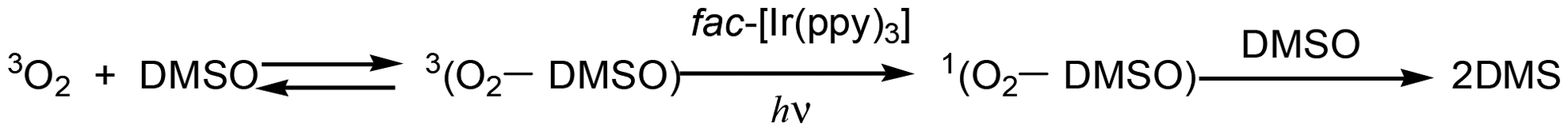DOI:
10.1039/D4DT01822B
(Frontier)
Dalton Trans., 2024,
53, 15782-15786
Colour/luminescence changes of transition metal complexes induced by gaseous small molecules for monitoring reaction progress and environmental changes
Received
24th June 2024
, Accepted 4th August 2024
First published on 8th August 2024
Abstract
Transition metal complexes act as monitoring devices for reaction progress and environmental changes through their color/luminescence changes. In this paper, we focus on colour/luminescence changes induced by gaseous small molecules in the environment. The gradual decrease in O2 content in solution can be monitored by the luminescence enhancement of an Ir(III) complex in dimethyl sulfoxide during photoirradiation. CO2 in air can be captured by a Pt(II) complex in basic aqueous solution, resulting in a colour change from yellow to red to blue-green due to higher degree aggregate formation. Moisture in air induces colour/luminescence changes in Ru(II) and Ir(III) complex salts due to the sorption of H2O into hydrophilic channels in the crystal. Volatile organic compound vapours such as CHCl3 and CH2Cl2 change the purple colour of Pt(II) complex crystals to red and blue, respectively. The purple crystal can adsorb two CHCl3 molecules under ambient conditions but only one CH2Cl2 molecule.
Introduction
Transition metal complexes (TMCs) showing strong luminescence and intense colour have attracted great attention from researchers involved in luminescence/colour studies across many fields including chemistry, biology, materials science, etc., because of their applications in synthesis, catalysis, sensors, bio-imaging, and OLEDs. The colour and luminescence occasionally change upon exposure to small amounts of gaseous molecules such as O2, CO2, H2O, or volatile organic compounds (VOCs). These characteristic responses to gaseous molecules are useful for the fabrication of monitoring devices for reactive and toxic chemicals based on emission measurements with quick response and high sensitivity. For example, phosphorescence from an excited TMC is quenched by O2 to produce reactive oxygen species (1O2), which selectively attacks and kills cancer cells in photodynamic therapy (PDT).1 However, in aerated dimethyl sulfoxide (DMSO) solution, phosphorescence is enhanced rather than quenched, where O2 contributes not to phosphorescence quenching but to the oxidation reaction of the DMSO molecule.2 Another example is the colour change resulting from the CO2 insertion reaction of M–OH to M–CO3H,3 which proceeds spontaneously under ambient conditions, capturing CO2 from the air along with a remarkable colour change. Vapochromism is the most characteristic phenomenon of TMCs involved in colour and luminescence changes induced by gaseous small molecules.4 Some salts consisting of TMCs and counterions contain waters of crystallization, which are easily lost upon heating accompanied by a crystal colour change. Since the heated crystal retains its intrinsic crystal framework, the original crystal colour can be recovered by the sorption of H2O molecules upon exposure to moisture in the air. Additionally, exposure to VOCs can trigger colour/luminescence changes in the Pt(II) complex crystal. The quick response and high sensitivity in vapochromism are useful for monitoring environmental changes in our daily lives; for example, the Pt(II) complex crystal can provide an alert signal through colour/luminescence changes when VOCs are emitted from household products.4a,5
In this manuscript, we summarize our recent studies on the colour/luminescence changes of TMCs as monitoring devices for reaction progress and environmental changes in the following sections.
Emergence of NIR absorption by higher degree aggregate formation of the Pt(II) complex due to the capture of CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) 6
6
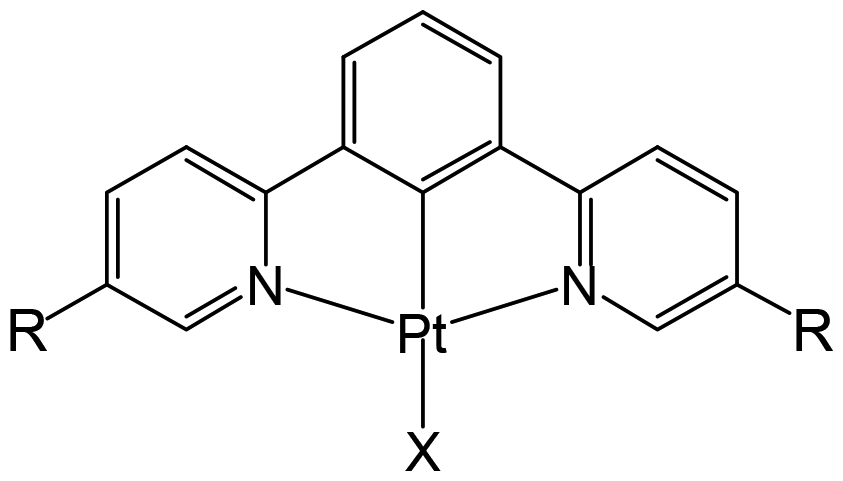
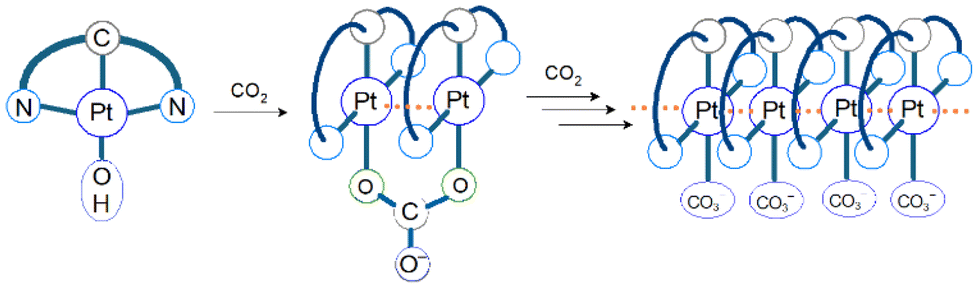
CO2 capture and storage (CCS) is an important concept for achieving net-zero emissions and realizing a sustainable society. Since the concentration of CO2 in the air is as low as 0.04%, the development of effective methods to capture CO2 from the air is highly desirable. The CO2 insertion reaction using TMCs offers an advantage for CCS because we can easily monitor the progress of the reaction through colour changes in response to CO2 insertion. The CO2 insertion induces aggregate formation in Pt(II) complex 1 (Fig. 1), accompanied by a colour change in aqueous solution from yellow to blue-green. The blue-green colour results from strong and broad absorption bands of the aggregate in the visible–near infrared (NIR) region assigned to the metal-to-ligand charge-transfer (MMLCT) transition. Recrystallization of 1 from sodium carbonate aqueous solution (Na2CO3 aq) provides both red and blue-green polymorphic crystals. The red colour of one of the polymorphs originates from the MMLCT transition of the Pt–Pt stacked dimer, where Pt(II) complex monomers are bridged by CO32− ions. For the blue-green colour of the other crystal, although X-ray crystallography has not yet succeeded, it is known that the crystal is constructed from arrays of complex 2 (–Pt–Pt–Pt–) connected by Pt–Pt interactions (Fig. 2). The formation process of the blue-green aggregate in the aqueous solution was monitored through transient absorption spectroscopy. A global analysis of the spectral changes provided evidence for the formation of a 32-mer of complex 2 in the blue-green solution.
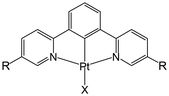 |
| | Fig. 1 Structure of 1 (R = COOH and X = OH−) and 2 (R = COO− and X = CO32−). | |
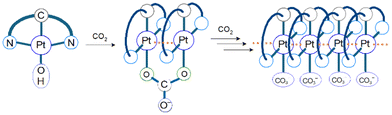 |
| | Fig. 2 An image of aggregation resulting from the reaction of complex 1 with CO2 in basic aqueous solution. The CO2 gas bubbling causes the aggregate formation. The degree of aggregation increases up to at least 32. | |
Emission enhancement of fac-[Ir(ppy)3] (ppyH = 2-phenylprydine) by consuming O2 in DMSO7
The phosphorescence of TMCs, such as fac-[Ir(ppy)3], consisting of heavy atoms is often quenched by O2 due to efficient triplet–triplet energy transfer, producing a strong oxidant, 1O2. Therefore, the photosensitization of phosphorescent TMCs has been applied in photodynamic therapy (PDT) for killing cancer cells and in new synthetic procedures involving oxidation reactions using reactive oxygen.10,11 Dimethyl sulfoxide (DMSO) has been widely used as a solvent in organic syntheses and PDTs because of its low toxicity, high polarity, and capability of dissolving many compounds. In aerated DMSO, photoirradiation gradually enhances the emission intensity of fac-[Ir(ppy)3] due to the prevention of quenching by O2, and the resultant intense-emission is suddenly quenched upon shaking the solution.2 The suppression of quenching during the photoirradiation is reasonably accounted for by the consumption of 3O2 through the formation of dimethyl sulfone (DMS) in the photosensitized reaction involving fac-[Ir(ppy)3]* and the 3(O2-DMSO) adduct. Since 3O2 rapidly reacts with DMSO under ambient conditions when 3O2 is dissolved in DMSO, photosensitization of fac-[Ir(ppy)3] potentially contributes to the activation of 3(O2-DMSO) to labile 1(O2-DMSO) rather than 3O2 to 1O2. The sudden drop in emission intensity upon shaking is accounted for by quenching from 3(O2-DMSO) formed from fresh 3O2 dissolved in the solution from the air or gas phase in the cuvette. DFT calculations supported that the formation of 1(O2-DMSO) is energetically more favourable than the formation of 1O2. We clarified the mechanism of a series of photochemical reactions in this system through kinetic analysis of DMS production along with the emission enhancement of fac-[Ir(ppy)3]. This analysis reproduced not only the production process of DMS but also the production/consumption processes of all species involved in the photosensitization, including transient fac-[Ir(ppy)3]* and 1(O2-DMSO). This DMS production via photosensitization can continue until all the 3O2 in the DMSO solution is completely consumed. It should be noted that the 1(O2-DMSO) adduct could participate in substrate oxidation in organic syntheses and in killing cells due to photosensitization using DMSO as a solvent (Scheme 1).
 |
| | Scheme 1 Reaction of O2 consumption by photosensitization of fac-[Ir(ppy)3]. | |
Detection of H2O through colour changes in TMC salt crystals8
TMC salts can detect moisture through changes in crystal colour in response to the sorption/desorption of H2O molecules into/from the crystal. For the demonstration of this vapochromic performance, it is required that the crystal exhibits a remarkable colour change upon attachment/detachment of H2O, possesses a robust crystal framework for the sorption/desorption cycle of H2O molecules, and has channels through which H2O molecules can move. For the crystal of fac-[Ir(tpy)2]I3·2H2O (tpy = 2,2′:6′,2′′-terpyridine), a reversible colour change between red and orange is observed in response to heating and cooling cycles. Since both intense colours originate from the I−-to-ligand charge-transfer (XLCT) transition, the colour change can be accounted for by the difference in the interaction between I− and the tpy ligand, resulting from the attachment/detachment of H2O to/from I−, respectively. Single crystal X-ray crystallography clarified that the Ir(III) complex possesses I− ions at distances of 2.9–3.1 Å from the closest H atoms of the tpy ligand, suggesting that the XLCT transition results from the interaction between the I− 5p orbital and tpy π* orbital. The two waters of crystallization, involved in the vapochromism, form a dimer structure, rO–O = 2.911 Å, by hydrogen bonding. The H2O dimer connects to one of the − ions with rO–I = 3.664 Å by hydrogen bonding and contacts another I− ion with rO–I = 3.747 Å, suggesting that the XLCT transition is easily changed by H2O desorption from the crystal. Thermogravimetric (TG) measurements revealed step-by-step H2O desorption from the crystal, and powder X-ray diffraction (PXRD) showed the robustness of the Ir(III) complex crystal framework during the sorption/desorption cycles. The H2O desorption takes place even at room temperature upon blowing dry nitrogen gas or exposure to organic solvent vapour, which results in a red coloration of the crystal. Conversely, when the red crystal of [Ir(tpy)2]I3 is exposed to H2O vapour, the crystal colour returns to orange even at ambient temperature. It is suggested that the hydrogen bonding of H2O to I− is too weak to keep H2O molecules in the vicinity of I−.
M[Ru(bpy)(CN)4] (M2+ = Ca2+ or Sr2+) acts as a moisture sensing device showing reversible colour and luminescence changes; both emission spectra of Ca2+ and Sr2+ salts are remarkably shifted to the longer wavelength region with increasing temperature from 296 to 500 K. An X-ray crystallographic study revealed that the crystals are constructed from linear chains of {[Ru(bpy)(CN)4][Ca(H2O)5]}n and {[Ru(bpy)(CN)4][Sr(H2O)6]}n. The chain –Ru–CN–M2+–NC–Ru– (Fig. 3) guarantees the robustness of the crystal during the sorption/desorption cycles of H2O. The hydrophilic channels between these chains contribute to the sorption/desorption of H2O into/from the crystal. Changes in the interaction of the M2+ ion with the equatorial CN ligand, depending on the number of H2O molecules attached to the M2+ ion, effectively contribute to colour/luminescence changes in the Ca2+ and Sr2+ salts. The results of TG measurements indicated that heating above 470 K enables the desorption of five H2O molecules for both salts, resulting in {[Ru(bpy)(CN)4][Ca]}n and {[Ru(bpy)(CN)4][Sr(H2O)]}n. The changes in the hydration structure around the M2+ ion regulate the shift of CN stretching modes, leading to a redshift in the ruthenium-to-bpy CT (MLCT) excited state. It should be noted that the colour/luminescence changes are caused by the changes in the MLCT transition energy modulated by the attachment/detachment of H2O to the M2+ ion but not the Ru(II) complex ion. The luminescence change based on the weak interaction between the M2+ ion and the CN ligand of Ru(II) complexes provides a novel design guideline for vapochromic materials.
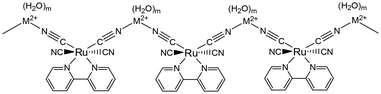 |
| | Fig. 3 Chain structure of {[Ru(bpy)(CN)4][M(H2O)m]}n. The number of H2O (m) depends on temperature. | |
Colour changes of the crystal upon exposure to halomethane vapour and selective sensing of CHCl39
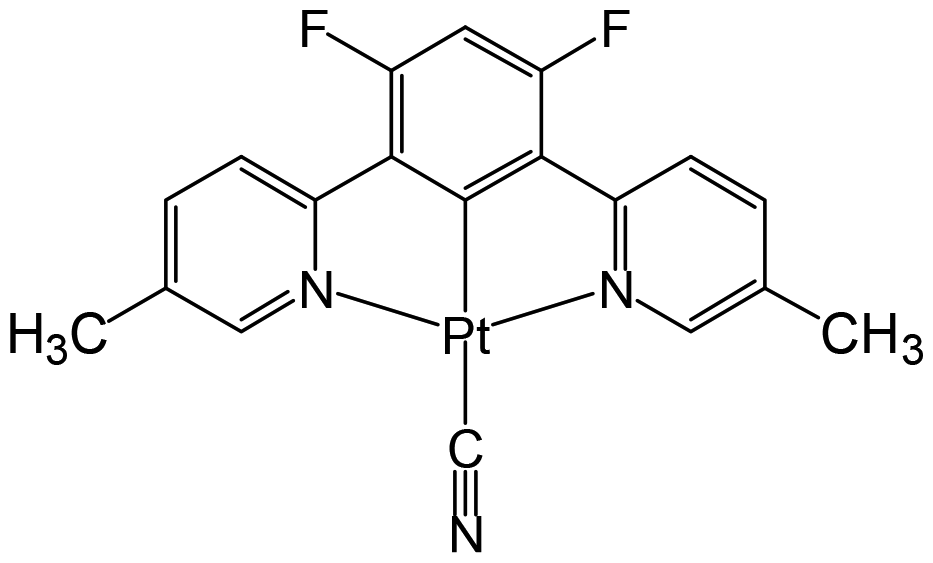
Pt(II) complex 3 (Fig. 4) is obtained as a red precipitate from CHCl3. However, surprisingly, when this precipitate is collected by suction filtration, the resultant material's colour rapidly changes to purple, and conversely, the purple colour changes to red upon exposure to CHCl3 vapour. The reversible colour-change between purple and red is accounted for by the vapochromic response of crystal 3 to CHCl3 vapour. Since the red and purple crystals emit red and NIR luminescence, respectively, we can perceive the vapochromism as an ON/OFF switch for red emission during the evacuation and exposure cycles of CHCl3 vapour. These characteristic features promise quick and highly sensitive detection of small amounts of CHCl3 vapour using crystal 3. X-ray crystallography of the red form revealed that two Pt(II) complexes are mutually twisted by ca. 180°, resulting in a head-to-tail arrangement of stacked dimers and the Pt–Pt units are aligned along the b-axis of the unit cell, forming infinite columnar structures. The CHCl3 molecules are stored in channels between these infinite –Pt–Pt–Pt– columns and thereby are desorption/sorption through the channels from/into the crystal. During the exposure to CHCl3 vapour, a step-by-step change from 3 to 3·CHCl3 to 3·2CHCl3 was observed. Unlike the M[Ru(bpy)(CN)4] and [Ir(tpy)2]I3 systems, the peak patterns in PXRD obtained for the crystals are different from one another, suggesting that the crystal framework consisting of complex 3 is soft and easily altered during the sorption/desorption cycles of CHCl3. When purple crystal 3 is exposed to CH2Cl2 vapour, which is chemically very similar to CHCl3, the colour of the crystal changes to blue (Fig. 5). This behaviour suggests that crystal 3 has the capability to discriminate CHCl3 from chemically similar CH2Cl2. A change in the PXRD pattern of crystal 3 upon increasing the CH2Cl2 vapour pressure up to 50 kPa showed a two-step peak change due to the sorption of CH2Cl2 similar to when exposed to CHCl3 vapour. Therefore, the difference between CHCl3 and CH2Cl2 in the vapochromic behaviour of crystal 3 under ambient conditions is concluded to be due to the difference in the number of molecules in the crystal; the purple crystal 3 can adsorb two CHCl3 molecules under ambient conditions but just one CH2Cl2 molecule (Fig. 5).
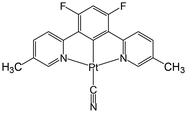 |
| | Fig. 4 Structure of complex 3. | |
 |
| | Fig. 5 Colour and luminescence variations of complex 3 in the solid state. (Top) The purple crystal of complex 3 changes to red and blue upon exposure to CHCl3 and CH2Cl2 vapours, respectively. (Bottom) The red crystal containing CHCl3 emits red luminescence, whereas both blue and purple crystals emit invisible NIR luminescence. This figure has been reproduced from ref. 8 with permission from the Royal Society of Chemistry, copyright 2022. | |
Conclusion
We presented changes in the colour/luminescence of TMCs induced by small molecules, enabling the visualization of the progress of chemical reactions involving these small molecules and changes in the surrounding atmosphere. A water-soluble Pt(II) complex reacts with CO2, resulting in higher degree aggregate formation and showing broad absorption across the visible to NIR region. Although we estimate that the degree of aggregation increases up to 32, the structure of the aggregate remains unknown. Recently, to identify the structure and morphology of the supramolecular assembly of Pt(II) complexes, measurements of circularly polarized luminescence have been conducted.4b However, studies on the structure of supramolecular assemblies or aggregates of Pt(II) complexes are still limited. We showed that the enhancement/quenching of emission from fac-[Ir(ppy)3] can be used in a monitoring device for tracking the progress of desired reactions in PDT. Moreover, we found that the O2-DMSO adduct contributes to the catalytic formation of DMS by excited fac-[Ir(ppy)3]. Therefore, further investigation into the O2-DMSO adduct should be performed for understanding the fundamental photodynamic effects of TMC phosphors dissolved in DMSO. For TMC salt crystals, the robustness of the crystal framework and the presence of hydrophilic channels are significant for repeatable sorption/desorption cycles of H2O, and the reversible colour/luminescence changes result from the indirect interaction of H2O with the TMC and the weak bonding of H2O to the counterion. We should clarify and understand the photophysics of the indirect interactions contributing to the colour/luminescence changes of TMCs. For molecular crystals of Pt(II) complexes, the “softness” of the crystal is important for vapochromism; the Pt(II) complex easily changes the intermolecular arrangement and Pt–Pt distance responsible for the coloration of the crystal. Although substantial knowledge on changes in the crystal structure upon applying mechanical stress or grinding has been accumulated,4c further investigation into the “softness” of the crystal is needed to clarify and understand the vapochromism of Pt(II) complexes. Since the Pt(II) complex crystal can discriminate CHCl3 from CH2Cl2, it is expected to be a detection device for specific molecules in a variety of VOCs. This may contribute to further improvements in sensors for VOCs and toxic chemicals using TMCs.4a It is also significant for the development of next-generation luminophores that both crystals containing CH2Cl2 and those without any crystal solvent emit NIR luminescence, as luminescence in the NIR II window is available for in vivo bio-imaging in deeper regions.12 Furthermore, it should be noted that the NIR emission wavelength can be modulated by changing the Pt–Pt interaction sensitive to stimuli for the environment, ancillary ligands such as Cl− and CN−, and the number and kind of small molecules in the crystal.
Data availability
The data that support the findings of this manuscript are openly available in ChemSusChem., 2023, 16, e202301174 at https://doi.org/10.1002/cssc.202301174 (section 1), J. Phys. Chem. B, 2021, 125, 9260–9267 at https://doi.org/10.1021/acs.jpcb.1c03753 (section 2), Dalton Trans., 2022, 51, 7068–7075 at https://doi.org/10.1039/d2dt00368f, Dalton Trans., 2022, 51, 1474–1480 at https://doi.org/10.1039/D1DT03666A (section 3), and Dalton Trans., 2022, 51, 15830–15841 at https://doi.org/10.1039/D2DT02360A (section 4).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The author acknowledges Dr S. Hattori for assisting with the experimental work and helpful discussion. This work was supported by JSPS KAKENHI (No. JP21K14647).
References
-
(a) A. Zamora, G. Vigueras, V. Rodríguez, M. D. Santana and J. Ruiz, Coord. Chem. Rev., 2018, 360, 34–76 CrossRef;
(b) X. Li, J. Wu, L. Wang, C. He, L. Chen, Y. Jiao and C. Duan, Angew. Chem., Int. Ed., 2020, 59, 6420–6427 CrossRef PubMed;
(c) Y. Wu, S. Li, Y. Chen, W. He and Z. Guo, Chem. Sci., 2022, 13, 5085–5106 RSC.
-
(a) S. Wan and W. Lu, Angew. Chem., Int. Ed., 2017, 56, 1784–1788 CrossRef PubMed;
(b) S. Wan, J. Lin, H. Su, J. Dai and W. Lu, Chem. Commun., 2018, 54, 3907–3910 RSC.
-
(a) Y. Hayashi, S. Kita, B. S. Brunschwig and E. Fujita, J. Am. Chem. Soc., 2003, 125, 11976–11987 CrossRef CAS;
(b) D. H. Gibson, X. Yin, H. He and M. S. Mashuta, Organometallics, 2003, 22, 337–346 CrossRef;
(c) D. H. Gibson and X. Yin, Chem. Commun., 1999, 1411–1412 RSC.
-
(a) O. S. Wenger, Chem. Rev., 2013, 113, 3686–3733 CrossRef PubMed;
(b) M. J. Bryant, J. M. Skelton, L. E. Hatcher, C. Stubbs, E. Madrid, A. R. Pallipurath, L. H. Thomas, C. H. Woodall, J. Christensen, S. Fuertes, T. P. Robinson, C. M. Beavers, S. J. Teat, M. R. Warren, F. Pradaux-Caggiano, A. Walsh, F. Marken, D. R. Carbery, S. C. Parker, N. B. McKeown, R. Malpass-Evans, M. Carta and P. R. Raithby, Nat. Commun., 2017, 8, 1800 CrossRef;
(c) E. Li, K. Jie, M. Liu, X. Sheng, W. Zhu and F. Huang, Chem. Soc. Rev., 2020, 49, 1517–1544 RSC.
-
(a) M. J. Bryant, S. Fuertes, L. E. Hatcher, L. H. Thomas and P. R. Raithby, Faraday Discuss., 2023, 244, 411–433 RSC;
(b) B. Li, Y. Li, M. H.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2021, 143(51), 21676–21684 CrossRef CAS;
(c) P. Yu, D. Peng, L.-H. He, J.-L. Chen, J.-Y. Wang, S.-J. Liu and H.-R. Wen, Inorg. Chem., 2022, 61, 254–264 CrossRef CAS;
(d) P. Jia, X. He, J. Yang, X. Sun, T. Bu, Y. Zhuang and L. Wang, Sens. Actuators, B, 2023, 374, 132803 CrossRef CAS;
(e) K. M. Fürpaß, L. M. Peschel, J. A. Schachner, S. M. Borisov, H. Krenn, F. Belaj and N. C. Mösch-Zanetti, Angew. Chem., Int. Ed., 2021, 60, 13401–13404 CrossRef.
- H. Mita, H. Shingo, T. Sasaki, S. Takamizawa and K. Shinozaki, ChemSusChem, 2023, 16, e202301174 Search PubMed.
- S. Hattori, S. Hirata and K. Shinozaki, J. Phys. Chem. B, 2021, 125, 9260–9267 CrossRef PubMed.
-
(a) S. Hattori, M. Kondo, A. Sekine and K. Shinozaki, Dalton Trans., 2022, 51, 7068–7075 RSC;
(b) S. Hattori, T. Nagai, A. Sekine, T. Otsuka and K. Shinozaki, Dalton Trans., 2022, 51, 1474–1480 RSC.
- S. Hattori, T. Nakano, N. Kobayashi, Y. Konno, E. Nishibori, T. Galica and K. Shinozaki, Dalton Trans., 2022, 51, 15830–15841 RSC.
-
(a) V. K. Soni, H. S. Hwang, Y. K. Moon, S.-W. Park, Y. You and E. J. Cho, J. Am. Chem. Soc., 2019, 141, 10538–10545 CrossRef CAS PubMed;
(b) D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef;
(c) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
-
(a) M. Martínez-Alonso, C. G. Jones, J. D. Shipp, D. Chekulaev, H. E. Bryant and J. A. Weinstein, J. Biol. Inorg. Chem., 2024, 29, 113–125 CrossRef PubMed;
(b) R. E. Doherty, I. V. Sazanovich, L. K. McKenzie, A. S. Stasheuski, R. Coyle, E. Baggaley, S. Bottomley, J. A. Weinstein and H. E. Bryant, Sci. Rep., 2016, 6, 22668 CrossRef CAS PubMed.
-
(a) I. Melendo, S. Fuertes, A. Martín and V. Sicilia, Inorg. Chem., 2024, 63, 5470–5480 CrossRef;
(b) R. J. Salthouse, P. Pander, D. S. Yufit, F. B. Dias and J. A. G. Williams, Chem. Sci., 2022, 13, 13600–13610 RSC;
(c) H. J. Park, C. L. Boelke, P. H.-Y. Cheong and D.-H. Hwang, Inorg. Chem., 2022, 61, 5178–5183 CrossRef PubMed;
(d) T. Li, C. Li, Z. Ruan, P. Xu, X. Yang and P. Yuan, ACS Nano, 2019, 13, 3691–3702 CrossRef PubMed;
(e) C. Li, G. Chen, Y. Zhang, F. Wu and Q. Wang, J. Am. Chem. Soc., 2020, 142, 14789–14804 CrossRef CAS PubMed;
(f) Y. Sun, F. Ding, Z. Zhou, C. Li, M. Pu, Y. Xu, Y. Zhan, X. Lu, H. Li, G. Yang, Y. Sun and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 1968–1973 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a