
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUpcycling of plastic membrane industrial scraps and reuse as sorbent for emerging contaminants in water†
Sara
Khaliha‡
a,
Francesca
Tunioli‡
a,
Luca
Foti
a,
Antonio
Bianchi
a,
Alessandro
Kovtun
 a,
Tainah Dorina
Marforio
bc,
Massimo
Zambianchi
a,
Cristian
Bettini
a,
Elena
Briñas
de,
Ester
Vázquez
de,
Letizia
Bocchi
f,
Vincenzo
Palermo
ag,
Matteo
Calvaresi
bc,
Maria Luisa
Navacchia
a and
Manuela
Melucci
*a
a,
Tainah Dorina
Marforio
bc,
Massimo
Zambianchi
a,
Cristian
Bettini
a,
Elena
Briñas
de,
Ester
Vázquez
de,
Letizia
Bocchi
f,
Vincenzo
Palermo
ag,
Matteo
Calvaresi
bc,
Maria Luisa
Navacchia
a and
Manuela
Melucci
*a
aInstitute for Organic Synthesis and Photoreactivity (ISOF), National Research Council of Italy (CNR), Via P. Gobetti 101, I-40129 Bologna, Italy. E-mail: manuela.melucci@isof.cnr.it
bDepartment of Chemistry ‘G. Ciamician’, Alma Mater Studiorum – University of Bologna, Via Selmi 2, 40126 Bologna, Italy
cCenter for Chemical Catalysis – C3 Alma Mater Studiorum – University of Bologna, Via Selmi 2, 40126 Bologna, Italy
dDepartment of Organic Chemistry, Faculty of Science and Chemistry Technologies, University of Castilla-La Mancha (UCLM), 13071, Ciudad, Spain
eRegional Institute of Applied Scientific Research (IRICA), University of Castilla-La Mancha, 13071, Ciudad Real, Spain
fMedica Spa, Via Degli Artigiani, 41036 Medolla, Modena, Italy
gChalmers University of Technology, Chalmersplatsen 4, 41296 Göteborg, Sweden
First published on 2nd April 2024
Abstract
Scraps obtained as waste of the industrial production of polysulfone and polysulfone–graphene oxide hollow fiber membranes (PSU-HF and PSU–GO-HF, respectively) were converted into granular materials and used as sorbents of several classes of emerging and standard water contaminants, such as drugs, heavy metal ions, and a mixture of per- and poly-fluoroalkyl substances (PFASs). The millimetric sized granules (PSU and PSU–GO, respectively) outperformed granular activated carbon (GAC), the industrial sorbent benchmark, in the adsorption of lead, diclofenac, and PFOA from tap water. Adsorption mechanism insight was achieved by molecular dynamics simulations, demonstrating the key role of graphene oxide (GO) on PSU–GO material performance. With respect to GAC, PSU–GO adsorption capacity was two times higher for diclofenac and PFOA and ten times higher for lead. Material safety was assessed by surface enhanced Raman spectroscopy, excluding GO nanosheets leaching, and combined potability test. Overall, our work proves that scrap conversion and reuse is a valuable strategy to reduce plastic industrial waste disposal and to integrate standard technology for enhanced water purification.
Water impactWaste derived from the industrial production of polysulfone hollow fibers (PSU-HF) and PSU–graphene oxide hollow fibers (PSU–GO-HF) can be converted into high-value adsorbent materials. Safe and innovative granules are manufactured from such production scraps and are exploited in the purification of drinking water, targeting the removal of emerging contaminants, such as PFASs. Molecular dynamics simulations were performed to highlight the adsorption mechanism. PSU–GO granules exhibited superior performance in the removal of lead, PFOA, and diclofenac, with respect to granular activated carbon (GAC). |
Introduction
The last seventy years have seen a fiftyfold increase in the production of chemicals, which is expected to triple again by 2050.1 Such chemicals are applied in thousands of industrial and civil products, and it is extremely challenging to introduce safe and sustainable technologies for their removal from the environment. The saturation limit capacity for some of these chemicals (e.g. per- and poly-fluoroalkyl substances, PFASs) has already been reached,2–4 calling for the urgent adoption of risk-mitigation actions and the development of new remediation strategies. Currently, great attention is focused on the removal of ‘emerging contaminants’ (ECs), i.e. pharmaceuticals, cosmetics, and pesticides, from water sources. Adsorption on granular activated carbon (GAC) is the most exploited technology to remove ECs from water in potabilization plants5,6 and more than 5 million tons of GAC are produced annually for water treatment applications.7 The environmental impact of GAC production, activation, transport, regeneration, and disposal is almost incalculable. Moreover, GAC shows poor adsorption performance for several classes of ECs, such as short chain PFASs and other small polar molecules, or metal and heavy metal ions.8,9 Finding alternative materials and strategies to replace GAC and to widen removal selectivity and efficiencies toward ECs is extremely challenging.In the last decade, new materials and technologies have been proposed as adsorbents of various class of contaminants. These include biochar,10 metal–organic frameworks11 and graphene related nanomaterials.12,13 Among them, graphene oxide (GO) exhibited remarkable maximum adsorption capacities (Qmax) for several organic pollutants with respect to GAC, i.e. for the ofloxacin antibiotic, Qmax of 650 mg gGO−1vs. 95 mg gGAC−1 was reported.12
Given the large global request of materials for water treatment, sustainability issues related to their production should be considered when proposing new solutions. In this respect, sorbents derived from industrial wastes are particularly interesting.
Due to their abundance and easy processability, plastic waste deriving from the production of polyethylene (PE), polypropylene (PP), polyvinylchloride (PVC), polystyrene (PS), and polyethylene terephthalate (PET) have been widely investigated. It has been shown that they can adsorb a wide range of pollutants, including toxic, hydrophobic, persistent, and bio-accumulative substances, such as polycyclic aromatic hydrocarbons (PAHs), polychlorinated biphenyls (PCBs), dichloro-diphenyl-trichloroethanes (DDTs), heavy metals, and others.14–16
In this regard, our group recently reported on the conversion of plastic waste derived from the industrial production of polysulfone hollow fiber (PSU-HF) membranes into porous granules.17 PSU-HF are largely exploited membranes for the production of ultrafiltration modules for biomedical filtration,18 gas separation,19 water disinfection,20 and nanomaterials purification.12,21–25
Their graphene oxide modified version (PSU–GO-HF), has further expanded their application range to drinking water purification thanks to the simultaneous filtration and adsorption properties, enabled by GO nanosheets.26,27
The production of commercial PSU-HF and PSU–GO-HF modules requires a hot-wire cutting process to cut the as-spun hollow fibers bundle to fit the final cartridge size (Fig. 1a). The process creates PSU–GO-HF scraps (about 10% of the total mass produced, Fig. 1b), which must be disposed, with consequent economic and environmental costs. It has been estimated that the current yearly production of hollow fiber membranes is approaching the hundreds of thousand tons scale and due to the increasing number of applications (i.e. ultrafiltration, membrane contactors, pervaporation, microfiltration, reverse osmosis, forward osmosis, pressure retarded osmosis, and many other liquid/liquid or liquid/solid separation), the hollow fiber membrane global market projections foresee an annual growth rate of 14.3% from 2023 to 2030, reaching USD 1.76 billion by 2030, meaning also a massive increase of the scraps byproducts.28
 | ||
| Fig. 1 a) Industrial hot-wire cutting of hollow fiber bundles, generating membrane scraps, b) PSU–GO-HF scraps. | ||
We demonstrated that PSU-HF and PSU–GO-HF membrane scraps, from here named PSU and PSU–GO, could be converted into granular porous materials with high potential for drinking water treatment and excellent adsorption capacity toward emerging contaminants, including PFASs.
The selectivity of PSU and PSU–GO toward drugs (i.e. ofloxacin, carbamazepine, and diclofenac),29–31 PFASs (i.e. (CF)3–(CF)13, where (CF)n indicates the number of fluorinated carbons),32–34 and heavy metals (i.e. U, V, Cr, As, Cu, and Pb),35–37 chosen for environmental relevance,38 was studied. Moreover, adsorption capacity tests were performed on three selected contaminants of environmental concern, i.e. diclofenac (DCF),29–31 perfluorooctanoic acid (PFOA)32–34 and lead (Pb).35–37
Production scale-up, which enabled automatic grinding of scraps precursors, allowed the validation of PSU and PSU–GO in standard sized commercial cartridges. Evaluation of such cartridges under domestic tap working conditions, in comparison to commercial standard technologies (GAC and hollow fibers ultrafiltration modules), was also performed.
