
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA high-resolution structural characterization and physicochemical study of how a peptoid binds to an oncoprotein MDM2†
Marin
Yokomine
 a,
Jumpei
Morimoto
*a,
Yasuhiro
Fukuda
a,
Takumi
Ueda
b,
Koh
Takeuchi
b,
Koji
Umezawa
cd,
Hideo
Ago
e,
Hiroaki
Matsuura
e,
Go
Ueno
e,
Akinobu
Senoo‡
a,
Satoru
Nagatoishi
f,
Kouhei
Tsumoto
afg and
Shinsuke
Sando
*af
a,
Jumpei
Morimoto
*a,
Yasuhiro
Fukuda
a,
Takumi
Ueda
b,
Koh
Takeuchi
b,
Koji
Umezawa
cd,
Hideo
Ago
e,
Hiroaki
Matsuura
e,
Go
Ueno
e,
Akinobu
Senoo‡
a,
Satoru
Nagatoishi
f,
Kouhei
Tsumoto
afg and
Shinsuke
Sando
*af
aDepartment of Chemistry and Biotechnology, Graduate School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: jmorimoto@chembio.t.u-tokyo.ac.jp; ssando@chembio.t.u-tokyo.ac.jp
bGraduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan
cDepartment of Biomedical Engineering, Graduate School of Science and Technology, Shinshu University, 8304 Minami-Minowa, Kami-Ina, Nagano, 399-4598, Japan
dDepartment of Biomolecular Innovation, Institute for Biomedical Sciences, Shinshu University, 8304 Minami-Minowa, Kami-Ina, Nagano, 399-4598, Japan
eRIKEN SPring-8 Center, 1-1-1 Kouto, Sayo, Hyogo 679-5148, Japan
fDepartment of Bioengineering, Graduate School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
gInstitute of Medical Science, The University of Tokyo, 4-6-1 Shirokanedai, Minato-ku, Tokyo 108-8639, Japan
First published on 19th April 2024
Abstract
Peptoids are a promising drug modality targeting disease-related proteins, but how a peptoid engages in protein binding is poorly understood. This is primarily due to a lack of high-resolution peptoid–protein complex structures and systematic physicochemical studies. Here, we present the first crystal structure of a peptoid bound to a protein, providing high-resolution structural information about how a peptoid binds to a protein. We previously reported a rigid peptoid, oligo(N-substituted alanine) (oligo-NSA), and developed an oligo-NSA-type peptoid that binds to MDM2. X-ray crystallographic analysis of the peptoid bound to MDM2 showed that the peptoid recognizes the MDM2 surface predominantly through the interaction of the N-substituents, while the main chain acts as a scaffold. Additionally, conformational, thermodynamic, and kinetic analysis of the peptoid and its derivatives with a less rigid main chain revealed that rigidification of the peptoid main chain contributes to improving the protein binding affinity. This improvement is thermodynamically attributed to an increased magnitude of the binding enthalpy change, and kinetically to an increased association rate and decreased dissociation rate. This study provides invaluable insights into the design of protein-targeting peptoids.
Introduction
Peptides are attracting attention as a drug modality that combines the advantages of small molecules with those of antibody drugs. These advantages include favorable pharmacokinetic properties due to the smaller molecular size compared to antibodies, and high binding affinity and specificity attributed to the larger surface area relative to small molecules. However, their low membrane permeability and poor proteolytic resistance limit the utility of peptides as clinically useful drugs.Peptides with various functional groups on amide nitrogens are called “peptoids” and have been shown to exhibit high membrane permeability1 and proteolytic resistance.2 Since oligo(N-substituted glycine) (oligo-NSG, Fig. 1A) was proposed as the first peptoid in 1992,3 peptoids are attracting a lot of attention in drug discovery. However, the main chain of NSG-type peptoids is intrinsically flexible, making it challenging to obtain ligands capable of binding to biomolecules with high affinity (Fig. 1A). Thus, researchers have been making efforts to constrain the conformation of the peptoid main chain, which has led to the discovery of peptoids that bind to proteins with high affinity.4–9 However, no high-resolution structures of a peptoid bound to a protein have ever been reported, and, consequently, detailed information on how a peptoid binds to a protein and how the rigidity of a peptoid contributes to the binding remains unclear.
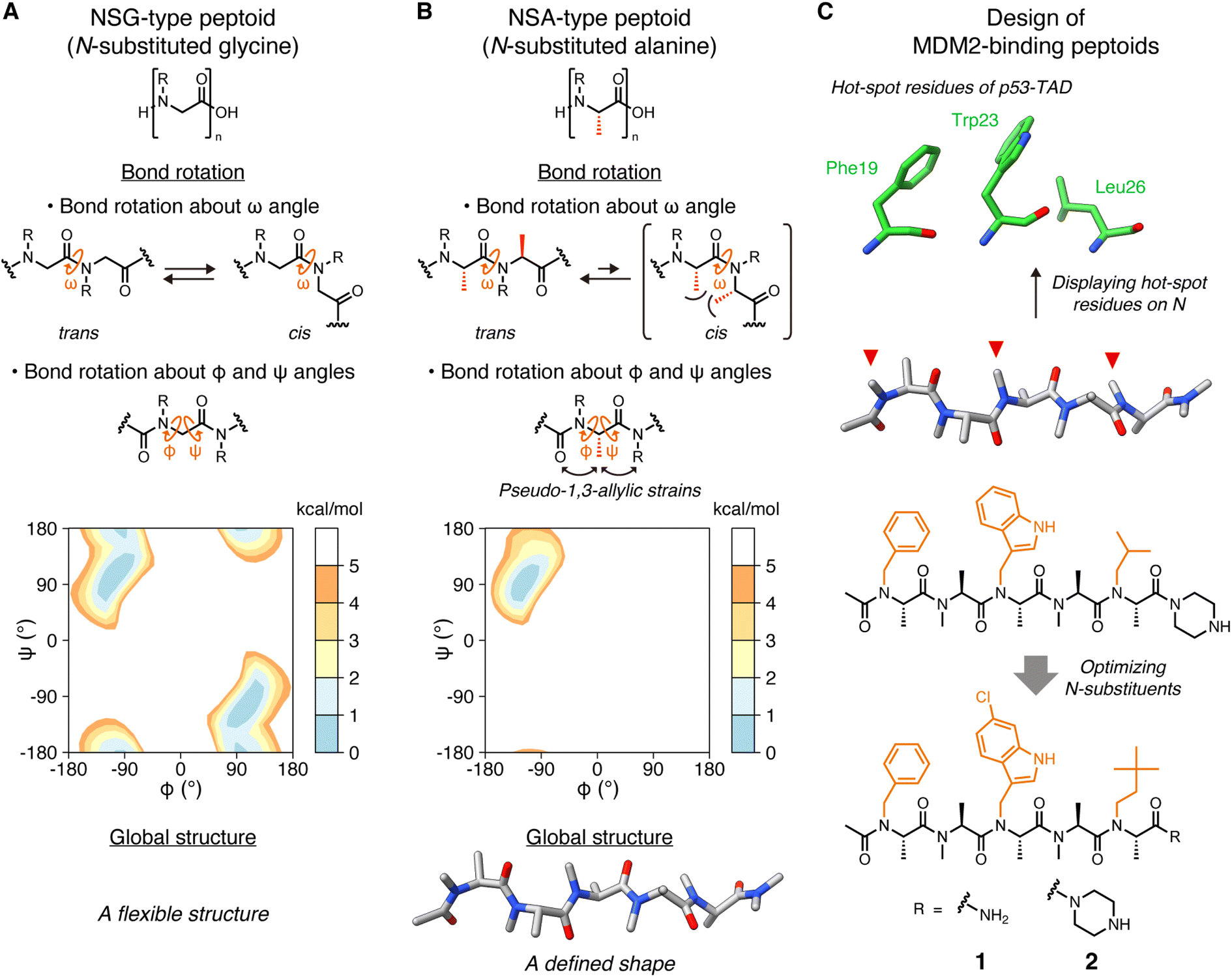 | ||
| Fig. 1 Structures of peptoids and the design of MDM2-binding peptoids (A) Chemical structures of NSG-type peptoid and the bond rotations about ω, φ, and ψ angles. The Ramachandran-type energy landscape of acetyl-N-methylglycine dimethylamide as a minimal model of NSG-type peptoid generated by density functional theory calculations is shown.6 (B) Chemical structures of NSA-type peptoid and the bond rotations about ω, φ, and ψ angles restricted by steric effects. The Ramachandran-type energy landscape of acetyl-N-methylalanine dimethylamide as a minimal model of NSA-type peptoid is shown.6 A predicted oligomer conformation is shown at the bottom. (C) Hot-spot residues of p53-TAD, namely Phe19, Trp23, and Leu26 (PDB ID 1YCR),13 the design flow of MDM2-binding peptoids, and chemical structures of peptoids 1 and 2 that bear functional N-substituents (orange) corresponding to the three hot-spot residues of p53-TAD.6,10,11 | ||
