
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesigning rock salt phase enriched surface in Mn-based partially disordered spinel cathode materials for mitigating degradation in Li-ion batteries†
Hyoi
Jo‡
a,
Changju
Lee‡
a,
HyeongJun
Nam
a,
Jee Ho
Ha
a,
Nyung Joo
Kong
a,
Kyojin
Ku
 b,
Seok Ju
Kang
a and
Sung-Kyun
Jung
*a
b,
Seok Ju
Kang
a and
Sung-Kyun
Jung
*a
aDepartment of Energy Engineering, School of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea
bDepartment of Materials Science and Engineering, Hanbat National University, N8-316, 125 Dongseodaero, Yuseong-gu, 34158, Daejoen, Republic of Korea
First published on 7th June 2024
Abstract
Mn-based materials show potential as next-generation candidates for lithium-ion battery (LIB) cathode materials owing to their cost-effectiveness, high energy density, and high power density. However, during repetitive charging/discharging processes, these materials undergo cation migration, structural evolution, and phase transition, resulting in sluggish kinetics, substantial voltage decay, and capacity degradation. Herein, we present a Mn-based partially disordered spinel cathode material decorated with a cation-disordered rock salt (DRX) phase-enriched surface to mitigate this voltage and capacity degradation. We demonstrate that a DRX-rich surface layer successfully suppresses the degradation associated with the unfavorable phase transition from the cubic spinel to tetragonal spinel phase. Instead, by harnessing a gradual phase transition from the rock salt to cubic spinel phase concentrated on the surface region, the capacity and voltage increase, delaying the degradation and leading to improved capacity retention. Our findings suggest a strategic approach to exploit Mn-based cathode materials for developing LIBs with superior cyclability and further highlight the potential of controlling the spatial distribution of each phase to enhance the battery performance in multi-phasic cathode materials.
 Sung-Kyun Jung | Sung-Kyun Jung is an assistant professor of the School of Energy and Chemical Engineering at Ulsan National Institute of Science and Technology (UNIST) in the Republic of Korea. He completed his PhD in 2018 at the Department of Materials Science and Engineering of Seoul National University (SNU), where he received his BSc and MS degrees. Since 2018, he was a staff researcher at Samsung Advanced Institute of Technology (SAIT) until May 2021, before moving to UNIST. His research laboratory focuses on materials and interface design for high energy density rechargeable batteries based on advanced characterization techniques using in situ and synchrotron X-ray analysis. |
Introduction
Despite the surge in demand for lithium-ion batteries (LIBs) in the transportation and industrial sectors to address climate change and global warming concerns, expansion of their supply has faced challenges due to the high cost compared to consumer expectations.1,2 As the cathode materials are the core components governing the energy density and cost of current LIBs,3–7 there is a pressing need to develop cost-effective cathode materials that do not rely on the expensive transition metals such as Co and Ni commonly used in commercial cathodes. To support the sustainable expansion of LIBs, it is thus essential to explore alternative cathode materials that are based on earth-abundant transition metals with high energy density.8–11Among the various potential cathode materials, Mn-based compounds, being earth-abundant transition metals, have garnered attention as promising candidates for next-generation battery cathodes owing to their high energy density and cost-effectiveness. Mn-rich cation-disordered rock salt (DRX) materials are regarded as a frontrunner because of their high capacity enabled by the ability to harness multi-cation and anion redox despite the sluggish lithium-diffusion kinetics.12,13 The introduction of partial cation ordering such as a spinel phase into DRX is known to improve the kinetics by providing easier access to a lithium percolating network with a low migration energy barrier via a non-transition-metal face-shared network (0-TM network).3,14,15 Therefore, a strategy to design partial spinel rock-salt cathode materials (Sp-DRXs), also known as partially disordered spinels, has been applied to improve the sluggish kinetics; however, the low capacity retention of Mn-based Sp-DRXs remained within 30 cycles, which is still regarded as an unresolved issue.16,17
Mn-based cathode materials universally face challenges associated with structural evolution and phase transitions induced by the cation migration during cycling, leading to poor cycling stability and sluggish kinetics.18–23 Mn-based materials in layered and orthorhombic phases are well known to transform into spinel and rock-salt phases during cycling.24–26 For ordered spinel LiMn2O4, a detrimental two-phase reaction occurs, resulting in the transition to a tetragonal spinel phase near 3 V.27–29 As this structural reorganization can result in degradation of the capacity retention and lithium-diffusion kinetics, strategies have been proposed to suppress it. Substituting oxygen anions (O2−) with fluorine anions (F−) shifts the charge compensation towards Mn-redox contributions rather than oxygen redox,30–32 inhibiting phase changes stemming from irreversible oxygen evolution. Additionally, doping with immobile transition metals (TMs) has been proposed to inhibit the migration of Mn ions.33 In the spinel structure of LiMn2O4, a strategy has been developed to enhance the accessibility to solid-solution reactions over two-phase reactions by forming 16c/16d disorder.15,17,34 However, despite these efforts, complete suppression of these phase transitions has not yet been achieved. Given the recent report that Sp-DRX exists not only as a single phase but also as a nanocomposite with a nanodomain of rock salt and spinel phases,35 different phase transitions in each phase could complexly occur within the Sp-DRX materials during electrochemical cycling. Therefore, if these phase transitions can be strategically utilized, they could provide a path toward Mn-based cathode materials with long cycle life.
Recently, it has been reported that the increase of the cycle stability can be achieved depending on structural evolution behavior on the surface during charge/discharge cycles.36 Li-excess DRX cathodes such as Li1.2Ni0.333Ti0.333Mo0.133O2 (LNTMO) and Li1.2Mn0.6Nb0.2O2 (LMNO) show different phase evolution on particle surfaces after cycling. Both materials experience cation densification on their surfaces due to irreversible oxygen evolution caused by charging and discharging, resulting in different phase evolutions: LMNO evolves into a spinel structure on the surface, whereas LNTMO maintains its DRX structure. The maintained DRX structure results in significant overpotential, whereas the spinel structure exhibits relatively stable cycling performance. In addition, the in situ formed partially disordered spinel phase from DRX is reported to allow stable electrochemical cycling and enhanced rate capability.33,37 Building upon previous studies, designing materials by effectively utilizing phase transitions occurring during cycling could be one approach to design high-performance Mn-based cathode materials.
Herein, we propose a strategy to design partially disordered Mn-based spinel cathode materials with a rock-salt-phase-enriched surface to maintain a stable capacity by inducing favorable phase transitions during cycling. We demonstrate that by prioritizing phase transitions from a DRX-like phase to spinel-like phases at the surface, the detrimental two-phase reaction from a cubic spinel to tetragonal spinel phase can be successfully delayed. Simple manipulation of the spatial distribution, specifically by allocating the desirable phase to the surface, such that the surface phase transition precedes that in the bulk, is verified to effectively enhance the capacity retention. We suggest the importance of inducing favorable phase transitions initially in Mn-based materials to enable superior cycling performance.
Results and discussion
Mn-based partially disordered spinel cathode with rock-salt-phase-enriched surface
A partially disordered spinel cathode was synthesized by targeting the Li-rich spinel compositions (Li1+xMn2O4).17 The structure of Li–Mn–O compounds is significantly affected by the Li![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mn:O atomic ratio. When the Li:Mn:O ratio is 1:2:4, a spinel structure is formed, whereas a DRX structure is favored at a ratio of 1:1:2.38 Utilizing these characteristics, it has been reported that when the Li content is between 1 and 2 in the Mn2O4 framework, a mixed phase of spinel and DRX (Sp-DRX) is formed.35 Here, we synthesized a partially disordered spinel cathode material with a composition of 2:3:6 (1.33:2:4) (thus between 1:2:4 and 1:1:2) through mechanochemical reactions (Li1+xMn2O4, x = 0.33). A Li:Mn ratio of 1.332:2.017 was determined by inductively coupled plasma mass spectrometry (ICP-MS), close to our target values. The particle size of the synthesized Li1.33Mn2O4 cathode material ranged from 200 to 500 nm with uniform distribution of Mn and O at the particle level, as confirmed by energy-dispersive X-ray spectroscopy (EDS) (Fig. 1a and S1†). Rietveld refinement of the X-ray diffraction (XRD) pattern in Fig. 1b shows that Li1.33Mn2O4 is composed of spinel and DRX-like phases belonging to the Fd
:Mn:O atomic ratio. When the Li:Mn:O ratio is 1:2:4, a spinel structure is formed, whereas a DRX structure is favored at a ratio of 1:1:2.38 Utilizing these characteristics, it has been reported that when the Li content is between 1 and 2 in the Mn2O4 framework, a mixed phase of spinel and DRX (Sp-DRX) is formed.35 Here, we synthesized a partially disordered spinel cathode material with a composition of 2:3:6 (1.33:2:4) (thus between 1:2:4 and 1:1:2) through mechanochemical reactions (Li1+xMn2O4, x = 0.33). A Li:Mn ratio of 1.332:2.017 was determined by inductively coupled plasma mass spectrometry (ICP-MS), close to our target values. The particle size of the synthesized Li1.33Mn2O4 cathode material ranged from 200 to 500 nm with uniform distribution of Mn and O at the particle level, as confirmed by energy-dispersive X-ray spectroscopy (EDS) (Fig. 1a and S1†). Rietveld refinement of the X-ray diffraction (XRD) pattern in Fig. 1b shows that Li1.33Mn2O4 is composed of spinel and DRX-like phases belonging to the Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m and Fmm space groups, with phase fractions of 69.58% and 30.42%, respectively. Both phases were confirmed to exist as a composite through transmission electron microscopy (TEM) analysis (Fig. 1c). Fast Fourier transform (FFT) patterns of the high-resolution TEM (HR-TEM) image in the marked yellow and blue areas revealed repetitive interplanar distances, corresponding to spinel and DRX-like phases, respectively. Notably, DRX-like phase patterns were predominantly observed on the surface, whereas spinel-like structures were mainly observed in the bulk, as clearly confirmed by the azimuthal integration of the FFT patterns for selected surface, bulk, and overall area (Fig. 1d). The pattern on the surface was consistent with the DRX-like phase with the absence of the (111) plane peak of the spinel structure; meanwhile, the spinel-like structure was observed in the bulk and overall images. These results imply a characteristic phase distribution where the DRX-like phases are rich on the surface and the spinel-like phases are rich in the bulk of Li1.33Mn2O4. Mechanochemical synthesis can increase the surface energy of a material, which can affect to alter its structure, chemical composition, and chemical reactivity through a milling process.39 Therefore, DRX-like phase rich surface could be induced by mechanochemical synthesis. Fig. S2† demonstrates that the phase discrepancy between the surface and the bulk observed in the pristine state was also present in various multi-particles.
