
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
3D printing polymerizable eutectics via RAFT polymerization†
Nathaniel
Corrigan
 *a,
Alexandra L.
Mutch
b,
Cyrille
Boyer
*a and
Stuart C.
Thickett
*b
*a,
Alexandra L.
Mutch
b,
Cyrille
Boyer
*a and
Stuart C.
Thickett
*b
aCluster for Advanced Macromolecular Design (CAMD) and Australian Centre for NanoMedicine (ACN), School of Chemical Engineering, University of New South Wales, Sydney, NSW 2052, Australia. E-mail: n.corrigan@unsw.edu.au; cboyer@unsw.edu.au
bSchool of Natural Sciences (Chemistry), University of Tasmania, Hobart, Tasmania 7005, Australia. E-mail: stuart.thickett@utas.edu.au
First published on 16th July 2024
Abstract
Over the past five years, there has been a notable increase in the application of three-dimensional (3D) printing techniques mediated by reversible addition–fragmentation chain-transfer (RAFT) polymerization. This increasing interest is due in part to the associated benefits that RAFT 3D printing systems provide, including tighter control over macromolecular network structures and the ability to easily prepare multifunctional materials. In parallel, deep eutectic solvents that feature polymerizable components, called polymerizable eutectics, have also been gaining attention for their outstanding properties and ease of manufacture of functional polymer materials. In this work, we develop polymerizable eutectic resins that contain RAFT agents and are suitable for application to visible-light induced vat 3D printing. The combination of the polymerizable eutectic components and a Z-connected bis-RAFT agent in the resin provides the resulting materials with excellent properties derived from both the eutectic components and the degenerative chain transfer mechanism of RAFT polymerization. While the base (non-RAFT) polymerizable eutectic materials display high modulus and adhesive strength on their own, the inclusion of RAFT agents provides materials with higher adhesive strength while retaining their high strength to higher temperatures. Moreover, the selection of N-isopropylacrylamide as one of the eutectic components provides these materials with reversible thermoresponsive behavior in water. The high shape fidelity and ease of preparation of these materials could be of potential use in the design of 3D printable biomaterials and actuators.
Introduction
Deep eutectic solvents (DESs) are an interesting class of solvents that provide several advantages compared to traditional organic solvents, including lower flammability, excellent solvation capacity, and ease of recoverability, and in some cases, high electrical and ionic conductivity.1–5 These excellent properties have led to application driven research of DESs in fields as diverse as energy, biochemistry, and chemical separations.6,7 DESs are typically binary or ternary mixtures containing hydrogen bond donating and accepting components, where the mixture has a significantly lower melting point compared to the individual components. As an extension of these systems, researchers have recently developed DESs where at least one component is polymerizable, so called polymerizable eutectics (PEs).2,8–10 In these cases, the mixture serves as both the reactant and the reaction solvent, which enables the straightforward fabrication of structurally interesting polymer materials, including thermoresponsive hydrogels, molecularly imprinted polymers, and highly porous polymer scaffolds.11–13More recently, reversible-deactivation radical polymerization (RDRP) has been used to prepare polymer materials, both using DESs as reaction solvents3,14–18 and using PE mixtures.9,12,19 Compared to conventional radical polymerization, RDRP provides benefits such as tight control over polymer molecular weights and molecular weight distributions, and the ability to repetitively reactivate polymerization to form (multi)block copolymers.20–23 Importantly, eutectic-based RDRP systems retain the benefits of both the DES and RDRP, including molecular weight control and block copolymer formation, as well as the ability to recover solvent components and fabricate structurally tailored materials. In addition, PEs typically have higher viscosity compared to their compositionally analogous counterparts prepared in traditional solvents, which can provide increased polymerization rates while still retaining a high degree of control over the polymerization.11,24,25
In parallel with these developments, RDRP systems have also been engineered for application to light-based three-dimensional (3D) printing.26–31 In several examples, the rich history of RDRP has been leveraged to prepare 3D printed materials with advanced functionalities, most frequently via reversible addition–fragmentation chain-transfer (RAFT) polymerization. Notable examples of materials made through RAFT polymerization 3D printing include self-healing materials,32 materials with tuneable surface properties,33 and nanostructured composite34 or fully organic35 materials with outstanding properties.36 The ability to reversibly reactivate dormant thiocarbonylthio (RAFT agent) functionalities during and after 3D printing allows polymer network rearrangement or secondary polymer chain growth processes in the presence of additional monomers.37 Furthermore, at a fundamental level, the inclusion of RAFT agents in 3D printing resins regulates the polymerization mechanism and provides polymer networks with more homogeneous distributions of crosslinking junction points, in turn providing tighter control over mechanical properties.38,39
Given the developments in both PEs and RDRP-mediated 3D printing approaches, we sought to apply this combination to determine the feasibility of 3D printing with PE mixtures that include RAFT agents, and to investigate the resulting material properties. While there are a few examples of 3D printing using eutectic mixtures,40–43 mostly extrusion-based 3D printing, this research area remains largely unexplored despite the prospective benefits for manufacturing functional materials. In this work, we prepared PE resins which contained N-isopropylacrylamide (NIPAm) and acrylamide (Am) as the major components, with the inclusion of N,N-methylenebisacrylamide (MBAm) as difunctional crosslinking monomer and a Z-connected bis-RAFT agent44 2,2′-(carbonothioylbis(thio))bis(2-methylpropanoic acid) (BisPAT) to regulate chain growth (Fig. 1a). Following formation of the PE from the solid starting components, which is achieved by straightforward heating of the mixtures, a visible light photoinitiator was added and the resins were applied to a commercially available light-based 3D printer (Fig. 1b). The resulting materials were characterized by several mechanical testing techniques, and the reversible thermoresponsive swelling properties were also analyzed. Altogether, these materials were easily printable and showed high-shape fidelity without significant optimization, with the resulting properties displaying benefits from both the use of a PE and the inclusion of RAFT agents in the resins (Fig. 1c).
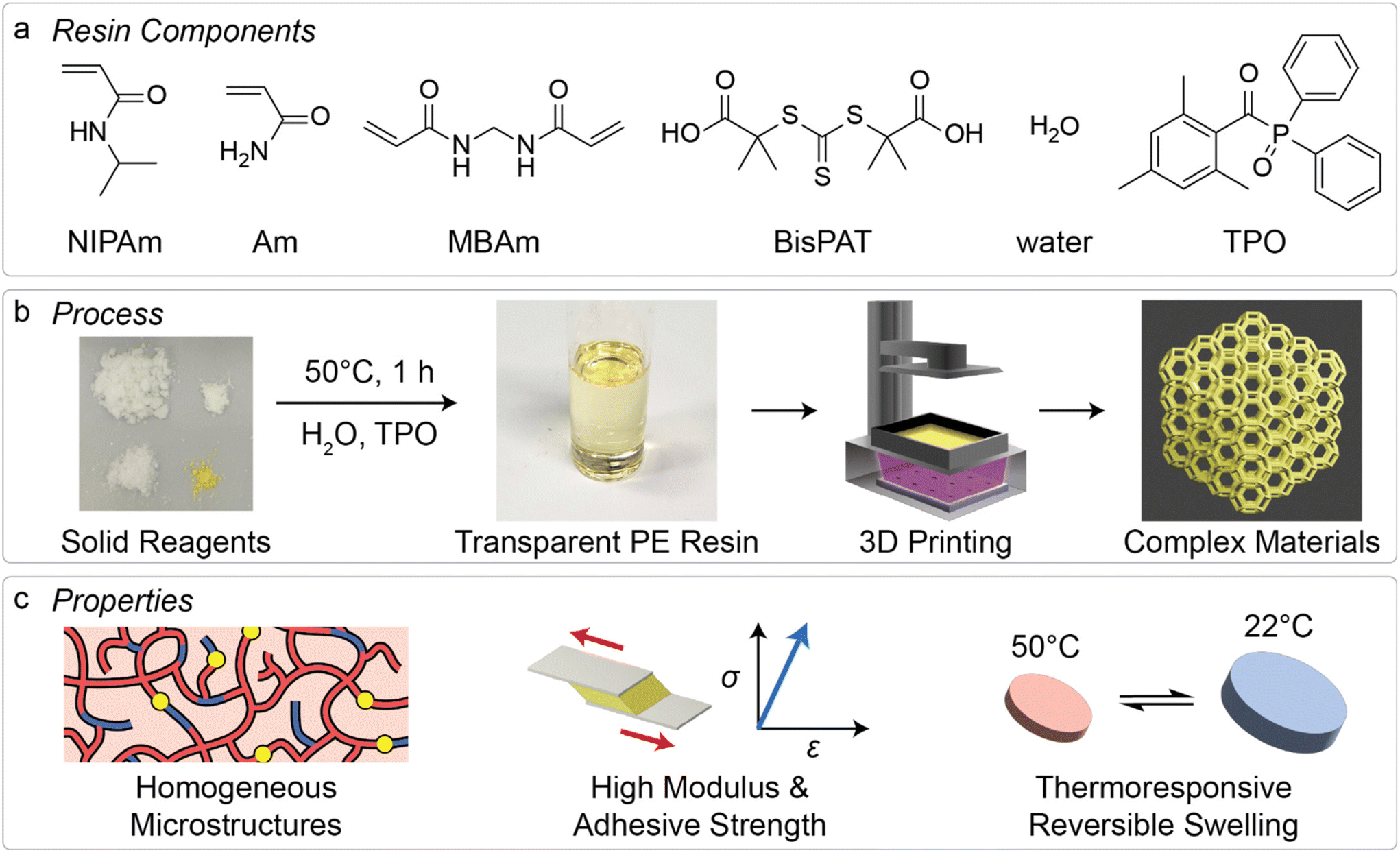 | ||
| Fig. 1 3D printing PE materials via RAFT polymerization. (a) Chemicals used in resin formulations in this work; (b) schematic of 3D printing process using commercial light-based 3D printers; and (c) favorable material properties for 3D printed PE materials, including homogeneous network structures, high adhesive strength and shear modulus, and reversible thermoresponsive swelling. | ||
Results and discussion
Resin development
As a base PE mixture, we selected NIPAm and Am as hydrogen bond donor and hydrogen bond acceptor. PE mixtures featuring NIPAm and Am have previously shown to have melting points below room temperature at molar ratios of NIPAm![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Am = 2:1 and 3:1, which represents a significant depression in melting point compared to the individual components (Tm, NIPAm = 64.0 °C; Tm, Am = 84.5 °C).12 Based on this work, and the potential to use the thermoresponsive behavior of PNIPAm,45 a PE mixture was formulated using a molar ratio of NIPAm:Am = 3:1 (mass ratio NIPAm:Am ≈ 4.8:1), and with 10 wt% water overall to aid in dissolution of all prospective resin components. Previous studies have shown that addition of small quantities of water do not significantly affect the stability of the eutectic, with the significant hydrogen bonding in these mixtures being maintained under these conditions.46–49 Due to its strong hydrogen bonding interactions, water is expected to play a role in stabilizing these hydrogen bonded eutectic mixtures. These mixtures can thus be considered a ternary eutectic mixture due to the molar ratio of NIPAm:Am:H2O = of 3:1:2.5. This mixture was stirred at 50 °C in an oil bath for 1 h to provide the liquid PE mixture (ESI Methods†). To provide a resin capable of crosslinking during 3D printing, 4 wt% overall of MBAm was added; this mixture is referred to as the base PE mixture. The viscosity of the base PE mixture was analyzed and determined to be 14.5 ± 0.3 mPa s (ESI Methods†). The freezing point of the base mixture was determined to be 6 °C, taken as the onset of the exothermic transition upon cooling during differential scanning calorimetry (ESI Methods and Fig. S1†). The freezing point of this mixture is well below the melting points of both the NIPAm and Am monomers, as expected for a eutectic solvent mixture.
:Am = 2:1 and 3:1, which represents a significant depression in melting point compared to the individual components (Tm, NIPAm = 64.0 °C; Tm, Am = 84.5 °C).12 Based on this work, and the potential to use the thermoresponsive behavior of PNIPAm,45 a PE mixture was formulated using a molar ratio of NIPAm:Am = 3:1 (mass ratio NIPAm:Am ≈ 4.8:1), and with 10 wt% water overall to aid in dissolution of all prospective resin components. Previous studies have shown that addition of small quantities of water do not significantly affect the stability of the eutectic, with the significant hydrogen bonding in these mixtures being maintained under these conditions.46–49 Due to its strong hydrogen bonding interactions, water is expected to play a role in stabilizing these hydrogen bonded eutectic mixtures. These mixtures can thus be considered a ternary eutectic mixture due to the molar ratio of NIPAm:Am:H2O = of 3:1:2.5. This mixture was stirred at 50 °C in an oil bath for 1 h to provide the liquid PE mixture (ESI Methods†). To provide a resin capable of crosslinking during 3D printing, 4 wt% overall of MBAm was added; this mixture is referred to as the base PE mixture. The viscosity of the base PE mixture was analyzed and determined to be 14.5 ± 0.3 mPa s (ESI Methods†). The freezing point of the base mixture was determined to be 6 °C, taken as the onset of the exothermic transition upon cooling during differential scanning calorimetry (ESI Methods and Fig. S1†). The freezing point of this mixture is well below the melting points of both the NIPAm and Am monomers, as expected for a eutectic solvent mixture.
To explore the potential feasibility for 3D printing and to screen different variations of this base PE mixture, we performed droplet polymerization on glass slides using the light source from a light-based 3D printer (Anycubic Photon S, 405 nm light array, I0 = 0.81 mW cm−2 at print surface). For this exploration, we selected a type I photoinitiator, diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide (TPO), at concentrations of 1 or 2 wt% overall. In addition to these two non-RAFT agent containing resins, we formulated four other resins which included 1 or 2 wt% TPO and 1 wt% of either 4-((((2-carboxyethyl)thio)carbonothioyl)thio)-4-cyanopentanoic acid (CTCPA) or BisPAT. These RAFT agents were selected as they contain carboxylic acid groups at both ends of the RAFT agent which provided solubility in the base PE mixture. Briefly, resins which contained no RAFT agents were able to be fully cured in less than 30 s, while the resin with 1 wt% BisPAT and 2 wt% TPO was effectively cured in 60 s (Table S1†). Resins containing 1 wt% BisPAT and 1 wt% TPO, or 1 wt% CTCPA with either 1 or 2 wt% TPO required longer irradiation times to provide solidified materials and were not considered further. Based on these results, we developed 3 resins to be applied to 3D printing which contained either 0, 0.5, or 1.0 wt% BisPAT, with all resins containing 2 wt% TPO within the base PE mixture. Using these resins, we performed kinetic analysis of droplet polymerization, using attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopy. For this analysis, 15 μL droplets of the resins were irradiated with 0.81 mW cm−2, 405 nm light (ESI Methods†) then measured by ATR-FTIR spectroscopy. The disappearance of the peak from 1403–1433 cm−1, corresponding to the ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 scissoring mode of the vinyl monomers was monitored, and the conversion of these resins was determined (ESI Fig. S2 and S3†). Under these open-to-air conditions, the resin without RAFT agent reached high conversion after 20 s, while the 0.5 wt% and 1.0 wt% BisPAT resins gelled around 30 s and 60 s, respectively. The resins containing BisPAT required longer cure times, due to both the RAFT polymerization mechanism (vide infra) and the absorption of 405 nm light by the thiocarbonylthio RAFT agent species.50
CH2 scissoring mode of the vinyl monomers was monitored, and the conversion of these resins was determined (ESI Fig. S2 and S3†). Under these open-to-air conditions, the resin without RAFT agent reached high conversion after 20 s, while the 0.5 wt% and 1.0 wt% BisPAT resins gelled around 30 s and 60 s, respectively. The resins containing BisPAT required longer cure times, due to both the RAFT polymerization mechanism (vide infra) and the absorption of 405 nm light by the thiocarbonylthio RAFT agent species.50
3D printing and material adhesion
The three resins were then applied to 3D printing using layer thicknesses of 50 μm and cure times of 15, 30, or 45 s per layer for the resins containing 0, 0.5, or 1.0 wt% BisPAT, respectively. While these cure times are slightly lower than the kinetic analysis via ATR-FTIR, this can be attributed to the 3D printing process being less influenced by oxygen inhibition from air. Simple disks were initially printed to check the overall feasibility of 3D printing using these resins. These disks were post-cured under 405 nm irradiation for 15 min (ESI Methods†). The double bond conversions were also measured before and after post-curing, with all materials showing high conversions before and after post-curing (conversion >94% in all cases, ESI Fig. S4 and Table S2†). As shown in ESI Fig. S5,† the materials printed using resins both with and without RAFT agents were printed into well-defined disks, which indicated a successful 3D printing process. However, when trying to remove simple objects from the build stage, we observed an extremely high adhesion between the 3D printed materials and the build stage. Moreover, the adhesion anecdotally became stronger with the inclusion of RAFT agents in the resin, with the 1.0 wt% BisPAT materials proving exceedingly difficult to remove from the build stage without breaking the printed object. The high baseline adhesion of these materials is attributed the use of a PE formulation. Indeed, previous PE materials have demonstrated very high adhesion.40 However, to the best of our knowledge, an increased adhesion in network copolymers due to the incorporation of RAFT agents has not been previously observed.To explore the adhesive properties of these resins more closely, we performed lap shear testing using aluminum laps to mimic the aluminum alloy used in our 3D printer build stage. For these experiments, the photoinitiator was replaced with a thermal initiator, azobisisobutyronitrile, and the resins were thermally cured between the laps using a heat gun (ESI Methods†). The results of the lap shear testing are shown in Fig. 2 and confirmed the observation of increasing adhesion with increasing BisPAT concentration. The stress at break for the aluminum laps was 797, 972, and 1353 kPa for the samples containing 0, 0.5, or 1.0 wt% BisPAT, respectively. These values are on the same order as previously disclosed PE materials40 and also “instant” cyanoacrylate adhesives.51
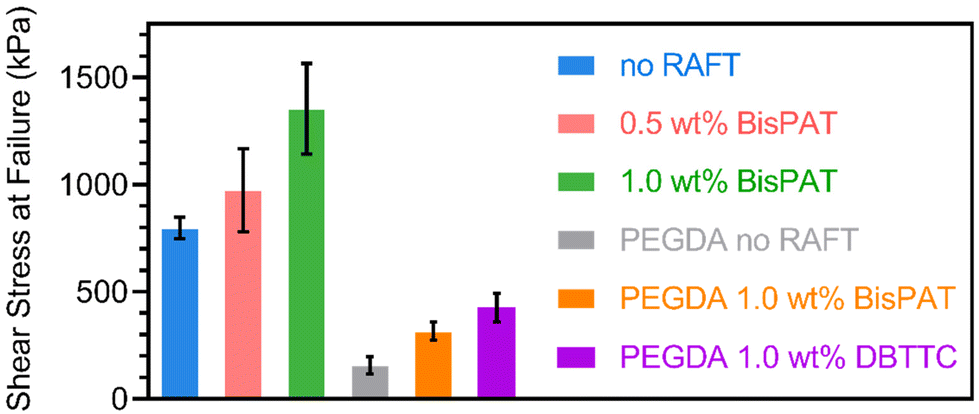 | ||
| Fig. 2 Shear stress at failure for polymer network materials cured between aluminum laps. No-RAFT samples (blue) were prepared with the base PE mixture; 0.5 wt% samples (red) were prepared using the base PE mixture and 0.5 wt% BisPAT; 1.0 wt% samples (green) were prepared using the base PE mixture and 1.0 wt% BisPAT; PEGDA no RAFT samples (grey) were prepared with pure PEGDA 250; PEGDA 1.0 wt% BisPAT samples (orange) were prepared with pure PEGDA 250 and 1.0 wt% BisPAT overall; PEGDA 1.0 wt% DBTTC samples (purple) were prepared with pure PEGDA 250 and 1.0 wt% DBTTC overall. All samples also contained 2 wt% TPO overall. | ||
Subsequently, we tested the adhesion between a non-PE resin composed of poly(ethylene glycol) diacrylate (PEGDA 250, average Mn = 250 g mol−1), a common crosslinking monomer used in 3D printing, and 1 wt% of either BisPAT or dibenzyl trithiocarbonate (DBTTC). DBTTC was selected as it is a Z-connected bis-RAFT agent with a similar structure to BisPAT, albeit with benzyl, rather than propanoic acid leaving groups (ESI Fig. S6†). PEGDA-based resins were required as a control here, as DBTTC and other non-acidic RAFT agents were not readily soluble in the base PE mixture. Very interestingly, inclusion of either BisPAT or DBTTC led to a significant increase in the shear stress at failure compared to the no RAFT system, indicating that the increased adhesion is not specific to the acidic BisPAT RAFT agent, and instead is likely to be a result of using RAFT polymerization with Z-connected bis-RAFT agents.
We propose that the increase in adhesion is ascribed to the enhanced network relaxation that occurs during RAFT polymerization to form the PE network. As noted previously, allowing polymer chain relaxation during the formation of adhesives can increase adhesion by reducing the internal stress within the polymer network.52,53 It has been well documented that using degenerate transfer radical polymerization, including with Z-connected bis-RAFT agents, can reduce internal stress within polymer networks.54–58 For the RAFT agents used in this work, degenerate transfer between a propagating radical and a dormant Z-connected trithiocarbonate polymer chain can lead to network reconfiguration, which promotes stress relaxation by allowing chains to adopt a more preferred configuration. A schematic of this process is shown in the ESI Fig. S7.† As noted previously, the build-up of internal stress (shrinkage stress) within polymer networks increases faster at higher double bond conversions,59–61 however, the reduction in internal stress when using addition–fragmentation chain transfer mechanism also occurs most prominently during the same stage of the reaction.58,62,63 In these addition–fragmentation chain transfer network syntheses, the final internal stress is frequently reduced by >75% compared to analogous non-adaptable networks. Notably, in these previous systems, the time scales for polymerization were on the order of seconds (typically in the range of 10–100 s after an inhibition period),58,62,63 which further supports the ability of our 3D printed systems to relieve internal stress via this mechanism in this time frame. In addition, network copolymer synthesis mediated by RAFT polymerization is associated with a delayed onset of gelation due to the formation of lower molecular weight branched copolymers in the early stages of the reaction.44,55 Rapid polymerization, as in the case of the no-RAFT system, does not allow enough time for viscous flow of the polymer chains to relieve the stress formed from shrinkage of the resins.61,64 For the systems containing RAFT agent, as the gel point is delayed there is additional time for the polymer chains to relax internal stress via viscous flow prior to gelation and fixation of the network structure. The reduction in internal stress via both these mechanisms ultimately leads to greater adhesive strength.
While the increased adhesion may be favorable in some applications, it was initially problematic in our 3D printing system. To more easily remove the PE materials from the build stage after 3D printing, a layer of sacrificial pre-polymer (PEGDA 250) was printed onto the build stage prior to printing with PE resins. After printing, the PE materials were easily removed and the PEGDA layer was peeled away from the 3D printed materials prior to analysis (ESI Methods†). Using this process, we 3D printed a range of relatively simple shapes for characterization of the material mechanical properties, specifically rectangular prisms for dynamic mechanical analysis (DMA), thin cylinders (disks) for analysis of swelling behavior, and dog-bone shaped pieces for tensile testing.
Material mechanical properties
To examine the properties of the 3D printed PE materials, we first performed dynamic mechanical analysis (DMA). For the samples analyzed by DMA, the materials were first dried under vacuum at 50 °C for 72 h. Samples that were not dried produced irregular results in DMA at temperatures higher than ∼65 °C. Fig. 3 shows the results from DMA, with all samples showing high room temperature shear modulus values above 3 GPa, and modulus at the rubbery plateau (∼195 °C) in the range of 10 MPa (Table S3†). While the shear modulus was in the same range at high and low temperatures, the tanδ curves showed significantly different behavior for materials 3D printed with 0, 0.5 and 1.0 wt% of BisPAT in the resin. For the no RAFT sample, there were two clearly visible peaks in the tanδ curve, one at ∼130 °C and the other at ∼152 °C, which is discussed below. The onset of the increase in tanδ, corresponding to the start of the glass transition, occurred at ∼85 °C, with the broad double peak tailing off around 195 °C. In comparison, the samples which contained BisPAT showed narrower tanδ peaks and were unimodal, however, the peak of the tanδ curve was observed at roughly the same temperature for the 0.5 wt% BisPAT system (glass transition temperature, Tg ≈ 154 °C) and marginally higher for the 1.0 wt% BisPAT system (Tg ≈ 158 °C). For the 0.5 and 1.0 wt% BisPAT samples, the onset of the glass transition occurred at ∼95 and ∼105 °C, respectively, and the tanδ peak again tailed off around 195 °C. The increase in the onset temperature of the glass transition for the materials prepared using RAFT agents indicates they retain their glassy high-modulus state at higher temperatures than the corresponding no-RAFT PE sample.
 | ||
| Fig. 3 Shear modulus (G′) and tanδ for 3D printed materials. Materials prepared using: (a) PE resin with no additional RAFT agent; (b) PE resin with 0.5 wt% BisPAT; (c) PE resin with 1.0 wt% BisPAT. Blue points correspond to shear modulus (G′) and red points correspond to tanδ. Figures show representative samples. | ||
While previous network copolymers synthesized via RAFT polymerization have shown narrower tanδ peaks in comparison to those made via free-radical polymerization,65 the appearance of two distinct tanδ peaks is more likely due to two compositionally different copolymers within the material. Materials with two phase-separated domains have previously shown similar effects, where the Tg of each individual polymer is observed in the DMA tanδ curve.66 In this case, for the sample prepared without RAFT agent, the two tanδ peaks occur around 130 and 152 °C; the low temperature tanδ can be ascribed to copolymers rich in NIPAm (Tg, NIPAm ≈ 135 °C (ref. 67–69)) while the higher temperature tanδ peak corresponds to a more mixed copolymer featuring both NIPAm and Am (Tg, Am ≈ 194 °C (ref. 67, 70 and 71)).
Based on these differences, the second low temperature Tg peak observed in the sample without RAFT agent may point to differences in the network microstructure compared to the RAFT samples. As noted by Chiklis and Grasshoff, the reactivity ratios for NIPAm (r1) and Am (r2) are r1 = 0.50 and r2 = 1.00, respectively, indicating that Am will be preferentially incorporated into the polymer chain over NIPAm.72 As such, in the early stages of the polymerization, polymers will have a slightly higher Am content compared to the feed ratio. Later in the reaction the polymer will have higher NIPAm content compared to the feed ratio. Previous literature on reactivity ratios in uncontrolled radical copolymerization versus RAFT copolymerization have noted that RAFT polymerization either has no effect on the reactivity ratios, or further increases the incorporation of the higher reactivity monomer (Am in this case).73–75 To verify that we have some compositional drift in our ternary PE system, 1H NMR was used to track the conversions of the NIPAm and Am units during photopolymerization for the no RAFT and 1.0 wt% BisPAT systems under 405 nm irradiation. Full details on this experiment are provided in the ESI Methods.† It was observed that the Am was preferentially incorporated in the polymers in the early stages of this model reaction, as shown in ESI Fig. S8, S9, and Tables S4, S5.† The experimentally determined reactivity ratios76 for NIPAm (r1) and Am (r2) were r1 = 0.55 and r2 = 1.83 for the no-RAFT system, and r1 = 0.51 and r2 = 1.97 for the system containing 1.0 wt% BisPAT. These values support the preferential incorporation of Am in the early stages of the reaction, and the formation of NIPAm-rich polymers at the later stages of the reaction. Despite the high Am reactivity, the polymers at the start of the polymerization still contain reasonable mole fractions of NIPAm due to its 3-fold concentration in the initial mixture (ESI Table S5†).
Considering that the high weight fraction of NIPAm in these mixtures, it follows then that composition of chains generated in the later stages of the reaction will have a very high proportion of NIPAm. For PE materials prepared using RAFT polymerization, however, the prominent low temperature Tg corresponding to a relatively pure NIPAm phase was not observed. A shoulder on the low temperature side of the tanδ curve was observed for both the 0.5 and 1.0 wt% BisPAT samples around 135 °C, however, the intensity of this peak was comparatively low. Given the reactivity ratios from literature and experiments which favor the early incorporation of Am in both RAFT and non-RAFT systems, we attribute the unification of the Tg in the BisPAT containing PE materials to differences in the chain growth process for the RAFT network copolymerization compared to the no-RAFT system.
At the early stages of the reaction in uncontrolled radical polymerization of multivinyl monomers, highly crosslinked nanogels are formed due to rapid polymerization and intramolecular cyclization reactions. These nanogels undergo intermolecular crosslinking later in the reaction to form the final network that features high heterogeneity in terms of density of crosslinking points.77 In addition, these polymer chains cannot be reactivated for further chain growth. As such, in our PE system we are likely to see isolated nanogels which contain a disproportionately high concentration of Am units within the polymer chains. Comparatively, RAFT polymerization of multivinyl monomers is associated with a delayed onset of gelation, due to the formation of lower molecular weight branched polymers in the early stages of the reaction, which combine later in the reaction to form a copolymer network with a more homogeneous distribution of crosslink junctions.44,78,79 Importantly, the chains produced in the earlier stages of the reaction, which have a higher proportion of Am units, are dormant chains which contain trithiocarbonate units within the polymer backbone. When degenerate chain transfer between propagating macroradicals and dormant polymers occurs, the polymer main chain is fragmented and the network can relax. This process supports the diffusion of Am rich polymer strands away from their original locations within the material. A schematic representation of this process is shown in Fig. 4. Altogether, the degenerate chain transfer process leads to network materials with more homogeneous microstructures.
 | ||
| Fig. 4 Schematic of network copolymer formation in the no-RAFT and BisPAT PE 3D printing systems. (a) No-RAFT starting materials form: (b) high crosslink density nanogels in the early stages of the reaction that are rich in Am due to the NIPAm–Am reactivity ratios; (c) later in the no-RAFT reaction the nanogels are crosslinked with the resulting copolymer chains showing a higher relative NIPAm composition. (d) Starting materials in the systems containing BisPAT: (e) form more highly branched polymer chains prior to gelation which still contain a high Am content, but also contain trithiocarbonate units within the main chains; (f) further reaction and degenerative chain transfer allows the main chains to fragment and react further with NIPAm in the later stages of the reaction. The relaxation of the Am rich copolymer chains effectively increases the homogeneity of the Am–NIPAm composition throughout the network. | ||
Following DMA analysis, we performed tensile testing on our 3D printed samples. As shown in ESI Fig. S10 and Table S6,† samples with 0.5 wt% BisPAT showed slightly higher yield stress (σy = 24.2 MPa) and elongation at break (εb = 0.7 mm mm−1), but slightly lower Young's modulus values (E′ = 143.2 MPa), compared to the sample prepared without RAFT (σy = 20.9 MPa, εb = 0.6 mm mm−1, E′ = 160.7 MPa). Overall, this provided materials with increased tensile toughness (11.4 MJ m−3 for 0.5 wt% BisPAT vs. 7.7 MJ m−3 for the no-RAFT sample). Further increasing the BisPAT concentration to 1 wt% provided materials with noticeably reduced Young's modulus (E′ = 79.7 MPa) and yield stress (σy = 7.2 MPa), but with greater extensibility (εb = 0.9 mm mm−1). The reduction in yield stress ultimately provided materials with a reduced tensile toughness of 5.2 MJ m−3. This decrease in modulus and increase in extensibility has been previously observed in other RAFT mediated 3D printing systems, particularly at higher RAFT agent concentrations.32,39 The chain transfer mechanism decreases the concentration of physical entanglements, providing more flexible materials with lower Young's modulus.78
In a final examination of the 3D printed PE material properties, swelling tests were performed on 3D printed PE disks (ESI Fig. S11†). Disks with dimensions of 8 × 1 mm (d × t) were 3D printed and post-cured, then placed in vials filled with water at room temperature (ESI Methods†). The mass of the disks was monitored periodically over 24 h to determine the rate of water uptake, as well as the water content absorbable at equilibrium. As shown in Fig. 5a, at room temperature (22 °C) the PE materials swelled significantly over 24 h, with the water uptake of the no-RAFT, 0.5 wt% and 1.0 wt% BisPAT systems reaching 2.23, 2.66 and 2.69 g g−1, respectively. In comparison, when the disks were swelled at 50 °C, which is above the lower critical solution temperature (LCST) of NIPAm,45 a volume phase transition occurs with the resulting water uptake at equilibrium significantly reduced (Fig. 5b). The water uptake at 50 °C was 1.03, 1.14 and 1.16 g g−1 for the no-RAFT, 0.5 wt% and 1.0 wt% BisPAT materials, respectively. At both temperatures, the materials containing RAFT agents showed higher water uptake at equilibrium, however, there was essentially no difference between the 0.5 wt% and the 1.0 wt% systems. In any case, due to the high NIPAm content in these PE materials, there was an obvious thermoresponsive swelling behavior for all three materials, with lower equilibrium water absorption above the LCST, as expected. As these BisPAT materials also contain carboxylic acid end-groups, swelling was performed to equilibrium in a pH = 1.0 solution to check the behavior when the carboxylic acids were protonated. In all materials, the swelling at equilibrium was reduced compared to the neutral pH swelling samples, with the no RAFT samples swelling the least (ESI Table S7†). This supports the physical network structure as the dominant factor in changing the swelling behavior of these materials.
 | ||
| Fig. 5 Water uptake in 3D printed PE materials prepared using various BisPAT concentrations. (a) Water uptake over 24 h at 22 °C and at (b) 50 °C; (c) kinetics of water uptake in the early stages of the swelling experiments for PE materials swelled at 22 °C, and at (d) 50 °C. Shaded error lines in all graphs represent the 99% confidence intervals of the fits. Graphs shown in a and b were fitted with a one-phase (exponential) decay, while graphs shown in c and d were fitted using the Korsmeyer and Peppas model (details in ESI†). | ||
To examine the differences in swelling rates and network behavior for each of these systems, we used the mass transfer model applied by Korsmeyer, Peppas and coworkers for solute transport in hydrogels: Mt/M∞ = K·tn, where Mt and M∞ are the mass at time t and at equilibrium, respectively, K is a complex term representing the diffusion constant, and n is an exponent which details the mass transport behavior.80 Full details of the modelling are shown in the ESI Methods.†Fig. 5c and d shows the differences in the swelling rate during the early stages of the experiments. For the samples swelled at 22 °C, there was essentially no difference in the swelling behavior, with each sample showing analogous preexponential factors and exponents from the model fitting (ESI Table S8†). The exponents from this fit were between 0.49 and 0.53, indicating typical Fickian mass transport behavior.80–82
In contrast, the materials swelled at 50 °C showed variable swelling rates relative to their equilibrium water content, with increasing RAFT concentrations providing swelling profiles with lower exponents (ESI Table S8†). In particular, the exponents for the no-RAFT, 0.5 wt% and 1.0 wt% BisPAT samples were 0.64, 0.56, and 0.54, respectively. Although the exponents for the 0.5 wt% and 1.0 wt% samples were slightly higher at 50 °C than at 22 °C, they were still close to 0.5, indicating mass transport behavior close to Fickian. However, the exponent for the no-RAFT sample (n = 0.64) was significantly higher, which indicates anomalous behavior in between Fickian and Case II transport, where both polymer relaxation and diffusion processes affect the overall mass transport.81 The higher exponent here indicates that there is a greater contribution of chain relaxation during the swelling of the no-RAFT samples, which is attributed to the solvation of collapsed PNIPAm rich domains above the LCST. The higher exponent here is also in alignment with the proposed network structures in Fig. 4, where the no-RAFT samples have domains richer in NIPAm compared to the BisPAT samples and require additional chain relaxation to reach equilibrium swelling.
3D printed lattices and reversible swelling behavior
PNIPAm network materials have gained great interest in several fields, particularly in biomedicine for tissue engineering scaffolds,83 biomolecule separation matrices,84,85 and drug delivery materials,86 among others.87,88 This has been largely driven by the ability to reversibly expand and contract these materials via external temperature control around physiological temperatures. To demonstrate the ability for the 3D printed PE materials to undergo multiple cycles of reversible volume change, we swelled 3D printed PE samples containing 0.5 wt% BisPAT at 22 °C and 50 °C and measured their mass after each cycle. Fig. 6 shows the corresponding sawtooth plot, indicating the average water uptake for these materials after each hot (50 °C) and cold (22 °C) swelling cycle (ESI methods†). After 7 cycles of cold swelling and 7 cycles of hot swelling, the gels still had water uptake close to the values in the first cycle, demonstrating the reversible thermoresponsive nature of these materials. | ||
| Fig. 6 Sawtooth plot showing the reversible thermoresponsive behavior of 3D printed materials prepared using a resin containing 0.5 wt% BisPAT. Points in the blue shaded regions represent samples swelled at 22 °C and sample points in the red shaded regions represent samples swelled at 50 °C. | ||
Aside from the notable properties of these thermoresponsive PE materials, their ability to be rapidly manufactured via 3D printing brings some additional benefits, most notably the ability to quickly form functional materials with relatively unconstrained geometries. To demonstrate the effectiveness of these resins in 3D printing, we used a 0.5 wt% BisPAT resin to prepare a complex lattice structure that would be extremely tedious to prepare using conventional manufacturing techniques. The designed lattice model is shown in Fig. 7a and features a 4 × 4 × 4 unit-cell array; the targeted overall dimensions were 30 × 30 × 30 mm and the target strut thickness was ∼850 μm. The 3D printing of this model was performed using identical parameters to the previous 3D printing, i.e., 30 s layer cure times and 50 μm layers, and with four bottom layers printed for 45 s each. As shown in Fig. 7b and c, the lattice was successfully printed with high-shape fidelity, with struts clearly printed and each unit cell readily observable. Both the overall dimensions and the strut thickness was the same as targeted, within measurement error (within 200 μm overall and 100 μm for strut thickness) but the four bottom layers were slightly overcured; these deviations should be readily rectified by adjusting the printing conditions. Regardless, without optimization of the 3D printing conditions, clearly well-defined 3D printed lattices were successfully produced.
 | ||
| Fig. 7 3D printing complex lattice structures prepared using a PE resin containing 0.5 wt% BisPAT. (a) designed model file; (b) material after 3D printing, isolation, and washing; (c) close up of lattice showing finer details of the struts; (d) 3D printed lattice after swelling in water for 24 h at 22 °C; (e) selected photos from the swelling of the 3D printed PE lattice, showing a monotonic increase in overall dimensions over the first 72 min of this procedure. Scale: the designed lattice model dimensions were 30 × 30 × 30 mm, which closely matched (<200 μm deviation in all axes) the printed dimensions for the material shown in b. The dimensions of the swelled lattice shown in d were 48.3 × 48.1 × 47.7 mm (l, w, h). | ||
Given the previous demonstration of effective swelling in these PE materials, we performed a final test to demonstrate the swelling capability of the 3D printed lattice. The dimensions of the lattice were taken before and after swelling to equilibrium at 22 °C, and as shown in Fig. 7d, the complex lattice prepared using the PE resin was effectively swelled in water, retaining the overall shape of the 3D printed material. A video of the early stages of this swelling process are included in the ESI,† with selected frames from ∼15 min intervals shown in Fig. 7e. The dimensions of the swelled 3D printed lattice were 48.3 × 48.1 × 47.7 mm (l, w, h), with the slightly lower height of this material attributed to the defects on the bottom layers which slightly restricted the expansion in this direction.
The success of the 3D printing process with these PE resins can be attributed to both the PE formulation and the inclusion of RAFT agents in these resins. Compared to NIPAm solutions, PE mixtures have higher viscosity which can provide faster polymerization.11,24,25 This is especially important in RAFT polymerization-based 3D printing, where the delayed onset of gelation can often induce the need for longer cure times. While the cure time in the current work is not exceptionally low, these resins are prepared with very low crosslinker concentrations (<4 wt% overall of MBAm), which is significantly lower than typical formulations. During the 3D printing process, the high adhesion and modulus of these materials also circumvents issues associated with excessive peel force which can often lead to 3D printing failure. Finally, the inclusion of RAFT agents which absorb at 405 nm act as photoblockers, restricting erroneous curing outside of the desired region.
Conclusion
In this work, polymerizable eutectic resins featuring NIPAm and Am as monomers and a Z-connected bis-RAFT agent (BisPAT) as chain transfer agent were applied to light-based 3D printing to prepare thermoresponsive network copolymers. These resins displayed high adhesion, with the adhesive strength increasing with increasing RAFT agent concentration, which was attributed to the increased polymer relaxation in these systems. Mechanical property data also showed that these PE materials were rigid and strong, with room temperature shear modulus exceeding 3 GPa. The inclusion of RAFT agents also affected the thermomechanical properties of the resulting materials, with a unification of the glass transition temperature observed for the materials printed with BisPAT. The degenerate chain transfer process and polymer relaxation during 3D printing with RAFT agents provided materials with more homogeneous microstructures, which was also supported by analysis of the swelling behavior, both above and below the volume phase transition temperature of these materials. The reversibility of this thermoresponsive behavior was also demonstrated by repetitive cycling at 22 °C and 50 °C. Finally, the capabilities of these resins in 3D printing were demonstrated via the production of geometrically complex lattice structures with high-shape fidelity compared to designed models. The high shape flexibility imparted by the 3D printing process (e.g., the complex lattice structure), coupled with the thermoresponsive behavior of these materials, tailors these 3D printed materials to prospective applications in both healthcare and sensing. For healthcare, personalized 3D materials (e.g., wound dressings) may be printed to interface with geometrically arbitrary parts of the human body. Materials loaded with drug would then release to the affected areas. For sensing applications, actuating materials based on temperature changes could be used to design sensors that require intricate parts or geometries.Altogether, the 3D printing resins developed in this work showed a range of favorable properties due in part to both the PE and the RAFT polymerization mechanism with the inclusion of Z-connected bis-RAFT agents. The high-shape fidelity of these materials, combined with high strength makes 3D printing straightforward. This work should inform the design of future 3D printing system using RAFT agents and polymerizable eutectics, especially for those with focus on applications in thermoresponsive materials.
Data availability
Data has been deposited at UNSWorks, an Open Access institutional repository which enables UNSW researchers to make their research outputs freely available and accessible. A doi indicator will be provided after the data has been accepted by the UNSWorks (data submitted, acceptance in progess as of 28th June 2024).Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. C. is a recipient of an Australian Research Council Discovery Early Career Researcher Award (DE240100917). C. B. is the recipient of an Australian Research Council - Australian Laureate Fellowship (project number FL220100016) funded by the Australian Government. S. C. T. is the recipient of an Australian Research Council Future Fellowship (FT220100096). The authors also express their gratitude to Eh Hau Pan for providing technical support and maintaining the necessary instruments in the School of Chemical Engineering at UNSW. The authors would like to thank the NMR facility within the Mark Wainwright Analytical Centre (MWAC) at UNSW for providing and maintaining the necessary instruments.References
- A. P. Abbott, D. Boothby, G. Capper, D. L. Davies and R. K. Rasheed, J. Am. Chem. Soc., 2004, 126, 9142–9147 CrossRef CAS.
- J. D. Mota-Morales, R. J. Sánchez-Leija, A. Carranza, J. A. Pojman, F. del Monte and G. Luna-Bárcenas, Prog. Polym. Sci., 2018, 78, 139–153 CrossRef CAS.
- L. Quirós-Montes, G. A. Carriedo, J. García-Álvarez and A. Presa Soto, Green Chem., 2019, 21, 5865–5875 RSC.
- H. Zhang, T. Gao, L. Jiang, X. Meng, J. Wang, N. Ma, H. Wei and X. Zhang, Eur. Polym. J., 2022, 173, 111266 CrossRef CAS.
- L. C. Tomé, L. Porcarelli, J. E. Bara, M. Forsyth and D. Mecerreyes, Mater. Horiz., 2021, 8, 3239–3265 RSC.
- Y. Hariyanto, Y. K. Ng, Z. Z. Siew, C. Y. Soon, A. C. Fisher, L. Kloyer, C. W. Wong and E. W. C. Chan, Energy Fuels, 2023, 37, 18395–18407 CrossRef CAS.
- H. Zhang, Y. Wang, Y. Zhou, K. Xu, N. Li, Q. Wen and Q. Yang, Talanta, 2017, 170, 266–274 CrossRef CAS.
- J. D. Mota-Morales, M. C. Gutiérrez, I. C. Sanchez, G. Luna-Bárcenas and F. del Monte, Chem. Commun., 2011, 47, 5328–5330 RSC.
- Y. Nahar, M. K. Stanfield, A. C. Bissember and S. C. Thickett, Polym. Chem., 2023, 14, 2724–2733 RSC.
- J. D. Mota-Morales, M. C. Gutiérrez, M. L. Ferrer, I. C. Sanchez, E. A. Elizalde-Peña, J. A. Pojman, F. D. Monte and G. Luna-Bárcenas, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1767–1773 CrossRef CAS.
- Y. Nahar, J. Horne, V. Truong, A. C. Bissember and S. C. Thickett, Polym. Chem., 2021, 12, 254–264 RSC.
- Y. Nahar, P. Wei, C. Cipriani, A. Khodabandeh, A. C. Bissember, E. B. Pentzer and S. C. Thickett, ACS Appl. Polym. Mater., 2022, 4, 8429–8440 CrossRef CAS.
- N. Ndizeye, S. Suriyanarayanan and I. A. Nicholls, Polym. Chem., 2019, 10, 5289–5295 RSC.
- P. V. Mendonça, M. S. Lima, T. Guliashvili, A. C. Serra and J. F. J. Coelho, Polymer, 2017, 132, 114–121 CrossRef.
- V. A. Pereira, P. V. Mendonça, J. F. J. Coelho and A. C. Serra, Polymer, 2022, 242, 124586 CrossRef CAS.
- C.-Y. Li and S.-S. Yu, Macromolecules, 2021, 54, 9825–9836 CrossRef CAS.
- J. Wang, J. Han, M. Y. Khan, D. He, H. Peng, D. Chen, X. Xie and Z. Xue, Polym. Chem., 2017, 8, 1616–1627 RSC.
- V. A. Pereira, T. C. Rezende, P. V. Mendonça, J. F. J. Coelho and A. C. Serra, Green Chem., 2020, 22, 6827–6835 RSC.
- P. Qi, S. Yuan, Y. Liu, R. Dong, A. Song and J. Hao, Adv. Opt. Mater., 2023, 11, 2300861 CrossRef CAS.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 111, 101311 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985–1076 CrossRef CAS.
- K. Matyjaszewski and N. V. Tsarevsky, J. Am. Chem. Soc., 2014, 136, 6513–6533 CrossRef CAS.
- S. Bednarz, M. Fluder, M. Galica, D. Bogdal and I. Maciejaszek, J. Appl. Polym. Sci., 2014, 131, 40608 CrossRef.
- S. Bednarz, K. Półćwiartek, J. Wityk, B. Strachota, J. Kredatusová, H. Beneš, A. Wesołowska-Piętak and G. Kowalski, Eur. Polym. J., 2017, 95, 241–254 CrossRef CAS.
- V. A. Bobrin, J. Zhang, N. Corrigan and C. Boyer, Adv. Mater. Technol., 2023, 8, 2201054 CrossRef CAS.
- Z. Zhang, N. Corrigan, A. Bagheri, J. Jin and C. Boyer, Angew. Chem., Int. Ed., 2019, 58, 17954–17963 CrossRef CAS PubMed.
- B. Zhao, J. Li, Y. Xiu, X. Pan, Z. Zhang and J. Zhu, Macromolecules, 2022, 55, 1620–1628 CrossRef CAS.
- A. Bagheri, K. E. Engel, C. W. A. Bainbridge, J. Xu, C. Boyer and J. Jin, Polym. Chem., 2020, 11, 641–647 RSC.
- B. J. Green and C. A. Guymon, Addit. Manuf., 2019, 27, 20–31 CAS.
- X. Wu, B. Gross, B. Leuschel, K. Mougin, S. Dominici, S. Gree, M. Belqat, V. Tkachenko, B. Cabannes-Boué, A. Chemtob, J. Poly and A. Spangenberg, Adv. Funct. Mater., 2022, 32, 2109446 CrossRef CAS.
- Z. Zhang, N. Corrigan and C. Boyer, Angew. Chem., Int. Ed., 2022, 61, e202114111 CrossRef CAS.
- C. W. A. Bainbridge, M. Neradt, A. Avzalova, N. Broderick and J. Jin, Macromol. Mater. Eng., 2023, 2300347, DOI:10.1002/mame.202300347.
- V. A. Bobrin, H. G. Hackbarth, Y. Yao, N. M. Bedford, J. Zhang, N. Corrigan and C. Boyer, Adv. Sci., 2023, 10, 2304734 CrossRef CAS.
- V. A. Bobrin, K. Lee, J. Zhang, N. Corrigan and C. Boyer, Adv. Mater., 2022, 34, 2107643 CrossRef CAS PubMed.
- K. Lee, N. Corrigan and C. Boyer, Angew. Chem., Int. Ed., 2023, 62, e202307329 CrossRef CAS PubMed.
- C. W. A. Bainbridge, N. Broderick and J. Jin, Polym. Chem., 2021, 12, 5017–5026 RSC.
- X. Pan, J. Li, Z. Li, Q. Li, X. Pan, Z. Zhang and J. Zhu, Angew. Chem., Int. Ed., 2024, 63, e202318564 CrossRef CAS PubMed.
- X. Shi, J. Zhang, N. Corrigan and C. Boyer, Polym. Chem., 2022, 13, 44–57 RSC.
- J. L. de Lacalle, A. Gallastegui, J. L. Olmedo-Martínez, M. Moya, N. Lopez-Larrea, M. L. Picchio and D. Mecerreyes, ACS Macro Lett., 2023, 12, 125–132 CrossRef CAS.
- G. C. Luque, M. L. Picchio, A. P. S. Martins, A. Dominguez-Alfaro, N. Ramos, I. del Agua, B. Marchiori, D. Mecerreyes, R. J. Minari and L. C. Tomé, Adv. Electron. Mater., 2021, 7, 2100178 CrossRef CAS.
- M. L. Picchio, A. Gallastegui, N. Casado, N. Lopez-Larrea, B. Marchiori, I. del Agua, M. Criado-Gonzalez, D. Mantione, R. J. Minari and D. Mecerreyes, Adv. Mater. Technol., 2022, 7, 2101680 CrossRef CAS.
- M. Liu, G. Zhang, Y. Hu, C. Bo, Y. Dai, L. Hu, G. Zhu and Y. Zhou, Green Chem., 2024 10.1039/D3GC02662K.
- G. Moad, Polym. Int., 2015, 64, 15–24 CrossRef CAS.
- A. Halperin, M. Kröger and F. M. Winnik, Angew. Chem., Int. Ed., 2015, 54, 15342–15367 CrossRef CAS.
- I. Delso, C. Lafuente, J. Muñoz-Embid and M. Artal, J. Mol. Liq., 2019, 290, 111236 CrossRef CAS.
- T. El Achkar, S. Fourmentin and H. Greige-Gerges, J. Mol. Liq., 2019, 288, 111028 CrossRef CAS.
- S. Rozas, C. Benito, R. Alcalde, M. Atilhan and S. Aparicio, J. Mol. Liq., 2021, 344, 117717 CAS.
- L. Sapir and D. Harries, J. Chem. Theory Comput., 2020, 16, 3335–3342 CrossRef CAS PubMed.
- T. G. McKenzie, Q. Fu, E. H. H. Wong, D. E. Dunstan and G. G. Qiao, Macromolecules, 2015, 48, 3864–3872 CrossRef CAS.
- S. Ebnesajjad and A. H. Landrock, Adhesives Technology Handbook, William Andrew, 2014 Search PubMed.
- V. Basin, Prog. Org. Coat., 1984, 12, 213–250 CrossRef CAS.
- F. Awaja, M. Gilbert, G. Kelly, B. Fox and P. J. Pigram, Prog. Polym. Sci., 2009, 34, 948–968 CrossRef CAS.
- T. F. Scott, A. D. Schneider, W. D. Cook and C. N. Bowman, Science, 2005, 308, 1615–1617 CrossRef CAS PubMed.
- S. Schoerpf, Y. Catel, N. Moszner, C. Gorsche and R. Liska, Polym. Chem., 2019, 10, 1357–1366 RSC.
- T. F. Scott, R. B. Draughon and C. N. Bowman, Adv. Mater., 2006, 18, 2128–2132 CrossRef.
- N. Sowan, H. B. Song, L. M. Cox, J. R. Patton, B. D. Fairbanks, Y. Ding and C. N. Bowman, Adv. Mater., 2021, 33, 2007221 CrossRef CAS.
- C. R. Fenoli, J. W. Wydra and C. N. Bowman, Macromolecules, 2014, 47, 907–915 CrossRef CAS.
- H. Lu, J. W. Stansbury and C. N. Bowman, Dent. Mater., 2004, 20, 979–986 CrossRef CAS.
- R. R. Braga and J. L. Ferracane, J. Dent. Res., 2002, 81, 114–118 CrossRef CAS PubMed.
- R. R. Braga, R. Y. Ballester and J. L. Ferracane, Dent. Mater., 2005, 21, 962–970 CrossRef CAS.
- D. Leung and C. N. Bowman, Macromol. Chem. Phys., 2012, 213, 198–204 CrossRef CAS.
- C. J. Kloxin, T. F. Scott and C. N. Bowman, Macromolecules, 2009, 42, 2551–2556 CrossRef CAS.
- B. S. Dauvillier, A. J. Feilzer, A. J. De Gee and C. L. Davidson, J. Dent. Res., 2000, 79, 818–823 CrossRef CAS PubMed.
- H. Fang and C. A. Guymon, Polymer, 2022, 256, 125197 CrossRef CAS.
- X. Shi, Y. Yao, J. Zhang, N. Corrigan and C. Boyer, Small, 2023, 2305268, DOI:10.1002/smll.202305268.
- R. G. Sousa, W. F. Magalhães and R. F. S. Freitas, Polym. Degrad. Stab., 1998, 61, 275–281 CrossRef CAS.
- W. Yin, H. Yang and R. Cheng, Eur. Phys. J. E: Soft Matter Biol. Phys., 2005, 17, 1–5 CrossRef CAS.
- A. Saeed, D. M. R. Georget and A. G. Mayes, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5848–5855 CrossRef CAS.
- L. Xu, L. Che, J. Zheng, G. Huang, X. Wu, P. Chen, L. Zhang and Q. Hu, RSC Adv., 2014, 4, 33269–33278 RSC.
- I. Janigová, K. Csomorová, M. Stillhammerová and J. Bartoň, Macromol. Chem. Phys., 1994, 195, 3609–3614 CrossRef.
- C. K. Chiklis and J. M. Grasshoff, J. Polym. Sci., Part B: Polym. Phys., 1970, 8, 1617–1626 CAS.
- A. Feldermann, A. A. Toy, H. Phan, M. H. Stenzel, T. P. Davis and C. J. P. Barner-Kowollik, Polymer, 2004, 45, 3997–4007 CrossRef CAS.
- J. J. Benvenuta-Tapia, J. A. Tenorio-López and E. Vivaldo-Lima, Macromol. React. Eng., 2018, 12, 1800003 CrossRef.
- U. Capasso Palmiero, A. Chovancová, D. Cuccato, G. Storti, I. Lacík and D. Moscatelli, Polymer, 2016, 98, 156–164 CrossRef CAS.
- B. S. Beckingham, G. E. Sanoja and N. A. Lynd, Macromolecules, 2015, 48, 6922–6930 CrossRef CAS.
- Y. Gu, J. Zhao and J. A. Johnson, Angew. Chem., Int. Ed., 2020, 59, 5022–5049 CrossRef CAS.
- R. Henkel and P. Vana, Macromol. Chem. Phys., 2014, 215, 182–189 CrossRef CAS.
- Q. Yu, S. Xu, H. Zhang, Y. Ding and S. Zhu, Polymer, 2009, 50, 3488–3494 CrossRef CAS.
- R. W. Korsmeyer, R. Gurny, E. Doelker, P. Buri and N. A. Peppas, Int. J. Pharm., 1983, 15, 25–35 CrossRef CAS.
- D. Hariharan and N. A. Peppas, Polymer, 1996, 37, 149–161 CrossRef CAS.
- R. W. Korsmeyer and N. A. Peppas, J. Membr. Sci., 1981, 9, 211–227 CrossRef CAS.
- A. Galperin, T. J. Long and B. D. Ratner, Biomacromolecules, 2010, 11, 2583–2592 CrossRef CAS PubMed.
- S. Sugiura, W. Imano, T. Takagi, K. Sakai and T. Kanamori, Biosens. Bioelectron., 2009, 24, 1135–1140 CrossRef CAS PubMed.
- Q. Xiao, Y. Cui, Y. Meng, F. Guo, X. Ruan, G. He and X. Jiang, Sep. Purif. Technol., 2022, 294, 121224 CrossRef CAS.
- J. C. Garbern, A. S. Hoffman and P. S. Stayton, Biomacromolecules, 2010, 11, 1833–1839 CrossRef CAS.
- M. Patenaude and T. Hoare, ACS Macro Lett., 2012, 1, 409–413 CrossRef CAS.
- K. Nagase, T. Okano and H. Kanazawa, Nano-Struct. Nano-Objects, 2018, 16, 9–23 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lp00144c |
| This journal is © The Royal Society of Chemistry 2024 |