqn (1), the data obtained in this study were fitted to a typical Michaelis–Menten curve (Fig. 4f and g) within the relevant concentration range using a nonlinear least-squares fitting method to determine the catalytic parameters (Km and Vmax). Km is an indication of the affinity of an enzyme for its substrate; a lower Km value suggests a higher affinity between the two. Vmax is the maximum rate of conversion into the product. As the concentrations of H2O2 and TMB increased, the absorbance values at 652 nm also increased (represented as velocity, Fig. 4f and g, respectively). At higher concentrations of TMB and H2O2, there was no inhibition in the catalysis process. Additionally, Lineweaver–Burk double-reciprocal plots were generated (Fig. 4h and i) to estimate the affinity of the nanozyme (FeSiNPs) to the substrate (TMB). The Km and Vmax values were calculated to be 0.060 mM and 0.672 × 10−8 M s−1 for H2O2, and 7.143 mM and 1.075 × 10−8 M s−1 for TMB, respectively. Based on the obtained results, it can be deduced that FeSiNPs have a higher affinity towards H2O2 (lower Km) than TMB. In comparison with the other previously reported nanozymes, the Km value for H2O2 obtained for FeSiNPs in this work was amongst the lowest observed, whereas the calculated Vmax value was comparable to the other nanozymes (Table S5, ESI†). A lower Km value indicates a higher affinity of a nanozyme towards its substrate (H2O2 in this case). Therefore, in comparison to the other nanozyme listed in Table S5 (ESI†), FeSiNPs used in this work exhibited a higher affinity towards the H2O2 substrate. The catalytic efficiency of FeSiNPs, taking only the Fe component as the catalytic species into consideration, was calculated to be 6.35 × 10−4 s−1 for H2O2 and 1.02 × 10−3 s−1 for TMB.
Next, we systematically investigated the peroxidase-mimicking activities of the P(St-stat-nBA)/FeSiNP nanocomposite films with different FeSiNP loadings. Films were prepared with similar thickness to eliminate the potential impact of thickness on the catalytic performance (Fig. 5a). First, we investigated unstretched films to obtain their ‘baseline’ performance and determine the impact of different FeSiNP loadings. As shown in Fig. 5b–d, there was a direct correlation between FeSiNP loading and catalytic performance (absorbance values). As expected, increasing the amount of FeSiNPs led to a dose-dependent increase in the catalytic performance, which can potentially be explained by the presence of FeSiNPs in the crevices on the film surface as observed in the SEM analysis (Fig. 3). The control neat P(St-stat-nBA) without FeSiNPs under the same experimental conditions exhibited absorbance values comparable to the buffer control (which was the baseline value ≡ 0), indicating that FeSiNPs present in the composite films were indeed functioning as nanozymes (Fig. S8, ESI†). Therefore, it can be deduced that P(St-stat-nBA)/FeSiNP films have potential to function as a nanozyme biosensor by making FeSiNPs available for catalysis. Considering the high stretchability of these P(St-stat-nBA)/FeSiNP films, we subsequently explored the possibility of tuning the catalytic performance by simply stretching them to different extents (50% and 100%). With increased stretching from 0 to 100%, a gradual increase in absorbance of the blue product (TMBox; a quantitative measure of catalytic performance) was observed for P(St-stat-nBA)/FeSiNP (5 wt%) and P(St-stat-nBA)/FeSiNP (20 wt%) films, whereas a sharp and more linear increase in absorbance was obtained for P(St-stat-nBA)/FeSiNP (10 wt%).