m and Fmm space groups, with phase fractions of 69.58% and 30.42%, respectively. Both phases were confirmed to exist as a composite through transmission electron microscopy (TEM) analysis (Fig. 1c). Fast Fourier transform (FFT) patterns of the high-resolution TEM (HR-TEM) image in the marked yellow and blue areas revealed repetitive interplanar distances, corresponding to spinel and DRX-like phases, respectively. Notably, DRX-like phase patterns were predominantly observed on the surface, whereas spinel-like structures were mainly observed in the bulk, as clearly confirmed by the azimuthal integration of the FFT patterns for selected surface, bulk, and overall area (Fig. 1d). The pattern on the surface was consistent with the DRX-like phase with the absence of the (111) plane peak of the spinel structure; meanwhile, the spinel-like structure was observed in the bulk and overall images. These results imply a characteristic phase distribution where the DRX-like phases are rich on the surface and the spinel-like phases are rich in the bulk of Li1.33Mn2O4. Mechanochemical synthesis can increase the surface energy of a material, which can affect to alter its structure, chemical composition, and chemical reactivity through a milling process.39 Therefore, DRX-like phase rich surface could be induced by mechanochemical synthesis. Fig. S2† demonstrates that the phase discrepancy between the surface and the bulk observed in the pristine state was also present in various multi-particles.
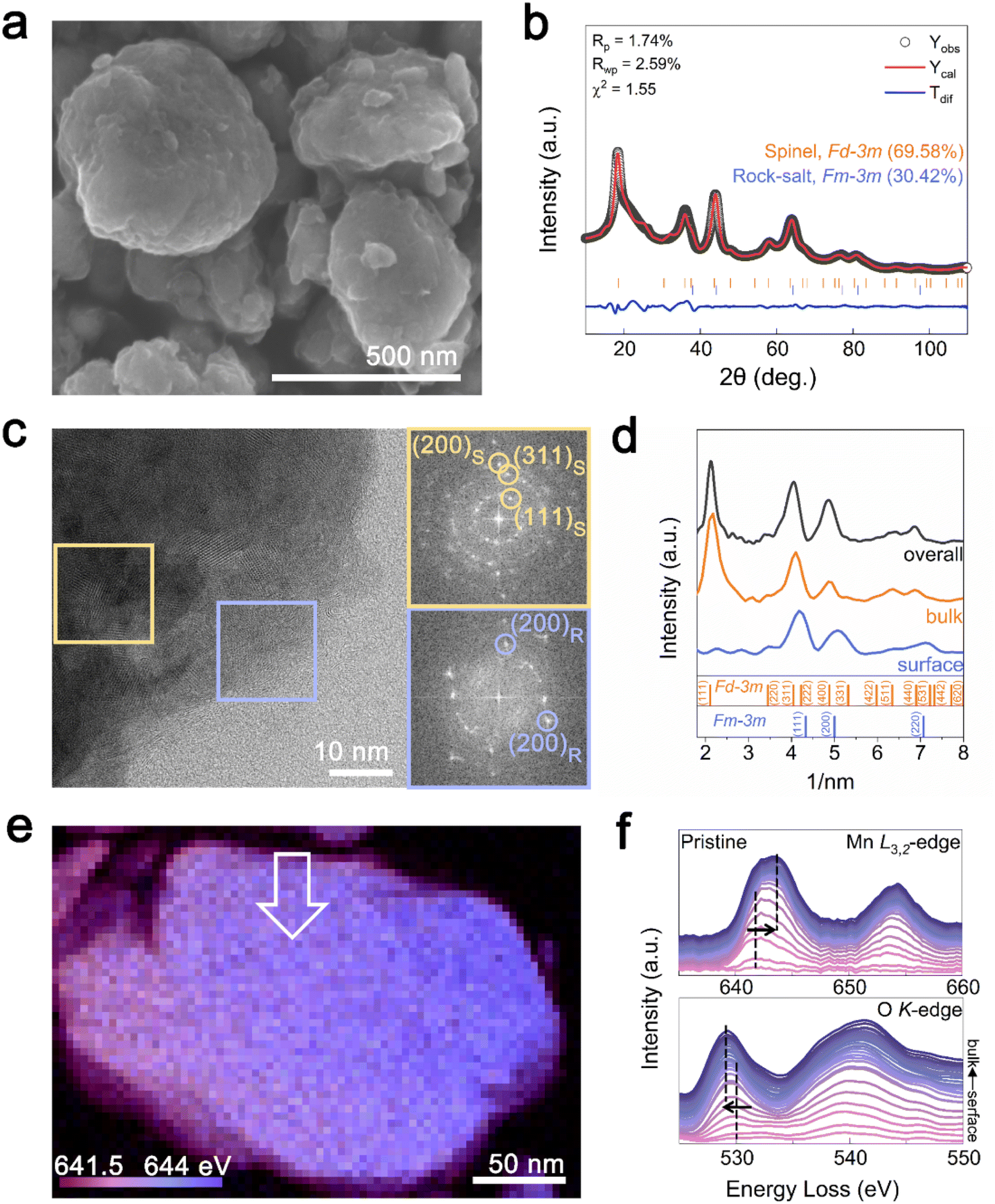 | ||
| Fig. 1 Partially disordered spinel cathode (Sp-DRX, Li1.33Mn2O4) with rock-salt-phase-enriched surface. (a) SEM image of Li1.33Mn2O4. (b) Rietveld refinement of XRD data (λ = 1.5406 Å) of Li1.33Mn2O4. (c) TEM images and FFT patterns of Li1.33Mn2O4. The yellow and blue boxes indicate areas where the spinel and rock-salt phases are predominantly present, respectively. (d) Azimuthal integration of FFT patterns for surface, bulk, and overall areas. (e) STEM-EELS mapping images of Li1.33Mn2O4 for the energy distribution of the Mn L3-edge peak position. (f) EELS spectra of Mn L3,2-edge and O K-edge for area indicated by the white arrow. | ||
To establish the distribution of both phases at the particle level, we examined the oxidation state of Mn ions using electron energy loss spectroscopy (EELS) mapping (Fig. 1e). Purple and blue represent the Mn L3-edge at low (641.5 eV) and high (644 eV) energies, respectively. The bulk of the particle was found to be more oxidized than the surface, whereas the surface exhibited a relatively reduced state compared with that of the bulk. To compare changes from the surface to the bulk, EELS spectra of the Mn L3,2-edge and O K-edge were investigated following the arrows marked in the scanning TEM(STEM)-EELS images (Fig. 1f). The Mn L3-edge at the surface (∼4 nm) was at 642 eV, whereas in the bulk (∼50 nm), it shifted to 644 eV, and the pre-edge of the O K-edge also shifted to lower energy with increasing distance from the surface.
