
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spin-crossover cobalt(II) complexes exhibiting temperature- and concentration-dependent optical changes in solution†
Naoki
Izumiyama
a,
Shun
Fujii
a,
Kiichi
Kato
b,
Ryuya
Tokunaga
c,
Shinya
Hayami
 cd and
Manabu
Nakaya
*ab
cd and
Manabu
Nakaya
*ab
aDepartment of Material Science, Graduate School of Science, Josai University, 1-1 Keyakidai, Sakado, Saitama 350-0295, Japan. E-mail: nakaya@josai.ac.jp
bDepartment of Chemistry, Faculty of Science, Josai University, 1-1 Keyakidai, Sakado, Saitama 350-0295, Japan
cDepartment of Chemistry, Graduate School of Science and Technology, Kumamoto University, 2-39-1 Kurokami, Chuo-ku, Kumamoto 860-8555, Japan
dInstitute of Industrial Nanomaterials (IINa), Kumamoto University, 2-39-1 Kurokami, Chuo-ku, Kumamoto 860-8555, Japan
First published on 9th May 2024
Abstract
This work investigated the spin states of the cobalt(II) complexes [Co(L1)2](X)2 (1·X; L1 = 4′-(4-N,N′-diphenylaminophenyl)-2,2′:6′,2′′-terpyridine, X = PF6, BPh4) and [Co(L2)2](X)2 (2·X; L2 = 4′-(4-N,N′-dimethylaminophenyl)-2,2′:6′,2′′-terpyridine, X = PF6, BPh4) in the solid state and in solution. In the solid state, 1·PF6 and 2·PF6, both containing smaller PF6− counter anions, showed gradual spin-crossover. In contrast, 1·BPh4 and 2·BPh4 remained in the high-spin state over the temperature range of 5–400 K due to a lower degree of molecular cooperativity. Each of the cobalt(II) complexes exhibited effects of temperature and concentration on their absorption spectra that were related to the spin states in various organic solvents. This work provides new insights into the spectroscopic properties resulting from the spin states of cobalt(II) complexes in solution.
Introduction
Spin-crossover (SCO) is a phenomenon in which a metal center converts its spin state between a low-spin (LS) and a high-spin (HS) state in response to an external stimulus such as temperature, light, pressure or guest molecules. SCO is also highly sensitive to structural changes that can act as triggers to induce this behavior.1–4 Spin conversion is seen as a promising approach to the creation of next-generation molecular devices.5–7 In particular, SCO cobalt(II) compounds switch their spin states between the LS (S = 1/2) and HS (S = 3/2) states. For these transitions, ΔSspin = R[ln(2S + 1)HS − ln(2S + 1)LS] = 5.8 J K−1 mol−1, which is smaller than the values for iron(II) (13.4 J K−1 mol−1) or iron(III) (9.1 J K−1 mol−1) SCO compounds. On this basis, cobalt(II) compounds can exhibit SCO behavior induced by fewer extreme external stimuli compared with those required for iron(II) or iron(III) complexes. Motivated by this, various SCO cobalt(II) systems have been developed so far.8–12 Above all, the spin transitions of cobalt(II) complexes can be induced by relatively minor changes to ligands, with the majority of work to date still involving terpyridine ligand derivatives.13–18Although SCO phenomena are associated with the electronic structures of single molecules, they can be observed in solutions or soft polymer matrices.19–25 The softening of the molecular structure and/or of molecular assemblies as a result of molecular design through chemical modification offers a means of tailoring the characteristics of functional molecular materials. As an example, the effects of softness on the spin conversion of SCO polymers have been investigated. Even so, studies of the SCO properties of cobalt(II) compounds in solution remain limited.
The present work demonstrates the synthesis of terpyridine cobalt(II) complexes possessing dynamic substituents based on diamine derivatives: [Co(L1)2]X2 (1; L1 = 4′-(4-N,N′-diphenylaminophenyl)-2,2′:6′,2′′-terpyridine) and [Co(L2)2]X2 (2; L2 = 4′-(4-N,N′-dimethylaminophenyl)-2,2′:6′,2′′-terpyridine). In this study, tetraphenylborate (BPh4−) or hexafluorophosphonium (PF6−) were employed as the counter anions (X−) to give 1·X or 2·X (X = BPh4 or PF6) (Scheme 1). The effects of temperature and concentration on spectroscopic features related to the spin states of 1·X and 2·X in various organic solvents are discussed, as are the spin states in the solid state.
 | ||
| Scheme 1 Molecular structures of complexes 1·X and 2·X (X = BPh4 or PF6). Red arrows indicate rotatable bonds in the terpyridine ligand derivatives. | ||
Results and discussion
Single-crystal and powder X-ray analysis
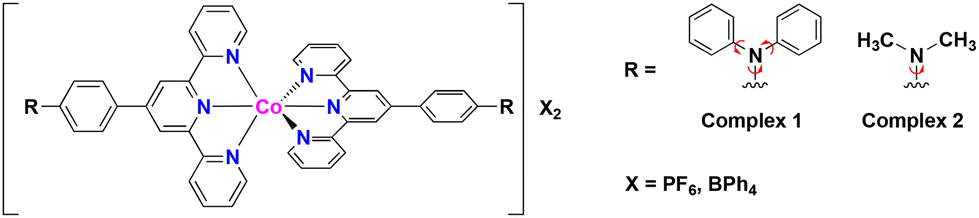
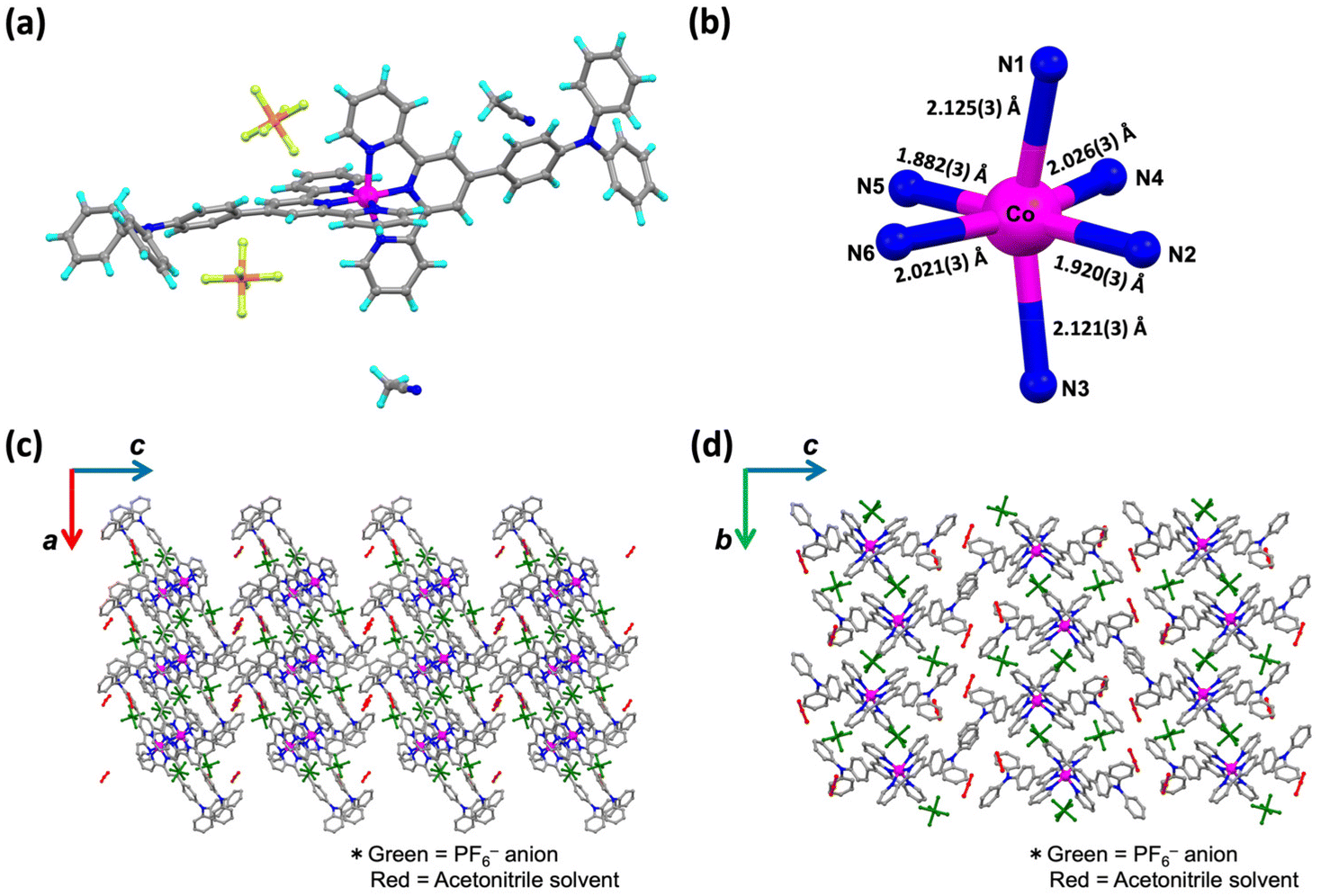