Experimental
Materials
Ofloxacin (OFLOX), diclofenac (DCF), benzophenone-4 (BP4), carbamazepine (CBZ), bisphenol A (BPA), benzophenone-3 (BP3), rhodamine B (RhB), and caffeine (CAF) were purchased from Sigma-Aldrich (DE) and used without further purification (Fig. S1, ESI†).PFASs standard mixture (CH3CN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O 9:1, 200 μg mL−1) were purchased from Agilent Technologies (Santa Clara, CA, USA) (Fig. S2, ESI†). Ethanol absolute anhydrous was purchased from Carlo Erba Reagents (Val-de-Reuil, Cedex, FR).
:H2O 9:1, 200 μg mL−1) were purchased from Agilent Technologies (Santa Clara, CA, USA) (Fig. S2, ESI†). Ethanol absolute anhydrous was purchased from Carlo Erba Reagents (Val-de-Reuil, Cedex, FR).
Metal salts were purchased from CPA chem Ltd. (BG) as UO2(OOCCH3)2, NH4VO3, Cr(NO3)3, H3AsO4, Cu(NO3)2, Pb(NO3)2, Cd(NO3)2, Ni (NO3)2 in HNO3 2% solution. Nitric acid (≥89.0%) was purchased from Honeywell (FR). Granular activated carbon (GAC) was purchased from CABOT Norit Spa (Ravenna, IT, Norit), product reference: GAC 830 AF (MB index min 240 mg g−1, BET surface area >1000 m2 g−1, details in Table S1, ESI†). To remove sub-millimetric particles, GAC was washed with deionized water at a mild flux, then dried overnight in an oven at 40 °C.
PSU and PSU–GO scraps and empty cartridges and PSU-HF and PSU–GO-HF modules were provided by Medica SpA.
Preparation of granules and cartridges assembly
PSU and PSU–GO granules were prepared by manual or mechanical grinding of commercial PSU-HF and PSU–GO-HF,7 coextruded with a 3.5% content of GO with respect to PSU weight (Fig. S3a and S3b, ESI†).27 For mechanical grinding, a commercial blade grinder (Ceramic Instruments Srl, IT, sieve cut-off = 2 mm, Fig. S18, ESI†), with a production capacity of 0.75 kg h−1, was used.The specific surface area of the granules measured by N2 adsorption (Brunauer–Emmett–Teller model) was in the range of 23–26 m2 g−1 for both materials.39
Small prototype cartridges (14 mm diameter, 65 mm length, dead volume 6 mL, empty bed contact time (EBCT) = 0.5 min, bed volume = 0.01 L) were filled with PSU granules, PSU–GO granules, or GAC (Fig. S4a–c, ESI†). The final weight of material in the cartridges was 0.4 g for PSU, 0.73 g for PSU–GO, and 2.3 g for GAC. These cartridges were used for the lab scale test reported in Fig. 3 and 5. For pilot plant testing (Fig. 6), commercial standard sized and reusable cartridges (49 mm diameter, 250 mm length, dead volume 250 mL, EBCT = 0.14 min, bed volume = 0.5 L) were filled with 33 g of PSU–GO mechanically grinded granules or 33 g of PSU granules or 130 g of GAC (Fig. S4d–f, ESI†). The different material weights reflected the need to maintain consistent EBCT for all adsorbents and to ensure cartridges volume fulfilling.
Adsorption bench-scale test
A tap water solution of eight heavy metals and metalloids (i.e. Pb, Cu, Cd, Ni, Cr(III), As(V), V, and U) at a final concentration of 100 μg L−1 each was prepared and then filtered on the cartridges using the filtration set up in Fig. S5, ESI.† Samples were collected every 250 mL. Each fraction was immediately acidified with 1% HNO3 and analyzed by ICP-MS (details in ESI,† section 4).A solution of eight emerging contaminants, including OFLOX, DCF, BP4, CBZ, BPA, BP3, RhB, and CAF (structures in Fig. S1, ESI†), at 0.5 mg L−1 each, was prepared and then filtered. Samples were collected every 100 mL and analyzed by HPLC-UV (details in the ESI,† section 4).
A solution of fourteen PFASs with alkyl chains in the range (CF)3–(CF)13 (structures in Fig. S2, ESI†) with concentration of 0.5 μg L−1 each in tap water was prepared and filtered on the tested cartridges. Samples were collected after 0.5 L and 1 L of filtration and analyzed by UPLC-MS/MS (Waters ACQUITY® UPLC H-Class PLUS – XEVO TQS Micro mass detector, details in the ESI,† section 4).
In each case, the total filtered volume of water was 1 L and samples were collected in polypropylene test tubes.
Filtration on PSU, PSU–GO, and GAC small cartridges was carried out at a constant flow of 20 mL min−1, corresponding to an EBCT = 0.5 min (set up in Fig. S5, ESI†).
New cartridges were used for each class of contaminants, and all tests were carried out in duplicate, with results reported as the mean value with standard deviation.
Bench-scale loading curves test on DCF, PFOA, and Pb
Experiments were carried out by flowing the spiked tap water through PSU, PSU–GO, and GAC small prototype cartridges (20 mL min−1, EBCT = 0.5 min) and by sampling aliquots at predefined intervals for further analyses and quantification of the contaminant. The experiments were carried out until cartridge saturation was reached (meaning when input concentration equals output concentration, CIN = COUT) or until the removal was about 50% of the initial value. Filtration set up is reported in Fig. S5, ESI.† The initial concentration was CIN = 100 μg L−1 (Pb), 1 mg L−1 (DCF) and 1 μg L−1 (PFOA). The concentration was chosen as the lowest possible in accordance to our detection limits and in good correlation with the maximum concentration found in water (i.e., 50 μg L−1 Pb,40 836 μg L−1 DCF41 and 5–25 μg L−1 PFOA42).New cartridges were used for each contaminant, and all tests were carried out in duplicate, with results reported as the mean value with standard deviation. Details of the protocol used for quantification are reported in the ESI† (section 4).
Cartridge integrity, regeneration and reuse
For GO leaching studies on PSU–GO cartridges 5 L of ultrapure water were filtered at 100 mL min−1 and fractions were collected after each liter. Finally, 10 L were recirculated for 1 h at 100 mL min−1. At the end of the experiment, 11 L of water were filtered. Samples were analyzed by surface-enhanced Raman spectroscopy (SERS). Details of the protocol and of the analysis are reported in ESI† (section 5, Fig. S6, S7 and Table S5).The release of adsorbed contaminants from exhausted cartridges was studied by flowing 1 L of fresh tap water in saturated cartridges at 20 mL min−1. The final concentration of DCF, PFOA, and Pb was analyzed by UV-vis, UPLC-MS/MS, and ICP-MS, respectively.
Regeneration experiments were performed on PSU–GO cartridges previously used for PFOA loading curve (Fig. 5) and then washed by using mQ water/EtOH (1 L) at different ratios (70:30 → 50:50 → 0:100 v/v),34 flowed at 20 mL min−1.
After washing, a solution of PFOA (2 L, 1 μg L−1) was flowed at 20 mL min−1.
Pilot-plant adsorption tests
Adsorption tests were performed on commercial standard sized cartridges already suitable for point-of-use applications and filled by PSU (33 g), PSU–GO (33 g), and GAC (130 g). Other comparative experiments were done by using PSU-HF and PSU–GO-HF commercial ultrafiltration modules.Experiments were performed using a pilot plant directly connected to the tap (flow rate about 3 L min−1, EBCT = 0.14 min in non-continuous sampling mode). Further details on the pilot plant set-up are reported in the ESI† (section 11). Tap water solution of Pb (CIN = 30 μg L−1) and PFOA (CIN = 0.5 μg L−1) were used. For each contaminant a new cartridge was used.
Results and discussion
Material preparation and characterization
Optical microscopy and Scanning Electron Microscopy (SEM) of PSU and PSU–GO, prepared by manual cutting of PSU-HF and PSU–GO-HF, showed granules with sizes in the range of 300 μm-2 mm (Fig. 2). | ||
| Fig. 2 Images of PSU (a) and PSU–GO (b) granules, and SEM images at different magnifications of PSU (c and e) and PSU–GO (d and f). | ||
The cutting process preserved the inner lumen size (250–300 μm), wall section thickness (about 50 μm), inner wall skin porosity (5–80 nm), and outer wall porosity (5–10 μm) of the pristine hollow fibers. Finger-like pore channels in the section of the fibers were also preserved (Fig. 2e and f).
