 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurveying the scope of aromatic decarboxylations catalyzed by prenylated-flavin dependent enzymes†
Anushree
Mondal
a,
Pronay
Roy
a,
Jaclyn
Carrannanto
a,
Prathamesh M.
Datar
a,
Daniel J.
DiRocco
a,
Katherine
Hunter
a and
E. Neil G.
Marsh
 *ab
*ab
aDepartment of Chemistry, University of Michigan, Ann Arbor, Michigan, USA. E-mail: nmarsh@umich.edu
bDepartment of Biological Chemistry, University of Michigan, Ann Arbor, Michigan, USA
First published on 5th June 2024
Abstract
The prenylated-flavin mononucleotide-dependent decarboxylases (also known as UbiD-like enzymes) are the most recently discovered family of decarboxylases. The modified flavin facilitates the decarboxylation of unsaturated carboxylic acids through a novel mechanism involving 1,3-dipolar cyclo-addition chemistry. UbiD-like enzymes have attracted considerable interest for biocatalysis applications due to their ability to catalyse (de)carboxylation reactions on a broad range of aromatic substrates at otherwise unreactive carbon centres. There are now ∼35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 protein sequences annotated as hypothetical UbiD-like enzymes. Sequence similarity network analyses of the UbiD protein family suggests that there are likely dozens of distinct decarboxylase enzymes represented within this family. Furthermore, many of the enzymes so far characterized can decarboxylate a broad range of substrates. Here we describe a strategy to identify potential substrates of UbiD-like enzymes based on detecting enzyme-catalysed solvent deuterium exchange into potential substrates. Using ferulic acid decarboxylase (FDC) as a model system, we tested a diverse range of aromatic and heterocyclic molecules for their ability to undergo enzyme-catalysed H/D exchange in deuterated buffer. We found that FDC catalyses H/D exchange, albeit at generally very low levels, into a wide range of small, aromatic molecules that have little resemblance to its physiological substrate. In contrast, the sub-set of aromatic carboxylic acids that are substrates for FDC-catalysed decarboxylation is much smaller. We discuss the implications of these findings for screening uncharacterized UbiD-like enzymes for novel (de)carboxylase activity.
000 protein sequences annotated as hypothetical UbiD-like enzymes. Sequence similarity network analyses of the UbiD protein family suggests that there are likely dozens of distinct decarboxylase enzymes represented within this family. Furthermore, many of the enzymes so far characterized can decarboxylate a broad range of substrates. Here we describe a strategy to identify potential substrates of UbiD-like enzymes based on detecting enzyme-catalysed solvent deuterium exchange into potential substrates. Using ferulic acid decarboxylase (FDC) as a model system, we tested a diverse range of aromatic and heterocyclic molecules for their ability to undergo enzyme-catalysed H/D exchange in deuterated buffer. We found that FDC catalyses H/D exchange, albeit at generally very low levels, into a wide range of small, aromatic molecules that have little resemblance to its physiological substrate. In contrast, the sub-set of aromatic carboxylic acids that are substrates for FDC-catalysed decarboxylation is much smaller. We discuss the implications of these findings for screening uncharacterized UbiD-like enzymes for novel (de)carboxylase activity.
Introduction
Enzyme-catalyzed decarboxylation and carboxylation reactions are both ubiquitous and essential to metabolism. Decarboxylase enzymes are also of increasing interest as “green” catalysts capable of catalyzing stereo- and regio-specific reactions under mild conditions.1,2 Decarboxylases have been used as components in engineered biosynthetic pathways to produce commodity chemicals such as acrylamide and styrene;3 bi-functional alcohols, e.g. 1,4-butanediol and 1,3-propanediol; isobutanol, which shows promise as a biofuel;4 and high-value fine chemicals and pharmaceuticals.1,2 Under favorable conditions, decarboxylases can function as carboxylases and hydratases, leading to interest in using them to capture CO2 and to produce chiral alcohols through stereospecific hydration of double bonds.2Decarboxylation reactions typically are associated with high transition state energies due the build-up of negative charge on the α-carbon, so nature has evolved a wide range of cofactors that function as electron sinks to stabilize the negatively charged transition state.5 Prenylated flavin mononucleotide (prFMN) is the most recently discovered decarboxylation cofactor,6,7 in which the isoalloxazine ring system is modified by the addition of a 6-membered ring bridging the N5 and C6 positions. The additional ring is derived from an isoprene unit that is added to the flavin by a specialized prenyl-transferase; depending upon the organism, the prenyl donor is either dimethylallyl phosphate (DMAP) or dimethylallyl pyrophosphate (DMAPP). prFMN-dependent decarboxylases are collectively known as the UbiD family of decarboxylases, and the prenyl-transferases as UbiX, after the archetypal enzymes identified in the biosynthesis of ubiquinone in many bacteria.8