Syntheses of the complexes 1·X and 2·X are described in the Experimental section of the ESI.† The crude samples were characterized by elemental analysis. Single crystals suitable for single-crystal X-ray diffraction (SC-XRD) analysis of 1·PF6·2CH3CN, 2·PF6 and 2·BPh4 were obtained from acetonitrile solutions by the diffusion method with diethyl ether. A single crystal of 1·BPh4·solv (solv = 2.75CH3CN·0.8CH3OH·0.25H2O) was obtained by recrystallization from mixed solution of acetonitrile and methanol. SC-XRD data for all complexes were obtained at 100 K.The crystal structures of 1·X and 2·X are displayed in Fig. 1a, 2a, S3a and S5a.† The lattice parameters for each cobalt(II) complex are summarized in Table S1† and complete structural data are available from the CCDC. All measurements were conducted at 100 K. The cobalt(II) complexes each exhibited Jahn–Teller distortion, as expected for cobalt(II) ions, to give a tetragonally distorted coordination environment for the [CoN6] metal center (Fig. 1b, 2b, S3b and S5b†). In this structure, the cobalt(II) to apical pyridine donor bonds were elongated or shrunken relative to those occupying the equatorial positions. The local structural features of the [CoN6] centers were examined with regard to the spin state (Table 1) as discussed later on.
 | ||
| Fig. 1 (a) Crystal structure of 1·PF6·2CH3CN. (b) Coordination environment of the [CoN6] core and the Co–N bond length. Crystal packing of 1·PF6·2CH3CN along (c) the ac plane and (d) the bc plane. Green and red molecules are PF6− counter anions and acetonitrile solvent molecules, respectively. | ||
 | ||
| Fig. 2 (a) Crystal structure of 1·BPh4·solv. One acetonitrile solvent was located with disorder. (b) Coordination environment of the [CoN6] core and the Co–N bond length. Crystal packing of 1·BPh4·solv along (c) the ac plane and (d) the bc plane. Green and red molecules are BPh4− counter anions and solvent molecules, respectively. | ||
| Co–N(ave.) (Å) | Σ (°) | φ (°) | |
|---|---|---|---|
| 1·PF 6 | 2.016(3) | 90.4(1) | 176.9(1) |
| 1·BPh 4 | 2.026(2) | 99.0(7) | 174.6(7) |
| 2·PF 6 (for Co1) | 2.008(1) | 94.7(4) | 178.1(4) |
| 2·PF 6 (for Co2) | 2.028(1) | 95.0(4) | 177.1(4) |
| 2·BPh 4 | 2.002(3) | 82.5(1) | 177.3(1) |
From a detailed analysis of 1·PF6·2CH3CN, one PF6− anion (displayed as P2) is about 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 disordered (Fig. S1†). However, the 15% electron density disorder was quite weak and fuzzy, and was technically difficult to treat as a disorder. It is aware that to assume PF6− anion as 100% occupancy is wrong by about 15% occupancy, but for technical reasons and not to induce a misleading for the stoichiometric ratio, here would like to be left it as 100% occupancy. Fig. 1b and c present the crystal packing structure of 1·PF6·2CH3CN. Fig. S2† demonstrates the selected intermolecular interactions observed in the molecular assembly of 1·PF6·2CH3CN. In this compound, CH–π interactions (2.850–2.892 Å) and π–π interactions (3.368–3.387 Å) were formed between adjacent pairs of molecules in the a- and b-axis directions, giving two-dimensional (2D) assemblies in the ab plane. PF6− counter anions interact with [Co(L1)2]2+ units through hydrogen bonding (2.338–2.624 Å). Solvent molecules in the c-axis direction were located between these 2D assemblies. The shortest distance between the cobalt(II) metal centers (that is, the Co–Co distance) is herein used as an indicator of the extent of molecular cooperativity associated with solid-state SCO behavior. In 1·PF6·2CH3CN, the shortest Co–Co distance is 8.784 Å. Diagrams of the 1·BPh4·solv molecular assemblies are provided in Fig. 2b and c. The selected intermolecular interactions observed in the molecular assembly of 1·BPh4·solv are displayed in Fig. S3.† In this compound, pairs of [Co(L1)2]2+ cations interact with one another through CH–π interactions (2.623–2.887 Å) and π–π interactions (3.425 Å). 1·BPh4·solv was found to have a Co–Co distance of 8.306 Å. Similar molecular assemblies produced by intermolecular π–π interactions, CH–π interactions and N⋯H interactions were also observed in the case of 2·PF6 and 2·BPh4 (Fig. S4–S7†). Note that the SC-XRD data quality of 2·PF6 was not so good, resulting in the high R value of 12.85%. 2·PF6 was composed of two cobalt(II) centers (expressed as Co1 and Co2 in Fig. S4†) unlike other cobalt(II) complexes. In the crystal packing structure of 2·PF6, intermolecular hydrogen bonds (2.426–2.662 Å) between [Co(L2)2]2+ units and PF6− counter anions as well as π–π interactions (3.716 Å) and CH–π interactions (2.812–2.875 Å) were observed. In the case of 2·BPh4, BPh4− anions surround a [Co(L2)2]2+ cation through CH–π interactions (2.598–2.861 Å). Due to the larger BPh4− anions, interactions between [Co(L2)2]2+ units were rarely produced, where only a few interactions, such as CH–π interactions and N⋯H interactions, were observed (2.549–2.861 Å). The shortest Co–Co distances for 2·PF6 and 2·BPh4 were 8.763 and 11.474 Å, respectively.