ATR FT-IR and TGA analyses on PSU and PSU–GO showed almost identical features, likely due to the low percentage of GO in the matrix (Fig. S8 and S9, ESI†). TGA curves displayed similar profiles with a slight increase in decomposition temperature for PSU–GO (536 °C vs. 528 °C, and 657 °C vs. 647 °C for PSU and PSU–GO, respectively, Fig. S9, ESI†). The extensive characterization of PSU–GO fibers (before the manual cutting) was reported in our previous work, including SEM, liquid–liquid displacement porometry, contact angle and Raman confocal microscopy. In particular, Raman spectra revealed homogeneous distribution of GO sheets within the hollow fiber, with no evidence of aggregation.27
Bench-scale adsorption selectivity tests
The selectivity of PSU and PSU–GO was studied under flow conditions on mixtures of heavy metals and different classes of organic contaminants in comparison to GAC. Removal results were normalized with respect to the sorbent weight and are shown in Fig. 3 (results in % removal and full data are reported in Fig. S10–S13, ESI†). PSU–GO showed higher selectivity with respect to PSU for all tested metals, in particular toward Pb (103 μg g−1vs. 48 μg g−1), Cu (90 μg g−1vs. 8 μg g−1) and Cr (58 μg g−1vs. 27 μg g−1), while GAC showed negligible adsorptions for all considered metals (Fig. 3b). The observed trend suggests a mechanism, primarily driven by electrostatic interactions between the metal ions and the negatively charged GO surface, as already highlighted in the literature.43 Indeed, the affinity of PSU–GO follows the order Pb(II) > Cu(II) > Cr(III), which well correlates with the electronegativity of the metals (2.3, 1.9, and 1.6, respectively).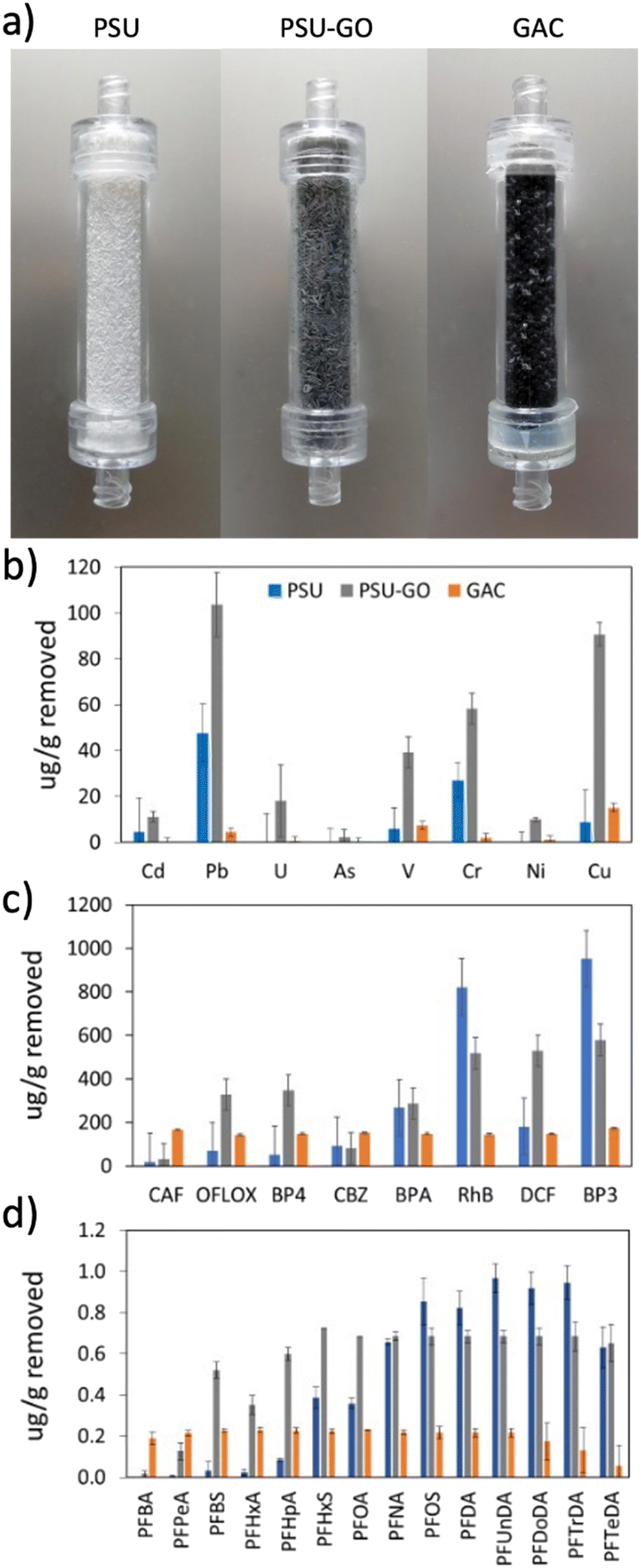 | ||
| Fig. 3 a) PSU, PSU–GO and GAC cartridges and adsorption selectivity on b) heavy metals, c) organic contaminants, and d) PFASs. | ||
With respect to organic contaminants, PSU–GO showed higher selectivity for OFLOX, BP4, and DCF than GAC and PSU (Fig. 3c). On the other hand, the removal of RhB and BP3 was slightly higher for PSU than that of PSU–GO.
With respect to PFASs, PSU–GO showed higher selectivity, compared to GAC, for PFASs with a chain length >(CF)3. PSU showed comparable performance to PSU–GO for >(CF)8. GAC was the only sorbent able to capture perfluorobutyric acid (PFBA, (CF)3) and perfluoropentanoic acid (PFPeA, (CF)4), with a removal >99%, which decreased down to 40% with longer chain length (Fig. 3d).
PFAS adsorption mechanism
Due to the critical environmental relevance of PFASs, and to the higher performance of PSU–GO with respect to PSU on their removal (Fig. 3d), we investigate the adsorption mechanism of PFAS on GO.The adsorption trend of PSU–GO as a function of the n-octanol/water partition coefficient (logKow) of each molecule (expressing the hydrophobicity) for carboxylate PFASs is plotted in Fig. 4a.
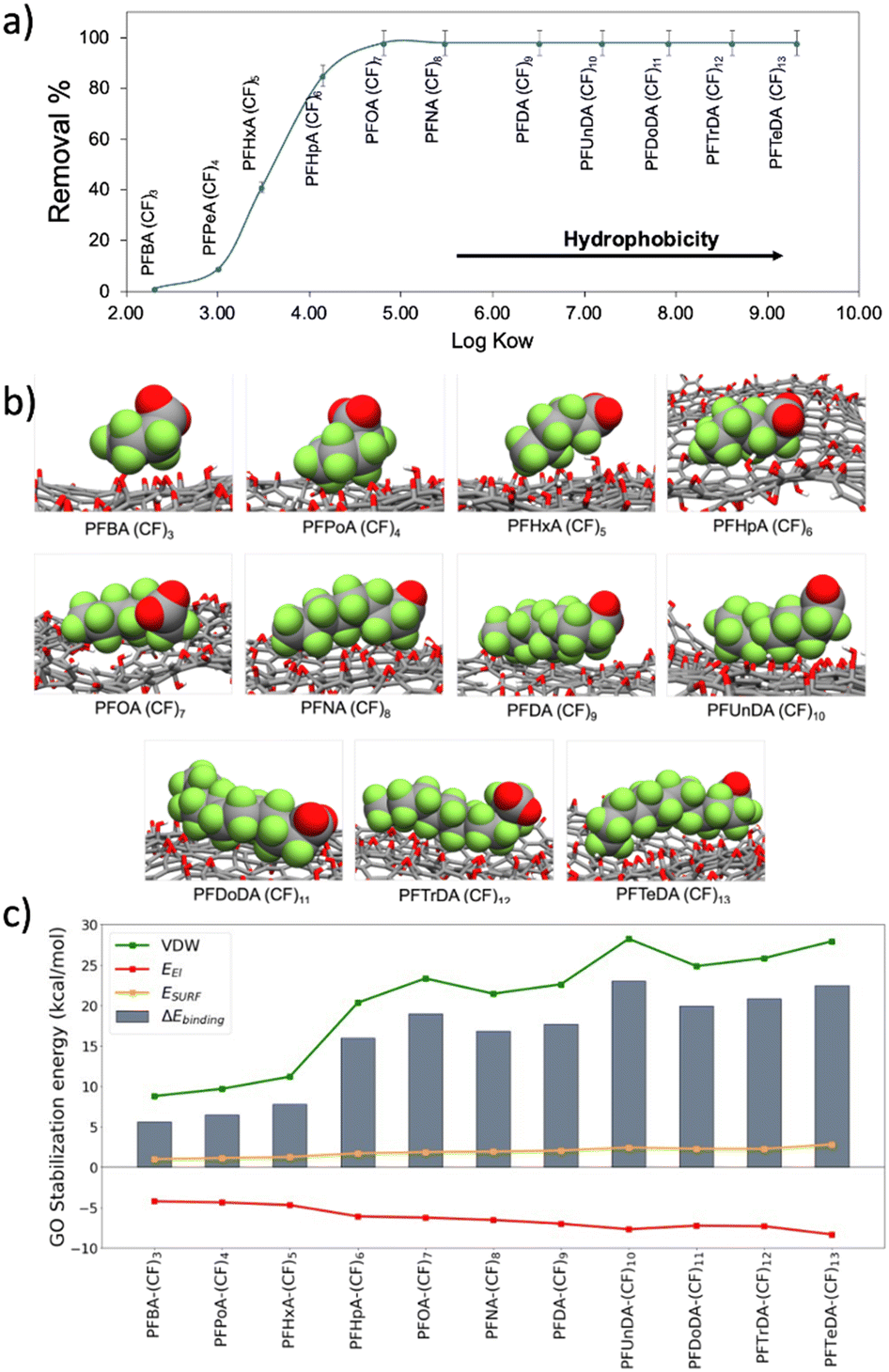 | ||
| Fig. 4 a) Trend of removal vs. logKow of carboxylates PFASs ((CF)3–(CF)13); b) adsorption of PFASs of different chain lengths on GO nanosheets (representative snapshots taken from MD simulations); c) energy components of the ΔEbinding for PFASs of different lengths with GO. Total binding energy (ΔEbinding, grey bars), van der Waals interactions (EvdW, green line), nonpolar solvation (Enonpolar solvation, yellow line), electrostatic terms (Eel, red line). | ||
The removal efficiency increased with the hydrophobicity of contaminant molecule (see Table S6, ESI†). According to previous studies,34,44,45 the two driving forces that need to be considered in PFASs adsorption are electrostatic repulsion and hydrophobic interaction. The comparison between the removals of sulfonate and carboxylate PFASs with same amount of CF ((CF)4: PFBS vs. PFPeA; (CF)6: PFHxS vs. PFHpA; (CF)8: PFOS vs. PFNA) highlights that i) there is a correlation between the number of CF groups and the removal capacity, and ii) due to a higher hydrophobicity of the sulfonate group, sulfonate PFASs are better adsorbed than the carboxylate ones by both PSU and PSU–GO, (Fig. S14 and Table S7, ESI†).
