
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSub-100 nm carriers by template polymerization for drug delivery applications
P. K.
Hashim
 *ab and
Shimaa Said Mohamed Ali
Abdrabou
c
*ab and
Shimaa Said Mohamed Ali
Abdrabou
c
aResearch Institute for Electronic Science, Hokkaido University, Kita 20, Nishi 10, Kita-ku, Sapporo, Hokkaido 001-0020, Japan. E-mail: hashim@es.hokudai.ac.jp
bGraduate School of Life Science, Hokkaido University, Kita 10, Nishi 8, Kita-ku, Sapporo, Hokkaido 060-0810, Japan
cDepartment of Pediatrics, Faculty of Medicine and Graduate School of Medicine, Hokkaido University, Sapporo, Japan
First published on 29th February 2024
Abstract
Size-controlled drug delivery systems (DDSs) have gained significant attention in the field of pharmaceutical sciences due to their potential to enhance drug efficacy, minimize side effects, and improve patient compliance. This review provides a concise overview of the preparation method, advancements, and applications of size-controlled drug delivery systems focusing on the sub-100 nm size DDSs. The importance of tailoring the size for achieving therapeutic goals is briefly mentioned. We highlight the concept of “template polymerization”, a well-established method in covalent polymerization that offers precise control over molecular weight. We demonstrate the utility of this approach in crafting a monolayer of a polymer around biomolecule templates such as DNA, RNA, and protein, achieving the generation of DDSs with sizes ranging from several tens of nanometers. A few representative examples of small-size DDSs that share a conceptual similarity to “template polymerization” are also discussed. This review concludes by briefly discussing the drug release behaviors and the future prospects of “template polymerization” for the development of innovative size-controlled drug delivery systems, which promise to optimize drug delivery precision, efficacy, and safety.
 P. K. Hashim | Dr P. K. Hashim is an assistant professor at research institute for electronic science (RIES), Hokkaido University, Japan. He received MS in Organic Chemistry from Aligarh Muslim University in 2008 and PhD from Hokkaido University in Biological Science in 2012 under the supervision of Professor Nobuyuki Tamaoki. He then worked as project researcher in the group of Professor Takuzo Aida at the University of Tokyo until 2021 and handled several projects including nanomedicine and protein supramolecular assembly. His research interests include molecular chirality, photochromism, protein nanoparticles, RNA delivery and photopharmacology. |
 Shimaa Said Mohamed Ali Abdrabou | Dr Shimaa Abdrabou is an assistant professor at the Department of Pediatrics, Hokkaido University, Japan. She earned her bachelor's degree in Medicine and Surgery from Menoufia University, Egypt, and subsequently worked as a pediatrician at the Egyptian Ministry of Health. She successfully pursued both MSc and PhD degrees in Pediatrics from Hokkaido University's Graduate School of Medicine in 2017 and 2022, respectively. She actively engaged in medical societies like ESID and JSAID. Her research focuses on the pathophysiology of inborn errors of immunity. Her goal is to provide new insights into disease pathophysiology and develop effective treatment strategies through innovative drug delivery systems. |
1. Introduction
Drugs, whether naturally occurring or synthesized, play a crucial role in the detection, diagnosis, and treatment of diseases. These drugs exhibit a range of characteristics, primarily determined by their solubility in water.1,2 They can be classified as hydrophobic (e.g., paclitaxel, cisplatin, and methotrexate) or hydrophilic (e.g., gemcitabine, L-asparaginase, and antibodies).3–6 Furthermore, drugs may possess different electrical charges, being either negatively charged (as seen in gene therapeutics like DNA, messenger RNA, and short interfering RNA),7,8 positively charged (e.g., doxorubicin or DOX),9 or electrically neutral (e.g., cisplatin), depending on the chemical nature of their components. Drugs can also be categorized as small molecules, with low molecular weights below 900 Daltons (e.g., DOX and cisplatin), or as high molecular weight molecules (e.g., DNA, RNA, and proteins).10 These structural characteristics play a vital role in defining drugs' functions. Small molecule drugs typically initiate their action by binding to specific genes or proteins, often leading to off-target effects due to their lack of specificity.11 Conversely, macromolecule drugs, such as genes and proteins, tend to exhibit high specificity, binding to their target sites within intracellular components, and influencing disease-causing elements with precision. Both small molecule and macromolecule drugs find utility in cancer therapy.12,13 Notably, over 150 anticancer drugs have received approval from the US Food and Drug Administration for the treatment of various cancer types.14–16 This collective array of drugs has significantly expanded the therapeutic options available for cancer patients.1.1. DDS size matters
In a broad context, a DDS can be described as a formulation, device, or a carrier designed to encapsulate a drug and transport it to the specific disease site while minimizing unintended targeting of other tissues or cells.17,18 In a conceptual framework, commercially available drug forms like tablets, capsules, suspensions, and powders represent one category of DDSs, often referred to as “macro-DDSs” due to their noticeable dimensions.19,20 Another category, “micro-DDSs,” boasts an average diameter ranging from 1 to 300 micrometers, examples of which include microemulsions and microsphere depots.21 Additionally, “nano-DDSs,” characterized by sizes in the nanometer range, just slightly larger than that of the drug molecules themselves, holds the potential to address many of the limitations associated with macro-/micro-DDS methods.22,23 In drug delivery, the size of particles or carriers used to transport medications plays a pivotal role in determining the effectiveness and safety of the treatment.24 Some of the parameters that the size of a DDS affects are (1) bioavailability and efficacy: the size of drug carriers, such as nanoparticles or liposomes, greatly influences the bioavailability of the drug. Smaller carriers can penetrate tissues and cells more effectively, enhancing drug uptake and improving therapeutic efficacy.25,26 (2) Targeted delivery: controlling the size of drug carriers enable the precise targeting of specific tissues or cells. Nanoscale drug carriers can be designed to accumulate in tumors or other disease sites, reducing off-target effects.27–29 (3) Drug release rates: the size of drug delivery systems can impact the rate at which drugs are released within the body. Smaller carriers tend to release drugs more slowly, resulting in sustained drug levels, while larger carriers may release drugs more rapidly.30,31 (4) Avoiding immune response: particles that are too large may be quickly recognized and cleared by the immune system. Designing drug carriers with an appropriate size can help avoid immune system clearance, extending their circulation time. (5) Toxicity and side effects: the size of particles can affect their toxicity. Small nanoparticles may have a higher tendency to accumulate in certain organs, potentially leading to toxicity, while large particles can induce inflammation. (6) Patient comfort and compliance: smaller drug delivery systems often lead to more comfortable and less invasive administration methods. This can improve patient compliance, particularly in the case of long-term treatment.32–341.2. Methods to control the size of DDSs
Several methods are employed to control the size of drug delivery systems, depending on the specific system and application. Some common methods are, though not strictly in the nanometer size range.31 Crystallization: this method involves controlling the rate of crystallization of the drug or carrier material to determine the particle size.35 Nanoprecipitation: in this method, the drug and carrier are dissolved in a water-miscible solvent, which is then rapidly injected into an aqueous phase.36 Emulsion techniques: this method involves emulsion solvent evaporation in which a drug and carrier solution is emulsified in a non-solvent. In emulsion solvent diffusion, the drug and carrier are dissolved in a solvent, which is then dispersed into a non-solvent.37,38 In all these physical methods, the size of the formed nanparticles is influenced by factors such as temperature, the rate of solvent injection or diffusion and the choice of solvents. Template-based methods: in these methods, a template or a scaffold is used to control the size and shape of the drug delivery system. This can include using porous templates, sacrificial templates, and biomolecule templates such as DNA/protein or molecular imprinting techniques.1.3. Ideal size range for best performance
“Nanomedicine” represents highly precise medical interventions occurring at the nanoscale level, aimed at advancing the fields of disease detection, diagnosis, and treatment.39,40 A significant component of nanomedicine focuses on DDSs, featuring nano-sized, soft nanoparticles like liposomes, lipid nanoparticles, polymeric nanoparticles, polymer–drug conjugates, dendrimers, and hydrogels, alongside hard nanoparticles such as carbon nanotubes, gold nanoparticles, and metal–organic frameworks.29,41–45 Among these, nanocarriers based on polymers offer several distinct advantages such as easy synthetic methods, tunable chemical and physical properties, incorporation of both hydrophilic and hydrophobic drugs and relatively higher stability than those based on liposomes. Biodegradable units can be easily integrated into polymers that greatly contribute to the controlled drug release from the nanocarrier in a sustainable manner.46–48 Many of the distinct types of DDSs developed in the sub-100 nm size range contain polymer segments. For instance, polymeric nanoparticles are typically made from biocompatible polymers such as PLGA (poly(lactic-co-glycolic acid)) or chitosan.49–54 A related category is polymeric micelles formed by the self-assembly of amphiphilic block copolymers.52 Their core–shell structure can carry hydrophobic drugs in the core while remaining water-soluble. Highly branched polymers (dendrimers) have a compact architecture with size below 100 nm and can encapsulate drugs within their branches.55 Another category is polymeric networks (nanogels) that are known for their high water content and biocompatibility.56–58 Polymeric nanocapsules constitute a special type with a hollow core and a polymeric shell. They can encapsulate both small molecular and bio-macromolecular drugs, and a polymeric shell containing stimuli-responsive segments facilitates controlled drug release.59,60Other two types of DDSs that come under the category of sub-100 nm size use small unilamellar liposomes61 and metal nanoparticles. By suitable preparation methods, the size of metal nanoparticles can be tuned with one or more drugs conjugated on their surface.62–65 Utilizing the physical properties of core metals (e.g., gold and iron) and exchangeable molecular ligands, metal nanoparticles are also useful for combination therapy where diagnosis and treatment are conducted using a single delivery system. However, metal nanoparticles tend to agglomerate in an aqueous medium and often show poor biodegradability causing toxicity.
In this review, our focus is on the polymer-based nanocarriers with an emphasis on unique polymerization techniques employed in the development of DDSs with dimensions below 100 nm. We introduce a concept of “template polymerization”, a well-established method in covalent polymerization that offers precise control over molecular weight. We demonstrate the utility of this approach in constructing a monolayer of a polymer around a biomolecule template, resulting in the generation of a DDS with sizes ranging from several tens of nanometers. Each section is divided based on the unique polymerization strategies developed using the biomolecule templates. Additionally, we discuss important categories of small size DDSs, which share a conceptual similarity to “template polymerization”. RNA-based therapeutics, particularly small interfering RNA (siRNA) used for gene silencing and gene therapy, are becoming more prominent, and receiving substantial attention.66 Effective RNA delivery necessitates the use of a DDS with dimensions below 100 nm. RNA-based lipid nanoparticles recently developed for COVID-19 vaccines were of sub-100 nm size.67,68 Thus, our review predominantly centers on DDSs involving biomacromolecule drugs, such as siRNA, though DDSs can be applied to both small molecule and macromolecule drugs.
The necessary requirements for biomacromolecule drugs (e.g., nucleic acid and protein) are efficient drug loading into a nanocarrier via appropriate charge neutralization, protection against degradation, stability in bloodstream and drug release at the intended site of action along with the overall size of the carrier/drug complex below 100 nm. Conventional synthesis of nanocarriers without guided by a template can only produce large size (>200 nm) polydisperse polymeric nanoparticles due to a random interaction between the long polymer and the biomacromolecule drug. In this regard, the template polymerization method is highly advantageous for constructing a small size delivery system. This method involves a precise interaction between a polymer and a template drug and hence the nanocarrier size is controlled by the number of interaction moieties and polymerization.
2. Template polymerization
Template polymerization is a specialized technique within the field of polymer chemistry that involves the use of a template or a pre-existing structure to guide and control the polymerization process69 (Fig. 1). This method allows for the precise and ordered formation of polymers with specific structures, shapes, and properties. Templates include porous materials, molecular scaffolds, or even biological structures like DNA and protein.70–76 Template polymerization has a wide range of applications, including the synthesis of nanomaterials, molecular imprinting for sensors and separation, and the creation of functional materials with tailored properties.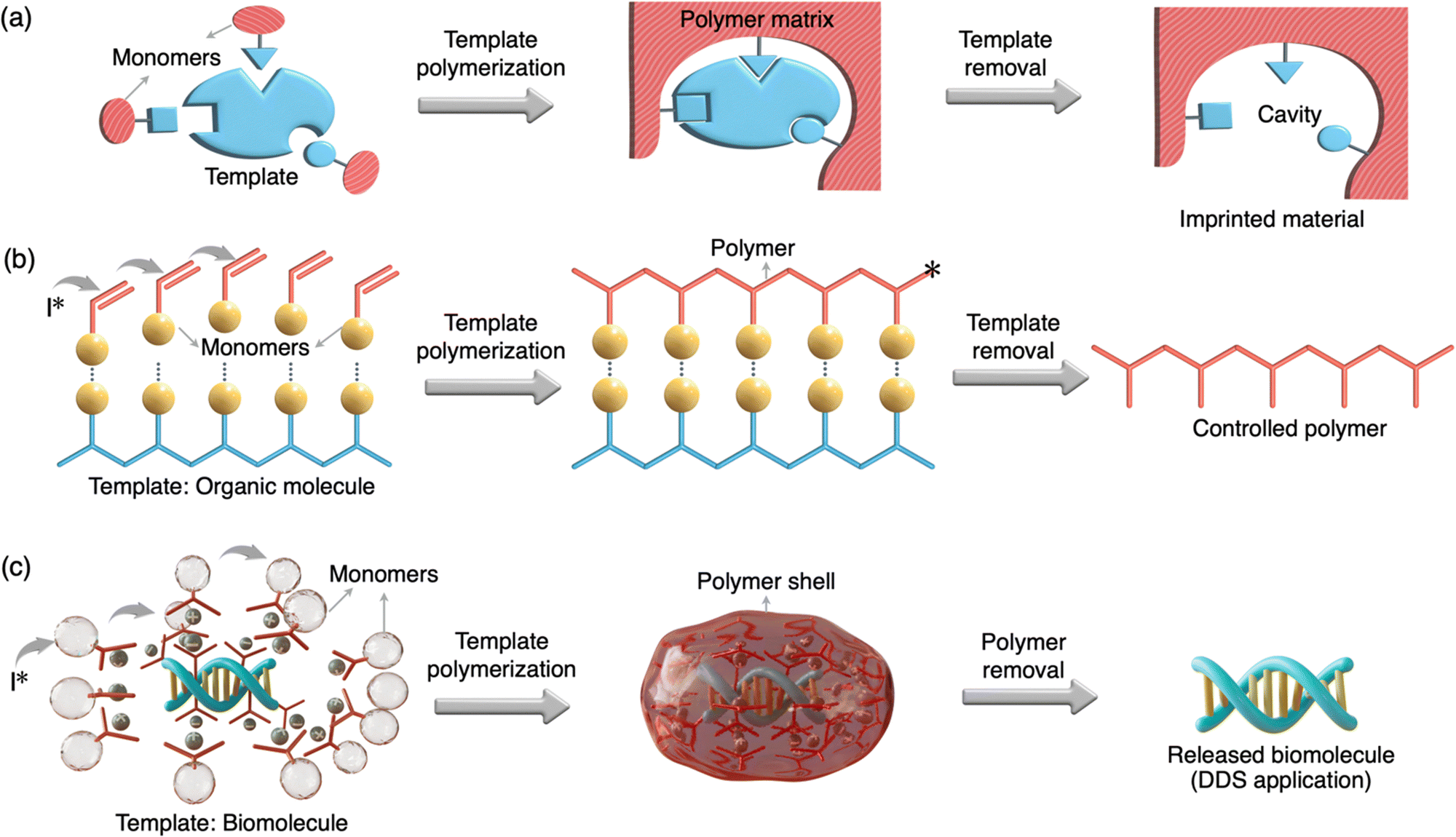 | ||
| Fig. 1 Scheme showing the concept of molecularly imprinted polymers and template polymerization. (a) Monomers and cross linkers bind to a template before polymerization. The template can be removed resulting in an imprinted material with a cavity. (b) Monomers non-covalently adhere onto a template before polymerization by an initiator. The template can be removed resulting in a controlled polymer. (c) Cationic monomers adhere onto anionic phosphate groups of a biomolecule (nucleic acid/protein) template before polymerization by an initiator. The polymer envelope can be cleaved to release the biomolecule template. | ||
2.1. Classification of template polymerization
Molecularly imprinted polymers (MIPs) are a notable class of template polymerization.77 In MIPs, a template (an imprinting compound such as a small molecule of <900 Da molecular weight) forms a complex with designed monomers and crosslinkers which then undergo a radical/photo-polymerization process. Then, the template is typically removed, leaving behind a polymer structure with well-defined cavities and selective binding sites (Fig. 1(a)). MIPs are designed to recognize and selectively bind to target molecules, making them useful in areas like drug delivery, catalysis, sensing, and chromatography. A related category is biomolecularly imprinted polymers (BMIPs), which are essentially the same as MIPs except the large dimension and functionality of the template (e.g., protein, heparin, virus etc).78 Although several methods for the preparation of BMIPs are reported, imprinted polymeric scaffolds for biologically relevant macromolecules (e.g., nucleic and protein) are highly limited. This may be due to the challenges associated with BMIPs such as solvent incompatibility, dynamic conformations of biomacromolecules and difficulty to remove the template from the polymeric matrix. Another category is a DNA-templated organic synthesis (DTS) where reactive building blocks conjugated on the single-stranded DNAs react only at the increased local concentration of reactants attained by DNA hybridization.79,80 The use of a DNA template to control the synthesis of organic compounds has a similarity to the ribosomal peptide synthesis in living systems in which peptide bond formation is directed by base-pairing between transfer RNAs and an mRNA template. Importantly, the DTS method is advantageous for the construction of sequence-controlled oligomers and macrocycles. The biomolecule-templated polymerization (BTP) method developed for the construction of sub-100 nm DDS is clearly different from the other templated strategies discussed above in terms of its preparation, type of templates, and applications (Fig. 1 and Table 1).| MIP | BMIP | DTS | TP | BTP | |
|---|---|---|---|---|---|
| Templates | Small molecule | Biomacromolecule | DNA | Small molecule | DNA, siRNA, and protein |
| Outcomes | Imprinted cavities | Imprinted cavities | Organic synthesis | Organic polymer | Polymer shell |
| Primary interactions | Covalent or non-covalent | Covalent or non-covalent | Covalent | Non-covalent | Non-covalent |
| Applications | Biosensors separations | Nanomolding photolithography | Tool for non-natural material discovery | Controlled polymer synthesis | Drug delivery system (DDS) |
2.2. Biomolecule-templated polymerization
This method involves using natural or engineered biomolecules as templates to guide the synthesis and assembly of polymers or other materials with precise shapes, sizes, and functionalities.81–83 The use of biomolecule templates has gained prominence in recent years due to their potential in creating complex, functional, and biocompatible materials such as DNA origami. Using biomolecule templates allows precise control over the size, shape, and functional properties of the resulting materials. This control can be crucial in tailoring materials for specific applications such as construction of ultra-small size DDSs. For instance, a monomer unit with a cation moiety can non-covalently bind to the anionic moieties of a nucleic acid/protein template (Fig. 1(c)). Polymerization initiated by an initiator can direct the polymerization only on the template surface resulting in a length-controlled polymer/template complex. With an appropriate design, a polymer can also be cleaved with bio-stimuli such as glutathione releasing the template. While biomolecule templates offer significant advantages, there are challenges related to stability, template removal, and scalability. The template and the polymerization process must be carefully chosen to address these challenges. The following sections discuss the construction and application of sub-100 nm DDSs based on unique polymerization by using biomolecule templates such as DNA, siRNA and proteins.3. DNA-templated condensation
This is a technique used in materials science and nanotechnology to create highly organized and controlled structures through the self-assembly of molecules in the presence of DNA templates.84–86 This approach takes advantage of the specific and complementary base pairing of DNA to guide the arrangement of other molecules, such as nanoparticles or polymers. In DNA-templated condensation, DNA strands are used as a molecular scaffold. The DNA can be single-stranded or double-stranded, depending on the desired structure. Single-stranded DNA is often used for the self-assembly of nanoparticles and other small molecules. By designing DNA sequences with specific complementary regions, researchers can control the assembly of materials. For example, nanoparticles functionalized with complementary DNA sequences will bind to each other precisely, creating ordered structures. DNA templated condensation has applications in biology and biotechnology, including the self-assembly of DNA origami structures for drug delivery, gene therapy, and biosensing. DNA-templated condensation can also facilitate the organization of polymers.87 By using DNA as a guiding framework, the arrangement of monomers can be controlled, leading to subsequent polymerization, resulting in the creation of structured nanoscale objects. This methodology finds application in the realm of biology for constructing a sub-100 nm DDS with DNA as a macromolecular drug. Early experiments conducted by Hagstrom and colleagues revealed that template polymerization, involving cationic monomers adhered to the template surface, could aid in condensing DNA into nanoparticles, each measuring less than 150 nm in diameter (Fig. 2(a)).88 Notably, this DNA condensation occurred when over 90% of the negative charges on the phosphate groups were neutralized by counter ions. This neutralization and DNA condensation during template polymerization likely hindered further template-dependent polymerization by preventing the monomer interaction with the DNA and the growth of polymer chains. Consequently, during the final stages of template polymerization, extensive aggregation of charge-neutralized DNA particles took place, as the surplus free DNA (bearing a negative charge) was neutralized by the growing polymeric counterions, and an excess of polymeric cationic charges could not be generated. Consequently, template polymerization facilitated the formation of individual uncharged particles (with a zeta potential close to 0 mV), as evidenced in the electron microscopy images shown in Fig. 2a. The inclusion of a monomer containing poly(ethylene glycol) (PEG) prevented the aggregation of these particles. The incorporation of PEG into the polymer provided steric stabilization for the DNA particles, even though the zeta potential was close to zero.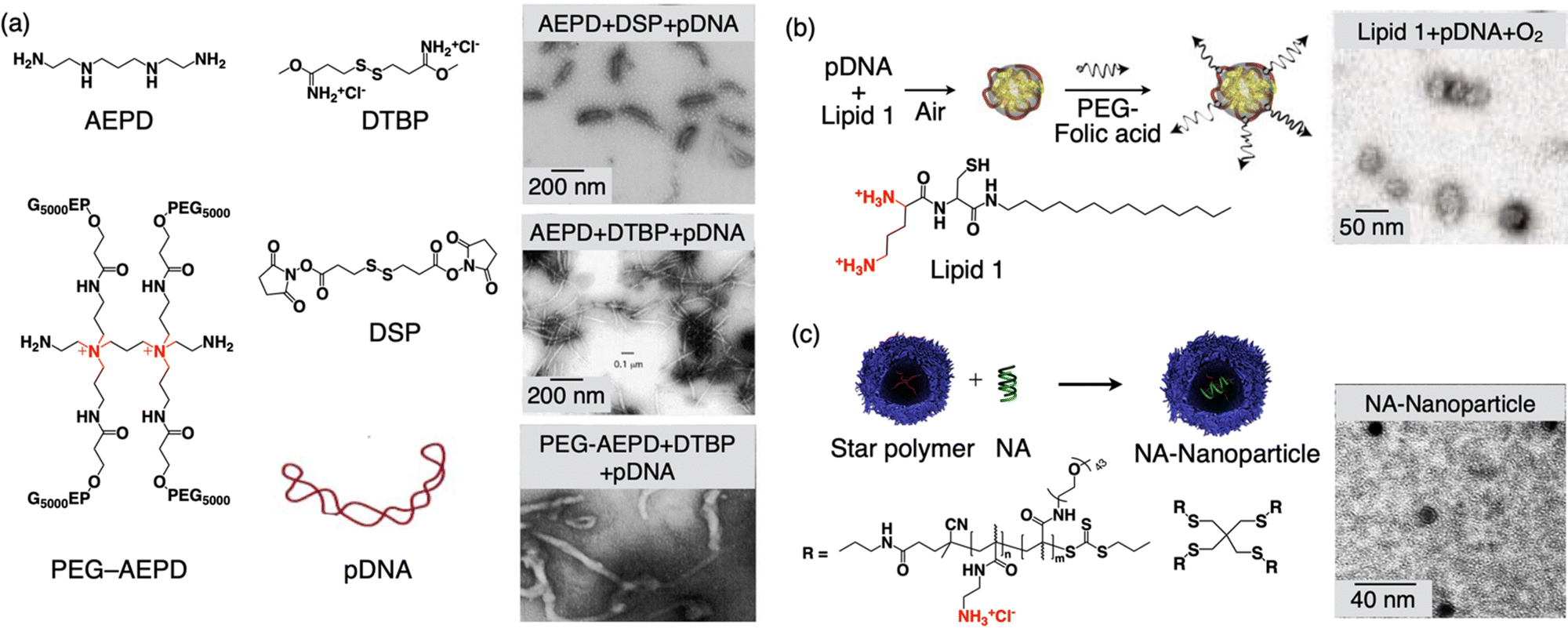 | ||
| Fig. 2 Schematic illustration of DNA templated condensation. (a) Molecular structures of the monomers for templated condensation along with the TEM images of formed nanoobjects. (b) Formation of 30-nm size nanoparticles via template-assisted oxidative polymerization of a cysteine-containing lipid along with the TEM image of the nanoparticle. (c) Molecular structures of the star polymer, the formation of NA-nanoparticles by complexation with nucleic acid (NA), and a TEM image of the NA-nanoparticle. Reproduced with permission ref. 88 Copyright 1998, Oxford University Press; ref. 89 Copyright 2003, John Wiley and Sons; and ref. 91 Copyright 2014, American Chemical Society. | ||
This illustrates that template polymerization can be harnessed to create non-aggregating, uncharged particles. The inclusion of AEPD-PEG caused the particles to adopt ‘worm’-like structures, as illustrated in Fig. 2(a). The complexes formed via template polymerization can also be utilized to deliver DNA into mammalian cells. Importantly, it was demonstrated that the DNA within these particles retains its biological activity and can express foreign genes within the cells.
In a separate study, researchers devised a universal method for the monomolecular condensation of genes, resulting in a monodisperse and stable population of particles measuring 30 nm in diameter.89,90 This technique takes advantage of the condensation of DNA by a cationic cysteine-based detergent. This interaction is reversible and can be employed to drive the system toward the maximum number of particles in an entropic manner. Subsequently, particle stability is achieved through template-assisted oxidative dimerization of the detergent into a gemini lipid (Fig. 2(b)). Leveraging their small size, the researchers demonstrated the cellular uptake of these DNA-containing nanoparticles, with a focus on targeting cancer cells. To maximize efficiency, they modified the monomolecular DNA particles with a folate-PEG motif and a disulfide linkage that could be cleaved within the cytoplasm.
The fabrication of nucleic acid nanoparticles with precise control over their small size and uniformity is challenging. Wang and colleagues have introduced an approach involving core–shell star polymers capable of forming complexes with 2, 16, and 53 molecules of oligonucleic acid (NA), leading to the creation of nanoparticles with diameters measuring 15, 23, and 30 nm, respectively (Fig. 2(c)).91 These nanoparticles exhibit a high degree of uniformity (PDIs < 0.08), have neutral zeta-potentials, and maintain colloidal stability for extended periods in phosphate-buffered saline. The process for generating these nanoparticles is remarkably straightforward and can be easily adapted by individuals without extensive expertise in drug delivery, highlighting its accessibility and potential for broad applications.
4. siRNA-templated polymerization
This technique combines the principles of siRNA, which is used to silence specific genes, with polymerization to create functional nanoparticles with therapeutic applications.92 The siRNA molecules contain anionic phosphate groups that are complementary to the cationic monomers, which allow them to act as templates for polymerization. Polymerization, often initiated through chemical reactions, occurs in the presence of siRNA templates. The monomers polymerize in a manner that is guided by the siRNA's anionic charges on the surface. As a result, the polymer forms in a pattern that mimics the siRNA's structure. The synthesized polymer nanoparticles can deliver therapeutic siRNA to target cells for gene silencing. While siRNA templated polymerization offers unique advantages, challenges include optimizing the reaction conditions, ensuring siRNA stability, and validating the effectiveness of gene silencing and drug delivery. The biocompatibility and potential toxicity of the synthesized polymer nanoparticles need to be carefully evaluated, especially for in vivo applications.4.1. Nanocapsules
Chen and colleagues introduced an innovative siRNA delivery technique based on template polymerization.93 This approach involves the creation of a degradable polymer shell encapsulating a single siRNA molecule, tailored to have the desired size and charge. The core of this single siRNA nanocapsule platform begins with the assembly of a positively charged monomer, a crosslinker, and a neutral monomer, and the arrangement of these molecules around the negatively charged siRNA's surface. This arrangement occurs through electrostatic interactions and hydrogen bonding in Step I (Fig. 3(a)). Following this, room-temperature radical polymerization in an aqueous solution takes place, resulting in the formation of a thin polymer shell around each siRNA molecule in Step II. This shielding effectively shields the siRNA from degradation by ribonucleases and exposure to serum. To facilitate the dispersion of the nanocapsules, a hydrophilic monomer (terminator) is introduced, while the crosslinker is included to ensure the polymer structure's stability in a serum environment (pH ∼ 7.4). Importantly, transmission electron microscopy (TEM) images of the siRNA nanocapsules showed an average diameter of 20 nm (Fig. 3(a), TEM image). Given that the average hydrodynamic diameter of a double-stranded siRNA (comprising 21 base pairs) is 4.2 nm, each nanocapsule likely contains only a single siRNA molecule. Notably, these nanocapsules are designed to degrade at a pH below 6, facilitating release within the acidic endosomes. This unique responsive design enables the nanocapsules with remarkable stability in serum (at pH ∼ 7.4), promotes effective endosomal escape through the proton-sponge effect and cation-mediated membrane destabilization, and release of the siRNA into the cytoplasm as the polymer shell degrades (Fig. 3(a), CLSM image).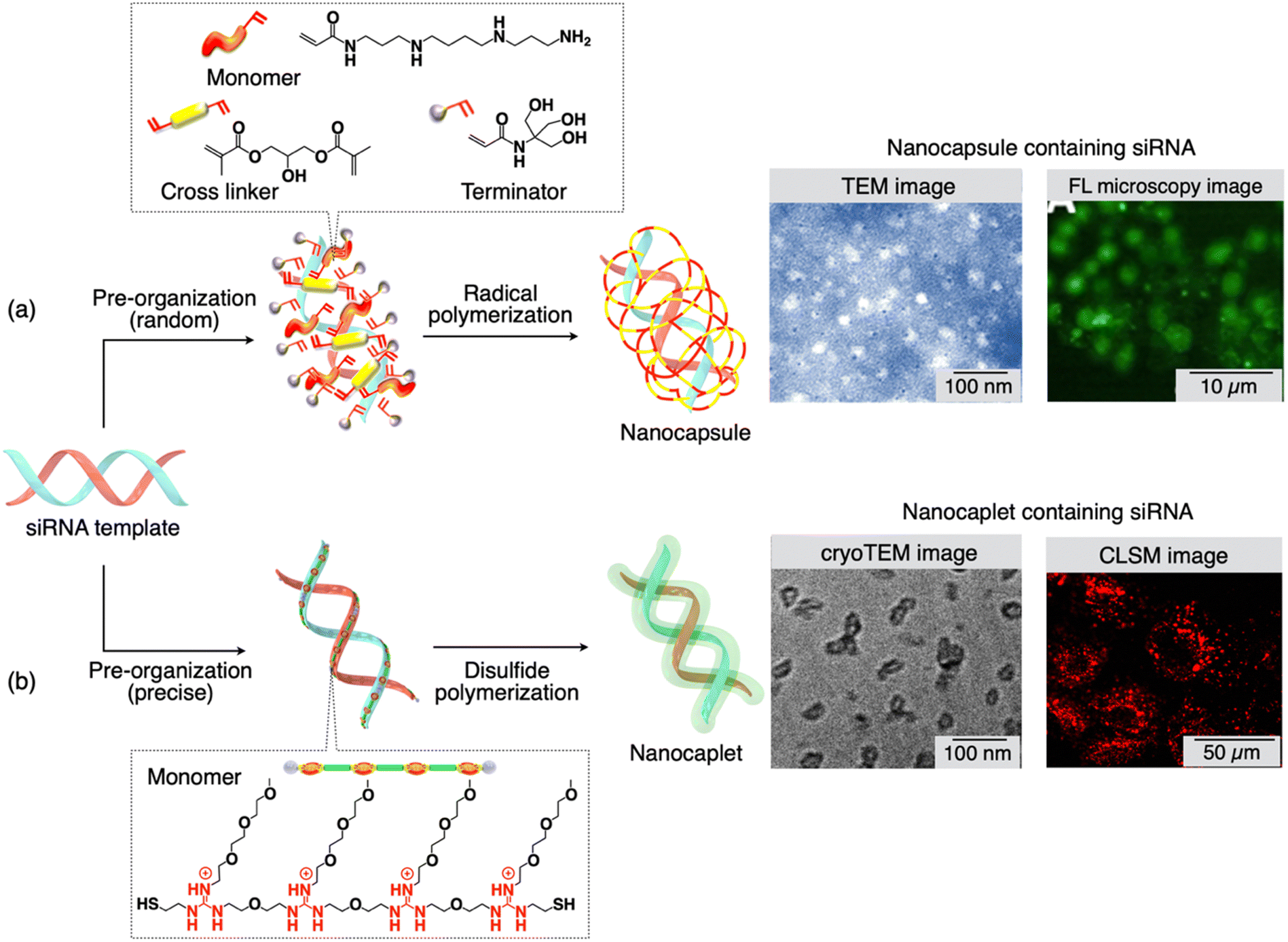 | ||
| Fig. 3 (a) and (b) Scheme showing the molecular structure of monomers and cross linkers, and the synthesis of nanocapsules/nanocaplets containing siRNA. Monomers first pre-organize on the siRNA template followed by in situ polymerization. TEM images show the small size of nanocapsules/nanocaplets, and microscopy images show the presence of siRNA-nanocapsules/nanocaplets in cancer cells. Reproduced with permission ref. 93 Copyright 2012, American Chemical Society: ref. 94 Copyright 2015, American Chemical Society. | ||
4.2. Nanocaplets
A novel technique, recently developed by Aida and colleagues, utilizes template polymerization, capable of producing siRNA/carrier conjugates with a compact size of 7 ± 2 nm.94,95 The pivotal concept within this method involves a water-soluble telechelic dithiol monomer (TEGGu4) that carries multiple guanidinium ions (Gu+). In the presence of siRNA as a template, the TEGGu4 monomer forms several ‘salt-bridges’ with the phosphate ion segments of siRNA. This interaction enables a disulfide-mediated polymerization through the terminal thiol moieties, resulting in the creation of a fine polymer layer around the siRNA template (Fig. 3(b)). The alkoxy spacer between adjacent Gu+ ions, the number of Gu+ ions in a monomer, and the ethylene glycol segment within the monomer play critical roles in achieving the ‘template’ effect. In contrast, the Gu-monomer, lacking the ethylene glycol segment, which contains only one heptaethylene glycol moiety between two Gu+ ions, fails to adhere to the siRNA template and forms nanocaplets.These nanocaplets consist of multiple disulfide bonds that can be cleaved by reductants. In the presence of substances like glutathione or dithiothreitol, siRNA is swiftly released from the nanocaplets due to the disulfide bond breakage. The average diameter of the siRNA-containing ‘nanocaplets,’ determined from dynamic light scattering, fluorescence correlation spectroscopy, and transmission electron microscopy experiments (Fig. 3(b), TEM image) was 7 ± 2 nm. The compact size of these nanocaplets proves advantageous facilitating their cellular uptake. To illustrate, when a dye-appended siRNA-nanocaplet was exposed to Hep3B cancer cells and examined through confocal laser scanning microscopy, it displayed intense fluorescence originating from the dye (Fig. 3(b), CLSM image). Given the highly reductive intracellular environment, primarily due to the presence of glutathione (8 mM), these nanocaplets can be cleaved within the cells to release siRNA, effectively reducing the expression of a target gene. An experiment involving luciferase-expressing Hep3B cells validated the siRNA's knockdown activity. The compact siRNA/carrier conjugates hold great potential for delivering siRNA to locations that conventional drug delivery systems may struggle to access. It is also possible to functionalize the siRNA-nanocaplet with targeting moieties for specific applications.
For achieving transcellular delivery of siRNA into deep tissues, the carrier is preferred to be as small as possible and needs to be reductively cleavable in the cytoplasm. Furthermore, it should be active for transcytosis (consecutive endocytosis/exocytosis events). Combining all these characteristics into a single delivery system and achieving siRNA delivery into tissues deeper than 40 μm is extremely challenging. For this purpose, Aida et al. developed a siRNA-containing nanocaplet appended with transferrin (Tf) units (TfNanocaplet), which can deeply deliver siRNA into tissues at a depth of up to nearly 70 μm, unprecedentedly (Fig. 4(a)).66
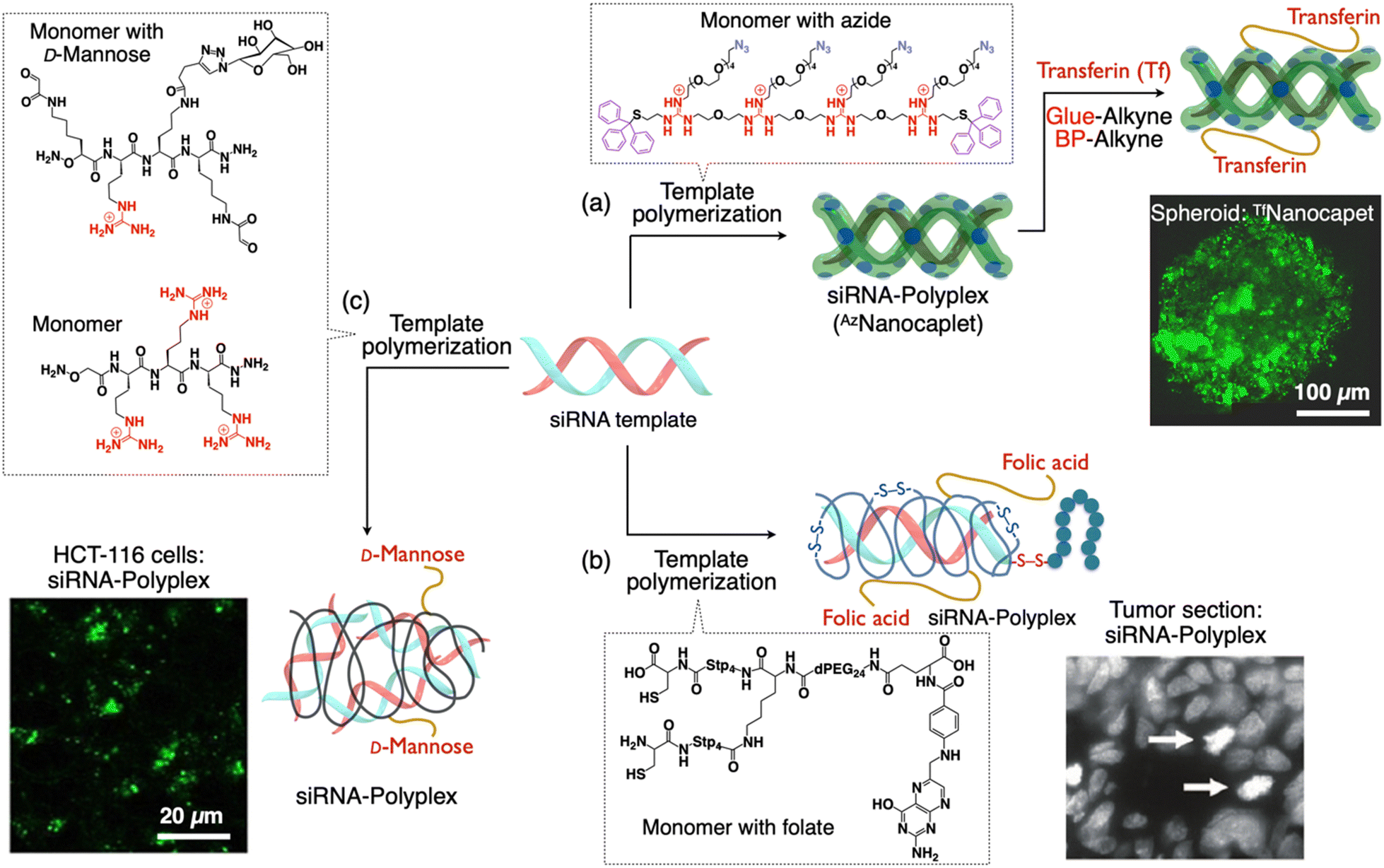 | ||
| Fig. 4 Scheme showing the formation of sub-100 nm siRNA-polyplexes with targeting moieties on their periphery via template polymerization. (a) Synthesis of a siRNA-nanocaplet with transferrin (Tf) units (TfNanocaplet). CLSM image of the Hep3B spheroid obtained after incubation of the TfNanocaplet for 3 days. (b) Synthesis of a polymer shell around siRNA with folate units. Histochemical analysis of gene silencing (tumor sections) by detection of aster formation (white arrows). (c) Synthesis of a siRNA-polyplex with mannose units from dynamic covalent polymers. CLSM image showing the internalized siRNA within HCT-116 cells. Reproduced with permission from ref. 66 Copyright 2019, American Chemical Society; ref. 98 Copyright 2012, American Chemical Society; and ref. 99 Copyright 2021, John Wiley and Sons. | ||
To synthesize a TfNanocaplet, they first prepared a siRNA-containing reactive nanocaplet (AzNanocaplet) by oxidative polymerization of AzGu, an azide (N3)-appended telechelic monomer. Then, the AzNanocaplet was allowed to react with a guanidinium (Gu+)-appended bioadhesive dendron followed by a benzophenone (BP) derivative to obtain an intermediate conjugate. Subsequently, this conjugate was incubated with Tf and exposed to UV light for covalent immobilization of the attached Tf units by reacting with the BP units. siRNA encapsulation within the nanocaplet and its release in response to a reducing agent were confirmed by agarose gel electrophoresis. The average hydrodynamic size obtained from DLS and electron microscopy was ∼20 nm (observed by TEM imaging). The TfNanocaplet efficiently entered living cells as evident from the confocal imaging of TfNanocaplet-treated Hep3B cells. When the Tf units were replaced with bovine serum albumin (BSA), the cellular uptake was negligible despite its comparable size and surface charges of BSA or Transferrin appended on the nanocaplet indicating the transferrin receptor-mediated endocytosis mechanism for the cellular entry. To investigate the transcytosis mechanism, they prepared a 3D-cultured Hep3B spheroid with an average diameter of ∼500 μm and incubated with the TfNanocaplet. After 3 days of incubation, the spheroid fluoresced throughout its cross sections of microscopy (CLSM) imaging at a depth of 50 μm (Fig. 4(a)). Importantly, even at a depth of 70 μm, the TfNanocaplet reached the central part of the cross-section, indicating the deep permeation of the TfNanocaplet. Moreover, the TfNanocaplet eventually transfers siRNA into the cytoplasm and causes RNA interference and gene knockdown, as evidenced by the suppression of target luciferase gene expression in Hep3B cells.
4.3. Polymer shell
While classical cationic polymers like polyethylenimine (PEI) have demonstrated their potential in nucleic acid transport, they still fall short in terms of efficiency compared to their viral counterparts. Despite significant advancements in the field of polymer-based carriers for DDSs the increased complexity of these next-generation polycationic polymers has given rise to structures that are notably diverse and polydisperse.96 Adding to the complexity, the polyplexes formed are often highly polydisperse and larger in size, resulting in mixtures of nanoparticles, including polyplex aggregates.97 To address these challenges, researchers have developed libraries of precisely defined polycationic structures. These libraries shed light on the advantages of using carriers with well-defined structures that exhibit clear structure–activity relationships. In this pursuit, Wagner and his team have introduced a novel strategy that involves the precise incorporation of functional targeting, shielding, and endosomolytic domains into siRNA polyplexes (Fig. 4(b)).98 This approach offers a highly controlled and well-defined framework, enhancing the potential for effective drug delivery and gene silencing. They synthesized a multifunctional carrier system through solid phase supported chemistry and showed effective delivery of siRNA both in vitro and in vivo. This sequence-defined assembly comprises a meticulously crafted cationic (oligoethanamino)-amide core. At its ends, it is capped with two cysteine moieties, facilitating bio-reversible polyplex stabilization. In the center, a well-defined polyethylene glycol chain is attached, terminated with a folate receptor-specific ligand (folic acid) for the targeted cell interaction. When combined with an endosomolytic influenza peptide–siRNA conjugate, these elements give rise to nanoscale functional polyplexes measuring 6 nm in hydrodynamic diameter.To affirm the importance of each constituent within the carrier system for specific and efficient gene silencing, extensive validation was conducted. The resulting nanosized polyplexes displayed stability under in vivo conditions, receptor-specific cell targeting, and effective silencing of the EG5 gene in receptor-positive tumors. The nanoscale dimensions of these particles can be precisely adjusted based on oligomer design, ranging from 5.8 to 8.8 nm in diameter. Thanks to a complete surface charge shielding and remarkable stability, these polyplexes demonstrate excellent in vivo tolerability, with no accumulation in non-targeted tissues such as the liver, lung, or spleen. Furthermore, due to their small size, siRNA polyplexes are efficiently eliminated through renal clearance. This study showcased the ability to create highly functional yet molecularly uniform delivery systems through the practical approach of solid-phase synthesis. This method not only permits the establishment of well-defined structure–activity relationships but also streamlines the process of producing these systems in a reproducible and scalable manner. The strategy outlined in this study has the potential to be extended to develop various nucleic acid carriers featuring alternative targeting ligands (including peptides), variations in shielding levels and types, diverse polymeric backbones, or unique endosomal escape domains.
Dynamic covalent libraries offer a unique platform for exploring new chemical systems, allowing the adaptive emergence of bioactive assemblies through template effects. Ulrich et al. investigated dynamic covalent libraries consisting of complementary bifunctional cationic peptides (Fig. 4(c)).99 These libraries gave rise to a wide range of structures, from macrocycles to polymers. Notably, polymers are typically formed only at high concentrations. However, they found an interesting phenomenon in which siRNA serves as a template, guiding the creation of dynamic covalent polymers at low concentrations, primarily driven by electrostatic interactions. By incorporating a glycosylated building block, they demonstrated that this templated polymerization extends to the multivalent presentation of carbohydrate ligands for efficient cellular uptake and cell-selective siRNA delivery. They showed that a D-mannose ligand on the peptide building block promoted cell-uptake of a siRNA-templated polymer through binding to mannose receptors on the cell membrane and knocked down the target gene expression.
5. Polymer escort/anchor
An alternative approach to deliver siRNA involves direct covalent modifications of the 5′- and/or 3′-termini of siRNA with various entities such as lipid groups, small molecules, nanostructured DNA, or poly(ethylene glycol) (PEG). Modifying siRNA with linear PEG or brush PEG has been achieved through disulfide formation or Michael-type addition reactions between the thiol and maleimide groups.100,101 While the disulfide linkage allows for the release of the siRNA duplex after cellular uptake, the generation of redox-sensitive thiols and disulfides that may undergo undesired side reactions or premature degradation presents challenges in the synthesis and purification of polymer–siRNA conjugates. Matyjaszewski and Das et al. have developed a simple method for creating siRNA–polymer constructs that serve as standalone siRNA delivery vehicles (Fig. 5(a)).102 This involves post-synthetic click conjugation of polymers to the passenger strand of an siRNA duplex, followed by annealing with the complementary guide strand, resulting in siRNA where one strand includes terminal polymer escorts. The polymer escorts serve a dual purpose by providing protection against nucleases and aiding in the cellular uptake of siRNA. These self-transfecting polymer-escort siRNAs are effective tools in RNA interference (RNAi) and have proven successful in the knockdown of both reporter and endogenous genes (Fig. 5(b)).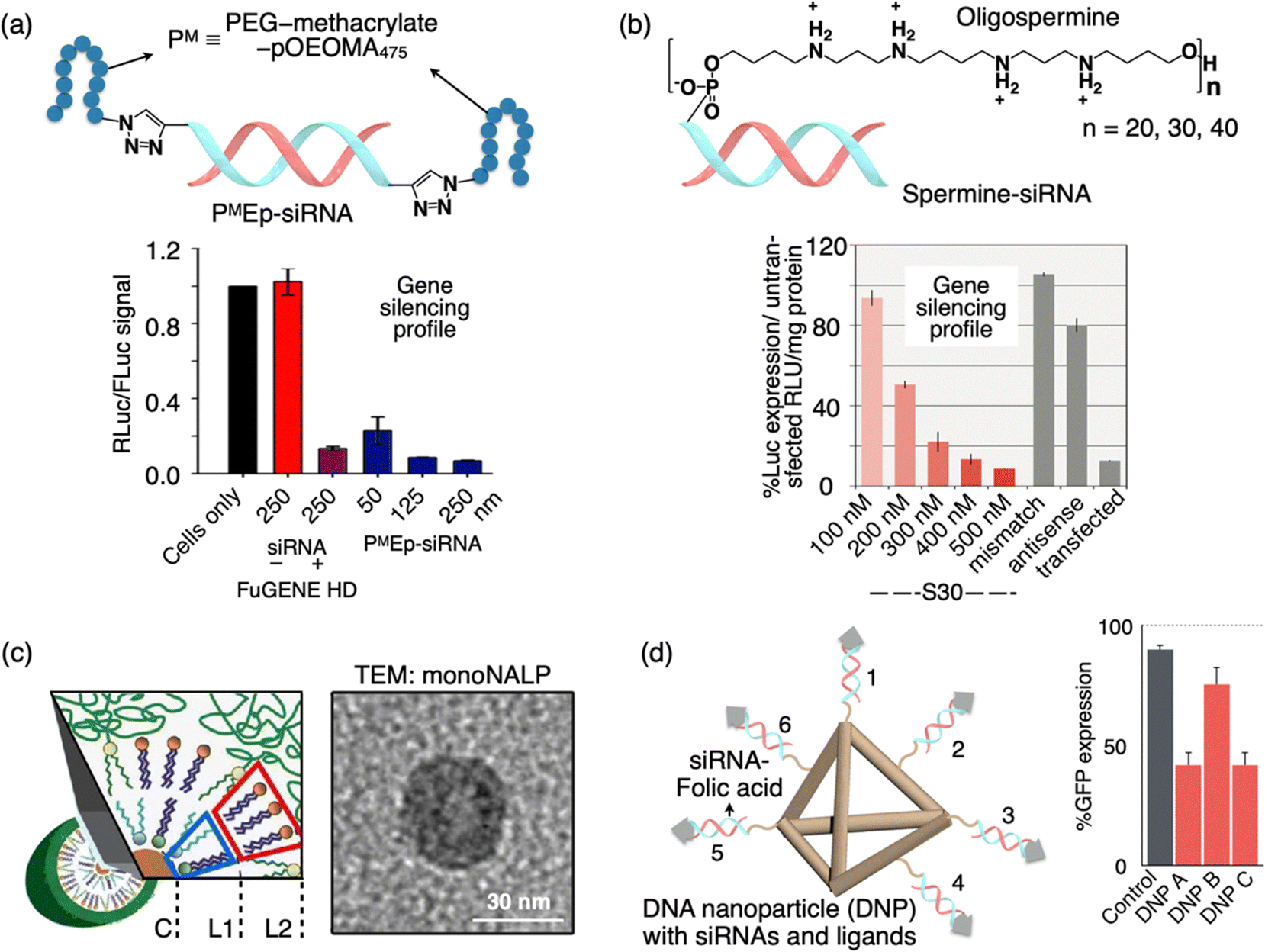 | ||
| Fig. 5 Schematic illustration of a sub-100 DDS formed by the polymer escort method or nucleic acid assembly. (a) PMEp-siRNA is formed by the click reaction of bisalkyne-siRNA and azido-PM (PM stands for the PEG-methacrylate-pOEOMA475 polymer). Graph showing the gene silencing activity of PMEp-siRNA DDS in comparison to the FuGENE HD positive control sample. (b) Spermine-siRNA along with the chemical structure of 5′-spermine. Graph showing the luciferase gene silencing by spermine-siRNA DDS in comparison to the mismatch/antisense negative control sample. (c) Scheme and TEM image of a mono-NALP: the inner monolayer consisting of the lipid composition (L1, L2) is combined to encapsulate short ds-DNA. (d) Structure of DNA tetrahedron nanoparticles (DNPs) with siRNA-folic acid (FA, gray arrowhead) overhangs. Graph showing the GFP gene silencing by DNPs having different orientations of the FA ligand (DNP A, FA on 1, 2, and 3; DNP B, FA on 1,3 and 4; DNP C, 1,3 and 5). Reproduced with permission from ref. 102 Copyright 2013, American Chemical Society; ref. 103 Copyright 2009, American Chemical Society; ref. 104 Copyright 2012, American Chemical Society. | ||
Molecular conjugates of siRNA with minimum overall changes in the structural properties are expected to exhibit a more conventional pharmacological profile and achieve a better balance between efficiency and inflammation compared to drug delivery vectors with siRNA encapsulated in it. Behr and colleagues developed a stepwise automated synthesis method for oligodeoxyribonucleotide-oligospermines (Fig. 5(b)).103 This approach allows the incorporation of various numbers of spermine residues at any position along an oligonucleotide. They demonstrated that these oligonucleotide conjugates can efficiently enter cells depending on the ratio between the anionic charge from oligonucleotides and the cationic charge from oligosperimines. They also showed the application of this method for siRNA delivery. By conjugating 30 spermines to the 5′ end of the siRNA sense strand, they achieved endogenous gene silencing in A549Luc cells. The luciferase expression knockdown was concentration-dependent (S30, red bars). Importantly, the knockdown was not observed with a shorter (S20 tail) or (S40 tail) oligospermine conjugated control using the “naked” S0 siRNA without spermine, or an S30 siRNA with a mismatch sequence (gray bar), did not show gene knockdown (Fig. 5(b)). Although these molecular siRNA drugs may exhibit slightly lower in vitro efficacy compared to cationic lipid formulations, they have the potential to surpass nanoparticles in vivo, where extracellular diffusion presents a significant challenge.
6. Assembled nucleic acid particles
Ra![[d with combining umlaut]](https://www.rsc.org/images/entities/char_0064_0308.gif) ler et al. developed a novel class of lipid-based nanoparticles known as mono-nucleic acid lipid particles (mono-NALPs) (Fig. 5(c)).104 These particles are composed of individual short double-stranded oligonucleotides or single siRNA molecules, each encased within a closed shell made up of a cationic–zwitterionic lipid bilayer. Additionally, they are equipped with an outer polyethylene glycol (PEG) shield for added stability and functionality. The formation of these particles is achieved through a self-assembly process via solvent exchange. This process involves mixing nucleic acid with four specific lipid components, namely DOTAP, DOPE, DOPC, and DSPE-PEG(2000). Using fluorescence correlation spectroscopy, the authors monitored the transformation of short double-stranded oligonucleotides or siRNA, in conjunction with lipids, into monodisperse particles that are approximately 30 nm in diameter. Further investigations through small-angle neutron and X-ray scattering, as well as transmission electron microscopy, have confirmed the micelle-like core–shell structure of these particles (Fig. 5(c)). The PEGylated lipid shell plays a crucial role in protecting the nucleic acid core from degradation by nucleases. It also provides steric stabilization to the mono-NALPs, preventing their disassembly in collagen networks and hinders nonspecific binding to cells. This innovative platform demonstrates promising potential for various applications in drug delivery and nucleic acid therapeutics. Anderson and colleagues have demonstrated the efficacy of self-assembled DNA tetrahedral nanoparticles (DNPs) with precisely defined dimensions in delivering siRNAs into cells and effectively silencing target genes within tumors (Fig. 5(d)).105 The production of monodisperse nanoparticles is achieved through the self-assembly of complementary DNA strands. Given the programmable nature of DNA strands, this approach allows for precise control over the size of the nanoparticles and the spatial arrangement and density of cancer-targeting ligands, such as peptides and folate, on the nanoparticle surface. Their study revealed that optimal siRNA delivery into cells requires a minimum of three folic acid molecules per nanoparticle, and effective gene silencing only occurs when the ligands are appropriately oriented in space (Fig. 5(d)). Notably, robust gene silencing was observed following the administration of these DNA-based nanoparticles, both through intratumor and systemic injections into KB xenograft tumors, with no discernible immune response. These versatile DNA-based particles hold potential for therapeutic applications in various tissues, as their size and ligand composition can be tailored to specific needs.
ler et al. developed a novel class of lipid-based nanoparticles known as mono-nucleic acid lipid particles (mono-NALPs) (Fig. 5(c)).104 These particles are composed of individual short double-stranded oligonucleotides or single siRNA molecules, each encased within a closed shell made up of a cationic–zwitterionic lipid bilayer. Additionally, they are equipped with an outer polyethylene glycol (PEG) shield for added stability and functionality. The formation of these particles is achieved through a self-assembly process via solvent exchange. This process involves mixing nucleic acid with four specific lipid components, namely DOTAP, DOPE, DOPC, and DSPE-PEG(2000). Using fluorescence correlation spectroscopy, the authors monitored the transformation of short double-stranded oligonucleotides or siRNA, in conjunction with lipids, into monodisperse particles that are approximately 30 nm in diameter. Further investigations through small-angle neutron and X-ray scattering, as well as transmission electron microscopy, have confirmed the micelle-like core–shell structure of these particles (Fig. 5(c)). The PEGylated lipid shell plays a crucial role in protecting the nucleic acid core from degradation by nucleases. It also provides steric stabilization to the mono-NALPs, preventing their disassembly in collagen networks and hinders nonspecific binding to cells. This innovative platform demonstrates promising potential for various applications in drug delivery and nucleic acid therapeutics. Anderson and colleagues have demonstrated the efficacy of self-assembled DNA tetrahedral nanoparticles (DNPs) with precisely defined dimensions in delivering siRNAs into cells and effectively silencing target genes within tumors (Fig. 5(d)).105 The production of monodisperse nanoparticles is achieved through the self-assembly of complementary DNA strands. Given the programmable nature of DNA strands, this approach allows for precise control over the size of the nanoparticles and the spatial arrangement and density of cancer-targeting ligands, such as peptides and folate, on the nanoparticle surface. Their study revealed that optimal siRNA delivery into cells requires a minimum of three folic acid molecules per nanoparticle, and effective gene silencing only occurs when the ligands are appropriately oriented in space (Fig. 5(d)). Notably, robust gene silencing was observed following the administration of these DNA-based nanoparticles, both through intratumor and systemic injections into KB xenograft tumors, with no discernible immune response. These versatile DNA-based particles hold potential for therapeutic applications in various tissues, as their size and ligand composition can be tailored to specific needs.
7. Polymer micelles
Kataoka et al. a pioneer in the field of polymer-based nanomedicine, developed polyion complex (PIC) micelles with diameters of several tens of nanometers with an extremely narrow size distribution.106,107 A PIC can be prepared simply by mixing a pair of block copolymers with the same degree of polymerization (DP) in aqueous media. A PIC has a core–shell architecture with the PIC core surrounded by a poly(ethylene glycol) (PEG) corona. They have developed a wide variety of PIC micelles for DDSs including siRNA delivery in vivo.52 Some of the techniques went up to clinical trials for practical use in humans. Although the formation mechanism is not a templated process, the size of polymeric micelles was less than 100 nm. Readers may refer to an excellent review related to this topic.108 A special category within PIC is unit PIC (uPIC), which form as an intermediate assembly with the minimal association number for charge neutralization.109 Compared to conventional micelles, uPIC formulation is smaller in size in the range of 10–50 nm. For instance, a siRNA-loaded uPIC constructed by charge-stoichiometric association between siRNA and PEG-b-poly(L-lysine) (PEG-PLL) with a well-defined degree of polymerization exhibited a hydrodynamic size of 12 nm (Fig. 6(a)).110 In this case, the structures (or size and association number) of siRNA-loaded uPICs can be precisely controlled by the MW of PEG and the degree of polymerization of the PLL polymer in the block catiomer.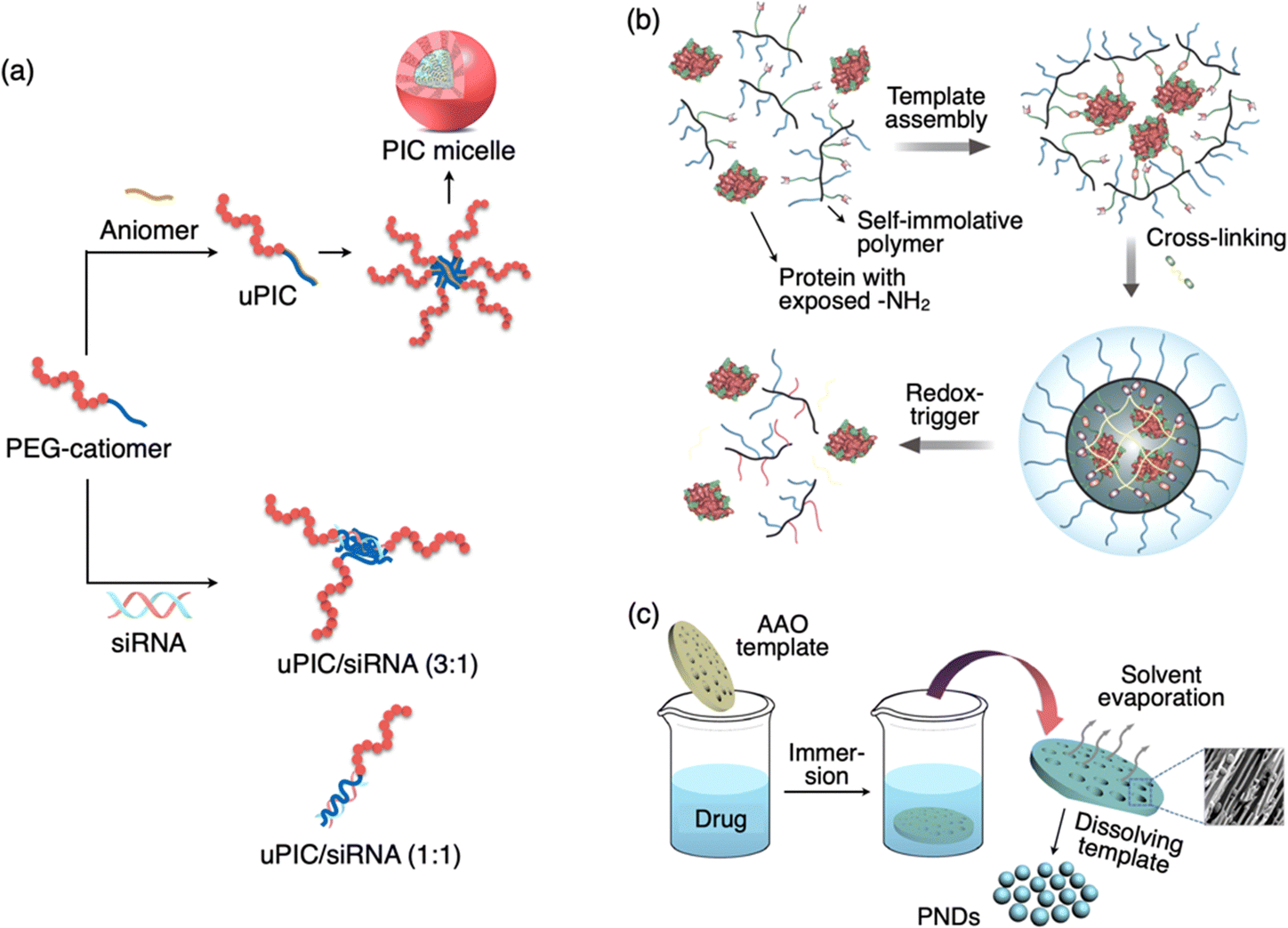 | ||
Fig. 6 Schematic illustration of a sub-100 DDS of PIC micelle, protein polymer complex, and pure nanodrugs. (a) Formation of PIC micelles via electrostatic interaction between the PEG-catiomer and aniomer resulting in a uPIC followed by its secondary assembly. Formation of a small-size uPIC/siRNA complex by stoichiometric (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or 1:1) charge neutralization of PEG-catiomer and siRNA. (b) Formation of a covalent polymer network around the protein template and the redox-triggered release of protein. (c) Formation of pure nanodrugs with the assistance of the AAO template. A SEM image of PND nanoparticles in the pores of the template is shown. Reproduced with permission from ref. 114 Copyright 2017, American Chemical Society; ref. 115 Copyright 2015, American Chemical Society. :1 or 1:1) charge neutralization of PEG-catiomer and siRNA. (b) Formation of a covalent polymer network around the protein template and the redox-triggered release of protein. (c) Formation of pure nanodrugs with the assistance of the AAO template. A SEM image of PND nanoparticles in the pores of the template is shown. Reproduced with permission from ref. 114 Copyright 2017, American Chemical Society; ref. 115 Copyright 2015, American Chemical Society. | ||
8. Protein-templated polymerization
The study of intracellular protein trafficking is an emerging field with promising applications in fundamental cell biology and biological therapeutics. To establish a reliable and enduring delivery approach, it is essential to ensure both the effective protection of the cargo and the ability to reverse the conjugation process without compromising activity. Covalent conjugation of a polymer and a protein is one of the techniques widely investigated for DDS applications.111–113 Thayumanavan et al. developed a unique method based on protein-templated polymer self-assembly (Fig. 6(b)).114 This approach enables the creation of a protective covering around the proteins and subsequently allows for their traceless release within the cytosol. The fundamental design concept is that an initial interaction occurs between the side-chain functionalities of a random copolymer and the numerous functional groups exposed on the surface of a target protein. This interaction prompts a few polymer chains to arrange themselves around the protein. Subsequently, this covalent capture event serves as a template for the formation of a polymer sheath around the protein through a cross-linking process involving the polymer's side chains.9. Drug only nanoparticles
Pure nanodrugs (PNDs), nanoparticles composed entirely of drug molecules, have emerged as promising candidates for the next generation of nanodrugs. However, the conventional preparation method, involving reprecipitation, encounters significant challenges, such as low production rates, relatively large particle sizes, and batch-to-batch variations. Lee et al. developed a novel, versatile, and precisely controlled approach for making PNDs through a template-assisted method, where an anodized aluminum oxide (AAO) template is simply dipped in a drug solution (Fig. 6(c)).115 Using this innovative technique, they have produced PNDs containing an anticancer drug (VM-26) with hydrodynamic size below 20 nm. This template-assisted method exhibits significantly enhanced potential for large-scale production in comparison to the conventional reprecipitation technique, making it an asset for future clinical applications. Furthermore, this method proves to be readily adaptable for a wide range of hydrophobic biomolecules without the necessity for custom molecular modifications. Additionally, it can be extended to fabricate all-in-one nanostructures incorporating various functional agents.10. Drug release behavior
The effectiveness of a DDS is often determined by its method of administration, behavior within bodily fluids, and its specific interactions at the tissue level within diseased areas. An ideal DDS should maintain stable circulation within the bloodstream, preventing premature drug release, avoiding nonspecific clearance by reticuloendothelial system (RES) organs/cells,116 and ultimately accumulate at the intended site of action. This accumulation can occur through the enhanced permeability and retention (EPR) effect or targeted delivery methods, leading to either internalization into target cells or the release of drugs in their proximity.117 Sub-100 nm DDSs can significantly decrease the renal clearance, thanks to their size remaining below the threshold limit (6.5 nm) crucial for kidney glomerular filtration. Moreover, the size of DDSs plays a pivotal role in their extravasation to tumor sites. For example, sub-100 nm DDSs can extravasate into hyper-vascularized tumors, such as Doxil® accumulation observed in Kaposi's sarcoma.118 Another important factor is the controlled drug release which can be achieved by introducing stimuli-sensitive segments within the DDS. Diseased sites often exhibit distinct endogenous stimuli compared to healthy tissues. For instance, lowered interstitial pH values (healthy tissue ∼7.4 versus tumor interstitial space ∼6.5–7.2), redox potentials (glutathione concentration in an intracellular milieu ∼2–10 mM versus an extracellular milieu ∼2–20 μM), and ATP levels (intracellular space ∼3 mM versus extracellular space ∼0.4 μM).119,120 Additionally, external stimuli, such as electric and magnetic fields, variations in temperature, light pulses (laser beams), ultrasound induction, and mechanical force, can also serve as triggers for controlled drug release in DDSs.121In principle, template polymerized DDSs are engineered to be large enough to evade renal filtration yet small enough to permeate leaky vasculature in tumor regions. Unlike non-templated polymeric carriers, template polymerized DDSs are molecularly defined, existing as a singular uniform population in terms of size. Although detailed biodistribution profiles and drug release data in animal models are lacking for many examples discussed in Section 4, cellular/tissue-level experiments have showed drug release and subsequent pharmacological effects. For instance, a template polymerized DDS (∼20 nm) with targeting groups on its surface exhibited deep tissue permeation of up to 70 μM in a tumor spheroid model.66 It internalized into cells, released drugs within the cell cytoplasm triggered by glutathione chemical stimuli, and suppressed the expression of target genes. In animal cancer model, siRNA delivery by ultrasmall polymeric carrier (<10 nm) functionalized with targeting ligands demonstrated gene silencing.98 In another example, a DNA nanoparticle (∼28 nm) with defined size, shape, and surface ligands showed how the number and spatial orientation of targeting ligands impacted drug action.105 Varying the proximity of folate ligands influenced gene silencing efficacy in HeLa cells. Animal models employing this delivery system exhibited prolonged blood circulation (∼24 min half-life), selective tumor accumulation, and effective gene silencing. Another DDS where uPICs conjugated onto a gold nanoparticle template (overall size 30–50 nm) showed prolonged blood circulation (∼30 min half-life), significant tumor accumulation, and efficient antitumor effects in animal models.122 Further research is necessary in this field of sub-100 nm DDSs to elucidate the critical role of small size, their physical attributes (such as hard or soft core), and surface properties in accumulating at diseased sites and governing the rate of drug release.
11. Conclusions and outlook
In this review, we have discussed various methodologies devised for the creation of sub-100 nm-sized DDSs. We have provided a brief overview of the advantages of small-sized DDSs and elaborated on why size plays a crucial role in drug delivery applications. A substantial body of experimental evidence suggests that sub-100 nm nanoparticles represent the optimal size for DDSs. Notably, we introduced a unique technique called “template polymerization,” a well-established approach in covalent polymerization, which enables precise control over molecular weight. This approach entails the creation of a monolayer of a polymer around a biomolecule template, resulting in the production of nanoparticles within the range of several tens of nanometers. We have also discussed various examples of sub-100 nm DDSs designed for nucleic acid delivery, where most polymeric carriers are synthesized by utilizing nucleic acids (DNA or RNA) as templates for polymerization, promoting the organization of monomers on the template surface. While other categories of DDSs, such as polymer escorts, polymer shells, and polymeric micelles, do not strictly adhere to the “template polymerization” concept, there are conceptual similarities in their design. For instance, both polymer escort and polymer shell methods involve enveloping a layer of a polymer around nucleic acids, resulting in a very compact DDS.While most research has centered on double-stranded nucleic acids (DNA or RNA) as templates for the “template polymerization” method, single-stranded nucleic acids can also serve as templates, albeit with potential challenges in designing monomers that can pre-organize on their flexible, single chains. Another promising avenue is the utilization of proteins as templates, an area that remains relatively unexplored. We have identified just one example where authors mention protein-templated polymerization. The field holds vast potential due to the wide variety of proteins with cationic/anionic surface charges, allowing for the design of monomers linked to polymerizable units with cationic/anionic properties. Furthermore, the “template polymerization” method proves valuable for post-functionalizing DDSs with minimal size increase and for controlling the number of functionalities.
Templated polymerization techniques may pose challenges. For instance, in the case of a protein template, the surface charge distribution is non-uniform and hence may hinder the uniform polymerization and the stability of the resulting polymer/protein complex. However, the surface charge of a protein can be regulated by introducing suitable amino acids via genetic engineering. Another potential issue is related to template release for therapeutic effects. Endogenous stimuli can cleave a long polymer to an oligomer or a monomer, but the subsequent dissociation of the template from the fragmented polymer is also necessary. To ensure complete release, the interaction between the monomer and the template should be non-covalent, however designing such a monomer for “template polymerization” is challenging in terms of repeating cationic species (multivalency), dispersibility and competitive polymerization both in solution and on the template. Despite the progress made, we believe that the full potential of “template polymerization” in DDSs remains untapped, and we anticipate the development of new molecular designs to address the challenges in the realm of nanomedicine.123
Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. K. H. acknowledges “FY2023 SOUSEI Support Program for Young Researchers” by Hokkaido University and 9th Hokkaido University Interdepartmental Symposium Research Grant Silver Award.Notes and references
- A. Dahan, J. M. Miller and G. L. Amidon, AAPS J., 2009, 11, 740–746 CrossRef CAS PubMed.
- A. E. Nel, L. Mädler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- S. S. Dunn, S. Tian, S. Blake, J. Wang, A. L. Galloway, A. Murphy, P. D. Pohlhaus, J. P. Rolland, M. E. Napier and J. M. DeSimone, J. Am. Chem. Soc., 2012, 134, 7423–7430 CrossRef CAS PubMed.
- K. Ganesan, Y. Wang, F. Gao, Q. Liu, C. Zhang, P. Li, J. Zhang and J. Chen, Pharmaceutics, 2021, 13, 1829 CrossRef CAS PubMed.
- K. Zhang, L. Hao, S. J. Hurst and C. A. Mirkin, J. Am. Chem. Soc., 2012, 134, 16488–16491 CrossRef CAS PubMed.
- G. Tiwari, R. Tiwari, B. Sriwastawa, L. Bhati, S. Pandey, P. Pandey and S. K. Bannerjee, Int. J. Pharm. Investig., 2012, 2, 2–11 CrossRef PubMed.
- K. Paunovska, D. Loughrey and J. E. Dahlman, Nat. Rev. Genet., 2022, 23, 265–280 CrossRef CAS PubMed.
- K. A. High and M. G. Roncarolo, NEJM, 2019, 381, 455–464 CrossRef CAS PubMed.
- M. Kurtoglu and T. J. Lampidis, Mol. Nutr. Food Res., 2009, 53, 68–75 CrossRef CAS PubMed.
- N. Martinho, T. C. B. Santos, H. F. Florindo and L. C. Silva, Front. Physiol., 2018, 9, 1898 CrossRef PubMed.
- Q. Zhu and G. K. E. Scriba, J. Pharm. Biomed. Anal., 2018, 147, 425–438 CrossRef CAS PubMed.
- J. Wang, Y. Li and G. Nie, Nat. Rev. Mater., 2021, 6, 766–783 CrossRef CAS PubMed.
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS PubMed.
- T. Olivier, A. Haslam and V. Prasad, JAMA Network Open, 2021, 4, e2138793 CrossRef PubMed.
- R. S. Riley, C. H. June, R. Langer and M. J. Mitchell, Nat. Rev. Drug Discovery, 2019, 18, 175–196 CrossRef CAS PubMed.
- E. Pérez-Herrero and A. Fernández-Medarde, Eur. J. Pharm. Biopharm., 2015, 93, 52–79 CrossRef PubMed.
- T. M. Allen and P. R. Cullis, Science, 2004, 303, 1818–1822 CrossRef CAS PubMed.
- E. G. Stanzl, B. M. Trantow, J. R. Vargas and P. A. Wender, Acc. Chem. Res., 2013, 46, 2944–2954 CrossRef CAS PubMed.
- R. Mogaki, P. K. Hashim, K. Okuro and T. Aida, Chem. Soc. Rev., 2017, 46, 6480–6491 RSC.
- M. Kawai, H. Higuchi, M. Takeda, Y. Kobayashi and N. Ohuchi, BCR, 2009, 11, R43 CrossRef PubMed.
- D. J. Gary, H. Lee, R. Sharma, J. S. Lee, Y. Kim, Z. Y. Cui, D. Jia, V. D. Bowman, P. R. Chipman, L. Wan, Y. Zou, G. Mao, K. Park, B. S. Herbert, S. F. Konieczny and Y. Y. Won, ACS Nano, 2011, 5, 3493–3505 CrossRef CAS PubMed.
- S. Padhi, N. Hassan, P. Jain, M. Singh, S. Mohapatra and Z. Iqbal, in Advanced Drug Delivery Strategies for Targeting Chronic Inflammatory Lung Diseases, ed. D. K. Chellappan, K. Pabreja and M. Faiyazuddin, Springer Singapore, Singapore, 2022, pp. 281–302 Search PubMed.
- P. Huang, X. Wang, X. Liang, J. Yang, C. Zhang, D. Kong and W. Wang, Acta Biomater., 2019, 85, 1–26 CrossRef CAS PubMed.
- J. Gao, J. M. Karp, R. Langer and N. Joshi, Chem. Mater., 2023, 35, 359–363 CrossRef CAS PubMed.
- J. Dolai, K. Mandal and N. R. Jana, ACS Appl. Nano Mater., 2021, 4, 6471–6496 CrossRef CAS.
- S. D. Perrault, C. Walkey, T. Jennings, H. C. Fischer and W. C. W. Chan, Nano Lett., 2009, 9, 1909–1915 CrossRef CAS PubMed.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- M. M. Schmidt and K. D. Wittrup, Mol. Cancer Ther., 2009, 8, 2861–2871 CrossRef CAS PubMed.
- A. Zielińska, F. Carreiró, A. M. Oliveira, A. Neves, B. Pires, D. N. Venkatesh, A. Durazzo, M. Lucarini, P. Eder, A. M. Silva, A. Santini and E. B. Souto, Molecules, 2020, 25, 3731 CrossRef PubMed.
- C. Ju, R. Mo, J. Xue, L. Zhang, Z. Zhao, L. Xue, Q. Ping and C. Zhang, Angew. Chem., Int. Ed., 2014, 53, 6253–6258 CrossRef CAS PubMed.
- W. Jiang, B. Y. Kim, J. T. Rutka and W. C. Chan, Opin. Drug Delivery, 2007, 4, 621–633 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- O. C. Farokhzad and R. Langer, Adv. Drug Delivery Rev., 2006, 58, 1456–1459 CrossRef CAS PubMed.
- B. Daglar, E. Ozgur, M. E. Corman, L. Uzun and G. B. Demirel, RSC Adv., 2014, 4, 48639 RSC.
- Y. Gao, J. Wang, Y. Wang, Q. Yin, B. Glennon, J. Zhong, J. Ouyang, X. Huang and H. Hao, Curr. Pharm. Des., 2015, 21, 3131–3139 CrossRef CAS PubMed.
- M. Kumar, R. S. Bishnoi, A. K. Shukla and C. P. Jain, Prev. Nutr. Food Sci., 2019, 24, 225–234 CAS.
- R. Jenjob, T. Phakkeeree, F. Seidi, M. Theerasilp and D. Crespy, Macromol. Biosci., 2019, 19, 1900063 CrossRef PubMed.
- C. C. L. Cheung, I. Monaco, N. Kostevšek, M. C. Franchini and W. T. Al-Jamal, Colloids Surf., B, 2021, 198, 111453 CrossRef CAS PubMed.
- S. Soares, J. Sousa, A. Pais and C. Vitorino, Front. Chem., 2018, 6, 360 CrossRef PubMed.
- H. Guerrero-Cázares, S. Y. Tzeng, N. P. Young, A. O. Abutaleb, A. Quiñones-Hinojosa and J. J. Green, ACS Nano, 2014, 8, 5141–5153 CrossRef PubMed.
- N. Uchida, Polym. J., 2023, 55, 829–836 CrossRef CAS PubMed.
- M. Alle, G. Sharma, S.-H. Lee and J.-C. Kim, J. Nanobiotechnol., 2022, 20, 222 CrossRef PubMed.
- B. D. Chithrani, A. A. Ghazani and W. C. W. Chan, Nano Lett., 2006, 6, 662–668 CrossRef CAS PubMed.
- R. Tenchov, R. Bird, A. E. Curtze and Q. Zhou, ACS Nano, 2021, 15, 16982–17015 CrossRef CAS PubMed.
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 16071 CrossRef CAS PubMed.
- P. K. Hashim, J. Bergueiro, E. W. Meijer and T. Aida, Prog. Polym. Sci., 2020, 105, 101250 CrossRef CAS.
- W. B. Liechty, D. R. Kryscio, B. V. Slaughter and N. A. Peppas, Annu. Rev. Chem. Biomol. Eng., 2010, 1, 149–173 CrossRef CAS PubMed.
- Y. K. Sung and S. W. Kim, Biomater. Res., 2020, 24, 12 CrossRef CAS PubMed.
- A. K. Maparu, P. Singh, B. Rai, A. Sharma and S. Sivakumar, Nanotechnology, 2022, 33, 49 CrossRef PubMed.
- A. B. Popov, F. Melle, E. Linnane, C. González-López, I. Ahmed, B. Parshad, C. O. Franck, H. Rahmoune and F. M. Richards, Nanoscale, 2022, 14, 6656–6669 RSC.
- H. Lee, H. Fonge, B. Hoang, R. M. Reilly and C. Allen, Mol. Pharmaceutics, 2010, 7, 1195–1208 CrossRef CAS PubMed.
- H. Cabral, Y. Matsumoto, K. Mizuno, Q. Chen, M. Murakami, M. Kimura, Y. Terada, M. R. Kano, K. Miyazono, M. Uesaka, N. Nishiyama and K. Kataoka, Nat. Nanotechnol., 2011, 6, 815–823 CrossRef CAS PubMed.
- Y. Hua, Y. Su, H. Zhang, N. Liu, Z. Wang, X. Gao, J. Gao and A. Zheng, Drug Delivery, 2021, 28, 1342–1355 CrossRef CAS PubMed.
- S. M. Matalqah, K. Aiedeh, N. M. Mhaidat, K. H. Alzoubi, Y. Bustanji and I. Hamad, Curr. Drug Targets, 2020, 21, 1613–1624 CrossRef CAS PubMed.
- C. M. Paleos, D. Tsiourvas, Z. Sideratou and L. A. Tziveleka, Expert Opin. Drug Delivery, 2010, 7, 1387–1398 CrossRef CAS PubMed.
- V. Korzhikov-Vlakh and T. Tennikova, Adv. Biochem. Eng./Biotechnol., 2021, 178, 99–146 CrossRef CAS PubMed.
- S. Hajebi, N. Rabiee, M. Bagherzadeh, S. Ahmadi, M. Rabiee, H. Roghani-Mamaqani, M. Tahriri, L. Tayebi and M. R. Hamblin, Acta Biomater., 2019, 92, 1–18 CrossRef CAS PubMed.
- M. Molina, M. Asadian-Birjand, J. Balach, J. Bergueiro, E. Miceli and M. Calderón, Chem. Soc. Rev., 2015, 44, 6161–6186 RSC.
- A. Tewabe and A. Abate, J. Multidiscip. Healthcare, 2021, 14, 1711–1724 CrossRef PubMed.
- S. Deng, M. R. Gigliobianco, R. Censi and P. Di Martino, Nanomaterials, 2020, 10, 847 CrossRef CAS PubMed.
- Q. Zhong and H. Zhang, Methods Mol. Biol., 2023, 2622, 49–56 CrossRef CAS PubMed.
- V. Chandrakala, V. Aruna and G. Angajala, Emerg. Mater., 2022, 5, 1593–1615 CAS.
- F. Y. Kong, J. W. Zhang, R. F. Li, Z. X. Wang, W. J. Wang and W. Wang, Molecules, 2017, 22, 1445 CrossRef PubMed.
- C. K. Kim, P. Ghosh and V. M. Rotello, Nanoscale, 2009, 1, 61–67 RSC.
- K. Niikura, N. Iyo, Y. Matsuo, H. Mitomo and K. Ijiro, ACS Appl. Mater. Interfaces, 2013, 5, 3900–3907 CrossRef CAS PubMed.
- A. Kohata, P. K. Hashim, K. Okuro and T. Aida, J. Am. Chem. Soc., 2019, 141, 2862–2866 CrossRef CAS PubMed.
- D. V. Parums, Med. Sci. Monit., 2021, 27, e934625 Search PubMed.
- M. G. Thompson, J. L. Burgess, A. L. Naleway, H. L. Tyner, S. K. Yoon, J. Meece, L. E. W. Olsho, A. J. Caban-Martinez, A. Fowlkes, K. Lutrick, J. L. Kuntz, K. Dunnigan, M. J. Odean, K. T. Hegmann, E. Stefanski, L. J. Edwards, N. Schaefer-Solle, L. Grant, K. Ellingson, H. C. Groom, T. Zunie, M. S. Thiese, L. Ivacic, M. G. Wesley, J. M. Lamberte, X. Sun, M. E. Smith, A. L. Phillips, K. D. Groover, Y. M. Yoo, J. Gerald, R. T. Brown, M. K. Herring, G. Joseph, S. Beitel, T. C. Morrill, J. Mak, P. Rivers, K. M. Harris, D. R. Hunt, M. L. Arvay, P. Kutty, A. M. Fry and M. Gaglani, MMWR, 2021, 70, 495–500 CAS.
- S. Połowiński, Template Polymerization, in Encyclopedia of Polymer Science and Technology, ed. H. F. Mark, 4th edn, Wiley Publishing, 2003, p. 465 Search PubMed.
- R. McHale, J. P. Patterson, P. B. Zetterlund and R. K. O'Reilly, Nat. Chem., 2012, 4, 491–497 CrossRef CAS PubMed.
- G. Pasparakis, N. Krasnogor, L. Cronin, B. G. Davis and C. Alexander, Chem. Soc. Rev., 2010, 39, 286–300 RSC.
- R. E. Kleiner, Y. Brudno, M. E. Birnbaum and D. R. Liu, J. Am. Chem. Soc., 2008, 130, 4646–4659 CrossRef CAS PubMed.
- D. M. Rosenbaum and D. R. Liu, J. Am. Chem. Soc., 2003, 125, 13924–13925 CrossRef CAS PubMed.
- Y.-J. Kim, H. Uyama and S. Kobayashi, Polym. J., 2004, 36, 992–998 CrossRef CAS.
- P. Walde and Z. Guo, Soft Matter, 2011, 7, 316–331 RSC.
- H. T. Van de Grampel, Y. Y. Tan and G. Challa, Macromolecules, 1990, 23, 5209–5216 CrossRef CAS.
- P. Zahedi, M. Ziaee, M. Abdouss, A. Farazin and B. Mizaikoff, Polym. Adv. Technol., 2016, 27, 1124–1142 CrossRef CAS.
- K. Haupt and K. Mosbach, Chem. Rev., 2000, 100, 2495–2504 CrossRef CAS PubMed.
- Z. J. Gartner, B. N. Tse, R. Grubina, J. B. Doyon, T. M. Snyder and D. R. Liu, Science, 2004, 305, 1601–1605 CrossRef CAS PubMed.
- R. K. O’Reilly, A. J. Turberfield and T. R. Wilks, Acc. Chem. Res., 2017, 50, 2496–2509 CrossRef PubMed.
- J. Lin, M. Surin, D. Beljonne, X. Lou, J. L. J. van Dongen and A. P. H. J. Schenning, Chem. Sci., 2012, 3, 2732–2736 RSC.
- J. M. Priegue, D. N. Crisan, J. Martínez-Costas, J. R. Granja, F. Fernandez-Trillo and J. Montenegro, Angew. Chem., Int. Ed., 2016, 55, 7492–7495 CrossRef CAS PubMed.
- J. García Coll and S. Ulrich, ChemBioChem, 2023, 24, e202300333 CrossRef PubMed.
- M. Ouyang, J.-S. Remy and F. C. Szoka, Bioconjugate Chem., 2000, 11, 104–112 CrossRef CAS PubMed.
- D. L. McKenzie, E. Smiley, K. Y. Kwok and K. G. Rice, Bioconjugate Chem., 2000, 11, 901–909 CrossRef CAS PubMed.
- T. Blessing, J.-S. Remy and J.-P. Behr, J. Am. Chem. Soc., 1998, 120, 8519–8520 CrossRef CAS.
- D. Soundara Manickam, H. S. Bisht, L. Wan, G. Mao and D. Oupicky, JCR, 2005, 102, 293–306 CAS.
- V. S. Trubetskoy, V. G. Budker, L. J. Hanson, P. M. Slattum, J. A. Wolff and J. E. Hagstrom, Nucleic Acids Res., 1998, 26, 4178–4185 CrossRef CAS PubMed.
- G. Zuber, L. Zammut-Italiano, E. Dauty and J. P. Behr, Angew. Chem., Int. Ed., 2003, 42, 2666–2669 CrossRef CAS PubMed.
- C. Chittimalla, L. Zammut-Italiano, G. Zuber and J.-P. Behr, J. Am. Chem. Soc., 2005, 127, 11436–11441 CrossRef CAS PubMed.
- A. Sizovs, X. Song, M. N. Waxham, Y. Jia, F. Feng, J. Chen, A. C. Wicker, Y. Yu and J. Wang, J. Am. Chem. Soc., 2014, 136, 234–240 CrossRef CAS PubMed.
- X. Jia, L. Wang and J. Du, Nano Res., 2018, 11, 5028–5048 CrossRef CAS.
- M. Yan, M. Liang, J. Wen, Y. Liu, Y. Lu and I. S. Chen, J. Am. Chem. Soc., 2012, 134, 13542–13545 CrossRef CAS PubMed.
- P. K. Hashim, K. Okuro, S. Sasaki, Y. Hoashi and T. Aida, J. Am. Chem. Soc., 2015, 137, 15608–15611 CrossRef CAS PubMed.
- P. K. Hashim, K. Okuro and T. Aida, JCR, 2017, 259, e101 Search PubMed.
- M. E. Davis, Z. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS PubMed.
- C. Boyer, J. Teo, P. Phillips, R. B. Erlich, S. Sagnella, G. Sharbeen, T. Dwarte, H. T. Duong, D. Goldstein, T. P. Davis, M. Kavallaris and J. McCarroll, Mol. Pharmaceutics, 2013, 10, 2435–2444 CrossRef CAS PubMed.
- C. Dohmen, D. Edinger, T. Fröhlich, L. Schreiner, U. Lächelt, C. Troiber, J. Rädler, P. Hadwiger, H.-P. Vornlocher and E. Wagner, ACS Nano, 2012, 6, 5198–5208 CrossRef CAS PubMed.
- N. Laroui, M. Coste, D. Su, L. M. A. Ali, Y. Bessin, M. Barboiu, M. Gary-Bobo, N. Bettache and S. Ulrich, Angew. Chem., Int. Ed., 2021, 60, 5783–5787 CrossRef CAS PubMed.
- K. Gunasekaran, T. H. Nguyen, H. D. Maynard, T. P. Davis and V. Bulmus, Macromol. Rapid Commun., 2011, 32, 654–659 CrossRef CAS PubMed.
- K. Q. Luo and D. C. Chang, BBRC, 2004, 318, 303–310 CrossRef CAS PubMed.
- S. E. Averick, E. Paredes, S. K. Dey, K. M. Snyder, N. Tapinos, K. Matyjaszewski and S. R. Das, J. Am. Chem. Soc., 2013, 135, 12508–12511 CrossRef CAS PubMed.
- M. Nothisen, M. Kotera, E. Voirin, J.-S. Remy and J.-P. Behr, J. Am. Chem. Soc., 2009, 131, 17730–17731 CrossRef CAS PubMed.
- S. Rudorf and J. O. Rädler, J. Am. Chem. Soc., 2012, 134, 11652–11658 CrossRef CAS PubMed.
- H. Lee, A. K. R. Lytton-Jean, Y. Chen, K. T. Love, A. I. Park, E. D. Karagiannis, A. Sehgal, W. Querbes, C. S. Zurenko, M. Jayaraman, C. G. Peng, K. Charisse, A. Borodovsky, M. Manoharan, J. S. Donahoe, J. Truelove, M. Nahrendorf, R. Langer and D. G. Anderson, Nat. Nanotechnol., 2012, 7, 389–393 CrossRef CAS PubMed.
- A. Harada and K. Kataoka, Science, 1999, 283, 65–67 CrossRef CAS PubMed.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113–131 CrossRef CAS PubMed.
- H. Cabral, K. Miyata, K. Osada and K. Kataoka, Chem. Rev., 2018, 118, 6844–6892 CrossRef CAS PubMed.
- H. J. Kim, Y. Yi, A. Kim and K. Miyata, Biomacromolecules, 2018, 19, 2377–2390 CrossRef CAS PubMed.
- K. Hayashi, H. Chaya, S. Fukushima, S. Watanabe, H. Takemoto, K. Osada, N. Nishiyama, K. Miyata and K. Kataoka, Macromol. Rapid Commun., 2016, 37, 486–493 CrossRef CAS PubMed.
- E. M. Pelegri-O'Day, E. W. Lin and H. D. Maynard, J. Am. Chem. Soc., 2014, 136, 14323–14332 CrossRef PubMed.
- A. Beloqui, S. R. Mane, M. Langer, M. Glassner, D. M. Bauer, L. Fruk, C. Barner-Kowollik and G. Delaittre, Angew. Chem., Int. Ed., 2020, 59, 19951–19955 CrossRef CAS PubMed.
- C. A. Stevens, K. Kaur and H. A. Klok, Adv. Drug Delivery Rev., 2021, 174, 447–460 CrossRef CAS PubMed.
- K. Dutta, D. Hu, B. Zhao, A. E. Ribbe, J. Zhuang and S. Thayumanavan, J. Am. Chem. Soc., 2017, 139, 5676–5679 CrossRef CAS PubMed.
- J. Zhang, Y. Li, F.-F. An, X. Zhang, X. Chen and C.-S. Lee, Nano Lett., 2015, 15, 313–318 CrossRef CAS PubMed.
- D. E. Owens, 3rd and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef PubMed.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 CAS.
- A. Gabizon, R. Catane, B. Uziely, B. Kaufman, T. Safra, R. Cohen, F. Martin, A. Huang and Y. Barenholz, Cancer Res., 1994, 54, 987–992 CAS.
- K. Miyata, N. Nishiyama and K. Kataoka, Chem. Soc. Rev., 2012, 41, 2562–2574 RSC.
- W. Sun and Z. Gu, Expert Opin. Drug Delivery, 2016, 13, 311–314 CrossRef PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- Y. Yi, H. J. Kim, P. Mi, M. Zheng, H. Takemoto, K. Toh, B. S. Kim, K. Hayashi, M. Naito, Y. Matsumoto, K. Miyata and K. Kataoka, JCR, 2016, 244, 247–256 CAS.
- P. K. Hashim and A. Dirisala, Diagnosis and Therapy: Nanomaterials inchemotherapy, in Biotechnology in Cancer, 1st edition, ed. S. Kumar and A. R. Girija, Jenny Standford Publishing, 2022, p. 66 Search PubMed.
| This journal is © The Royal Society of Chemistry 2024 |