:15 disordered (Fig. S1†). However, the 15% electron density disorder was quite weak and fuzzy, and was technically difficult to treat as a disorder. It is aware that to assume PF6− anion as 100% occupancy is wrong by about 15% occupancy, but for technical reasons and not to induce a misleading for the stoichiometric ratio, here would like to be left it as 100% occupancy. Fig. 1b and c present the crystal packing structure of 1·PF6·2CH3CN. Fig. S2† demonstrates the selected intermolecular interactions observed in the molecular assembly of 1·PF6·2CH3CN. In this compound, CH–π interactions (2.850–2.892 Å) and π–π interactions (3.368–3.387 Å) were formed between adjacent pairs of molecules in the a- and b-axis directions, giving two-dimensional (2D) assemblies in the ab plane. PF6− counter anions interact with [Co(L1)2]2+ units through hydrogen bonding (2.338–2.624 Å). Solvent molecules in the c-axis direction were located between these 2D assemblies. The shortest distance between the cobalt(II) metal centers (that is, the Co–Co distance) is herein used as an indicator of the extent of molecular cooperativity associated with solid-state SCO behavior. In 1·PF6·2CH3CN, the shortest Co–Co distance is 8.784 Å. Diagrams of the 1·BPh4·solv molecular assemblies are provided in Fig. 2b and c. The selected intermolecular interactions observed in the molecular assembly of 1·BPh4·solv are displayed in Fig. S3.† In this compound, pairs of [Co(L1)2]2+ cations interact with one another through CH–π interactions (2.623–2.887 Å) and π–π interactions (3.425 Å). 1·BPh4·solv was found to have a Co–Co distance of 8.306 Å. Similar molecular assemblies produced by intermolecular π–π interactions, CH–π interactions and N⋯H interactions were also observed in the case of 2·PF6 and 2·BPh4 (Fig. S4–S7†). Note that the SC-XRD data quality of 2·PF6 was not so good, resulting in the high R value of 12.85%. 2·PF6 was composed of two cobalt(II) centers (expressed as Co1 and Co2 in Fig. S4†) unlike other cobalt(II) complexes. In the crystal packing structure of 2·PF6, intermolecular hydrogen bonds (2.426–2.662 Å) between [Co(L2)2]2+ units and PF6− counter anions as well as π–π interactions (3.716 Å) and CH–π interactions (2.812–2.875 Å) were observed. In the case of 2·BPh4, BPh4− anions surround a [Co(L2)2]2+ cation through CH–π interactions (2.598–2.861 Å). Due to the larger BPh4− anions, interactions between [Co(L2)2]2+ units were rarely produced, where only a few interactions, such as CH–π interactions and N⋯H interactions, were observed (2.549–2.861 Å). The shortest Co–Co distances for 2·PF6 and 2·BPh4 were 8.763 and 11.474 Å, respectively.
At higher temperature, the crystallinity of each complex was too low to allow SC-XRD analysis. Therefore, powder X-ray diffraction (PXRD) analyses were conducted for the structural discussion (Fig. S8†). The PXRD patterns of the crystalline powder samples (red solid line) were consistent with the simulated patterns obtained from the SC-XRD analysis (black solid line). Blue solid lines indicate the PXRD patterns of each sample after annealing at 100 °C in vacuo. Except for that of 1·BPh4·solv, all the patterns are almost the same as that of the freshly crystallised one. The PXRD patterns for the annealed 2·BPh4 sample showed a slight peak shift, in which the overall molecular packing motif of the compound is the same, but the cell size was found to be slightly changed after desolvation. As for the annealed 1·BPh4, the disappearance of some peaks at lower angles and peak broadening at higher angles were observed, in which the molecular packing structure might be different before/after the annealing due to the desolvation.