The binding energy (ΔEbinding) between PFASs and GO is obtained by the sum of three energetic terms: electrostatic interactions, van der Waals interactions, and surface energy (Fig. 4c). As the PFASs chain elongates, the ΔEbinding with GO increases, well reproducing the experimental trend. The driving forces controlling the adsorption process are the van der Waals (VDW) interactions, originated between the perfluoroalkyl chains and the GO sheet. VDW contribution is hydrophobic in nature and strongly depends on the adsorbate chain length: the longer the PFAS chain, the stronger the interaction with GO.
Additionally, the surface energy ESURF contribution (hydrophobic effect) assists the binding with an almost constant value among the different PFASs, even if in terms of magnitude ESURF is smaller than the VDW interactions. The surface energy term originates from the hydrophobic perfluoroalkyl chain of the PFAS that interact with the hydrophobic surface of the GO instead of interacting with water, with which the interaction is unfavorable. While VDW and ESURF contributions favor the adsorption process, the electrostatic term (Eel) is detrimental to the binding. This term takes into consideration i) the Coulombic repulsion between the negatively charged GO (ζpotential = −43.1 ± 2.4 mV) and the negatively charged carboxylate of PFASs, and ii) the polar solvation term. The hydrophilic portions of PFASs are forcedly desolvated upon the formation of the complex with GO, causing an overall destabilization of the system.
Altogether, these results confirm that, as previously reported in literature,34 when the hydrophobic interactions (van der Waals plus hydrophobic effect) overcome the electrostatic repulsion between PFASs and GO, the binding of PFASs, and their consequent removal, occurs.
Bench-scale adsorption capacity tests
Based on the selectivity observed in previous experiments (Fig. 3), we selected one contaminant per each class (i.e., DCF, PFOA and Pb) and tested the adsorption capacity of PSU–GO, PSU and GAC. Fig. 5 shows the results in removal % vs. bed volume while results are expressed as a function of output concentration (COUT), or cumulative μg of contaminant removed on g of sorbent (μg g−1) are reported in the ESI† (Fig. S15). Experiments were carried at EBCT = 0.5 min, until saturation conditions occurred (i.e., COUT = CIN) or when the adsorption capacity was half of the initial value.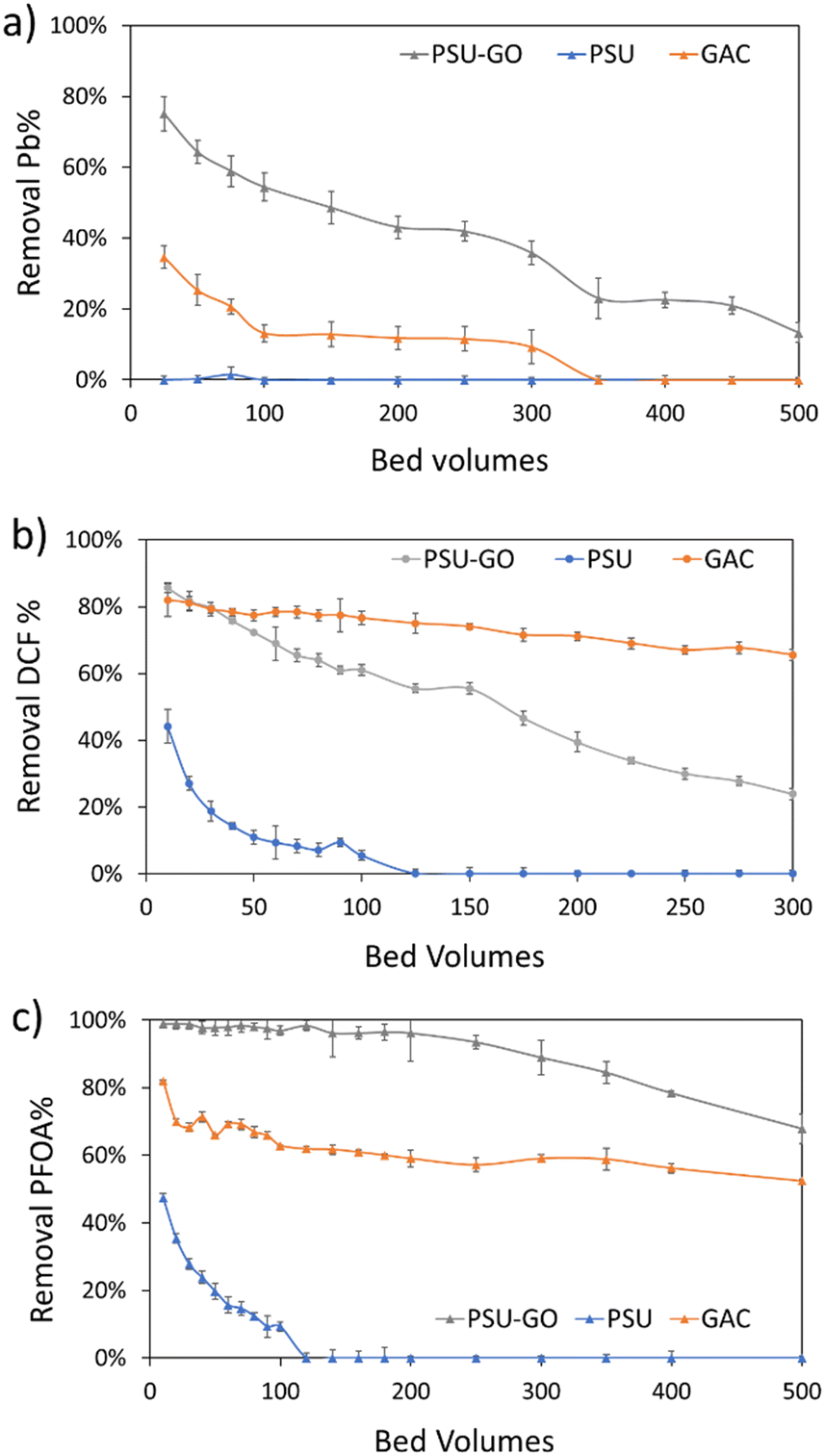 | ||
| Fig. 5 Loading curves of a) Pb, b) DCF, and c) PFOA expressed as removal % vs. bed volumes of PSU (blue lines), PSU–GO (grey lines) and GAC (orange lines). Full results are reported in Fig. S15, ESI.† | ||
In Fig. 5, PSU–GO adsorbed Pb with maximum removal approaching values in the range 75–43%, after 500 bed volumes, while PSU was ineffective, and GAC saturated after 100 bed volumes (Fig. 5a). Similarly, PSU–GO showed higher adsorption capacity than PSU (Fig. 5b) toward DCF, and no saturation was observed even though the adsorption capacity decreases faster than for GAC.
