
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Counterion chemistry of 5-halo (X: Cl, Br, I)-uracil derived carbon nitride: unlocking enhanced photocatalytic performance†
Toshali
Bhoyar
 a,
B. Moses
Abraham
b,
Akanksha
Gupta
c,
Dong Jin
Kim
d,
Nilesh R.
Manwar
e,
Kedhareswara Sairam
Pasupuleti
f,
Devthade
Vidyasagar
*agh and
Suresh S.
Umare
*a
a,
B. Moses
Abraham
b,
Akanksha
Gupta
c,
Dong Jin
Kim
d,
Nilesh R.
Manwar
e,
Kedhareswara Sairam
Pasupuleti
f,
Devthade
Vidyasagar
*agh and
Suresh S.
Umare
*a
aMaterials and Catalysis Laboratory, Department of Chemistry, Visvesvaraya National Institute of Technology (VNIT), South Ambazari Road, Nagpur 440010, India. E-mail: ssumare1965@gmail.com; vidyasagar.devtade@gmail.com
bDepartment of Chemical Engineering, Indian Institute of Technology Kanpur, Kanpur-208016, India
cDepartment of Chemistry, Bar-Ilan University, Ramat Gan 5290002, Israel
dSchool of Energy Engineering, Kyungpook National University, Buk-gu, Daegu, 41566, Republic of Korea
eInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland
fDepartment of Physics, Chungnam National University, 99 Daehak-ro, Yuseong-gu, Daejeon 34134, Republic of Korea
gDepartment of Chemistry, Indian Institute of Technology Hyderabad, Kandi, Sangareddy, 502285, India
hInstitute for Environmental and Climate Technology, Korea Institute of Energy Technology (KENTECH), Naju 58330, Korea
First published on 2nd December 2023
Abstract
The performance of modified polymeric carbon nitride (PCN) photocatalysts exceeds that of their bulk counterparts, owing to their distinct advantages and enhanced features. In this work, we present the synthesis methodology for halide-molecular doped PCN through a single molecule precursor approach by combining π-aromatic uracil and halide as a dopant and counterions, respectively. We systematically investigated the effect of halide ions (X−: Cl−, Br−, and I−) on the photoactivity of 5-halouracil derived PCN (XUCN). Our results demonstrate that halide counterions significantly enhance the photocatalytic activity of XUCN for hydrogen peroxide and hydrogen production (126.6 μmol h−1 and 0.563 mmol h−1 g−1), compared to the undoped bulk PCN (48.3 μmol h−1 and 0.260 mmol h−1 g−1). The charge-carrier analysis and structural analysis of synthesized XUCN catalysts suggest that the improvement in photoactivity is due to the synergistic interactions of uracil and halide ions, which enhance the charge-carrier lifetime (1.51 to 3.08 ns), generate additional catalytic sites (10.6 to 83.3 m2 g−1), and extend light absorption (450 to 480 nm). Theoretical investigations reveal high structural stability for XUCN with favourable adsorption energies to bind with reactive oxygen species. The new insights provided in this study can have important implications for future design and synthesis of metal-free PCN with improved photoactivity via single molecule precursor doping.
Introduction
Polymeric carbon nitride (PCN) is a promising metal-free photocatalyst for numerous solar-powered energy and environmental applications, due to its ideal bandgap and attractive physicochemical features.1–3 However, the inherent limitations of bulk PCN-based catalysts, including rapid recombination of photogenerated electron–hole pairs, a limited number of surface-active sites, inadequate absorption of visible light (λ < 450 nm), and slow reaction kinetics, have hindered their overall photoactivity.4 A plethora of strategies have been previously reported to address these challenges. For example, hetero-elements (metal or non-metal) or π-molecular doping can be applied to modulate the surface and bulk characteristics of PCN. Likewise, π-molecular doping of PCN with aromatic functional groups including phenyl,5 pyridine,6 pyrimidine,7 pyrazine,8 thiazole,9 benzothiazole,10 caffeine,11 nucleotides (adenine, guanine, cytosine, uracil, and thymine),12 and thiophene,13 is known to tailor the photo response and enhance photoactivity. This approach enables tuning of electronic energy levels, exciton migration behaviour, and microstructure of PCN, ultimately enhancing its photocatalytic performance.6,14Incorporating non-metals such as F, Cl, Br, or I through elemental doping is another effective way to influence the electronic band structure of the PCN framework by reconstructing the corresponding highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO).15 For example, Wang et al. created an extended aromatic heterocycle via iodine doping, which generated impurity energy levels near the valence band (VB) edge, resulting in an enhanced optical response and hydrogen evolution activity.16 Similarly, PCN doped with F, Cl, and Br has demonstrated a significant surge in photocatalytic hydrogen production, and photooxidation behaviour.14,17,18 Theoretical studies using density functional theory (DFT) calculations have shown that the redistribution of molecular levels depends on the electronegativity of the halogen atom.19 The F-atom constitutes the HOMO because of its higher electronegativity, while Cl, Br, and I-atoms are involved in the LUMO. Consequently, doped PCN exhibits varying optical responses depending on the specific halogen atom used. In this regard, Li et al. produced a binary potassium–halogen (K–X) doped carbon nitride and found that the existence of C–X and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N cooperatively accelerated carrier dynamics to enhance overall photocatalytic activity.20 Nonetheless, the effect of single-component halogen atom doping on the optoelectronic behaviour of PCN has not yet proposed to support the theoretical predictions. Previous studies anticipated that an I-atom, with the least electronegativity amongst the other halogen atoms, possesses higher ability to interact with the π-electrons of PCN.16,19 However, the formation of a stable I-doped PCN structure is thermodynamically and geometrically unfavourable due to the large atomic size of the I-atom. On the other hand, a F-atom is more likely to be doped in PCN due to its smaller size and higher electronegativity, but it has a low promotional effect on the optical response.17 Of all, Br-doping is ideal for the PCN framework and can greatly alter its optoelectronic properties.14 Nevertheless, a relative study explaining the effect of halide ions (Cl−, Br− and I−) on the optical response and photo-exciton dynamics has been rarely undertaken. Additionally, the concept of combining both non-metal and π-molecular systems into a PCN framework is expected to have a noteworthy impact.
N cooperatively accelerated carrier dynamics to enhance overall photocatalytic activity.20 Nonetheless, the effect of single-component halogen atom doping on the optoelectronic behaviour of PCN has not yet proposed to support the theoretical predictions. Previous studies anticipated that an I-atom, with the least electronegativity amongst the other halogen atoms, possesses higher ability to interact with the π-electrons of PCN.16,19 However, the formation of a stable I-doped PCN structure is thermodynamically and geometrically unfavourable due to the large atomic size of the I-atom. On the other hand, a F-atom is more likely to be doped in PCN due to its smaller size and higher electronegativity, but it has a low promotional effect on the optical response.17 Of all, Br-doping is ideal for the PCN framework and can greatly alter its optoelectronic properties.14 Nevertheless, a relative study explaining the effect of halide ions (Cl−, Br− and I−) on the optical response and photo-exciton dynamics has been rarely undertaken. Additionally, the concept of combining both non-metal and π-molecular systems into a PCN framework is expected to have a noteworthy impact.
The present work aims to explore the impact of halogen atoms and π-molecular doping on the optoelectronic characteristics and photocatalytic performance of PCN, which has not been extensively studied. Herein, we present a comprehensive study of the synthesis, characterization, and photoactivity of 5-halouracil-derived PCN-based photocatalysts, with the aim of addressing the existing challenges and exploring new avenues for their applications in solar-driven processes.
Experimental
Materials and methods
| H2O2 + 3I− + 2H+ → I3− + 2H2O | (1) |
The RhB degradation experiments were performed in the same photoreactor setup. In a typical experiment, 100 mg of catalyst was dispersed in a 100 mL RhB (10 mg L−1) solution and stirred continuously for 30 min in the dark. The reaction mixture was then irradiated, and sample aliquots were collected at pre-decided time intervals. For analysis, the aliquot was collected and centrifuged to remove the residual catalyst. The resultant supernatants were then analysed using a UV-visible spectrophotometer at a wavelength of 554 nm.