Magnetic properties in the solid state
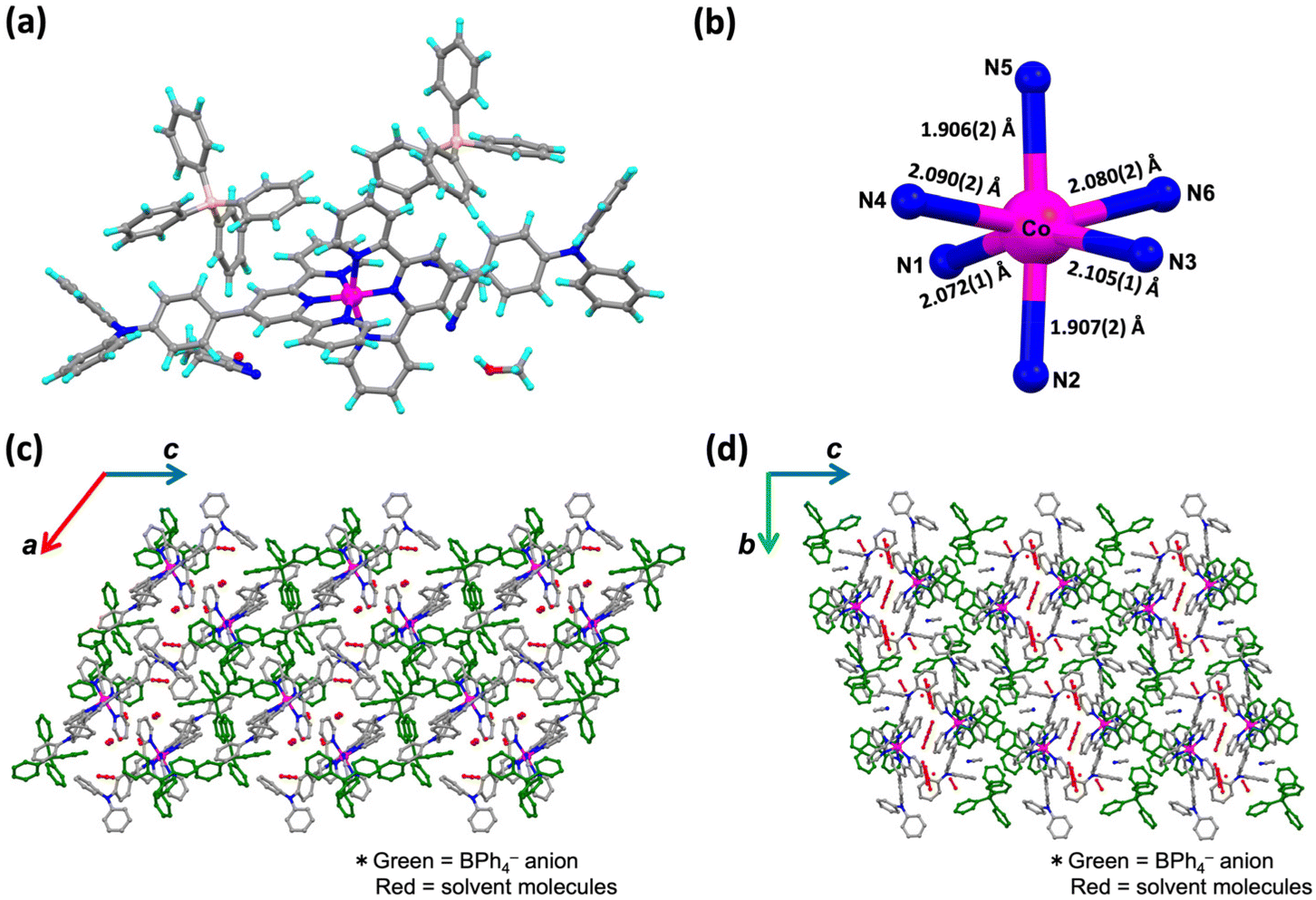
The effect of temperature on the spin states of 1·X and 2·X in the solid state was investigated using a superconducting quantum interference device with a scan rate of 5 K min−1 (Fig. 3). Magnetic susceptibilities are evaluated by generating plots of χmT versus T, where χm is the molar magnetic susceptibility and T is the temperature. The solvated complexes, 1·PF6·2CH3CN and 1·BPh4·solv, showed similar SCO behaviour after desolvation (when kept at 400 K for 2 h in the SQUID system). Here, the slight differences in the PXRD signals were found not to be effective on the SCO behavior. Thus, in Fig. 3, all SCO behaviors are described in terms of the desolvated state. The χmT value of 1·PF6 at 5 K was 0.53 cm3 K mol−1, which is consistent with the typical LS state of cobalt(II) ions in an octahedral coordination environment.26 Upon heating to 400 K, SCO behavior was observed to gradually occur (Fig. 3a). On reaching 400 K, the χmT value of 1·PF6 was 1.36 cm3 K mol−1, of which ca. 40% of the LS domain still remained even at 400 K. Then, the sample was cooled from 400 to 5 K, whereupon the plots followed the same route as the heating process. The χmT value of 1·BPh4 at 5 K was 0.79 cm3 K mol−1, implying that this complex was partially in the HS state at that temperature (Fig. 3b). On heating, the χmT value gradually increased up to 2.08 cm3 K mol−1 at 400 K. This value is in the range typical of the HS state of cobalt(II) ions. That is, 1·BPh4 also showed the gradual onset of SCO behavior over the temperature range of 5–400 K but was occupied by HS domains over most of this range. Similar behaviors were also observed in 2·PF6 and 2·BPh4. The former showed gradual onset of SCO along with a change in χmT from 0.39 cm3 K mol−1 at 5 K to 1.30 cm3 K mol−1 at 400 K. There were no significant differences between the data acquired during heating and cooling processes. Unlike 2·PF6, which exhibited gradual SCO behavior, 2·BPh4 had a χmT value of 1.07 cm3 K mol−1 at 5 K and so was partially in the HS state, as was also the case for 1·BPh4. Then, the χmT value increased up to 2.22 cm3 K mol−1 at 400 K, from which SCO behavior in 2·BPh4 could be elucidated. | ||
| Fig. 3 χ m T vs. T plots for (a) 1·PF6, (b) 1·BPh4, (c) 2·PF6 and (d) 2·BPh4 for the heating process (red) and cooling process (blue) between 5 and 400 K. Black solid line is the simulated curve fit by the values of the regular solution model. | ||
These results were assessed by considering the cooperativity of the complexes. Spin transition (ST) behaviors can be generally categorized based on the cooperative factor C value: hysteretic (C > 1) and abrupt (C ≈ 1) ST behaviour and gradual SCO behaviour with a reduced cooperative factor (C < 1). Such cooperative factor (C) values are estimated by fitting the experimental χmT versus T plots using the regular solution model (eqn (1)–(3) in the ESI†).27 On this basis, the C value for 1·PF6 was estimated to be 0.37. This value is congruent with the gradual SCO behavior of 1·PF6, which was not sufficient to produce abrupt spin transition behavior. Conversely, a C value that satisfied the fitting model for 1·BPh4 was almost zero, in agreement with the more HS-like gradual SCO behaviour of 1·BPh4. That is, the cooperativity of 1·BPh4 was almost zero. Similar results were obtained for 2·PF6 and 2·BPh4 with a C value of 0.27 determined for 2·PF6 and almost zero for 2·BPh4. The C values of 0.37 for 1·PF6 and 0.27 for 2·PF6 are enough to understand the “gradual” SCO behavior, not “abrupt” behavior, in which the slight difference of 0.1 in the cooperativity did not significantly influence the SCO behaviors.