With regards to PFOA, PSU–GO adsorption capacity remained almost constant even after 500 bed volumes (Fig. 5c), outperforming GAC and PSU.
Table 1 summarizes the total amount of contaminant (i.e., Pb, DCF, PFOA) removed, normalized per gram of sorbent. In the case of Pb, the mass removed by PSU–GO was 10 times higher than that obtained with GAC, while PSU showed negligible adsorption. The amount of DCF and PFOA globally removed by PSU–GO was 2 and 6 times higher than the amount adsorbed by GAC and PSU, respectively. This evidence supports our previous study showing that the SSA for N2 measured by BET is not representative of the sorbent capacity in the liquid phase (SSA for N2 being 23 m2g−1vs. 1000 m2g−1).39
| Contaminant | Adsorption capacity (mass of contaminant/mass of adsorbent; μg g−1) | ||
|---|---|---|---|
| PSU | GAC | PSU–GO | |
| Pb | 1.1 | 21.5 | 230.1 |
| DCF | 389.8 | 951.6 | 2400.2 |
| PFOA | 1.1 | 3.2 | 6.1 |
To date, the best sorption performances for Pb, DCF and PFOA have been achieved by using carbonaceous materials, including i) GAC (PFOA 112 mg g−1,34 DCF 6.85 mg g−1,46 Pb 58 mg g−147), ii) GO (PFOA 0.4 mg g−1,48 DCF 128 mg g−1,49 Pb 55.80 mg g−150), iii) carbon-nanotubes (Pb 97 mg g−1,51 PFOA 124 mg g−152) or iv) nanocomposites, such as modified graphene aerogel (Pb 368 mg g−1,53,54 PFOA 1575 mg g−155). However, it should be noted that the above-mentioned materials and performance were estimated from batch experiments and related adsorption isotherms at the equilibrium time (not under flow as in this work), carried out in ultrapure water (not tap drinking water as in this work) and with contact times of hours (rather than seconds as in this work).
Overall, these discrepancies prevent a proper and direct comparison of our results with the literature. To overcome this issue, we characterized GAC and PSU/PSUGO-HF standard cartridges in the same experimental conditions of our materials.
Materials integrity, regeneration and reuse
We investigated the potential leaching of GO nanosheets from PSU–GO cartridges by surface-enhanced Raman spectroscopy (SERS) analysis of filtered water. This methodology is based on the deposition of the analyte on a SERS active substrate based on gold nanoparticles and allows the quantification of GO down to 0.1 μg L−1.56 No significant differences were found between tap water, used as control, and the PSU–GO treated water samples (Fig. S7 and Table S5, ESI†), indicating that no release of GO occurred. In addition, chemical and biological water potability was verified on tap water after filtration (Table S8, ESI†).Moreover, stable adsorption of contaminants was tested by washing the saturated cartridges with fresh tap water and measuring the concentration of the analytes in the washing solution.
Releases lower than 8% for Pb, 6% for DCF, and 1.5% for PFOA (Fig. S16a–c, ESI†) were found.
Finally, given the importance of cartridge regeneration, we carried out some preliminary regeneration tests on cartridges saturated with PFOA.
To this aim, the cartridge was washed with ultrapure H2O/EtOH solution at different ratios and the amount of PFOA recovery under different conditions was estimated. The best recovery in terms of maximum amount recovered (2.1 μg, 45.3%) was achieved by using a solution at 70:30 v/v ratio (ultrapure H2O/EtOH).
The washed cartridge was then used for a second filtration cycle and adsorption capacity are reported in Fig. S17, ESI.† Both cycles showed adsorption efficiency of about 98% suggesting that it is possible to regenerate and reuse PSU–GO cartridges. Further studies on different contaminants will be addressed to fully assess the possibility of reuse for these materials.
Granules production upscale and pilot-plant tests
The scraps grinding process was upscaled by using a commercial blade mechanical grinder with steel blades and production capability of 0.75 kg h−1 (Fig. S18a and b, ESI†). Chromium release from the blades during the grinding was excluded by dedicated tests, with released Cr(III) < 5 ng g−1 (section 11, ESI†).The size of the granules was in the range 0.3–2 mm (due to the grinder cut-off) and a standard sized commercial cartridge was filled with the obtained granules (Fig. S18c, ESI†).
Due to the mechanical stress applied during the grinding process, the granules displayed a flattened and partially opened structure in comparison to manually ground granules, which exhibited a homogeneous tubular shape (Fig. S19, ESI†). However, despite the different morphology, the granules showed adsorption performance very similar to those obtained by manual grinding (Fig. S20, ESI†).
Commercial standard cartridges were filled with PSU–GO granules (Fig. S4, ESI†) and characterized in a pilot plant test on Pb removal.
As shown in Fig. 6b, PSU showed negligible adsorption of Pb (total removal about 8 μg g−1), while PSU–GO removed up to 250 μg g−1, with the highest removal within the first 100 L treated (Fig. 6b and S21, ESI†). Remarkably, comparable adsorption capacity was obtained with small and larger cartridges (230 μg g−1vs. 250 μg g−1), despite the different concentration of Pb (100 μg L−1vs. 30 μg L−1) and EBCT (0.5 min vs. 0.14 min). GAC was not tested since no Pb adsorption was observed in the lab test. In addition, we compared the granule adsorption performance on Pb to the performance of standard commercially available PSU-HF and PSU–GO-HF cartridges, which are the precursors of the granules. As shown in Fig. 6b, neither PSU granules nor PSU-HF removed Pb. On the contrary, PSU–GO granules and PSU–GO HF showed high Pb removal capacity with values of 195 μg g−1 and 202 μg g−1, respectively (treated volume 420 L) suggesting that i) granules and HF are characterized by the same adsorption selectivity and capacity and ii) the adsorption of Pb is exclusively promoted by GO.