The adsorption energy between the surface and adsorbates (Eads) is computed using eqn (2).
| Eads = Etotal − Esurface − Eadsorbate | (2) |
| ΔG = ΔEH + ΔEZPE − TΔSH | (3) |
Results and discussion
Structure and optical features of 5-halouracil derived PCN
The thermal co-polymerization of PCN precursors with 5-halouracil yields the doping of π-aromatic uracil and halide ions (X: Cl−, Br−, and I−) into the core structure of PCN, as depicted in Fig. 1A.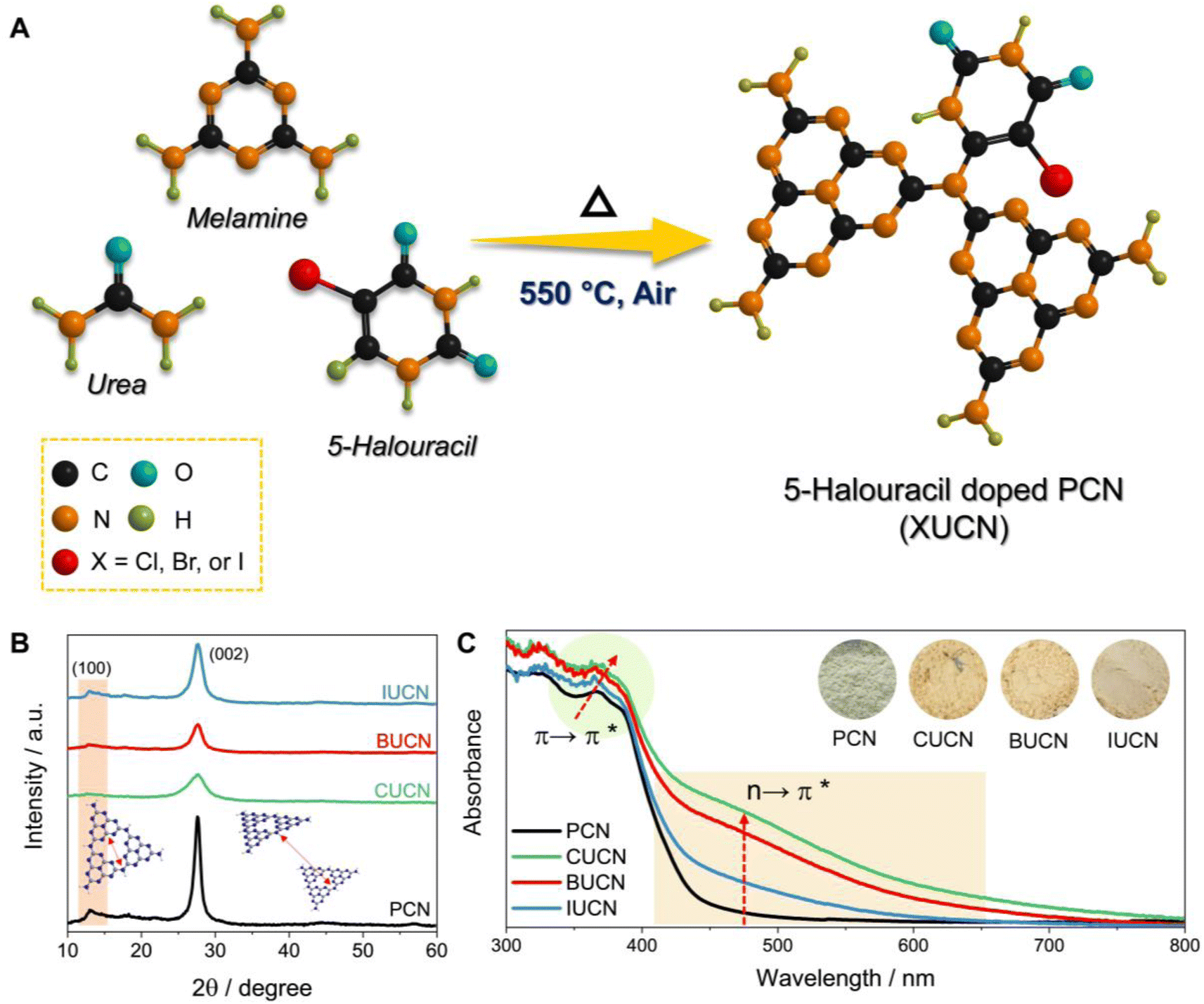 | ||
| Fig. 1 Structure and optical properties. (A) Schematic illustration of the preparation of a 5-halouracil doped polymeric carbon nitride catalyst (XUCN), (B) powder X-ray diffraction (XRD) profiles, and (C) UV-visible diffuse reflectance spectra (UV-vis DRS) with digital photographs of powder catalyst as inset. | ||
The incorporation of the 5-halouracil moiety into the PCN framework induces structural modifications at the molecular level, thereby giving rise to altered physico-chemical properties. Hence, the investigation of alteration in the crystal structure of XUCN samples was conducted through powder X-ray diffraction (XRD) analysis. Pristine PCN has shown a low angle diffraction peak at 13.0°, which is due to the intraplanar stacking of the heptazine motif assigned to the (100) crystal plane (Fig. 1B). While peak at the higher angle, 27.7° corresponds to the heptazine motifs in melon stacked over one another. An analogous diffraction pattern was observed for the modified CUCN, BUCN, and IUCN samples. However, after the incorporation of 5-halouracil moieties, a notable decrease in the diffraction peak intensity was observed for XUCN samples. This indicates the distortion in the symmetrical planar structure of PCN. On comparing the relative intensity of diffraction peaks, CUCN exhibited the lowest peak intensity followed by BUCN and IUCN. This observation suggests that amongst all the 5-halouracils, the 5-chlorouracil (CU) moiety have the highest degree of interaction and influence on the crystalline architecture of the resultant PCN framework. This could be ascribed to the higher C–Cl bond strength and lower atomic radius of the chlorine atom. The larger atomic radius of bromine and iodine in comparison to chlorine hinders the probability of doping. In addition to the decreased diffraction intensity, the full width at half maxima (β) corresponding to the (002) peak increased in the order PCN < IUCN < BUCN < CUCN. The broadening of ‘β’ indicates a decrease in the crystallite size. These findings affirm that the 5-halouracil moieties have a significant impact on PCN's crystal structure.
The XUCN samples further examined via Fourier transform-infrared spectroscopy (FTIR) revealed a distinctive out-of-plane bending mode at 810 cm−1, which corresponds to the tri-s-triazine rings. (Fig. S1†), while the stretching modes of an aromatic C–N heterocycle were observed in the range of 1000–1800 cm−1. In addition, broad bands in the region of 3000–3500 cm−1 were observed corresponding to the stretching vibration of terminal –NH/–NH2 and O–H bonds. The FTIR spectra indicate that the XUCN samples retain the basic carbon nitride structural features. Furthermore, a notable change in the intensity of bands was observed after incorporation of 5-halouracils. Specifically, the absorption bands at 810 and 1000–1800 cm−1 that correspond to tri-s-triazine groups and the C–N heterocycle are much weaker for CUCN, showing altered heptazine structural connections. The FTIR result combined with XRD substantiates that the incorporation of 5-halouracil moieties into PCN rendered structural changes and disorderness in the planar carbon nitride structure. To support the incorporation of a 5-halouracil moiety into the PCN backbone, solid-state cross polarized magic angle spinning (CP-MAS) 13C nuclear magnetic resonance (NMR) spectra of the PCN and BUCN catalyst were recorded. The 13C CP-MAS NMR spectrum of PCN shows two distinct signals at 164.8 and 156.4 ppm, that are assigned to the sp2 hybridized carbon atom in the N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and C–(N)3 linkage of the heptazine core, respectively (Fig. S2A†).33 In analogy with PCN, BUCN showcases similar characteristics signals to that of PCN with a slight shift. The shift in the major NMR signals could be due to the shielding effect of the electron withdrawing groups of the attached uracil moiety.34 However, in addition to the representative signals corresponding to PCN, BUCN demonstrates additional signals affirming structural distortion and changes in the local carbon environment of BUCN due to the incorporation of 5-bromouracil moieties into the PCN backbone. The weak signals located at nearly 167.1, 166.4, and 159.5 ppm are ascribed to the C3, C4, and C5 carbon atoms in the BUCN structure as shown in Fig. S2B and C.† These findings show the successful incorporation of 5-bromouracil moieties into PCN. The elemental analysis of the samples further revealed no considerable change in the C
N and C–(N)3 linkage of the heptazine core, respectively (Fig. S2A†).33 In analogy with PCN, BUCN showcases similar characteristics signals to that of PCN with a slight shift. The shift in the major NMR signals could be due to the shielding effect of the electron withdrawing groups of the attached uracil moiety.34 However, in addition to the representative signals corresponding to PCN, BUCN demonstrates additional signals affirming structural distortion and changes in the local carbon environment of BUCN due to the incorporation of 5-bromouracil moieties into the PCN backbone. The weak signals located at nearly 167.1, 166.4, and 159.5 ppm are ascribed to the C3, C4, and C5 carbon atoms in the BUCN structure as shown in Fig. S2B and C.† These findings show the successful incorporation of 5-bromouracil moieties into PCN. The elemental analysis of the samples further revealed no considerable change in the C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N:H values of XUCN catalysts in comparison to those of bulk PCN (Table S1†). The lack of measurable changes in elemental analysis of the catalyst composition could be due to insufficient doping concentration, stable catalyst structure, and surface vs. bulk level doping.
:N:H values of XUCN catalysts in comparison to those of bulk PCN (Table S1†). The lack of measurable changes in elemental analysis of the catalyst composition could be due to insufficient doping concentration, stable catalyst structure, and surface vs. bulk level doping.
The ultraviolet-visible diffuse reflectance spectra (UV-vis DRS) of bulk PCN show an optical absorption edge at 450 nm, which is due to the intrinsic π → π* electronic transition (Fig. 1C), caused by the π-conjugated aromatic framework and orderly packing of the heptazine tectonics in the polymeric structure.35 On the other hand, the XUCN catalyst demonstrated extended visible light response (Fig. S3A†). By simply increasing the dopant concentration from 10 to 100 mg, the light absorption behaviour of XUCN samples shows a remarkable change (Fig. 1C). Importantly, as compared to bulk PCN, XUCN catalysts have significantly longer visible light absorption above 450 nm. The tailored optical response can be observed by the colour change from pale yellow to brownish yellow (inset of Fig. 1C). This is due to the newly emerged shoulder absorption peak between 450 and 800 nm, which is ascribed to the n → π* electronic transition involving lone pair electrons on edge nitrogen and halogen atoms.36 This characteristic n → π* transition is spatially forbidden in PCN because of the perfectly symmetric and planar structural units. The appearance of n → π* transition is an indication of distortion in the planar and symmetric heptazine structure of XUCN catalysts. Furthermore, there is a noticeable change in visible-light absorption intensity corresponding to the n → π* electronic transition, with an increased intensity in the order IUCN < BUCN < CUCN. This observation implies that uracil moieties with halogen functionalities induce n → π* electronic transition and greatly expands the photo-response. CUCN exhibited the most prominent enhancement in the visible light response and n → π* electronic transition, which can be ascribed to the higher electronegativity and effective Cl-doping in the CUCN structure. The Tauc plot revealed a narrowed bandgap with the addition of 5-halouracil dopants (Fig. S3B†). Overall, pristine PCN has an estimated bandgap of 2.71 eV, whereas those of CUCN, BUCN, and IUCN are 2.57, 2.61, and 2.66 eV, respectively. Furthermore, it was observed that the CUCN and BUCN catalysts evinced the presence of mid-gap states, which were positioned above the valence band with transition energies of 1.81 and 1.88 eV, respectively (as depicted in the inset of Fig. S3B†). Conversely, neither PCN nor IUCN show the existence of mid-gap states due to their symmetrical planar structures and the absence of a shoulder peak, which can be attributed to the relatively low I-doping effect.37 Theoretical explanation from a previous study suggests that doped halogen atoms contribute to the conduction band (CB) of the PCN electronic structure.19 Due to the change in electronegativity of halogens, the configuration of CB experiences changes in the electronic band structure. Therefore, the reduced bandgap of XUCN catalysts is attributed to the different levels of doping and extended π-conjugated aromatic network.38
X-ray photoelectron spectroscopy (XPS) was applied to comprehend the chemical composition and bonding in PCN catalysts before and after 5-halouracil doping. The deconvoluted C 1s spectrum of PCN contained three peaks located at a binding energy value of 288.4, 285.5, and 284.8 eV, corresponding to the characteristic N–CN linkage of the aromatic heptazine ring of PCN, C–NHx (x = 1, 2) groups, and adventitious carbon (C–C/CC), respectively (Fig. 2A).39 On the other hand, four peaks were deconvoluted for all XUCN catalysts, of which three are consistent with those of PCN. The additional featured peak at 287.9 eV is assigned to the signature CO groups of the doped 5-halouracil moieties.40 Compared to PCN, a noticeable increase in the peak area at 284.8 eV was also observed, which is an indication of increased adventitious carbon contribution. This is mainly because of the C–C/CC bonds of 5-halouracil moieties attached to the heptazine core of PCN. The high-resolution N 1s XPS spectra of PCN and XUCN catalysts were fitted with identical peaks indicating that the nitrogen environment remains unaffected by the doping of 5-halouracils (Fig. S4†).41 The O 1s spectrum of PCN demonstrated two peaks located at 533.4 and 531.8 eV ascribed to the surface adsorbed water and oxygen molecules, respectively (Fig. 2B).42 Apart from the signature peaks, a third new peak appeared for the XUCN catalyst, which is assigned to the CO group confirming the incorporation of the 5-halouracil moiety. To confirm the existence of halogen functionalities in the doped structure, the high-resolution spectrum of the corresponding doped halogen atom was obtained (Fig. 2C). The high-resolution Br 3d spectra of the BUCN catalyst is deconvoluted into three peaks located at 70.9, 68.4, and 66.9 eV. The two peaks at 70.9 and 68.4 eV can be assigned to the C–Br 3d3/2 and C–Br 3d5/2 covalent bonds, respectively,43,44 while the third peak at 66.9 eV is likely from the ionic C–Br+ 3d5/2 bond.45 The existence of ionic or semi-ionic bonds contributes significantly to improving the catalytic activity. This is due to the fact that these bonds serve as either electron acceptors or donors inducing the polarization of a π-electron cloud on polymeric network for effective charge transfer between halogen and carbon atoms.46 This leads to higher conductivity as well as modified electronic properties. Furthermore, the Cl 2p XPS spectrum of CUCN is fitted with three peaks at 205.2, 201.3, and 196.9 eV allocated to the C–Cl 2p1/2, C–Cl 2p3/2, and C–Cl+ 2p3/2 bonds, respectively.18 These finding indicate successful retention of bromine and chlorine in the BUCN and CUCN structures, respectively. Contrastingly, no peaks corresponding to iodine were detected in high resolution I 3d spectra of IUCN which is attributed to the cleavage of the fragile I–C bond during annealing and instability of the I–C type network.47 Overall, XPS results confirm that the incorporation of 5-halouracil moieties into the triazine network stabilizes the bonding structures based on bromine and chlorine.
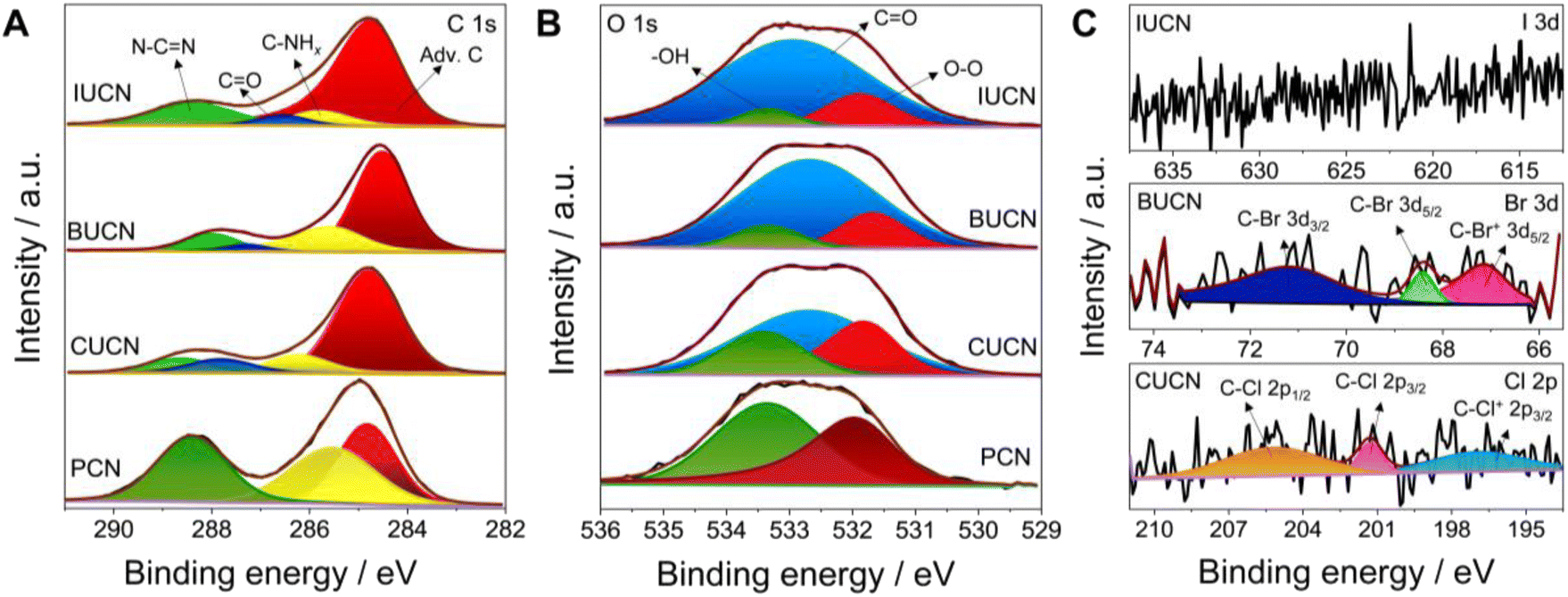 | ||
| Fig. 2 Deconvoluted high-resolution X-ray photoelectron spectrum for (A) C 1s, (B) O 1s, and (C) Cl 2p, Br 3d, and I 3d of PCN and XUCN (X = Cl, Br, or I) samples. | ||
Comprehensive analysis of morphology, microstructure, and electronic properties
The distinct variations in morphology and microstructure of modified XUCN were examined by field emission scanning electron microscopy (FE-SEM) and transmission electron microscopy (TEM). PCN exhibited 2D bulk sheet-like morphology with hardly any pores on the surface (Fig. S5 and S6†). In contrast, CUBU, BUCN, and IUCN catalysts demonstrated a porous broken-layered structure (Fig. 3A). It can be observed that CUCN shows a relatively higher order of porosity in comparison to BUCN and IUCN. Here, the high porosity of CUCN can be attributed to a higher degree of interaction of chlorine compared to bromine and iodine due to high electronegativity, small atomic size, and possible vaporization of chlorine gas. Later, energy dispersive spectroscopy (EDS) employed to map the constituent elements revealed the uniformly dispersed C, N, O, and the corresponding halogen elements (Cl, Br, and I on CUCN, BUCN, and IUCN, respectively) on the surface of the XUCN matrix (Fig. S7–S9†). Furthermore, the constituent weight percentage was estimated by using EDX spectra. The EDX spectra of CUCN and BUCN revealed the presence of 0.2 and 0.1 wt% of chlorine and bromine, respectively (Fig. S7 and S8†). However, due to low doping probability, iodine could not be detected in the EDX spectrum of IUCN (Fig. S9†). The porous layered structure of XUCN catalysts was further visualized by using the TEM images. However, the high-resolution TEM (HR-TEM) images of all the materials demonstrated a hollow tubular type microstructure feature at the edge (Fig. 3B). The observed phenomenon can be attributed to the alteration of the molecular van der Waals force resulting from the inclusion of halouracil, which facilitates the bending and flexibility of the edges. The results support the substantial impact of 5-halouracil moieties on the morphological and microstructural characteristics of the modified PCN. The porous nature of the XUCN materials is further confirmed by using the nitrogen adsorption–desorption isotherm, which indicates a micro/mesoporous structure with slit-shaped pores as evidenced by a Type IV isotherm and typical H3 hysteresis loop.48 The measured surface property parameters are presented in Table S2.† The specific surface area (SBET) values of XUCN samples are higher as compared to those of their PCN counterparts and follow the order PCN < IUCN < BUCN < CUCN (inset of Fig. 3C, Table 1). The highest surface area of CUCN can be attributed to the higher degree of interaction between reactant molecules and smaller atomic radius of Cl. Moreover, the pore radius (Dr) and pore volume (DV) values of XUCN samples exhibit a significant increase compared to those of PCN. Hence, XUCN samples provide large active surface area to reactant molecules by exposing more surface-active sites. Next, the effect of halogen doping on the dissociation and migration of photo-excitons was systematically investigated using room temperature photoluminescence (PL) and time-resolved photoluminescence (TR-PL) spectroscopy. A significant quenching in PL intensity is observed after 5-halouracil doping in PCN, indicating that the doping enhances the separation of photogenerated charge carriers (Fig. 3D). The TR-PL spectra of PCN and XUCN catalysts exhibit triexponential decay, indicative of multiple relaxation (Fig. 3E, Table S3†). The estimated average lifetime (τavg) of PCN, CUCN, BUCN, and IUCN is found to be 1.51, 1.63, 1.94, and 3.08 ns, respectively (Table 1). The increased lifetime for the XUCN catalysts indicates enhanced possibility of photoexcitons to participate in the photocatalytic reaction before recombination.4 Further to be noted that the τavg of XUCN catalysts decreases with decreasing halogen atom size, highlighting the influence of the halogen atom on the electron lifetime decay.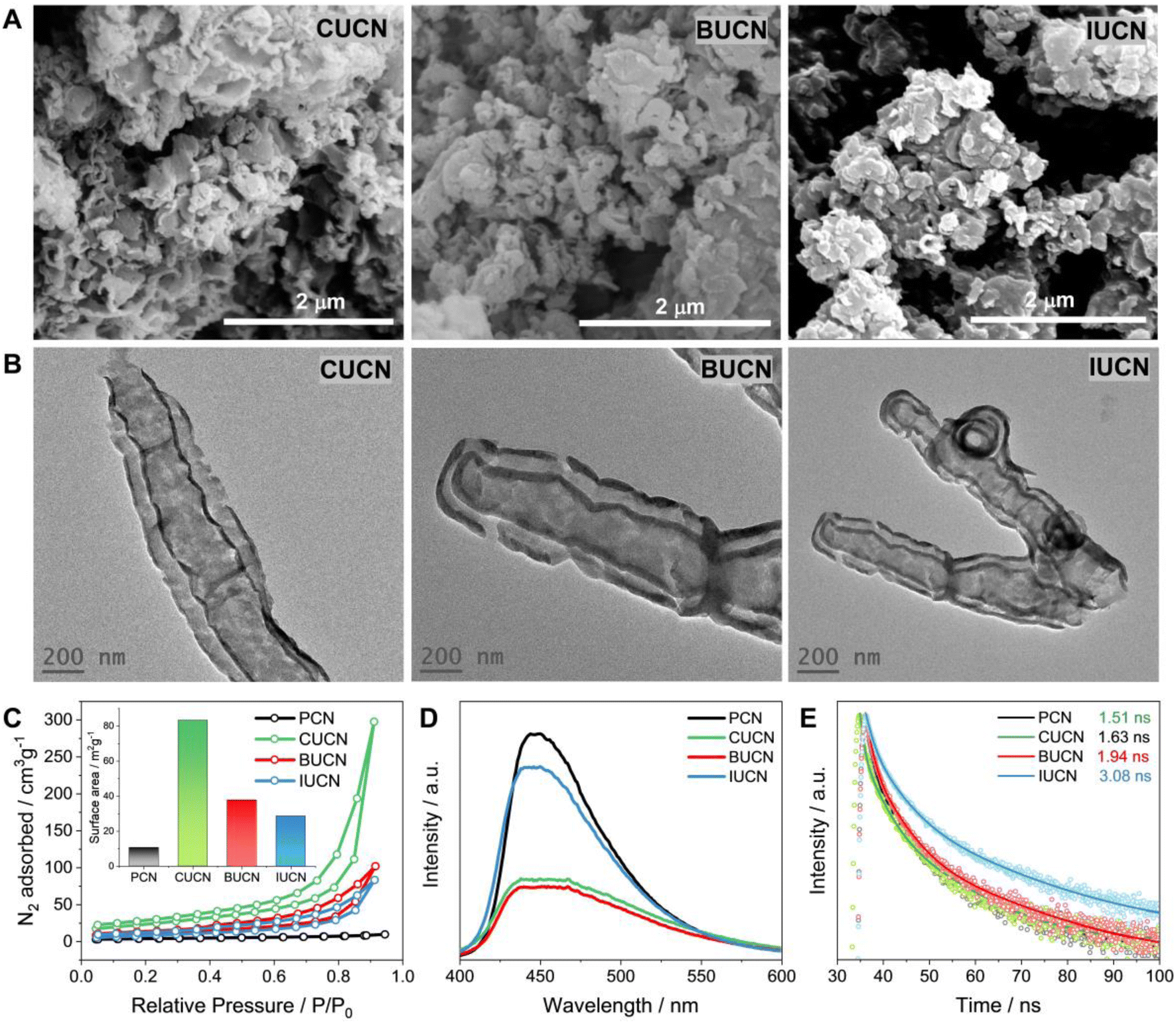 | ||
| Fig. 3 Morphology and optical properties. (A) Field emission scanning electron microscopy (FE-SEM) images (B) transmission electron microscopy (TEM) images, (C) nitrogen adsorption–desorption isotherm (the calculated surface are values are in the inset), (D) room temperature photoluminescence (PL) spectra, and (E) time-resolved photoluminescence (TR-PL) spectra of PCN, and XUCN (X = Cl, Br, or I) samples. | ||
| Catalysts | E g (eV) | S BET (m2 g−1) | D v (cm3 g−1) | D r (nm) | τ avg (ns) |
|---|---|---|---|---|---|
| a Bandgap estimated from UV-DRS measurements. b BET specific surface area calculated from the N2 adsorption–desorption isotherm. c BJH pore volume. d BJH pore radius. e Average lifetime of photoexcitation. | |||||
| PCN | 2.71 | 11 | 0.013 | 1.23 | 1.5 |
| CUCN | 2.57 | 83 | 0.155 | 1.63 | 1.6 |
| BUCN | 2.61 | 38 | 0.084 | 1.64 | 1.9 |
| IUCN | 2.66 | 29 | 0.061 | 1.62 | 3.1 |
The paramagnetic properties of PCN and XUCN were probed through room temperature electron paramagnetic resonance (EPR) spectroscopy. PCN exhibited a Lorentzian line with a Lande g-factor of 2.0036 (Fig. S10†) indicating delocalized unpaired electrons that are generated on the sp2-carbon atom of the π-conjugated aromatic rings.49 In contrast, the XUCN catalysts showed a Lorentzian line which shifted to a lower g-value of 1.9873. This shift in the g-value provides crucial information regarding the intrinsic properties of the paramagnetic centre, chemical structure (localized or delocalized structure), and spin states.50 The high and low g-values represent a delocalized and localized π-conjugated structure, respectively. The downshifted g-value EPR signals of CUCN, BUCN, and IUCN, indicate the formation of a localized π-conjugated structure. Furthermore, compared to PCN, XUCN catalysts demonstrated intensified Lorentzian lines in the order PCN < IUCN < CUCN < BUCN. This increase in intensity implies the enhanced generation of unpaired electrons over the XUCN catalyst, with BUCN showing superiority in producing unpaired electrons over the others. Overall, the EPR results exemplify that 5-halouracil doping in PCN results in the formation of localized π-conjugated structures that generate more unpaired electrons.
Boosting PCN photocatalytic activity through 5-halouracil doping
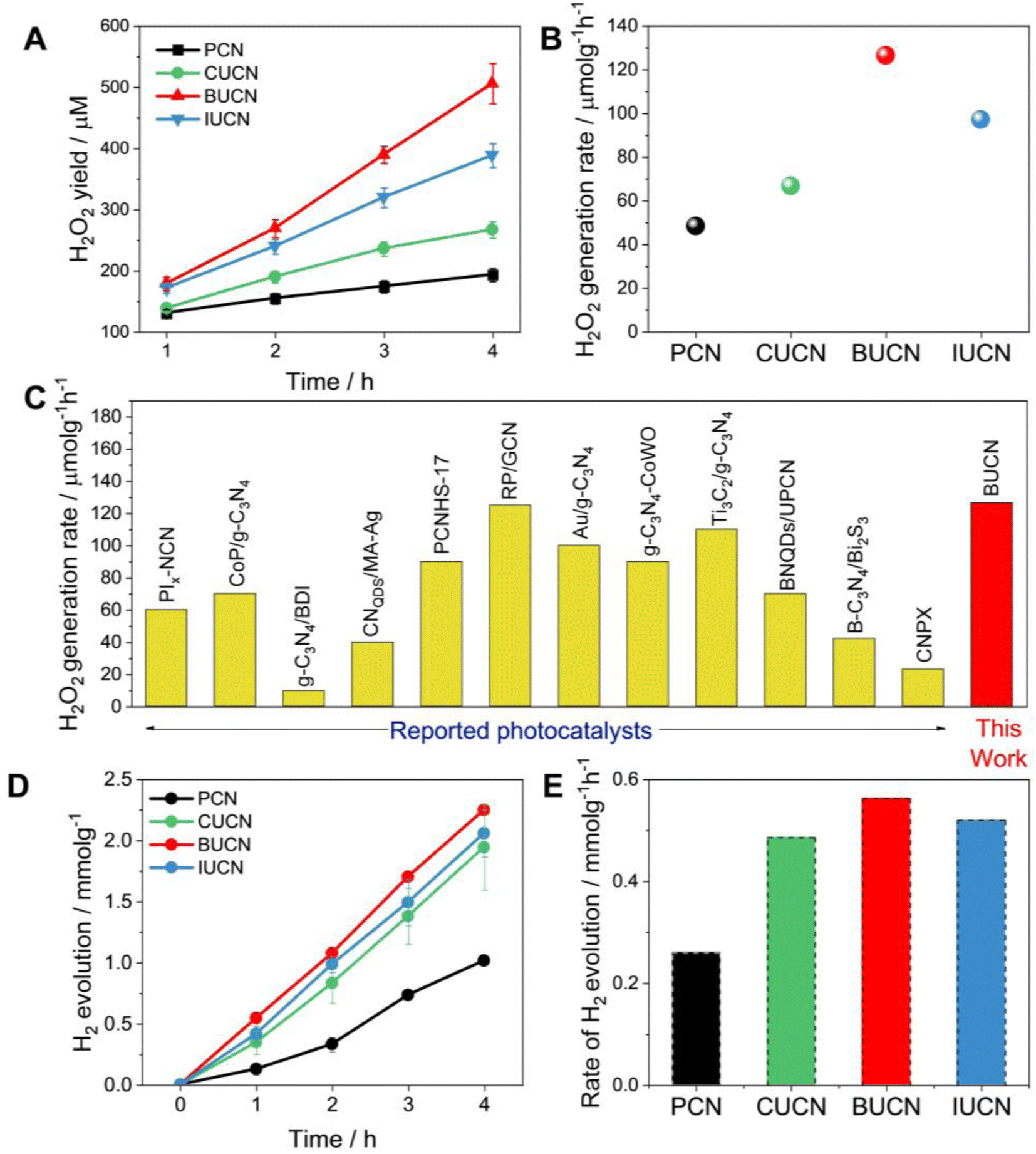 | ||
| Fig. 4 Photocatalytic performance. (A) photocatalytic H2O2 production as a function of irradiation time under visible light (λ > 450 nm) with 10 vol% isopropanol, (B) rate of H2O2 generation over PCN and XUCN catalysts, (C) a comparison of the H2O2 rate production with state-of-art PCN-based catalysts; more details can be found in the Table S4, ESI,† (D) photocatalytic H2 production and the corresponding (E) H2 production rate constants over PCN and XUCN catalysts. | ||
The photocatalytic H2 evolution ability of PCN and XUCN catalysts was evaluated under visible light irradiation (λ > 420 nm), employing 1 wt% Pt as a co-catalyst and 10% TEOA as a sacrificial donor. Like the high H2O2 production activity, the XUCN catalysts showcased a significant enhancement in the photocatalytic H2 generation activity compared to PCN after a reaction time of 4 h (Fig. 4D). Notably, the BUCN catalyst displayed the highest H2 production rate of 0.563 mmol g−1 h−1, nearly three times that of PCN (0.260 mmol g−1 h−1) (Fig. 4E). Furthermore, the apparent quantum yield (AQY) for H2 generation was calculated to be 2.62% for the BUCN catalyst at 420 nm. All the XUCN catalysts outperformed the hydrogen generation capabilities of a bare PCN catalyst. The optical, textural, and dynamic studies suggest that the increased photocatalytic activity of the XUCN catalysts compared to PCN can be ascribed to their broad optical response, and relatively higher specific surface area due to their porous structure. Additionally, the intimate interaction between the doped uracil and halide ions extends the π-conjugated system, thereby facilitating the mobility of photo-excitons. To note, the BUCN catalyst exhibited the highest PL quenching ability compared to the CUCN and IUCN catalysts. Despite the higher surface area exhibited by CUCN, BUCN demonstrated a higher photocatalytic H2O2 yield. This discrepancy is likely due to the valence electrons of Br-atoms being less electronegative than those of Cl-atoms, resulting in greater delocalization and interaction with the π-electron cloud of PCN. Conversely, doped I-atoms, despite possessing less electronegativity than Br-atoms, fail to interact with the π-electron cloud of PCN due to their large atomic radius, resulting in a lower doping probability. These factors collectively contribute to the enhanced photocatalytic activity of the BUCN catalyst. The findings underscore the important role of halogen ions in the photocatalytic ORR activity for H2O2 production and hydrogen generation.
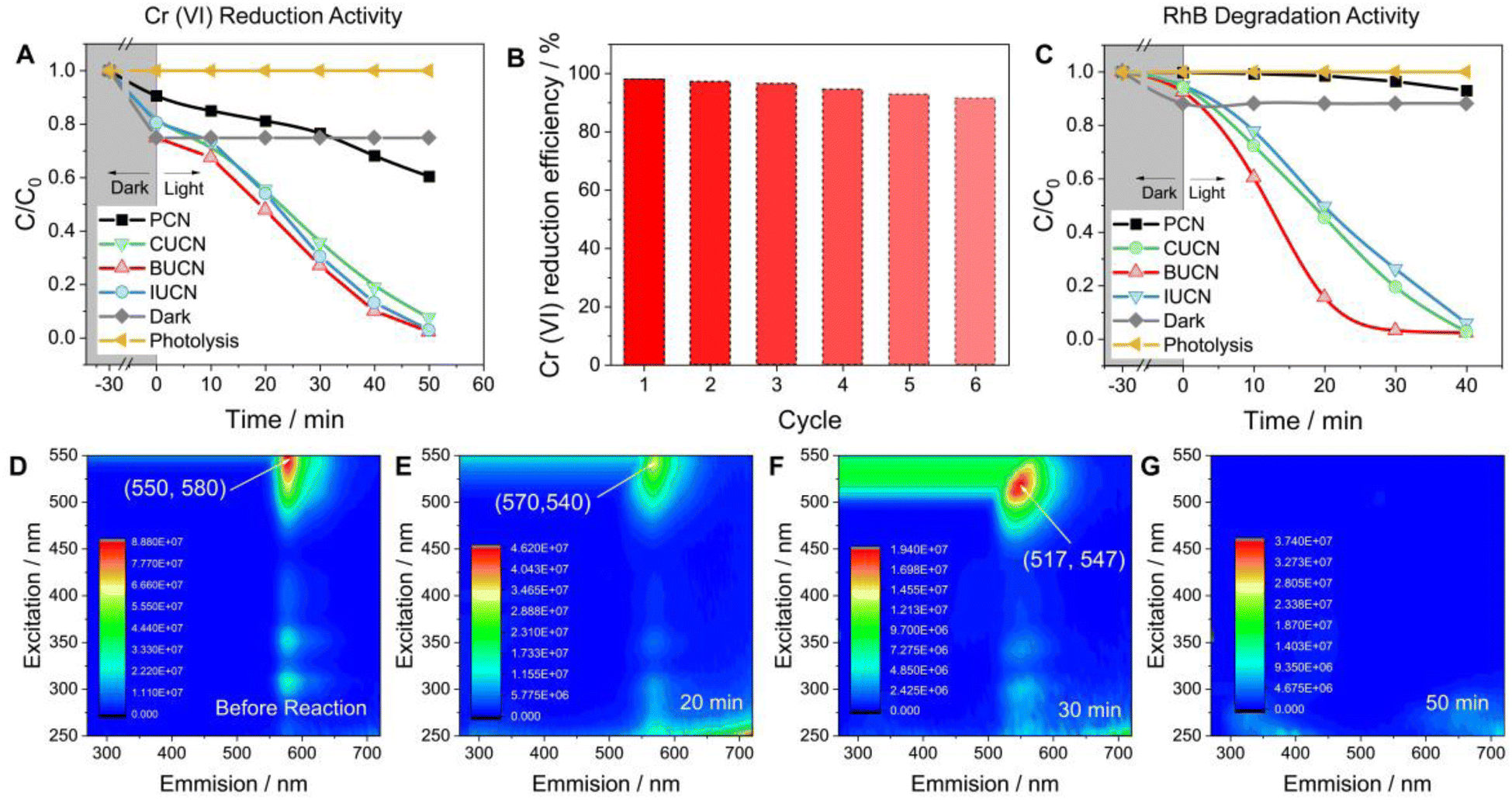 | ||
| Fig. 5 Photocatalytic removal of aquatic pollutants: (A) photocatalytic Cr(VI) reduction performance of PCN and XUCN catalysts (reaction conditions: 20 mg of catalyst, 100 mL Cr(VI) solution of 10 mg L−1), (B) cyclic durability test of an optimized BUCN catalyst for Cr(VI) reduction during repetitive six cycles, (C) relative decrease in rhodamine B (RhB) concentration as a function of time over PCN and XUCN photocatalysts under visible light irradiation, and the corresponding 3D excitation-emission (3D EEM) matrix of (D) untreated RhB stock solution (before reaction), and photocatalytically treated RhB solution with a BUCN catalyst after (E) 20 min, (F) 30 min, and (G) 50 min of visible light irradiation, respectively. | ||
The XUCN photocatalyst demonstrated a higher rate of photooxidative degradation of rhodamine B (RhB, 20 mg L−1) compared to the bare PCN counterpart (Fig. 5C). BUCN exhibited the highest degradation rate constant of 0.116 min−1 (0.002 min−1 for PCN) (Fig. S18†). Furthermore, the control experiments suggested a completely photooxidative RhB degradation process on the catalyst's surface. To investigate the photocatalytic degradation process and dissolved organic matter in residual RhB solution after illumination, 3D excitation emission matrix (3D-EEM) fluorescence spectroscopy was used (Fig. 5D–G). The 3D-EEM of untreated RhB solution (stock) revealed a characteristic fluorescence peak centred at an excitation/emission value of 550/580 nm.52 As the photocatalytic reaction progressed, the intensity of the peak decreased significantly after 20 min, indicating the predominant decolouration process of RhB and generation of photo-transformation intermediates.53 After 30 min of irradiation, the peak was blue shifted to 517/547 nm, which was attributed to the dissociation of large organic molecules into smaller fragments, cleavage of RhB's chomophoric structure, and elimination of functional groups.54 This observation suggests the N-deethylation process of RhB molecules and fragmentation of N-deethylated intermediates into small molecules. Interestingly, the peak disappeared after 50 min of irradiation, indicating the complete decomposition of RhB molecules and their mineralization into CO2 and H2O.55
Overall, the photocatalytic experiments reveal a higher photocatalytic activity for the prepared BUCN catalyst compared to the PCN and other XUCN catalysts (Table 2). The excellent photoactivity of the BUCN catalyst can be ascribed to its physicochemical properties as discussed in the previous section.
| Catalyst | H2O2 production rate (μmol h−1) | H2 evolution rate (mmol h−1 g−1) | Rate constant | |
|---|---|---|---|---|
| Cr(VI) reduction | RhB degradation | |||
| PCN | 48.3 | 0.260 | 0.008 | 0.002 |
| CUCN | 66.7 | 0.486 | 0.047 | 0.052 |
| BUCN | 126.6 | 0.563 | 0.068 | 0.116 |
| IUCN | 97.2 | 0.520 | 0.061 | 0.043 |
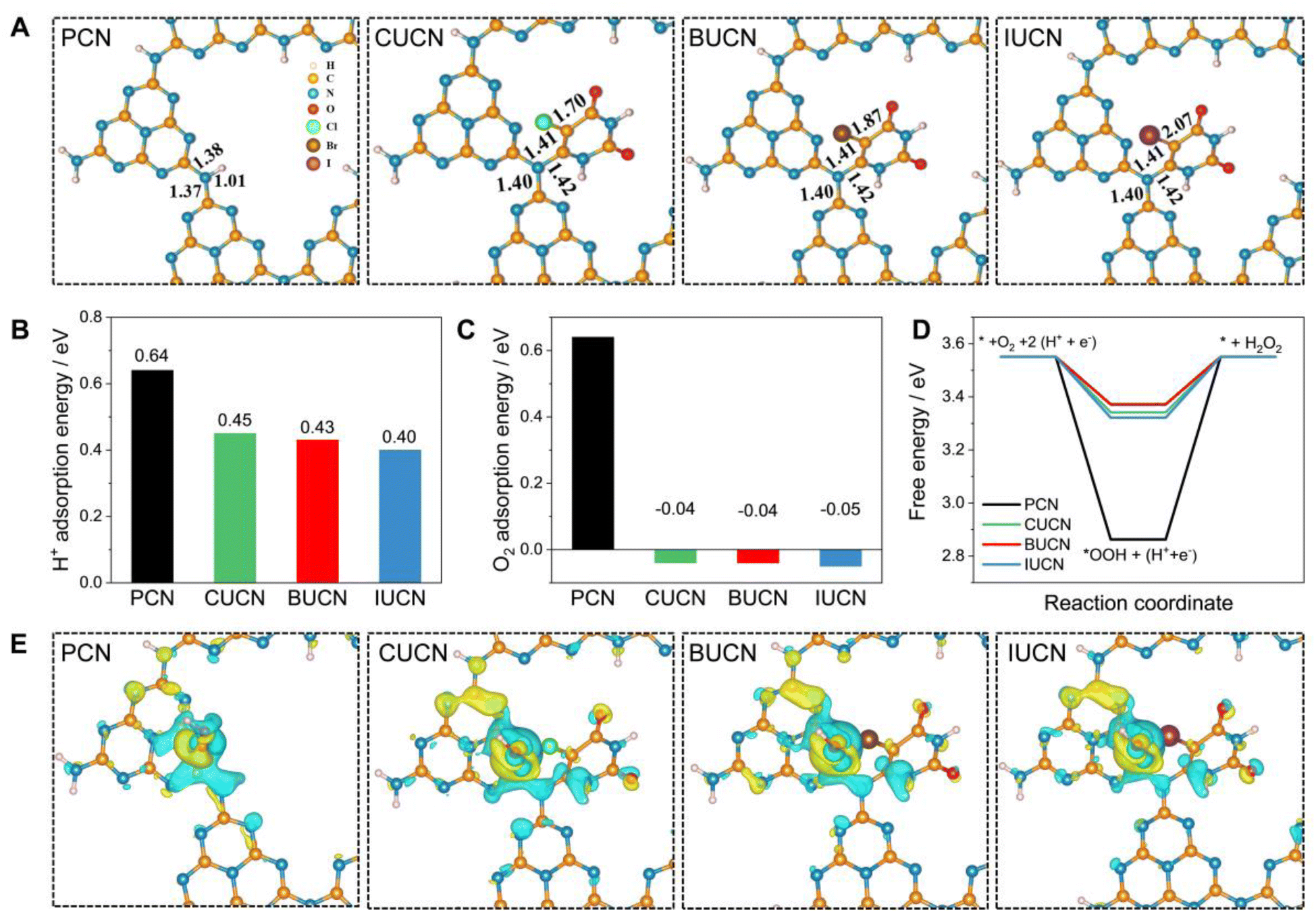 | ||
| Fig. 6 Theoretical analysis: (A) optimized structures of PCN, and XUCN configurations, (B) proton (H+) adsorption and (C) oxygen (O2) adsorption energies on pristine PCN and halouracil doped PCN, and (D) free energy profile of photocatalytic O2 reduction to hydrogen peroxide (H2O2) over PCN and XUCN catalysts at U = 0.7 V, and (E) charge density difference of OOH* species adsorbed on PCN and XUCN catalysts (the yellow and blue iso-surface represents electron accumulation and depletion regions, respectively). | ||
| Sample | C–N bond length (Å) | C–X bond length (Å) | H+ adsorption energy (eV) | O2 adsorption energy (eV) | ΔG*OOH (eV) |
|---|---|---|---|---|---|
| PCN | 1.37 | — | 0.64 | 0.64 | 2.86 |
| CUCN | 1.40 | 1.70 | 0.45 | −0.04 | 3.34 |
| BUCN | 1.40 | 1.87 | 0.43 | −0.04 | 3.37 |
| IUCN | 1.40 | 2.07 | 0.40 | −0.05 | 3.32 |
The electron spin resonance (ESR) spin trapping experiment was performed to gain further insights into the generation of reactive oxidative species (ROS), ˙O2− and ˙OH. Under light irradiation the ESR spectra of PCN and XUCN catalysts exhibited characteristic four-line signals using 5,5-dimethyl-1-pyrroline N-oxide (DMPO) with a peak intensity ratio of 1:2:2:1, indicating the generation of ˙OH radicals on PCN and XUCN (Fig. S22A†). Remarkably, the XUCN catalysts showed heightened signal intensity with respect to PCN. Later, the spin trapping experiment by adding DMSO along with DMPO effectively split the ESR signal into radical adducts indicating the generation of ˙O2−. This finding indicates that XUCN catalysts are more capable in the generation of ˙O2− than bulk PCN (Fig. S22B†).
The presence of bromide ions (Br−) in XUCN catalysts has a significant impact on the photocatalytic mechanism. The Br− acts as electron acceptors and enhance the generation of ROS, such as ˙OH radicals and ˙O2− , leading to improved photocatalytic performance. This is supported by the heightened signal intensity observed in the ESR spectra of XUCN catalysts compared to those of PCN, indicating increased generation of ˙OH radicals and ˙O2−. Thus, the presence of Br− ions contributes to the overall efficiency of the photocatalytic process and enhances the degradation performance of the catalyst.
Based on the experimental and theoretical calculations, a plausible mechanism on the BUCN catalyst is depicted in Scheme 1. Under light irradiation, BUCN absorbs photons and generates electron–hole pairs. The reconstructed electronic energy levels cause the electron–hole couples to dissociate rapidly. On the other hand, the high surface area and low adsorption energy enable better adsorption of H+ and O2 on the BUCN surface. The synergy between photo-exciton dissociation and efficient adsorption behaviour promotes 2e− O2 reduction to generate H2O2. The O2 can accept one electron to generate ˙O2− and then ˙O2− takes another electron to generate H2O2. According to this observation and in conjunction with previous reports, photogenerated electrons reduce O2 to generate H2O2 (eqn (4)).
| O2 + 2e− + 2H+ → H2O2 | (4) |
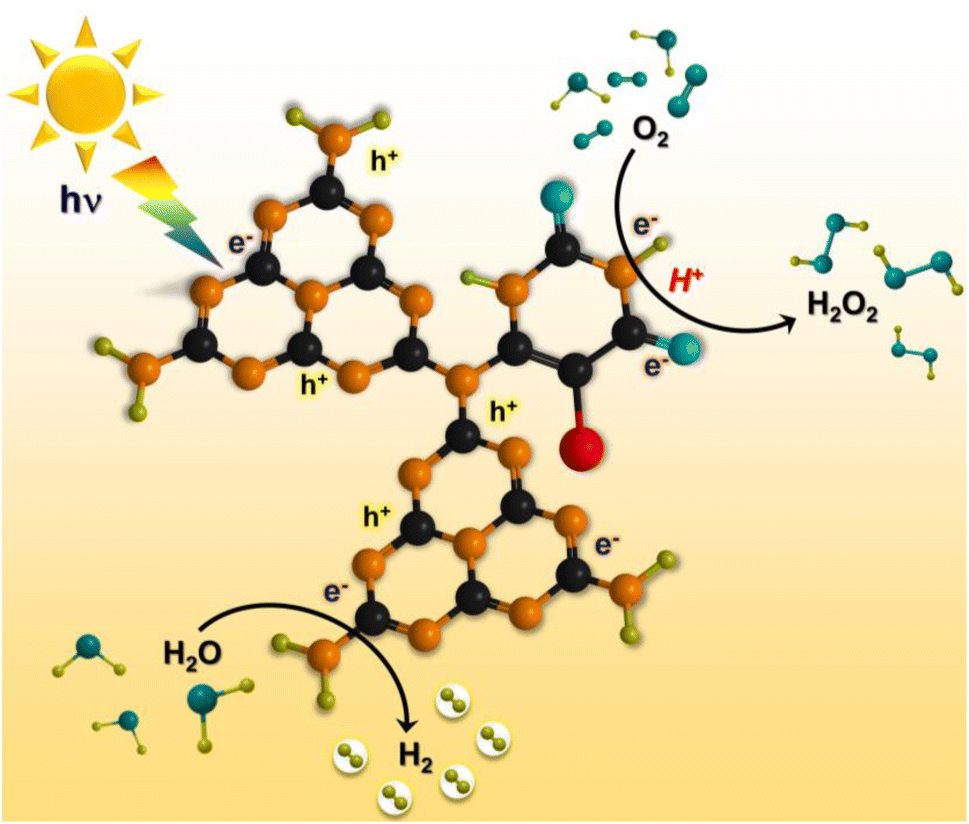 | ||
| Scheme 1 Schematic illustration of photocatalytic H2O2 production and hydrogen generation (the black, orange, blue, green, and red spheres represent C, N, O, H, and halogen ions). | ||
Conclusion
In summary, 5-halouracil derivates were used to incorporate both a π-conjugated organic moiety and halogen atoms into the PCN framework. 5-halouracil doped PCN (XUCN, X = Cl, Br, or I) exhibited a significantly extended visible-light response above 450 nm, tailored the surface topological features, and accelerated the exciton dissociation. The photoactivity assessment revealed that the BUCN catalyst exhibits H2O2 and hydrogen generation rate of 126.6 μmol h−1 and 0.563 mmol g−1 h−1, which is much higher than that of pristine PCN. In addition, BUCN exhibited over 95% Cr(VI) reduction activity in 50 min of irradiation and achieved complete mineralization of RhB dye into CO2 and H2O in 50 min of visible light irradiation. Furthermore, theoretical investigation unveiled the low H+ and O2 adsorption energy for BUCN that accelerated the H2O2 and hydrogen generation performance. The experimental results in conjunction with theoretical calculations clarify the boosted photocatalytic activity of the BUCN catalyst. We believe the results provided here will motivate the use of the one-step dual doping approach to systematically tweak the optoelectronic characteristics of PCN and accomplish superior photocatalytic performance for both energy and environmental applications.Author contributions
T. B. and V. D. conceptualized the project. T. B. developed experimental design and methodology. S. S. U. supervised the project. T. B., B. M. A., A. G., D. J. K., N. M., D. V., and S. S. U. investigated and formally analysed the results. T. B. wrote the original draft and B. M. A., A. G., D. J. K., N. M., K. S. P., D. V., and S. S. U. revised and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
TB would like to thank the Director, VNIT, Nagpur, for the doctoral fellowship. We would like to thank the Department of Science and Technology Fund for Improvement of Science and Technology Infrastructure (DST-FIST) through project no. SR/FST/CSI-279/2016(C) for providing the instrumentation facility at the Department of Chemistry, VNIT, Nagpur. We thank SAIF IIT Madras and IIT Bombay for the central instrumentation facility. BMA would like to thank the HPC centre, IIT Kanpur for providing the computational facilities. NRM is thankful to PASIFIC postdoctoral funding received from the European Union's Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement No. 847639 and from the Ministry of Education and Science.Notes and references
- Y. Wang, S. Z. F. Phua, G. Dong, X. Liu, B. He, Q. Zhai, Y. Li, C. Zheng, H. Quan and Z. Li, Chem, 2019, 5, 2775–2813 CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS.
- D. Vidyasagar, T. Bhoyar, G. Singh and A. Vinu, Macromol. Rapid Commun., 2021, 42, 2000676 CrossRef CAS.
- Q. Zhou, Y. Guo, Z. Ye, Y. Fu, Y. Guo and Y. Zhu, Mater. Today, 2022, 58, 100–109 CrossRef CAS.
- Y. Wang, K. Wang, F. Dai, K. Zhang, H. Tang, L. Wang and J. Xing, Nat. Commun., 2022, 13, 6495 CrossRef CAS.
- T. Bhoyar, D. J. Kim, B. M. Abraham, S. Tonda, N. R. Manwar, D. Vidyasagar and S. S. Umare, Appl. Catal., B, 2022, 310, 121347 CrossRef CAS.
- L. Heymann, S. C. Bittinger and C. Klinke, ACS Omega, 2018, 3, 17042–17048 CrossRef CAS.
- T. Zhou, J. Shi, G. Li, B. Liu, B. Hu, G. Che, C. Liu, L. Wang and L. Yan, J. Colloid Interface Sci., 2023, 632, 285–298 CrossRef CAS PubMed.
- D. Cruz, J. Garcia Cerrillo, B. Kumru, N. Li, J. Dario Perea, B. V. K. J. Schmidt, I. Lauermann, C. J. Brabec and M. Antonietti, J. Am. Chem. Soc., 2019, 141, 12322–12328 CrossRef CAS.
- C. Zhu, T. Wei, Y. Wei, L. Wang, M. Lu, Y. Yuan, L. Yin and L. Huang, J. Mater. Chem. A, 2021, 9, 1207–1212 RSC.
- T. Jordan, N. Fechler, J. Xu, T. J. K. Brenner, M. Antonietti and M. Shalom, ChemCatChem, 2015, 7, 2826–2830 CrossRef CAS.
- C. Yang, B. Wang, L. Zhang, L. Yin and X. Wang, Angew. Chem., Int. Ed., 2017, 56, 6627–6631 CrossRef CAS.
- J. Shi, M. Tai, J. Hou, Y. Qiao, C. Liu, T. Zhou, L. Wang and B. Hu, Chem. Eng. J., 2023, 456, 141029 CrossRef CAS.
- Z.-A. Lan, G. Zhang and X. Wang, Appl. Catal., B, 2016, 192, 116–125 CrossRef CAS.
- A. Gupta, T. Bhoyar, B. M. Abraham, D. J. Kim, K. S. Pasupuleti, S. S. Umare, D. Vidyasagar and A. Gedanken, ACS Appl. Mater. Interfaces, 2023, 15, 18898–18906 CrossRef CAS.
- G. Zhang, M. Zhang, X. Ye, X. Qiu, S. Lin and X. Wang, Adv. Mater., 2014, 26, 805–809 CrossRef CAS.
- Y. Wang, Y. Di, M. Antonietti, H. Li, X. Chen and X. Wang, Chem. Mater., 2010, 22, 5119–5121 CrossRef CAS.
- C. Liu, Y. Zhang, F. Dong, A. H. Reshak, L. Ye, N. Pinna, C. Zeng, T. Zhang and H. Huang, Appl. Catal., B, 2017, 203, 465–474 CrossRef CAS.
- B. Zhu, J. Zhang, C. Jiang, B. Cheng and J. Yu, Appl. Catal., B, 2017, 207, 27–34 CrossRef CAS.
- Q.-H. Zhu, Z. Chen, L.-N. Tang, Y. Zhong, X.-F. Zhao, L.-Z. Zhang and J.-H. Li, Int. J. Hydrogen Energy, 2019, 44, 27704–27712 CrossRef CAS.
- T. Bhoyar, N. Saraswat, M. V. Jyothirmai, A. Gupta, S. K. Malla, J. Park, D. Vidyasagar and S. S. Umare, ACS Appl. Nano Mater., 2023, 6, 3484–3496 CrossRef CAS.
- T. Bhoyar, D. J. Kim, B. M. Abraham, A. Gupta, N. Maile, N. R. Manwar, S. Tonda, D. Vidyasagar and S. S. Umare, iScience, 2022, 25, 105567 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188 CrossRef.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2004, 25, 1463–1473 CrossRef CAS PubMed.
- B. M. Abraham, J. Phys. Chem. Solids, 2021, 149, 109777 CrossRef CAS.
- B. M. Abraham, Mater. Today Commun., 2021, 26, 101901 CrossRef CAS.
- S. Siahrostami, A. Verdaguer-Casadevall, M. Karamad, D. Deiana, P. Malacrida, B. Wickman, M. Escudero-Escribano, E. A. Paoli, R. Frydendal and T. W. Hansen, Nat. Mater., 2013, 12, 1137–1143 CrossRef CAS PubMed.
- A. Verdaguer-Casadevall, D. Deiana, M. Karamad, S. Siahrostami, P. Malacrida, T. W. Hansen, J. Rossmeisl, I. Chorkendorff and I. E. Stephens, Nano Lett., 2014, 14, 1603–1608 CrossRef CAS.
- T. Bhoyar, D. Vidyasagar and S. S. Umare, Mater. Today Commun., 2020, 24, 101119 CrossRef CAS.
- Z. Li, Y. Zhou, Y. Zhou, K. Wang, Y. Yun, S. Chen, W. Jiao, L. Chen, B. Zou and M. Zhu, Nat. Commun., 2023, 14, 5742 CrossRef CAS.
- S. An, G. Zhang, K. Li, Z. Huang, X. Wang, Y. Guo, J. Hou, C. Song and X. Guo, Adv. Mater., 2021, 33, 2104361 CrossRef CAS.
- G. Zhang, A. Savateev, Y. Zhao, L. Li and M. Antonietti, J. Mater. Chem. A, 2017, 5, 12723–12728 RSC.
- D. Jang, S. Choi, N. H. Kwon, K. Y. Jang, S. Lee, T.-W. Lee, S.-J. Hwang, H. Kim, J. Kim and S. Park, Appl. Catal., B, 2022, 310, 121313 CrossRef CAS.
- B. Zhai, H. Li, G. Gao, Y. Wang, P. Niu, S. Wang and L. Li, Adv. Funct. Mater., 2022, 2207375 CrossRef CAS.
- X. Zhan, H. Wang, G. Zhou, L. Chen, Y. Sun, Y. Zhao, J. Liu and H. Shi, ACS Appl. Mater. Interfaces, 2021, 13, 12118–12130 CrossRef CAS PubMed.
- M. Zhang, J. Wen, S. Zhang and Y. Zhai, Sep. Purif. Technol., 2021, 257, 117984 CrossRef CAS.
- Q. Gu, P. P. Jiang, Y. Shen, K. Zhang, P. T. Wai and A. Haryono, Mol. Catal., 2021, 504, 111441 CrossRef CAS.
- L. Chen, Y. Xu, Z. Yang, K. Zhang and B. Chen, Appl. Catal., B, 2020, 277, 119207 CrossRef CAS.
- M. Wang, Y. Zeng, G. Dong and C. Wang, Chin. J. Catal., 2020, 41, 1498–1510 CrossRef CAS.
- L. Shen, G. Lei, Y. Zheng, S. Liang, F. Liu, S. Wang, Y. Cao, Y. Xiao and L. Jiang, ChemCatChem, 2021, 13, 2386–2392 CrossRef CAS.
- J. Zheng, H.-T. Liu, B. Wu, C.-A. Di, Y.-L. Guo, T. Wu, G. Yu, Y.-Q. Liu and D.-B. Zhu, Sci. Rep., 2012, 2, 662 CrossRef.
- T. Ishizaki, Y. Wada, S. Chiba, S. Kumagai, H. Lee, A. Serizawa, O. L. Li and G. Panomsuwan, Phys. Chem. Chem. Phys., 2016, 18, 21843–21851 RSC.
- E. Prabakaran, T. Velempini, M. Molefe and K. Pillay, J. Mater. Res. Technol., 2021, 15, 6340–6355 CrossRef CAS.
- K. Liu, N. Zakharova, A. Adeyilola and L. Zeng, Energy Fuels, 2021, 35, 2183–2191 CrossRef CAS.
- W. Wang, H. Zhang, S. Zhang, Y. Liu, G. Wang, C. Sun and H. Zhao, Angew. Chem., Int. Ed., 2019, 58, 16644–16650 CrossRef CAS.
- P. Xia, B. Cheng, J. Jiang and H. Tang, Appl. Surf. Sci., 2019, 487, 335–342 CrossRef CAS.
- J. Shao, H. Fei, H. Li, L. Yang, M. Li, J. Gao, H. Liao and J. Lu, Sep. Purif. Technol., 2022, 297, 121515 CrossRef CAS.
- J. Yan, J. Liu, Y. Sun, D. Ding, C. Wang, L. Sun and X. Li, Inorg. Chem. Front., 2022, 9, 1423–1433 RSC.
- Y. Xie, Y. Dai, X. Yuan, L. Jiang, L. Zhou, Z. Wu, J. Zhang, H. Wang and T. Xiong, J. Colloid Interface Sci., 2018, 530, 493–504 CrossRef CAS.
- H. Guo, C.-G. Niu, X.-J. Wen, L. Zhang, C. Liang, X.-G. Zhang, D.-L. Guan, N. Tang and G.-M. Zeng, J. Colloid Interface Sci., 2018, 513, 852–865 CrossRef CAS.
- H. Wang, X. Yuan, Y. Wu, G. Zeng, W. Tu, C. Sheng, Y. Deng, F. Chen and J. W. Chew, Appl. Catal., B, 2017, 209, 543–553 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta04938h |
| This journal is © The Royal Society of Chemistry 2024 |