The pre-edge peak at the O K-edge primarily reflects the electron transition from the oxygen 1s orbital to the unoccupied d orbitals of Mn, and this transition is closely related to the chemical environment surrounding Mn. For manganese oxides, a higher oxidation state typically induces stronger ligand-field splitting, which lowers the energy levels of the unoccupied d orbitals. Consequently, electron transition can occur at lower energies, implying that the pre-edge peak of the O K-edge appears at lower energy loss.40,41 Therefore, the shift of the pre-edge towards lower energies from the surface to the bulk can be attributed to the oxidation of Mn ions. By using the intensity ratio of L3/L2 (Fig. S3†) to determine the relative oxidation states of Mn, the oxidation states at the bulk and surface are shown to be close to +3.04 and +3.48, respectively.41,42 From the surface to the bulk, the Mn L3,2 and O K signals exhibit a similar intensity trend without being either particularly abundant or deficient at any specific location (Fig. S4†). Therefore, the change of oxidation states appears to be attributed to the different crystal structure of the DRX (LiMn3+O2)-like phase rich on the surface and the spinel (LiMn3.5+2O4)-like phase rich in the bulk of Li1.33Mn2O4 depending on the lithium inhomogeneity. The difference observed in the EELS spectra between the surface and bulk was also noted in different local regions of the particle, suggesting that the DRX structure is uniformly rich on the surface (Fig. S5 and S6†).
Surface evolution from the DRX-like phase to the spinel-like phase in initial cycles
For the initial electrochemical cycling of Li1.33Mn2O4, phase evolution from the rock-salt phase to spinel phase was observed at the surface. First, the electrochemical performance was evaluated within a voltage range of 1.5 to 4.6 V at 60 °C at a current density of 50 mA g−1. In Fig. 2a, the initial five cycles showed not only an increase in capacity from 278 to 293 mA h g−1 but also the distinct evolution of voltage plateaus at ∼3 V and ∼4 V. These changes in the electrochemical profile are strongly reminiscent of Li+ insertion into tetrahedral (8a) and octahedral (16c) sites in the spinel structure.33,37 To clarify the suspected structural changes, XRD analysis was conducted on electrodes discharged to 2.85 V after cycling for comparison with the pristine state with the same amount of Li in the cathode. The XRD pattern in Fig. 2b shows that the peaks corresponding to the spinel structure became significantly more pronounced, especially at 18.8°, 36.3°, and 57.9° (marked with yellow stars) with an increase in cycling. This structural transition was further confirmed by Raman spectroscopy, where Mn–O bond vibrations can be categorized into three types based on their vibrational frequencies (Fig. 2c). The broad peak observed in the low-frequency range of 200–400 cm−1 is related to the Mn–O stretching vibration mode (Ag) of the halite structure. The subtle peaks at 450–500 cm−1 are assigned to Eg and F2g, the characteristic vibrational modes observed in spinel structures. The high-frequency peaks in the range of 600–700 cm−1 correspond to the symmetric Mn–O stretching vibrations (A1g).43–45 Upon cycling of the battery, the stretching vibration characteristics of Mn–O in the rock-salt structure decrease, whereas the peak characteristics of the spinel structure increase, which is consistent with the XRD results.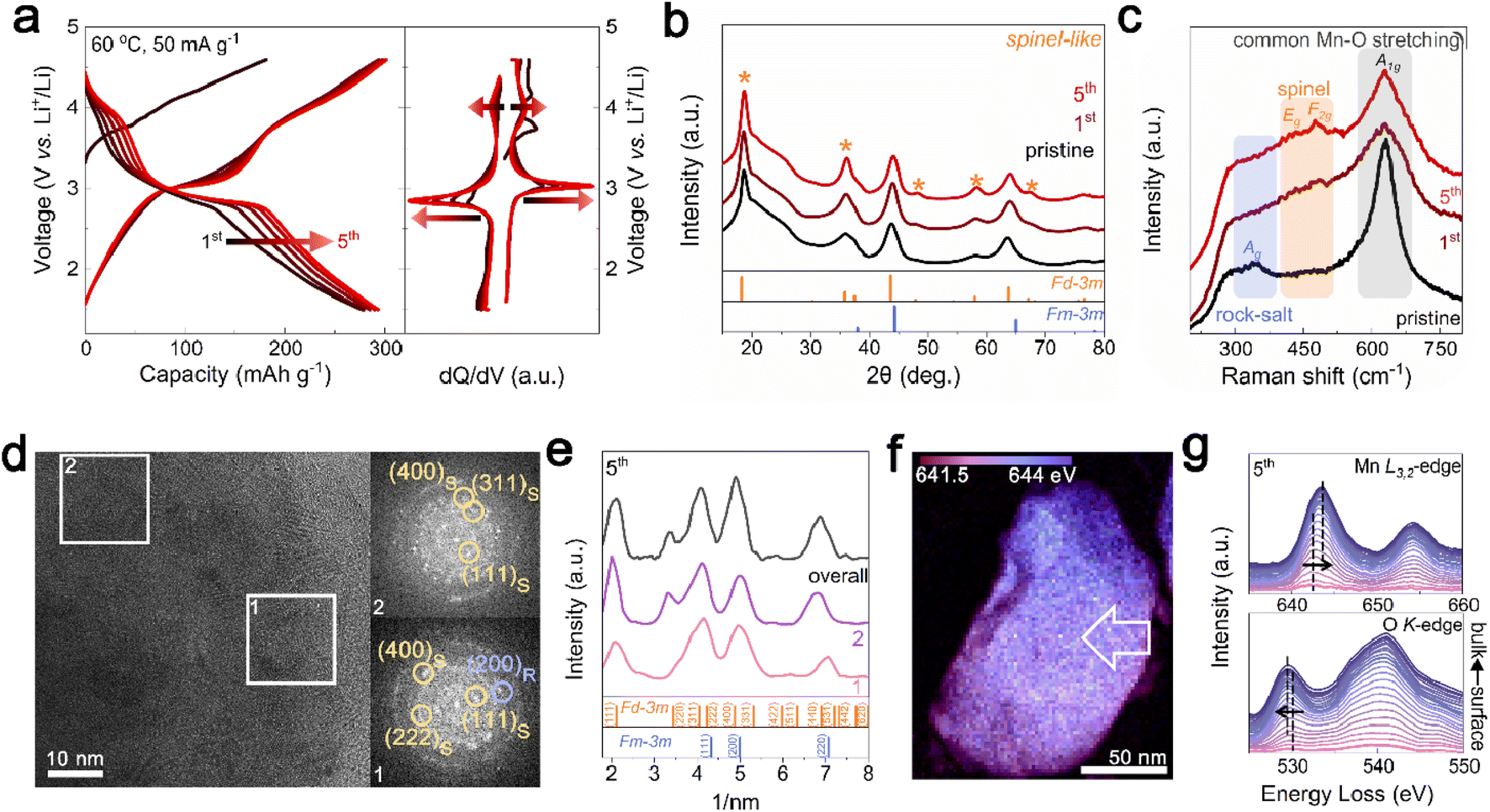 | ||
| Fig. 2 Gradual phase transition from rock salt to spinel phase at the surface of Li1.33Mn2O4. (a) Electrochemical profile of Li1.33Mn2O4 at 60 °C and a current density of 50 mA g−1 with differential analysis of voltage profile. (b) Ex situ XRD patterns at 2.85 V discharged state of the pristine electrode and after 1st and 5th cycle. (c) Raman spectra for pristine electrode and after 1st and 5th cycle. Each shaded region indicates the structural characteristics of Mn–O stretching related to rock salt and spinel structure. (d) TEM images and FFT patterns at 2.85 V discharge state of 5th cycle. (e) Azimuthal integration of FFT pattern for surface, bulk, and overall images. (f) STEM-EELS images of the 2.85 V discharged state after the 5th cycle with the energy distribution of the Mn L3-edge peak position. (g) EELS spectra of Mn L3,2-edge and O K-edge peak of area indicated by the white arrow. | ||
This gradual phase transition from the rock-salt to spinel phase, which is advantageous for the voltage and capacity, including the lithium-diffusion kinetics,33,37 is mainly observed at the surface region. Fig. 2d presents TEM images of the electrode as discharged to 2.85 V in the 5th cycle. In contrast to the pristine state, where the DRX-like phase was abundant on the surface, the FFT patterns for both regions 1 and 2, representing the surface and bulk, respectively, showed the presence of the spinel structure. Azimuthal integration of the FFT patterns revealed consistent patterns of the spinel structure regardless of surface and bulk area, indicating the phase transition of the DRX-like phase on the surface to spinel-like structures (Fig. 2e and S7†). This surface structural evolution also affected the energy distribution of the Mn L3-edge. In contrast to the energy distribution of the pristine state observed in Fig. 1e, the 5th cycle showed a relatively uniform energy distribution (Fig. 2f). To identify the spatial energy distribution difference, EELS spectra for the Mn L3,2-edge and O K-edge were examined following the arrows marked in the STEM-EELS images (Fig. 2g and S8†). Unlike the pristine state, where there was a 1.75 eV gap in the Mn L3-edge energy between the surface and bulk, the 5th cycled sample exhibited a reduced energy gap of 0.75 eV, and there was almost no shift in the pre-edge of the O K-edge. Based on the XRD and TEM analysis, the reduction in energy difference observed at the 5th cycle is attributed to the homogenization of the phase between the surface and the bulk.
Mitigated voltage decay and capacity degradation with structural evolution in Li1.33Mn2O4
Designing the surface with a DRX-like phase led to a phase transition to a spinel-like structure in the early cycles, increasing the capacity and voltage. The capacity retention of Li1.33Mn2O4 was maintained at 96.5% even after 60 cycles (Fig. 3a). In the case of the average discharge voltage, it increases up to 2.85 V due to the evolution of the voltage plateau in the initial cycles, but a slight decrease is observed after 10 cycles. To observe the subsequent changes in capacity and voltage and how they were correlated with the structural evolution, the analysis was conducted at the 10th cycle (both the capacity and voltage increased), the 30th cycle (the capacity was maintained, but the voltage decreased), and the 60th cycle (both the capacity and voltage decreased) (Fig. 3a and S9†). The electrochemical profiles across different cycles show a clear evolution (Fig. 3b). The 10th cycle exhibited distinct voltage plateaus at 3 V and 4 V; however, the 4 V plateau became steeper at the 30th cycle, whereas the discharge capacity was maintained. After the 60th cycle, severe voltage fade near 3 V and a shortened 4 V plateau led to a reduction in the average voltage (2.68 V) and total capacity (287.50 mA h g−1) compared to those of the 10th cycle (2.85 V, 297.85 mA h g−1).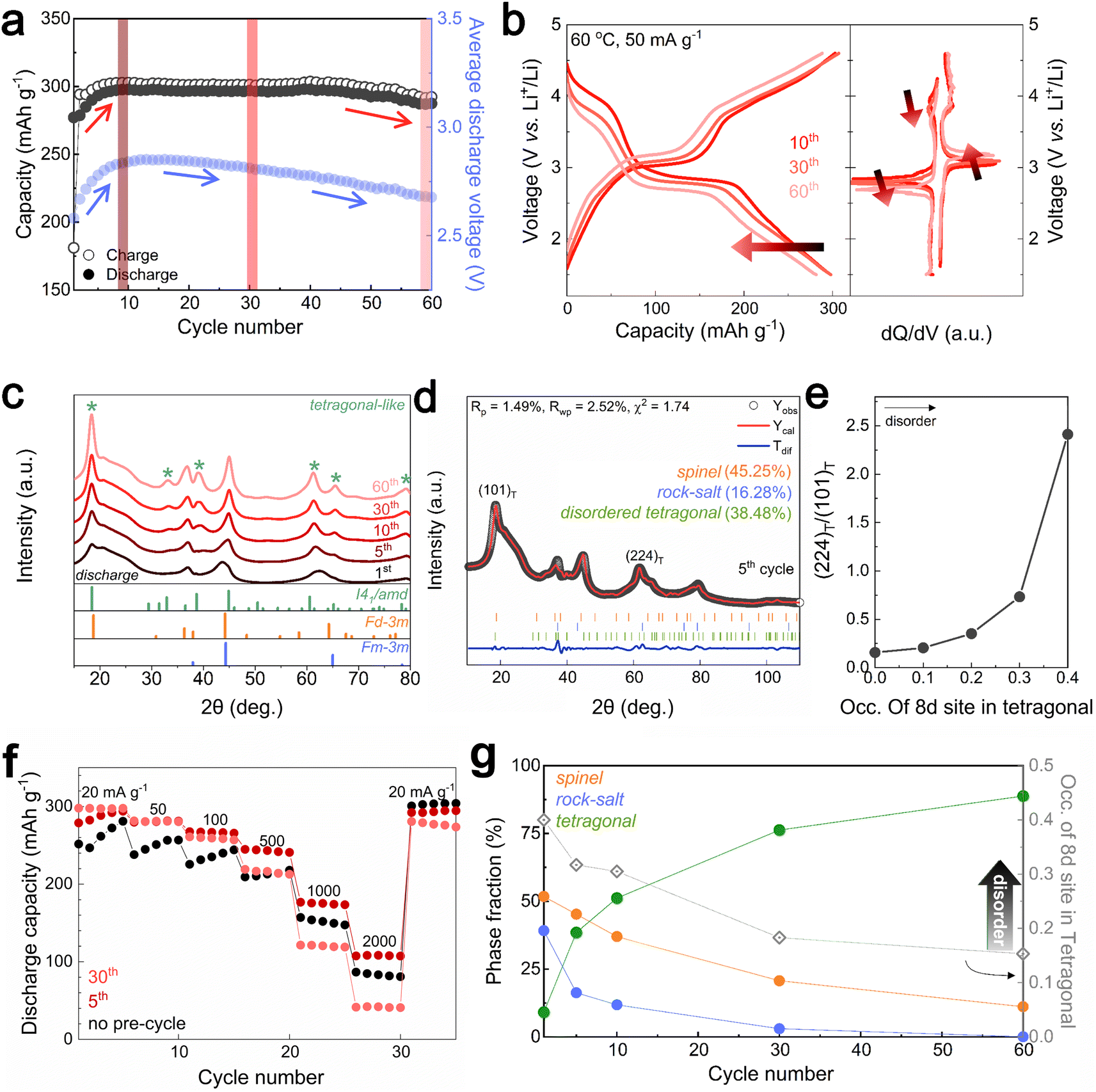 | ||
| Fig. 3 Structural evolution of Li1.33Mn2O4 mitigating voltage decay and capacity degradation. (a) Cycling performance of Li1.33Mn2O4. (b) Electrochemical profile after the 10th, 30th, and 60th cycle at 60 °C and a current density of 50 mA g−1. The differential analysis of voltage profile. (c) Ex situ XRD patterns at discharge state of various cycles. (d) Rietveld refinement of the XRD data (λ = 1.5406 Å) for discharged-state electrode after the 5th cycle. (e) Intensity ratio of (101) and (224) planes according to 8c/8d disorder of tetragonal phase. (f) Rate performance comparison of the pristine electrode and after 5th and 30th cycles. (g) Phase fraction of the discharge state at various cycles with occupancy in 8d site for tetragonal spinel structure. | ||
Therefore, we performed ex situ XRD analysis of the discharged state at various cycles to determine whether detrimental structural evolution occurred (Fig. 3c). It is well established that spinel LiMn2O4 undergoes severe phase transitions to tetragonal spinel, inducing disproportionation reactions and Jahn–Teller distortions as the Mn oxidation state approaches trivalence, potentially leading to significant capacity loss.46–51 In contrast to LiMn2O4 spinel, Li1.33Mn2O4 exhibited solid-solution behavior with a slight shift of the diffraction peak position in the initial cycle without two-phase transition of the tetragonal phase (Fig. S10†). This behavior may result from the 16c/16d disorder of Sp-DRX that can break the 16d Mn ordering, which is responsible for the collective Jahn–Teller distortion in ordered spinel, thereby increasing the accessibility to solid-solution behavior.15,17,34 In addition, these results could stem from not only disordering in the spinel but also the spinel phase induced in DRX by cycling. From the 5th cycle discharge state, XRD peaks near 33.1°, 39.0°, 61.2°, and 64.5° emerged (marked with green stars), indicating a tetragonal-like structure. However, Rietveld refinement of the 5th discharged state XRD pattern showed a better fit for a disordered tetragonal model over an ordered one (Fig. 3d and S11†). Ordered and disordered tetragonal structures show similar XRD patterns; however, as disorder increases, the intensity of the (224)Tetra peak becomes more dominant than that of the (101)tetra peak (Fig. 3e). Disordered structures generally have higher structural flexibility than ordered structures. This can improve the reversible reactivity by diversifying Li-ion migration pathways and lowering the energy barrier within the structure.34,52–57 To further verify the effect of the evolution of the disordering in the tetragonal phase, the rate performance was compared after various cycles (Fig. 3f and S12†). The performance after the 5th cycle was superior to that without pre-cycling, which corroborates the positive impact of the phase transition from the initial DRX structure to a spinel with an excellent Li-ion diffusion path and high electrical conductivity,58,59 as well as the evolution of a disordered tetragonal phase. However, the rate performance deteriorated after the 30th cycle due to the phase transition from the spinel to an ordered tetragonal-like phase, a well-known detrimental two-phase reaction. Thus, despite the initial observation of a tetragonal-like phase, the maintenance of the capacity until the 30th cycle may be attributed to the disordering of the tetragonal phase (Fig. 3g and S13†).
Spatial phase evolution and distribution in the region of cycle degradation
TEM analysis was performed to understand the cycling degradation process of Li1.33Mn2O4 from a spatial perspective. Fig. 4a and e present TEM images of the electrode after the 30th cycle, where voltage decay occurs, and after the 60th cycle, where both the voltage and capacity have decayed, respectively. To discern how the phase evolution progresses from the pristine state, analysis of the FFT patterns and their integration were performed on the surface, bulk, and overall area of electrodes discharged to 2.85 V.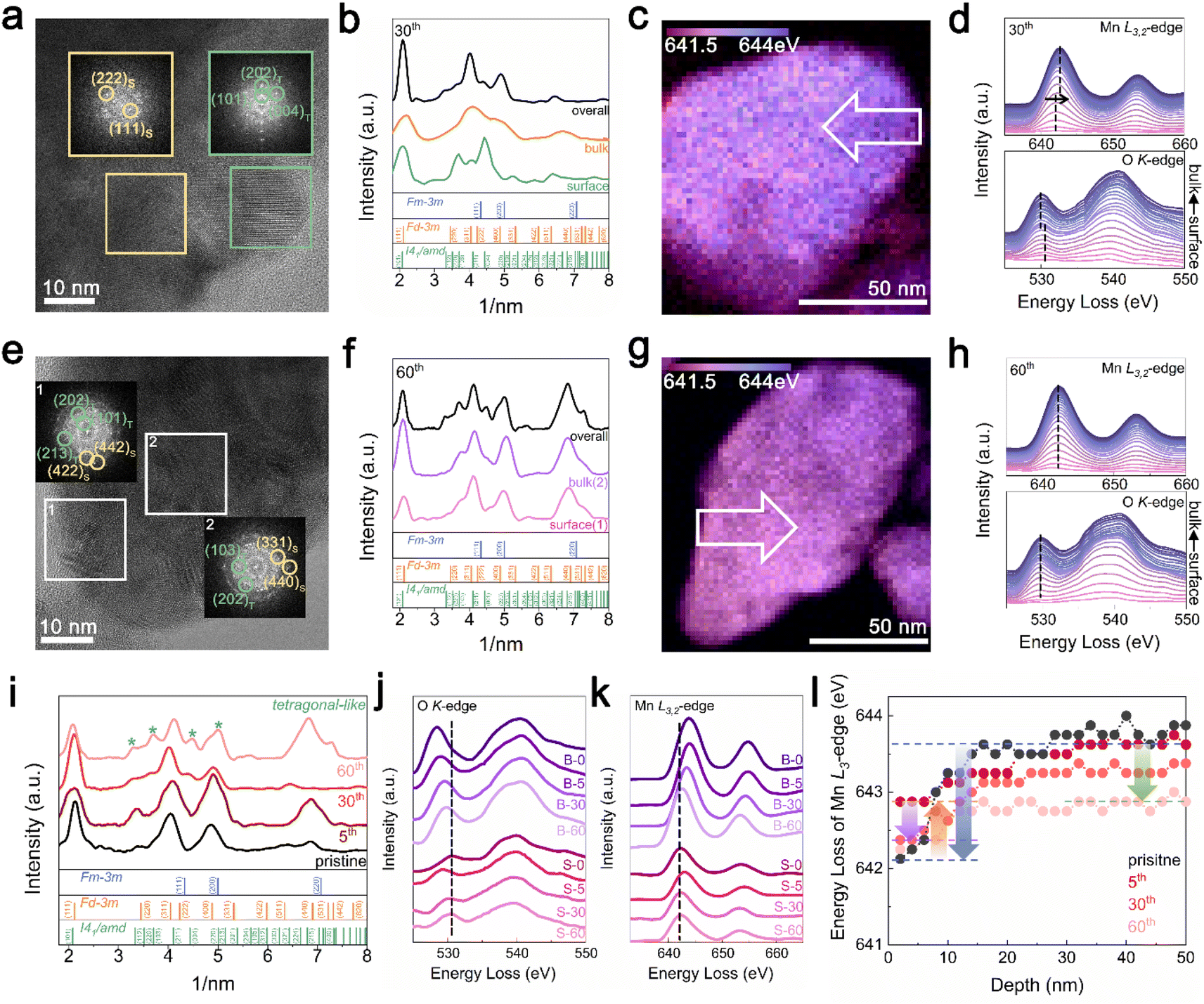 | ||
| Fig. 4 Phase evolution and spatial distribution of Li1.33Mn2O4 upon electrochemical cycling. TEM images and FFT patterns at 2.85 V discharge state of (a) 30th and (e) 60th cycle. (b, and f) Azimuthal integration of FFT pattern for surface, bulk, and overall images. STEM-EELS images of 2.85 V discharged state of (c) 30th and (g) 60th cycle for the energy distribution of the Mn L3-peak. (d, and h) EELS spectra of Mn L3,2-edge and O K-edge peak of area indicated by the white arrow. (i) Azimuthal integration of FFT pattern for overall images at various cycles. EELS spectra of (j) O K-edge peak and (k) Mn L3,2-edge at surface and bulk in various cycles. (l) Mn L3-edge energy according to depth in various cycles. | ||
For the electrode after the 30th cycle, peaks corresponding to the (202)tetra and (004)tetra planes at 0.24 and 0.23 nm, respectively, were identified on the surface, indicating that the surface region evolved into a tetragonal phase belonging to I41/amd. Although characteristic patterns of the tetragonal spinel phase were observed in the bulk and overall regions, the patterns of the cubic spinel phase still predominated (Fig. 4a and b). Because the 4 V voltage plateau is primarily associated with Li insertion into the 8a site of the cubic spinel phase,34,60,61 the predominant presence of the cubic spinel phase in the bulk aligns with the maintenance of capacity despite the slightly reduced electrochemical profile at the 4 V plateau. However, for the electrode after the 60th cycle, the copresence of the FFT pattern of tetragonal and cubic spinel phases was observed across both the surface and bulk (Fig. 4e and f). The propagation of the tetragonal-phase evolution from the surface to the bulk of the particles upon electrochemical cycling was universally observed in various particles and regions (Fig. S14 and S15†).
In addition, as the structural evolution occurs, the oxidation state of Mn consistently changed (Fig. 4c, d, g and h). The reduced state of surface relative to that of the bulk was exhibited by the energy gap between the surface and bulk for both the Mn L2,3-edge (1.00 eV) and O K-edge (0.50 eV) spectra after the 30th cycle. In addition, both the surface and bulk exhibited nearly the same oxidation state after 60 cycles; this trend was consistently observed in different regions of the particles depending on the electrochemical cycling (Fig. S16† and 17).
Fig. 4i presents integrated FFT patterns for the TEM image including the surface and bulk, indicating that these regions are at the same lithiation state across various cycles. The pattern in the pristine state comprised peaks for both DRX and spinel-like phases, with the spinel-like phase pattern becoming more pronounced after the 5th cycle. After further cycling, the 30th cycle showed the pattern for the tetragonal-like phase, which became increasingly distinct as cycling progressed. As the spectra of the O K-edge and Mn L3,2-edge are affected not only by the Mn oxidation state but also by the crystal structure, the observed changes in energy loss between the surface and bulk for various cycles can be explained by considering the structural changes observed through XRD and TEM analyses. Fig. 4j compares the O K-edge spectra at the surface and in the bulk across different cycles. Although there was an energy difference in the pre-edge region of the O K-edge between the surface and bulk for the pristine and 30th cycles, the 5th and 60th cycles showed similar energies. This trend observed throughout the cycling process was also reflected in the Mn L3,2-edge spectra (Fig. 4k and l). In the pristine state, the presence of a DRX-like phase on the surface led to a noticeable difference in the energy of the Mn L3,2-edge between the bulk and surface (blue arrow). However, with the formation of the spinel-like phase by the 5th cycle, the energy of the Mn L3,2-edge on the surface shifted to higher energy compared to that in the pristine state, thereby diminishing the energy gap between the surface and bulk, as indicated by the yellow arrow. In the 30th cycle, the formation of the tetragonal-like phase on the surface shifted the Mn L3,2-edge energy back to a lower position (purple arrow), regenerating an energy gap with the bulk. As cycling progressed, the formation of the tetragonal phase within the bulk increased, shifting the energy to a lower energy position (green arrow), thereby resulting in an energy similar to that of the surface. The Mn L3/L2 intensity ratio, which can be used to compare the relative Mn oxidation states, was analyzed for both the surface and bulk in each cycle, serving as evidence for the phase evolution induced by cycling (Fig. S3†).
Discussion
Mn-based cathode materials have been regarded as undergoing inevitable detrimental phase transitions, which hinder the Li+ diffusion or reversible capacity delivery. These phase transitions lead to cycle degradation with accompanying issues such as irreversible oxygen evolution, Mn-ion migration, disproportionation reactions, and Jahn–Teller distortions. Strategies to mitigate these structural changes in Mn-based cathode materials, such as doping with immobile metals, substituting with F− ions, and creating partial 16c/16d disorder, have been suggested; however, these strategies have failed to fully suppress the phase transitions.15,17,30–34 We suggest that strategically utilizing the cycle-induced phase transition occurring in Mn-based cathode materials can delay capacity degradation. A partially disordered spinel Li1.33Mn2O4 material, rich in a DRX-like phase on the surface, exhibits an increase in voltage and capacity during the initial cycles due to the phase transition from the DRX-like to spinel-like phase. Furthermore, the spinel phase formed from DRX is partially disordered, leading to solid-solution reactions predominating near 3 V. In addition, the formation of a disordered tetragonal phase that is gradually formed in the discharge state maintains capacity retention by delaying the two-phase reaction of the ordered tetragonal-like phase evolution from the surface to the bulk (Fig. 5).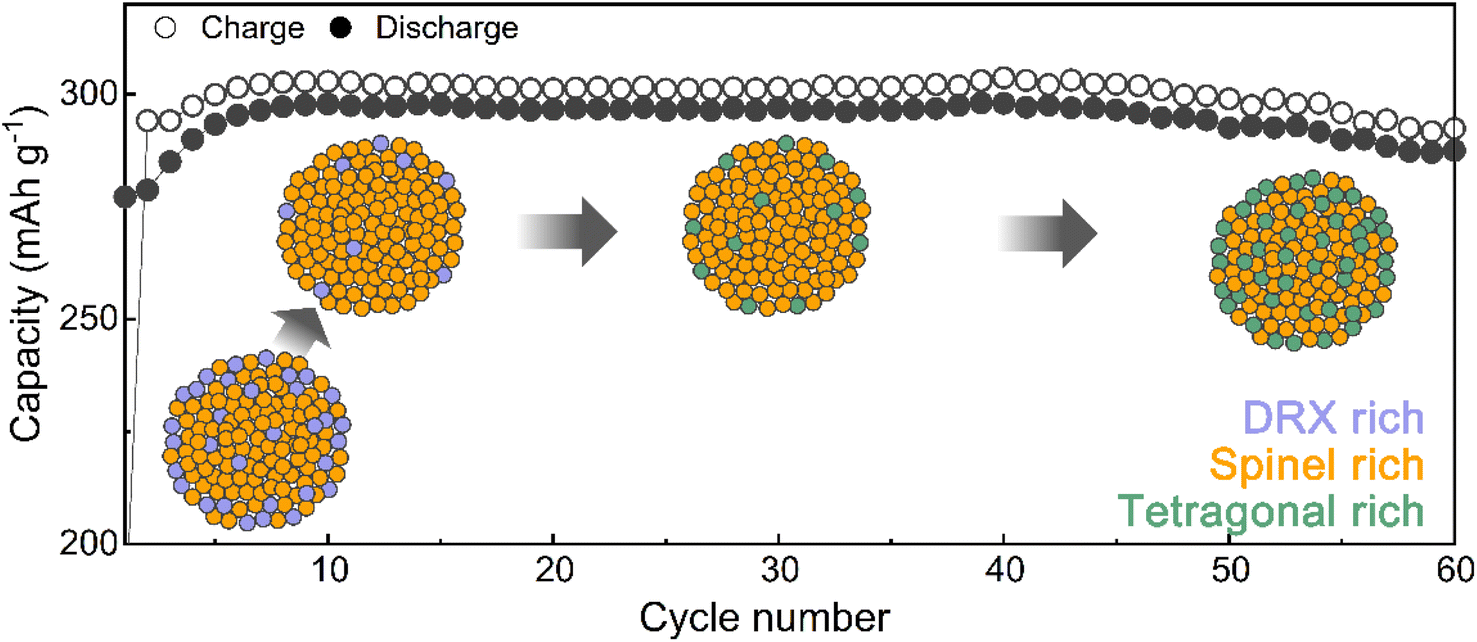 | ||
| Fig. 5 Phase evolution mechanism in Li1.33Mn2O4 with DRX-rich surface mitigating voltage decay and capacity degradation during electrochemical cycling. | ||
The viability of designing surfaces to induce favorable phase transitions was confirmed for Li1.26Mn1.88O4, which has a less DRX-like phase on the surface compared to Li1.33Mn2O4. Compared to Li1.33Mn2O4, which consists of a spinel-like phase and a DRX-like phase in a ratio of approximately 7:3, Li1.26Mn1.88O4 contains these phases in a 8:2 ratio (Fig. S18†). The target composition was verified by determining the Li:Mn ratio of 1.258:1.887 through ICP-MS analysis. Based on TEM images and STEM-EELS analysis, Li1.26Mn1.88O4 is shown to contain a DRX-like phase on the surface; however, this phase is less abundant than in Li1.33Mn2O4 (Fig. S19–S21†). Consequently, Li1.26Mn1.88O4, which has a higher proportion of spinel-like phases, initially possesses a higher specific capacity than Li1.33Mn2O4. However, due to facile transition from DRX-like to spinel-like phases, it undergoes rapid degradation and exhibits poorer cycle stability compared to Li1.33Mn2O4 (63% capacity retention after 50 cycles, Fig. S22†). It is may be due to the easier phase transition to tetragonal phase for Li1.26Mn1.88O4 compared to Li1.33Mn2O4 (Fig. S23†). This finding demonstrates the effectiveness of designing the surface with a suitably rich DRX layer and suggests that by efficiently utilizing structural transitions, the onset of detrimental structural changes can be delayed in Sp-DRXs.
Our strategy resulted in excellent capacity retention of 96.5% even after 60 cycles; nevertheless, capacity fade still occurred over long-term cycling (Fig. S22†). In Mn-based cathode materials, capacity degradation occurs not only due to structural changes but also for various other reasons, including Mn dissolution, irreversible oxygen evolution, and side reactions with the electrolyte. Although oxygen evolution was not observed in the differential electrochemical mass spectroscopy (DEMS) measurements conducted across various cycles for Li1.33Mn2O4 (Fig. S24†), continuous Mn-ion dissolution was noted (Fig. S25†). Moreover, after 30 cycles, a rapid increase in the presence of LixPFyOz, a decomposition product of the LiPF6 salt, was observed in XPS O 1s, F 1s, and P 2p spectra (Fig. S27†).9,62,63 These results suggest that the severe capacity decay observed during long-term cycling may stem from serious side reactions with the electrolyte, which is more prominently observed in a high-voltage environment (Fig. S28†). Moreover, since sluggish diffusion kinetics can lead to rapid capacity decay, it is also necessary to design materials with appropriate particle size and crystallinity (Fig. S29†).37 Therefore, concerted efforts to overcome the issue of capacity fade over long-term cycling are crucial in developing high-performance DRX materials.
Conclusions
Our research demonstrates that strategic manipulation of structural transitions in Mn-based cathode materials can substantially enhance the performance of lithium-ion batteries (LIBs). We developed a novel Mn-based Sp-DRX electrode engineered to initiate favorable early phase transitions, effectively delaying detrimental transitions to extend battery life. This design strategy concentrates on enriching the surface with DRX-like phase, thereby leveraging the transition to a partially disordered spinel-like phase and postponing the shift to a tetragonal-like phase for improved capacity and cycle stability. This approach is corroborated by the improved cycling performance of Li1.33Mn2O4, which achieved a notable capacity of 297.85 mA h g−1 and retained 98.4% of this capacity after 50 cycles. The comparative analysis with Li1.26Mn1.88O4, which exhibits faster degradation due to its lower DRX phase content and a thinner DRX surface layer, further highlights the importance of surface design in material performance. These results emphasize the significance of controlling phase transitions to improve the electrochemical performance of Mn-based cathodes. Integrating these insights with material design strategies to mitigate degradation could lead to the development of high-performance Mn-based materials for LIBs, offering a promising direction for future research and application.Experimental
Synthesis of nanocomposite Li1.33Mn2O4 and Li1.26Mn1.88O4
Li1.33Mn2O4 and Li1.26Mn1.88O4 were synthesized via high-energy planetary ball-milling. A 1:1 molar ratio mixture of lithium peroxide (Li2O2, 95% purity, Thermo Fisher) and manganese (II, III) oxide (Mn3O4, 97% purity, Aldrich) was sealed within an Ar-filled environment. The prepared powder mixture underwent high-energy ball milling (Pulverisette 7 premium line) at 530 rpm for 48 h to synthesize Li1.33Mn2O4. To synthesize Li1.26Mn1.88O4, lithium carbonate (Li2CO3, 99% purity, Alfa Aesar) and manganese dioxide (MnO2, 99.9% purity, Alfa Aesar) were first calcined at 800 °C to produce lithium manganese oxide (Li2MnO3). This powder was then high-energy ball-milled at 530 rpm for 48 h in a 1:4 molar ratio with lithium manganese oxide (LiMn2O4, >99% purity, Aldrich). The high-crystallinity H_Li1.33Mn2O4 and H_Li1.26Mn1.88O4 were prepared by only changing the ball milling conditions (milled at 275 rpm for 48 hours) within the same synthesis process.
Electrochemistry
For fabrication of the cathode film, the synthesized powder was mixed with 10 wt% of graphite at 300 rpm for 6 h. The prepared active material (70 wt%), conductive agent (20 wt%, Super P), and binder (10 wt%, polyvinylidene fluoride) were dispersed in N-methyl-2-pyrrolidone (NMP) using a planetary centrifugal mixer (AR-100, Thinky). The slurry was cast onto Al foil to create a cathode film with a loading level of ∼4 mg cm−2. Coin cells (CR2032) were assembled in an Ar atmosphere using a 1 M LiPF6 solution in a 1:1 volume ratio of ethylene carbonate and dimethyl carbonate (Enchem) as the electrolyte. Glass microfiber filters (Whatman) and Li metal foil (FMC) were employed as the separator and anode material, respectively. Electrochemical analysis was conducted at 60 °C within the voltage range of 1.5–4.6 V at a current density of 50 mA g−1 utilizing a battery testing system (WBCS 3000, WonATech). DEMS was performed to monitor the gas evolution during galvanostatic charge and discharge cycles between 1.5 and 4.6 V at a current density of 20 mA g−1 at room temperature. The detection of gas evolution was scheduled at 5 min intervals.
Characterization
XRD patterns of the sample were collected at the 6D UNIST-PAL beamline located at the Pohang Accelerator Laboratory (PAL) across a 2θ range of 10°–110°. For the ex situ XRD patterns, cells that reached specific voltages were disassembled, and the electrodes were retrieved and then washed in diethyl carbonate. Samples were collected from the electrodes and sealed in capillaries. The wavelength of the X-ray beam was 1.5406 Å, and all the XRD patterns were recorded with an exposure time of approximately 2 min. To eliminate noise, outliers, and especially spikes caused by high-energy X-rays in the measured XRD data, each sample was measured twice to obtain the XRD patterns. Rietveld refinement of the XRD data was performed using the FullProf software.SEM images were obtained at 10 kV using field-emission scanning electron microscopy (SU8220 Cold FE-SEM, Hitachi) to observe the morphology and size of the samples. Local crystallographic features were characterized using TEM analysis. For ex situ analysis, a specific lithiated electrode was disassembled in a glove box and washed using dimethyl carbonate (DMC). The sample, separated from the aluminum current collector, was immersed in DMC and dispersed using ultrasonic treatment. The TEM sample grids were maintained in a vacuum state until being inserted into the transmission electron microscope to minimize exposure to air during sample transfer. STEM-EELS measurements were conducted at 160 kV using a Cs-corrected JEM-ARM300F (JEOL). For elemental mapping in STEM-EELS, the energy dispersion was set to 0.25 eV per channel. To minimize electron-beam damage to the sample and stabilize the electron beam before analysis, beam showering was performed for approximately 20 min.
Raman spectroscopy was performed for structural analysis using a confocal Raman spectrometer (Alpha300R, WITec) with a 532 nm laser at 10 mW power. Each spectrum represents the average of spectra collected from three different spots on the sample. Time-of-flight secondary ion mass spectrometry (TOF-SIMS) was employed to measure the dissolved Mn ions on the Li–metal anode throughout cycling. TOF-SIMS analysis involved a pulsed Bi3+ ion beam (25 keV) operating in high current mode for surface spectroscopy, and the measurement area was set at 500 × 500 μm2. Ex situ XPS (K-Alpha, ThermoFisher) with Al Kα (hν = 1486.6 eV) radiation was employed to identify side reactions with the electrolyte. All the XPS spectra were calibrated based on the C–C peak (284.8 eV). Sample transfer of all the samples was performed in a vacuum atmosphere to suppress reaction with air and moisture before XPS analysis. The composition of the synthesized material was measured using inductively coupled plasma mass spectrometry (ICP-MS, Agilent ICP-MS 7700S).
Author contributions
H. Jo, C. Lee, and S.-K. Jung conceived the research design. H. Jo, C. Lee, H. N., and S.-K. Jung were responsible for the manuscript preparation. H. Jo and C. Lee conducted the TEM analysis. J. H. Ha and N. J. Kong performed the DEMS measurements and analysis. All the authors contributed to the discussion of this work. S.-K. Jung supervised the project.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This research was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (No. 2022R1C1C1006575); a grant of the Basic Research Program funded by the Korea Institute of Machinery and Materials (No. NK243H). This work was supported by the 2021 Research Fund (1.210130.01) of UNIST (Ulsan National Institute of Science & Technology).References
- X. T. Wang, Y. Yang, J. Z. Guo, Z. Y. Gu, E. H. Ang, Z. H. Sun, W. H. Li, H. J. Liang and X. L. Wu, J. Mater. Sci. Technol., 2022, 102, 72–79 CrossRef CAS
.
- M. Du, J. Z. Guo, S. H. Zheng, Y. Liu, J. L. Yang, K. Y. Zhang, Z. Y. Gu, X. T. Wang and X. L. Wu, Chin. Chem. Lett., 2023, 34, 107706 CrossRef CAS
- W. Li, E. M. Erickson and A. Manthiram, Nat. Energy, 2020, 5, 26–34 CrossRef CAS
- R. Schmuch, R. Wagner, G. Hörpel, T. Placke and M. Winter, Nat. Energy, 2018, 3, 267–278 CrossRef CAS
- D. Andre, S. J. Kim, P. Lamp, S. F. Lux, F. Maglia, O. Paschos and B. Stiaszny, J. Mater. Chem. A, 2015, 3, 6709–6732 RSC
- S. T. Myung, F. Maglia, K. J. Park, C. S. Yoon, P. Lamp, S. J. Kim and Y. K. Sun, ACS Energy Lett., 2017, 2, 196–223 CrossRef CAS
- M. Greenwood, M. Wentker and J. Leker, J. Power Sources Adv., 2021, 9, 100055 CrossRef CAS
- H. Li, R. Fong, M. Woo, H. Ahmed, D. H. Seo, R. Malik and J. Lee, Joule, 2022, 6, 53–91 CrossRef CAS
- G. Lim, D. Shin, K. H. Chae, M. K. Cho, C. Kim, S. S. Sohn, M. Lee and J. Hong, Adv. Energy Mater., 2022, 13, 2202049 CrossRef
- H. Zhao, W. Y. A. Lam, L. Sheng, L. Wang, P. Bai, Y. Yang, D. Ren, H. Xu and X. He, Adv. Energy Mater., 2022, 12, 1–36 Search PubMed
- E. A. Olivetti, G. Ceder, G. G. Gaustad and X. Fu, Joule, 2017, 1, 229–243 CrossRef
- Y. Yue, N. Li, Y. Ha, M. J. Crafton, B. D. McCloskey, W. Yang and W. Tong, Adv. Funct. Mater., 2021, 31, 1–8 Search PubMed
- Y. He, W. Xiang, G. Lv, Y. J. Zhong, Y. Song, K. Zhou, Z. Wu, J. Zhang, E. Yang and X. Guo, Chem. Eng. J., 2021, 417, 128189 CrossRef CAS
- J. Lee, A. Urban, X. Li, D. Su, G. Hautier and G. Ceder, Science, 2014, 343, 519–522 CrossRef CAS PubMed
- H. Ji, J. Wu, Z. Cai, J. Liu, D. H. Kwon, H. Kim, A. Urban, J. K. Papp, E. Foley, Y. Tian, M. Balasubramanian, H. Kim, R. J. Clément, B. D. McCloskey, W. Yang and G. Ceder, Nat. Energy, 2020, 5, 213–221 CrossRef CAS
- Y. shan Jiang, F. da Yu, W. Ke, L. Deng, Y. Xia, X. yu Li, L. fang Que, N. Zhang, L. Zhao and Z. bo Wang, Adv. Funct. Mater., 2023, 33, 2213615 CrossRef
- Z. Cai, H. Ji, Y. Ha, J. Liu, D. H. Kwon, Y. Zhang, A. Urban, E. E. Foley, R. Giovine, H. Kim, Z. Lun, T. Y. Huang, G. Zeng, Y. Chen, J. Wang, B. D. McCloskey, M. Balasubramanian, R. J. Clément, W. Yang and G. Ceder, Matter, 2021, 4, 3897–3916 CrossRef CAS
- Y. Gao, J. Ma, Z. Wang, G. Lu and L. Chen, Phys. Chem. Chem. Phys., 2017, 19, 7025–7031 RSC
- M. Wagemaker, F. G. B. Ooms, E. M. Kelder, J. Schoonman, G. J. Kearley and F. M. Mulder, J. Am. Chem. Soc., 2004, 126, 13526–13533 CrossRef CAS PubMed
- E. S. Lee, K. W. Nam, E. Hu and A. Manthiram, Chem. Mater., 2012, 24, 3610–3620 CrossRef CAS
- W. Hua, S. Wang, M. Knapp, S. J. Leake, A. Senyshyn, C. Richter, M. Yavuz, J. R. Binder, C. P. Grey, H. Ehrenberg, S. Indris and B. Schwarz, Nat. Commun., 2019, 10, 5365 CrossRef PubMed
- A. R. Armstrong, M. Holzapfel, P. Nová, C. S. Johnson, S.-H. Kang, M. M. Thackeray and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 8694–8698 CrossRef CAS PubMed
- H. Koga, L. Croguennec, M. Ménétrier, P. Mannessiez, F. Weill, C. Delmas and S. Belin, J. Phys. Chem. C, 2014, 118, 5700–5709 CrossRef CAS
- J. Zheng, P. Xu, M. Gu, J. Xiao, N. D. Browning, P. Yan, C. Wang and J. G. Zhang, Chem. Mater., 2015, 27, 1381–1390 CrossRef CAS
- M. Freire, N. V. Kosova, C. Jordy, D. Chateigner, O. I. Lebedev, A. Maignan and V. Pralong, Nat. Mater., 2016, 15, 173–177 CrossRef CAS PubMed
- B. R. Wygant, L. C. Merrill, K. L. Harrison, A. A. Talin, D. S. Ashby and T. N. Lambert, Adv. Sci., 2022, 9, 2105803 CrossRef CAS PubMed
- T. Erichsen, B. Pfeiffer, V. Roddatis and C. A. Volkert, ACS Appl. Energy Mater., 2020, 3, 5405–5414 CrossRef CAS
- T. Okumura, Y. Yamaguchi, M. Shikano and H. Kobayashi, J. Mater. Chem. A, 2014, 2, 8017–8025 RSC
- B. Song, G. M. Veith, J. Park, M. Yoon, P. S. Whitfield, M. J. Kirkham, J. Liu and A. Huq, Chem. Mater., 2019, 31, 124–134 CrossRef CAS
- I. Hwang, E. Ahn and Y. Tak, Int. J. Electrochem. Sci., 2014, 9, 5454–5466 CrossRef
- F. Wang, P. Zuo, Z. Xue, Y. Liu, C. Wang and G. Chen, ACS Energy Lett., 2024, 9, 1249–1260 CrossRef CAS PubMed
- L. Li, Z. Lun, D. Chen, Y. Yue, W. Tong, G. Chen, G. Ceder and C. Wang, Adv. Funct. Mater., 2021, 31, 2101888 CrossRef CAS
- Z. Cai, B. Ouyang, H. M. Hau, T. Chen, R. Giovine, K. P. Koirala, L. Li, H. Ji, Y. Ha, Y. Sun, J. Huang, Y. Chen, V. Wu, W. Yang, C. Wang, R. J. Clément, Z. Lun and G. Ceder, Nat. Energy, 2024, 9, 27–36 CrossRef CAS
- T. Chen, J. Yang, L. Barroso-Luque and G. Ceder, ACS Energy Lett., 2023, 8, 314–319 CrossRef CAS
- E. Lee, T. U. Wi, J. Park, S. W. Park, M. H. Kim, D. H. Lee, B. C. Park, C. Jo, R. Malik, J. H. Lee, T. J. Shin, S. J. Kang, H. W. Lee, J. Lee and D. H. Seo, Adv. Mater., 2023, 35, 2208423 CrossRef CAS PubMed
- D. H. Kwon, J. Lee, N. Artrith, H. Kim, L. Wu, Z. Lun, Y. Tian, Y. Zhu and G. Ceder, Cell Rep. Phys. Sci., 2020, 1, 100187 CrossRef
- J. Ahn, R. Giovine, V. C. Wu, K. P. Koirala, C. Wang, R. J. Clément and G. Chen, Adv. Energy Mater., 2023, 13, 2300221 CrossRef CAS
- R. C. Longo, F. T. Kong, K. C. Santosh, M. S. Park, J. Yoon, D. H. Yeon, J. H. Park, S. G. Doo and K. Cho, Phys. Chem. Chem. Phys., 2014, 16, 11233–11242 RSC
- C. Xu, S. De, A. M. Balu, M. Ojeda and R. Luque, Chem. Commun., 2015, 51, 6698–6713 RSC
- H. Kurata and C. Colliex, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 2102–2108 CrossRef CAS PubMed
- H. Tan, J. Verbeeck, A. Abakumov and G. Van Tendeloo, Ultramicroscopy, 2012, 116, 24–33 CrossRef CAS
- Z. L. Wang, J. S. Yin and Y. D. Jiang, Micron, 2000, 31, 571–580 CrossRef CAS PubMed
- Y. Wei, K. B. Kim and G. Chen, Electrochim. Acta, 2006, 51, 3365–3373 CrossRef CAS
- P. Legutko, T. Jakubek, W. Kaspera, P. Stelmachowski, Z. Sojka and A. Kotarba, Top. Catal., 2017, 60, 162–170 CrossRef CAS
- C. M. Julien and M. Massot, Mater. Sci. Eng., B, 2003, 100, 69–78 CrossRef
- M. M. Thackeray and K. Amine, Nat. Energy, 2021, 6, 566 CrossRef CAS
- J. Zheng, M. Gu, J. Xiao, P. Zuo, C. Wang and J. G. Zhang, Nano Lett., 2013, 13, 3824–3830 CrossRef CAS PubMed
- F. Jiao, J. Bao, A. H. Hill and P. G. Bruce, Angew. Chem., Int. Ed., 2008, 47, 9711–9716 CrossRef CAS PubMed
- S. Zhang, W. Deng, R. Momen, S. Yin, J. Chen, A. Massoudi, G. Zou, H. Hou, W. Deng and X. Ji, J. Mater. Chem. A, 2021, 9, 21532–21550 RSC
- N. Labyedh, F. Mattelaer, C. Detavernier and P. M. Vereecken, J. Mater. Chem. A, 2019, 7, 18996–19007 RSC
- C. D. Amos, M. A. Roldan, M. Varela, J. B. Goodenough and P. J. Ferreira, Nano Lett., 2016, 16, 2899–2906 CrossRef CAS PubMed
- H. H. Heenen, C. Scheurer and K. Reuter, Nano Lett., 2017, 17, 3884–3888 CrossRef CAS PubMed
- A. Abdellahi, A. Urban, S. Dacek and G. Ceder, Chem. Mater., 2016, 28, 3659–3665 CrossRef CAS
- J. Lee, D. A. Kitchaev, D. H. Kwon, C. W. Lee, J. K. Papp, Y. S. Liu, Z. Lun, R. J. Clément, T. Shi, B. D. McCloskey, J. Guo, M. Balasubramanian and G. Ceder, Nature, 2018, 556, 185–190 CrossRef CAS PubMed
- Z. Lun, B. Ouyang, D. H. Kwon, Y. Ha, E. E. Foley, T. Y. Huang, Z. Cai, H. Kim, M. Balasubramanian, Y. Sun, J. Huang, Y. Tian, H. Kim, B. D. McCloskey, W. Yang, R. J. Clément, H. Ji and G. Ceder, Nat. Mater., 2021, 20, 214–221 CrossRef CAS PubMed
- H. Ji, A. Urban, D. A. Kitchaev, D. H. Kwon, N. Artrith, C. Ophus, W. Huang, Z. Cai, T. Shi, J. C. Kim, H. Kim and G. Ceder, Nat. Commun., 2019, 10, 592 CrossRef CAS PubMed
- C. Zuo, Z. Hu, R. Qi, J. Liu, Z. Li, J. Lu, C. Dong, K. Yang, W. Huang, C. Chen, Z. Song, S. Song, Y. Yu, J. Zheng and F. Pan, Adv. Energy Mater., 2020, 10, 2000363 CrossRef CAS
- J. Reed, G. Ceder and A. Van Der Ven, Electrochem. Solid-State Lett., 2001, 4, 78–81 CrossRef
- A. Urban, J. Lee and G. Ceder, Adv. Energy Mater., 2014, 4, 1400478 CrossRef
- H. Xia, Z. Luo and J. Xie, Prog. Nat. Sci.: Mater. Int., 2012, 22, 572–584 CrossRef
- R. Hendriks, D. M. Cunha, D. P. Singh and M. Huijben, ACS Appl. Energy Mater., 2018, 1, 7046–7051 CrossRef CAS PubMed
- Q. Li, Y. Wang, X. Wang, X. Sun, J. N. Zhang, X. Yu and H. Li, ACS Appl. Mater. Interfaces, 2020, 12, 2319–2326 CrossRef CAS PubMed
- H. Liu, A. J. Naylor, A. S. Menon, W. R. Brant, K. Edström and R. Younesi, Adv. Mater. Interfaces, 2020, 7, 2000277 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Table S1–S4, Fig. S1–29. See DOI: https://doi.org/10.1039/d4ta02173h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |