
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoluminescence mechanism of red emissive carbon dots from a diaminobenzoic acid isomer†
Yunpeng
Liu
ab,
Haojie
Ding
a,
Shulan
Zhang
a,
Mujing
Qu
a,
Jiaxin
Duan
a,
Henglong
Dai
a and
Huili
Li
 *ab
*ab
aEngineering Research Center for Nanophotonics & Advanced Instrument, Ministry of Education, School of Physics and Electronic Science, East China Normal University, Shanghai 200241, P. R. China. E-mail: hlli@phy.ecnu.edu.cn
bChongqing Key Laboratory of Precision Optics, Chongqing Institute of East China Normal University, Chongqing 401120, P. R. China
First published on 23rd November 2023
Abstract
Red emissive carbon dots (RCDs), as nanoscale luminescent particles, hold great promise for bioimaging and full-color displays. However, the luminescence principle of RCDs remains rather elusive due to the poor understanding of the structure–function relationship. To address this, green to red emissive CDs were synthesized from o-phenylenediamine (OPD) as well as isomers of 2,3-diaminobenzoic acid (23DBA) and 3,4-diaminobenzoic acid (34DBA), respectively, by a simple one-step solvothermal method. The similar conjugated structure and pure composition help to reveal the luminescence mechanism of CDs. Experimental and computational studies confirmed that molecular states should be responsible for the fluorescence of CDs. The emission color variations originated from the different molecular fluorophores. Quinoxalino[2,3-b]phenolizine-2,3-diamine (QXPDA) was dominant for the red emission of OPD-based RCDs. Similarly, for DBA-based RCDs, carboxyl groups can provide an acidic environment and act as a catalyst, and new molecular fluorophores like QXPDAA-1 and QXPDAA-2 with different carboxyl group positions were demonstrated to be formed. Meanwhile, the different positions (para- and ortho-position) of –COOH on QXPDA affected the intramolecular charge transfer (ICT), thereby lowering the photon transition band gap, and further promoting red emission. Finally, transient absorption (TA) spectra confirm that the multiple-peak emission of the DBA-based RCDs originated from the single luminescence center. This study is a potential reference for the controlled synthesis of RCDs from other aromatic precursors.
1. Introduction
Carbon dots (CDs), a class of quasi-spherical nanoparticles, owing to their distinctive optical and electrical properties, provide a new perspective for carbon-based luminescent materials since their accidental discovery in 2004.1–8 In particular, red emissive CDs (RCDs) with low cytotoxicity, weak auto-fluorescence, and deep-tissue penetration have recently shone as promising imaging agents in biomedical applications.9–16 Unfortunately, the controversial photoluminescence (PL) origin still hinders the controllable design and preparation of efficient RCDs.17–21So far, the following three categories of PL mechanisms have been widely accepted: carbon core state, surface state, and molecular state.22–25 Among them, the molecular state has gradually attracted much more attention because some specific molecular fluorophores have been successively discovered in related studies on CDs.26,27 For example, imidazo[1,2-a]pyridine-7-carboxylic acid (IPCA) and 4-hydroxy-1H-pyrrolo[3,4-c]pyridine-1,3,6(2H,5H)-trione (HPPT) had been found in blue and green emissive CDs synthesized from non-conjugated precursor molecules such as citric acid (CA).28–30 These molecular fluorophores aggregated via hydrogen bonding either in ordered crystals or in amorphous forms, and finally the CD like structures and optical properties were obtained.9,31 By contrast, preparation of RCDs with excellent optical properties is usually much more difficult because it often suffers from low PL quantum yield (PLQY), very complicated purification processes, and limited precursors.32,33
According to reports, the precursors consistently used for the synthesis of high-performance RCDs are aromatic compounds, particularly o-phenylenediamine (OPD) because it has a simple conjugated structure and self-doping (i.e., N-doping) behavior.34,35 In general, CDs synthesized with OPD as a single precursor give green-yellow fluorescence. The molecular fluorophore named 2,3-diaminophenazine (DAP) had been demonstrated to be dominant for the green-yellow emission, which further confirmed the luminescence mechanism of the molecular states.19 Subsequently, several research groups successfully synthesized OPD-based RCDs with similar emission peaks by adding low concentrations of strong acidic reagents or other precursors, such as dopamine, ionic liquids and dicyandiamide, in the hydrothermal process.36–39 By performing systematic and complex purification, a large amount of quinoxalino[2,3-b]phenolizine-2,3-diamine (QXPDA) molecular fluorophore was found to be present in the RCDs, accounting for 80% of the total mass. When QXPDA was attached to the surface of the carbon core and aggregated, green and blue emission was observed.9 However, Zhong and coworkers supposed that the DAP fluorophore on the surface of CDs was directly responsible for the molecular state fluorescence of CDs. Its protonation changed the emission of CDs from yellow to red.37 In addition, the Sun group used OPD and catechol (CAT) as precursors to prepare RCDs. The PL mechanism was considered as the molecular state fluorophore of 5,14-dihydroquinoxalino[2,3-b]phenazine (DHQP).35 Table S1 (ESI†) summarizes the synthesis and PL properties of OPD-based RCDs reported in recent years. It can be found that although more and more studies attribute the red luminescence of OPD-based CDs to molecular states, the chemical structure of fluorophores is controversial due to the introduction of too many variables in the synthesis process. Therefore, the simple synthesis of RCDs with high PLQY by using the single molecular structure of the precursor without any other additives is very necessary but still lacking. The implementation of this kind of research will help to disclose the defined chemical structures of RCDs and promote the understanding of the PL mechanism of RCDs.
To this end, two RCDs (named RCDs-1 and RCDs-2) were synthesized from isomers of 2,3-diaminobenzoic acid (23DBA) and 3,4-diaminobenzoic acid (34DBA) by a simple one-step solvothermal method. Diaminobenzoic acid (DBA) isomers were chosen as the precursors due to the following reasons: (1) their conjugated structures with two amino groups are similar to OPD, serving as both carbon and nitrogen sources; (2) the introduction of the carboxyl group can provide an acidic environment which is usually required for synthesizing OPD-based RCDs while avoiding too many variables; (3) many previous reports have found that the fluorescence transfer from blue to red can be achieved by adjusting the surface oxygen-containing functional groups of CDs, especially electron-accepting groups like carboxyl; (4) different positions of carboxyl groups on the benzene ring should affect the luminescence properties of CDs. As a comparison, green-yellow-emissive CDs (GYCDs) were synthesized under the same conditions by using only OPD as a precursor. Based on the results of structural and optical characterization, it is confirmed that PL of DBA-based CDs originates from the conjugated molecular fluorophores. As the main oxidation product of the reactant OPD, molecular fluorophore DAP dominates the green-yellow emission. After acid-assisted catalysis, molecular fluorophore DAP can laterally polymerize to form molecular fluorophore QXPDA. Similarly, for DBA-based RCDs, carboxyl groups can provide an acidic environment and act as a catalyst, and molecular fluorophores like QXPDAA-1 and QXPDAA-2 with carboxyl groups were formed. They give bright red emission. The effect of the position of the carboxyl groups on PL properties further indicates that the luminescence mechanism of DBA-based RCDs should belong to the molecular state.
2. Materials and methods
2.1 Chemicals
OPD (≥99%), 23DBA (≥97%) and 34DBA (≥97%) were purchased from Adamas Reagent, Ltd. Absolute ethyl alcohol (≥99.5%) was purchased from Shanghai Titan Scientific Co., Ltd. CCK-8 was purchased from Dojindo (Kumamoto, Japan). Dulbecco's modified Eagle's medium (DMEM), 4',6-diamidino-2-phenylindole (DAPI) and phosphate-buffered saline (PBS) were obtained from Hyclone (Thermo Fisher Scientific, Waltham, Massachusetts, USA). Trypsin/ethylene diamine tetraacetic acid (EDTA) solution, penicillin and streptomycin, were obtained from Gibco BRL (Carlsbad, CA, USA). Human immortalized epidermal cells (Hacat) and 4Ti were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China).2.2 Synthesis of the CDs
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 mins, and then treated by filtration (0.22 μm mixed cellulose esters filter) and dialysis (24 h, molecular weight cut-off: 500 Da). Finally, purified GYCDs were concentrated and lyophilized for storage.
000 rpm for 10 min again. The time-consuming dialysis process was completely avoided. Finally, the purified products were concentrated and lyophilized for storage, named RCDs-1 and RCDs-2, respectively.
000 rpm for 10 mins, and then treated by filtration (0.22 μm mixed cellulose esters filter) and dialysis (24 h, molecular weight cut-off: 500 Da). Finally, purified GYCDs were concentrated and lyophilized for storage.
000 rpm for 10 min again. The time-consuming dialysis process was completely avoided. Finally, the purified products were concentrated and lyophilized for storage, named RCDs-1 and RCDs-2, respectively.
2.3 Structural and compositional characterization
Transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) images of as-prepared CDs were collected by using a JEOL JEM 2100. The chemical environments, elemental compositions and functional groups of the samples were analyzed by an X-ray photoelectron spectrometer (XPS, Thermo ESCALAB 250XI) and a Fourier transform infrared spectrometer (FTIR, Nicolet, Magna-IR 750). The Raman spectrum was measured by using laser confocal micro-Raman spectroscopy (LabRAM Aramis) with a 532 nm laser beam as the excitation source. Proton nuclear magnetic resonance (1H-NMR) was analyzed by nuclear magnetic resonance spectroscopy (600MHz, Avance III, Germany).2.4 Optical characterization
An ultraviolet/visible (UV/Vis) spectrophotometer (Hitachi U-3900) was adopted to measure the absorption spectra of the CDs. PL spectra and PLQY were measured using a fluorescence spectrophotometer (Horiba Jobin Yvon, FluoroMax-4) containing an integrating sphere unit (Horiba Jobin Yvon, F3029). The PL lifetime was measured using FLS1000 (Edinburgh Instrument). Transient absorption (TA) spectra with a spectral range from 430 to 750 nm were acquired on a TA spectrometer (Helios-EOS Fire, Ultrafast Systems) with a 1 kHz Ti:sapphire amplifier (Astrella, Coherent Inc.). Samples were held in a 2 mm fused silica cuvette. The instrument response function was determined to be B120 fs by measuring solvent responses under the same experimental conditions. All measurements were performed at RT.2.5 Cell cytotoxicity assessment and bioimaging
Cell toxicity test: the toxicity of as-prepared RCDs-1 to 4T1 and Hacat cells was assessed by CCK-8 kit. First, the cells (4T1 or Hacat) were seeded in 96 well plates at the density of 104 cells per well and cultured for 24 h at 37 °C in a humidified atmosphere with 5% CO2. Subsequently, the fresh media containing as-synthesized RCDs-1 with various concentrations from 0 to 100 μg mL−1 were prepared, and the cells were exposed in fresh media for another 24 or 48 h under 37 °C with 5% CO2. Finally, the relative cellular viability was assessed via collecting the absorbance at 520 nm on a microplate reader (Thermo Multiskan MK3).Cell bio-imaging: first, the cells (4T1 or Hacat) were placed in DMEM + 10% FBS and cultured for 12 h (5% CO2 at 37 °C), and then the medium was replaced with fresh DMEM containing 100 μg mL−1 RCDs-1. The cells were allowed to incubate for 4 h. Subsequently, unreacted RCDs-1 was removed by washing with PBS. Finally, an inverted fluorescence microscope (Leica DMI3000B) was applied to observe the fluorescence images of the cells.
2.6 Computational details
The ground-state geometric structures of CDs were fully optimized by density functional theory (DFT) at B3LYP functional with 6-31G(d) basis set. The distribution of frontier molecular orbital was calculated on the optimized molecules. The emission characteristics were simulated via TD-DFT with CAM-B3LYP at 6-31G(d) basis set. All calculations were performed by using the Gaussian 09 program.3. Results and discussion
As shown in Fig. 1a, three types of CDs (GYCDs, RCDs-1, RCDs-2) were obtained by the simple one-step solvothermal treatment of the mixture with the corresponding precursor (OPD, 23DBA, 34DBA) and absolute ethanol. GYCDs were fabricated as a reference. Interestingly, unlike the water- and ethanol-soluble GCDs, RCD-1 and R-CD-2 are highly hydrophobic. RCDs are strongly hydrophobic but can dissolve well in ethanol. Therefore, pure solid-state RCDs can be easily and quickly obtained by precipitation in water and centrifugation. It does not require any additional and complicated purification steps, for example, time-consuming dialysis and column chromatography, can facilitate large-scale production for future commercial applications. RCDs from DBA precursor in ethanol (1 mg L−1) form a transparent red solution under daylight and emit a bright red fluorescence under a 365 nm lamp, whereas YGCDs emit a dull yellow-green light (Fig. 1b). This implies that the introduction of DBA not only achieves a red shift of the emission peak but also improves the luminescence intensity.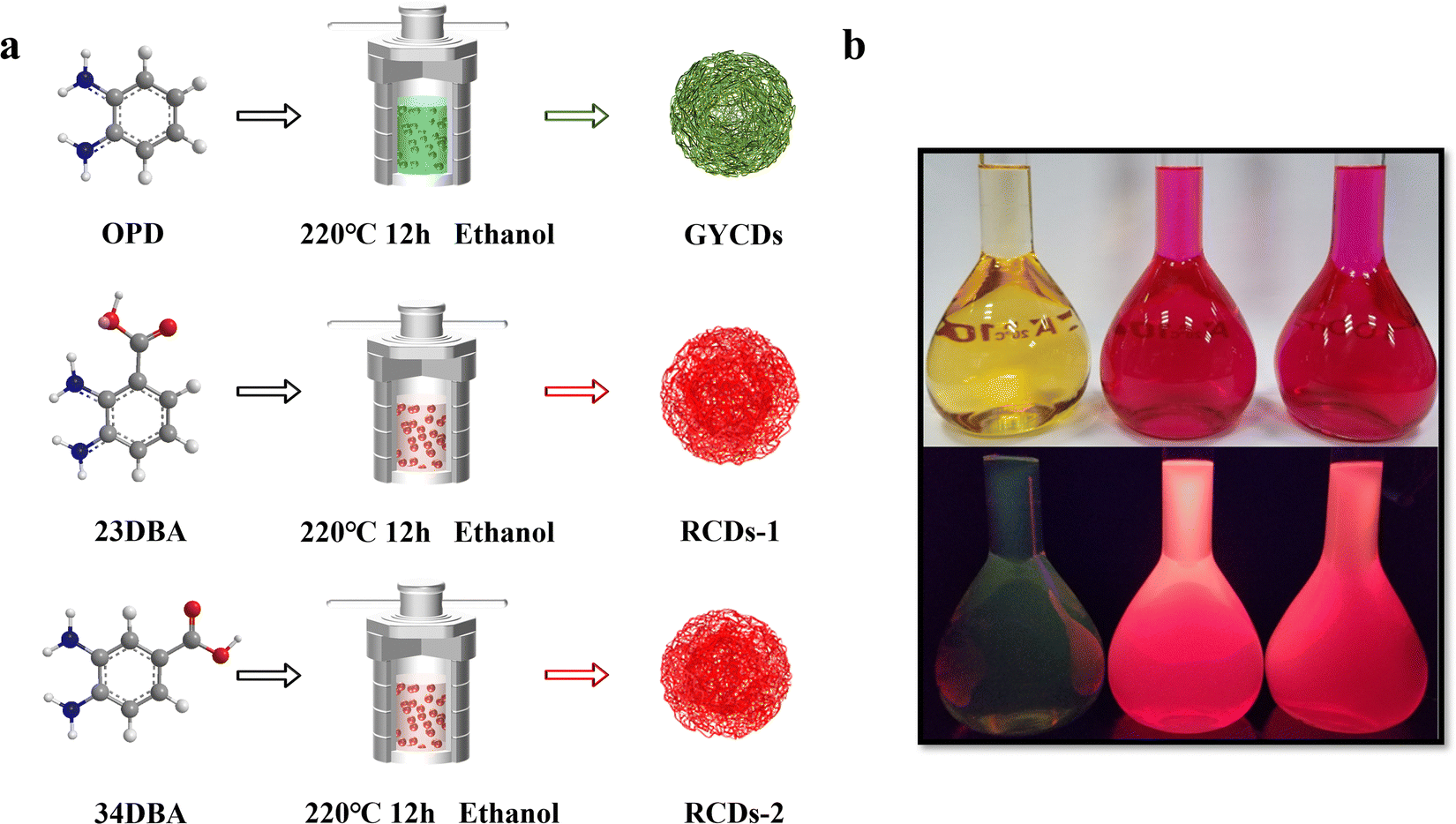 | ||
| Fig. 1 (a) Schematic for the synthesis of GYCDs, RCDs-1, and RCDs-2 by solvothermal treatment of OPD, 23DBA, and 34DBA, respectively. (b) Photographs of GYCDs (left), RCDs-1 (middle), and RCDs-2 (right) diluted in ethanol (1 mg L−1) under daylight (top) and under 365 nm UV light (bottom). | ||
The normalized fluorescence spectrum of GYCDs displays a single-peak emission at 539 nm with a full width at half maximum (FWHM) of 77 nm (Fig. 2a). The shape and position of emission spectra exhibit an excitation-independent characteristic, while the maximum emission intensity is observed at 420 nm excitation, as shown in Fig. S1a (ESI†). Combined with the previous reported literatures,9,19,37 the green-yellow emission peaked at 539 nm from OPD-based CDs is attributed to the DAP molecular fluorophore. It can be seen from Fig. S2 (ESI†) that the emission characteristics irrespective of intensity between GYCDs and DAP are fully consistent under different excitation wavelengths, further validating that DAP is the luminescence origin of GYCDs. The corresponding UV-vis absorption spectrum for OPD-based CDs is categorically separated into three regions (Fig. 2b). The deep-UV absorption bands peaked at 220 and 239 nm are assigned to the n–σ* transitions from C–NH/C–OH. The UV absorption peak at 293 nm is ascribed to the π–π* transitions of the conjugated phenazine ring structure including of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN. The visible absorption band peaked at 433 nm should be from the conjugated structure between the phenazine ring and the –NH2 groups.19 All the absorption bands basically match with the DAP structure (Fig. S3, ESI†).
C and CN. The visible absorption band peaked at 433 nm should be from the conjugated structure between the phenazine ring and the –NH2 groups.19 All the absorption bands basically match with the DAP structure (Fig. S3, ESI†).
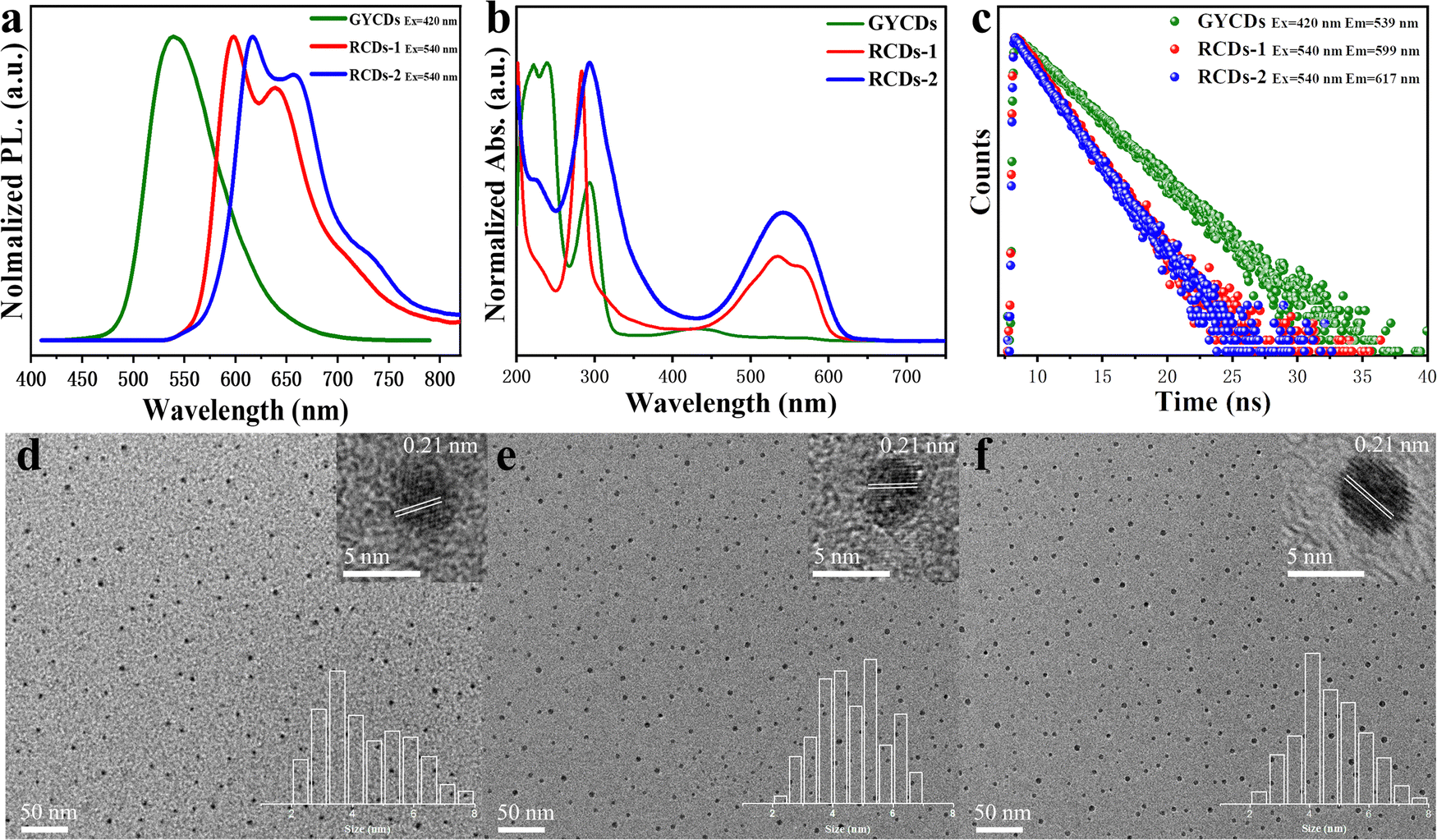 | ||
| Fig. 2 (a) Normalized emission spectra, (b) UV-vis absorption spectra, (c) PL decay curves, TEM images of (d) GYCDs, (e) RCDs-1, and (f) RCDs-2. The insets in the upper and bottom right corner of the TEM image give the corresponding HRTEM and statistical size distributions, respectively. | ||
However, just replacing the OPD precursor with DBA isomers without changing any other reaction conditions, the resultant DBA-based CDs exhibit markedly different multiple emissions from 550 to 800 nm and a large redshift of more than 60 nm. RCDs-1 from 23DBA precursor gives the maximum intensity at 599 nm with the other two shoulders at around 640 and 709 nm. However, RCDs-2 from the 34DBA precursor with a different –COOH group position shows the maximum intensity at 617 nm with the other two shoulders at around 658 and 727 nm. Three emission peaks simultaneously redshift by 18 nm. Obviously, the redshift of 18 nm is attributed to the change in carboxyl group position, which suggests that the luminescence origin of the DBA-based CDs should also belong to the molecular states, and moreover, two molecular fluorophores are isomers. Similar to GYCDs, under different excitation wavelengths, both RCDs exhibit very strong excitation independent fluorescence (Fig. S1b and c, ESI†). Mondal and coworkers reported that the purified QXPDA molecular fluorophore from as-synthesized red emissive CDs owned excitation independent emission with an intensity maximum at around 605 nm along with two progression peaks at around 650 nm and 710 nm.9 Through comparison, it is not difficult to find that the emission characteristics between as-prepared RCDs in our work and the reported molecular fluorophore are almost identical except for the shift of several nanometers. Therefore, it can be inferred that the red fluorescence of DBA-based CDs should come from a kind of molecular fluorophore that has the same structure as QXPDA but contains a carboxyl functional group. Their UV-vis absorption spectra (Fig. 2b) consisted of three peaks which locate at 227 nm, 285 nm, and 534 nm for RCDs-1 and 226 nm, 292 nm, and 542 nm for RCDs-2, respectively. Obviously, the absorption intensities of RCDs in UV and visible regions are higher than that of GYCDs, implying a larger conjugated structure and much more –NH2 and –COOH groups in RCDs.29 The resultant GYCDs, RCDs-1, and RCDs-2 in ethanol under the corresponding optimum excitation wavelength of 420 nm, 540 nm, and 540 nm exhibit the absolute PLQY of 8.4%, 33.7%, and 34.4%, respectively. What is particularly exciting is that the efficiencies of RCDs are almost identical and much higher than GYCDs, which is rather unconventional. All these spectral features indicate that the function of carboxyl groups is completely identical to the added strong acids such as sulfuric acid or nitric acid during the hydrothermal synthesis of OPD-based RCDs. It can provide an acidic environment to promote the red emission of CDs and simultaneously avoid the introduction of other impurity ions. Moreover, carboxyl groups can form hydrogen bonds to enhance the rigidity of aromatic domains, thereby suppressing energy losses from nonradiative vibrational and rotational relaxation processes. Finally, the PLQY of RCDs is improved markedly.40
The steady state PL lifetimes of three CDs were measured by a single photon counting method and are shown in Fig. 2c. Three CDs in ethanol solution have similar single-exponential decay curves. The calculated lifetime is about 4.2, 2.4, and 2.3 ns for GYCDs, RCDs-1, and RCDs-2, respectively, implying the singlet state nature of PL emission and only one radiative transition channel. It should be pointed out that the lifetimes of 4.2 ns for GYCDs and 2.3–2.4 ns for RCDs are very close to 4.64 ns of the reported DAP and 2.15 ns of QXPDA molecular fluorophores.9,19 Therefore, we can further speculate that molecular fluorophore is responsible for the fluorescence of CDs. TEM images reveal that as-prepared three CDs are apparently spherical and uniformly dispersed with the average particle size of 4.39 ± 0.28, 4.63 ± 0.38, and 4.71 ± 0.35 nm for GYCDs, RCDs-1, and RCDs-2, respectively (Fig. 2d–f). The HRTEM images demonstrate the high crystallinity with a lattice spacing of 0.21 nm corresponding to the (100) plane of graphene along the [001] direction (upper right insets in Fig. 2d–f).41 In fact, there is growing evidence that the crystallinity and size of CDs do not significantly correlate with their luminescent properties. The ordered arrangement of some organic molecules even results in the formation of crystalline structures.9,17,26,40,42,43
The above observations motivate us to carry out a systematic analysis of the chemical structures and compositions of as-prepared RCDs. FTIR and XPS spectra were characterized and given in Fig. 3. As a comparison, the results of OPD-based GYCDs are also presented simultaneously. Similar to the previous reports concerning the fluorescent molecule of QXPDA, no obvious G/D bands appear in Raman spectroscopic data for each sample, which are typically observed in graphitic CDs (Fig. 3a).9 Rather, a few sharp transitions at 1219–1231 cm−1, 1365–1380 cm−1 and 1458–1470 cm−1 attributed to N–H bending, C–N and CN stretching, respectively, are observed. These data demonstrate that the dots observed under the TEM should be an aggregated structure of the organic compound. The higher intensities of the C–N and CN stretching peaks for RCDs-1 and RCDs-2 compared to GYCDs imply that they consist of a larger N-containing sp2 hybridized structure. On the other hand, all three CDs fractions display four similar characteristic FTIR regions (Fig. 3b) at 3448–3346 cm−1 (N–H stretching from primary amine), 1628–1585 cm−1 (CN stretching), 1522-1455 cm−1 (CC stretching from aromatic ring), and 1318–1201 cm−1 (C–N stretching), which matches well with Raman data. While, unlike GYCDs, RCDs exhibit the evident carboxylic vibrations (CO) at 1705 and 1710 cm−1, indicating that the carboxyl groups in DBA precursor are retained at different positions in RCDs-1 and RCDs-2. The XPS survey spectra in Fig. 3c show that all three CDs contain only three elements of C (284.6-284.8 eV), N (398.5–399.9 eV), and O (531.6–533.2 eV). As expected, RCDs have a higher O content than GYCDs, which should relate to the carboxyl groups in the DBA precursor. In the high-resolution C1s spectrum of GYCDs, signals of C–C/CC groups at 284.6 eV and C–N/CN groups at 286.2 eV are observed (Fig. 3d). A distinct CO bonding peak does not appear due to the lowest 1.2 at% O element. While the C1s spectra of RCDs-1 and RCDs-2 are deconvoluted into three components, corresponding to C–C/CC groups at 284.7–284.9 eV, C–N/CN groups at 286.0–286.3 eV, and CO groups at 288.6–288.7 eV, further confirming the presence of carboxyl groups (Fig. 3e and f). At the same time, the O 1s high-resolution spectra of RCDs-1 and RCDs-2 are deconvoluted into two peaks of CO (531.6 eV) and C–O (533.2 eV), respectively (Fig. S4, ESI†). The high-resolution N1s spectra of three CDs are the sum of two similar sublevels at 398.5 and 399.6 eV corresponding to pyridinic nitrogen and amino, respectively (Fig. 3g–i). Furthermore, three CDs were investigated by 1H NMR, as shown in Fig. S5 (ESI†). Specifically, in 1H NMR spectra, the chemical shifts at 6.32–8.85 ppm were almost the same as DAP.19 Among them, 7.36–8.85 ppm were ascribed to the vibration of H in the benzene ring, and 7.10–7.25 ppm belonged to the vibration of H in the –NH2. Compared with the 1H NMR spectrum of GYCDs, the 1H NMR spectra of both RCDs display the additional chemical shifts at 9.62–10.13 ppm, implying that the conjugation of RCDs-1 and RCDs-2 might be longer.35 It should be noted that it is difficult to guarantee the absolute purity of collected luminophores with the present simple purification process, so the characterization results can only be qualitative rather than quantitative analysis. Finally, the above results can be summarized in the following four aspects: (1) the amino groups in the precursors do not form graphitized N, but tend to form conjugated molecular fluorophores via CN; (2) the red fluorescence from DBA-based CDs should be attributed to molecular fluorophore; (3) the composition of the red-emissive molecular fluorophores is similar to the yellow-emissive DAP molecular fluorophore, but the conjugated structure may be longer; (4) red-emissive molecular fluorophores synthesized from 23DBA acid and 34DBA could still be isomer with different –COOH position.
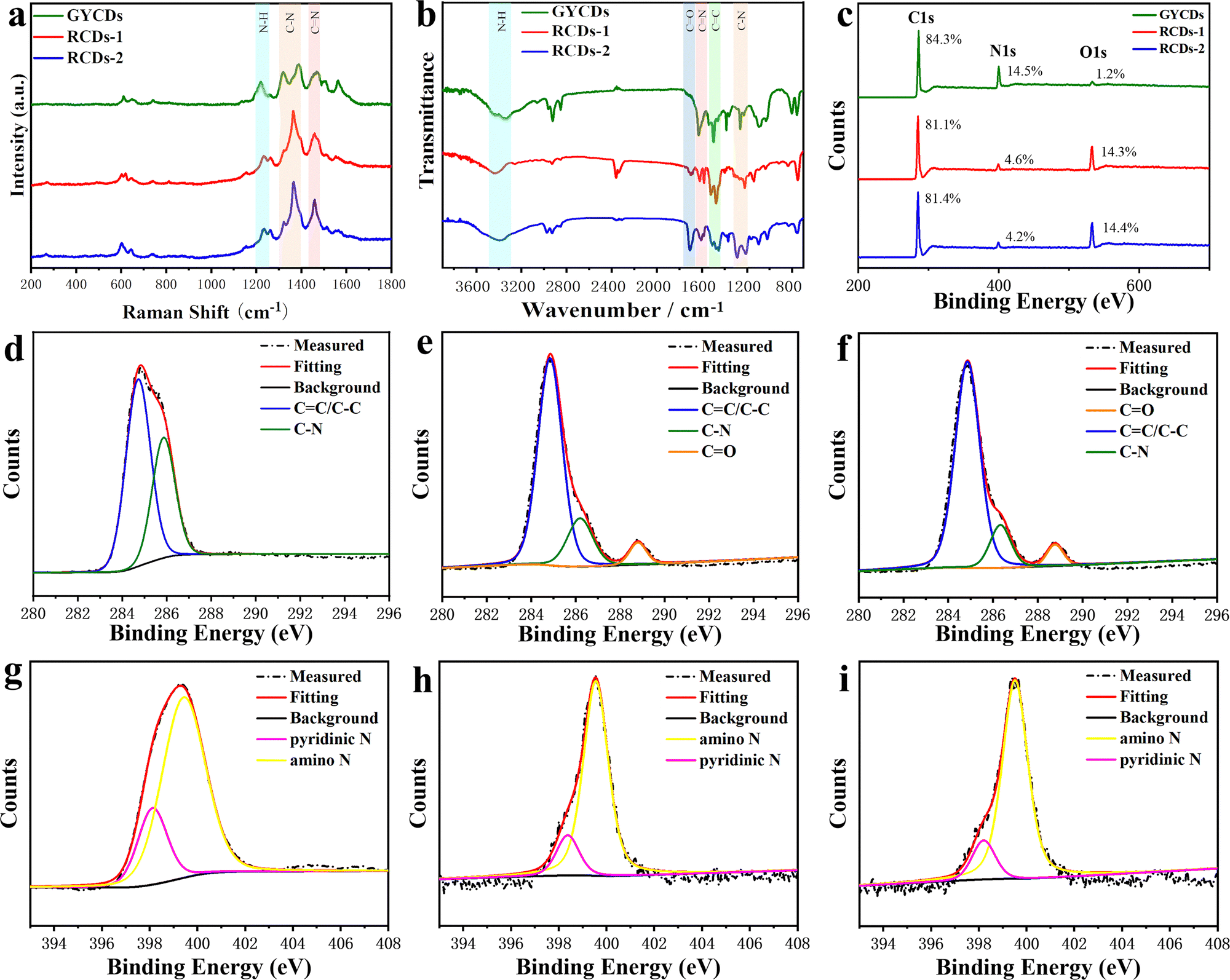 | ||
| Fig. 3 (a) Raman, (b) FTIR, and (c) XPS spectra of three CDs; high-resolution C1s spectra of (d) GYCDs, (e) RCDs-1, and (f) RCDs-2; high-resolution N1s spectra of (g) GYCDs, (h) RCDs-1, and (i) RCDs-2. | ||
Based on the above structural analysis and previous reports, we propose a possible formation mechanism for DBA-based RCDs, as shown in Fig. 4a. When OPD is used as a precursor, two OPD molecules are condensed together under oxidizing conditions to generate yellow-green-emissive DAP molecular fluorophore. After the addition of sulfuric acid or nitric acid during the hydrothermal process, OPD and DAP were further polymerized horizontally to form a molecular fluorophore of QXPDA which is responsible for the red emission of OPD-based RCDs.32 In our work, the OPD precursor is replaced by DBA, the introduced –COOH groups act as an acid-assisted catalyst and simultaneously locate at different positions of polymerized QXPDA to form new molecular fluorophores. We named them QXPDAA-1 and QXPDAA-2, respectively. Molecular fluorophores could be further cross-linked and polymerized together through hydrogen bonding to form spherical rigid structures with crystalline or non-crystalline properties.40 We seek to explore the PL mechanism of these molecular fluorophores through numerous DFT calculations. The geometrically optimized structures as well as the LUMO and HOMO distributions of the four molecular fluorophores are shown in Fig. 4b. It can be seen that the calculated band gaps and corresponding maximum emission peaks for DAP (λem = 531 nm), QXPDAA-1 (λem = 598 nm) and QXPDAA-2 (λem = 605 nm) are basically consistent with the experimentally synthesized GYCDs (λem = 539 nm), RCDs-1 (λem = 599 nm) and RCDs-2 (λem = 617 nm), demonstrating that the fluorescence emissions of as-prepared DBA-based RCDs should originate from these molecular fluorophores. The calculated bandgaps of the four molecular fluorophores by DFT are 3.55, 2.33, 2.28, and 2.26 eV, respectively. The energy gaps of QXPDA, QXPDAA-1 and QXPDAA-2 are much lower than DAP, indicating that increasing the conjugate length of fluorescent molecules can effectively reduce the energy gap to promote the redshift of emission. In addition, the introduction of electron accepting groups (-COOH) can also reduce the energy gap of molecular fluorophores to a small extent. The energy gap of QXPDAA-2 with para-carboxyl group is smaller than that of QXPDAA-1 with ortho-carboxyl group, which means that the further the spatial distance between the carboxyl group and the amino group, the lower the energy gap of the molecular fluorophores. For the HOMO and LUMO orbitals of DAP and QXPDA, the electron clouds are mainly located on the amino groups and the whole π-conjugated skeleton structure. For QXPDAA-1 and QXPDAA-2, the HOMO orbitals are mainly clustered on the whole π-conjugated skeleton structure and around the amino groups, while a part of the electron clouds of LUMO orbitals tend to be around the carboxyl groups with typical induced intermolecular charge transfer (ICT) characteristics.44,45 The introduction of –COOH allows QXPDAA-1 and QXPDAA-2 to form the so-called D–π–A structure, which induces stronger ICT from the donor (D) to the acceptor (A) via π-bridges (π) upon photoexcitation. ICT mechanisms have recently been studied and identified as the main origin of the attractive optical properties observed in organic dye-based D–π–A systems.12
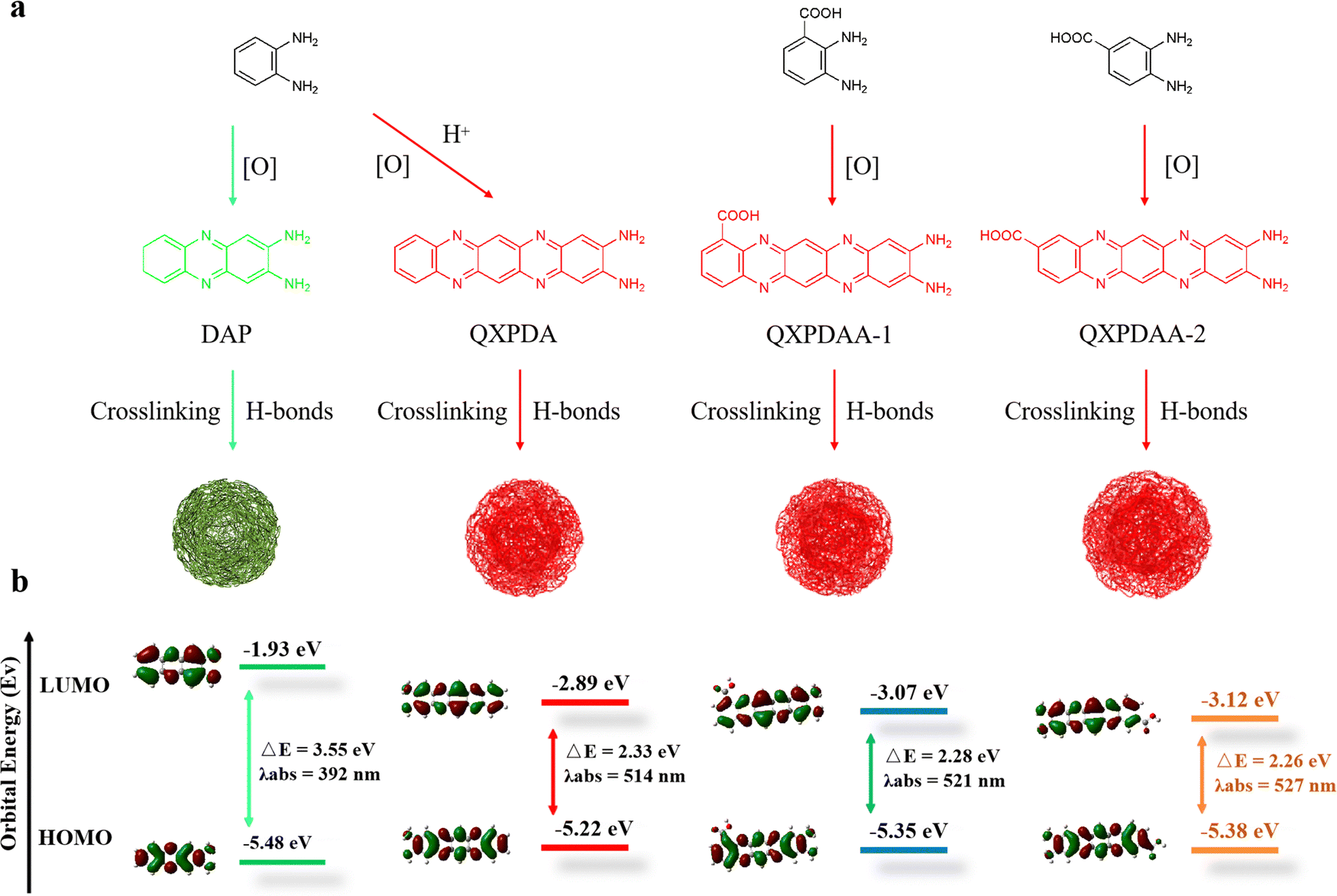 | ||
| Fig. 4 (a) Schematic diagram of the preparation of the proposed DAP, QXPDA, QXPDAA-1 and QXPDAA-2. (b) Theoretical calculations of DAP, QXPDA, QXPDAA-1 and QXPDAA-2 at the (TD-DFT) B3LYP/6-31G (d, p) level. HOMO: the highest occupied molecular orbital, LUMO: the lowest unoccupied molecular orbital. | ||
To further scrutinize the difference in the transient carrier dynamics in three CDs, femtosecond TA spectroscopy measurements were carried out at 430 nm and 500 nm excitation for GYCDs and RCDs, respectively. The obtained results are presented in Fig. 5. The strong negative (blue) features correspond to ground state bleaching (GSB) and stimulated emission (SE), while the weak positive (red) signal corresponds to the excited state absorption (ESA).46 GYCDs exhibit a single ESA peaked at 480 nm, and the SE signal is too weak to be clearly observed (Fig. 5a). As shown in Fig. 5b and c, there are two similar contributions to the TA signals of RCDs-1 and RCDs-2. One is ESA centered around 480 nm and the other is SE band ranging from 580 to 730 nm, which is consistent with the steady-state PL spectra. No GSB is found in the TA data for all three CDs, possibly due to the spectral overlap with the more intense ESA.47 This speculation can be confirmed by the very faint blue signals centered at 450 nm of Fig. 5a and 450, 530 nm of Fig. 5b and c. To clarify the internal energy transfer process, the kinetic traces at different wavelengths as a function of delay time are displayed in Fig. 5d–f. Each SE decay curve for RCDs is fitted by a triple-exponential function with time constants 10–30 ps, 100–300 ps and a few nanoseconds (Table S2, ESI†). After excitation at 500 nm, the remaining hot carriers in the RCDs are scattered by optical phonons (the fast component τ1) and acoustic phonons (the second faster component τ2) to the lowest excited state, which belongs to ICT.48τ2 of RCDs-2 is lower than RCDs-1, implying a fast ICT for RCDs-2. Apparently, the long component (τ3) is attributed to the fluorescent radiation process via electron and hole recombination from the lowest excited state to the ground state. This value is already very close to the steady state PL lifetime in Fig. 2c, suggesting that the radiation recombination process dominates the lifetime of RCDs. The similar kinetic traces at different wavelengths indicate that the carriers go through the same relaxation channel and there exists the single PL center for RCDs-1 and RCDs-2.
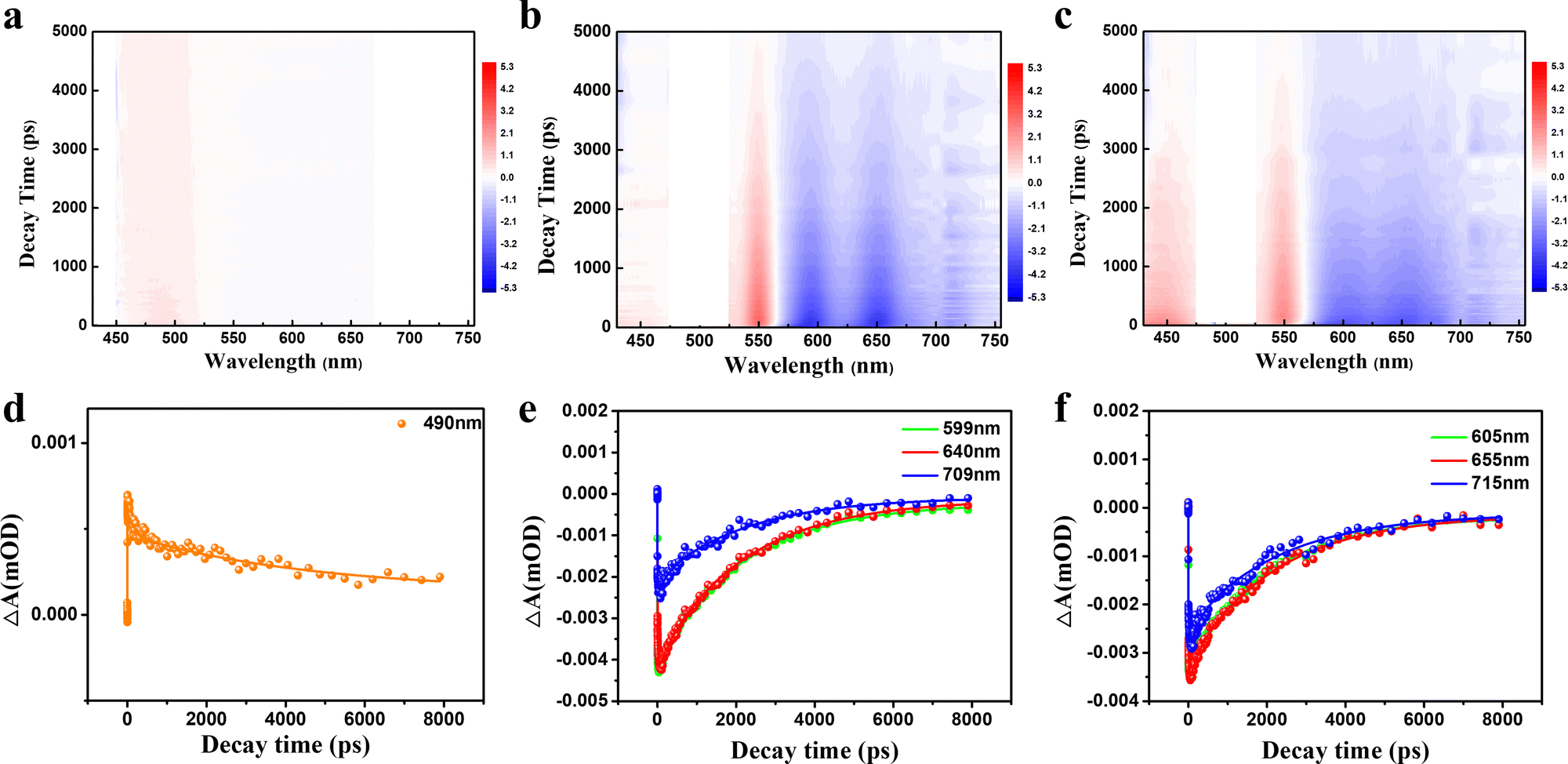 | ||
| Fig. 5 Broadband TA spectra of (a) GYCDs under 430 nm excitation, (b) RCDs-1 and (c) RCDs-2 excited by 500 nm; bleach signal kinetic traces and corresponding fitting curves of (d) GYCDs, (e) RCDs-1, and (f) RCDs-2 at different probe wavelengths. | ||
Finally, we examined the biocompatibility of as-prepared RCDs for bioimaging applications. Due to the extremely similar optical properties, RCDs-1 was chosen as a representative. Cytotoxicity experiments were performed by culturing 4T1 and Hacat cells in RCDs-1 solution. CCK-8 assay in Fig. 6a shows that even at a high dose of 100 μg mL−1 for RCDs-1 solution, the cell viability of more than 90.8% is still maintained after incubation for 48 h, indicative of a little toxicity and good biocompatibility of as-prepared RCDs. Confocal fluorescence images of the biological samples were recorded and are shown in Fig. 6b and c. Images of 4T1, Hacat cells, and fluorescence signals from CDs blend well with each other. All the data suggest that the newly synthesized RCDs can find potential bioimaging applications with very high efficiency.
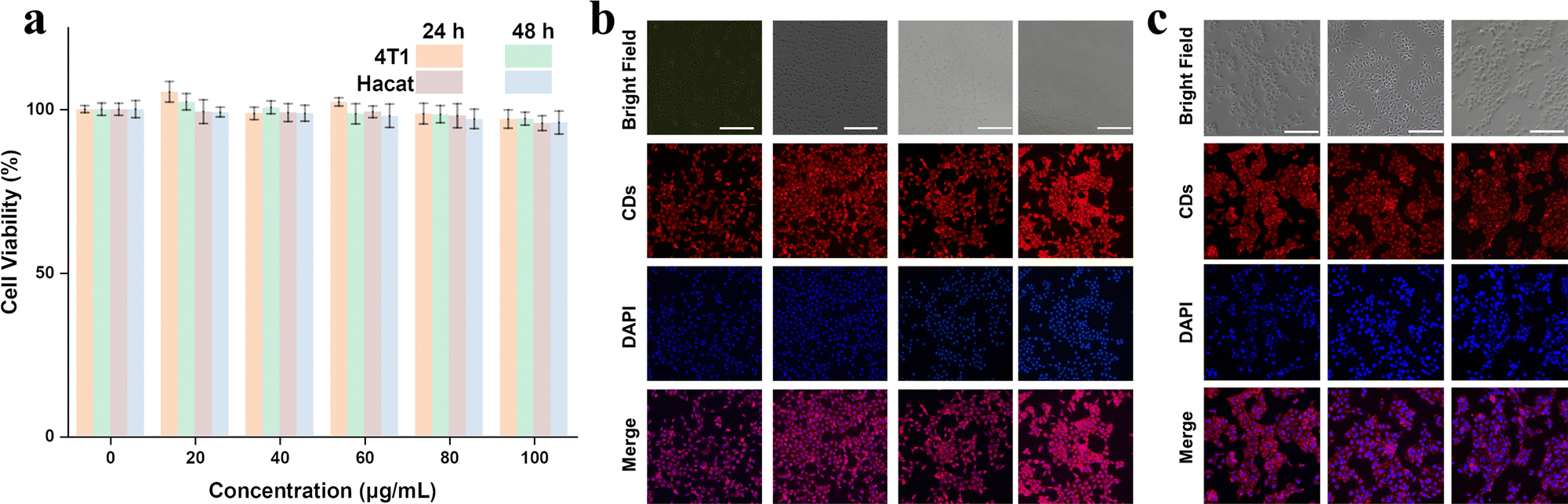 | ||
| Fig. 6 (a) The effect of the RCDs-1 concentration on cell viability and confocal images of (b) 4T1 cells and (c) Hacat cells using 100 μg mL−1 RCDs-1 as a red-emitting marker. | ||
4. Conclusions
In summary, for the first time, red-emissive CDs were synthesized from the single DBA precursor without any other additions by a simple one-step solvothermal method in absolute ethyl alcohol solution. The as-prepared RCDs are strongly hydrophobic but can dissolve well in ethanol. The detailed investigations reveal that red fluorescence actually originates from molecular fluorophores, QXPDAA-1 and QXPDAA-2, which are isomers containing –COOH at different positions on the structure of the QXPDA molecular fluorophore. The carboxyl groups provide an acidic environment to promote the formation of molecular fluorophores QXPDAA-1 and QXPDAA-2, and simultaneously avoid the introduction of other impurity ions. The theoretical calculations based on DFT further confirm the origin of red emission from QXPDAA-1 and QXPDAA-2. The different positions of the carboxyl group in the OXPDA molecule can cause an emission shift of about 18 nm. A high PLQY of 34.4% was achieved. Furthermore, the as-prepared RCDs are highly efficient for potential bioimaging applications. The obtained results in this work provide further evidence for disclosing the PL mechanism of CDs and guide the future development of high-performance RCDs to some extent.Author contributions
This manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 12274136), the Shanghai Municipal Natural Science Foundation (No. 19ZR1415400, 18ZR1411000 and 18ZR1411900), the Chongqing Municipal Natural Science Foundation (No. 33606015, the Chongqing Key Laboratory of Precision Optics, Chongqing Institute of East China Normal University, Chongqing 401120, China), and the Joint Institute of Advanced Science and Technology, East China Normal University, Shanghai 200062, People's Republic of China (No. 40500-20105-222053).References
- Q. Zhang, F. Wang, R. Wang, J. Liu, Y. Ma, X. Qin and X. Zhong, Adv. Sci., 2023, 10, 2207566 CrossRef CAS PubMed.
- F. Yuan, Y.-K. Wang, G. Sharma, Y. Dong, X. Zheng, P. Li, A. Johnston, G. Bappi, J. Z. Fan, H. Kung, B. Chen, M. I. Saidaminov, K. Singh, O. Voznyy, O. M. Bakr, Z.-H. Lu and E. H. Sargent, Nat. Photonics, 2019, 14, 171–176 CrossRef.
- B. Wang, H. Wang, Y. Hu, G. I. N. Waterhouse and S. Lu, Nano Lett., 2023, 23, 8794–8800 CrossRef CAS.
- P. Innocenzi and L. Stagi, Nano Today, 2023, 50, 101837 CrossRef CAS.
- Y. Shi, W. Su, F. Yuan, T. Yuan, X. Song, Y. Han, S. Wei, Y. Zhang, Y. Li, X. Li and L. Fan, Adv. Mater., 2023, 35, 2210699 CrossRef CAS PubMed.
- H. Bai, X. Jin, Z. Cheng, H. Zhou, H. Wang, J. Yu, J. Zuo and W. Chen, Adv. Compos. Hybrid Mater., 2023, 6, 62 CrossRef CAS.
- H. Wang, L. Ai, H. Song, Z. Song, X. Yong, S. Qu and S. Lu, Adv. Funct. Mater., 2023, 33, 2303756 CrossRef CAS.
- C. Ji, W. Xu, Q. Han, T. Zhao, J. Deng and Z. Peng, Nano Energy, 2023, 114, 108623 CrossRef CAS.
- N. Soni, S. Singh, S. Sharma, G. Batra, K. Kaushik, C. Rao, N. C. Verma, B. Mondal, A. Yadav and C. K. Nandi, Chem. Sci., 2021, 12, 3615–3626 RSC.
- L. Tang, L. Ai, Z. Song, L. Sui, J. Yu, X. Yang, H. Song, B. Zhang, Y. Hu, Y. Zhang, Y. Tian and S. Lu, Adv. Funct. Mater., 2023, 33, 2303363 CrossRef CAS.
- H. Ding, X.-X. Zhou, J.-S. Wei, X.-B. Li, B.-T. Qin, X.-B. Chen and H.-M. Xiong, Carbon, 2020, 167, 322–344 CrossRef CAS.
- H. Jia, Z. Wang, T. Yuan, F. Yuan, X. Li, Y. Li, Z. A. Tan, L. Fan and S. Yang, Adv. Sci., 2019, 6, 1900397 CrossRef PubMed.
- K. Yuan, X. Zhang, X. Li, R. Qin, Y. Cheng, L. Li, X. Yang, X. Yu, Z. Lu and H. Liu, Chem. Eng. J., 2020, 397, 125487 CrossRef CAS.
- H. Sun, P. Xia, H. Shao, R. Zhang, C. Lu, S. Xu and C. Wang, J. Colloid Interface Sci., 2023, 646, 932–939 CrossRef CAS.
- T. Liu, G. Yin, Z. Song, J. Yu, X. Yong, B. Zhang, L. Ai and S. Lu, ACS Mater. Lett., 2023, 5, 846–853 CrossRef CAS.
- Y. Shi, Z. Wang, T. Meng, T. Yuan, R. Ni, Y. Li, X. Li, Y. Zhang, Z. A. Tan, S. Lei and L. Fan, J. Am. Chem. Soc., 2021, 143, 18941–18951 CrossRef CAS PubMed.
- C. J. Reckmeier, J. Schneider, Y. Xiong, J. Häusler, P. Kasák, W. Schnick and A. L. Rogach, Chem. Mater., 2017, 29, 10352–10361 CrossRef CAS.
- M. Alafeef, I. Srivastava, T. Aditya and D. Pan, Small, 2023 DOI:10.1002/smll.202303937.
- L. Cao, M. Zan, F. Chen, X. Kou, Y. Liu, P. Wang, Q. Mei, Z. Hou, W.-F. Dong and L. Li, Carbon, 2022, 194, 42–51 CrossRef CAS.
- S. Xue, P. Li, L. Sun, L. An, D. Qu, X. Wang and Z. Sun, Small, 2023, 19, 2206180 CrossRef CAS PubMed.
- X. Yang, L. Ai, J. Yu, G. I. N. Waterhouse, L. Sui, J. Ding, B. Zhang, X. Yong and S. Lu, Sci. Bull., 2022, 67, 1450–1457 CrossRef CAS.
- A. Sharma, T. Gadly, S. Neogy, S. K. Ghosh and M. Kumbhakar, J. Phys. Chem. Lett., 2017, 8, 1044–1052 CrossRef CAS PubMed.
- N. V. Tepliakov, E. V. Kundelev, P. D. Khavlyuk, Y. Xiong, M. Y. Leonov, W. Zhu, A. V. Baranov, A. V. Fedorov, A. L. Rogach and I. D. Rukhlenko, ACS Nano, 2019, 13, 10737–10744 CrossRef CAS.
- Y. Wang, Y. Liu, X. Hao, X. Zhou, H. Peng, Z. Shen, I. I. Smalyukh, X. Xie and B. Yang, Adv. Mater., 2023, 35, 2303680 CrossRef CAS.
- B. Zhao, H. Ma, H. Jia, M. Zheng, K. Xu, R. Yu, S. Qu and Z. A. Tan, Angew. Chem., Int. Ed., 2023, 62, e202301651 CrossRef CAS PubMed.
- V. Strauss, H. Wang, S. Delacroix, M. Ledendecker and P. Wessig, Chem. Sci., 2020, 11, 8256–8266 RSC.
- F. Ehrat, S. Bhattacharyya, J. Schneider, A. Löf, R. Wyrwich, A. L. Rogach, J. K. Stolarczyk, A. S. Urban and J. Feldmann, Nano Lett., 2017, 17, 7710–7716 CrossRef CAS.
- A. Sciortino, F. Ferrante, N. Mauro, G. Buscarino, L. Sciortino, G. Giammona, M. Cannas, D. Duca and F. Messina, Carbon, 2021, 173, 454–461 CrossRef CAS.
- M. Righetto, F. Carraro, A. Privitera, G. Marafon, A. Moretto and C. Ferrante, J. Phys. Chem. C, 2020, 124, 22314–22320 CrossRef CAS.
- Y. Wang, Y. Li and L. Feng, J. Phys. Chem. Lett., 2020, 11, 10439–10445 CrossRef CAS.
- B. Zhi, X. Yao, M. Wu, A. Mensch, Y. Cui, J. Deng, J. J. Duchimaza-Heredia, K. J. Trerayapiwat, T. Niehaus, Y. Nishimoto, B. P. Frank, Y. Zhang, R. E. Lewis, E. A. Kappel, R. J. Hamers, H. D. Fairbrother, G. Orr, C. J. Murphy, Q. Cui and C. L. Haynes, Chem. Sci., 2021, 12, 2441–2455 RSC.
- C. Ji, Q. Han, Y. Zhou, J. Wu, W. Shi, L. Gao, R. M. Leblanc and Z. Peng, Carbon, 2022, 192, 198–208 CrossRef CAS.
- Z. Zhu, Y. Zhai, Z. Li, P. Zhu, S. Mao, C. Zhu, D. Du, L. A. Belfiore, J. Tang and Y. Lin, Mater. Today, 2019, 30, 52–79 CrossRef CAS.
- B. Wang, Z. Wei, L. Sui, J. Yu, B. Zhang, X. Wang, S. Feng, H. Song, X. Yong, Y. Tian, B. Yang and S. Lu, Light: Sci. Appl., 2022, 11, 172 CrossRef CAS PubMed.
- P. Li, S. Xue, L. Sun, X. Zong, L. An, D. Qu, X. Wang and Z. Sun, Light: Sci. Appl., 2022, 11, 298 CrossRef CAS PubMed.
- Z. Yan, T. Chen, L. Yan, X. Liu, J. Zheng, F. D. Ren, Y. Yang, B. Liu, X. Liu and B. Xu, Adv. Sci., 2023, 10, 2206386 CrossRef CAS PubMed.
- Q. Zhang, R. Wang, B. Feng, X. Zhong and K. Ostrikov, Nat. Commun., 2021, 12, 6856 CrossRef CAS PubMed.
- W. Xu, Q. Han, C. Ji, F. Zeng, X. Zhang, J. Deng, C. Shi and Z. Peng, Small, 2023 DOI:10.1002/smll.202304123.
- Y. Ma, L. Wu, X. Ren, Y. Zhang and S. Lu, Adv. Funct. Mater., 2023 DOI:10.1002/adfm.202305867.
- L. Vallan, E. P. Urriolabeitia, F. Ruipérez, J. M. Matxain, R. Canton-Vitoria, N. Tagmatarchis, A. M. Benito and W. K. Maser, J. Am. Chem. Soc., 2018, 140, 12862–12869 CrossRef CAS PubMed.
- L. Jiang, H. Ding, M. Xu, X. Hu, S. Li, M. Zhang, Q. Zhang, Q. Wang, S. Lu, Y. Tian and H. Bi, Small, 2020, 16, 2000680 CrossRef CAS.
- B. Bartolomei, A. Bogo, F. Amato, G. Ragazzon and M. Prato, Angew. Chem., Int. Ed., 2022, 61, e202200038 CrossRef CAS.
- M. Shamsipur, A. Barati, A. A. Taherpour and M. Jamshidi, J. Phys. Chem. Lett., 2018, 9, 4189–4198 CrossRef CAS PubMed.
- I. Srivastava, J. S. Khamo, S. Pandit, P. Fathi, X. Huang, A. Cao, R. T. Haasch, S. Nie, K. Zhang and D. Pan, Adv. Funct. Mater., 2019, 29, 1902466 CrossRef.
- H. A. Nguyen, I. Srivastava, D. Pan and M. Gruebele, ACS Nano, 2020, 14, 6127–6137 CrossRef CAS.
- Y. Liu, J. H. Lei, G. Wang, Z. Zhang, J. Wu, B. Zhang, H. Zhang, E. Liu, L. Wang, T. M. Liu, G. Xing, D. Ouyang, C. X. Deng, Z. Tang and S. Qu, Adv. Sci., 2022, 9, 2202283 CrossRef CAS.
- C. Liu, L. Bao, M. Yang, S. Zhang, M. Zhou, B. Tang, B. Wang, Y. Liu, Z.-L. Zhang, B. Zhang and D.-W. Pang, J. Phys. Chem. Lett., 2019, 10, 3621–3629 CrossRef CAS PubMed.
- M. Imran, A. M. El-Zohry, C. Matt, M. Taddei, S. Doria, L. Bussotti, P. Foggi, J. Zhao, M. Di Donato, O. F. Mohammed and S. Weber, J. Mater. Chem. C, 2020, 8, 8305–8319 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00836c |
| This journal is © The Royal Society of Chemistry 2024 |