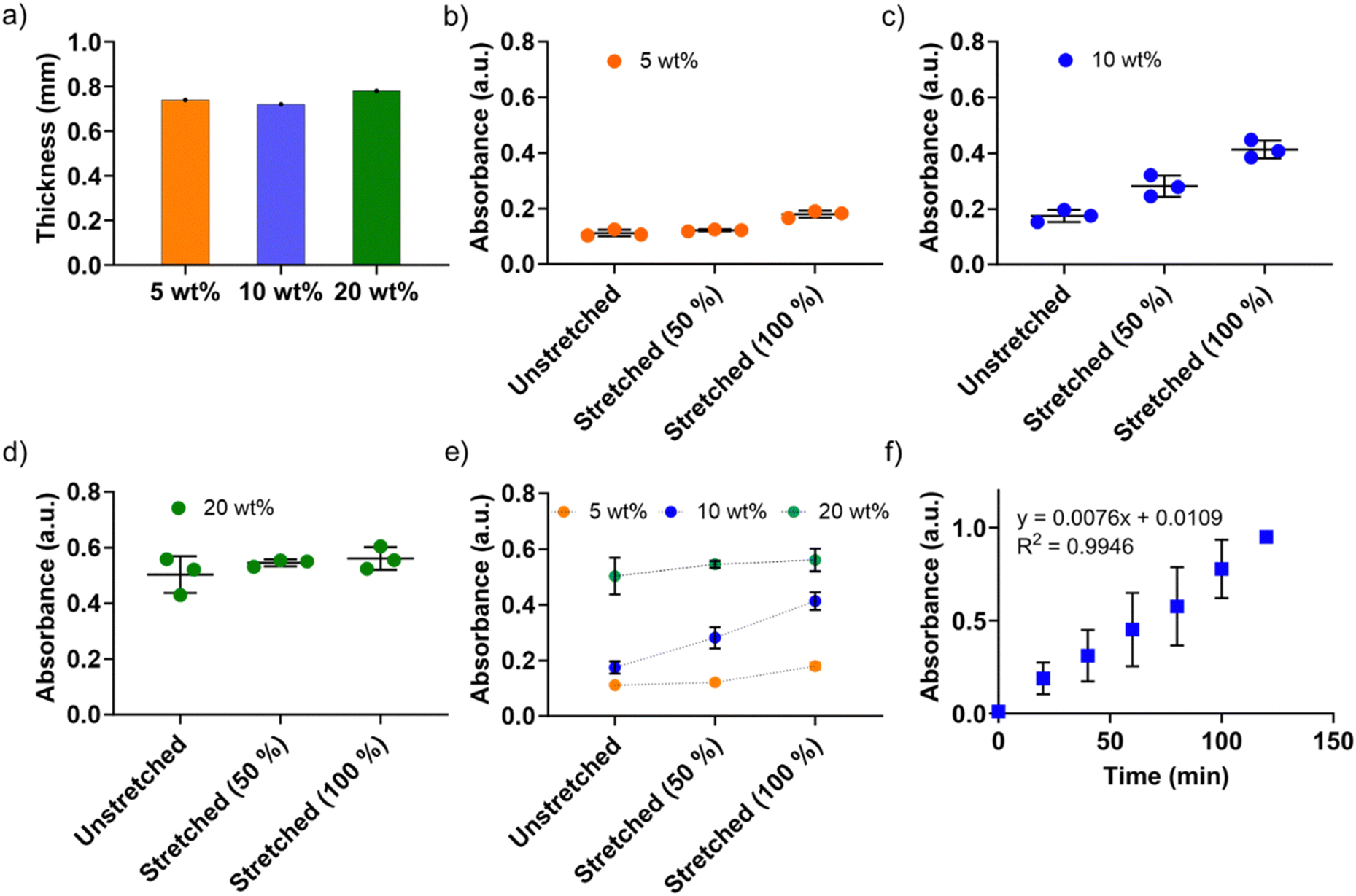 | ||
| Fig. 5 (a) Thickness of P(St-stat-nBA)/FeSiNP films, (b)–(d) peroxidase enzyme-mimicking nanozyme activity of P(St-stat-nBA)/FeSiNP (5 wt%, 10 wt%, and 20 wt%, respectively) films under unstretched and stretched conditions at different stretching lengths, (e) combined peroxidase enzyme-mimicking nanozyme activity, (f) catalytic activity of P(St-stat-nBA)/10 wt% FeSiNP films over a 2 h reaction period. | ||
In the case of 5 wt% FeSiNP, almost all FeSiNPs present on the film surface (and crevices) participate in catalysis under unstretched conditions, and therefore, stretching induced only a marginal increase in the catalytic activity (Fig. 5b). It is hypothesised that 5 wt% FeSiNPs loading is quite low in nanocomposite films, and despite stretching, a very small amount of new surface or embedded FeSiNPs becomes available for catalysis. If this is the case, one would expect the absorbance values to improve with increasing amounts of FeSiNPs loading, which is what is observed under all conditions (0, 50 and 100% stretching). In the case of 20 wt%, the amount of FeSiNPs is too high, resulting in a stronger catalytic response compared to lower NP loadings but also causing greater NP agglomeration throughout the film. This agglomeration may limit the number of previously unexposed NPs embedded within the film from becoming available under stretching conditions, resulting in only a marginal increase in catalytic performance (Fig. 5d). In the case of 10 wt% FeSiNP-loaded films, we believe that the NP concentration is optimal in these films such that despite some level of NP agglomeration, previously unexposed NPs continuously become available for catalysis when stretched from 0, 50 to 100% (Fig. 5c). Overall, these results highlight that the developed nanocomposite films provide a platform with unprecedented tuneability in catalytic performance by simple stretching (Fig. 5e).
To investigate the durability in terms of long-term performance, we selected the 10 wt% FeSiNP-loaded nanocomposite film under a 100% stretched condition and performed the catalytic response experiment for 2 h. As shown in (Fig. 5f), a linear catalytic response was observed (R2 = 0.994) with time. A plausible explanation for this observation could be that the FeSiNPs that are present deeper inside the films increasingly get exposed to the reagents with time to show the catalytic activity. If there are enough FeSiNPs available on the film surface to continually drive catalysis with time, one would expect the absorbance to plateau and not continue to increase with time. These data highlight that the developed films can function as a long-lasting continuous monitoring platform.
On-demand catalytic response
To further validate stretching-mediated tuneability in the catalytic performance, we stretched P(St-stat-nBA)/FeSiNP (10 wt%) films at different time points during the catalytic reaction. We incubated films in the reaction buffer containing H2O2 and TMB for 18 h and recorded the absorbance value of ∼0.2, which was considered as a starting point before stretching the films. The unstretched film exhibited a marginal reduction in the catalytic performance with time (Fig. 6). These values at individual time points were considered as a ‘baseline’ to compare the change in catalytic response as a result of film stretching. Next, we stretched films after 0, 20, and 80 min of incubation and recorded the change in absorbance values compared to the respective baseline values. | ||
| Fig. 6 On-demand activation of peroxidase enzyme-mimicking activity by stretching the P(St-stat-nBA)/FeSiNP (10 wt%) films at different time points: (a) 0 min, (b) 20 min, (c) 80 min; (d) plot showing combined stretching effects at different time points. | ||
All films exhibited a significant increase in the absorbance values when stretched, and the increased absorbance values were similar in all stretched films regardless of whether the film was stretched at 0, 20, and 80 min after incubation (a marginally lower response was observed for 80 min stretched films). These data highlight the tuneability of the developed nanozyme-loaded films, i.e. activity can be enhanced on-demand or can be refreshed during its performance life. Taken together, we propose the developed film to be a highly tuneable platform with prolonged functional activity where the catalytic functionality can be made active by simply stretching the nanocomposite film.
Conclusions
The aim of the present work has been to fabricate nanozyme-loaded tunable polymer nanocomposite films for on-demand, responsive, and sustained peroxidase-mimicking activity. P(St-stat-nBA) films containing iron oxide-loaded porous silica nanozymes (FeSiNP) were prepared using miniemulsion polymerisation where FeSiNP functions as a peroxidase-mimic. The nanozyme-loaded films exhibited nanozyme (FeSiNP) concentration-dependent catalytic activity with higher performance observed with increasing nanoparticle loading. When stretched, the catalytic performance increased with the extent of stretching from 0 to 100%. Furthermore, the films exhibited a sustained and linear response with time, confirming their ability to facilitate continuous detection. On-demand switching at different time points revealed successful activation by a sharp increase in the catalytic performance simply by stretching the 10 wt% FeSiNPs-loaded films. Overall, the developed nanocomposite films demonstrated responsive and tuneable peroxidase-mimicking activity. This study has for the first time demonstrated tuneable on-demand activatable films for enzyme-mimicking activity, thus setting up a platform for the future development of smart sensors for various diagnostic applications.Data availability
Data for this article are available in the UNSW public repository at https://doi.org/10.26190/unsworks/30229.Conflicts of interest
No conflict to declare.Acknowledgements
V. A. acknowledges the National Health and Medical Research Council (NHMRC), Australia, for an Early Career Fellowship (GNT1139060) and UNSW Safety Net Fellowship. The authors acknowledge the facilities and the scientific and technical assistance of Microscopy Australia at the Electron Microscope Unit (EMU), and the Solid State & Elemental Analysis Unit within the Mark Wainwright Analytical Centre (MWAC) at UNSW Sydney.References
- V.-D. Doan, V.-C. Nguyen, A.-T. Nguyen and T.-D. Nguyen, Spectrochim. Acta, Part A, 2022, 268, 120709 CrossRef CAS PubMed.
- S. Naderizadeh, A. Shakeri, H. Mahdavi, N. Nikfarjam and N. Taheri Qazvini, Starch/Staerke, 2019, 71, 1800027 CrossRef.
- M. Ma, H. Wang, T. Zhang, X. Wang, Z. Xu, R. Zhang, X. Ma and F. Shi, ACS Omega, 2023, 8, 35779–35790 CrossRef CAS PubMed.
- S. C. Saccà, S. Vernazza, E. L. Iorio, S. Tirendi, A. M. Bassi, S. Gandolfi and A. Izzotti, Prog. Brain Res., 2020, 256, 151–188 Search PubMed.
- D. S. Barreiro, R. N. Oliveira and S. R. Pauleta, Coord. Chem. Rev., 2023, 485, 215114 CrossRef CAS.
- T. Wang, X. Bi, L. Wang, M. Liu, W. Y. William, Z. Zhu and N. Sui, J. Colloid Interface Sci., 2022, 607, 470–478 CrossRef CAS.
- J. E. Giaretta, H. Duan, S. Farajikhah, F. Oveissi, F. Dehghani and S. Naficy, Sens. Actuators, B, 2022, 371, 132483 CrossRef CAS.
- J. E. Giaretta, H. Duan, F. Oveissi, S. Farajikhah, F. Dehghani and S. Naficy, ACS Appl. Mater. Interfaces, 2022, 14, 20491–20505 CrossRef CAS.
- Q. Rasheed, H. Ajab, M. Farooq, S. A. Shahzad and A. Yaqub, Appl. Nanosci., 2022, 12, 2841–2855 CrossRef CAS.
- Y. Huang, E. Su, J. Ren and X. Qu, Nano Today, 2021, 38, 101205 CrossRef CAS.
- A. Deshwal, K. Saxena, G. Sharma, F. A. Sheikh, C. S. Seth and R. M. Tripathi, Int. J. Biol. Macromol., 2023, 128272 Search PubMed.
- Y. Wang, L. Xu and W. Xie, Microchem. J., 2019, 147, 75–82 CrossRef CAS.
- Y. Liu, J. Yan, Y. Huang, Z. Sun, H. Zhang, L. Fu, X. Li and Y. Jin, Front. Bioeng. Biotechnol., 2022, 9, 790849 CrossRef.
- Q. Wang, H. Wei, Z. Zhang, E. Wang and S. Dong, TrAC, Trends Anal. Chem., 2018, 105, 218–224 CrossRef CAS.
- P. Wang, T. Wang, J. Hong, X. Yan and M. Liang, Front. Bioeng. Biotechnol., 2020, 8, 15 CrossRef PubMed.
- G. Saleem, X. Chen, R. Gu, M. Qasim, M. Usama and N. Rajput, Nanotechnol. Rev., 2022, 11, 2575–2583 CrossRef CAS.
- X. Liu, Y. Gao, R. Chandrawati and L. Hosta-Rigau, Nanoscale, 2019, 11, 21046–21060 RSC.
- M. K. Masud, J. Na, M. Younus, M. S. A. Hossain, Y. Bando, M. J. A. Shiddiky and Y. Yamauchi, Chem. Soc. Rev., 2019, 48, 5717–5751 RSC.
- X. Yu, Y. Wang, J. Zhang, J. Liu, A. Wang and L. Ding, Adv. Healthcare Mater., 2023, 2302023 Search PubMed.
- F. Attar, M. G. Shahpar, B. Rasti, M. Sharifi, A. A. Saboury, S. M. Rezayat and M. Falahati, J. Mol. Liq., 2019, 278, 130–144 CrossRef CAS.
- B. Das, J. L. Franco, N. Logan, P. Balasubramanian, M. I. Kim and C. Cao, Nanomicro Lett., 2021, 13, 1–51 Search PubMed.
- M. K. Masud, J. Kim, M. M. Billah, K. Wood, M. J. A. Shiddiky, N.-T. Nguyen, R. K. Parsapur, Y. V. Kaneti, A. A. Alshehri, Y. G. Alghamidi, K. A. Alzahrani, M. Adharvanachari, P. Selvam, M. S. A. Hossain and Y. Yamauchi, J. Mater. Chem. B, 2019, 7, 5412–5422 RSC.
- N. Alizadeh and A. Salimi, J. Nanobiotechnol., 2021, 19, 1–31 CrossRef.
- M. Liang and X. Yan, Acc. Chem. Res., 2019, 52, 2190–2200 CrossRef CAS PubMed.
- Y. Huang, J. Ren and X. Qu, Chem. Rev., 2019, 119, 4357–4412 CrossRef CAS PubMed.
- H. Dong, W. Du, J. Dong, R. Che, F. Kong, W. Cheng, M. Ma, N. Gu and Y. Zhang, Nat. Commun., 2022, 13, 5365 CrossRef CAS PubMed.
- P. Šálek, A. Golunova, J. Dvořáková, E. Pavlova, H. Macková and V. Proks, Mater. Lett., 2020, 269, 127610 CrossRef.
- R. Bhattacharjee, S. Tanaka, S. Moriam, M. K. Masud, J. Lin, S. M. Alshehri, T. Ahamad, R. R. Salunkhe, N.-T. Nguyen, Y. Yamauchi, M. S. A. Hossain and M. J. A. Shiddiky, J. Mater. Chem. B, 2018, 6, 4783–4791 RSC.
- K. Boriachek, M. K. Masud, C. Palma, H.-P. Phan, Y. Yamauchi, M. S. A. Hossain, N.-T. Nguyen, C. Salomon and M. J. A. Shiddiky, Anal. Chem., 2019, 91, 3827–3834 CrossRef CAS PubMed.
- S.-M. You, J.-S. Park, K. Luo, K.-B. Jeong, H. J. Adra and Y.-R. Kim, Carbohydr. Polym., 2021, 267, 118164 CrossRef CAS.
- I. Safarik, J. Prochazkova, M. A. Schroer, V. M. Garamus, P. Kopcansky, M. Timko, M. Rajnak, M. Karpets, O. I. Ivankov and M. V. Avdeev, ACS Appl. Mater. Interfaces, 2021, 13, 23627–23637 CrossRef CAS.
- S. A. Geleto, A. M. Ariti, B. T. Gutema, E. M. Abda, A. A. Abiye, S. M. Abay, M. L. Mekonnen and Y. A. Workie, ACS Omega, 2023, 8, 48764–48774 CrossRef CAS PubMed.
- X. Liu, T. Gao, H. Liu, Y. Fang and L. Wang, J. Exp. Nanosci., 2022, 17, 75–85 CrossRef CAS.
- R. Satvekar, S. Rohiwal, A. Tiwari, A. Raut, B. Tiwale and S. Pawar, Mater. Res. Express, 2015, 2, 015402 CrossRef CAS.
- B. H. Hui and M. N. Salimi, IOP Conf. Ser.: Mater. Sci. Eng., 2020, 743, 012036 CAS.
- J. Ndayishimiye, Y. Cao, T. Kumeria, M. A. T. Blaskovich, J. R. Falconer and A. Popat, J. Mater. Chem. B, 2021, 9, 7145–7166 RSC.
- J. M. Asua, Prog. Polym. Sci., 2002, 27, 1283–1346 CrossRef CAS.
- Y. Fadil, S. C. Thickett, V. Agarwal and P. B. Zetterlund, Prog. Polym. Sci., 2022, 125, 101476 CrossRef CAS.
- K. Edgehouse, M. Escamilla, L. Wang, R. Dent, K. Pachuta, L. Kendall, P. Wei, A. Sehirlioglu and E. Pentzer, J. Colloid Interface Sci., 2019, 541, 269–278 CrossRef CAS.
- S. Che Man, S. C. Thickett, M. R. Whittaker and P. B. Zetterlund, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 47–58 CrossRef CAS.
- L. Delafresnaye, P.-Y. Dugas, P.-E. Dufils, I. Chaduc, J. Vinas, M. Lansalot and E. Bourgeat-Lami, Polym. Chem., 2017, 8, 6217–6232 RSC.
- Y. Yin, J. Wang, T. Li, J. P. Hill, A. Rowan, Y. Sugahara and Y. Yamauchi, ACS Nano, 2021, 15, 13240–13248 CrossRef CAS PubMed.
- X. Qiao, P.-Y. Dugas, V. Prevot and E. Bourgeat-Lami, Polym. Chem., 2020, 11, 3195–3208 RSC.
- N. Maslekar, R. A. M. Noor, R. P. Kuchel, Y. Yao, P. B. Zetterlund and V. Agarwal, Nanoscale Adv., 2020, 2, 4702–4712 RSC.
- V. Agarwal, Y. Fadil, A. Wan, N. Maslekar, B. N. Tran, R. A. Mat Noor, S. Bhattacharyya, J. Biazik, S. Lim and P. B. Zetterlund, ACS Appl. Mater. Interfaces, 2021, 13, 18338–18347 CrossRef CAS.
- Y. Fadil, L. N. Dinh, M. O. Yap, R. P. Kuchel, Y. Yao, T. Omura, U. A. Aregueta-Robles, N. Song, S. Huang and F. Jasinski, ACS Appl. Mater. Interfaces, 2019, 11, 48450–48458 CrossRef CAS PubMed.
- L. N. Dinh, B. N. Tran, V. Agarwal and P. B. Zetterlund, ACS Appl. Polym. Mater., 2022, 4, 1867–1877 CrossRef CAS.
- S. W. Zhang, R. Liu, J. Q. Jiang and H. Y. Bai, Prog. Org. Coat., 2009, 65, 56–61 CrossRef CAS.
- H. T. Ip, L. Liu, L. Hong and T. Ngai, Colloids Surf., A, 2022, 654, 130104 CrossRef CAS.
- T. G. Fox Jr and P. J. Flory, J. Appl. Phys., 1950, 21, 581–591 CrossRef.
- M. G. Saborio, N. Maslekar, Y. Yao, P. B. Zetterlund and V. Agarwal, ACS Appl. Nano Mater., 2023, 6, 2413–2420 CrossRef CAS.
- B. N. Tran, S. Bhattacharyya, Y. Yao, V. Agarwal and P. B. Zetterlund, ACS Appl. Nano Mater., 2021, 4, 12461–12471 CrossRef CAS.
- Y. Luo, X. Wang, Y. Zhu, B.-G. Li and S. Zhu, Macromolecules, 2010, 43, 7472–7481 CrossRef CAS.
- Z. Wang, R. Zhang, X. Yan and K. Fan, Mater. Today, 2020, 41, 81–119 CrossRef CAS.
- T. Lin, L. Zhong, L. Guo, F. Fu and G. Chen, Nanoscale, 2014, 6, 11856–11862 RSC.
- B. Yuan, H.-L. Chou and Y.-K. Peng, ACS Appl. Mater. Interfaces, 2022, 14, 22728–22736 CrossRef CAS.
- E. Karaseva, Y. P. Losev and D. Metelitsa, Russ. J. Bioorg. Chem., 2002, 28, 128–135 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb00755g |
| This journal is © The Royal Society of Chemistry 2024 |