Oligo(N-substituted alanine) (oligo-NSA, Fig. 1B) was recently reported as a rigid peptoid, the main chain of which stably forms an extended shape in water.6,8 The main chain of the NSA-type peptoid is conformationally constrained by steric repulsion at the monomer level. The main chain structure of a peptoid is defined by three dihedral angles: ω, φ, and ψ. For ω angles, the trans configurations (ω = 180°) are favored in the NSA-type peptoid due to the steric repulsion between two methyl groups on the α-carbons on neighboring residues in the cis configurations (ω = 0°) (Fig. 1B). Also, for φ and ψ angles, steric effects, known as pseudo-1,3-allylic strains induced by the methyl group on the α-carbon restrict rotation about the φ and ψ angles (Fig. 1B); therefore, an NSA-type peptoid stably forms an extended shape (Fig. 1B). Using this NSA-type peptoid as a scaffold, we developed the MDM2-binding peptoid by displaying three functional groups on amide nitrogens of an NSA-type peptoid, mimicking three hot-spot residues of p53, MDM2-binding protein.6,10 Furthermore, by optimizing three N-substituents, NSA-type peptoid 1, which has a high binding affinity to MDM2, was successfully obtained (Fig. 1C).11
Here, we report the crystal structure of the peptoid 1 derivative bound to MDM2. This is the first high-resolution structure of a peptoid oligomer bound to a protein. In addition, we describe the investigation of how the rigidity of a peptoid contributes to its binding affinity to a protein based on the thermodynamic and kinetic parameters of the interaction between MDM2 and a series of peptoid 1 derivatives with different degrees of rigidity.
Results and discussion
Crystal structure of the peptoid bound to MDM2
To determine the crystal structure of the peptoid–MDM2 complex, we conducted crystallization screening. To facilitate the crystallization process, the C-terminal amide of NSA-type peptoid 1 was replaced with piperazine (NSA-type peptoid 2, Fig. 1C), which improves the aqueous solubility.12 Our previous study suggests that the difference in the C-terminal structure does not significantly affect the binding affinity of the peptoid to MDM2.11 From the crystals obtained from the solution of 2 and MDM2, we successfully determined the high-resolution structure of the complex. The resolution of the solved structure was 1.35 Å (Fig. 2A, left and Table S1†). Although there have been reports about crystal structures of peptides containing a peptoid residue or peptoid monomers bound to a protein, to the best of our knowledge, this is the first high-resolution structure of a peptoid oligomer bound to a protein. As a comparison, the crystal structure of the MDM2 complexed with the transactivation domain of p53 (p53-TAD) reported by Kussie and co-workers (PDB ID 1YCR)13 is shown in Fig. 2A, right. The X-ray crystallographic analysis revealed that the first, third, and fifth N-substituents of 2 mimic the spatial arrangement of the three hot-spot residues of p53-TAD (Phe19, Trp23, Leu26) as designed. Consequently, 2 binds to MDM2 primarily through van der Waals interactions made by these N-substituents (Fig. 2A–C). This is consistent with the previous observations that modifications to these N-substituents largely changed the binding affinity.6,10,11 The buried surface area of the complex is 996 Å2. It is smaller than that of the peptidic binder, p53-TAD (PDB ID 1YCR,13 1477 Å2) but larger than that of the small molecule binder, Nutlin-3a (PDB ID 4J3E,14 825 Å2). Regarding the main chain of 2 in the crystal structure, all dihedral angles are in the expected ranges. Specifically, all the amide bonds are in trans configuration, and all the observed φ and ψ angles are in the low-energy region of the previously reported Ramachandran-type energy landscape of a minimal model structure of the NSA-type peptoid (Fig. 2D).6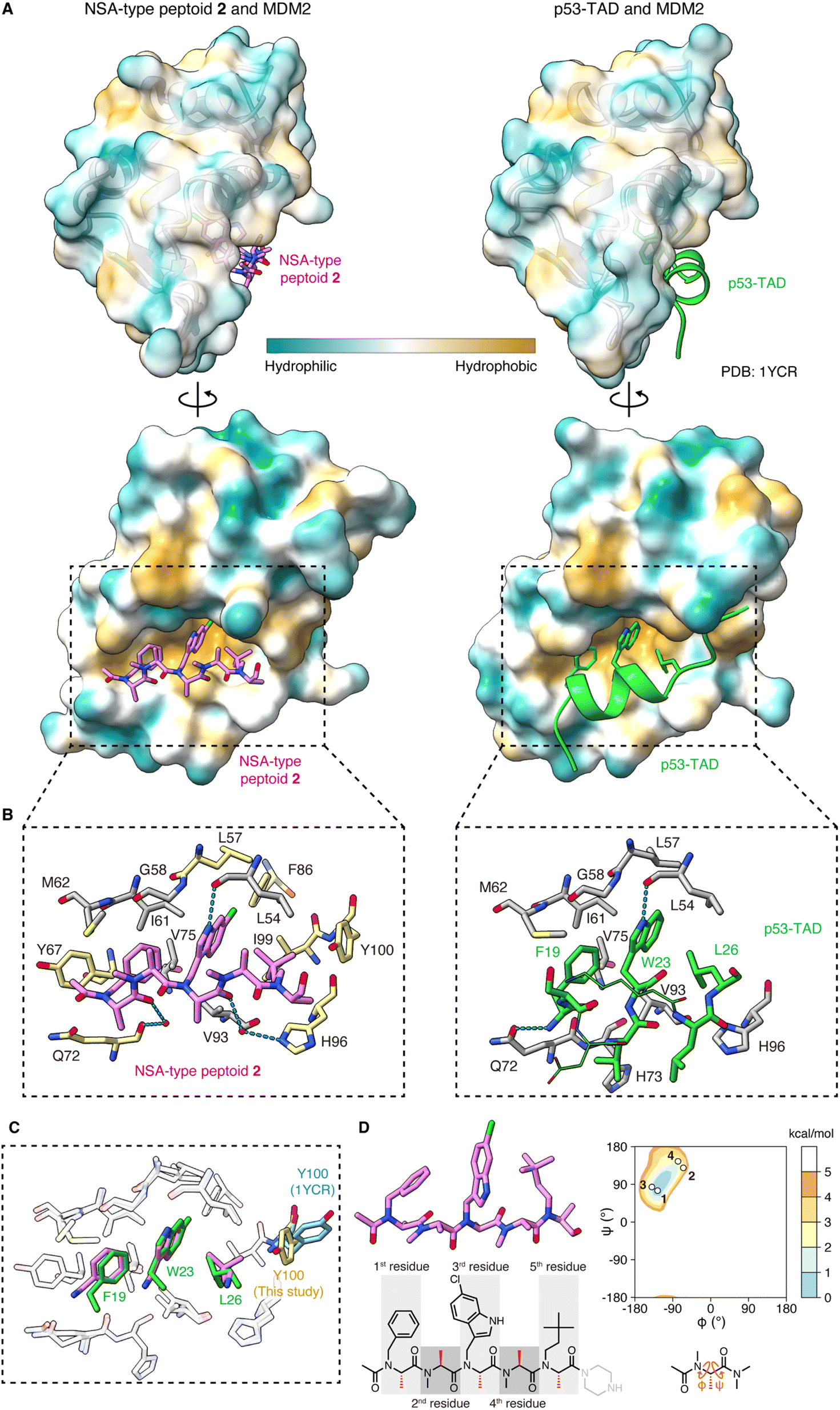 | ||
| Fig. 2 Crystal structure of peptoid 2 bound to MDM2 (A) crystal structure of 2 (pink, left) and p53-TAD (light green, right) (PDB ID 1YCR) bound to MDM2. The surface color of MDM2 represents the hydrophobicity. (B) A close-up view of the interface between (left) 2 (pink) and MDM2 (gray and yellow) or (right) p53-TAD (light green) and MDM2 (gray). Only the MDM2 residues that make van der Waals contacts with 2 or Phe19–Leu26 of p53-TAD are shown. In the left figure, the MDM2 residues also shown in the right figure are colored gray and the unique ones in the left are colored yellow. The hydrogen bond is shown as a cyan dashed line. (C) Overlay of the crystal structure of 2 (pink) complexed with MDM2 (white) and p53-TAD (light green) complexed with MDM2 (white) (PDB ID 1YCR), aligned by MDM2. Only MDM2 residues in van der Waals contact with 2 or Phe19–Leu26 of p53-TAD are shown. The Tyr100 residue of MDM2, which is in different orientations in the two structures, is also shown (yellow for the MDM2–2 complex and cyan for the MDM2–p53-TAD complex). (D) The structure of 2 observed in the crystal and its dihedral angles plotted on the Ramachandran-type energy landscape of the minimal model structure of NSA-type peptoid shown below the landscape.6 The ψ angle of the 5th residue could not be determined due to the absence of the electron density of N of the C-terminal structure. | ||
The structure of MDM2 in the complex with 2 closely resembles its structure in the complex with p53-TAD. This similarity is evidenced by a small root-mean-square deviation of 0.63 Å between the α-carbons in the two MDM2 structures. The orientations of the MDM2 side chains around the binding interface are also almost identical in the two structures. As an exception, the orientation of the Tyr100 side chain was significantly different. While the Tyr100 side chain orients outward from the p53-TAD-binding cleft when bound to p53-TAD (colored in cyan in Fig. 2C),15 it orients inward when bound to 2 (colored in yellow in Fig. 2C). The difference in MDM2 structure reflects that 2 is smaller than p53-TAD and indicates that 2 can bind to MDM2 even when the Tyr100 side chain orients inward.
A more detailed illustration of the interactions between MDM2 and 2 is shown in Fig. 2B. The N-substituents of 2 form intensive interactions with MDM2. The first, third, and fifth N-substituents of 2 engage in van der Waals interactions with MDM2 that are analogous to the interactions observed with the three hot-spot residues of p53-TAD.13 N–H of the 6-chloroindolylmethyl group forms a hydrogen bond with the main chain carbonyl oxygen of Leu54 in MDM2, as seen between the indole N–H of Trp23 in p53-TAD and the main chain carbonyl oxygen of Leu54 in MDM2 (dotted line in Fig. 2B). In addition to these analogous interactions, the third and fifth N-substituents of 2 form interactions with MDM2 that are not observed with the corresponding hot-spot residues of p53-TAD. 6-Cl of the 6-chloroindolylmethyl group sticks deep into the pocket of the Trp23 binding site of MDM2, forming van der Waals interactions with Leu57, Phe86, and Ile99. As a result, 6-Cl enhances the shape complementarity of the peptoid with the MDM2 surface. The neohexyl group on the fifth residue also has a unique interaction with MDM2. While two of its three terminal methyl groups are located at the same position as the two terminal methyl groups of the Leu26 of p53-TAD, the third terminal methyl group forms van der Waals interactions with the Tyr100 side chain of MDM2 (Fig. 2B). Altogether, the non-proteogenic structures of the N-substituents of 2 enable them to engage in more intensive interactions with MDM2 compared to the corresponding hot-spot residues of p53-TAD. These interactions likely contribute significantly to the strong affinity of the peptoid.
In the complex with MDM2, the main chain atoms (N, Cα, C, O) of 2 serve as a scaffold and do not form extensive interactions with MDM2. van der Waals interactions between the main chain atoms of 2 and MDM2 were only observed between the amide nitrogen of the first residue and Gln72 of MDM2 (Fig. 2B, left). While the amide proton of Phe19 of p53-TAD forms a hydrogen bond with the Gln72 side chain of MDM2 (dotted line in Fig. 2B, right), 2 does not form a hydrogen bond with the Gln72 side chain because of the lack of amide hydrogens in peptoid main chain. The carbonyl oxygens of the first and fourth residues of 2 form water-mediated hydrogen bonds with the main chain carbonyl oxygen of Gln72 and Hε2 of His96 of MDM2 (dotted lines in Fig. 2B, left), although the indirect hydrogen bonds may not strongly contribute to the 2–MDM2 binding.
Interactions are also found between MDM2 and the terminal structures of 2. Specifically, the methyl group and the carbonyl carbon of the N-terminal acetyl group in 2 make van der Waals contacts with the Tyr67 side chain and Gln72 side chain of MDM2, respectively (Fig. 2B, left). On the other hand, the absence of electron density in the crystal structure suggests that the C-terminal structure is disordered and does not interact with MDM2.
In summary, 2 binds to MDM2 primarily through the interactions mediated by its N-substituents that mimic three hot-spot residues of p53-TAD, while its main chain acts as a scaffold for positioning the N-substituents. It is intriguing that the peptoid realizes high affinity to protein, although the peptoid main chain lacks amide hydrogens and the ability to provide hydrogen bond donors for interaction with protein. The structural study illustrated that the lack of the main chain interactions by a peptoid can be compensated for by extensive interactions mediated by the N-substituents including non-proteinogenic structures.
Design of peptoid 2 derivatives with different degrees of rigidity and evaluation of their conformational dynamics
We previously showed that replacements of NSA residues to NSG residues in an NSA-type peptoid dramatically reduce the binding affinity to MDM2, which suggests the significant contribution of the main chain rigidity to the binding affinity of the NSA-type peptoid to MDM2.6 To understand how the conformational rigidity of the peptoid affects its binding affinity to MDM2, we designed 2 derivatives with different degrees of rigidity and evaluated the binding affinity to MDM2 of these derivatives. The main chain rigidity of an NSA-type peptoid can be reduced by replacing some residues with NSG residues. The high-resolution crystal structure showed that all methyl groups on the α-carbons of 2 in the crystal structure are exposed to the solvent and do not directly interact with MDM2 (Fig. S1†). Therefore, we believe that the rigidity of 2 can be reduced without affecting the direct interactions with MDM2 by replacing some of the NSA residues of 2 with NSG residues in which the methyl groups on the α-carbons are removed from NSA residues.As peptoids with reduced rigidity, we designed NSA/G-type peptoids 3 and S1–S4 by replacing one of the five NSA residues in 2 with an NSG residue with the same N-substituent structure as the original NSA residue (Fig. 3A and Table S2†). Similarly, we designed NSA/G-type peptoid 4 by replacing two NSA residues with NSG residues and NSG-type peptoid 5 by replacing all NSA residues with NSG residues (Fig. 3A). We synthesized the designed peptoids (2–5, S1–S4 with fluorescent labeling) and measured their binding affinities to MDM2 by fluorescence anisotropy (FA) assay (Table S2†). As a result, by replacing one NSA residue with an NSG residue, the binding affinity was reduced by 2- to 100-fold from 2, depending on the replacement position. 4, which contains two NSG residues, exhibited an 87-fold reduced binding affinity compared to 2. 5, wherein all five residues are NSGs, exhibited a more than 2000-fold reduction in binding affinity compared to 2. As expected, replacing NSA residues with NSG residues led to a decrease in binding affinity to MDM2, presumably because of the reduced conformational rigidity.
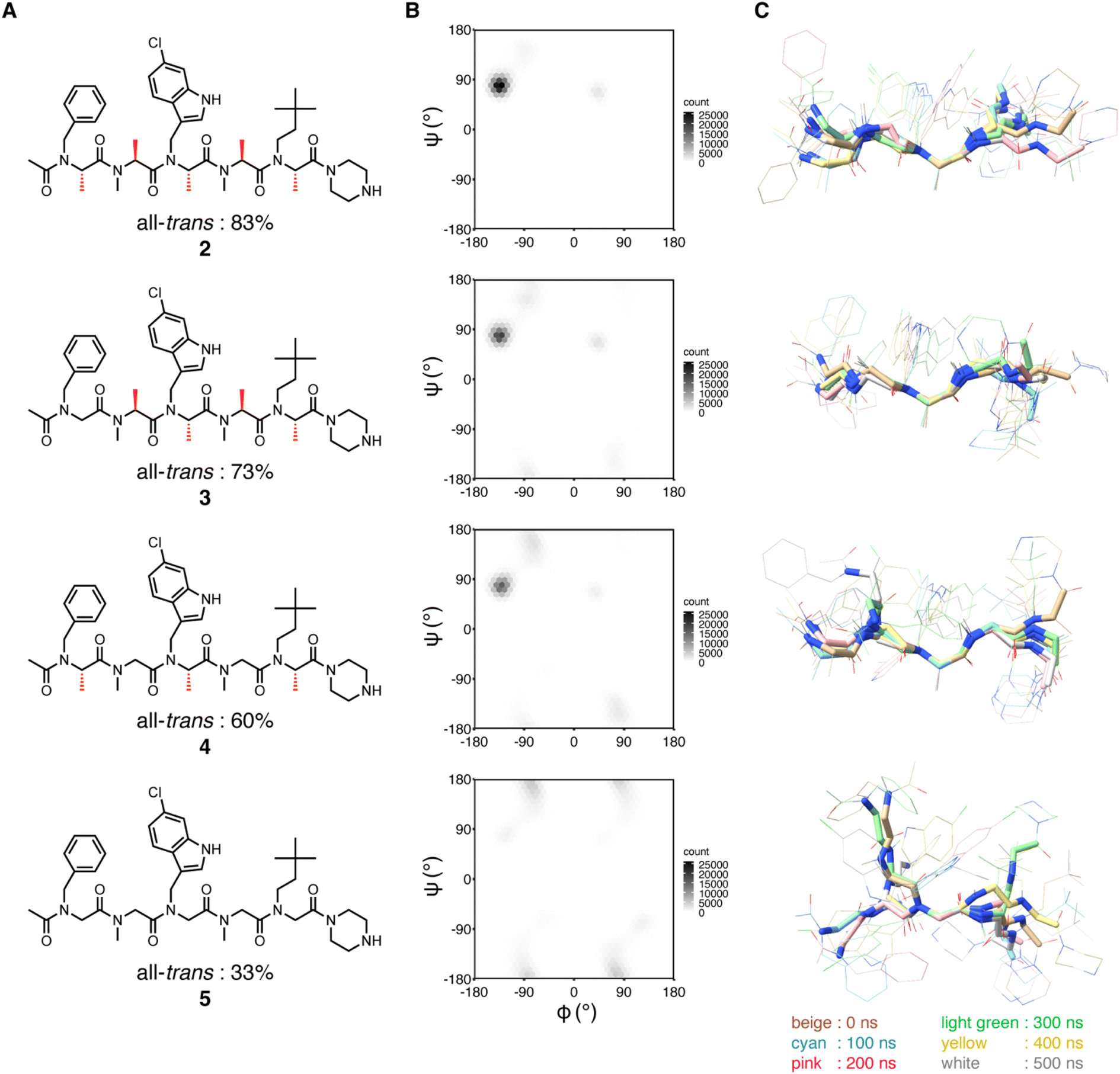 | ||
| Fig. 3 Structural analysis of peptoids (A) chemical structures of 2–5. The chemical structures of the peptoids with fluorescent labeling can be found in ESI.† The population percentage of the all-trans conformer determined by NMR spectroscopy in 20% DMSO-d6/D2O is shown below the structure. (B and C) MD simulations of 2–5. (B) Distribution of φ and ψ angles of all residues of 2–5 during simulations. The results of run 1–5 are integrated into a single plot. (C) Conformations at every 100 ns during simulations (run 1) were overlaid by the main chain atoms (N, Cα, and C) of the third residue. | ||
Based on the FA results, 2, 3 (16-fold lower binding affinity than 2), 4 (87-fold lower binding affinity than 2), and 5 (more than 2000-fold lower binding affinity than 2) were selected for conformational analysis to evaluate how the conformational rigidity of the peptoids differs with each other.
First, to investigate the rotational freedom of the dihedral angle ω of 2–5 in water, NMR analysis was performed in 20% DMSO-d6/D2O (Fig. 3A and S2–S18, and Tables S3–S6†). DMSO-d6 was included to increase the concentration of peptoids for the NMR analysis. While cis–trans isomerization of the amide bond between the N-terminal acetyl group and the first residue was found in all peptoids, trans configuration was found preferred for the amide bonds connecting two peptoid residues. The cis–trans ratio of each amide bond was dependent on the composition of NSA and NSG residues in a peptoid. In 2, the relative population of the all-trans conformer among the observed conformers was 83%, in which all five amide bonds in the peptoid were in the trans configuration. In 3, cis–trans isomerization was observed in the amide bonds between the second and third residues, as well as between the fourth and fifth residues, and consequently, the all-trans conformer population was 73%. This result is consistent with the fact that a peptoid residue with a bulky N-substituent is more likely to be in cis configuration.16,17 In 4, cis–trans isomerization was observed in the same amide bonds as in 3, and the all-trans conformer constituted only 60% of the population. In 5, cis–trans isomerization was observed in multiple amide bonds although the positions of the isomerized amides were not identified due to the complex signal pattern. Consequently, the population of the all-trans conformer of 5 was 33% (Fig. 3A). These results indicate that, for the dihedral angle ω, replacing the NSA residues with NSG residues increases the population of the cis configuration. As a result, while the NSA-type peptoid predominantly adopts a single conformation, NSA/G-type peptoids exist in an equilibrium of multiple conformations, with slower exchange rates than the chemical shift differences.
Next, the rotational freedom of the dihedral angles φ and ψ of 2–5 in water was evaluated. For the evaluation of the differences in φ and ψ angles, molecular dynamics (MD) simulations of each oligomer were performed for 500 ns (Fig. 3B and C). A conformation observed in the crystal structure of 2 bound to MDM2 was used as the initial conformation for the MD simulations. During the simulations, all NSA residues except for the C-terminal residues primarily form the extended shape constrained by pseudo-1,3-allylic strains. On the other hand, as for NSG residues where constraint by pseudo-1,3-allylic strains does not exist, the range of accessible φ and ψ angles is larger than that of NSA residues. Thus, replacing the NSA residues with NSG was suggested to broaden the range of accessible φ and ψ angles (Fig. 3B and S19†) and increase the flexibility of the overall oligomer structure (Fig. 3C, S20 and S21†).
The conformational analysis showed that NSA-type peptoid 2 primarily forms the extended shape observed in the crystal structure even in water. Therefore, peptoid 2 conformation in water can be considered preorganized for binding to MDM2. The analysis also suggests that replacing NSA residues with NSG residues reduces the population of the MDM2-binding conformation because the rotational restrictions about all dihedral angles, ω, φ, and ψ angles, get weakened.
Evaluation of the effect of the main chain rigidity of a peptoid on the binding to MDM2
Using peptoids 2–5, which vary in rigidity, the effect of peptoid rigidity on MDM2 binding was evaluated. Specifically, isothermal titration calorimetry (ITC) and surface plasmon resonance (SPR) measurements were conducted to analyze the binding affinities, thermodynamics, and kinetics of the interactions between 2–5 and MDM2 (Table 1). The KD values measured by the three assays were similar to each other, suggesting the reliability of the KD values. In addition, consistent with the results of the FA assay, the peptoids containing a higher number of NSG residues exhibited weaker binding affinities (Table 1).| Binding FA assaya | ITCb | SPRc | |||||
|---|---|---|---|---|---|---|---|
| K D (nM) | K D (nM) | ΔH (kcal mol−1) | −TΔS (kcal mol−1) | K D (nM) | k on (M−1 s−1) | k off (s−1) | |
a
K
D values between peptoids with fluorescent labeling and MDM2 determined by a direct binding FA assay. The structures of fluorescently-labeled peptoids (2–5–Sar3–EDA–Flu) are shown in ESI. The assay was conducted using 10 nM fluorescein-labeled compounds and MDM2 in PBS with 0.01% Tween 20 at 25 °C. The binding stoichiometry was assumed to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for the fitting of the binding curve. The KD value of 2 may be subject to some error since the concentration of fluorescent peptoid probe (10 nM) is close to the KD value.
b ITC profiles of the interaction between peptoids (2–5) and MDM2. The binding assay was conducted in PBS at 25 °C. 100, 200, or 500 μM solution of the peptoids in PBS was titrated into 10, 20, or 50 μM MDM2 solution in PBS. As a control experiment, each peptoid solution was titrated into PBS.
c Kinetic parameters measured by SPR analysis of MDM2 binding to immobilized peptoids on CM5 chip. The structures of the peptoids (2–5–Sar3–EDA) used for immobilization on the sensor chip are shown in ESI. 0.94–15 nM, 12.3–1000 nM, or 0.074–6 μM MDM2 was analyzed in PBS containing 0.01% Tween 20 at 10 °C.
d
K
D values determined by kinetic analysis.
e
K
D values determined by steady-state affinity analysis.
f N.D. means not determined. :1 for the fitting of the binding curve. The KD value of 2 may be subject to some error since the concentration of fluorescent peptoid probe (10 nM) is close to the KD value.
b ITC profiles of the interaction between peptoids (2–5) and MDM2. The binding assay was conducted in PBS at 25 °C. 100, 200, or 500 μM solution of the peptoids in PBS was titrated into 10, 20, or 50 μM MDM2 solution in PBS. As a control experiment, each peptoid solution was titrated into PBS.
c Kinetic parameters measured by SPR analysis of MDM2 binding to immobilized peptoids on CM5 chip. The structures of the peptoids (2–5–Sar3–EDA) used for immobilization on the sensor chip are shown in ESI. 0.94–15 nM, 12.3–1000 nM, or 0.074–6 μM MDM2 was analyzed in PBS containing 0.01% Tween 20 at 10 °C.
d
K
D values determined by kinetic analysis.
e
K
D values determined by steady-state affinity analysis.
f N.D. means not determined.
|
|||||||
| 2 | 8.6 | 17.5 | −9.99 | −0.590 | 9.5d, 9.6e | 6.9 × 106 | 0.065 |
| 3 | 140 | 318 | −7.78 | −1.09 | 418d, 350e | 5.37 × 105 | 0.225 |
| 4 | 750 | 1630 | −5.58 | −2.32 | N.D.d,f, 1400e | N.D.f | N.D.f |
| 5 | 21000 |
No binding | — | — | — | — | — |
From ITC measurements, thermodynamic parameters were determined (Table 1, center). The binding of 2 to MDM2 was enthalpy-driven (ΔH = −9.99 kcal mol−1, −TΔS = −0.590 kcal mol−1). This high enthalpic contribution to the MDM2 binding is considered derived from the high shape complementarity of the peptoid to the p53-TAD binding site of MDM2. The shape complementarity enables the intensive site-selective interactions, namely numerous van der Waals contacts and a hydrogen bond, leading to the enthalpy-driven binding.
The conformational rigidity of 2 is considered to contribute to the high enthalpic gain upon MDM2-binding. This is supported by observations that replacing NSA residues with NSG residues in the peptoid reduces the enthalpic contribution. Specifically, for 3 and 4, which contain one and two NSG residues, respectively, there was a decrease in the magnitude of the enthalpy change (ΔH), resulting in values of −7.78 kcal mol−1 for 3 and −5.58 kcal mol−1 for 4. One possible reason for this is that because the NSA/G-type peptoid has a flexible conformation even in the complex with MDM2, the binding stability of the peptoid–MDM2 complex is reduced compared with that for the NSA-type peptoid. To examine this hypothesis, we conducted MD simulations of the complexes of 2, 4, or 5 with MDM2. As a result, 2 most stably maintained the conformation similar to the crystal structure during the simulations and presented the N-substituents in the binding pocket of MDM2 (Fig. S22–S24†). This is consistent with the hypothesis. In addition to the difference in the conformational stability of peptoids in the complex with MDM2, the difference in conformational stability of peptoids when not bound to MDM2 also possibly contributes to the difference in binding enthalpy. The NSA-type peptoid 2 forms similar conformations whether it is bound to MDM2 or unbound. In contrast, the NSA/G-type peptoids can change their conformation to more enthalpically stable ones when they are not bound to MDM2 due to the rotational freedom of the dihedral angles (Fig. 2). Thus, the NSA/G-type peptoids are considered to require extra enthalpic cost to transition to the MDM2-bound conformation, resulting in the reduced contribution of favorable binding enthalpy. A common strategy for achieving an enthalpy gain is the introduction of functional groups to form additional interaction.18 However, our results indicate that conformational rigidification is also a viable strategy to gain enthalpy in protein-binding peptoid design.
In contrast to the enthalpic contribution, the conformational rigidification of the peptoid resulted not in an entropic advantage but rather a relative disadvantage. The entropic contribution (−TΔS) of 2, 3, and 4 were −0.590, −1.09, and −2.32 kcal mol−1, respectively. Generally, the increase in binding affinity achieved by conformational rigidification to preorganize a molecule for binding is considered a result of reduced entropy loss during protein binding.17,19–21 However, in the binding of MDM2 and NSA- or NSA/G-type peptoids evaluated in this study, the conformational rigidification did not lead to an entropic advantage. This observation contradicts the general consideration. It is possible that the changes in hydration entropy and conformational entropy of MDM2 have combined to cancel out the entropic benefit attributed to the preorganization of the peptoid.22–24 This was supported by the fact that, in MD simulations, MDM2 exhibits reduced fluctuations in complexes with 2, 4, or 5 compared to MDM2 alone (Fig. S25†). Although more extensive simulations may be preferable to explore the comprehensive energy landscape of the complex, the current MD results suggest that the overall entropy change of the system is influenced not solely by the conformational entropy of the peptoid, but also other factors.
From SPR analysis, kinetic parameters were determined (Table 1, right). The measurements were conducted at a low temperature (10 °C) to reduce the dissociation rate and facilitate the measurement of rapidly dissociating compounds. The association rate constant (kon) and the dissociation rate constant (koff) of the interaction between immobilized 2 and MDM2 were determined to be 6.9 × 106 M−1 s−1 and 0.065 s−1, respectively. For 3, in which an NSA residue was replaced with an NSG residue, the kon and koff were 5.37 × 105 M−1 s−1 and 0.225 s−1, showing a 13-fold slower binding rate and a 3.5-fold faster dissociation rate compared to 2. In the case of 4, the kinetic parameters could not be determined, possibly because the dissociation was too fast or there were multiple binding pathways. The comparison between 2 and 3 indicates that NSA-type peptoid 2, which has a rigid main chain, exhibits more favorable binding kinetics in both association and dissociation rates compared to NSA/G-type peptoid 3.
From the physicochemical parameters in the interaction between peptoids 2–5 and MDM2 (Table 1) and the results of the conformational analysis (Fig. 3), the higher binding affinity of NSA-type peptoid 2 compared with the NSA/G-type peptoids 3–5 can be attributed to the following two effects originated from the conformational rigidity of 2 (Fig. 4): (1) 2 adopts a conformation preorganized to that observed in its complex structure with MDM2, even when unbound. This preorganization facilitates a faster association with MDM2 compared to more flexible peptoids. This is evidenced by the higher association rate (kon) of 2 relative to 3 (1 in Fig. 4); (2) the rigidity of the main chain in 2 allows for more stable van der Waals interactions with MDM2, resulting in a higher energy barrier to dissociation compared to its derivatives with NSG-residues. This increased stability of the interaction is corroborated by the greater magnitude of enthalpy change observed in the binding of 2 compared to 3 and 4, and by the slower dissociation of 2 from MDM2 compared to 3 (2 in Fig. 4).
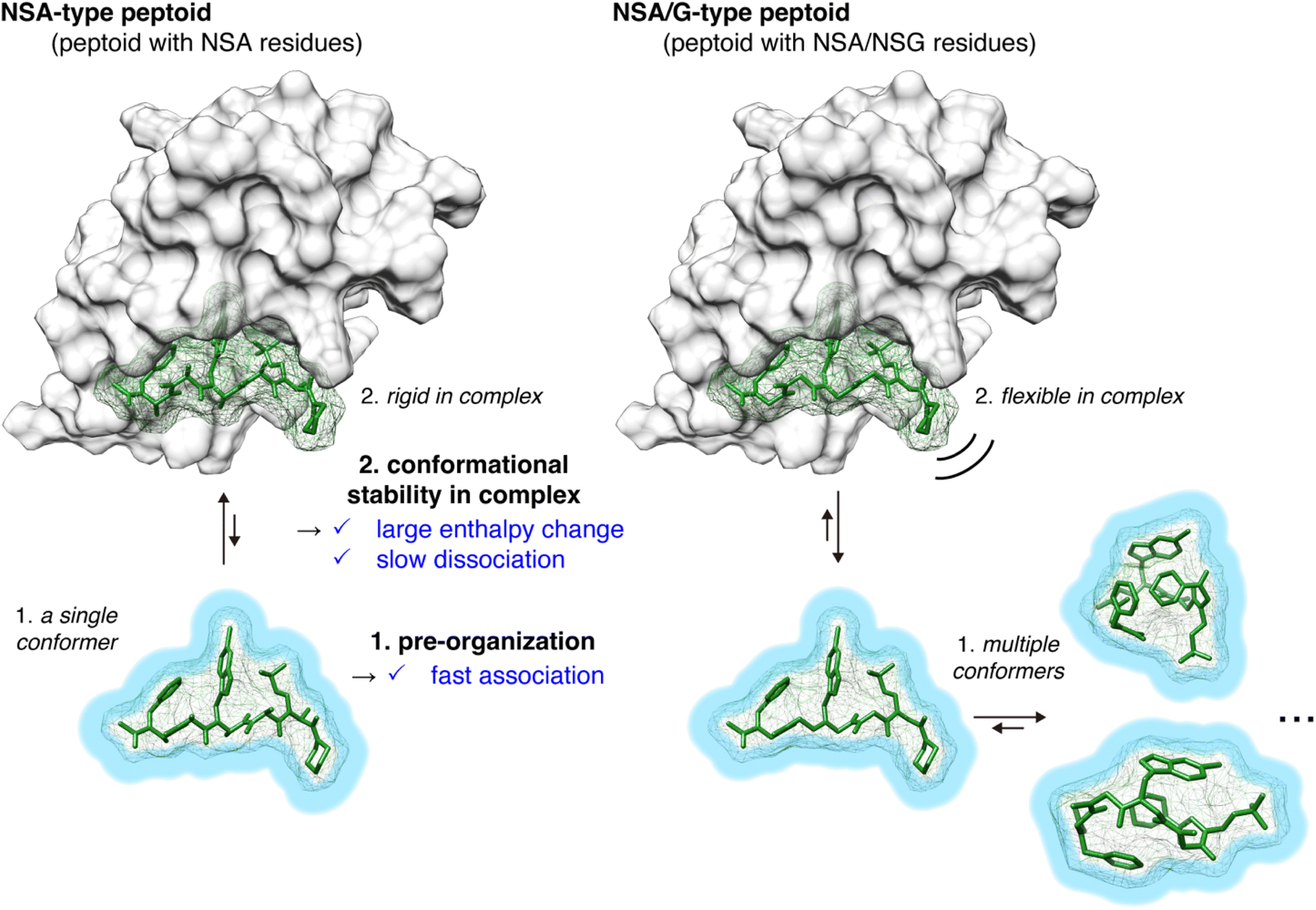 | ||
| Fig. 4 Plausible role of the peptoid conformational rigidity in enhancing protein-binding affinity. | ||
Conclusions
Using the highly rigid NSA-type peptoid and its derivatives, the binding mode of the peptoids to proteins was revealed at atomic resolution for the first time. Moreover, the role of the rigidity of NSA-type peptoids in protein binding was evaluated. The high-resolution structure and thermodynamic analysis of the peptoid–MDM2 complex revealed that the NSA-type peptoid binds to MDM2 in an enthalpy-driven manner, mainly due to the interaction formed by N-substituents on the MDM2 surface. Introducing non-proteogenic structures to the N-substituents increases shape complementarity with MDM2 compared to the corresponding hot-spot residues of p53-TAD, resulting in the stronger affinity of the peptoid relative to p53-TAD (KD = 600 nM).13 Comparison with NSA/G-type peptoids, which exhibit reduced rigidity, demonstrated that the increased conformational rigidity in an NSA-type peptoid contributes to an increase in both the rates of association and dissociation with MDM2. In addition, in a thermodynamic context, the increased rigidity was found to increase the magnitude of enthalpy change upon binding. Our findings validate the long-sought strategy of obtaining peptoids that bind strongly to proteins by conformational rigidification.Rigidification of the structure of peptide and peptidomimetic ligands is considered effective in improving binding affinity to proteins.25–27 However, systematic modulations of ligand rigidity without affecting the direct interaction between the ligand and the protein are difficult, making it challenging to clarify the role of rigidity in binding affinity.28 For example, in a previous study, “stapling”, involving a covalent cross-linking of two side chains, was applied to a peptidic ligand of MDM2 to increase the conformational rigidity and the role of the enhanced rigidity by the stapling was investigated.21,29 However, the role of the enhanced rigidity on binding remained ambiguous. This is because the introduced stapling moiety not only increased the rigidity but also directly influenced the electrostatic and van der Waals interactions with MDM2. Another study attempted to increase the binding affinity of an MDM2-binding peptoid by introducing Nα-chiral substituents4 which is known to increase peptoid rigidity. However, it was difficult to systematically modulate the peptoid's rigidity due to reduced aqueous solubility and synthetic difficulty associated with the introduction of the bulky rigidifying groups. Besides, the lack of a high-resolution structure of the peptoid–protein complex made it challenging to conduct a reliable discussion about the role of conformational rigidity on binding affinity. In the present study, the combined use of NSA and NSG, together with the high-resolution peptoid–protein complex structure, enabled a systematic study of the correlation between the rigidity of peptoid ligands and their binding affinity to MDM2 without affecting their direct interaction with MDM2. This approach has clarified the kinetic and thermodynamic effects induced by ligand rigidification, at least for the present interaction partners.
An NSA-type peptoid has strong rotational restrictions on all three main chain dihedral angles, ω, φ, and ψ, leading to higher conformational rigidity than an NSG-type peptoid. Considering that the rate of amide cis–trans isomerization is slow, the increased rotational restriction about ω angle in an NSA-type peptoid is assumed beneficial to lock a peptoid to the protein-binding conformation and thereby increase the binding affinity to the protein. However, the difference in the percentage of all-trans conformers among 2, 3, and 4 (83%, 73%, and 60% for 2, 3, and 4) is not high enough to solely explain the large differences in the binding affinity, namely a 16-fold difference between 2 and 3 and an 87-fold difference between 2 and 4. This suggests that not only the rotational freedom of ω but also the rotational freedom of φ and ψ, which change on short timescales, are important for the binding affinity of NSA-type peptoids. Therefore, when designing high-affinity peptoids, it is important to consider rotational control for all dihedral angles, ω, φ, and ψ, and introduction of NSA residues is a useful means for achieving this goal. These findings are expected to accelerate the development of bioactive peptoids, especially intracellular protein–protein interaction inhibitors that take advantage of the high membrane permeability of peptoids.
Data availability
All the data supporting the key findings in the paper is listed in the manuscript and ESI.†Author contributions
J. M. and S. S. conceived and directed the study. M. Y. synthesized peptoids and performed recombinant expression of MDM2, crystallization, FA assays, MD simulations, ITC analysis, and SPR analysis. Y. F. synthesized peptoids and performed recombinant expression of MDM2 and crystallization screening. T. U. and K. Takeuchi conducted NMR measurements and conformational analysis. K. U. assisted with MD simulations. H. A., H. M., and G. U. collected, processed, and refined X-ray data. A. S. assisted with crystallization screening. S. N. and K. Tsumoto assisted with X-ray crystallographic analysis and performed ITC analysis. M. Y., J. M., and S. S. wrote the manuscript with contributions from all authors.Conflicts of interest
The authors declare the following competing financial interest: the authors (J. M., Y. F., and S. S.) have filed a patent application (PCT/JP2020/27010). All other authors declare they have no competing interests.Acknowledgements
This research was partially supported by Research Support Project for Life Science and Drug Discovery (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under Grant Number JP23ama121001 and JP23ama121033. The computations were performed using Research Center for Computational Science, Okazaki, Japan (Project: 23-IMS-C071). We thank Dr Y. Kohuku at the University of Tokyo and Dr I. Shimada at RIKEN Center for Biosystems Dynamics Research for NMR measurements using high magnetic field NMR spectrometers. We also thank Dr K. Suzuki and Prof. S. Maeda at Hokkaido University for the discussion and advice for conformational sampling and MD simulations of peptoids. We also thank Mr M. Shirakawa at the University of Tokyo for help with crystallization. The pGEX-6P-2-MDM2 (17–125) was a gift from Gary Daughdrill (Addgene plasmid # 62063).30 Molecular graphics and analyses were performed with UCSF Chimera31 or UCSF ChimeraX,32–34 developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco. S. S. and K. U. acknowledge financial support from CREST (JPMJCR21N5), Japan Science and Technology Agency. J. M. acknowledges financial support from PRESTO (JPMJPR21AF), Japan Science and Technology Agency, and JSPS KAKENHI (JP23H02078). M. Y. acknowledges financial support from KAKENHI (JP23KJ0443). K. Tsumoto acknowledges financial support from JSPS KAKENHI (JP20H02531) and AMED (JP23ama121033). T. U. acknowledges financial support from MEXT/JSPS KAKENHI (JP23K18177, JP23H02618, JP21H05509, and JP20H03375). K. Takeuchi acknowledges financial support from JSPS KAKENHI (JP20K21494 and JP20H03378).Notes and references
- Y. U. Kwon and T. Kodadek, J. Am. Chem. Soc., 2007, 129, 1508–1509 CrossRef CAS PubMed.
- S. M. Miller, R. J. Simon, S. Ng, R. N. Zuckermann, J. M. Kerr and W. H. Moos, Drug Dev. Res., 1995, 35, 20–32 CrossRef CAS.
- R. J. Simon, R. S. Kania, R. N. Zuckermann, V. D. Huebner, D. A. Jewell, S. Banville, S. Ng, L. Wang, S. Rosenberg and C. K. Marlowe, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 9367–9371 CrossRef CAS PubMed.
- T. Hara, S. R. Durell, M. C. Myers and D. H. Appella, J. Am. Chem. Soc., 2006, 128, 1995–2004 CrossRef CAS PubMed.
- J. A. Schneider, T. W. Craven, A. C. Kasper, C. Yun, M. Haugbro, E. M. Briggs, V. Svetlov, E. Nudler, H. Knaut, R. Bonneau, M. J. Garabedian, K. Kirshenbaum and S. K. Logan, Nat. Commun., 2018, 9, 4396 CrossRef PubMed.
- J. Morimoto, Y. Fukuda, D. Kuroda, T. Watanabe, F. Yoshida, M. Asada, T. Nakamura, A. Senoo, S. Nagatoishi, K. Tsumoto and S. Sando, J. Am. Chem. Soc., 2019, 141, 14612–14623 CrossRef CAS PubMed.
- M. Oh, J. H. Lee, H. Moon, Y. J. Hyun and H. S. Lim, Angew. Chem., Int. Ed., 2016, 55, 602–606 CrossRef CAS PubMed.
- Y. Gao and T. Kodadek, Chem. Biol., 2013, 20, 360–369 CrossRef CAS PubMed.
- D. J. Trader, S. Simanski and T. Kodadek, J. Am. Chem. Soc., 2015, 137, 6312–6319 CrossRef CAS PubMed.
- M. Yokomine, J. Morimoto, Y. Fukuda, Y. Shiratori, D. Kuroda, T. Ueda, K. Takeuchi, K. Tsumoto and S. Sando, Angew. Chem., Int. Ed., 2022, 61, e202200119 CrossRef CAS PubMed.
- Y. Fukuda, M. Yokomine, D. Kuroda, K. Tsumoto, J. Morimoto and S. Sando, Chem. Sci., 2021, 12, 13292–13300 RSC.
- C. M. Darapaneni, P. J. Kaniraj and G. Maayan, Org. Biomol. Chem., 2018, 16, 1480–1488 RSC.
- P. H. Kussie, S. Gorina, V. Marechal, B. Elenbaas, J. Moreau, A. J. Levine and N. P. Pavletich, Science, 1996, 274, 948–953 CrossRef CAS PubMed.
- B. Vu, P. Wovkulich, G. Pizzolato, A. Lovey, Q. Ding, N. Jiang, J. J. Liu, C. Zhao, K. Glenn, Y. Wen, C. Tovar, K. Packman, L. Vassilev and B. Graves, ACS Med. Chem. Lett., 2013, 4, 466–469 CrossRef CAS PubMed.
- S. G. Dastidar, D. P. Lane and C. S. Verma, BMC Bioinf., 2009, 10, S6 CrossRef PubMed.
- J. R. B. Eastwood, E. I. Weisberg, D. Katz, R. N. Zuckermann and K. Kirshenbaum, Pept. Sci., 2023, 115, e24307 CrossRef CAS.
- D. Kalita, B. Sahariah, S. P. Mookerjee and B. K. Sarma, Chem.–Asian J., 2022, 17, e202200149 CrossRef CAS PubMed.
- G. G. Ferenczy and G. M. Keserű, Expert Opin. Drug Discovery, 2020, 15, 117–129 CrossRef CAS PubMed.
- G. R. Marshall and F. Ballante, Drug Dev. Res., 2017, 78, 245–267 CrossRef CAS PubMed.
- Y. Ge, S. Zhang, M. Erdelyi and V. A. Voelz, J. Chem. Inf. Model., 2021, 61, 2353–2367 CrossRef CAS PubMed.
- A. Maity, A. R. Choudhury and R. Chakrabarti, J. Chem. Inf. Model., 2021, 61, 1989–2000 CrossRef CAS PubMed.
- S. Leavitt and E. Freire, Curr. Opin. Struct. Biol., 2001, 11, 560–566 CrossRef CAS PubMed.
- M. L. Verteramo, O. Stenström, M. M. Ignjatović, O. Caldararu, M. A. Olsson, F. Manzoni, H. Leffler, E. Oksanen, D. T. Logan, U. J. Nilsson, U. Ryde and M. Akke, J. Am. Chem. Soc., 2019, 141, 2012–2026 CrossRef CAS PubMed.
- J. Wallerstein, V. Ekberg, M. M. Ignjatović, R. Kumar, O. Caldararu, K. Peterson, S. Wernersson, U. Brath, H. Leffler, E. Oksanen, D. T. Logan, U. J. Nilsson, U. Ryde and M. Akke, JACS Au, 2021, 1, 484–500 CrossRef CAS PubMed.
- P. Wójcik and Ł. Berlicki, Bioorg. Med. Chem. Lett., 2016, 26, 707–713 CrossRef PubMed.
- C. Martin, L. E. Gimenez, S. Y. Williams, Y. Jing, Y. Wu, C. Hollanders, O. Van Der Poorten, S. Gonzalez, K. Van Holsbeeck, S. Previti, A. Lamouroux, S. Zhao, D. Tourwé, R. C. Stevens, R. D. Cone and S. Ballet, J. Med. Chem., 2021, 64, 357–369 CrossRef CAS PubMed.
- V. Azzarito, K. Long, N. S. Murphy and A. J. Wilson, Nat. Chem., 2013, 5, 161–173 CrossRef CAS PubMed.
- J. E. DeLorbe, J. H. Clements, M. G. Teresk, A. P. Benfield, H. R. Plake, L. E. Millspaugh and S. F. Martin, J. Am. Chem. Soc., 2009, 131, 16758–16770 CrossRef CAS PubMed.
- A. V. Strizhak, O. Babii, S. Afonin, I. Bakanovich, T. Pantelejevs, W. Xu, E. Fowler, R. Eapen, K. Sharma, M. O. Platonov, V. V. Hurmach, L. Itzhaki, M. Hyvönen, A. S. Ulrich, D. R. Spring and I. V. Komarov, Org. Biomol. Chem., 2020, 18, 5359–5369 RSC.
- W. Borcherds, F. Theillet, A. Katzer, A. Finzel, K. M. Mishall, A. T. Powell, H. Wu, W. Manieri, C. Dieterich, P. Selenko, A. Loewer and G. W. Daughdrill, Nat. Chem. Biol., 2014, 10, 1000–1002 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- T. D. Goddard, C. C. Huang, E. C. Meng, E. F. Pettersen, G. S. Couch, J. H. Morris and T. E. Ferrin, Protein Sci., 2018, 27, 14–25 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, E. C. Meng, G. S. Couch, T. I. Croll, J. H. Morris and T. E. Ferrin, Protein Sci., 2021, 30, 70–82 CrossRef CAS PubMed.
- E. C. Meng, T. D. Goddard, E. F. Pettersen, G. S. Couch, Z. J. Pearson, J. H. Morris and T. E. Ferrin, Protein Sci., 2023, 32, e4792 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01540a |
| ‡ Present address: Kyushu University, Fukuoka, 812-8582, Japan. |
| This journal is © The Royal Society of Chemistry 2024 |