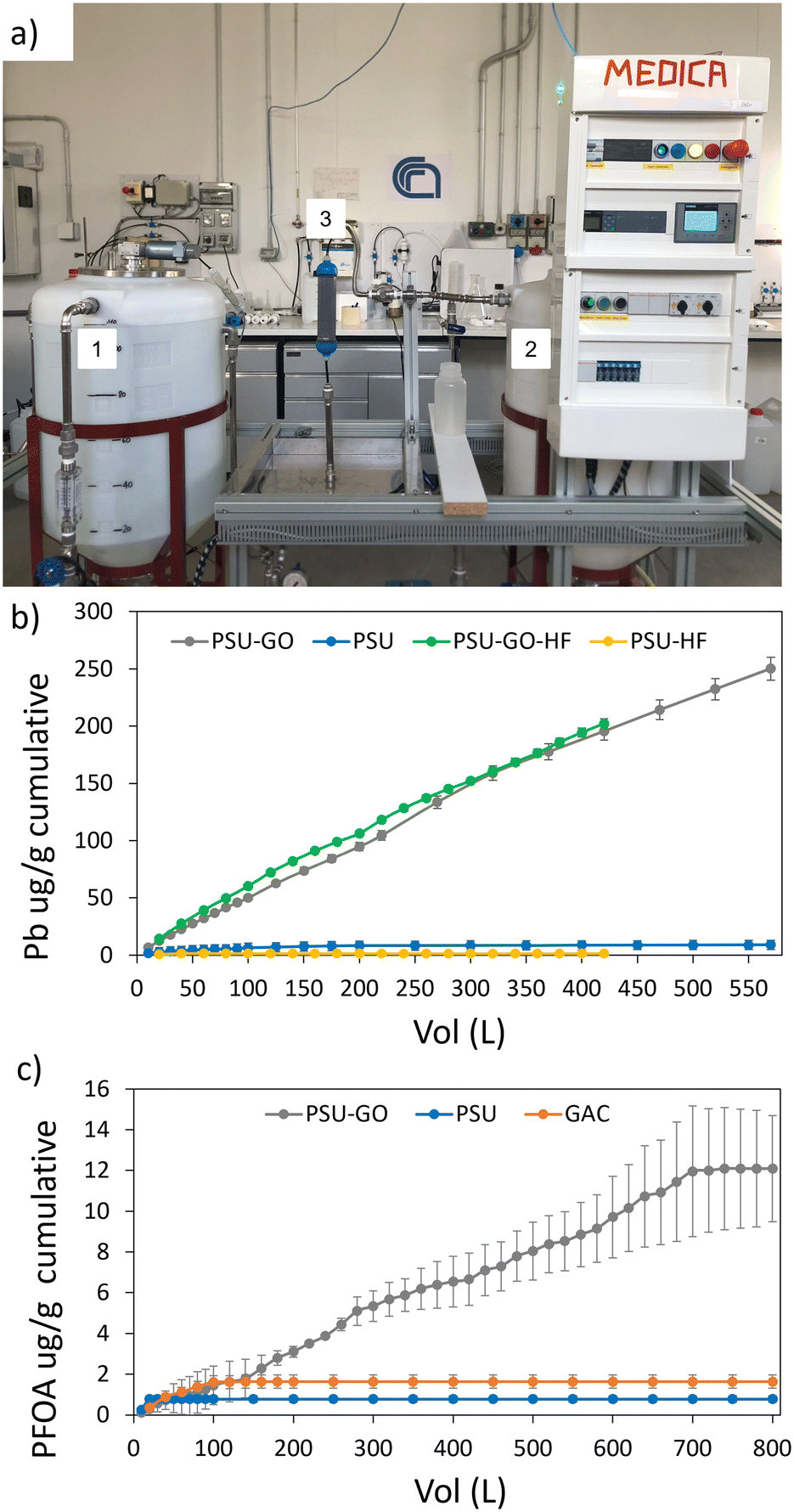 | ||
| Fig. 6 a) Set-up of the pilot plant used in this work. The pilot is connected directly to the tap. Spiked water in tank 1 (100 L capacity) is flowed through the cartridge (PSU–GO in the picture, filter 3) and filtered water is collected in tank 2 (100 L capacity). After 100 L are filtered, water is pumped from tank 2 to tank 1 (bypassing the cartridge) and the concentration is checked and adjusted to the target initial value. b) and c) Comparison between removal capacity on Pb and PFOA. | ||
In the same experimental setup, PSU and PSU–GO cartridges were tested on PFOA removal and compared to GAC and PSU cartridges (Fig. 6c). Remarkably, PSU–GO overcomes GAC and PSU in the adsorption of PFOA with maximum capacities of 12 μg g−1vs. 1.63 μg g−1 and 0.8 μg g−1, respectively.
Conclusions
In conclusion, we reported new sorbent materials derived from waste of the industrial production of polymeric hollow fiber membranes. The scraps were converted into granules (PSU and PSU–GO) through mechanical grinding and their adsorption properties toward selected water contaminants, including PFASs, were characterized.Cartridges of PSU and PSU–GO materials showed excellent adsorption properties toward several contaminants, higher than GAC, highlighting their potential for drinking water purification.
In general, with respect to GAC, PSU showed higher selectivity for BP3 and RhB and for PFASs with chain length > (CF)8. PSU–GO showed higher selectivity, compared to GAC, for Pb, Cu, Cr, OFLOX, BP4, DCF and for PFASs with chain length (CF)3 → (CF)13. Given the interest for PFASs removal and their structural similarity, the adsorption mechanism on GO was investigated by molecular dynamics simulation. Calculations showed that the GO active sites mainly drive the adsorption process and favor the removal of hydrophobic molecules.
In terms of adsorption capacities, PSU–GO removal of DCF and PFOA were more than 2 times higher than GAC and 6 times higher than PSU. Moreover, the maximum Pb removal capacity of PSU–GO was 10 times higher than that obtained with GAC.
A grinding scale up through an automatic grinder with a production capability close to 1 kg h−1 was demonstrated, allowing the fabrication and test of larger cartridges (commercial standard size) and treatment of water volumes up to 800 L.
Test performed under tap operational conditions showed that PSU–GO performances on Pb and PFOA are poorly affected by the flow rate and overcome GAC standard material.
Considering the massive global membrane production and the related mass of scrap byproducts, which is expected to further increase in the next few years, the approach herein described, and the suggested application could contribute to the reduction of plastic waste from the membrane producers.
Moreover, the granular materials obtained from the plastic scraps could be exploited in synergy with other standard technologies, including activated carbon sorption and membrane filtration. Studies in this direction are underway in situ in a municipal potabilization plant (Hera, Fe, Italy, Po River source) for drinking water production.
Author contributions
Sara Khaliha: methodology, investigation, data curation, and editing. Francesca Tunioli: methodology and investigation. Luca Foti: methodology and investigation. Antonio Bianchi: methodology, reviewing and editing. Alessandro Kovtun: investigation and formal analysis. Massimo Zambianchi: methodology and investigation. Cristian Bettini: methodology and investigation. Tainah Dorina Marforio: investigation, methodology, and formal analysis. Maria Luisa Navacchia: investigation and formal analysis. Elena Briñas: investigation. Ester Vázquez: methodology and investigation. Letizia Bocchi: conceptualization, methodology and resources. Vincenzo Palermo: conceptualization and validation. Matteo Calvaresi: methodology and investigation. Manuela Melucci: conceptualization, validation, and writing – original draft.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors gratefully acknowledge the support of this work by the projects Life-Remembrance, ENV/IT/001001 Life Resource and Environment LIFE20 ‘Give plastic wastes from the production of hollow-fiber membranes a second life’, and the project PNRR MUR project ECS_00000033_ECOSISTER. MM thanks M. Bergamini and M. Brunetti (Gruppo HERA Spa, Bologna, Italy) for providing samples of GAC used in their municipal plants.Notes and references
- L. Persson, B. M. Carney, A. C. D. Collins, S. Cornell, C. A. de Wit, M. Diamond, P. Fantke, M. Hassellov, M. Macleod, M. W. Ryberg, P. S. Jorgensen, P. Villarrubia-Gomez, Z. Wang and M. Zwicky Haushild, Outside the Safe Operating Space of the Planetary Boundary for Novel Entities, Environ. Sci. Technol., 2022, 56(3), 1510–1521, DOI:10.1021/acs.est.1c04158.
- T. Teymoorian, G. Munoz, S. Vo Duy, J. Liu and S. Sauvé, Tracking PFAS in Drinking Water: A Review of Analytical Methods and Worldwide Occurrence Trends in Tap Water and Bottled Water, ACS ES&T Water, 2023, 3, 246–261, DOI:10.1021/acsestwater.2c00387.
- F. Xiao, B. Deng, D. Dionysiou, T. Karanfil, K. O'Shea, P. Roccaro, Z. J. Xiong and D. Zhao, Cross-national challenges and strategies for PFAS regulatory compliance in water infrastructure, Nature Water, 2023, 1, 1004–1015, DOI:10.1038/s44221-023-00164-8.
- Scientists Letter: The World Health Organization should significantly revise or withdraw its draft PFAS drinking water guidelines, https://greensciencepolicy.org/docs/General/pfas-scientists-letter-to-who-20221110.pdf.
- G. Bertanza, G. U. Capoferri, M. Carmagnani, F. Icarelli, S. Sorlini and R. Pedrazzani, Long-term investigation on the removal of perfluoroalkyl substances in a full-scale drinking water treatment plant in the Veneto Region, Italy, Sci. Total Environ., 2020, 734, 139154 CrossRef CAS PubMed , https://www.sciencedirect.com/science/article/pii/S0048969720326711.
- Y. Zhang, X. Tan, R. Lu, Y. Tang, H. Qie, Z. Huang, J. Zhao, J. Cui, W. Yang and A. Lin, Enhanced Removal of Polyfluoroalkyl Substances by Simple Modified Biochar: Adsorption Performance and Theoretical Calculation, ACS ES&T Water, 2023, 3(3), 817–826, DOI:10.1021/acsestwater.2c00597.
- Activated Carbon Market Size, Share & Trends Analysis Report By Type (Powdered, Granular), By Application (Liquid Phase, Gas Phase) By End Use (Water Treatment, Air Purification), By Region, And Segment Forecasts, 2022–2030, Report 978-1-68038-073-6, 2020 Search PubMed.
- G. Crini and E. Lichtfouse, Advantages and disadvantages of techniques used for wastewater treatment, Environ. Chem. Lett., 2019, 17, 145–155, DOI:10.1007/s10311-018-0785-9.
- H. B. Quesada, T. P. de Araújo, D. T. Vareschini, M. de Barros, R. G. Gomes and R. Bergamasco, Chitosan, alginate and other macromolecules as activated carbon immobilizing agents: A review on composite adsorbents for the removal of water contaminants, Int. J. Biol. Macromol., 2020, 164, 2535–2549 CrossRef CAS PubMed.
- Q. Shi, W. Wang, H. Zhang, H. Bai, K. Liu, J. Zhang, Z. Li and W. Zhu, Porous biochar derived from walnut shell as an efficient adsorbent for tetracycline removal, Bioresour. Technol., 2023, 383, 129213 CrossRef CAS PubMed , https://www.sciencedirect.com/science/article/pii/S0960852423006399.
- S. Dhaka, R. Kumar, A. Deep, M. B. Kurade, S.-W. Ji and B.-H. Jeon, Metal–organic frameworks (MOFs) for the removal of emerging contaminants from aquatic environments, Coord. Chem. Rev., 2019, 380, 330–352 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S0010854518302261.
- S. Khaliha, T. D. Marforio, A. Kovtun, S. Mantovani, A. Bianchi, M. L. Navacchia, M. Zambianchi, L. Bocchi, N. Boulanger, A. Iakunkov, M. Calvaresi, A. V. Talyzin, V. Palermo and M. Melucci, Defective graphene nanosheets for drinking water purification: Adsorption mechanism, performance, and recovery, FlatChem, 2021, 29, 100283 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S2452262721000623.
- L. Zhao, J. Deng, P. Sun, J. Liu, Y. Ji, N. Nakada, Z. Qiao, H. Tanaka and Y. Yang, Nanomaterials for treating emerging contaminants in water by adsorption and photocatalysis: Systematic review and bibliometric analysis, Sci. Total Environ., 2018, 627, 1253–1263 CrossRef CAS PubMed , https://www.sciencedirect.com/science/article/pii/S0048969718303929.
- A. Bakir, S. J. Rowland and R. C. Thompson, Competitive sorption of persistent organic pollutants onto microplastics in the marine environment, Mar. Pollut. Bull., 2012, 64, 2782–2789 CrossRef CAS PubMed , https://www.sciencedirect.com/science/article/pii/S0025326X12004602.
- D. Brennecke, B. Duarte, F. Paiva, I. Caçador and J. Canning-Clode, Microplastics as vector for heavy metal contamination from the marine environment, Estuarine, Coastal Shelf Sci., 2016, 178, 189–195 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S027277141530158X.
- J. F. Provencher, S. Avery-Gomm, M. Liboiron, B. M. Braune, J. B. Macaulay, M. L. Mallory and R. J. Letcher, Are ingested plastics a vector of PCB contamination in northern fulmars from coastal Newfoundland and Labrador?, Environ. Res., 2018, 167, 184–190 CrossRef CAS PubMed.
- M. Zambianchi, A. Aluigi, M. L. Capobianco, F. Corticelli, I. Elmi, S. Zampolli, F. Stante, L. Bocchi, F. Belosi, M. L. Navacchia and M. Melucci, Polysulfone Hollow Porous Granules Prepared from Wastes of Ultrafiltration Membranes as Sustainable Adsorbent for Water and Air Remediation, Adv. Sustainable Syst., 2017, 1, 1700019 CrossRef , https://onlinelibrary.wiley.com/doi/abs/10.1002/adsu.201700019.
- C. Dumitriu, S. I. Voicu, A. Muhulet, G. Nechifor, S. Popescu, C. Ungureanu, A. Carja, F. Miculescu, R. Trusca and C. Pirvu, Production and characterization of cellulose acetate – titanium dioxide nanotubes membrane fraxiparinized through polydopamine for clinical applications, Carbohydr. Polym., 2018, 181, 215–223 CrossRef CAS PubMed , https://www.sciencedirect.com/science/article/pii/S0144861717312389.
- G. Li, W. Kujawski, R. Válek and S. Koter, A review - The development of hollow fibre membranes for gas separation processes, Int. J. Greenhouse Gas Control, 2021, 104, 103195 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S1750583620306204.
- V. K. Thakur and S. I. Voicu, Recent advances in cellulose and chitosan based membranes for water purification: A concise review, Carbohydr. Polym., 2016, 146, 148–165 CrossRef CAS PubMed , https://www.sciencedirect.com/science/article/pii/S0144861716302594.
- A. Kovtun, A. Bianchi, M. Zambianchi, C. Bettini, F. Corticelli, G. Ruani, L. Bocchi, F. Stante, M. Gazzano, T. D. Marforio, M. Calvaresi, M. Minelli, M. L. Navacchia, V. Palermo and M. Melucci, Core–shell graphene oxide–polymer hollow fibers as water filters with enhanced performance and selectivity, Faraday Discuss., 2021, 227, 274–290, 10.1039/C9FD00117D.
- S. Mantovani, S. Khaliha, L. Favaretto, C. Bettini, A. Bianchi, A. Kovtun, M. Zambianchi, M. Gazzano, B. Casentini, V. Palermo and M. Melucci, Scalable synthesis and purification of functionalized graphene nanosheets for water remediation, Chem. Commun., 2021, 57, 3765–3768, 10.1039/D1CC00704A.
- S. Khaliha, A. Bianchi, A. Kovtun, F. Tunioli, A. Boschi, M. Zambianchi, D. Paci, L. Bocchi, S. Valsecchi, S. Polesello, A. Liscio, M. Bergamini, M. Brunetti, M. L. Navacchia, V. Palermo and M. Melucci, Graphene oxide nanosheets for drinking water purification by tandem adsorption and microfiltration, Sep. Purif. Technol., 2022, 300, 121826 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S1383586622013818.
- S. Mantovani, S. Khaliha, T. D. Marforio, A. Kovtun, L. Favaretto, F. Tunioli, A. Bianchi, G. Petrone, A. Liscio, V. Palermo, M. Calvaresi, M. L. Navacchia and M. Melucci, Facile high-yield synthesis and purification of lysine-modified graphene oxide for enhanced drinking water purification, Chem. Commun., 2022, 58, 9766–9769, 10.1039/D2CC03256B.
- Y. Qiu, S. Depuydt, L.-F. Ren, C. Zhong, C. Wu, J. Shao, L. Xia, Y. Zhao and B. Van der Bruggen, Progress of Ultrafiltration-Based Technology in Ion Removal and Recovery: Enhanced Membranes and Integrated Processes, ACS ES&T Water, 2023, 3(7), 1702–1719, DOI:10.1021/acsestwater.2c00625.
- M. Zambianchi, M. Durso, A. Liscio, E. Treossi, C. Bettini, M. L. Capobianco, A. Aluigi, A. Kovtun, G. Ruani, F. Corticelli, M. Brucale, V. Palermo, M. L. Navacchia and M. Melucci, Graphene oxide doped polysulfone membrane adsorbers for the removal of organic contaminants from water, Chem. Eng. J., 2017, 326, 130–140 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S1385894717309026.
- M. Zambianchi, S. Khaliha, A. Bianchi, F. Tunioli, A. Kovtun, M. L. Navacchia, A. Salatino, Z. Xia, E. Briñas, E. Vázquez, D. Paci, V. Palermo, L. Bocchi, B. Casentini and M. Melucci, Graphene oxide-polysulfone hollow fibers membranes with synergic ultrafiltration and adsorption for enhanced drinking water treatment, J. Membr. Sci., 2022, 658, 120707 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S0376738822004525.
- G. V. Research, Hollow Fiber Filtration Market Size, Share & Trends Analysis Report By Membrane Material (Polysulfone), By Process (Single-use Hollow Fiber Membranes), By Technology, By Application, By End-users, By Region, And Segment Forecasts, 2023–2030, 2022, 150, https://www.grandviewresearch.com/industry-analysis/hollow-fiber-filtration-market-report?utm_source=prnewswire&utm_medium=referral&utm_campaign=HC_22-May-23&utm_term=pharmaceutical_filtration_market&utm_content=rl1 Search PubMed.
- N. Vieno and M. Sillanpää, Fate of diclofenac in municipal wastewater treatment plant — A review, Environ. Int., 2014, 69, 28–39 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S0160412014000944.
- N. M. Vieno, H. Härkki, T. Tuhkanen and L. Kronberg, Occurrence of Pharmaceuticals in River Water and Their Elimination in a Pilot-Scale Drinking Water Treatment Plant, Environ. Sci. Technol., 2007, 41, 5077–5084, DOI:10.1021/es062720x.
- U. Anand, B. Adelodun, C. Cabreros, P. Kumar, S. Suresh, A. Dey, F. Ballesteros and E. Bontempi, Occurrence, transformation, bioaccumulation, risk and analysis of pharmaceutical and personal care products from wastewater: a review, Environ. Chem. Lett., 2022, 20, 3883–3904, DOI:10.1007/s10311-022-01498-7.
- J. Glüge, M. Scheringer, I. T. Cousins, J. C. DeWitt, G. Goldenman, D. Herzke, R. Lohmann, C. A. Ng, X. Trier and Z. Wang, An overview of the uses of per- and polyfluoroalkyl substances (PFAS), Environ. Sci.: Processes Impacts, 2020, 22, 2345–2373, 10.1039/D0EM00291G.
- S. Valsecchi, M. Rusconi, M. Mazzoni, G. Viviano, R. Pagnotta, C. Zaghi, G. Serrini and S. Polesello, Occurrence and sources of perfluoroalkyl acids in Italian river basins, Chemosphere, 2015, 129, 126–134 CrossRef CAS.
- E. Gagliano, M. Sgroi, P. P. Falciglia, F. G. A. Vagliasindi and P. Roccaro, Removal of poly- and perfluoroalkyl substances (PFAS) from water by adsorption: Role of PFAS chain length, effect of organic matter and challenges in adsorbent regeneration, Water Res., 2020, 171, 115381 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S0043135419311558.
- M. N. Shahid, S. Khalid and M. Saleem, Unrevealing arsenic and lead toxicity and antioxidant response in spinach: a human health perspective, Environ. Geochem. Health, 2022, 44, 487–496, DOI:10.1007/s10653-021-00818-0.
- R. Nag and E. Cummins, Human health risk assessment of lead (Pb) through the environmental-food pathway, Sci. Total Environ., 2022, 810, 151168 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S004896972106246X.
- I. R. Chowdhury, S. Chowdhury, M. A. J. Mazumder and A. Al-Ahmed, Removal of lead ions (Pb2+) from water and wastewater: a review on the low-cost adsorbents, Appl. Water Sci., 2022, 12, 185, DOI:10.1007/s13201-022-01703-6.
- R. Tröger, H. Ren, D. Yin, C. Postigo, P. D. Nguyen, C. Baduel, O. Golovko, F. Been, H. Joerss, M. R. Boleda, S. Polesello, M. Roncoroni, S. Taniyasu, F. Menger, L. Ahrens, F. Yin Lai and K. Wiberg, What's in the water? – Target and suspect screening of contaminants of emerging concern in raw water and drinking water from Europe and Asia, Water Res., 2021, 198, 117099 CrossRef PubMed , https://www.sciencedirect.com/science/article/pii/S0043135421002979.
- A. Kovtun, M. Zambianchi, C. Bettini, A. Liscio, M. Gazzano, F. Corticelli, E. Treossi, M. L. Navacchia, V. Palermo and M. Melucci, Graphene oxide–polysulfone filters for tap water purification, obtained by fast microwave oven treatment, Nanoscale, 2019, 11, 22780–22787, 10.1039/C9NR06897J.
- C. Zamora-Ledezma, D. Negrete-Bolagay, F. Figueroa, E. Zamora-Ledezma, M. Ni, F. Alexis and V. H. Guerrero, Heavy metal water pollution: A fresh look about hazards, novel and conventional remediation methods, Environ. Technol. Innovation, 2021, 22, 101504 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S2352186421001528.
- M. S. Shamsudin, S. F. Azha and S. Ismail, A review of diclofenac occurrences, toxicology, and potential adsorption of clay-based materials with surfactant modifier, J. Environ. Chem. Eng., 2022, 10, 107541 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S2213343722004146.
- G. R. Johnson, M. L. Brusseau, K. C. Carroll, G. R. Tick and C. M. Duncan, Global distributions, source-type dependencies, and concentration ranges of per- and polyfluoroalkyl substances in groundwater, Sci. Total Environ., 2022, 841, 156602 CrossRef CAS PubMed , https://www.sciencedirect.com/science/article/pii/S0048969722036993.
- Z. Gao, T. J. Bandosz, Z. Zhao, M. Han and J. Qiu, Investigation of factors affecting adsorption of transition metals on oxidized carbon nanotubes, J. Hazard. Mater., 2009, 167, 357–365 CrossRef CAS PubMed , https://www.sciencedirect.com/science/article/pii/S0304389409000107.
- G. Hotová, V. Slovák, T. Zelenka, R. Maršálek and A. Parchaňská, The role of the oxygen functional groups in adsorption of copper (II) on carbon surface, Sci. Total Environ., 2020, 711, 135436 CrossRef , https://www.sciencedirect.com/science/article/pii/S0048969719354294.
- M. Adel, M. A. Ahmed, M. A. Elabiad and A. A. Mohamed, Removal of heavy metals and dyes from wastewater using graphene oxide-based nanomaterials: A critical review, Environ. Nanotechnol., Monit. Manage., 2022, 18, 100719 CAS , https://www.sciencedirect.com/science/article/pii/S2215153222000794.
- N. Genç, E. Durna and E. Erkişi, Optimization of the adsorption of diclofenac by activated carbon and the acidic regeneration of spent activated carbon, Water Sci. Technol., 2021, 83, 396–408 CrossRef PubMed.
- M. M. Johns, W. E. Marshall and C. A. Toles, Agricultural by-products as granular activated carbons for adsorbing dissolved metals and organics, J. Chem. Technol. Biotechnol., 1998, 71, 131–140 CrossRef CAS , https://www.scopus.com/inward/record.uri?eid=2-s2.0-0032006079&doi=10.1002%2f%28SICI%291097-4660%28199802%2971%3a2%3c131%3a%3aAID-JCTB821%3e3.0.CO%3b2-K&partnerID=40&md5=0eedaa6b22984e44f7e7e409022de473.
- S. Lath, D. Navarro, D. Losic, A. Kumar and M. McLaughlin, Sorptive remediation of perfluorooctanoic acid (PFOA) using mixed mineral and graphene/carbon-based materials, Environ. Chem., 2018, 15, 472–480 CrossRef CAS.
- A. C. S. Guerra, M. B. de Andrade, T. R. Tonial Dos Santos and R. Bergamasco, Adsorption of sodium diclofenac in aqueous medium using graphene oxide nanosheets, Environ. Technol., 2021, 42, 2599–2609 CrossRef CAS PubMed.
- M. G. Azam, M. H. Kabir, M. A. A. Shaikh, S. Ahmed, M. Mahmud and S. Yasmin, A rapid and efficient adsorptive removal of lead from water using graphene oxide prepared from waste dry cell battery, J. Water Process Eng., 2022, 46, 102597 CrossRef , https://www.sciencedirect.com/science/article/pii/S221471442200040X.
- N. Saeidi, M. Parvini and Z. Niavarani, High surface area and mesoporous graphene/activated carbon composite for adsorption of Pb(II) from wastewater, J. Environ. Chem. Eng., 2015, 3, 2697–2706 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S2213343715002547.
- F. Cao, L. Wang, Y. Yao, F. Wu, H. Sun and S. Lu, Synthesis and application of a highly selective molecularly imprinted adsorbent based on multi-walled carbon nanotubes for selective removal of perfluorooctanoic acid, Environ. Sci.: Water Res. Technol., 2018, 4, 689–700, 10.1039/C7EW00443E.
- L. Pan, Z. Wang, Q. Yang and R. Huang, Efficient Removal of Lead, Copper and Cadmium Ions from Water by a Porous Calcium Alginate/Graphene Oxide Composite Aerogel, Nanomaterials, 2018, 8, 957 CrossRef PubMed.
- N. Yousefi, X. Lu, M. Elimelech and N. Tufenkji, Environmental performance of graphene-based 3D macrostructures, Nat. Nanotechnol., 2019, 14, 107–119, DOI:10.1038/s41565-018-0325-6.
- D. Tian, D. Geng, W. Tyler Mehler, G. Goss, T. Wang, S. Yang, Y. Niu, Y. Zheng and Y. Zhang, Removal of perfluorooctanoic acid (PFOA) from aqueous solution by amino-functionalized graphene oxide (AGO) aerogels: Influencing factors, kinetics, isotherms, and thermodynamic studies, Sci. Total Environ., 2021, 783, 147041 CrossRef CAS PubMed , https://www.sciencedirect.com/science/article/pii/S0048969721021112.
- E. Briñas, V. J. González, M. A. Herrero, M. Zougagh, Á. Ríos and E. Vázquez, SERS-Based Methodology for the Quantification of Ultratrace Graphene Oxide in Water Samples, Environ. Sci. Technol., 2022, 56, 9527–9535, DOI:10.1021/acs.est.2c00937.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ew00900a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |