
Enhancing the performance of indoor organic photovoltaics through precise modulation of chlorine density in wide bandgap random copolymers†
Soyoung
Kim
ab,
Seon Joong
Kim
c,
Gayoung
Ham
d,
Ji-Eun
Jeong
 f,
Donghwa
Lee
g,
Eunho
Lee
g,
Hyungju
Ahn
h,
Hyojung
Cha
*de,
Jae Won
Shim
*c and
Wonho
Lee
*ab
f,
Donghwa
Lee
g,
Eunho
Lee
g,
Hyungju
Ahn
h,
Hyojung
Cha
*de,
Jae Won
Shim
*c and
Wonho
Lee
*ab
aDepartment of Polymer Science and Engineering, Kumoh National Institute of Technology, Gumi, Gyeongbuk 39177, Republic of Korea
bDepartment of Energy Engineering Convergence, Kumoh National Institute of Technology, Gumi, Gyeongbuk 39177, Republic of Korea. E-mail: 1holee@kumoh.ac.kr
cSchool of Electrical Engineering, Korea University, Seoul 02841, Republic of Korea. E-mail: jwshim19@korea.ac.kr
dDepartment of Energy Convergence and Climate Change, Kyungpook National University, Daegu 41566, Republic of Korea
eDepartment of Hydrogen and Renewable Energy, Kyungpook National University, Daegu 41566, Republic of Korea. E-mail: hcha@knu.ac.kr
fCenter for Advanced Specialty Chemicals, Korea Research Institute of Chemical Technology, Ulsan 44412, Republic of Korea
gDepartment of Chemical Engineering, Kumoh National Institute of Technology, Gumi, Gyeongbuk 39177, Republic of Korea
hPohang Accelerator Laboratory, Pohang, Gyeongbuk 37673, Republic of Korea
First published on 22nd December 2023
Abstract
We present highly efficient indoor organic photovoltaic (IOPV) devices based on a series of four wide-bandgap random copolymers, denoted as B30T70-XCl (X = 0, 2, 4 and 6). The absorption range of these copolymers efficiently covers the spectral range of indoor light sources, with a systematic decrease in the HOMO levels based on the number of chlorine atoms (0 > 2 > 4 > 6Cl). The introduction of Cl is an effective and cost-efficient strategy because of the simplicity of the synthesis. We use PC71BM as the electron acceptor, which not only effectively absorbs indoor light spectra, but also significantly reduces production costs compared with state-of-the-art non-fullerene acceptors (NFAs). Among the B30T70-XCl:PC71BM blends, the B30T70-2Cl-based devices exhibit optimized power conversion efficiencies (PCEs) with a high VOC, achieving a record-breaking PCE of 25.0% under fluorescent lamp (FL) illumination, compared with reported fullerene-based IOPVs. Through a comprehensive analysis of the energy levels, transient absorption dynamics, and blend morphology, we reveal that increasing the Cl density decreases the HOMO offset between the polymer donors and the PC71BM acceptor and induces a phase-separated blend morphology, critically impacting the performance of IOPVs by influencing the population of charge-separated states and charge transport behavior, respectively. The performance of these IOPVs based on wide-bandgap random copolymers and the PC71BM acceptor suggests that the development of such classical, low-cost photoactive layer blends holds promise for integration into low-power portable electronics and Internet-of-Things (IoT) sensors.
1. Introduction
The widespread use of the Internet-of-Things (IoT), enabling real-time data collection, analysis, and decision-making, requires the development of indoor energy harvesting devices for enhancing the quality of human life.1–7 Among various energyharvesting technologies, indoor organic photovoltaics (IOPVs), which utilize organic semiconductors as photoactive layers, have gained significant attention due to their light weight, flexibility, and cost-effectiveness for large-area manufacturing through solution processing.8–12 Notably, IOPVs offer the advantages of a high photon-to-electricity conversion efficiency under illumination by indoor light sources, thanks to their high shunt resistance and thin-film nature with vivid colors, making them appealing for indoor applications.10,13–15 Recent advancements in optimizing photoactive materials, buffer layers, and device architectures have yielded remarkable power conversion efficiencies (PCEs), exceeding 30% in IOPVs.16–18 However, despite their superior performance, IOPVs based on non-fullerene acceptors (NFAs) face challenges in penetrating the mass market primarily due to the high synthetic complexity (SC) of the photoactive layer materials, resulting in high manufacturing costs.19–21 This limits the suitability of IOPVs for low-power IoT applications that operate in low-light conditions, given the typically low power levels generated from indoor light sources (on the order of μW cm−2).16,17,19,20In contrast with solar energy, which covers a wide range of the UV, visible, and infrared (IR) regions with high irradiance (100 mW cm−2), indoor lighting sources such as fluorescent lamps (FLs) and light-emitting diodes (LEDs) emit light in a narrow range of the visible region (400–750 nm) with low intensity (100–1000 lx, 25–300 μW cm−2).22 To achieve high-performance IOPVs, two key considerations are crucial. Firstly, the absorption spectrum of the photoactive layer materials, consisting of electron donors and acceptors, should be well-matched with the emission spectrum of the indoor light source.23–25 Hence, both donors and acceptors need to have a wide bandgap to efficiently convert the narrow and low-intensity indoor light into electricity.17,22 Secondly, the magnitude of the open-circuit voltage (VOC) is influenced by the incident light power (VOC ∝ log(Pin)), resulting in lower VOC values under indoor lighting conditions compared to those under 1-Sun irradiance.26–28 For instance, VOC values are approximately 0.17 V lower under 300 lx indoor lighting, commonly found in living rooms.29 Therefore, developing photoactive materials with a high VOC is crucial, requiring the design of donor/acceptor combinations with deep highest occupied molecular orbital (HOMO) levels for donors and shallow lowest unoccupied molecular orbital (LUMO) levels for acceptors.22,30,31 Our previous study highlighted the effectiveness of simple-structured random copolymers, comprising only two conjugated moieties, 4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)benzo[1,2-b:4,5-b′]dithiophene (BDTT) and thieno[3,4-c]pyrrole-4,6-dione (TPD), for IOPV applications.25 Despite the typically poor performance of polymers containing TPD under 1-Sun conditions, TPD was selected due to its relatively low SC (18.3%) compared to that of other widely-known accepting moieties such as 1,3-bis(thiophen-2-yl)-5,7-bis(2-ethylhexyl)benzo-[1,2-c:4,5-c′]dithiophene-4,8-dione (BDD, 27.9%), dithieno[3′,2′:3,4;2′′,3′′:5,6]benzo[1,2-c][1,2,5]thiadiazole (DTBT, 49.6%), fluorinated thieno[3,4-b]thiophene (TT, 55.2%), fluorinated 2,1,3-benzothiadiazole (BT, 35.4%), etc.19,25 Furthermore, we used PC71BM as the electron acceptor, which not only effectively absorbs indoor light spectra but also significantly reduces production costs compared to state-of-the-art non-fullerene acceptors (NFAs). For example, fullerene acceptors exhibit a remarkably low SC, with PC71BM standing out at 17.4%, which ranks among the lowest SC values for electron acceptor materials (e.g., SC of Y6: 71.0%; SC of ITIC: 54.5%). Thus, the random copolymer:PC71BM blends demonstrated efficient light absorption across the entire spectra of FLs and LEDs, with a high VOC, leading to good IOPV performance.
It has been reported that introducing electron-withdrawing groups (EWGs) such as fluorine (F) and chlorine (Cl) into the thiophene unit of the side chain of the BDTT moiety is a simple and effective method that does not require extensive modifications to the polymer backbone.31–37 The introduction of these EWGs into the side chains of polymer donors leads to a downshift of the HOMO level, thereby enhancing the VOC under indoor lighting conditions, while minimally impacting the absorption spectra, ensuring that the polymer donors maintain their optimized absorption range for indoor lighting.33,38–40 Furthermore, non-covalent bonding interactions can enhance the crystallinity of the polymers, potentially increasing the JSC and FF.33,34,41–44 Among the halogens, Cl exhibits a higher dipole moment and possesses empty d-orbitals, enabling an efficient downshift of the HOMO level and enhanced intermolecular interactions.33,42,45,46 Importantly, Cl-attached thienyl side chains offer a more cost-effective synthesis route compared to F-attached side chains, requiring four less synthetic steps and thus reducing manufacturing costs.34,41,47–51 For example, when Cl atoms are introduced onto the thienyl side chains of BDTT, the SC increases from 30.4% (BDTT) to 34.2% (BDTT-Cl). In contrast, upon fluorination, the SC increases significantly to 60.01% (BDTT-F).52,53
In this study, we aim to further enhance the PCE of a B30T70 ([BDTT-BDTT]30-ran-[BDTT-TPD]70) random copolymer by incorporating electron-withdrawing Cl atoms into the side chains of the BDTT moiety. Through the synthesis of B30T70, B30T70-2Cl, B30T70-4Cl, and B30T70-6Cl polymer donors, we systematically varied the Cl density in the copolymer. As the Cl density increases, the absorption range changes minimally, thus preserving the optimized absorption range of the B30T70 copolymers for indoor lighting while gradually downshifting the HOMO level of the polymer donors: B30T70 (−5.14 eV), B30T70-2Cl (−5.34 eV), B30T70-4Cl (−5.43 eV), and B30T70-6Cl (−5.53 eV). Consequently, a remarkable PCE of 25.0% is achieved for the B30T70-2Cl:PC71BM-based device under FL illumination at 1000 lx, setting a new efficiency record for fullerene-based IOPVs. This achievement is attributed to energy level optimization and the excellent utilization of FL illumination. Notably, B30T70-2Cl shows a moderate performance under AM 1.5 G (7.8%) irradiation but exhibits a significant 3.2-fold increase in efficiency under indoor lighting compared to 1-Sun conditions. In contrast, devices based on B30T70-4Cl:PC71BM (12.5% under FL 1000 lx) and B30T70-6Cl:PC71BM (7.5% under FL 1000 lx) show a substantial decrease in efficiency. Through comprehensive analysis, including energy levels, transient absorption dynamics, and blend morphology, and more, we identified that monomolecular recombination resulting from an insufficient HOMO offset (ΔEHOMO) is the dominant factor leading to the decreased performance. Additionally, increasing the Cl density from B30T70 to B30T70-6Cl induces phase separation of the blend morphology, hindering charge transport. These findings offer valuable insights into the substantial impact of incorporating electron-withdrawing Cl atoms into IOPVs and pave the way for developing highly efficient and cost-effective devices for low-power IoT applications.
2. Results and discussion
2.1. Synthesis, physical, and optical properties of B30T70-XCl (X = 0, 2, 4 and 6) random copolymers
Our primary objective is to enhance the PCEs of IOPVs by precisely modulating the chemical structure of the side chains in the B30T70 random copolymer. Our previous report demonstrated that the parent polymer B30T70, synthesized through a Stille coupling reaction with 30% BDTT-BDTT and 70% BDTT-TPD units, exhibited excellent IOPV performance due to its ideal spectral match with both indoor FL and LED light sources.25 To systematically control the Cl density per repeating unit and potentially decrease the HOMO level while maintaining the absorption region of the B30T70 polymer donor, we synthesized four polymeric donors: B30T70, B30T70-2Cl, B30T70-4Cl, and B30T70-6Cl by introducing Cl into the thienyl group of the BDTT moiety (Fig. 1a). Briefly, we prepared Cl-free (BDTT-Br and BDTT-Sn) and chlorinated monomers (BDTTCl-Br and BDTTCl-Sn). These monomers were then combined in different combinations: B30T70-2Cl was synthesized using BDTTCl-Br, B30T70-4Cl was synthesized using BDTTCl-Sn, and B30T70-6Cl was synthesized using both the BDTTCl-Br and BDTTCl-Sn monomers. For a detailed description of the synthetic procedure, please refer to the Experimental section, as well as Scheme S1 and Table S1.† Successful modulation of the Cl density was verified by elemental analysis (EA). The results clearly showed that the Cl density systematically increased from B30T70 to B30T70-6Cl, and the measured values were in good agreement with the theoretical values (Table S2†). Additionally, the number average molecular weights (Mn) of the polymers were determined using gel permeation chromatography (GPC): the Mn values of B30T70, B30T70-2Cl, B30T70-4Cl, and B30T70-6Cl were 20, 41, 24, and 25 kg mol−1, respectively. It's worth noting that while all the polymers were synthesized using the same catalyst system, relatively diverse Mn values were obtained. GPC analysis of conjugated polymers can sometimes yield varying results because the instrument is calibrated using flexible polymer standards, and the solubility of conjugated polymers in the mobile phase is relatively poor.54 Nevertheless, it is essential to emphasize that all the polymers synthesized in this study exhibited sufficiently high molecular weights for a meaningful and valid comparison.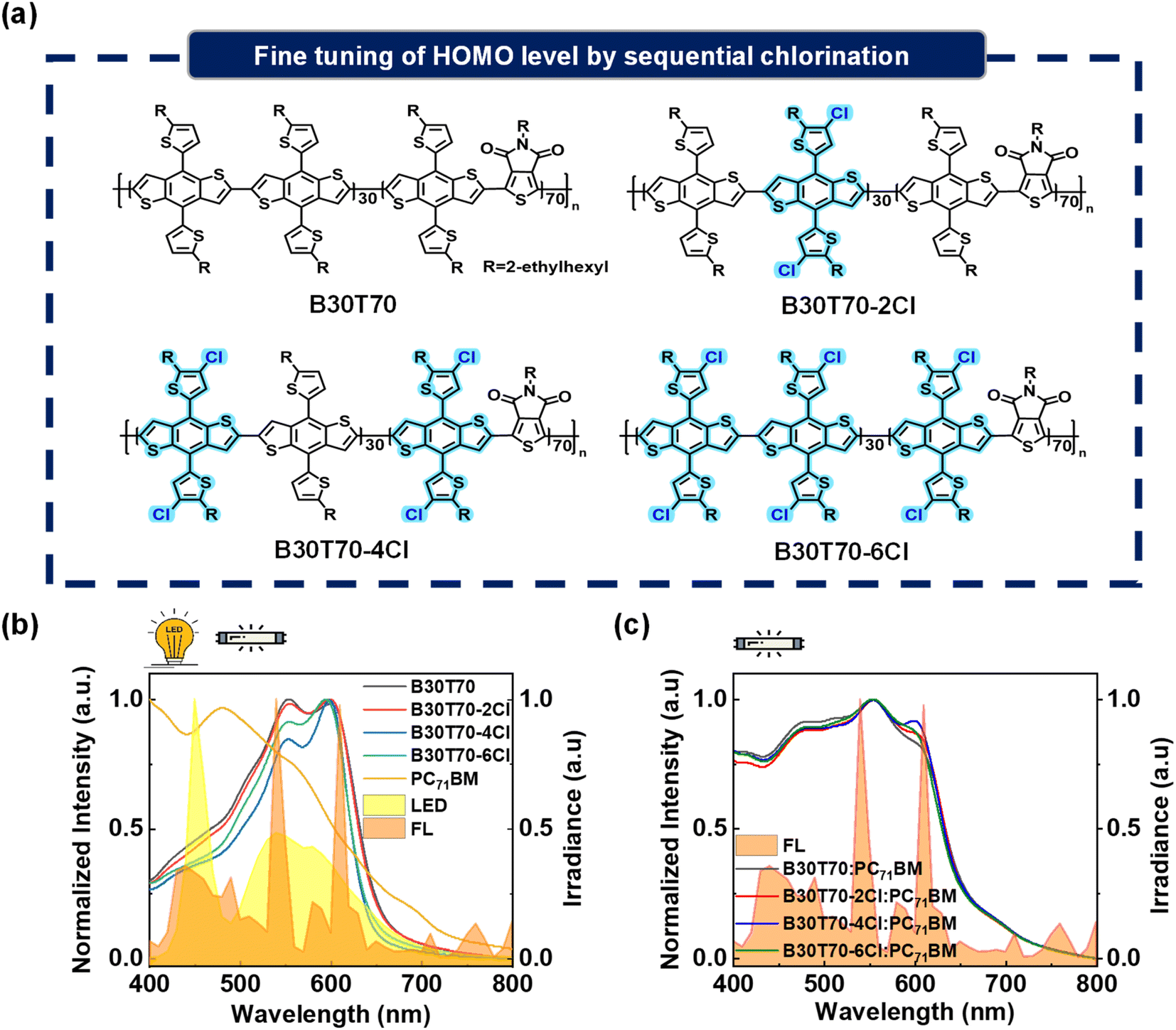 | ||
| Fig. 1 (a) Chemical structures of B30T70-XCl (X = 0, 2, 4 and 6), (b) absorption spectra of pristine polymer donors, acceptor PC71BM, and illumination spectra of indoor light sources (LED and FL), and (c) absorption spectra of blend films. | ||
We investigated the thermal properties of the random copolymers. All the polymers exhibited high stability, with a decomposition temperature (TD) above 400 °C, as evaluated through thermogravimetric analysis (TGA) (Fig. S1a†). Moreover, our previous study confirmed that the B30T70 random copolymer has an amorphous structure.25 Chlorination of the side chains in the random copolymers did not induce any crystalline properties. Differential scanning calorimetry (DSC) analysis revealed no melting (Tm) and crystallization temperatures (Tc) within a wide temperature range of 50–300 °C (Fig. S1b†).25,55 These findings validate the successful synthesis and thermal properties of the B30T70-XCl random copolymers, providing a solid basis for further investigation of their optoelectronic properties and device performance.
Achieving an absorption band suitable for indoor light sources is crucial for producing high-performance IOPVs. The effect of varying the density of Cl in the side chains of the random copolymers on the absorption characteristics was evaluated using UV-vis absorption spectroscopy. The absorption spectra of the pristine donor and acceptor in the film state are presented in Fig. 1b. The detailed values of parameters such as the absorption edge (λedge) and maxima (λmax) are provided in Table S3.† The introduction of Cl had no significant effect on the absorption region, suggesting that all polymers efficiently absorbed the emission range of FL and LED sources used in this study. Upon closer examination of the absorption spectra, we observed a slight increase in the bandgap from B30T70 (1.88 eV) and B30T70-2Cl (1.88 eV) to B30T70-4Cl (1.92 eV) and B30T70-6Cl (1.91 eV), accompanied by a very slight blue-shift in the λedge of approximately 12 nm. For B30T70-4Cl and B30T70-6Cl, we speculate that the substitution of the electron-withdrawing Cl substituent on the electron-donating moiety of BDTT somewhat suppresses the intramolecular charge transfer (ICT) interaction from the BDTT to TPD units. Unlike B30T70-4Cl and B30T70-6Cl, the absorption spectrum of B30T70-2Cl was similar to that of B30T70 because of the relatively low Cl density of B30T70-2Cl (Table S1†). Upon examining the absorption spectra of the B30T70-XCl (X = 0, 2, 4 and 6):PC71BM blend films shown in Fig. 1c, it is evident that all blend films exhibit sufficient absorption across a broad range in the visible region, with PC71BM complementing the insufficient absorption of the polymer donors at shorter wavelength. Although the longer-wavelength absorption of the pristine films of B30T70-4Cl and B30T70-6Cl was slightly blue-shifted, the λedge was the same for all the blend films due to the ability of PC71BM to absorb light up to 720 nm. In summary, although chlorination of the side chains induced a slight blue-shift in the absorption of the B30T70-4Cl and 6Cl polymers, all the blend films exhibited almost identical absorption profiles that effectively cover the range of indoor light sources owing to the complementary absorption of the B30T70-XCl polymer donors and the PC71BM acceptor.
2.2. Device performance under 1-Sun and indoor light sources
To investigate the effect of precise modulation of chlorination process within the B30T70 polymer donors on the performance of OPVs, we fabricated devices with an inverted structure of the ITO/ZnO/PEIE/polymers:PC71BM/MoOx/Ag. All polymers exhibited excellent solubility in chlorobenzene (CB) up to 50 mg mL−1. Consequently, the photoactive layers were deposited by dissolving the donor and acceptor mixture in CB with a weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (D:A) at a total concentration of 25 mg mL−1. Fig. 2a shows the J–V curves of the OPVs under AM 1.5 G; the photovoltaic parameters are summarized in Table 1. The device fabrication conditions are described in detail in the experimental section.
:2 (D:A) at a total concentration of 25 mg mL−1. Fig. 2a shows the J–V curves of the OPVs under AM 1.5 G; the photovoltaic parameters are summarized in Table 1. The device fabrication conditions are described in detail in the experimental section.
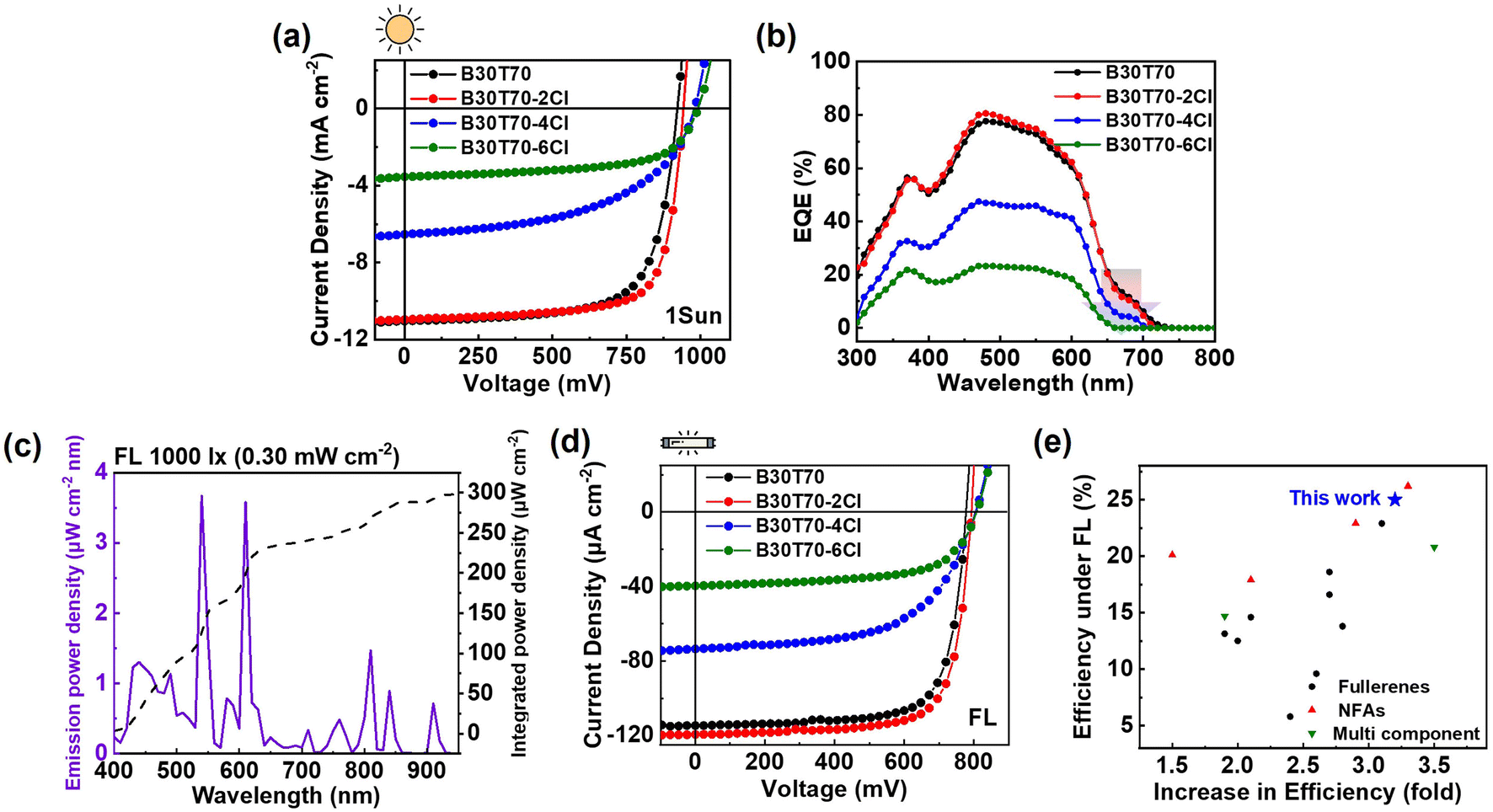 | ||
| Fig. 2 (a and d) J–V curves of B30T70-XCl (X = 0, 2, 4 and 6):PC71BM devices under illumination of 1-Sun and FL 1000 lx, respectively, and (b) corresponding EQE spectra of B30T70-XCl (X = 0, 2, 4 and 6):PC71BM. (c) Emission power density and integrated power density of FL 1000 lx illumination. (e) Comparison of PCE and increase in PCE of IOPVs under FL illumination. | ||
| Polymer | Condition | V OC (mV) | J SC (outdoor: mA cm−2, indoor: μA cm−2) | J cal (mA cm−2) | FF (%) | PCEavga (PCEmax) (%) |
|---|---|---|---|---|---|---|
| a Average PCE values obtained from 5 different devices. | ||||||
| B30T70 | AM 1.5 G | 914 ± 7 | 10.9 ± 0.3 | 10.97 | 71.2 ± 1.8 | 7.2 ± 0.1 (7.4) |
| B30T70-2Cl | 927 ± 10 | 11.0 ± 0.2 | 11.18 | 73.0 ± 1.2 | 7.5 ± 0.2 (7.8) | |
| B30T70-4Cl | 979 ± 6 | 6.7 ± 0.2 | 6.50 | 52.6 ± 1.2 | 3.4 ± 0.2 (3.6) | |
| B30T70-6Cl | 991 ± 2 | 3.6 ± 0.2 | 3.12 | 61.1 ± 1.3 | 2.2 ± 0.1 (2.4) | |
| B30T70 | FL 1000 lx (irradiance 0.30 mW cm−2) | 778 ± 15 | 117.2 ± 3.7 | — | 72.2 ± 1.8 | 22.4 ± 0.5 (22.9) |
| B30T70-2Cl | 797 ± 13 | 120.7 ± 1.0 | — | 74.9 ± 1.7 | 24.0 ± 1.0 (25.0) | |
| B30T70-4Cl | 811 ± 8 | 75.1 ± 1.7 | — | 60.7 ± 0.8 | 12.3 ± 0.2 (12.5) | |
| B30T70-6Cl | 814 ± 4 | 41.2 ± 1.2 | — | 64.6 ± 1.2 | 7.2 ± 0.2 (7.5) | |
The device based on the non-chlorinated B30T70 polymer donor exhibited a PCEavg of 7.2%, with a VOC of 914 mV, a JSC of 10.9 mA cm−2, and an FF of 71.2%. As the Cl density increased, a clear increasing trend in the VOC was observed: 914, 927, 979, and 991 mV for B30T70, B30T70-2Cl, B30T70-4Cl, and B30T70-6Cl, respectively. The B30T70- and B30T70-2Cl-based devices yielded similar JSC values of 10.9 and 11.0 mA cm−2, respectively. However, we observed a sudden decrease in the JSC for the B30T70-4Cl- and B30T70-6Cl-based devices, with values of 6.7 and 3.6 mA cm−2, respectively. Similarly, the FF value of the B30T70-4Cl devices (52.6%) was lower than that of the B30T70-2Cl-based devices (73%). Therefore, among the B30T70-XCl:PC71BM devices, the B30T70-2Cl-based device exhibited the highest PCEavg of 7.5%, whereas the B30T70-4Cl- and B30T70-6Cl-based devices had a poor PCEavg of 3.4% and 2.2%, respectively. The reason for the poor performance of the B30T70-4Cl and B30T70-6Cl blends, despite their high VOC, is discussed hereinafter. The external quantum efficiency (EQE) data are presented in Fig. 2b, and the calculated JSC values (Jcal) are summarized in Table 1. The EQEs of the B30T70-4Cl- and B30T70-6Cl-based devices decreased noticeably across the entire area compared to those of the B30T70- and B30T70-2Cl-based devices. In addition, the low-intensity peak in the 650–720 nm range is attributed to the photocurrent generated by PC71BM absorption, which was confirmed by PC71BM absorption spectrum in Fig. 1b. In this region, B30T70 and B30T70-2Cl exhibited similar EQE spectra, however, the peak intensities weakened with increasing Cl density. Consequently, for B30T70-6Cl, no EQE response was recorded in the range of 650–720 nm range. Comprehensive explanations of these phenomena are provided in the following sections.
The OPV performance under indoor light conditions using FL with an irradiance of 0.30 mW cm−2 was evaluated (Fig. 2c). The J–V curves and corresponding photovoltaic parameters are presented in Fig. 2d and Table 1, respectively. Overall, the trend was similar to that observed under 1-Sun illumination. The B30T70-2Cl-based device yielded a higher PCEavg of 24.0% compared to that of the B30T70-based device (22.4%), whereas the B30T70-4Cl (12.3%) and B30T70-6Cl (7.2%)-based devices exhibited considerably lower PCEs. The remarkable performance of the B30T70-2Cl-based device can be attributed to its perfect spectral match (Fig. 1c), as the blend effectively absorbed the illumination maxima of FL, and the deep HOMO level of the polymer. Notably, our IOPVs show a dramatic increase in PCE, approximately 3.2-fold, upon transitioning from 1-Sun to FL illumination; the moderate PCEmax of 7.8% for the B30T70-2Cl devices under 1-Sun increased to 25.0% under FL illumination (Table S4†). To quantify the efficiency of the B30T70-2Cl-based device, we compared its IOPV performance with that of previously reported devices evaluated under FL illumination (Fig. 2e and Table S5†). The B30T70-2Cl-based device achieved the second-highest efficiency among all types of IOPVs. Many recently reported high-efficiency solar cells are based on NFAs, which are not suitable for low-power IOPVs because of their high SC. A glance at the summary of the SC values in Table S6† shows that most NFAs have very high SC values; notable examples include Y6, ITIC, and IT-4F, with SC values of 71.0%, 54.5%, and 64.1%, respectively.19,21 In contrast, the fullerene acceptors PC61BM and PC71BM have much lower SC values of only 17.4%, making them far more suitable for IOPV applications. When considering only the fullerene acceptor-based devices, our B30T70-2Cl:PC71BM devices achieved the highest PCE under FL illumination (Fig. 2e). Moreover, our IOPVs demonstrated one of the most significant increases in the PCE (3.2-fold increase from 1-Sun to indoor conditions), ranking third in this aspect. We also conducted the same experiment with a commonly used LED light source (0.254 mW cm−2) (Fig. S2 and Table S7†). Similar to the results obtained under FL illumination, the B30T70-2Cl-based device achieved the highest efficiency (22.0%) among all analyzed devices. We also collected previous reports evaluated under LEDs and plotted them in Fig. S2c and Table S8.† Compared to that of devices based on all types of small-molecule acceptors, the PCE of the B30T70-2Cl:PC71BM devices demonstrated moderate efficiency. However, within the realm of fullerene-based devices, our device exhibited superior performance, ranking 2nd after the PDTBTBz-2Fanti:PC71BM device.56
Long-term stability of IOPVs under continuous illumination is crucial for practical applications, particularly when integrated into low-power IoT devices.22,57 To evaluate the stability of our devices, we conducted long-term lifetime tests under continuous LED illumination at 2000 lx (Fig. S3a†). The B30T70-2Cl:PC71BM-based devices exhibited no critical performance degradation for more than 300 h under these accelerated aging conditions, demonstrating their robustness and suitability for real-world applications. Moreover, to assess the feasibility of large-scale production, we fabricated and evaluated the performance of large-area devices with active areas of 1 cm2 (Fig. S3b, c and Table S9†). The B30T70-2Cl:PC71BM-based large-area devices achieved a high PCE of 21.5%, confirming the scalability and potential of these devices for practical implementation in indoor energy-harvesting applications.
2.3. Effects of Cl density on energy levels and blend morphology
Increasing the Cl density gradually down-shifted the HOMO levels of the polymer donors, leading to a higher VOC in the devices. However, a sharp decrease in the PCE was observed for the B30T70-4Cl- and B30T70-6Cl-based devices because of the severe decrease in the JSC and FF. Therefore, B30T70-2Cl showed the optimal efficiency, with a high VOC, JSC, and FF. Two factors plausibly contributed to the sudden decrease in the PCEs of B30T70-4Cl and B30T70-6Cl: (1) energy level mismatch between the polymer donors and PC71BM acceptor, and (2) unfavorable blend morphology.To examine the energy level alignment between the polymer donors and PC71BM acceptor for efficient exciton dissociation and charge separation, the frontier molecular orbital energy levels (HOMO and LUMO) of the polymer donors and PC71BM acceptor were measured using photoelectron spectroscopy in air (PESA) and cyclic voltammetry (CV).58,59Fig. 3a shows the PESA results, where the ionization energy (IE) of the polymers gradually increased with increasing Cl density owing to the electron-withdrawing nature of the Cl atom. Based on the PESA measurements, the HOMO/LUMO of the polymer donors and the PC71BM acceptor were calculated (Fig. 3b); the Eoptg values were used to estimate the LUMO levels. With an increase in the EWG density (Cl), the HOMO levels of the polymer donors were gradually down-shifted. The HOMO levels for B30T70, B30T70-2Cl, B30T70-4Cl, and B30T70-6Cl were −5.14, −5.34, −5.43, and −5.53 eV, respectively. This trend corresponds to the VOC observations in photovoltaic devices. In NFA-based OPVs, it has been reported that energy level offsets close to 0 do not significantly impact charge separation.60–66 However, in fullerene acceptor-based OPVs, an energy level offset of 0.3 eV or more is required for efficient charge separation.67–69 For our blend systems, ΔELUMO decreased in the following order: 0.67 eV (B30T70), 0.47 eV (B30T70-2Cl), 0.42 eV (B30T70-4Cl), and 0.31 eV (B30T70-6Cl). This indicates that all the blends have a sufficient driving force for the excitons generated from the polymers to dissociate into the PC71BM acceptor, as further supported by photoluminescence (PL) experiments. In contrast with ΔELUMO, a distinct transition point was observed for ΔEHOMO. The ΔEHOMO values for B30T70 and B30T70-2Cl were 0.51 eV and 0.31 eV, respectively, whereas the ΔEHOMO values for B30T70-4Cl was only 0.22 eV and further decreased to 0.12 eV for B30T70-6Cl. These trends were also confirmed by CV measurements (Fig. S4†). The PESA and CV measurements indicate that the low ΔEHOMO for B30T70-4Cl and B30T70-6Cl may not provide enough driving force for generating a charge-separated state, leading to a sharp drop in JSC and FF (Fig. 3c).61,67,69–72 As the HOMO levels of polymer donors decrease, the free energy difference between the singlet exciton and the charge-separated state, ΔGCS, also increases, resulting in insufficient excess thermal energy to overcome the binding energy of the exciton or charge transfer (CT) state.69 The decrease in ΔEHOMO may also hinder exciton dissociation through hole transfer from excitons generated by PC71BM absorption. These findings are supported by the EQE data (Fig. 2b), which show a significant drop in the intensity in the longer-wavelength region (650–720 nm), particularly for B30T70-6Cl, where the EQE nearly converges to 0. This suggests that excitons in PC71BM do not effectively form CT states or charge-separated states through hole transfer and are more prone to monomolecular recombination.
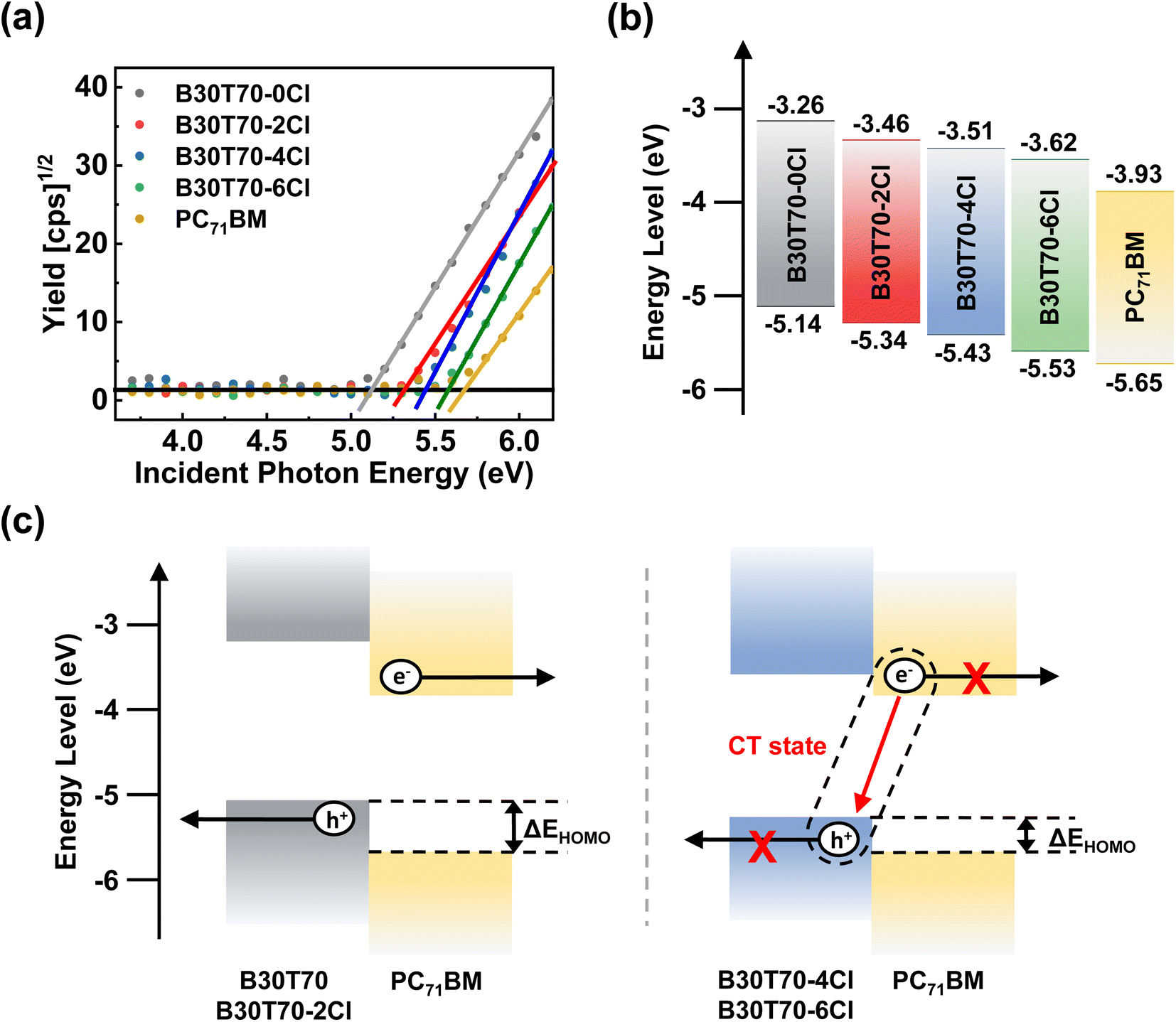 | ||
| Fig. 3 (a) PESA measurements of B30T70-XCl (X = 0, 2, 4 and 6) and PC71BM, along with (b) corresponding energy level diagram. (c) Schematics showing different populations of charge-separated states in B30T70 (or 2Cl) and B30T70-4Cl (or 6Cl) devices. | ||
Next, to investigate the effect of the blend morphology on the photovoltaic performance, we analyzed the atomic force microscopy (AFM) of the blend films. Fig. 4a shows that increasing Cl density induced phase-separated blend morphology, where a smooth morphology was observed for B30T70 and severe phase separation occurred in B30T70-6Cl. Accordingly, the root-mean-square (RMS) roughness increased gradually from B30T70 to B30T70-4Cl and then increased sharply for B30T70-6Cl. Severe phase separation typically reduces the donor/acceptor interface area, hindering exciton dissociation and charge transport.
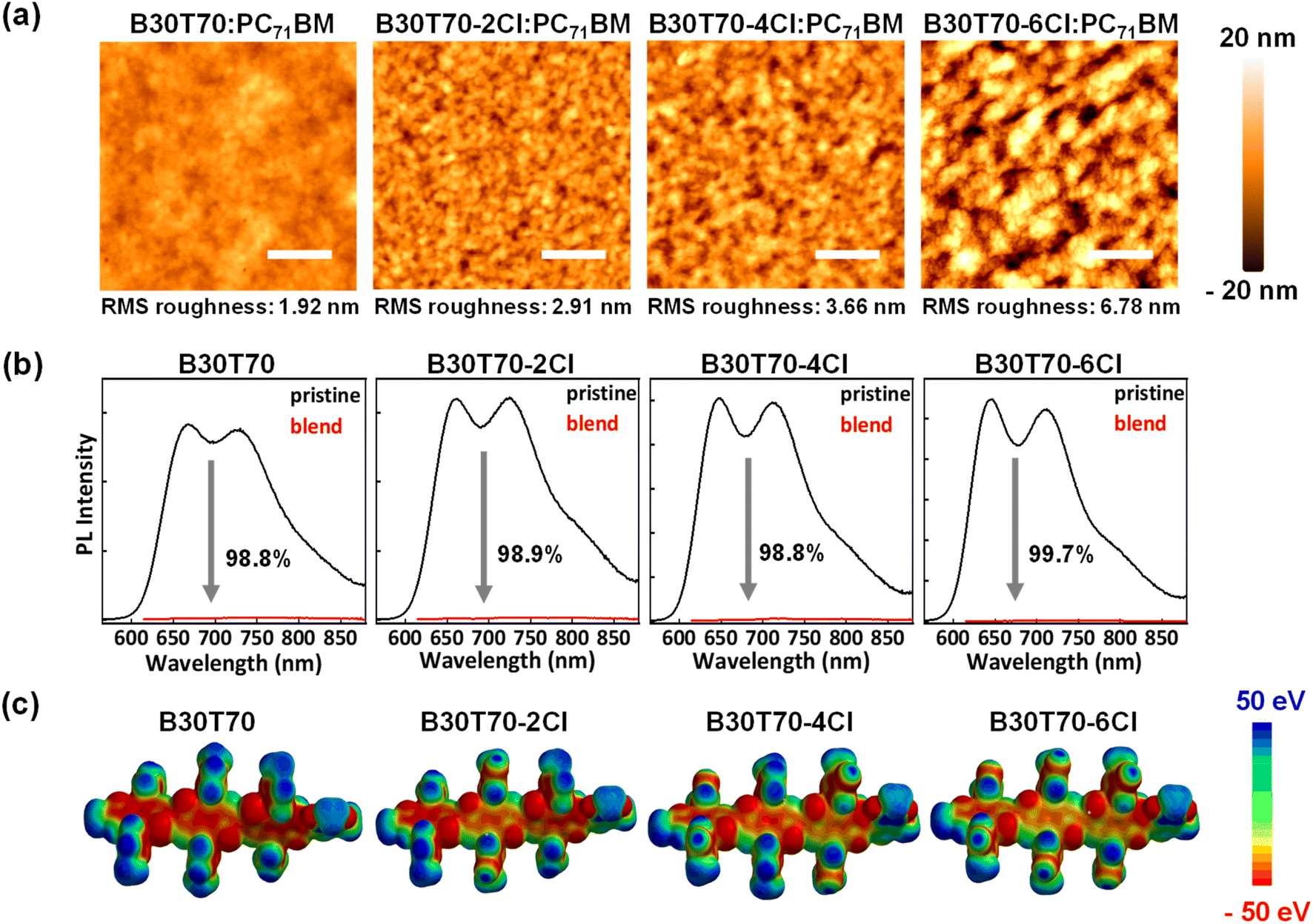 | ||
| Fig. 4 (a) AFM surface topographic images of B30T70-XCl:PC71BM blends (the scale bar in the AFM images is 500 μm), (b) PL quenching efficiency of B30T70-XCl:PC71BM blend films, and (c) ESP map of B30T70-XCl units. | ||
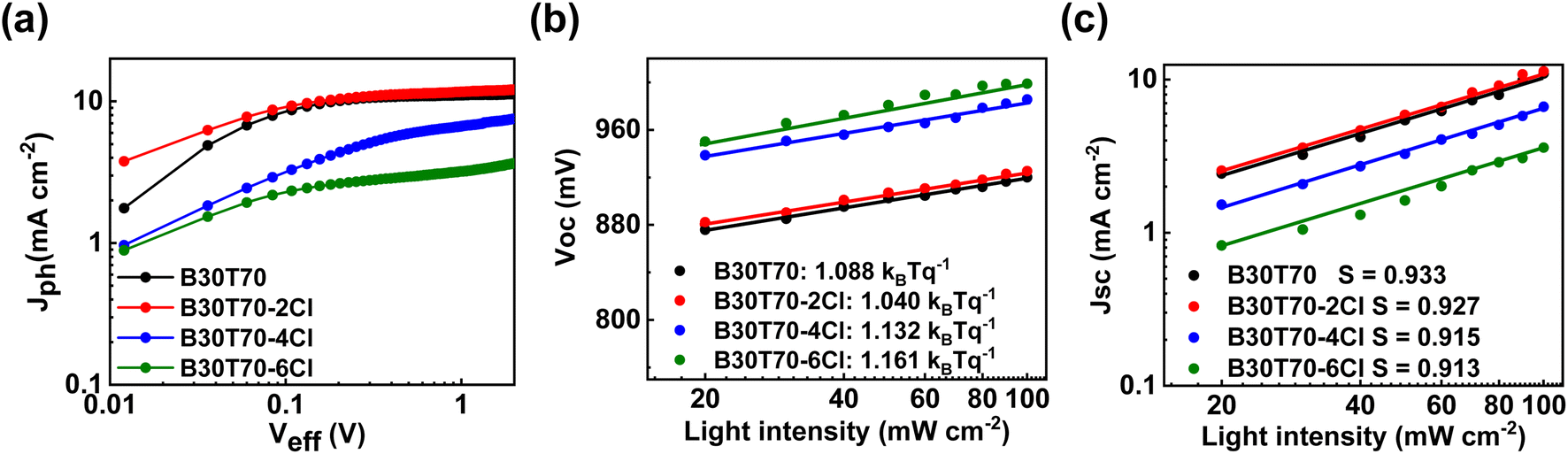
The different phase separation behaviors of the blend films can significantly affect the exciton dissociation efficiency. To confirm this, we measured the PL quenching efficiency of the B30T70-XCl:PC71BM blend films by comparing the PL intensities of the pristine and blend films (Fig. 4b).73–75 Generally, fullerene acceptors exhibit a notably low photoluminescence quantum yield (PLQY); therefore, PL quenching experiments can confirm the efficiency of exciton dissociation generated from absorption in the polymer donor.76,77 All pristine films exhibited Stokes-shifted PL spectra between 650 nm and 850 nm when excited at 600 nm. Interestingly, all of the blend films exhibited almost quenched spectra, with a PL quenching efficiency 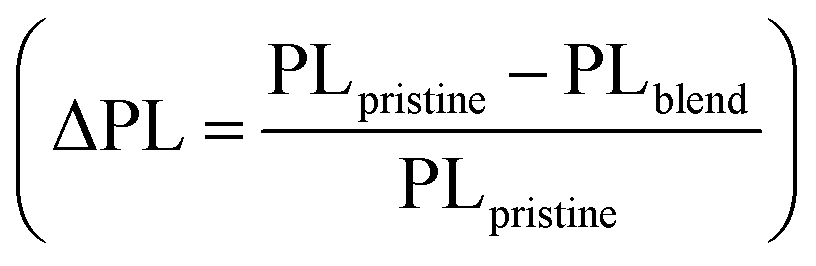 of over 98% for all four blends. This suggests that the phase separation that occurs with increasing Cl density does not adversely affect the exciton dissociation efficiency on the scale observed in this study. Therefore, the sharp decrease in the PCE observed for B30T70-4Cl (and 6Cl) is mainly due to the low driving force for generating charge-separated states from the CT state by the low energy level offset, while the highest PCE is achieved for B30T70-2Cl. Note that bimolecular recombination due to phase separation cannot be ruled out, as discussed hereinafter.
of over 98% for all four blends. This suggests that the phase separation that occurs with increasing Cl density does not adversely affect the exciton dissociation efficiency on the scale observed in this study. Therefore, the sharp decrease in the PCE observed for B30T70-4Cl (and 6Cl) is mainly due to the low driving force for generating charge-separated states from the CT state by the low energy level offset, while the highest PCE is achieved for B30T70-2Cl. Note that bimolecular recombination due to phase separation cannot be ruled out, as discussed hereinafter.
The degree of phase separation with different Cl densities can be attributed to two factors: (1) pre-aggregation of the random copolymers in the solution state prior to film-casting, induced by the Cl atoms and (2) different molecular interactions between the polymer donors and the PC71BM acceptor. Firstly, to determine the degree of the pre-aggregation of random copolymers, we measured the temperature-dependent absorption spectra of a dilute polymer solution in CB.78,79 Fig. S5† displays the temperature-dependent UV-vis absorption spectra of the random copolymers at temperatures from 30 to 110 °C. Overall, for all random copolymers, the 0–0 peak intensity decreased gradually as the temperature increased, indicating reduced intermolecular interactions of the polymer chains at higher temperatures. The decreasing trend of the 0–0 peak intensity depends on the presence or absence of Cl. Chlorinated polymers, B30T70-2Cl, 4Cl, and 6Cl, almost retained their 0–0 peak up to 110 °C, whereas for B30T70, the peak intensity decreased significantly. This difference suggests that the chlorinated polymers exhibit strong intermolecular aggregation induced by strong noncovalent interactions arising from the vacant d-orbitals of Cl.35,38,39,41 However, these results cannot fully explain the distinct phase separation observed among the B30T70-2Cl, 4Cl, and 6Cl polymers, as their temperature-dependent absorption spectra are very similar. Electrostatic potential (ESP) calculations were performed to further understand the weakening of the donor/acceptor molecular interactions with increasing Cl ratio in the polymer donor.80,81 By examining the ESP distributions on the molecular surfaces, we can assess the strength of the intermolecular interactions. We selected the BDTT-BDTT-BDTT-TPD units with methyl chains for easy calculation. Fig. 4c and S6† show the ESP maps of the random copolymers and PC71BM, respectively. It is clearly observed that the main backbone of the BDTT-BDTT units became positive with increasing Cl density because the Cl group attached to the side chains effectively withdraws electrons from the main backbone. Consequently, the molecular interactions with PC71BM, which tends to be positive overall as shown in Fig. S6,† gradually decreased with increasing Cl density, indicating that B30T70-6Cl:PC71BM can exhibit the most severely phase-separated morphology. Furthermore, the ESP results suggest that the increase in the Cl density causes the BDTT-BDTT backbone to become more positively charged, weakening its electron-donating ability to TPD. This interpretation is consistent with the blue-shifts in the absorption spectra of B30T70-4Cl and B30T70-6Cl (Fig. 1b). In summary, phase separation was larger in the blends with higher Cl density in the polymer donors. This is due to two factors: (1) solution phase pre-aggregation, which is more pronounced in the B30T70-2Cl, 4Cl, and 6Cl polymers, affecting the phase separation to some extent, and more majorly, (2) a decrease in the molecular interaction with PC71BM with increasing Cl density, inducing gradual phase separation.
2.4. Charge recombination analysis with varying Cl density
The results of the energy level and morphology studies indicate that an increase in the Cl density leads to a decrease in ΔEHOMO and induces a phase-separated blend morphology. We speculate that the excitons efficiently dissociate, regardless of the degree of phase separation, to form CT states. Nevertheless, the population of the charge-separated states varies depending on the Cl density in the random copolymers. The B30T70-4Cl and 6Cl blends, with the deep HOMO levels, may have a low driving force for producing charge-separated states from CT state. Additionally, the excitons in PC71BM did not effectively form CT states through hole transfer from PC71BM. Consequently, the B30T70-4Cl and 6Cl blends may exhibit high monomolecular recombination, thereby reducing the JSC and FF.82,83 To provide clear evidence for this hypothesis, we conducted a comprehensive study on the charge carrier dynamics in the active layers of photovoltaic devices using transient absorption (TA) spectroscopy. Here, we focused on the time-resolved behavior of the photoinduced charge carriers in the donor:acceptor blends. Specifically, we examined the charge separation yield resulting from four series of B30T70-XCl:PC71BM blend films, covering the time range from a few nanoseconds to 3 μs. The blend films were excited using 355 nm pump pulse and the relative optical density (ΔOD) was subsequently probed using white light after a specific delay time. Considering that the exciton lifetime is known to be within 1 ns, we monitored the CT state dynamics that arise from exciton dissociation.84 Understanding the kinetics of the CT state is crucial for optimizing the device performance, as the kinetics of the CT state directly affect the recombination kinetics of the free charge carriers and their efficient separation into free charges.85,86Fig. 5a–d presents the TA spectra of the B30T70-XCl:PC71BM blends, focusing on the carrier dynamics of the long-lived CT state with a ground state bleaching (GSB) at 620 nm, with the aim of elucidating the influence of CT state on the material energetics and charge separation. The B30T70-2Cl:PC71BM blend exhibited the highest charge generation yield within a few nanoseconds, indicative of favorable charge separation due to its sufficient ΔEHOMO (Fig. 5e). This can be attributed to the presence of long-lived carriers that mitigate monomolecular recombination in the CT state and facilitate effective charge separation. Conversely, we observed fast decay kinetics of the GSB at 620 nm for B30T70-4Cl:PC71BM and B30T70-6Cl:PC71BM, indicating severe CT state recombination and a poor charge separation yield. This behavior is primarily attributed to the deep HOMO levels of the B30T70-4Cl and 6Cl polymers, which are close to the HOMO of PC71BM, hampering effective charge separation.
 | ||
| Fig. 5 (a–d) TAS of B30T70-XCl:PC71BM films for time decays up to 500 ns and (e) transient absorption kinetics monitored with 620 nm probe wavelength. | ||
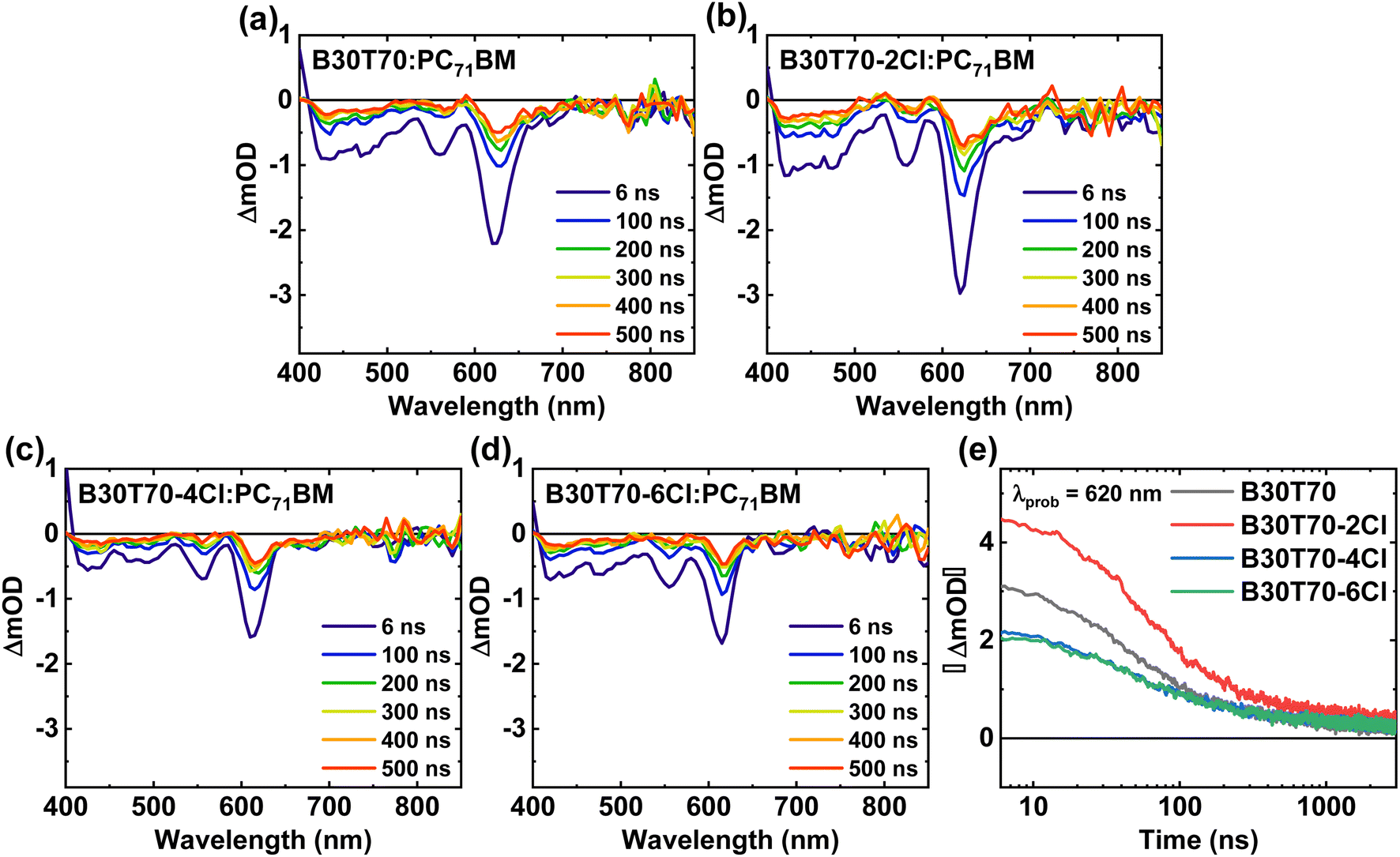
The TA spectra suggest that charge-separated states were not well formed in the B30T70-4Cl and 6Cl blends, and monomolecular recombination was dominant. To confirm this conjecture, the exciton dissociation probabilities (Pdiss, JSC/Jsat) were calculated by measuring the dependence of the photocurrent (Jph) on the effective voltage (Veff) (Fig. 6a).87,88 The calculated JSC, Jsat, and Pdiss values are summarized in Table S10.† The Jph of both B30T70- and B30T70-2Cl-based devices reached saturation at Veff = 1 V, resulting in Pdiss values of 0.98 and 0.94, respectively. This indicates that the excitons were effectively separated in these devices, leading to favorable charge transport and collection. On the other hand, for B30T70-4Cl and B30T70-6Cl, Jph did not saturate even when Veff was increased to 2 V, suggesting severe monomolecular recombination in these devices, resulting in a significantly lower JSC and FF.37,56 Based on the Pdiss data, the effect of the Cl density on the recombination was investigated by evaluating the dependence of VOC and JSC on the light intensity (Fig. 6b and c). The degree of monomolecular recombination can be inferred from the light intensity-VOC measurement, where a value of n close to 2 indicates increased monomolecular recombination.89,90 The n values for the devices employing B30T70 (1.088) and B30T70-2Cl (1.040) were close to unity, whereas those of the devices with B30T70-4Cl and B30T70-6Cl were higher at 1.132 and 1.161, respectively. This is attributed to significant CT state recombination in the B30T70-4Cl- and B30T70-6Cl-based devices owing to the inadequate energy level offset, which correlated with the trend of the Pdiss calculations. The light dependence of JSC was determined to estimate the degree of bimolecular recombination, where a S value below 1 indicates increased bimolecular recombination.34,56 The S values for B30T70, B30T70-2Cl, B30T70-4Cl, and B30T70-6Cl were 0.933, 0.927, 0.915, and 0.913, respectively, showing a slight decrease with increasing Cl density. This suggests that bimolecular recombination increased slightly with increasing Cl density, which can be explained by the phase separation observed in the AFM images. As the Cl density of the polymers increases, phase separation gradually occurs, resulting in the production of dead points in the donor/acceptor network, leading to the recombination of free holes in the donor phase and free electrons in the acceptor phase. However, it is difficult to consider the above described degree of bimolecular recombination as a significant difference that causes a drastic decrease in the efficiency of the B30T70-4Cl and B30T70-6Cl systems.
 | ||
| Fig. 6 (a) Jph–Veff curves under AM 1.5 G illumination. Light intensity dependence of (b) VOC and (c) JSC. | ||
2.5. Analysis of nanostructure and charge mobility
The crystallinity and mobility of the blend can also affect the photovoltaic performance; therefore, the nano-scale morphology of the blend films was investigated using two dimensional grazing incidence wide angle X-ray scattering (2D-GIWAXS). 2D-GIWAXS images of the blend film and the corresponding in-plane (IP) and out-of-plane (OOP) graphs are shown in Fig. S7.† As indicated by DSC, our random copolymer system has an amorphous state, which was also evident in the 2D-GIWAXS images. We observed the presence of lamellar (100) and broad π–π stacking (010) peaks of the polymer, indicating an overall highly disordered state.25,91 Hence, it is reasonable to conclude that charge transport in the blends predominantly occurs through local hopping in the amorphous regions, and the crystallinity of the polymer has limited impact on the PCEs. However, an interesting trend was observed when examining the line cut profile of the (100) peak. As the Cl density increased, the intensity of the (100) peak in the OOP direction also increased. This indicates that the local edge-on orientation becomes more pronounced with increasing Cl density, particularly in the B30T70-4Cl and B30T70-6Cl blend films. To evaluate the influence of this orientation change on the carrier mobility, the mobility of the pristine and blend films was measured using organic field-effect transistors (OFETs). The transfer curves are presented in Fig. S8,† and the corresponding mobilities are listed in Table S11.† The mobility values were not correlated with the face-on/edge-on orientation observed in the 2D-GIWAXS analysis. All devices exhibited mobilities in the range of 10−5 to 10−4 cm2 V−1 s−1, and in some devices, clear mobility values could not be determined. This is due to the highly disordered nature of all the B30T70-XCl polymers, where the majority of the material exists in an amorphous state with only localized regions of crystallinity. Therefore, it is speculated that the edge-on orientation in the localized regions observed by 2D-GIWAXS does not significantly affect the mobility. Based on the comprehensive analysis conducted thus far, the photovoltaic performance of the B30T0-XCl:PC71BM devices is primarily influenced by the energy level, which is linked to CT state recombination. The insufficient energy level offset in B30T70-4Cl and B30T70-6Cl leads to monomolecular recombination, ultimately causing a decrease in JSC. Moreover, the phase-separated blend morphology, driven by the increasing Cl density, affects the charge transport behavior and promotes bimolecular recombination rather than influencing the exciton dissociation efficiency through monomolecular recombination.3. Conclusions
We have achieved highly efficient IOPVs by synthesizing wide bandgap random copolymers of B30T70-XCl and systematically controlling the Cl density. The result demonstrated the significant impact of Cl substitution on the photovoltaic performance, achieved by controlling the Cl density in the random copolymers. Our blend system, B30T70-XCl:PC71BM, is well-suited for the illumination range of indoor light sources, such as FLs and LEDs. Among the different blend systems, the B30T70-2Cl-based devices exhibited optimized PCEs with a high VOC, and achieved the highest PCEs of 25.0% under FL 1000 lx conditions, surpassing those of other fullerene-based IOPVs. Importantly, these devices showed a substantial improvement of approximately 3.2-fold in PCE compared to that under 1 Sun irradiance. This remarkable enhancement is attributed to the wide bandgap nature of the random copolymers, which enables efficient light absorption and a high VOC. Furthermore, our comprehensive investigation of the effect of the Cl density on the photovoltaic performance sheds light on the underlying mechanisms. We found that increasing the Cl density in the polymer donors led to a decrease in the energy level offset (ΔEHOMO) between the donor and acceptor, resulting in a reduction in the driving force for charge separation. Furthermore, a high Cl density induced phase-separated blend morphology, adversely affecting charge transport and leading to bimolecular recombination. Consequently, these factors contributed to a lower JSC and FF, ultimately resulting in a sharp drop in the PCEs of the B30T70-4Cl and B30T70-6Cl devices compared with those of the B30T70 and B30T70-2Cl devices. Taken together, our study presents a highly efficient and cost-effective IOPV system based on random copolymers blended with PC71BM, which provides an ideal combination for indoor light harvesting. The present findings highlight the delicate balance between maximizing the PCE with a high VOC and the potential decrease in the PCEs owing to a diminished driving force for charge separation. These insights deepen our understanding of the structure–property relationships in IOPVs, offering valuable guidance for the design and optimization of high-performance, cost-effective devices.Author contributions
Soyoung Kim: conceptualization, investigation, visualization, writing – original draft, Seon Joong Kim: investigation, visualization, writing – original draft, Gayoung Ham: investigation, visualization, writing – original draft. Ji-Eun Jeong: investigation, Donghwa Lee: investigation, Eunho Lee: investigation, Hyungju Ahn: investigation, Hyojung Cha: writing – review & editing, Jae Won Shim: writing – review & editing, Wonho Lee: conceptualization, writing – review & editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. K. and S. J. K. contributed equally to this work. This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education (NRF-2020R1I1A306779). This work was also supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (NRF-2022R1A2C2009523 and NRF-2021R1A4A1031761). This work received support from Editage (https://www.editage.co.kr) for English language editing.References
- I. Lee and K. Lee, Bus. Horiz., 2015, 58, 431–440 CrossRef.
- Z. Wu, T. Cheng and Z. L. Wang, Sensors, 2020, 20, 11–21 Search PubMed.
- J. W. Matiko, N. J. Grabham, S. P. Beeby and M. J. Tudor, Mst, 2014, 25, 12002–12025 CAS.
- A. Al-Fuqaha, M. Guizani, M. Mohammadi, M. Aledhari and M. Ayyash, Comst, 2015, 17, 2347–2376 Search PubMed.
- Z. L. Wang, ACS Nano, 2013, 7, 9533–9557 CrossRef CAS.
- S. Siddiqui, D. Kim, E. Roh, L. T. Duy, T. Q. Trung, M. T. Nguyen and N. Lee, Nano Energy, 2016, 30, 434–442 CrossRef CAS.
- M. H. Zarifi, S. Deif and M. Daneshmand, Sens. Actuators, A, 2017, 261, 24–29 CrossRef CAS.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CrossRef CAS.
- A. J. Heeger, Adv. Mater., 2014, 26, 10–28 CrossRef CAS.
- L. Xie, W. Song, J. Ge, B. Tang, X. Zhang, T. Wu and Z. Ge, Nano Energy, 2021, 82, 105770 CrossRef CAS.
- H. Liu, M. Yu, C. Lee, X. Yu, Y. Li, Z. Zhu, C. Chueh, Z. Li and A. K. Y. Jen, Adv. Mater. Technol., 2021, 6, 2000960 CrossRef CAS.
- Z. Li, C. Chueh and A. K.-Y. Jen, Prog. Polym. Sci., 2019, 99, 101175 CrossRef CAS.
- S. Hwang and T. Yasuda, Polym. J., 2023, 55, 297–316 CrossRef CAS.
- X. Hou, Y. Wang, H. K. H. Lee, R. Datt, N. Uslar Miano, D. Yan, M. Li, F. Zhu, B. Hou, W. C. Tsoi and Z. Li, J. Mater. Chem. A, 2020, 8, 21503–21525 RSC.
- H. Yin, J. K. W. Ho, V. Piradi, S. Chen, X. Zhu and S. K. So, Small Methods, 2020, 4, 2000136 CrossRef CAS.
- L. Ma, Y. Chen, P. C. Y. Chow, G. Zhang, J. Huang, C. Ma, J. Zhang, H. Yin, A. M. Hong Cheung, K. S. Wong, S. K. So and H. Yan, Joule, 2020, 4, 1486–1500 CrossRef CAS.
- Y. Cui, Y. Wang, J. Bergqvist, H. Yao, Y. Xu, B. Gao, C. Yang, S. Zhang, O. Inganäs, F. Gao and J. Hou, Nat. Energy, 2019, 4, 768–775 CrossRef CAS.
- Z. Ding, R. Zhao, Y. Yu and J. Liu, J. Mater. Chem. A, 2019, 7, 26533–26539 RSC.
- R. Po, G. Bianchi, C. Carbonera and A. Pellegrino, Macromolecules, 2015, 48, 453–461 CrossRef CAS.
- F. Zhao, J. Zhou, D. He, C. Wang and Y. Lin, J. Mater. Chem. C, 2021, 9, 15395–15406 RSC.
- D. Luo, C. J. Brabec and A. K. K. Kyaw, Nano Energy, 2023, 114, 108661 CrossRef CAS.
- M. Jahandar, S. Kim and D. C. Lim, ChemSusChem, 2021, 14, 3449–3474 CrossRef CAS PubMed.
- J. K. W. Ho, H. Yin and S. K. So, J. Mater. Chem. A, 2020, 8, 1717–1723 RSC.
- Y. Cui, H. Yao, T. Zhang, L. Hong, B. Gao, K. Xian, J. Qin and J. Hou, Adv. Mater., 2019, 31, 1904512 CrossRef CAS PubMed.
- J. Kim, M. A. Saeed, S. H. Kim, D. Lee, Y. Jang, J. S. Park, D. Lee, C. Lee, B. J. Kim, H. Y. Woo, J. W. Shim and W. Lee, Macromol. Rapid Commun., 2022, 43, 2200279 CrossRef CAS.
- M. Mainville and M. Leclerc, ACS Energy Lett., 2020, 5, 1186–1197 CrossRef CAS.
- X. Zhou, H. Wu, U. Bothra, X. Chen, G. Lu, H. Zhao, C. Zhao, Q. Luo, G. Lu, K. Zhou, D. Kabra, Z. Ma and W. Ma, Mater. Horiz., 2023, 1, 566–575 RSC.
- H. K. H. Lee, Z. Li, J. R. Durrant and W. C. Tsoi, J. Mater. Chem. A, 2018, 6, 5618–5626 RSC.
- H. Yin, J. K. W. Ho, S. H. Cheung, R. J. Yan, K. L. Chiu, X. Hao and S. K. So, J. Mater. Chem. A, 2018, 6, 8579–8585 RSC.
- S. Yang, Z. Hsieh, M. L. Keshtov, G. D. Sharma and F. Chen, Sol. RRL, 2017, 1, 1700174 CrossRef.
- P. Chao, M. Guo, Y. Zhu, H. Chen, M. Pu, H. Huang, H. Meng, C. Yang and F. He, Macromolecules, 2020, 53, 2893 CrossRef CAS.
- S. Park, H. Ahn, J. Kim, J. B. Park, J. Kim, S. H. Im and H. J. Son, ACS Energy Lett., 2019, 5, 170 CrossRef.
- Q. Fan, T. Liu, W. Gao, Y. Xiao, J. Wu, W. Su, X. Guo, X. Lu, C. Yang, H. Yan, M. Zhang and Y. Li, J. Mater. Chem. A, 2019, 7, 15404–15410 RSC.
- Q. Fan, Q. Zhu, Z. Xu, W. Su, J. Chen, J. Wu, X. Guo, W. Ma, M. Zhang and Y. Li, Nano Energy, 2018, 48, 413–420 CrossRef CAS.
- A. Zeng, X. Ma, M. Pan, Y. Chen, R. Ma, H. Zhao, J. Zhang, H. K. Kim, A. Shang, S. Luo, I. C. Angunawela, Y. Chang, Z. Qi, H. Sun, J. Y. L. Lai, H. Ade, W. Ma, F. Zhang and H. Yan, Adv. Funct. Mater., 2021, 31, 2102413 CrossRef CAS.
- M. Zhang, X. Guo, S. Zhang and J. Hou, Adv. Mater., 2014, 26, 1118–1123 CrossRef CAS PubMed.
- X. Wang, H. Chen, J. Yuan, Q. Wei, J. Li, L. Jiang, J. Huang, Y. Li, Y. Li and Y. Zou, J. Mater. Chem. A, 2021, 9, 14752–14757 RSC.
- Q. Zhao, J. Qu and F. He, Adv. Sci., 2020, 7, 2000509 CrossRef CAS.
- H. Je, E. Shin, K. J. Lee, H. Ahn, S. Park, S. H. Im, Y. Kim, H. J. Son and S. Kwon, ACS Appl. Mater. Interfaces, 2020, 12, 23181 CrossRef CAS PubMed.
- H. Zhou, L. Yang, A. C. Stuart, S. C. Price, S. Liu and W. You, Angew. Chem., Int. Ed., 2011, 50, 2995–2998 CrossRef CAS PubMed.
- S. Zhang, Y. Qin, J. Zhu and J. Hou, Adv. Mater., 2018, 30, 1800868 CrossRef PubMed.
- Z. Liu, Y. Gao, J. Dong, M. Yang, M. Liu, Y. Zhang, J. Wen, H. Ma, X. Gao, W. Chen and M. Shao, J. Phys. Chem. Lett., 2018, 9, 6955 CrossRef CAS.
- P. Chao, Z. Mu, H. Wang, D. Mo, H. Chen, H. Meng, W. Chen and F. He, ACS Appl. Energy Mater., 2018, 1, 2365 CrossRef CAS.
- R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 4, 3496–3508 RSC.
- M. L. Tang, J. H. Oh, A. D. Reichardt and Z. Bao, J. Am. Chem. Soc., 2009, 131, 3733–3740 CrossRef CAS PubMed.
- Y. Li, J. Lin, X. Che, Y. Qu, F. Liu, L. Liao and S. R. Forrest, J. Am. Chem. Soc., 2017, 139, 17114 CrossRef CAS PubMed.
- B. Huang, Y. Cheng, H. Jin, J. Liu, X. Huang, Y. Cui, X. Liao, C. Yang, Z. Ma and L. Chen, Small, 2021, 17, 2104451 CrossRef CAS.
- H. Wang, P. Chao, H. Chen, Z. Mu, W. Chen and F. He, ACS Energy Lett., 2017, 2, 1971 CrossRef CAS.
- Q. Zhang, X. Song, R. Singh, S. Chung, Z. Zhou, Y. Lu, B. Zhang, K. Cho, W. Zhu and Y. Liu, Chem. Eng. J., 2022, 437, 135182 CrossRef CAS.
- H. Chen, Z. Hu, H. Wang, L. Liu, P. Chao, J. Qu, W. Chen, A. Liu and F. He, Joule, 2018, 2, 1623–1634 CrossRef CAS.
- S. J. Jeon, Y. W. Han and D. K. Moon, Sol. RRL, 2019, 3, 1900094 CrossRef.
- D. Yuan, F. Pan, L. Zhang, H. Jiang, M. Chen, W. Tang, G. Qin, Y. Cao and J. Chen, Sol. RRL, 2020, 4, 2000064 Search PubMed.
- J. Xiao, X. Jia, C. Duan, F. Huang, H. Yip and Y. Cao, Adv. Mater., 2021, 33, 2008158 CrossRef CAS.
- R. A. Fair, R. Xie, Y. Lee, R. H. Colby and E. D. Gomez, ACS Appl. Polym. Mater., 2021, 3, 4572 CrossRef CAS.
- W. Lee, H. Cha, Y. J. Kim, J. Jeong, S. Hwang, C. E. Park and H. Y. Woo, ACS Appl. Mater. Interfaces, 2014, 6, 20510 CrossRef CAS.
- Y. You, C. E. Song, Q. V. Hoang, Y. Kang, J. S. Goo, D. Ko, J. Lee, W. S. Shin and J. W. Shim, Adv. Funct. Mater., 2019, 29, 1901171 CrossRef.
- D. Lee, Y. Jang, J. Kim, S. Y. Jeong, H. Y. Woo, D. Lee, J. Kim, Y. Lee, C. Lee and W. Lee, J. Mater. Chem. A, 2023, 11, 308–317 Search PubMed.
- J. Bertrandie, J. Han, De Castro, S. P. Catherine, E. Yengel, J. Gorenflot, T. Anthopoulos, F. Laquai, A. Sharma and D. Baran, Adv. Mater., 2022, 34, 2202575 CrossRef CAS.
- K. Wang, Y. Firdaus, M. Babics, F. Cruciani, Q. Saleem, A. El Labban, M. A. Alamoudi, T. Marszalek, W. Pisula, F. Laquai and P. M. Beaujuge, Chem. Mater., 2016, 28, 2200–2208 CrossRef CAS.
- H. Yao, Y. Cui, D. Qian, C. S. Ponseca, A. Honarfar, Y. Xu, J. Xin, Z. Chen, L. Hong, B. Gao, R. Yu, Y. Zu, W. Ma, P. Chabera, T. Pullerits, A. Yartsev, F. Gao and J. Hou, J. Am. Chem. Soc., 2019, 141, 7743 CrossRef CAS PubMed.
- D. Qian, Z. Zheng, H. Yao, W. Tress, T. R. Hopper, S. Chen, S. Li, J. Liu, S. Chen, J. Zhang, X. Liu, B. Gao, L. Ouyang, Y. Jin, G. Pozina, I. A. Buyanova, W. M. Chen, O. Inganäs, V. Coropceanu, J. Bredas, H. Yan, J. Hou, F. Zhang, A. A. Bakulin and F. Gao, Nat. Mater., 2018, 17, 703 CrossRef CAS PubMed.
- S. Chen, Y. Wang, L. Zhang, J. Zhao, Y. Chen, D. Zhu, H. Yao, G. Zhang, W. Ma, R. H. Friend, P. C. Y. Chow, F. Gao and H. Yan, Adv. Mater., 2018, 30, 1804215 CrossRef PubMed.
- S. Li, L. Zhan, C. Sun, H. Zhu, G. Zhou, W. Yang, M. Shi, C. Li, J. Hou, Y. Li and H. Chen, J. Am. Chem. Soc., 2019, 141, 3073–3082 CrossRef CAS PubMed.
- H. Zhang, S. Li, B. Xu, H. Yao, B. Yang and J. Hou, J. Mater. Chem. A, 2016, 4, 18043–18049 RSC.
- H. Bin, Y. Yang, Z. Peng, L. Ye, J. Yao, L. Zhong, C. Sun, L. Gao, H. Huang, X. Li, B. Qiu, L. Xue, Z. Zhang, H. Ade and Y. Li, Adv. Energy Mater., 2018, 8, 1702324 CrossRef.
- Z. Zhang, W. Liu, T. Rehman, H. Ju, J. Mai, X. Lu, M. Shi, J. Zhu, C. Li and H. Chen, J. Mater. Chem. A, 2017, 5, 9649–9654 RSC.
- M. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A. Heeger and C. Brabec, Adv. Mater., 2006, 18, 789–794 CrossRef CAS.
- D. Veldman, S. C. J. Meskers and R. A. J. Janssen, Adv. Funct. Mater., 2009, 19, 1939–1948 CrossRef CAS.
- T. M. Clarke and J. R. Durrant, Chem. Rev., 2010, 110, 6736–6767 CrossRef CAS PubMed.
- C. Yang, J. Zhang, N. Liang, H. Yao, Z. Wei, C. He, X. Yuan and J. Hou, J. Mater. Chem. A, 2019, 7, 18889–18897 RSC.
- R. A. J. Janssen and J. Nelson, Adv. Mater., 2013, 25, 1847–1858 CrossRef CAS.
- J. Liu, S. Chen, D. Qian, B. Gautam, G. Yang, J. Zhao, J. Bergqvist, F. Zhang, W. Ma, H. Ade, O. Inganäs, K. Gundogdu, F. Gao and H. Yan, Nat. Energy, 2016, 1, 16089 CrossRef CAS.
- P. Chao, H. Wang, D. Mo, H. Meng, W. Chen and F. He, J. Mater. Chem. A, 2018, 6, 2942–2951 RSC.
- X. Liao, H. Pei, H. Zhao, Y. Cui, L. Li, X. Shi, P. Zhu, W. Ma, Y. Chen and A. K.-Y. Jen, Chem. Eng. J., 2022, 435, 135020 CrossRef CAS.
- K. Kawashima, Y. Tamai, H. Ohkita, I. Osaka and K. Takimiya, Nat. Commun., 2015, 6, 10085 CrossRef CAS PubMed.
- E. Zakhidov, M. Imomov, V. Quvondikov, S. Nematov, I. Tajibaev, A. Saparbaev, I. Ismail, B. Shahid and R. Yang, Appl. Phys. A, 2019, 125, 1–7 CrossRef CAS.
- S. Narra, S. Tsai, K. Awasthi, S. Rana, E. W. Diau and N. Ohta, J. Chin. Chem. Soc., 2021, 69, 140 CrossRef.
- H. Yao, Y. Li, H. Hu, P. C. Y. Chow, S. Chen, J. Zhao, Z. Li, J. H. Carpenter, J. Y. L. Lai, G. Yang, Y. Liu, H. Lin, H. Ade and H. Yan, Adv. Energy Mater., 2017, 8, 1800234 Search PubMed.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- L. Ma, S. Zhang, J. Wang, J. Ren, M. Gao, J. Zhang, T. Zhang, H. Yao, L. Ye and J. Hou, ACS Mater. Lett., 2021, 3, 1276 CrossRef CAS.
- Y. Xu, H. Yao, L. Ma, L. Hong, J. Li, Q. Liao, Y. Zu, J. Wang, M. Gao, L. Ye and J. Hou, Angew. Chem., 2020, 132, 9089 CrossRef.
- T. M. Clarke, F. C. Jamieson and J. R. Durrant, J. Phys. Chem. C, 2009, 113, 20934–20941 CrossRef CAS.
- S. Shoaee, M. P. Eng, E. Espíldora, J. L. Delgado, B. Campo, N. Martín, D. Vanderzande and J. R. Durrant, Energy Environ. Sci., 2010, 3, 971–976 RSC.
- S. D. Dimitrov, B. C. Schroeder, C. B. Nielsen, H. Bronstein, Z. Fei, I. Mcculloch, M. Heeney and J. R. Durrant, Polymers, 2016, 8, 14 CrossRef PubMed.
- Y. Dong, H. Cha, H. L. Bristow, J. Lee, A. Kumar, P. S. Tuladhar, I. Mcculloch, A. A. Bakulin and J. R. Durrant, J. Am. Chem. Soc., 2021, 143, 7599 CrossRef CAS PubMed.
- F. D. Eisner, M. Azzouzi, Z. Fei, X. Hou, T. D. Anthopoulos, T. J. S. Dennis, M. Heeney and J. Nelson, J. Am. Chem. Soc., 2019, 141, 6362 CrossRef CAS PubMed.
- J. Chen, C. Cui, Y. Li, L. Zhou, Q. Ou, C. Li, Y. Li and J. Tang, Adv. Mater., 2015, 27, 1035–1041 CrossRef CAS PubMed.
- V. D. Mihailetchi, J. Wildeman and P. Blom, Phys. Rev. Lett., 2005, 94, 126602 CrossRef CAS PubMed.
- R. Singh, C. L. Chochos, V. G. Gregoriou, A. D. Nega, M. Kim, M. Kumar, S. Shin, S. H. Kim, J. W. Shim and J. Lee, ACS Appl. Mater. Interfaces, 2019, 11, 36905 CrossRef CAS PubMed.
- J. Zhang, W. Liu, G. Zhou, Y. Yi, S. Xu, F. Liu, H. Zhu and X. Zhu, Adv. Energy Mater., 2019, 10, 1902398 Search PubMed.
- Z. Peng, L. Ye and H. Ade, Mater. Horiz., 2022, 9, 577–606 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta06624j |
| This journal is © The Royal Society of Chemistry 2024 |