These findings can be explained by considering the shortest Co–Co distances and the structure of the overall molecular assemblies as obtained from SC-XRD analyses. Specifically, 1·BPh4 and 1·PF6 had much shorter distances (8.306(5) and 8.784(8) Å). However, each pair of [Co(L1)2]2+ units were surrounded by larger BPh4− counter anions, such that the SCO sites were unable to work cooperatively. The 2·PF6 had a similar Co–Co distance (8.763(3) Å) to that of the 1·PF6 and also showed gradual SCO. The 2·BPh4 had a much longer distance (11.474(6) Å), indicating that lower molecular cooperativity resulted in the HS-like characteristics of this complex. Furthermore, interaction parameters Γ for each complex have been calculated based on eqn (3).†Γ for 1·PF6 was 2.36 kJ mol−1 and for 2·PF6 was 4.13 kJ mol−1, respectively. On the other hand, Γ values for 1·BPh4 and 2·BPh4 were almost zero (−0.82 × 10−2 kJ mol−1 and −0.25 kJ mol−1), which indicate that both complexes exhibited noncooperative SCO behavior.28
As results from structural features identified from SC-XRD analyses and studies based on the regular solution model, it can be revealed that the smaller PF6− counter ions induced some molecular cooperativity in both 1 and 2, thus stabilizing the LS state at lower temperatures and promoting gradual SCO in the solid state. However, the larger BPh4− counter ions isolated the SCO cation sites from one another and so decreased the molecular cooperativity, such that the HS state was stabilized.
Correlation between structure and spin state in the solid state
To assess the apparent spin states of the present complexes based on the local structures of the [CoN6] cores, the average bond lengths of Co–N coordination bonds and the distortion parameters Σ and φ were calculated (Table 1). Here, Σ is the sum of |90° − α| for the twelve cis-N–Co–N angles and φ is the inter-ligand angle for trans-N–Co–N.29,30 Hayami et al. previously reported correlation between the structure and magnetic properties of SCO cobalt(II) complexes.31 According to the report, the threshold value Σ for the LS–HS transition is approximately 95°. Thus, 1·PF6 with a Σ value of 90.4° was in the range associated with the LS state at 100 K, which is consistent with the magnetic susceptibility data. The 1·BPh4 (Σ = 99.0°) also satisfied this same structure–magnetic property correlation. 2·PF6 has two cobalt(II) centers (as mentioned in the section on SC-XRD). The Σ value for Co1 was 94.7° and for Co2 was 95.0°. Furthermore, the average Σ value was 94.6°, which allows us to understand that 2·PF6 was in the LS state at 100 K. The Co–N(ave.) distances of 2.008 Å and 2.028 Å also assisted with corroborating the LS state of the cobalt(II) ion. With the exception of 2·BPh4, the estimated spin states agreed with the structural data (in terms of the average bond lengths, Co–N(ave.)) reported for SCO cobalt(II) complexes.31 Only in the case of 2·BPh4 (Σ = 82.5°) did the data suggest the LS state is based on structural distortion even though the susceptibility data at 100 K indicated the HS state. This discrepancy could not be explained based on the present work.Correlation between spectroscopic properties and spin state in solution
The present complexes contained dynamic substituents at the ends of the terpyridine ligands and so it was expected that spectroscopic features might vary with the spin state in solution. Spin states in solution are sometimes directly ascertained using NMR.32,33 In the case of cobalt(II) complexes, there have been some reports concerning evaluations of changes in the spin state in solution based on NMR data.34,35 On the other hand, Hauser et al. reported that the molar absorption constant (ε) at 22600 cm−1 of a terpyridine cobalt(II) complex varied with temperature and that these variations were associated with changes in the spin state.36
Based on such prior work, spin states in solution of the present complexes were investigated using spectroscopy. Fig. 4a–c plots the temperature dependency of the molar absorption constant ε for 20 μM solutions of 1·BPh4 in acetonitrile (ACN), DMF and DMSO, respectively. The temperature was varied between 283 and 343 K. Note that the temperature of the DMSO solution was varied between 293 and 343 K due to the higher freezing point of DMSO. In all solvents, the maximum absorbance peak at 430 nm (23255 cm−1) can be attributed to metal-to-ligand charge-transfer (MLCT) and no significant peak shifts are seen with increases in the temperature. The peak around 360 nm is attributed to the π–π* transition based on the phenyl ring and pyridine ring of the terpyridine ligand.37 Although the absorbance based on the π–π* transition has also varied based on the temperature, concentration and solvent changes, it has nothing to do with the discussion of the spin state change. The absorbance of LS species is known to decrease with increases in temperature, in accordance with classical thermal SCO behavior. The insets provide 1/ε versus T plots that assist in understanding the increase in the HS proportion with increasing temperature. Although the spin conversion ratio could not be determined precisely, 1·BPh4 exhibited an LS to HS conversion in solution, as has been observed for other SCO cobalt(II) complexes in solution. The 1·PF6, 2·PF6 and 2·BPh4 showed similar spectroscopic changes at 430 nm associated with the temperature variations (Fig. S9–S11†).
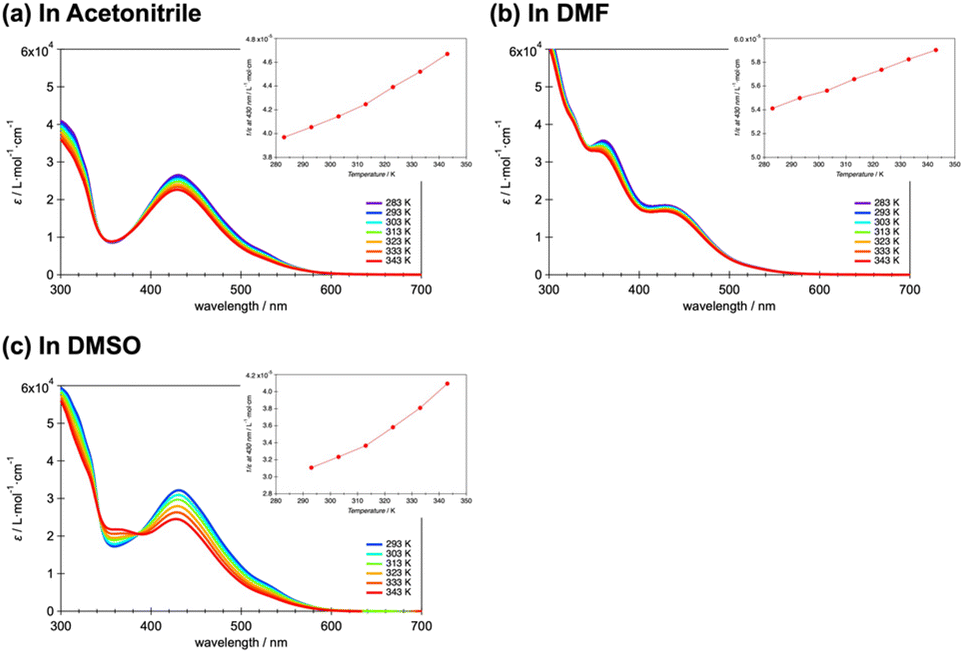 | ||
| Fig. 4 Effect of temperature on the molar adsorption constant (ε) of 1·BPh4 in (a) acetonitrile, (b) DMF and (c) DMSO. The temperature was varied from 283 to 343 K except for trials with DMSO, which used 293–343 K due to the higher freezing point of this solvent. The insets show plots of T vs. 1/ε. | ||
To the best of our knowledge, variations in spin state (as monitored herein based on spectroscopic parameters) with changes in concentration have not yet been reported. In Fig. 5a–c, the effect of concentration on the absorption spectra of 1·BPh4 in ACN, DMF and DMSO, respectively, is presented. Although ε is concentration-independent, the peak at 430 nm corresponding to the MLCT band increased as the concentration was increased from 2 to 50 μM. This is thought to be due to aggregation-induced absorption enhancement behavior.38,39 Unlike the decrease in ε with increasing temperature, implying conversion of the LS to HS state, these increases in ε with increases in concentration suggest a shift from HS to LS. This behavior can possibly be attributed to the aggregated molecular assemblies produced at higher concentrations, which likely exert pressure on the [CoN6] coordination cores. The insets show 1/ε versus T plots that assist in understanding the HS to LS conversion with increasing concentration. The 1·PF6, 2·PF6 and 2·BPh4 showed similar spectroscopic changes at 430 nm associated with the concentration changes (Fig. S12–S14†). Above a certain concentration, the peak changes were saturated, which indicates the limit of HS to LS conversion under concentration changes at room temperature.
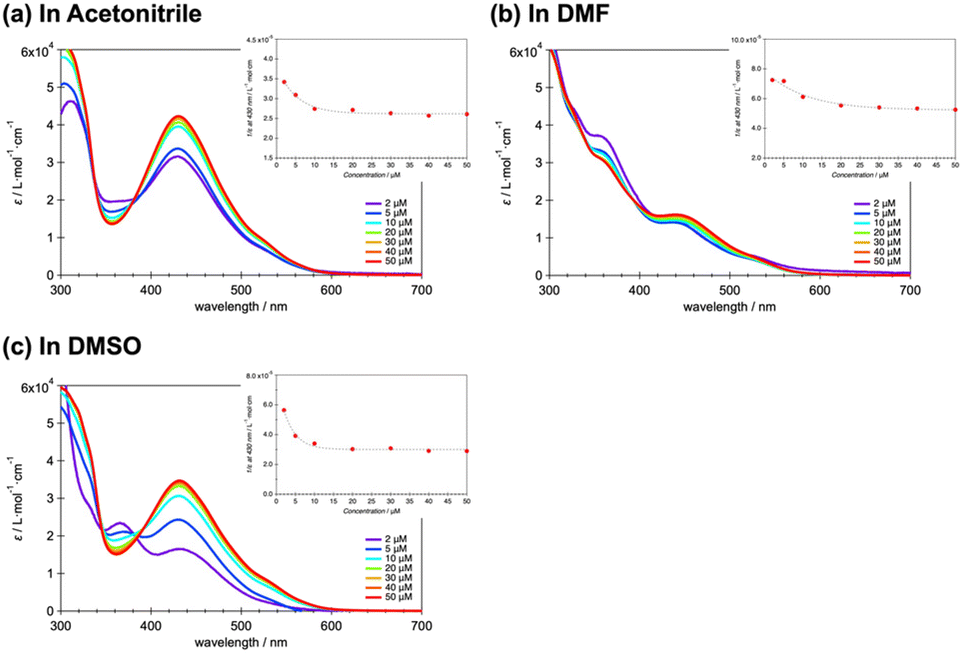 | ||
| Fig. 5 Effect of concentration on the molar adsorption constant (ε) of 1·BPh4 in (a) acetonitrile, (b) DMF and (c) DMSO. The complex concentration was varied from 2 to 50 μM. The insets show plots of 1/ε versus T. | ||
Influence of solvent viscosity on spectroscopic properties
Complexes 1·X and 2·X had dynamic di-substituted amine groups on the terpyridine ligands. These bulky ligands might be expected to affect the molecular motion and assembly of the complexes based on the viscosity of the solutions. In this work, the non-protonic solvents ACN, DMF and DMSO were used so as not to affect the amine sites on the ligands. The viscosities of ACN, DMF and DMSO at 20 °C are 0.34, 0.84 and 2.00 cP, respectively. Incidentally, water (H2O) exhibits 1.00 cP at 20 °C. Fig. 6a depicts a bar chart of maximum percentage of ε change of the respective cobalt(II) complexes in ACN, DMF and DMSO solution under different temperature changes. The degree to which ε is modified reflects the ease with which the spin state changes (from LS to HS, as seen in Fig. 4) with changes in temperature. As an example, the maximum ratio changes in ε of 1·BPh4 were 15.0% in ACN, 8.32% in DMF and 24.1% in DMSO, respectively, as the temperature was varied from 283 (293 for DMSO) to 343 K. Complex 2·X, having dimethyl amine substituents, showed linear increases in the percentage ε change. With some exceptions, the rate of change in ε increased with the viscosity of the solvent. The spin equilibration of the cobalt(II) ion in solution is known to be rapid but a higher viscosity value would likely slow down the structural change around the cobalt(II) centers (that is, would modify the coordination environment). This effect may have increased the relative proportion of complexes in the LS state at room temperature and increased the ratio of ε change.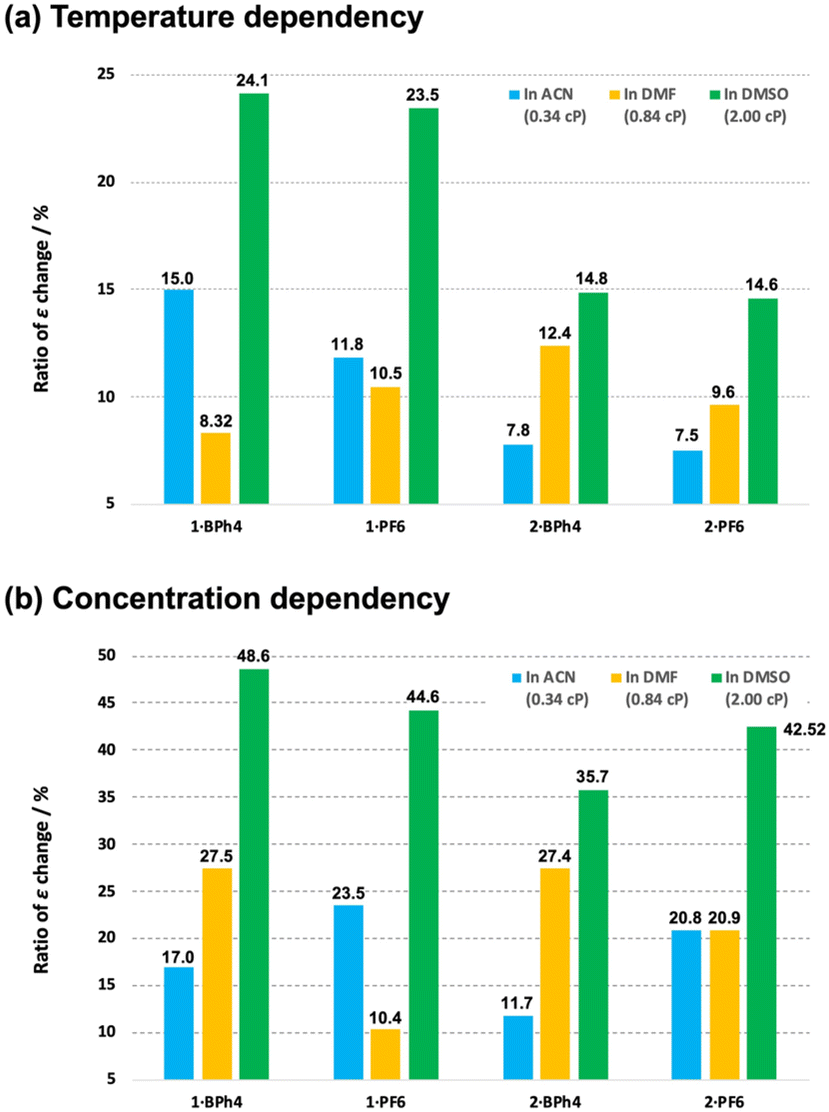 | ||
| Fig. 6 Bar chart of percentage change in the molar adsorption constant (ε) at 430 nm for the present cobalt(II) complexes in ACN (blue), DMF (orange) and DMSO (green) in response to changes in (a) temperature and (b) concentration. | ||
Fig. 6b shows a bar chart of maximum percentage of ε change of the respective cobalt(II) complexes in ACN, DMF and DMSO solution with variation of concentration. The extent to which ε varies reflects the ease with which the spin state changes (from HS to LS, as seen in Fig. 5) with concentration change. Compared with the temperature dependency, the extent of the change in ε is much larger here, suggesting that the HS to LS conversion is relatively easy to induce by concentration changes in solution. This may be because molecular aggregation stabilizes the LS state. In the present case, the rate of change in ε systematically increases as the solvent viscosity increases.
When the temperature was varied, systematic changes in ε were observed depending on the combination of [Co(L)2]2+ cations and counter anions. In contrast, with concentration dependence, systematic changes in ε of any cobalt(II) complex were observed depending on the solvent viscosity, but no regularity for the size of [Co(L)2]2+ or counter anion was obtained.
Conclusions
In conclusion, the present work discussed changes in the spin states of terpyridine-based cobalt(II) complexes with bulky substituents in the solid state and in solution. In the solid state, the rigidly assembled complex molecules did not have the flexibility expected from rotatable substituents and exhibited gradual SCO behaviors with low cooperative values. The spin state changes in solution were also evaluated and correlated with variations in ε. Although the temperature dependency of ε resembled the typical low-cooperative gradual SCO behavior in solution, the effect of concentration suggested an HS to LS transition accompanied by molecular aggregation. This study provides new insights into molecular design as an approach to controlling the spin states of cobalt(II) complexes in solution, which has thus far received minimal attention.Author contributions
M. N. designed and supervised the project. N. I., S. F., K. K. and M. N. performed all experimental work. N. I. and M. N. wrote the manuscript. R. T. and S. H. determined and evaluated the magnetic properties of the samples. All authors contributed to the manuscript and have given approval to the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a JSPS KAKENHI Grant-in-Aid for Early-Career Scientists (No. JP22K14695). M. N. also acknowledges financial support from the Iketani Science and Technology Foundation (ISTF). We are also thankful to Dr Kenji Yoza at Bruker Japan and Dr Mitsuaki Suzuki at Josai University for technical support for SC-XRD.References
- P. Gütlich, Y. Garcia and H. A. Goodwin, Chem. Soc. Rev., 2000, 29, 419–427 RSC.
- J. A. Real, A. B. Gaspar and M. C. Munoz, Dalton Trans., 2005, 2062–2079 RSC.
- A. Bousseksou, G. Molnμr and G. Matouzenko, Eur. J. Inorg. Chem., 2004, 4353–4369 CrossRef CAS.
- S. Brooker, Chem. Soc. Rev., 2015, 44, 2880–2892 RSC.
- O. Kahn and C. J. Martinez, Science, 1998, 279, 44 CrossRef CAS.
- Spin Crossover in Transition Metal Compounds I-III, ed. P. Gütlich and H. A. Goodwin, Springer-Verlag, Berlin, Heidelberg, Germany, 2004 Search PubMed.
- Spin-Crossover Materials, ed. M. A. Halcrow, Wiley, Oxford, United Kingdom, 2013 Search PubMed.
- R. A. Taylor, A. J. Lough and M. T. Lemaire, J. Mater. Chem. C, 2016, 4, 455–459 RSC.
- F. Fürmeyer, D. Münzberg, L. M. Carrella and E. Rentschler, Molecules, 2020, 25, 855 CrossRef PubMed.
- R. Akiyoshi, Y. Komatsumaru, M. Donoshita, S. Dekura, Y. Yoshida, H. Kitagawa, Y. Kitagawa, L. F. Lindoy and S. Hayami, Angew. Chem., Int. Ed., 2021, 60, 12717–12722 CrossRef CAS PubMed.
- V. García-López, N. Giaconi, L. Poggini, J. Calbo, A. Juhin, B. Cortigiani, J. Herrero-Martín, E. Ortí, M. Mannini, M. Clemente-León and E. Coronado, Adv. Funct. Mater., 2023, 33, 2300351 CrossRef.
- Y.-F. Deng, Y.-N. Wang, X.-H. Zhao and Y.-Z. Zhang, Dalton Trans., 2024, 53, 699–705 RSC.
- (a) S. Hayami, Y. Komatsu, T. Shimizu, H. Kamihata and Y. H. Lee, Coord. Chem. Rev., 2011, 255, 1981–1990 CrossRef CAS; (b) S. Hayami, M. Nakaya, H. Ohmagari, A. S. Alao, M. Nakamura, R. Ohtani, R. Yamaguchi, T. Kuroda-Sowa and J. K. Clegg, Dalton Trans., 2015, 44, 9345–9348 RSC; (c) M. Nakaya, W. Kosaka, H. Miyasaka, Y. Komatsumaru, S. Kawaguchi, K. Sugimoto, Y. Zhang, M. Nakamura, L. F. Lindoy and S. Hayami, Angew. Chem., Int. Ed., 2020, 59, 10658–10665 CrossRef CAS PubMed.
- D. Shao, L. Shi, F. X. Shen, X. Q. Wei, O. Sato and X. Y. Wang, Inorg. Chem., 2019, 58, 11589–11598 CrossRef CAS PubMed.
- T. Kanetomo, Z. Ni and M. Enomoto, Dalton Trans., 2022, 51, 5034–5040 RSC.
- C. Ni, J. C. Fettinger, G. J. Long and P. P. Power, Inorg. Chem., 2009, 48, 2443–2448 CrossRef CAS PubMed.
- M. G. Cowan, J. Olguin, S. Narayanaswamy, J. L. Tallon and S. Brooker, J. Am. Chem. Soc., 2012, 134, 2892–2894 CrossRef CAS PubMed.
- A. V. Vologzhanina, A. S. Belov, V. V. Novikov, A. V. Dolganov, G. V. Romanenko, V. I. Ovcharenko, A. A. Korlyukov, M. I. Buzin and Y. Z. Voloshin, Inorg. Chem., 2015, 54, 5827–5838 CrossRef CAS PubMed.
- C. Brady, J. J. McGarvey, J. K. McCusker, H. Toftlund and D. N. Hendrickson, Top. Curr. Chem., 2004, 235, 1–22 CrossRef CAS.
- P. N. Martinho, Y. Ortin, B. Gildea, C. Gandolfi, G. McKerr, B. O'Hagan, M. Albrecht and G. G. Morgan, Dalton Trans., 2012, 41, 7461–7463 RSC.
- S. Sundaresan and S. Brooker, Inorg. Chem., 2023, 62, 12192–12202 CrossRef CAS PubMed.
- S. Hayami, N. Motokawa, A. Shuto, R. Moriyama, N. Masuhara, K. Inoue and Y. Maeda, Polyhedron, 2007, 26, 2375–2380 CrossRef CAS.
- A. B. Gaspar and M. Seredyuk, Coord. Chem. Rev., 2014, 268, 41–58 CrossRef CAS.
- A. Enriquez-Cabrera, A. Rapakousiou, M. P. Bello, G. Molnár, L. Salmon and A. Bousseksou, Coord. Chem. Rev., 2020, 419, 213396 CrossRef CAS.
- R. Akiyoshi, R. Ohtani, L. F. Lindoy and S. Hayami, Dalton Trans., 2021, 50, 5065–5079 RSC.
- H. A. Goodwin, Spin Crossover in Cobalt(II) Systems, in Spin Crossover in Transition Metal Compounds II. Topics in Current Chemistry, Springer, Berlin, Heidelberg, 2004, vol. 234, pp. 23–47 Search PubMed.
- W. Nicolazzi and A. Bousseksou, C. R. Chim., 2018, 21, 1060–1074 CrossRef CAS.
- S. Vela and H. Paulsen, Inorg. Chem., 2018, 57, 9478–9488 CrossRef CAS PubMed.
- M. A. Halcrow, Chem. Soc. Rev., 2011, 40, 4119–4142 RSC.
- P. Guionneau, M. Marchivie, G. Bravic, J.-F. Létard and D. Chasseau, J. Mater. Chem., 2002, 12, 2546–2551 RSC.
- M. Nakaya, R. Ohtani, J. W. Shin, M. Nakamura, L. F. Lindoy and S. Hayami, Dalton Trans., 2018, 47, 13809–13814 RSC.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- B. Weber and F. A. Walker, Inorg. Chem., 2007, 46, 6794–6803 CrossRef CAS PubMed.
- A. A. Pavlov, G. L. Denisov, M. A. Kiskin, Y. V. Nelyubina and V. V. Novikov, Inorg. Chem., 2017, 56, 14759–14762 CrossRef CAS PubMed.
- Y. Pankratova, D. Aleshin, I. Nikovskiy, V. Novikov and Y. Nelyubina, Inorg. Chem., 2020, 59, 7700–7709 CrossRef CAS PubMed.
- C. Enachescu, I. Krivokapic, M. Zerara, J. A. Real, N. Amstutz and A. Hauser, Inorg. Chim. Acta, 2007, 360, 3945–3950 CrossRef CAS.
- L. Xiao, Y. Xu, M. Yan, D. Galipeau, X. Peng and X. Yan, J. Phys. Chem. A, 2010, 114, 9090–9097 CrossRef CAS PubMed.
- W. Wang, X. Sun, J. Qu, X. Xie, Z.-H. Qi, D. Hong, S. Jing, D. Zheng, Y. Tian, H. Ma, S. Yu and J. Ma, Phys. Chem. Chem. Phys., 2017, 19, 31443–31451 RSC.
- C. J. Zeman IV, G. Kang and K. L. Kohlstedt, ACS Appl. Mater. Interfaces, 2022, 14, 45644–45657 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2332743–2332746. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00433g |
| This journal is © The Royal Society of Chemistry 2024 |