This unusual modification of flavin mononucleotide (FMN) results in the formation of a nitrogen ylide at N5 of prFMN that is central to the cofactor’s ability to catalyze (de)carboxylation reactions at sp2-hybridized carbon atoms.9–12 prFMN-dependent decarboxylation appears to operate by two distinct mechanisms depending upon the substrate undergoing decarboxylation. In the first mechanism, a 1,3-dipolar cycloaddition reaction occurs between the nitrogen ylide of the cofactor and the double bond adjacent to the carboxyl-group. Formation of the cycloadduct provides a pathway for electrons to flow into the flavin nucleus in the following decarboxylation step (Fig. 1). Following decarboxylation, proton-transfer from a conserved glutamate residue generates a product-like cycloadduct, with the final step being cycloelimination of the product to regenerate the cofactor.7 This mechanism is best established for ferulic acid decarboxylase (FDC), the first prFMN-dependent enzyme to be discovered and the enzyme on which most mechanistic studies have focused.7,13–18 A second mechanism likely operates in enzymes that catalyze decarboxylation of electron-rich aromatic carboxylic acids, such as AroY.19 Here, electrophilic addition of the substrate to C1′ of prFMN provides a plausible route to facilitate decarboxylation of the aromatic ring (Fig. 1).19
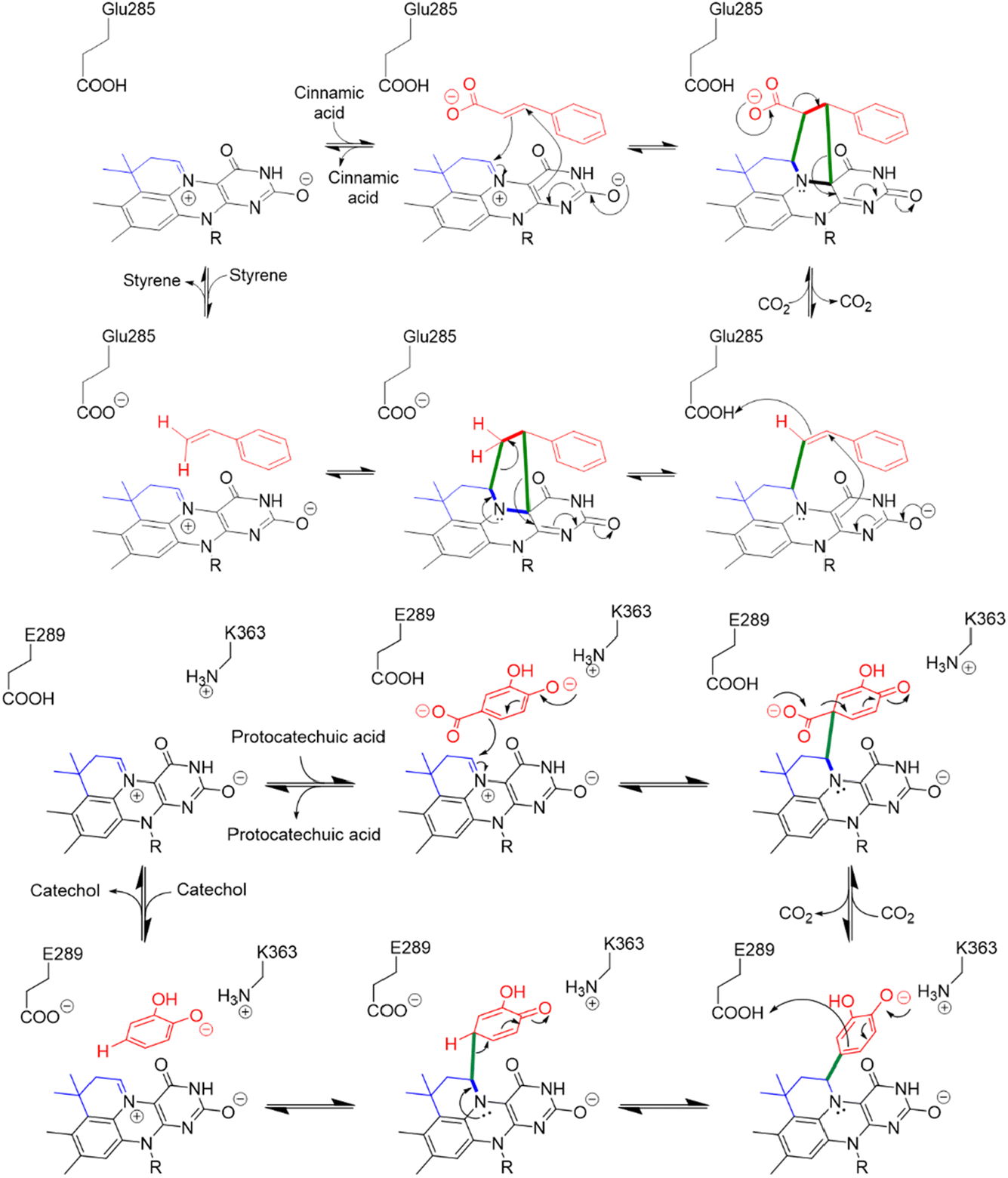 | ||
| Fig. 1 Divergent reaction mechanisms of prFMN dependent decarboxylases: (Top) 1,3-dipolar cycloaddition mechanism proposed for the decarboxylation of cinnamic acid by FDC. (Bottom) Electrophilic addition mechanism proposed for the decarboxylation of protocatechuic acid by AroY. | ||
Since their discovery, it has become apparent that prFMN-dependent (de)carboxylases are widely distributed in microbes,9,20 with ∼35000 putative decarboxylase sequences currently listed in the UbiD UniProt Pfam database. Only a relatively small number of prFMN-dependent enzymes have been biochemically characterized, but it appears that many are involved in the bacterial metabolism of aromatic hydrocarbons. This observation has generated intense interest in exploiting UbiD-like enzymes as selective and environmentally benign catalysts for (de)carboxylation reactions in industrial applications, with recent studies highlighting their potential utility for catalyzing C–C bond forming reactions.12,21,22
A key mechanistic feature of UbiD-like enzymes is their ability to catalyze the exchange of a proton in the product with water, which occurs by partial reversal of the reaction. We have previously demonstrated the exchange of solvent deuterium into both styrene and phenazine catalyzed by the prFMN-dependent enzymes FDC and phenazine-1-carboxylate decarboxylase (PhdA) respectively.17,23 Significantly, this exchange reaction can be observed under conditions in which no carboxylation products can be detected. We have used this property of prFMN-dependent enzymes to screen a range of small aromatic molecules for their ability to react with prFMN, and hence potentially to act as substrates for UbiD-like enzymes. Using FDC as a test case, we show that the enzyme can catalyze H/D exchange for a diverse range of compounds, implying that they react with the cofactor and thus may be substrates for UbiD-like enzymes.
Results and discussion
FDC, like many prFMN-dependent enzymes, exhibits a high degree of promiscuity in the substrates it will decarboxylate. In addition, it was previously found that mutating Ile330 in the substrate-binding cavity to Ser further extended the substrate range of FDC.24,25 Moreover, FDC is one of the easier UbiD-like enzymes to work with because it can be purified as the active holoenzyme. In contrast, many other UbiD-like enzymes require a complex reconstitution procedure to form the holoenzyme that needs enzymatically synthesized prFMN. For these reasons, we chose FDC as the model enzyme for our substrate-screening approach.Based on the structures of the carboxylic acids previously reported as substrates, we assembled a library of aromatic and heterocyclic compounds to test as substrates for FDC-catalyzed H/D exchange, which is shown in Fig. 2. Among these compounds we included non-physiological substrates that were previously reported to be decarboxylated by FDC, and for which one would expect to observe H/D exchange.
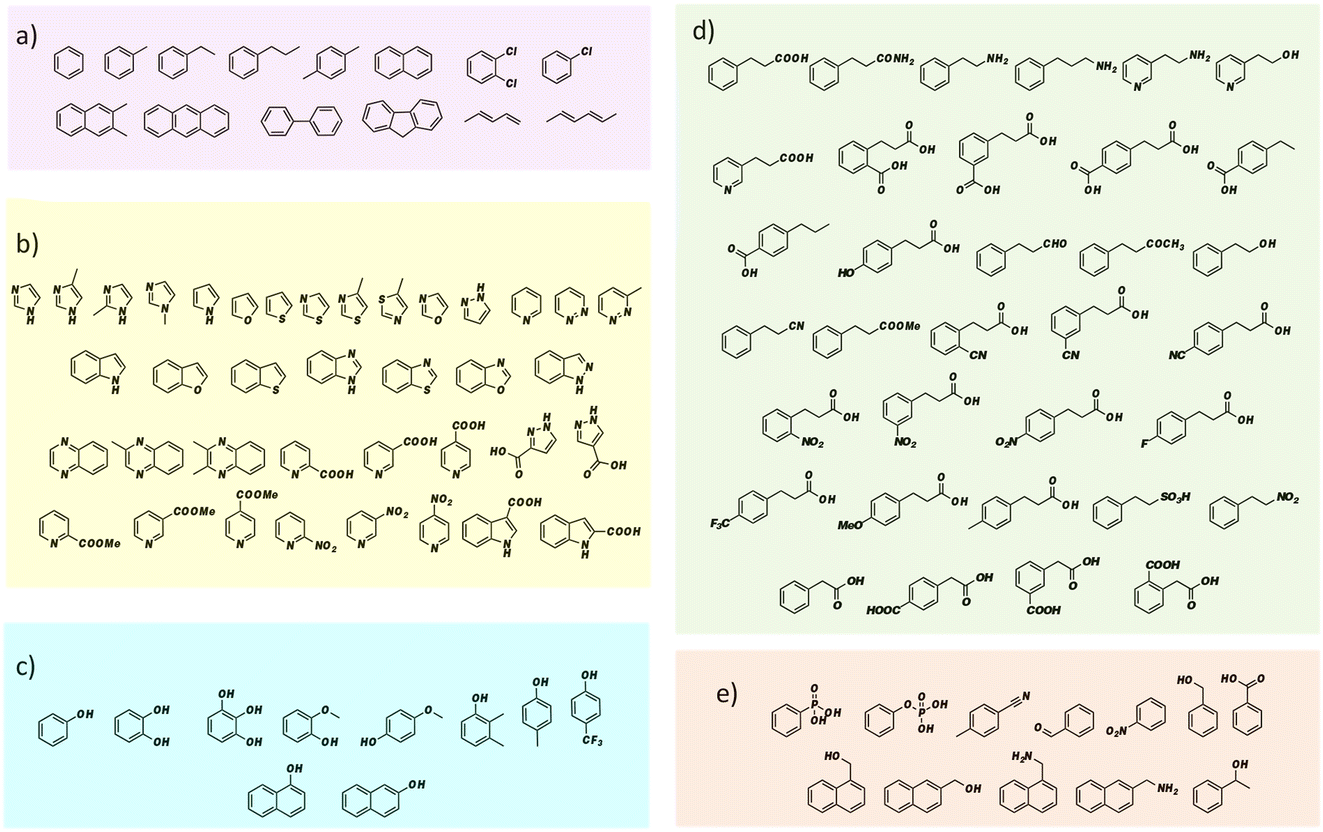 | ||
| Fig. 2 Substrate library for screening FDC-catalyzed solvent H/D exchange grouped by compound type: (a) aromatic hydrocarbon compounds; (b) heterocyclic compounds; (c) phenolic compounds; (d) compounds based on the cinnamic acid framework; (e) other aromatic compounds. | ||
In a typical exchange reaction, FDC or FDC-I330S, 10 μM, was incubated in buffered D2O (∼99 mol% deuterium; pD = 6.5) for 16–18 h at 30 °C with the substrate of interest at 2 mM concentration. Stock solutions of all the substrates were prepared in DMSO to enhance solubility, so that the final reactions typically contained 1–5% DMSO. For each compound examined, two control reactions were set up to account for non-enzymatic H/D exchange: in one reaction no enzyme was added; in the other, apo-FDC was added that had been recombinantly expressed and purified from an E. coli strain in which the endogenous ubiX gene had been deleted.17
The reactions were quenched by adding acetonitrile and their deuterium content analyzed by UHR-MS. To detect the generally low levels of deuterium exchange in these experiments, we used an Orbitrap Fusion Lumos Tribrid mass analyzer to provide sufficient resolution to separate the natural abundance 13C peak (M13C) from the peak due to 2H (a mass difference of 0.0029 amu). Under the experimental conditions used, we were confidently able to detect 2H incorporation (MD peak) with a lower limit of 0.03 mol%.
The compounds that showed significant H/D exchange, i.e. reliably above background levels, in our initial screening experiments are summarized in Fig. 3 and Table S1.† It is apparent that nitrogen-containing heterocycles predominate as hits in this screen. Benzimidazole showed the greatest deuterium incorporation, although this compound also undergoes significant non-enzymatic H/D exchange. Interestingly, other non-heterocyclic compounds that approximate the phenylacrylic acid framework e.g., phenylpropionamide, 4-carboxyphenylpropionic acid and 3-carboxyphenylpropionic acid also showed significant deuterium incorporation with respect to control reactions. Equally intriguing is the observation of H/D exchange with 2-phenylethylamine and 3-phenylpropylamine. It is logical to assume that H/D exchange occurs on the unactivated aromatic ring, with the amino functionality providing solubility, although the low levels of deuterium incorporation do not allow us to identify where in the molecule the isotope is introduced.
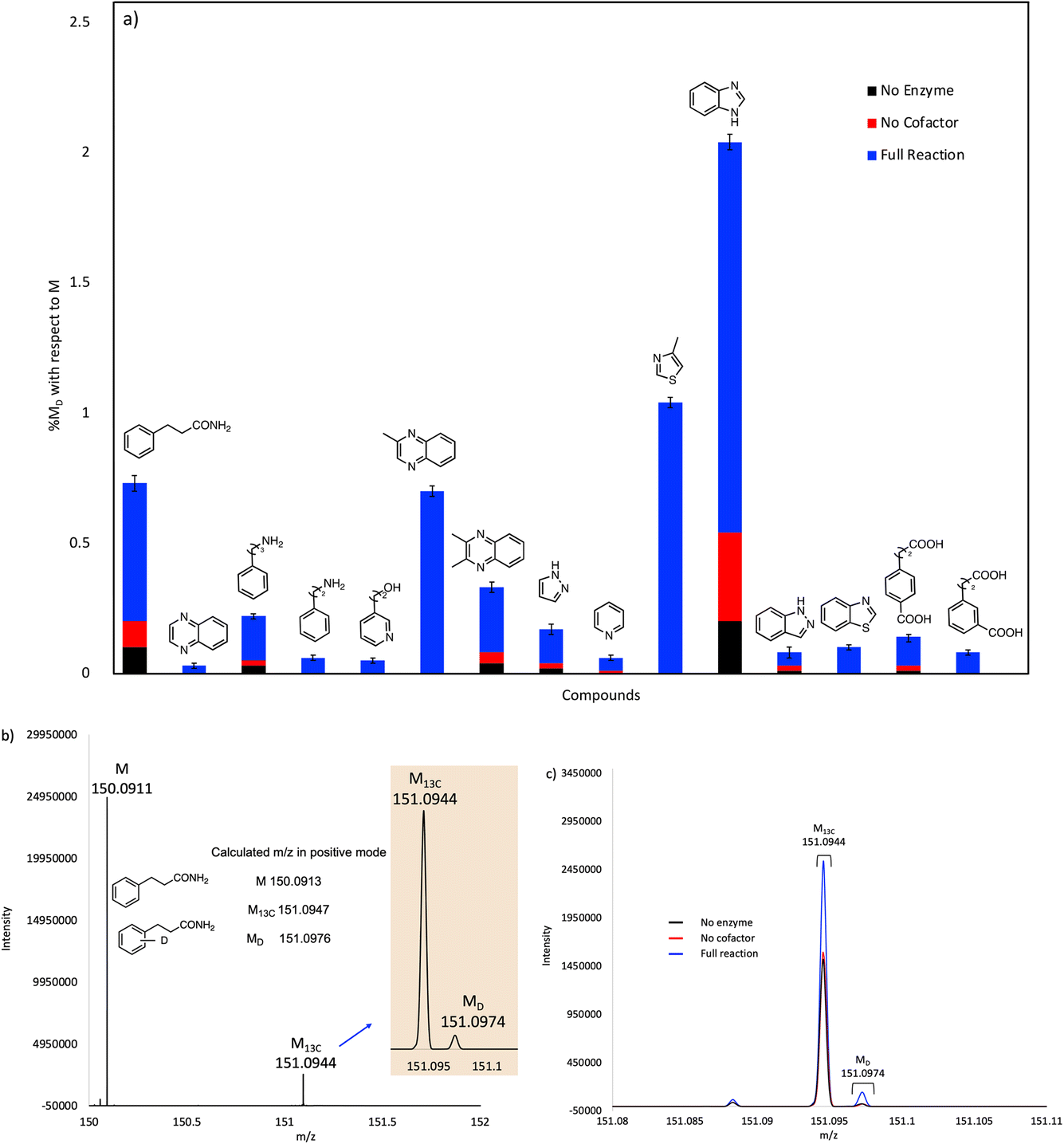 | ||
| Fig. 3 (a) Plot showing solvent deuterium incorporation in various substrates catalyzed by purified, holo-FDC. Mol% deuterium incorporations (% MD) are shown relative to molecular ion (M) peak for each compound. (b) Mass spectrum showing the calculated and experimental M, M13C and MD peaks for 3-phenylpropanamide substrate. (c) Mass spectra for 3-phenylpropanamide of the experimental reaction, no enzyme, and no cofactor controls overlayed for comparison. | ||
Our initial screening studies also revealed some limitations to this approach. Although the methodology provides a very sensitive test for whether a compound can be activated by prFMN, this approach requires that the non-enzymatic, background rate of deuterium exchange into the compound of interest is low or negligible and also that the compound can be analyzed by a suitable high resolution MS technique. These limitations prevented us from evaluating two classes of aromatic compounds as substrates for FDC. Thus, the unfunctionalized aromatic and unsaturated hydrocarbons, shown in Fig. 2a, although of significant interest as UbiD-like enzyme substrates, could not be reliably evaluated due to their low solubility and the technical difficulty of analyzing small amounts of these compounds by high-resolution MS (Fig. S1†). Even styrene, which we previously showed by NMR undergoes FDC-catalyzed deuterium exchange, could not be reliably detected by MS due to its unfavorable ionization properties. Phenolic compounds represent another class of interesting potential substrates that we were unable to analyze. Although phenols can be readily ionized and detected by electrospray MS, high rates of non-enzymatic H/D exchange, due to keto–enol tautomerism, obscured any potential enzymatically catalyzed H/D exchange (Fig. S1d†).
Our initial studies were performed using purified FDC, but we subsequently switched to using freeze-dried cell lysates prepared from E. coli strains over-expressing FDC and the cognate prenyl-transferase, PAD1, with the aim of increasing the degree of H/D exchange.25 Control reactions were set up in which substrates were incubated in buffered D2O under identical conditions to account for non-enzymatic H/D exchange. As a further control, lysates were prepared from a ΔubiX E. coli strain that expresses FDC but cannot make prFMN (Fig. S2 and S3†) to account for any potential non-prFMN dependent H/D exchange. Using this approach, the amount of H/D exchange observed in the experimental reactions increased significantly (Fig. 4 and Table S2†), whereas the amount of H/D exchange in the control reactions remained similar.
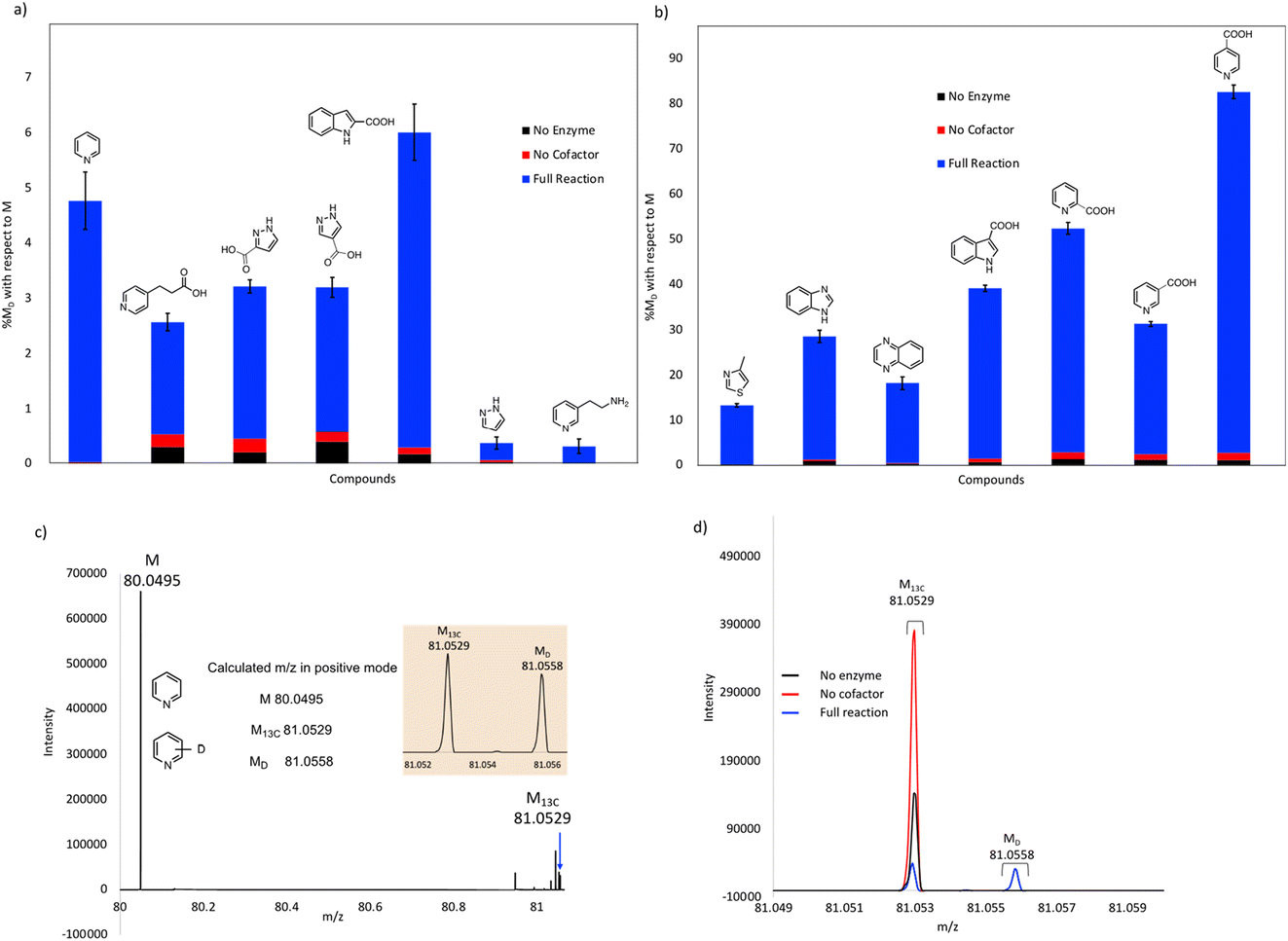 | ||
| Fig. 4 Exchange of deuterium into compounds catalyzed by FDC in lyophilized cell-free extracts. % deuterium incorporations (% MD) are shown relative to molecular ion (M) peak for each compound: (a) compounds showing less than 10% H/D exchange, (b) compounds showing greater than 10% H/D exchange. (c) Mass spectrum showing the calculated and obtained M, M13C and MD peaks for pyridine. (d) Mass spectra for pyridine of the experimental reaction, no enzyme, and no cofactor controls overlayed for comparison. | ||
As expected, compounds that were active with purified enzyme were also active with FDC present in crude cell lysates. It appears that other proteins in the cell lysate may stabilize FDC, resulting in higher levels of H/D exchange. In general, substrates containing N-based heterocycles showed significant deuterium incorporation. In particular, we observed that several heterocyclic carboxylic acids underwent moderate to high deuterium incorporation compared to other substrates. For example, pyridine 2-, 3-, and 4-carboxylic acids showed deuterium incorporations varying from ∼28 to 79% (see also Fig. S4†). In contrast, the corresponding methyl esters of these carboxylic acids and the 2-, 3- and 4-nitro pyridines did not show any deuterium exchange when tested as potential substrates. These results suggest that the carboxylate group may be important for binding and/or orientation of these substrates in the active site of FDC.
In a few cases, the use of cell lysates resulted in a sufficient increase in H/D exchange for the position of deuterium incorporation to be established by NMR. This is best illustrated for the case of indole. For this compound 1H NMR analysis clearly shows that the proton on C-3 of the indole ring is exchanged with solvent (Fig. 5). We note that indole-2-carboxylic acid has been reported as a substrate for FDC24,25 and that H/D exchange of indole has also been previously reported; however the NMR spectrum was mis-interpreted to assign the site of deuterium exchange to be C-2.25 Similarly, for the case of pyridine 4-carboxylic acid, NMR analysis indicated that the proton at C-3 exchanges with solvent (Fig. S5†). Based on these observations we tested both indole-2-carboxylic acid and indole-3-carboxylic acid as substrates for FDC (Fig. S6†). We found that FDC could catalyze the decarboxylation of both compounds, consistent with previous reports.26 This example encouraged us to examine whether carboxylic acid derivatives of other molecules that show H/D exchange in the screen would be substrates for FDC.
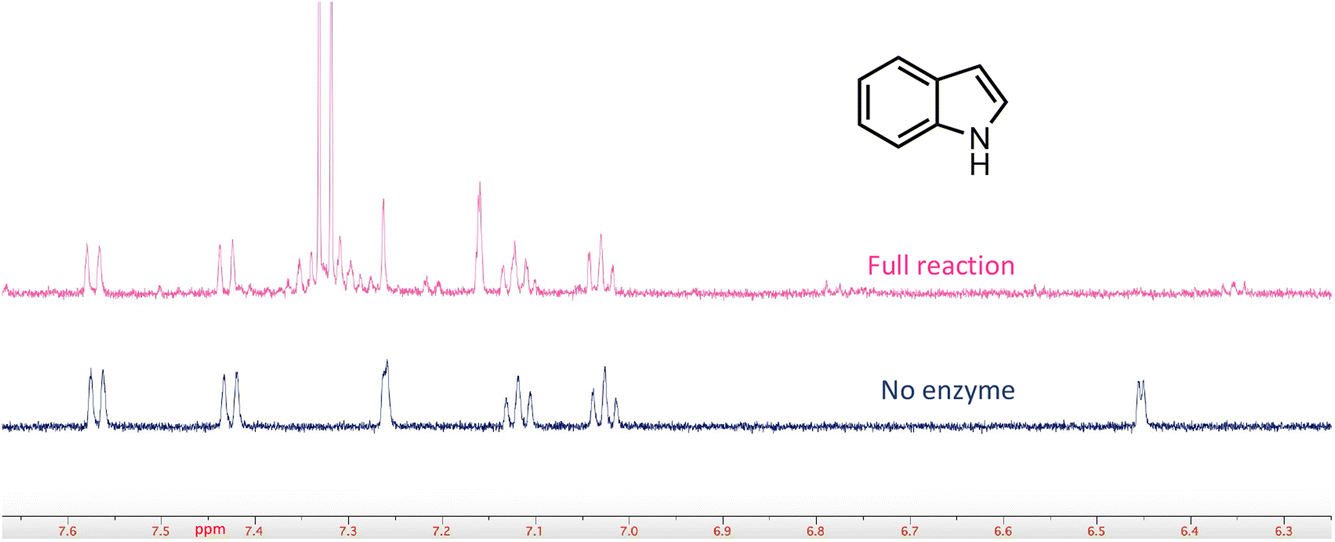 | ||
| Fig. 5 FDC-catalyzed H/D exchange at C-3 of indole monitored by 1H-NMR. | ||
To examine the scope of FDC-catalyzed decarboxylation we screened 60 commercially available carboxylic acid derivatives of the compounds described above that could potentially be substrates for FDC, based on our observation of H/D exchange in these compounds. The various compounds, shown in Fig. 6, were reacted with holo-FDC or holo-FDC-I330S mutant and the formation of decarboxylated products determined by HPLC. In a typical decarboxylation reaction, FDC or FDC-I330S, 10 μM, was incubated in 20 mM phosphate buffer (pH 6.5) for between 10 min and 18 h at room temperature or 30 °C with the substrate of interest at 2 mM concentration. Stock solutions of all the substrates were prepared in DMSO to enhance solubility, so that the final reactions typically contained 1–5% DMSO. Under the conditions of the reaction, decarboxylation should be strongly favored.
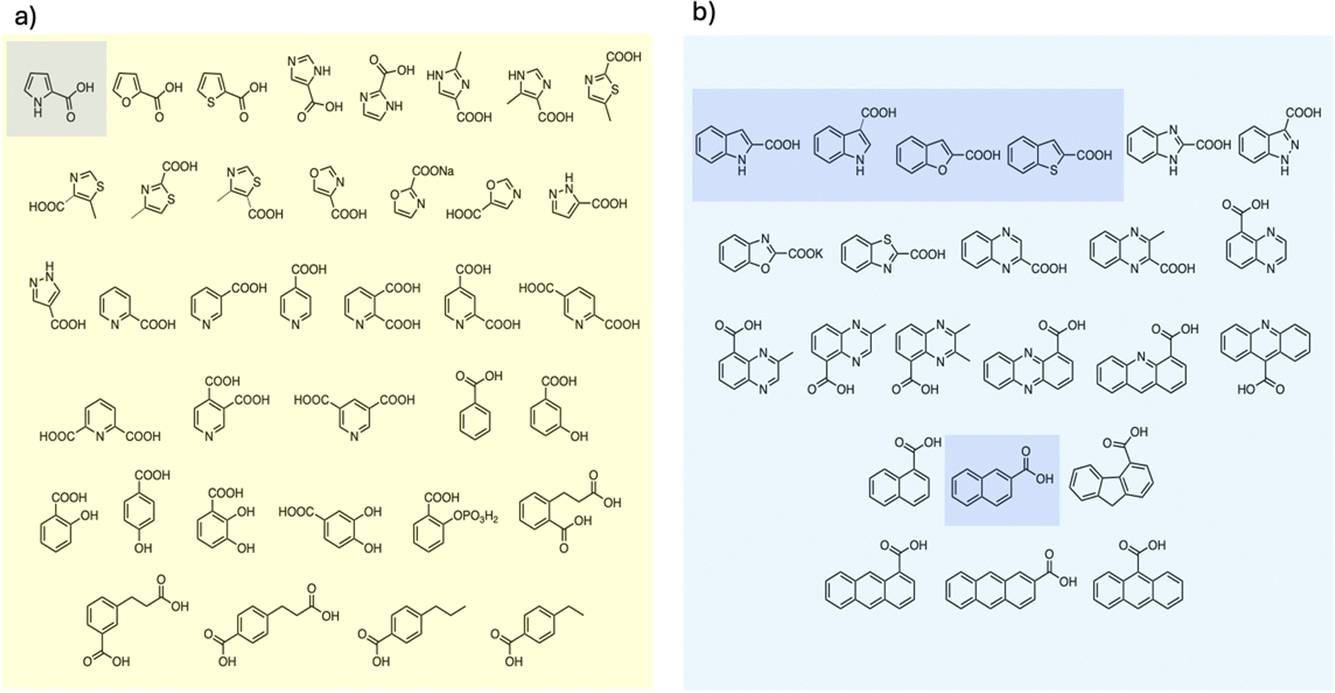 | ||
| Fig. 6 Carboxylic acids screened as substrates for (a) holo-FDC; (b) holo-FDC-I330S. Compounds showing detectable levels of decarboxylation are highlighted. | ||
Somewhat surprisingly, apart from the two indole carboxylic acids, only 4 of the carboxylic acids tested were decarboxylated by FDC at detectable levels: these compounds (highlighted in Fig. 6) were pyrrole-2-carboxylic acid, naphthalene-2-carboxylic acid; benzofuran-2-carboxylic acid and benzothiophene-2-carboxylic acid.24 Interestingly, although the pyridine carboxylic acids showed high levels of H/D exchange for all 3 substitution patterns, none of them were substrates for decarboxylation. We further tested all 6 of the possible pyridine dicarboxylic acid isomers, so, in principle, a carboxylate group would have to occupy the site of H/D exchange. However, we found no evidence that FDC catalyzed the decarboxylation of any of these compounds.
To our knowledge, this is the first study to compare the ability of substrates to react with prFMN, as assessed by H/D exchange, with the ability of the enzyme to decarboxylate the cognate carboxylic acid. The discrepancy between the relatively large number of molecules that undergo FDC-catalyzed H/D exchange – implying that they react to form covalent adducts with prFMN in the enzyme active site – compared with the relatively small subset of carboxylic acid derivatives that undergo decarboxylation is informative. This observation indicates that the requirements for (de)carboxylation are more stringent than simply the ability of the substrate to react with prFMN to form an activated substrate adduct. The enzyme presumably also needs to correctly orient the carboxylate group in the active site and facilitate its deprotonation to catalyze decarboxylation.
Two modes of catalysis are proposed for prFMN-dependent enzymes: the cycloaddition mechanism is more likely to operate with electron-poor aromatic systems (Fig. 1 top) whereas electron-rich aromatics favor the electrophilic addition mechanism (Fig. 1 bottom). Our experiments do not distinguish between these mechanisms, but we note that both electron-rich molecules, such as pyrroles and indoles, and electron-poor molecules such as pyridine carboxylic acids undergo H/D exchange suggesting that FDC supports both modes of reaction. Furthermore, the fact that FDC can catalyze H/D exchange with many small molecules such as diazole that lack an obvious resemblance to the native substrate for FDC, suggests that these molecules are intrinsically reactive towards prFMN. Presumably they bind non-specifically and sub-optimally in the hydrophobic active site of FDC, which would account for the low levels of H/D exchange. On the other hand, one might envision that these molecules could be efficient substrates for UbiD-like enzymes that had evolved to recognize them.
Some nitrate and sulfate reducing bacteria are able to grow on benzene as a sole carbon source under strictly anaerobic conditions.27 Labelling studies indicate that benzene is first converted to benzoic acid, which is a common metabolite in the bacterial fermentation of aromatic compounds. There is intriguing but indirect evidence from genomic and RNA transcript analysis that the carboxylation of benzene maybe catalyzed by a UbiD-like enzyme.28,29 It is therefore particularly interesting that a number of molecules that contain an unactivated benzene ring, e.g. 3-phenylpropionamide (Fig. 4), undergo FDC-catalyzed H/D exchange, presumably on the aromatic ring, albeit at low levels. (The technical limitations we encountered in analyzing benzene, described above, unfortunately prevented us from drawing any conclusions as to whether FDC catalyzes H/D exchange on this extremely stable aromatic system.) The observation of H/D exchange on otherwise inert aromatic compounds suggests that the UbiD enzymes might be further developed as catalysts to selectively carboxylate aromatic systems. Such transformations would be especially useful if the resulting carboxylic acid can be removed from the system by further chemical transformations, e.g. by reduction or amidation, as has been recently demonstrated with FDC.21
So far, a relatively small number of prFMN-dependent enzymes have been biochemically characterized to the extent that their substrates are known. However, there are ∼35000 sequences currently annotated as UbiD-like enzymes in the InterPro Pfam database. To better estimate the number of different UbiD-like (de)carboxylases we analyzed the entire UbiD Pfam using the Enzyme Similarity Network (ESN) tool30,31 developed by the Enzyme Function Initiative (Woese Institute for Genomic Biology, U. Illinois). ESN allows very large datasets to be analyzed and provides a useful guide to the potential diversity of function represented in the population.30,31 The sequence similarity network (SSN) generated from analysis of the UbiD Pfam is shown in Fig. 7, onto which are mapped the positions of known UbiD-like enzymes. It is evident from the SSN analysis that there are many clusters from which an enzyme has yet to be characterized. This observation suggests that UbiD-like enzymes may catalyze the (de)carboxylation of a significantly wider array of molecules than has so far been observed.
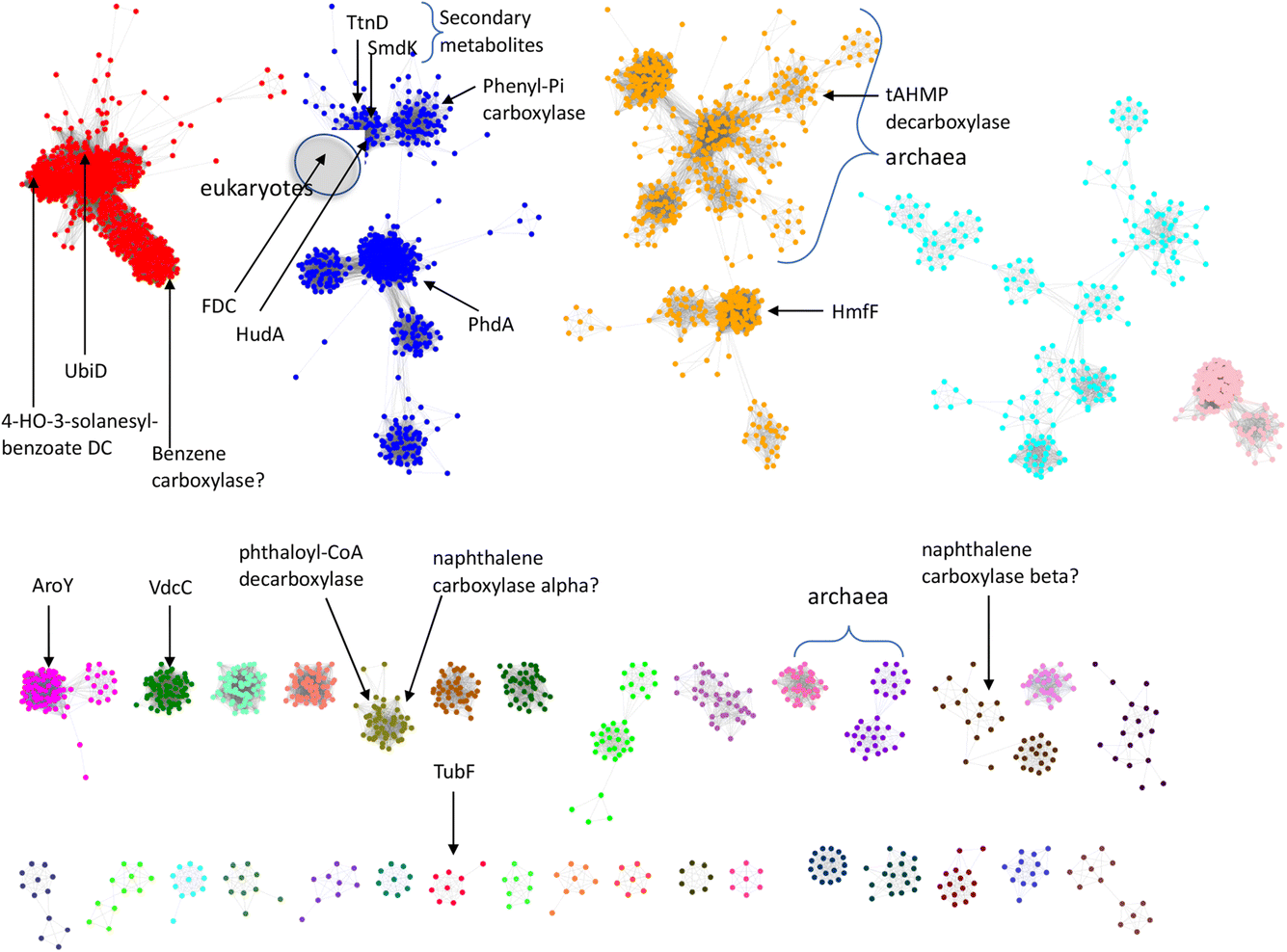 | ||
| Fig. 7 Sequence similarity network analysis of the UbiD Pfam, (edge threshold, E = 100) with locations indicated for UbiD enzymes with known or inferred catalytic activities. | ||
Lastly, we note that although all the UbiD-like enzymes so far characterized are (de)carboxylases, in principle the activation of unsaturated molecules by reaction with prFMN renders them reactive towards other electrophilic molecules. As such, there is the potential for engineering this family of carboxylases towards C–C bond forming reactions with electrophiles such as aldehydes, as has been achieved for some other classes of decarboxylases.2 Indeed, given the large and diverse collection of sequences represented in the UbiD Pfam, it is tempting to speculate that some of these enzymes may have evolved to catalyze reactions other than (de)carboxylations.
Conclusions
The experiments presented here demonstrate that the simple H/D solvent exchange reaction, that is a central mechanistic feature of prFMN-dependent enzymes, provides a very sensitive screen to examine the activation of small molecules by reaction with this cofactor. As a test case, we showed that FDC can catalyse H/D exchange of a very wide array of aromatic and heterocyclic molecules, many of which bear little resemblance to the natural substrate. This screening strategy is quite general and can, in principle, be applied to examine the substrate scope of any prFMN-dependent enzyme. Two caveats emerged from this study. Molecules with protons that can exchange non-enzymatically, e.g. phenolic compounds, cannot be reliably screened due to a high background rate of H/D exchange. Also, very hydrophobic molecules that have poor ionization characteristics, e.g. unfunctionalized hydrocarbons, proved unsuitable for analysis with the MS instrumentation at our disposal.A surprising finding is that although many molecules underwent H/D exchange, implying that they reacted with prFMN, very few molecules were substrates for decarboxylation. This observation implies that decarboxylation is functionally distinct from the formation of a substrate– or product–prFMN adduct. The constraints for carboxylic acids binding productively within the active site are presumably more stringent than those for the corresponding product molecule. This separation of the two halves of the catalytic cycle – decarboxylation and protonation – suggests that it may be possible to engineer UbiD-like enzymes to accept other electrophiles apart from CO2. Our SSN analysis suggests that there are many so far uncharacterized UbiD-like enzymes; these may prove valuable catalysts for (de)carboxylation reactions and possibly even catalyse other types of C–C bond forming reactions with suitable electrophiles.
Materials and methods
Substrates for screening in H/D exchange and decarboxylation reactions were purchased from Sigma-Aldrich, Enamine Building Blocks, Ambeed, TCI Chemicals, Fisher Scientific, 1Click Chemistry, Acros Organics, Combiblocks, Alfa Chemistry, Matrix Scientific, Santacruz Biotechnology and AK Scientific. IPTG and kanamycin sulfate were obtained from GoldBio and Thermo Scientific respectively. Histrap columns were obtained from Cytiva. 10 DG desalting columns were obtained from Bio-Rad. Amicon Ultra centrifugal filter devices (10000 MWCO) were obtained from Millipore. Centrifugal filters for protein filtering were obtained from VWR. Deuterium oxide, DMSO-d6 and acetonitrile-d3 were obtained from Cambridge Isotopic Laboratory. Kinetex LC-18 HPLC column (5 μm particle size, L × I.D. 250 mm × 4.6 mm) was purchased from Phenomenex. HPLC and LC-MS grade solvents (acetonitrile and formic acid) were purchased from Fisher Chemicals. HPLC grade trifluoroacetic acid was purchased from Millipore Sigma.
Overexpression and purification of holo-FDC, FDC-I330S mutant and apo-FDC
Expression and purification of recombinant holo-FDC from a recombinant E. coli expressing the S. cerevisiae FDC gene and a truncated form of the cognate prFMN synthase gene tPAD was performed as described previously.18 The FDC-I330S mutant was constructed by standard site-directed mutagenesis protocols using the Agilent quick change II XL kit. The primers used were forward primer: gacgaaacccatacgctgagcggtagcctg, reverse primer: caggctaccgctcagcgtatgggtttcgtc. FDC-I330S was overexpressed and purified using the same procedure as for holo-FDC. Apo-FDC was prepared from a recombinant E. coli strain from which the endogenous ubiX gene had been deleted as described previously.18Preparation of cell-free protein powders
E. coli strains overexpressing holo-FDC, holo-FDC-I330S mutant or apo-FDC were resuspended in 20 mM potassium phosphate, 500 mM potassium chloride, 10 mM imidazole, 5% glycerol, pH 7.5 (4 mL of buffer/1 g of cell pellet), EDTA-free protease inhibitor tablets (Roche), lysozyme (4 mg lysozyme/1 g of cell pellet), and Benzonase nuclease (Sigma-Aldrich). Cells were lysed by sonication (10 s pulses with 30 s pauses for a total of 15 min of sonication) and the resulting lysate clarified by centrifugation at 13000 rpm at 4 °C for 45 min. After centrifugation, the supernatant was lyophilized overnight to obtain the crude protein powder.25 The enzymatic activity of the powders was determined as described in the supplementary information, Fig. S7.†
MS analyses
All mass spectrometric data were acquired using a Thermo Scientific Lumos Tribrid MS instrument. The orbitrap analyzer was used with a heated-ESI probe and samples were analyzed in either positive or negative ion mode (positive ion spray voltage was 3500 V, negative ion spray voltage was 2500 V, sheath gas 5 Arb units, aux gas 4 Arb units, sweep gas 2 Arb units, ion transfer tube temperature 320 °C, vaporizer temperature 60 °C, scan type was MS scan, orbitrap resolution 240000, RF lens 30%, normalized AGC target 100%, direct injection flow 20 μL min−1). All MS data analysis was performed using the FreeStyle software package.
HPLC analyses
All HPLC analyses were performed on a Shimadzu Prominence LC-20AT series chromatography system equipped with a diode array detector. A Phenomenex kinetex C18 column (5 μm particle size, 250 × 4.6 mm) was used to obtain separation at a flow rate of 0.5 mL min−1 and a detection wavelength of 260 nm (unless specified otherwise). The mobile phase consisted of 0.1% trifluoroacetic acid in water (buffer A) and 0.1% trifluoroacetic acid in acetonitrile (buffer B). Different methods were employed for different analytes. HPLC method 1: 0 min – 50%B, 5 min – 50%B, 15 min – 95%B, 20 min – 95%B, 20.1 min – 50%B, 27 min – 50%B. HPLC method 2: 0 min – 5%B, 1 min – 55%B, 2 min – 55%B, 5 min – 55%B, 15 min – 60%B, 16 min – 95%B, 19 min – 95%B, 19.5 min – 5% B, 25 min – 5% B. HPLC method 3: 0 min – 5%B, 5 min – 5%B, 30 min – 100%B, 35 min – 100%B, 35.1 min – 5%B, 42 min – 5%B. Flow rate 0.4 mL min−1. HPLC method 4: 0 min – 10%B, 5 min – 10%B, 12 min – 100%B, 17 min – 100%B, 17.1 min – 10%B, 22 min – 10%B. Solvents, buffer A consisted of 10 mM Tris/Cl pH 7.2 in water and buffer B was 10 mM Tris/Cl pH 7.2 in 60% acetonitrile:40% water mixture.
H/D exchange reactions
Reactions were performed in D2O (∼99%) buffered with 20 mM potassium phosphate (pD 6.5) containing 10–20 μM holo-FDC, or holo-FDC-I330S mutant, with the substrate of interest at 2 mM. Stock solutions of all substrates were made up as 100× in DMSO. Reactions were incubated for 16–18 h at 30 °C with shaking at 1000 rpm. The reaction was quenched by addition of an equal volume of acetonitrile followed by centrifugation to remove the protein. The supernatant was analyzed by UHR-MS, as described before, to determine the deuterium content of the substrates. Control reactions were performed containing no enzyme or containing apo-FDC. H/D exchange reactions using cell-free extracts were performed similarly. In this case, lyophilized protein powder was dissolved in D2O (∼99%) buffered with 20 mM potassium phosphate (pD 6.5) to give holo-FDC, holo-FDC-I330S or apo-FDC concentrations of ∼60–70 μM.Decarboxylation reactions
Substrates were evaluated for decarboxylation by incubating them with purified holo-FDC or holo-FDC-I330S mutant (10 μM) in 20 mM potassium phosphate buffer (pH 6.5). Substrate were present at 2 mM final concentration, diluted from 100× stocks made up in DMSO. Reactions were performed at room temperature or 30 °C with shaking at 1000 rpm for varying times, ranging from 10 min to 18 h. Reactions were quenched by addition of acetonitrile followed by centrifugation to remove the protein. The supernatant was analyzed by HPLC to detect decarboxylated products or substrate consumption. Control reactions were performed containing no enzyme or containing apo-FDC.Abbreviations
| FMN | Flavin mononucleotide |
| prFMN | Prenylated FMN |
| FDC | Ferulic acid decarboxylase |
| SSN | Sequence similarity network |
| DMAP | Dimethylallyl phosphate |
| DMAPP | Dimethylallyl pyrophosphate |
| IPTG | Isopropyl-β-D-1-thiogalactopyranoside |
| tPAD | Truncated phenylacrylic acid decarboxylase |
| ESI | Electrospray ionization |
| APCI | Atmospheric pressure chemical ionization |
Author contributions
A. M., P. M. D. and E. N. G. M. conceptualized the experiments. A. M., J. C. and K. H. performed the experiments and analyzed the data. P. R. and D. J. D. provided technical advice and analysis. A. M. and E. N. G. M. wrote the manuscript.Conflicts of interest
The authors declare no conflicting interests.Acknowledgements
We thank Profs Kristina Hakansson and Brandon Ruotolo (University of Michigan) for advice on the MS analysis of substrates and products. This work was funded by grants CHE 2203729 and CHE 1904759 from the National Science Foundation to E. N. G. M.References
- J. D. Keasling, Science, 2010, 330, 1355–1358 CrossRef CAS PubMed.
- R. Kourist, J.-K. Guterl, K. Miyamoto and V. Sieber, ChemCatChem, 2014, 6, 689–701 CrossRef CAS.
- R. McKenna, B. Thompson, S. Pugh and D. R. Nielsen, Microb. Cell Fact., 2014, 13, 123 CrossRef PubMed.
- C. Somerville, H. Youngs, C. Taylor, S. C. Davis and S. P. Long, Science, 2010, 329, 790–792 CrossRef CAS PubMed.
- F. Jordan and H. Patel, ACS Catal., 2013, 3, 1601–1617 CrossRef CAS PubMed.
- M. D. White, K. A. Payne, K. Fisher, S. A. Marshall, D. Parker, N. J. Rattray, D. K. Trivedi, R. Goodacre, S. E. Rigby, N. S. Scrutton, S. Hay and D. Leys, Nature, 2015, 522, 502–506 CrossRef CAS PubMed.
- K. A. P. Payne, M. D. White, K. Fisher, B. Khara, S. S. Bailey, D. Parker, N. J. W. Rattray, D. K. Trivedi, R. Goodacre, R. Beveridge, P. Barran, S. E. J. Rigby, N. S. Scrutton, S. Hay and D. Leys, Nature, 2015, 522, 497–501 CrossRef CAS PubMed.
- R. Meganathan, FEMS Microbiol. Lett., 2001, 203, 131–139 CrossRef CAS PubMed.
- D. Leys, Curr. Opin. Chem. Biol., 2018, 47, 117–125 CrossRef CAS PubMed.
- V. Piano, B. A. Palfey and A. Mattevi, Trends Biochem. Sci., 2017, 42, 457–469 CrossRef CAS PubMed.
- D. Leys and N. S. Scrutton, Curr. Opin. Struct. Biol., 2016, 41, 19–26 CrossRef CAS PubMed.
- S. E. Payer, K. Faber and S. M. Glueck, Adv. Synth. Catal., 2019, 361, 2402–2420 CrossRef CAS PubMed.
- A. K. Kaneshiro, K. J. Koebke, C. Y. Zhao, K. L. Ferguson, D. P. Ballou, B. A. Palfey, B. T. Ruotolo and E. N. G. Marsh, Biochemistry, 2021, 60, 125–134 CrossRef CAS PubMed.
- S. S. Bailey, K. A. P. Payne, A. Saaret, S. A. Marshall, I. Gostimskaya, I. Kosov, K. Fisher, S. Hay and D. Leys, Nat. Chem., 2019, 11, 1049–1057 CrossRef CAS PubMed.
- S. S. Bailey, K. A. P. Payne, K. Fisher, S. A. Marshall, M. J. Cliff, R. Spiess, D. A. Parker, S. E. J. Rigby and D. Leys, J. Biol. Chem., 2018, 293, 2272–2287 CrossRef CAS PubMed.
- K. L. Ferguson, J. D. Eschweiler, B. T. Ruotolo and E. N. G. Marsh, J. Am. Chem. Soc., 2017, 139, 10972–10975 CrossRef CAS PubMed.
- K. L. Ferguson, N. Arunrattanamook and E. N. G. Marsh, Biochemistry, 2016, 55, 2857–2863 CrossRef CAS PubMed.
- F. Lin, K. L. Ferguson, D. R. Boyer, X. N. Lin and E. N. G. Marsh, ACS Chem. Biol., 2015, 10, 1137–1144 CrossRef CAS PubMed.
- S. E. Payer, S. A. Marshall, N. Bärland, X. Sheng, T. Reiter, A. Dordic, G. Steinkellner, C. Wuensch, S. Kaltwasser, K. Fisher, S. E. J. Rigby, P. Macheroux, J. Vonck, K. Gruber, K. Faber, F. Himo, D. Leys, T. Pavkov-Keller and S. M. Glueck, Angew. Chem., Int. Ed., 2017, 56, 13893–13897 CrossRef CAS PubMed.
- T. Annaval, L. Han, J. D. Rudolf, G. B. Xie, D. Yang, C. Y. Chang, M. Ma, I. Crnovcic, M. D. Miller, J. Soman, W. J. Xu, G. N. Phillips and B. Shen, ACS Chem. Biol., 2018, 13, 2728–2738 CrossRef CAS PubMed.
- G. A. Aleku, A. Saaret, R. T. Bradshaw-Allen, S. R. Derrington, G. R. Titchiner, I. Gostimskaya, D. Gahloth, D. A. Parker, S. Hay and D. Leys, Nat. Chem. Biol., 2020, 16, 1255–1260 CrossRef CAS PubMed.
- K. Kawanabe, R. Aono and K. Kino, J. Biosci. Bioeng., 2021, 132, 18–24 CrossRef CAS PubMed.
- P. M. Datar and E. N. G. Marsh, ACS Catal., 2021, 11, 11723–11732 CrossRef CAS.
- G. A. Aleku, A. Saaret, R. T. Bradshaw-Allen, S. R. Derrington, G. R. Titchiner, I. Gostimskaya, D. Gahloth, D. A. Parker, S. Hay and D. Leys, Nat. Chem. Biol., 2020, 16, 1255–1260 CrossRef CAS PubMed.
- L. Wang, Y. Lou, W. Xu, Z. Chen, J. Xu and Q. Wu, ACS Catal., 2022, 12, 783–788 CrossRef CAS.
- D. Gahloth, K. Fisher, K. A. Payne, M. Cliff, C. Levy and D. Leys, J. Biol. Chem., 2022, 298, 101771 CrossRef CAS PubMed.
- R. U. Meckenstock, M. Boll, H. Mouttaki, J. S. Koelschbach, P. C. Tarouco, P. Weyrauch, X. Y. Dong and A. M. Himmelberg, J. Mol. Microbiol. Biotechnol., 2016, 26, 92–118 CAS.
- S. Atashgahi, B. Hornung, M. J. van der Waals, U. N. da Rocha, F. Hugenholtz, B. Nijsse, D. Molenaar, R. van Spanning, A. J. M. Stams, J. Gerritse and H. Smidt, Sci. Rep., 2018, 8, 4490 CrossRef PubMed.
- I. Heker, G. Haberhauer and R. U. Meckenstock, Appl. Environ. Microbiol., 2023, 89, e01927 CrossRef PubMed.
- R. Zallot, N. Oberg and J. A. Gerlt, Biochemistry, 2019, 58, 4169–4182 CrossRef CAS PubMed.
- J. A. Gerlt, J. T. Bouvier, D. B. Davidson, H. J. Imker, B. Sadkhin, D. R. Slater and K. L. Whalen, Biochim. Biophys. Acta, Proteins Proteomics, 2015, 1854, 1019–1037 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4fd00006d |
| This journal is © The Royal Society of Chemistry 2024 |