DOI:
10.1039/D3DT04049F
(Perspective)
Dalton Trans., 2024,
53, 5320-5341
Stimuli-responsive luminescence from polar cyano/isocyano-derived luminophores via structural tailoring and self-assembly
Received
4th December 2023
, Accepted 5th February 2024
First published on 13th February 2024
Abstract
Polar cyano fragments and their isomeric isocyano counterparts have attracted great attention as stimuli-responsive luminescent materials in a wide range of fields including organic light-emitting diode devices, chemical fluorescent sensors, photoelectric semiconductors, anti-counterfeit products, etc., mainly because of their typical electron-deficient activity, noncovalent recognition ability, and variable coordination capacity. The electron-deficient and polar nature of these blocks have significant effects on the properties of the cyano/isocyano-based luminophore materials, especially concerning their condensed state-dependent electronic structures. Among them, donor–acceptor (D–A) derived unimolecular and co-assembled luminophores have attracted more attention because their large delocalized structures and noncovalent interaction recognition sites can rebuild the electronic transfer character in the aggregative state, thus endowing them with outstanding stimuli-responsive luminescent behavior via intermolecular and intramolecular charge transfer in polytropic morphologies. In this perspective paper, we give a brief introduction on stimuli-responsive organic and coordinated luminophores and the documented typical design concepts and applications in recent years. It is expected that this perspective article will not only summarize the recent developments of polar cyano/isocyano-derived luminophores and their coordination compounds via structural tailoring and self-assembly but also throw light on the future of the design of more sophisticated stimuli-responsive architectures and their versatile properties.
1. Introduction
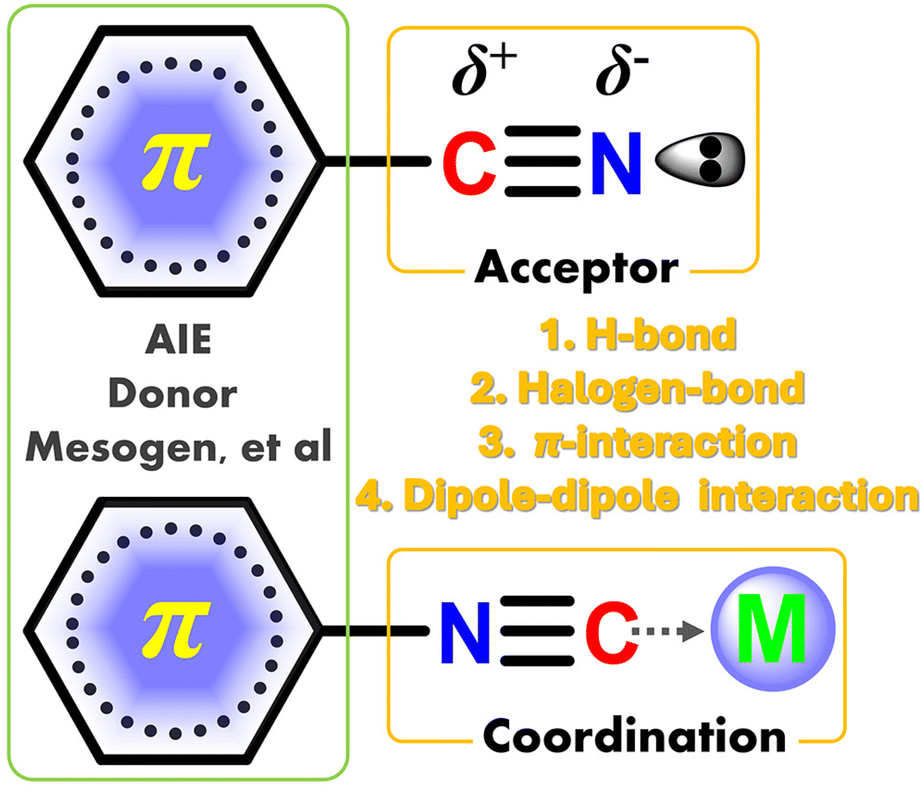
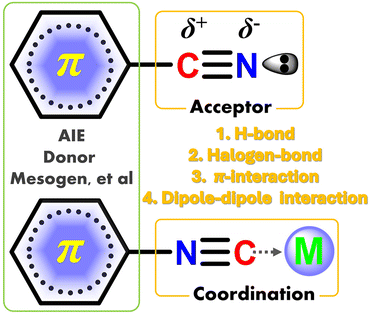
Stimuli-responsive luminescence (SRL) refers to the molecule that possesses the changed color of fluorescence/phosphorescence under external photo-excitation or force-triggered glow via contact/contactless stimuli operations.1–13 Among them, the first mentioned color-change behavior is usually defined as the mechanochromic luminescence (MCL),9 whereas the concurrent interest of force-triggered emission is described as triboluminescence (TL).7 The stimulative operations involve contact grinding, extrusion, heat, acid–base metathesis or contactless electric field, and light radiation. Up to now, polar cyano/isocyano derivatives are the most popular high-efficient luminescence species, which play significant roles in the fields of organic light-emitting diodes (OLEDs),11,14–21 biological imaging,22,23 ion recognition24,25 anti-counterfeit material,26 and many photocatalytic reactions.27,28 Benefiting from accessibility and structural transformation, the polar cyano/isocyano groups can be introduced into the conjugated molecular skeletons through many multifarious methods, such as redox, coupling, and condensation reactions.29–33 One of the most important features of cyano/isocyano units is their high electron deficiency instinct and asymmetric charge distribution of polar C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond and exposed lone–pair electron, which allows abundant supramolecular non-covalent interactions (NCIs) including π–π stacking, hydrogen bonding, coordination, halogen bonding, and dipole–dipole interactions (Scheme 1). Consequently, the NCIs in cyano/isocyano-based components can be easily introduced and regulated and their luminescence behavior is affected by many factors, like molecular conjugation, planarity, symmetry, steric hindrance, captodative host–guest effects, etc.
N bond and exposed lone–pair electron, which allows abundant supramolecular non-covalent interactions (NCIs) including π–π stacking, hydrogen bonding, coordination, halogen bonding, and dipole–dipole interactions (Scheme 1). Consequently, the NCIs in cyano/isocyano-based components can be easily introduced and regulated and their luminescence behavior is affected by many factors, like molecular conjugation, planarity, symmetry, steric hindrance, captodative host–guest effects, etc.
 |
| | Scheme 1 Schematic representation of structural features and NCIs scope for polar cyano/isocyano derived luminophores. | |
Based on the understanding of these structural factors together with the advanced self-assembly techniques, mechano-responsive luminescence can be realized through many facile molecular designs and various external stimulations. For instance, by combining modern synthetic chemistry, crystal engineering, and self-assembly tactics, the flexible molecular conformation and supramolecular morphology could be tailored by alteration in the external environment, e.g. temperature, solvent polarity, and stress. Those polymorphisms might change after stimulation, leading to the alternant mutations in charge/energy transfer and radiative processes. Moreover, stimuli-responsive luminescence induced by supramolecular interactions could be established by regulating the interactions among organic molecules in the aggregate state and even extending synchronously to coordination compounds. Notably, though many cyano/isocyano derivatives have been reported, the studies of their luminescence, especially the relationship between crystalline morphological prediction and practical mechano-responsive performance, remain largely limited. Thus, there is still much space for expansion in this area. In this perspective, we summarized the current research evolution in stimuli-responsive luminescence derived from cyano/isocyano-based organic molecules and coordination compounds, including MCL and TL after extrusion and grinding. The irradiation, thermal, electric field, and acid–base metathesis triggered luminescence switching have been discussed as well. This manuscript is not intended to be comprehensive for all stimuli-responsive luminescent emitters, where only cyano/isocyano derivatives are particularly focused and surveyed.
2. Discussion
2.1. Polar cyano-based stimuli-responsive luminescence
Early comprehensive reviews have been published on multiple types of stimuli-responsive emitters and their use in organic optoelectronics.3,7,9,10,34 Synthesis and applications of unique classes of AIE luminophores,1 noble–metal complexes,35–37 mesophase aggregates,38 and triboluminescent system7 have been summarised elsewhere. This perspective focuses specifically on the cyano/isocyano derivatives and recent advances in their design, synthesis, SRL properties, and applications, and provides critical consideration of the challenges and prospects for future developments. Overall, a recent overview of comprehensive mechano-responsive luminescence is provided, not only including popular organic matrices but also coordination compounds that feature additional optical characteristics.
2.2. General considerations of molecular design and synthesis
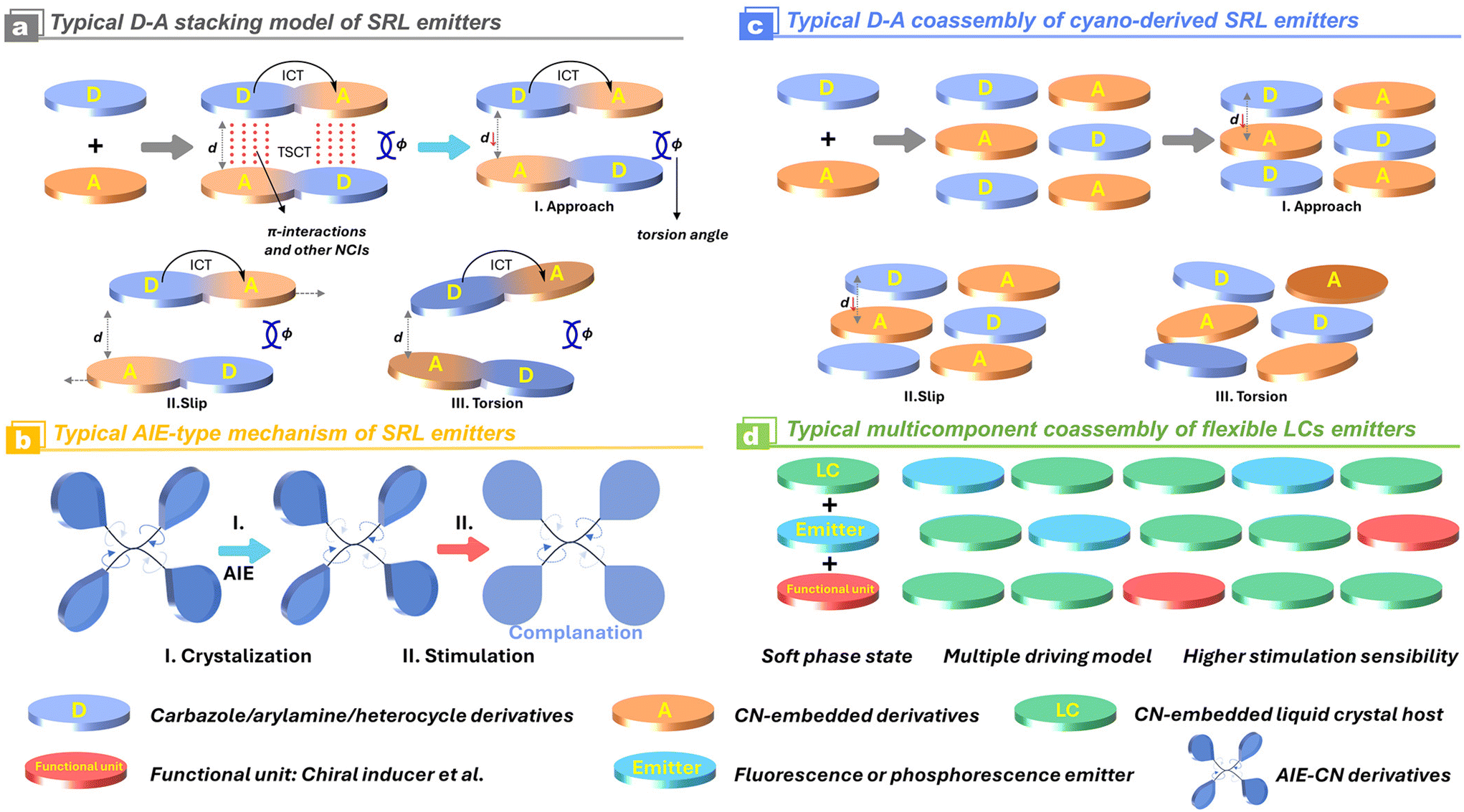
In consideration of the electron-withdrawing character of the cyano/isocyano groups, extensive efforts have been devoted to developing electron-separated D–A, D–π–A structures and their varieties (Scheme 3). The prevalent strategies for the construction of the efficient stimuli-responsive luminescence are based on the following considerations: (1) when the electron-deficient cyano/isocyano blocks are introduced into some electron donors, especially for asymmetrically nonplanar ones, the molecular conformation of corresponding luminophores could be significantly perturbated through preventing the tight packing of complex accumulation in solid; (2) if the parent cyano/isocyano based emission skeleton is fused with some bridging aggregation-induced emission (AIE) backbones like tetraphenylethylene (TPE), polyarylamine, carbazole derivatives, the resultant luminescence is usually considerably strong since the electron cloud is delocalized over the torsional nonplanar π-system as well as the intramolecular charge transfer (ICT); (3) abundant supramolecular NCIs have demonstrated to be very conducive to stabilizing the polymorphism because of the presence of versatile molecular conformation, stacking orientation, and host–guest interactions. The molecular planarity and packing fashion could be regulated and controlled by external stimulations, leading to the formation of extraordinary changes in intermolecular and intramolecular charge transfer during the excitation (Scheme 3). This process applies to both a single-molecular component and a multi-molecular host–guest system.
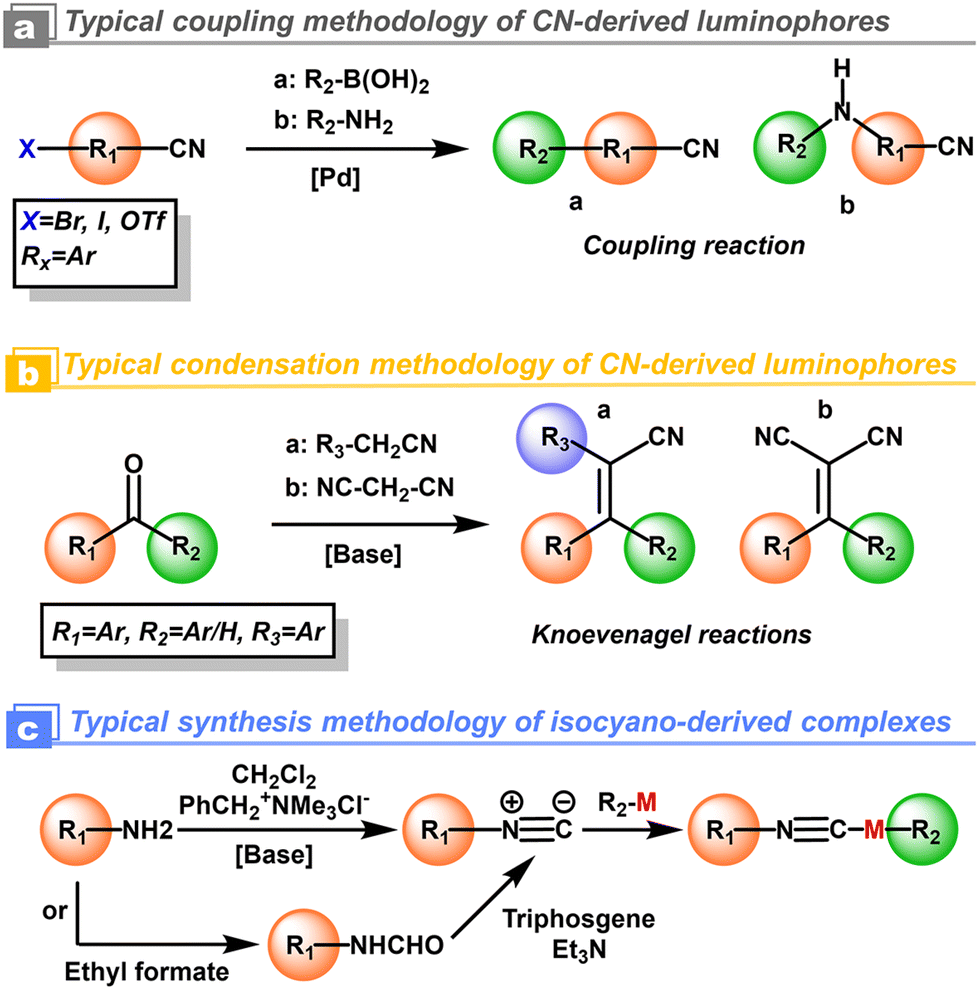
For the cyanation at the aryls, alkali metal salts, such as toxic NaCN and KCN, were used in the early years as cyanidation reagents. With the developments of transition-metal catalysis, CuCN and K4[Fe(CN)6] with low toxicity have been used to replace the alkali metal cyanides. The highly efficient cyanidation process and reagents are often combined with transition-metal catalysts i.e. [Pd], [Ni], and [Cu].32 Indeed, thanks to the current developments of modern synthetic chemistry and commercial availability, additional cyanidation is not required in most cases. The above-mentioned cyanation could be replaced by Suzuki coupling,33 Buchwald coupling,29 and Knoevenagel reactions from predesigned organic cyano-blocks and aromatic chromophores.31 As a comparison, the isocyano compounds could be prepared from amine and dihalocarbene by the Hofmann reaction (Scheme 2).30
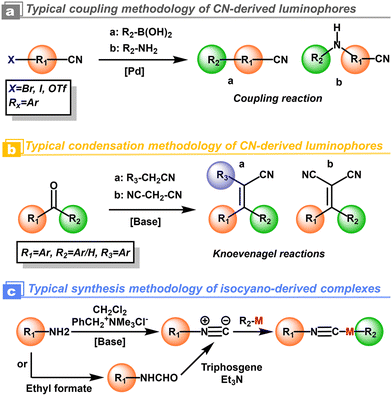 |
| | Scheme 2 Schematic representation of synthesis methodologies for polar cyano/isocyano-derived luminophores. Rx represents aryl groups. | |
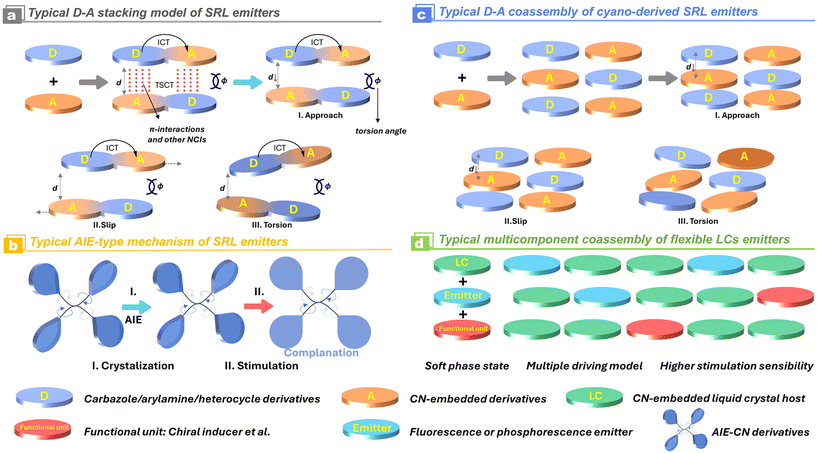 |
| | Scheme 3 Schematic representation of different construction methodologies for SRL materials and the variations of their molecular/supramolecular interactions. | |
2.3. Cyano-embedded luminophores and their SRL behavior
2.3.1. Strong electron donor fused skeletons.
Based on the above motivation about D–A and D–π–A structures triggered ICT and polymorphic capacity, strong electron donor fused skeletons (triphenylamine, carbazole, phenoxazine derivative donors, etc.) have attracted great interest in recent years due to the possibility of innovating luminescence properties derived from the charge transition state, such as energy harvesting pathway, modular modification, and conformational complexity. Compared with traditional fluorescent materials, many previous reports proved that suitable structures helped improve the exciton utilization of D–A structures like thermally activated delayed fluorescence (TADF) and organic phosphorescence.11,17
2.3.1.1. Carbazole donor fused derivatives.
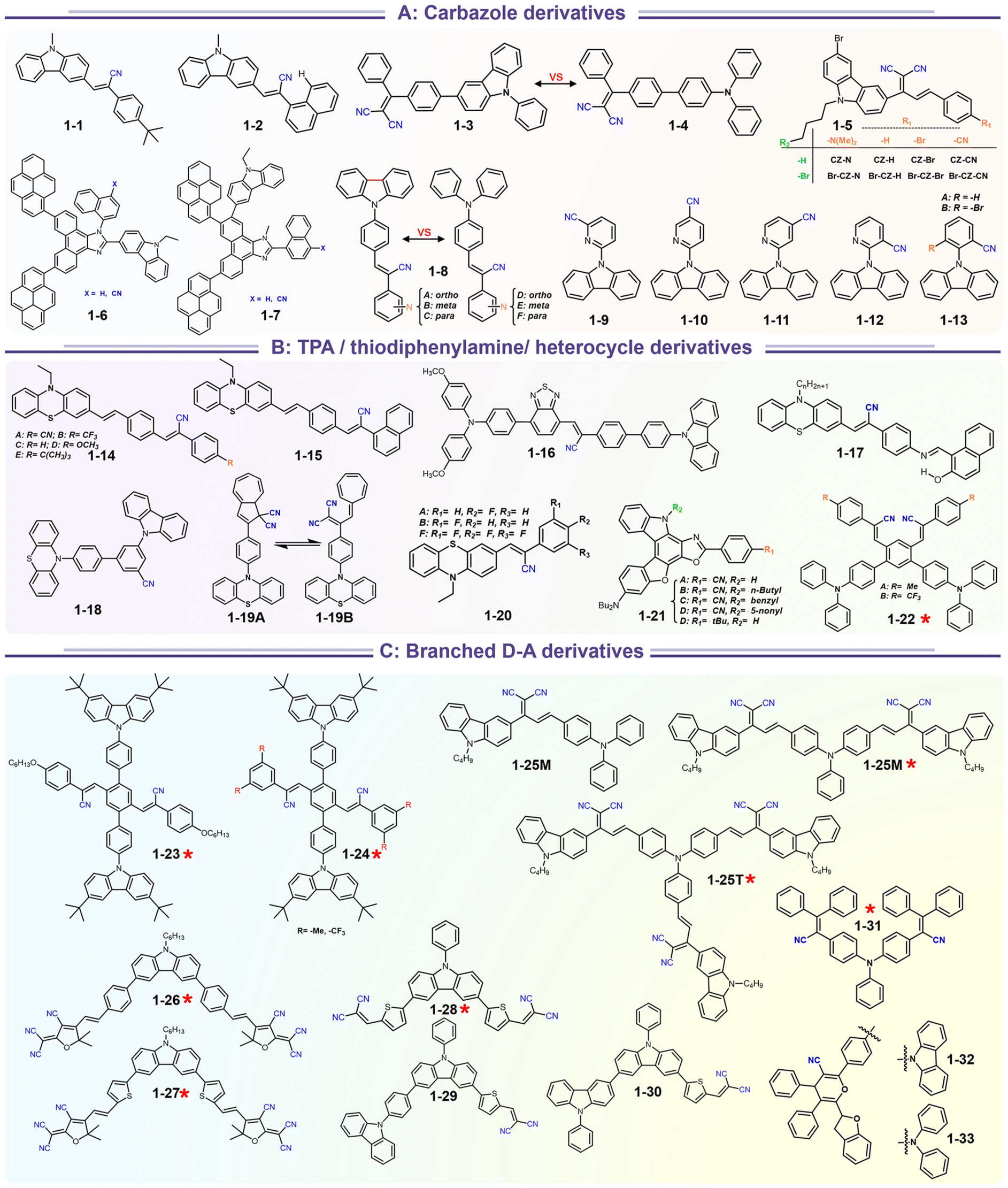
Carbazole and triphenylamine fused cyano derivatives represent the most critical families among mechano-responsive luminescence compounds. These compounds are easily acquired based on the combination of modular donors with acceptors via Suzuki coupling, Buchwald coupling, and Knoevenagel condensation. The most extensively studied integrations are those produced using cheap carbazole derivatives to build luminescent moieties (Scheme 4). As a consequence of the abundance of available carbazole blocks and aryl cyanide units, an appropriate design of the constituent building blocks allows the production of a variety of mechano-responsive skeletons with various stimuli-responsive behaviors.
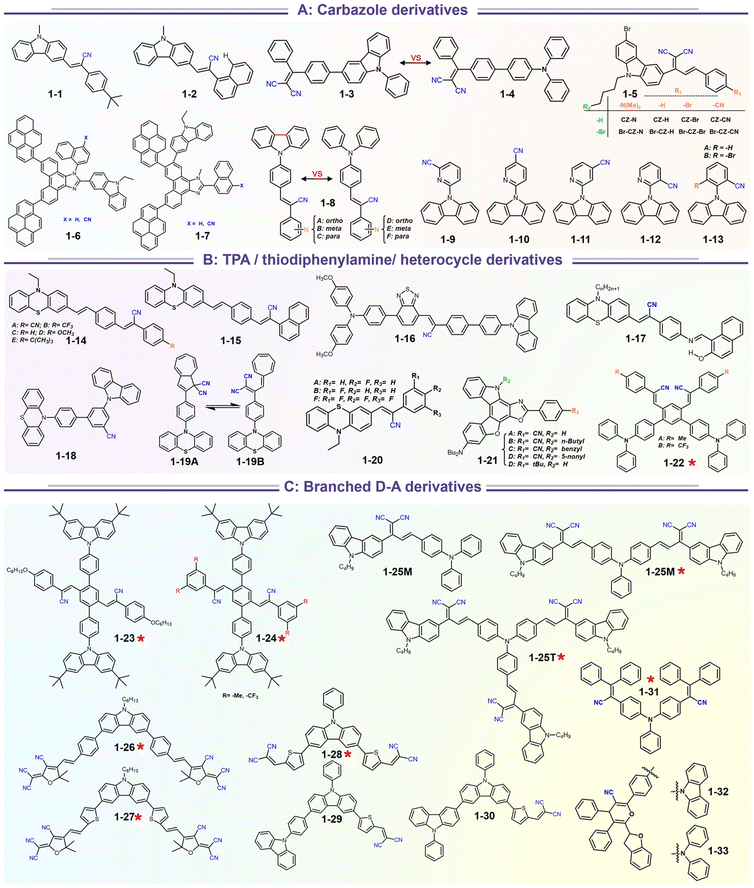 |
| | Scheme 4 Representative structures of SRL materials based on strong electron donor fused cyano derivatives. | |
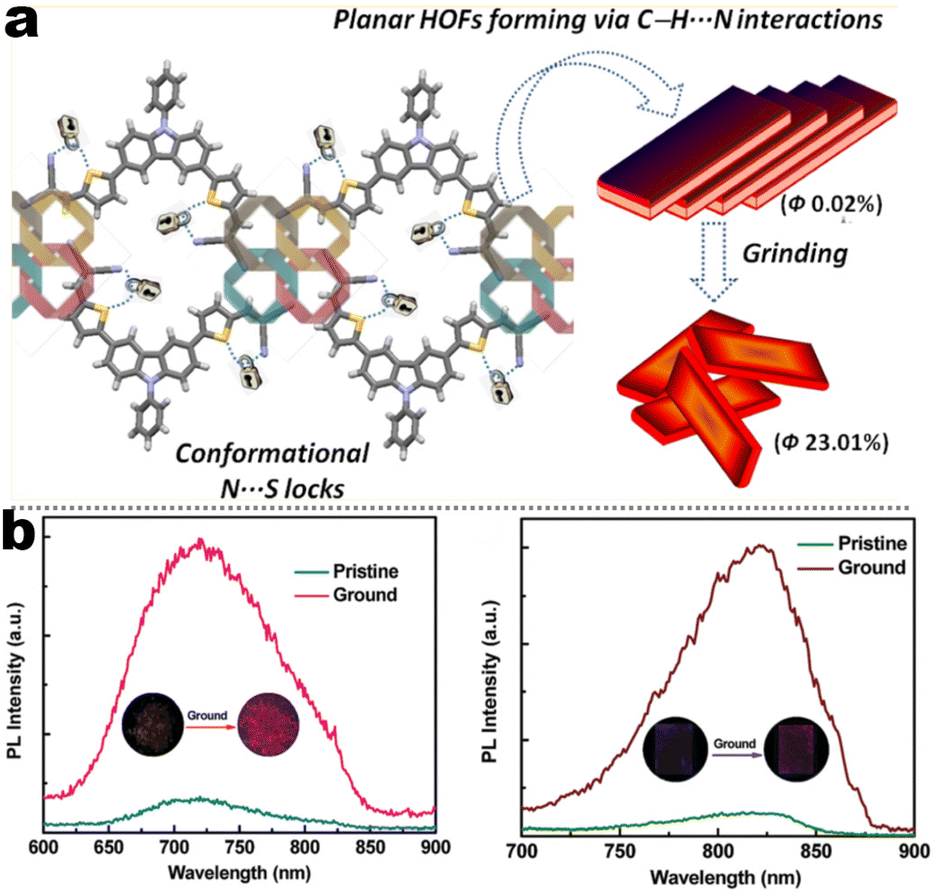
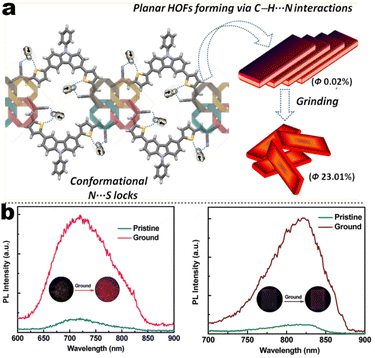
A recent search in the references has shown that a lot of carbazole-fused mechano-responsive skeletons have been investigated. There are two main kinds of divisions of structures: (1) the –CN unit was placed on the substituted aromatic ring (i.e.1-6 to 1-7, 1-9 to 1-13etc.); (2) the –CN was inserted in the alkene. The former was mainly derived from primitive CN-substituent substrates, and the popularity of the latter can be promoted by the modular Knoevenagel reaction. Among them, the most relevant compounds were nonplanar asymmetric structures (Scheme 4, marked by a red asterisk). In the above cases, the conjugation prolongation promoted the corresponding covalent-bond assisted intramolecular electron transfer and a bathochromic shift from ultraviolet (carbazole precursor) to the visible region. In this area, Xue et al. prepared two carbazole-fused monocyanoethene derivatives and described the crystallization-induced blue emission (433, 434 nm) and MCL (Δλmax for 1-1: 26 nm, 1-2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 nm). Single-crystal structure analysis indicated that the two molecules adopted more distorted conformation via π–π and CN⋯H–C hydrogen bonds in their packing. Two compounds exhibited reversible MCL behavior. Force stimuli promoted a larger bathochromic shift for p-tert-butylphenyl substitute than 2-naphthyl one because more pronounced planarization for the former existed after force-induced stacking in the crystal.39 In addition to monocyanoethene compounds, dicyanoethene compounds were also a simple but important family. The increased numbers of CN-units could enhance the electron deficiency and NCI contact sites in the compounds. Xiang et al. synthesized two AIE-active luminogens 1-3/1-4 using the classical Suzuki–Miyaura coupling and Knoevenagel condensation reactions.40 Both of them showed specific ICT characteristics and aggregation-induced emission enhancement (AIEE) properties. In addition, 1-3 and 1-4 exhibited different MCL behavior with wavelength changes of 27 and 57 nm, respectively. DFT calculations and experiments revealed that 1-4 possessed a stronger ICT degree and more twisted molecular conformation than those of 1-3, which endowed 1-4 with loose molecular packing and weak intermolecular interactions. These results indicated that the substituent effect not only affected the molecular orbital level but also the packing mode. This phenomenon has also been found in eight derivates (1-5) prepared by Kong's group, where the emission contrast of MCL reached up to 52 nm.41 For mixed aryl cyanide and traditional fluorophore ramified structures, Jayabharathi et al. introduced pyrene, carbazoles, and anthracyl cyanide into arylimidazole to study their mechanochromism and aggregation-induced emission (1-6/1-7). As same as most reports, powder X-ray diffraction (PXRD) revealed that the MCL (Δλmax = 64 nm) could be attributed to the morphological transformation of these compounds from crystalline to amorphous states.42 Subsequently, this group replaced the carbazole unit with TPEs, and the new compounds showed variable emission colors by varying water fractions which can be ascribed to the size effect of different aggregates.43 Moreover, the position effect represented a great influence on the molecular conformation and stacking fashion in different solid states. Anthony et al. presented another good model, where two sets of isomeric CZ/TPA-fused fluorophors with partially planar and propeller construction (1-8) have been described. Molecular aggregation studies indicated the formation of 1D nanostructures of nanoparticles was influenced by the evolution of NCIs with increasing water fraction and time. This evolution of nanostructures led to tunable fluorescence from green to red. The subtle structural change and formation of different crystal forms (polymorphs) resulted in huge fluorescence alteration between 514 and 644 nm (Δλmax = 130 nm). Solid-state structural studies showed that relatively weak intermolecular interactions in the crystal packing of 1-8A, 1-8C, and 1-8F resulted in the formation of different crystal polymorphs and varied molecular assemblies with tunable fluorescence.44 Those above reports were monotonous fluorescence and mono-responsive MCL. However, the multi-responsive and room temperature phosphorescence (RTP)/long afterglow was a crucial platform for obtaining applicative optical encryption. Thus, acid/base regulated protonation and deprotonation on pyridine and fluorescence shifts have also been utilized for demonstrating the self-erasable and rewritable RTP platforms based on promoted spin-forbidden transition. In Ma's work, they showed a force-stimulate and acid-responsiveness of pure organics with persistent phosphorescence at room temperature via simple isomerization in the D–A–A′ fused pyridine carbazoles (Fig. 1, 1-9 to 1-13).45,46 With the comprehensive D–A fused strategy, other more twisted symmetric structures have also been reported (Scheme 4, 1-25 to 1-31), such as the C2 symmetric dimers, C3 symmetric trimers, and helicates (1-21).47 These carbazole units typically occupied the terminals in molecules, and the molecules displayed linear or dendritic construction (1-6, 1-9 to 1-12, 1-25), leading to more accessibility for polymorphism, extra near-infrared MCL performance (1-25). In particular, Zang's group demonstrated three twisted donor–acceptor cruciform luminophores, which possessed AIE and MCL due to planar intramolecular charge transfer (PICT) under external force grinding (1-23, 1-24).48 Although its emission extended into the near-infrared region, no structural insights from single crystals could be obtained. To seek the improvement between NIR emission and MCL, Yang et al. developed three small oligomers based on incorporative CZ/TPA donors and multiple dicyano accepters (1-25). Only compound 1-25M displayed the crystallization-induced emission enhancement (CIEE) effect and a distinct bathochromic shift of fluorescence emission from the orange (λem = 598 nm) to the near-infrared section (λem = 643 nm) by mechanical grinding. However, the most bathochromic shift of fluorescence was found at the NIR region for dimeric 1-25D (peak at 695 nm) and trimeric 1-25T (peak at 689 nm) compounds, respectively.47 Another result also certified that the more mixed donors and D–A framework could facilitate the narrow band gap (Fig. 2, 1-16). This tendency has also been found in two CZ/thiophene donors joint organic luminophores with ultra-strong dipole moments of 16.1 (1-26) and 22.7 (1-27) Debye and remarkable MCL turn-on features at 822 nm (Fig. 3b). It should be noted that their NIR emission not only formed in solid but also in solution. Their solid luminescence could be enhanced after grounding, which might be caused by a disturbance in the dipole–dipole interactions in the solid state.49 In this area, our group has developed novel hydrogen-bonded organic frameworks (HOFs), where the cyano-based frameworks included alternant CZ and thiophene units (Fig. 3a, 1-28 to 1-30). The molecules adopted the V-shaped conformation with tunable symmetry, which was dependent on their substituted terminals. Importantly, these kinds of layered HOFs facilitated the deep-red emission in crystals and unusual blue-shifted MCL as well
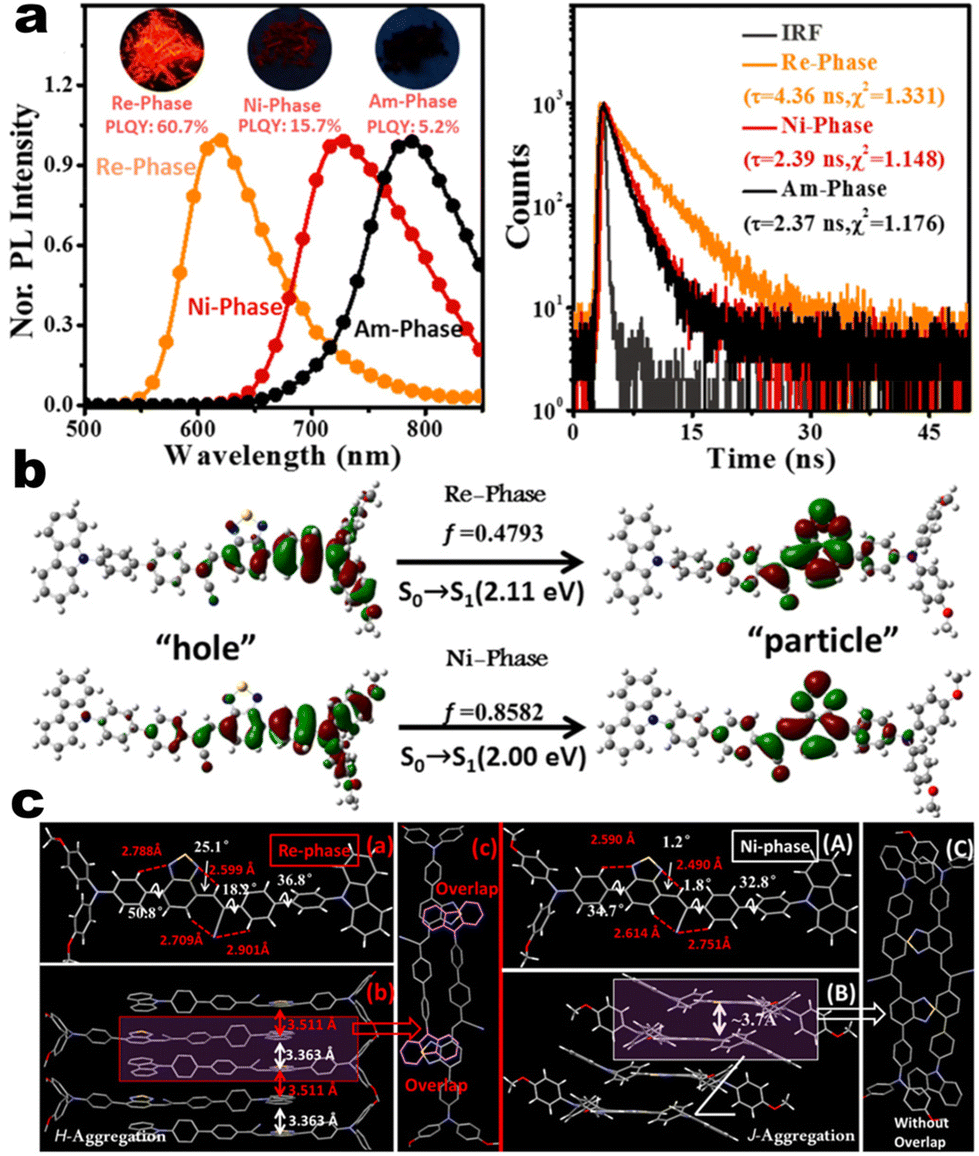
as turn-on switch in ground powder. PXRD analyses demonstrated that the close interlayer stacking was broken obviously, resulting in the hypochromatic shift of MCL. This study would inspire the exploration of high-contrast MCL materials by using the layer HOF-involved assembly.50 This type of hybrid donor/acceptor compounds have been further promoted by Zhang's group in the D–A–A–π–D′ system (Fig. 2, 1-16), showing ultra-high MCL contrast from red (615 nm) to NIR (775 nm). Crystal configuration analysis confirmed the polymorphism where the noncovalent conformational lock played key roles (Fig. 2).51
:15 nm). Single-crystal structure analysis indicated that the two molecules adopted more distorted conformation via π–π and CN⋯H–C hydrogen bonds in their packing. Two compounds exhibited reversible MCL behavior. Force stimuli promoted a larger bathochromic shift for p-tert-butylphenyl substitute than 2-naphthyl one because more pronounced planarization for the former existed after force-induced stacking in the crystal.39 In addition to monocyanoethene compounds, dicyanoethene compounds were also a simple but important family. The increased numbers of CN-units could enhance the electron deficiency and NCI contact sites in the compounds. Xiang et al. synthesized two AIE-active luminogens 1-3/1-4 using the classical Suzuki–Miyaura coupling and Knoevenagel condensation reactions.40 Both of them showed specific ICT characteristics and aggregation-induced emission enhancement (AIEE) properties. In addition, 1-3 and 1-4 exhibited different MCL behavior with wavelength changes of 27 and 57 nm, respectively. DFT calculations and experiments revealed that 1-4 possessed a stronger ICT degree and more twisted molecular conformation than those of 1-3, which endowed 1-4 with loose molecular packing and weak intermolecular interactions. These results indicated that the substituent effect not only affected the molecular orbital level but also the packing mode. This phenomenon has also been found in eight derivates (1-5) prepared by Kong's group, where the emission contrast of MCL reached up to 52 nm.41 For mixed aryl cyanide and traditional fluorophore ramified structures, Jayabharathi et al. introduced pyrene, carbazoles, and anthracyl cyanide into arylimidazole to study their mechanochromism and aggregation-induced emission (1-6/1-7). As same as most reports, powder X-ray diffraction (PXRD) revealed that the MCL (Δλmax = 64 nm) could be attributed to the morphological transformation of these compounds from crystalline to amorphous states.42 Subsequently, this group replaced the carbazole unit with TPEs, and the new compounds showed variable emission colors by varying water fractions which can be ascribed to the size effect of different aggregates.43 Moreover, the position effect represented a great influence on the molecular conformation and stacking fashion in different solid states. Anthony et al. presented another good model, where two sets of isomeric CZ/TPA-fused fluorophors with partially planar and propeller construction (1-8) have been described. Molecular aggregation studies indicated the formation of 1D nanostructures of nanoparticles was influenced by the evolution of NCIs with increasing water fraction and time. This evolution of nanostructures led to tunable fluorescence from green to red. The subtle structural change and formation of different crystal forms (polymorphs) resulted in huge fluorescence alteration between 514 and 644 nm (Δλmax = 130 nm). Solid-state structural studies showed that relatively weak intermolecular interactions in the crystal packing of 1-8A, 1-8C, and 1-8F resulted in the formation of different crystal polymorphs and varied molecular assemblies with tunable fluorescence.44 Those above reports were monotonous fluorescence and mono-responsive MCL. However, the multi-responsive and room temperature phosphorescence (RTP)/long afterglow was a crucial platform for obtaining applicative optical encryption. Thus, acid/base regulated protonation and deprotonation on pyridine and fluorescence shifts have also been utilized for demonstrating the self-erasable and rewritable RTP platforms based on promoted spin-forbidden transition. In Ma's work, they showed a force-stimulate and acid-responsiveness of pure organics with persistent phosphorescence at room temperature via simple isomerization in the D–A–A′ fused pyridine carbazoles (Fig. 1, 1-9 to 1-13).45,46 With the comprehensive D–A fused strategy, other more twisted symmetric structures have also been reported (Scheme 4, 1-25 to 1-31), such as the C2 symmetric dimers, C3 symmetric trimers, and helicates (1-21).47 These carbazole units typically occupied the terminals in molecules, and the molecules displayed linear or dendritic construction (1-6, 1-9 to 1-12, 1-25), leading to more accessibility for polymorphism, extra near-infrared MCL performance (1-25). In particular, Zang's group demonstrated three twisted donor–acceptor cruciform luminophores, which possessed AIE and MCL due to planar intramolecular charge transfer (PICT) under external force grinding (1-23, 1-24).48 Although its emission extended into the near-infrared region, no structural insights from single crystals could be obtained. To seek the improvement between NIR emission and MCL, Yang et al. developed three small oligomers based on incorporative CZ/TPA donors and multiple dicyano accepters (1-25). Only compound 1-25M displayed the crystallization-induced emission enhancement (CIEE) effect and a distinct bathochromic shift of fluorescence emission from the orange (λem = 598 nm) to the near-infrared section (λem = 643 nm) by mechanical grinding. However, the most bathochromic shift of fluorescence was found at the NIR region for dimeric 1-25D (peak at 695 nm) and trimeric 1-25T (peak at 689 nm) compounds, respectively.47 Another result also certified that the more mixed donors and D–A framework could facilitate the narrow band gap (Fig. 2, 1-16). This tendency has also been found in two CZ/thiophene donors joint organic luminophores with ultra-strong dipole moments of 16.1 (1-26) and 22.7 (1-27) Debye and remarkable MCL turn-on features at 822 nm (Fig. 3b). It should be noted that their NIR emission not only formed in solid but also in solution. Their solid luminescence could be enhanced after grounding, which might be caused by a disturbance in the dipole–dipole interactions in the solid state.49 In this area, our group has developed novel hydrogen-bonded organic frameworks (HOFs), where the cyano-based frameworks included alternant CZ and thiophene units (Fig. 3a, 1-28 to 1-30). The molecules adopted the V-shaped conformation with tunable symmetry, which was dependent on their substituted terminals. Importantly, these kinds of layered HOFs facilitated the deep-red emission in crystals and unusual blue-shifted MCL as well
as turn-on switch in ground powder. PXRD analyses demonstrated that the close interlayer stacking was broken obviously, resulting in the hypochromatic shift of MCL. This study would inspire the exploration of high-contrast MCL materials by using the layer HOF-involved assembly.50 This type of hybrid donor/acceptor compounds have been further promoted by Zhang's group in the D–A–A–π–D′ system (Fig. 2, 1-16), showing ultra-high MCL contrast from red (615 nm) to NIR (775 nm). Crystal configuration analysis confirmed the polymorphism where the noncovalent conformational lock played key roles (Fig. 2).51
 |
| | Fig. 1 (a) PL spectra of compounds from 1-9 to 1-12 at different states. (b) Crystal structures of 1-9, 1-10, 1-12. Reproduced from ref. 45 with permission. Copyright (2021), the Royal Society of Chemistry. | |
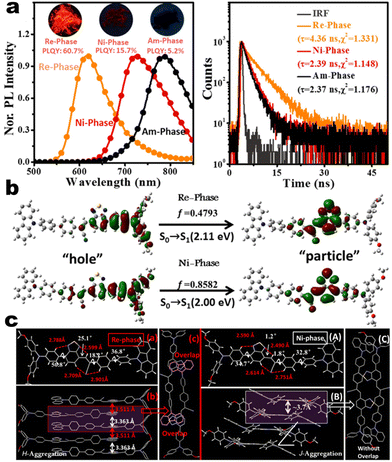 |
| | Fig. 2 (a) PL and decay spectra of compounds 1-16 at different states. (b) Natural transition orbitals of 1-16 in Re-phase and Ni-phase from S0 → S1. (c) Crystal structures and packing of Re-phase and Ni-phase. Reproduced from ref. 51 with permission. Copyright (2021), Wiley Online Library. | |
 |
| | Fig. 3 (a) HOF structure and structural transformation diagram of compound 1-28 at solid state. Reproduced from ref. 50 with permission. Copyright (2018), American Chemical Society. (b) PL spectra of compounds 1-26 and 1-27 at different states. Reproduced from ref. 49 with permission. Copyright (2018), the Royal Society of Chemistry. | |
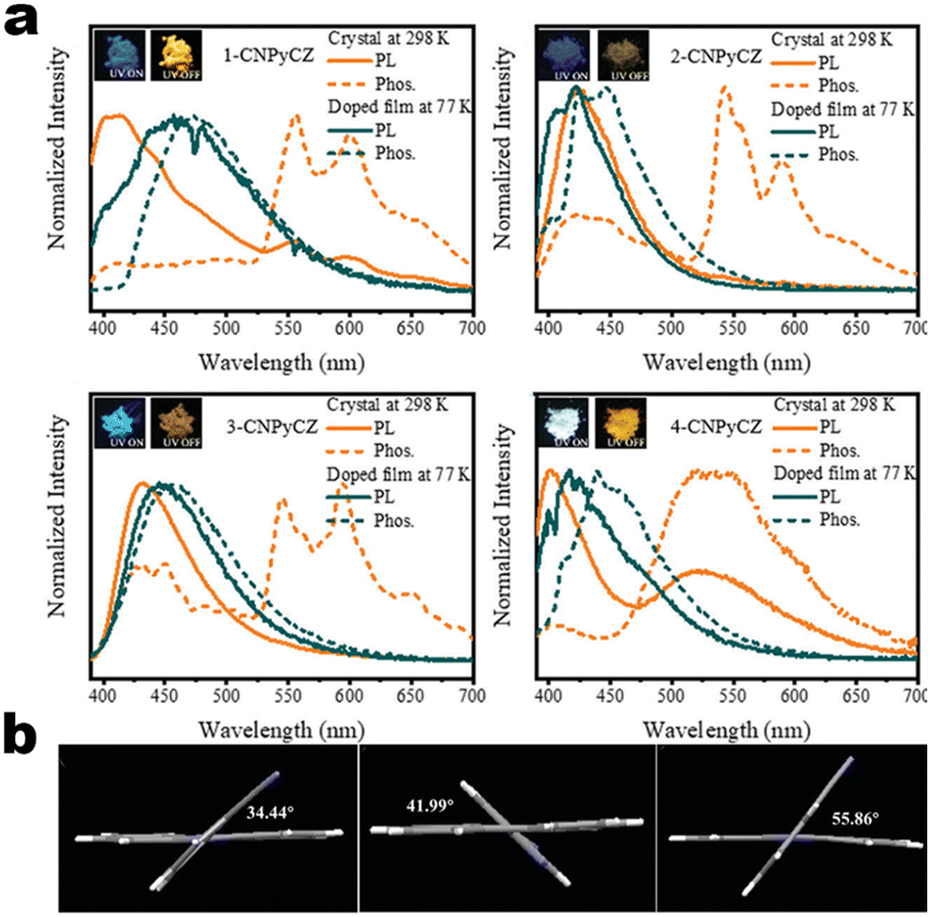
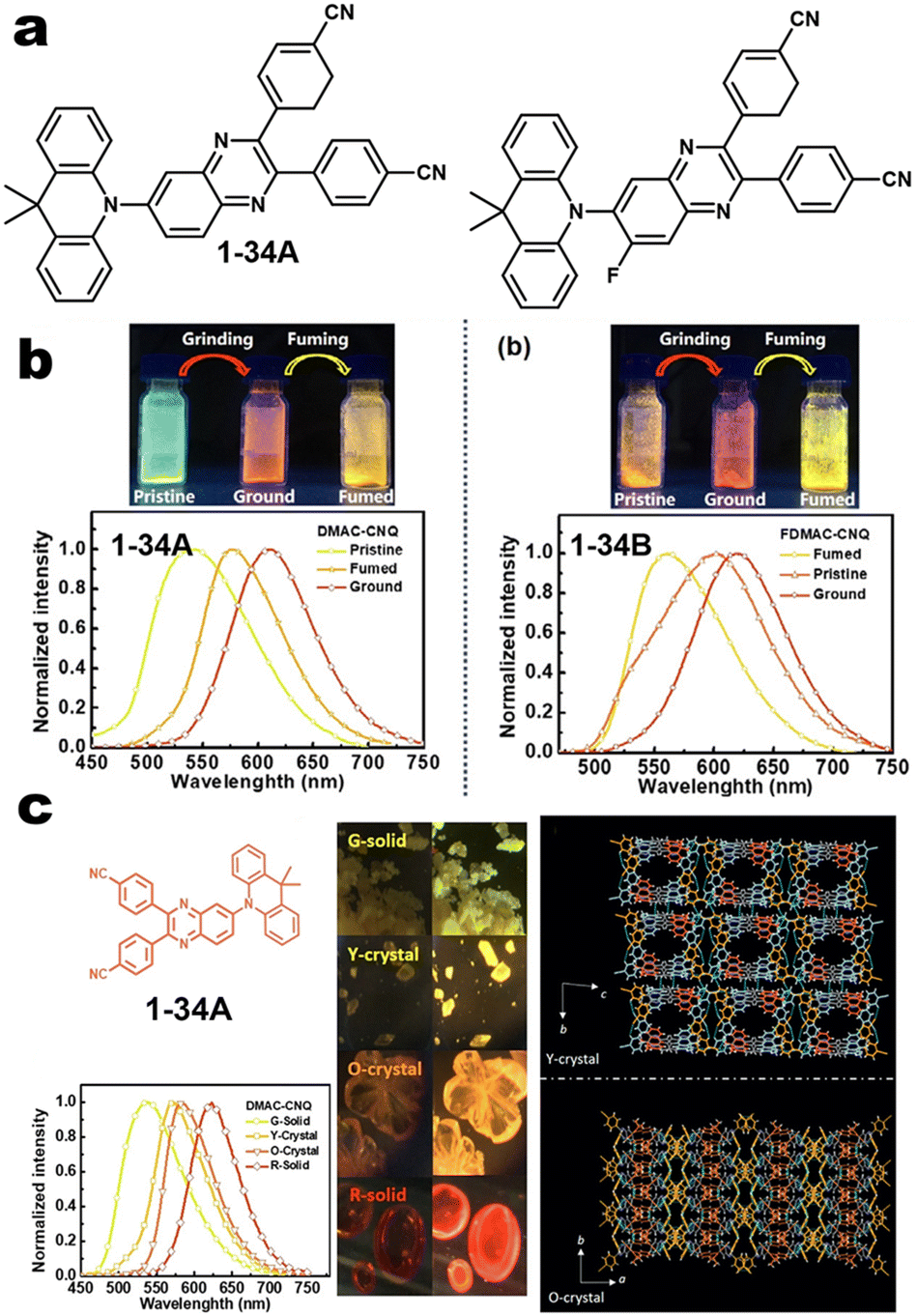
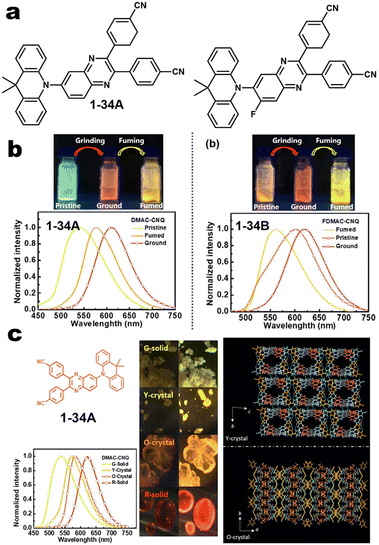 |
| | Fig. 4 (a and b) Chemical structures of 1-34A and 1-34B and their MCL pictures. Reproduced from ref. 60 with permission. Copyright (2023), American Chemical Society. (c) Crystal structure of 1-34A. Copyright (2020), Wiley Online Library. | |
Abundant chemical donors brought various kinds of emissive backbones. The CT intensity and optical band gap could be finely adjusted via manipulation of the molecular packing model to rebuild both molecular planarity and conformation. Despite electron donor-fused mechano-responsive skeletons in the field of MCL material being popular so far, they still present several issues. For instance, (1) they tend to decrease quantum efficiency in the ground powder for most cases because of the aggregation-induced quenching (ACQ) effect and energy gap law; (2) the absence of in-depth artificial intelligence prediction of photophysical properties in the different states and good tools of high-contrast emission (>150 nm); (3) multiple responses of polarize-related luminescence (linear-polarized or circular-polarized light) is still rare, although this could carry out additional photon information and operable encryption dimension in many applications.61,62
2.3.2. AIE matrix-fused SRL emitters.
Among the various well-developed stimuli-responsive fluorophors, TPE-based electron-rich skeletons have gained great attention for their interesting aggregation behavior, intense AIE effect, and potential applications in optoelectronic devices.63 Typically, propeller-liked TPE unit was conducive to varieties of NCIs and CT in D–A building blocks, solvent effect,12 and solvophobic interaction-induced physical encapsulation.64 These interactions shall induce single species into solid assemblies with the non-compact molecular arrangement, and some interesting behaviors, such as polymorphism, MCL, and TL, have been reported in recent years.
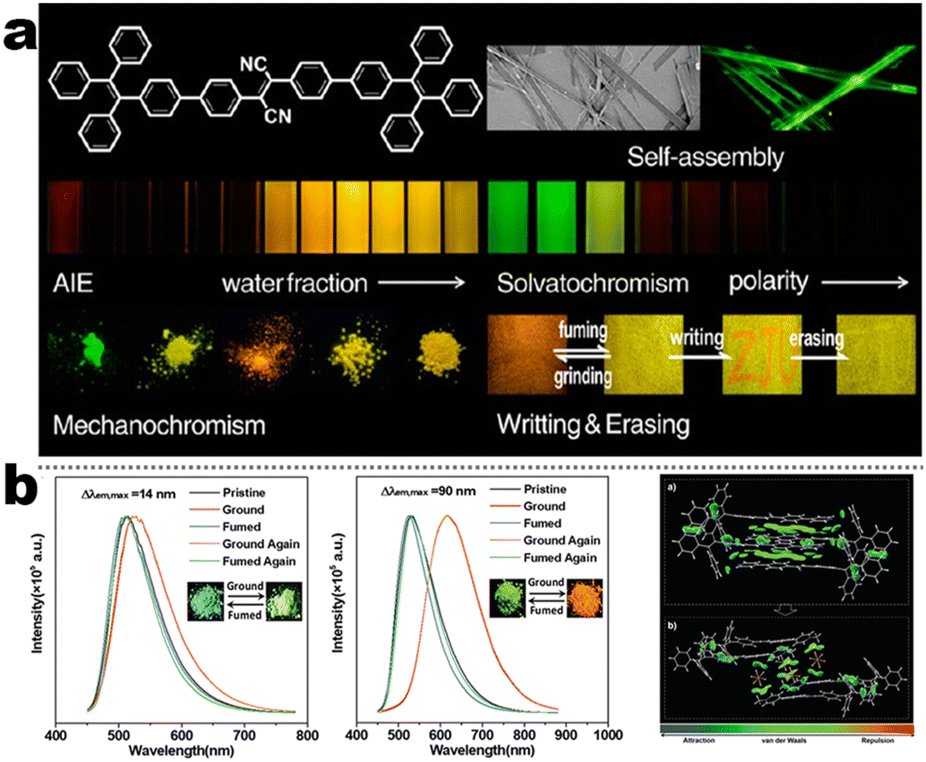
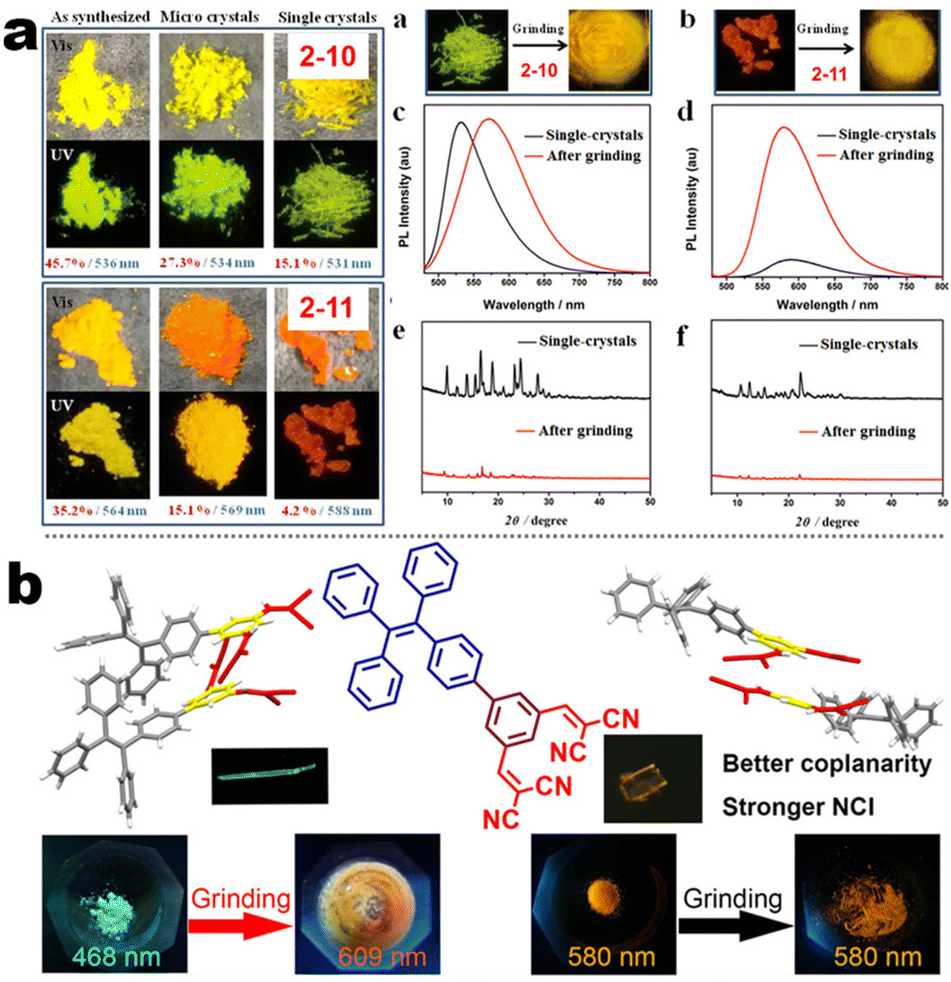
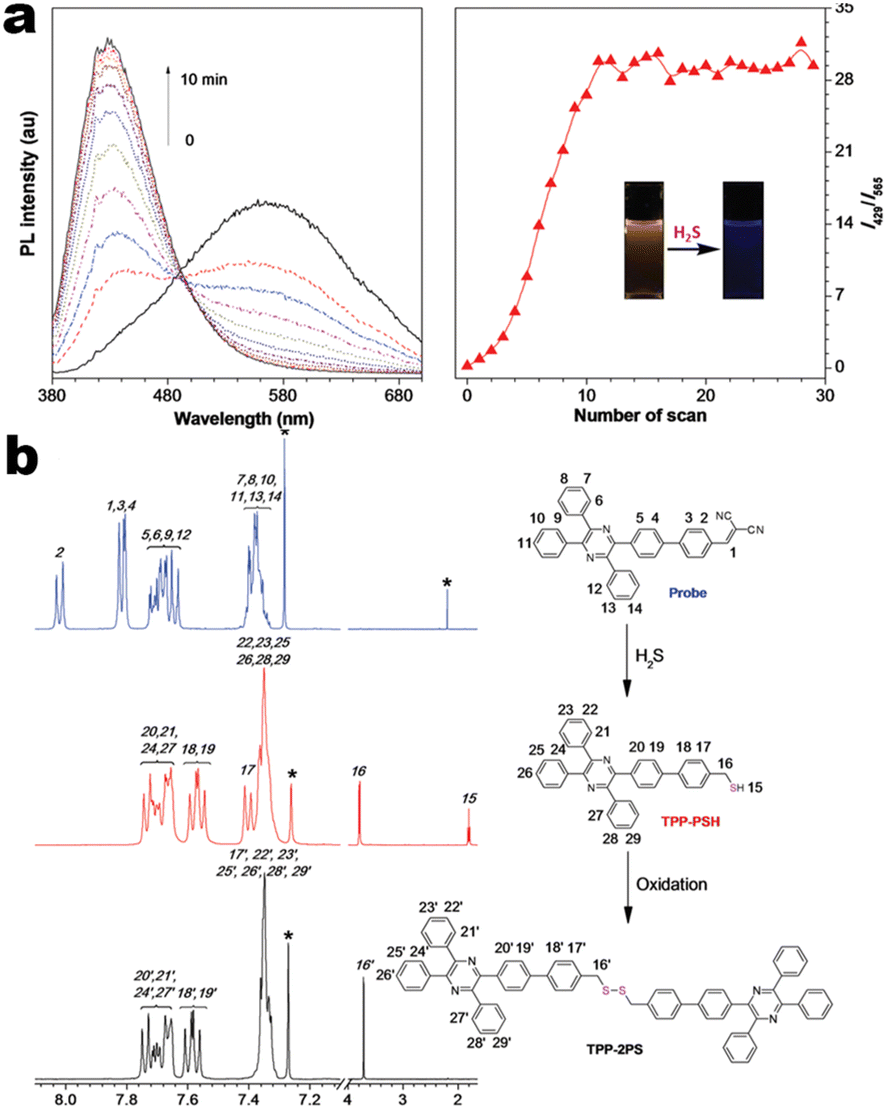
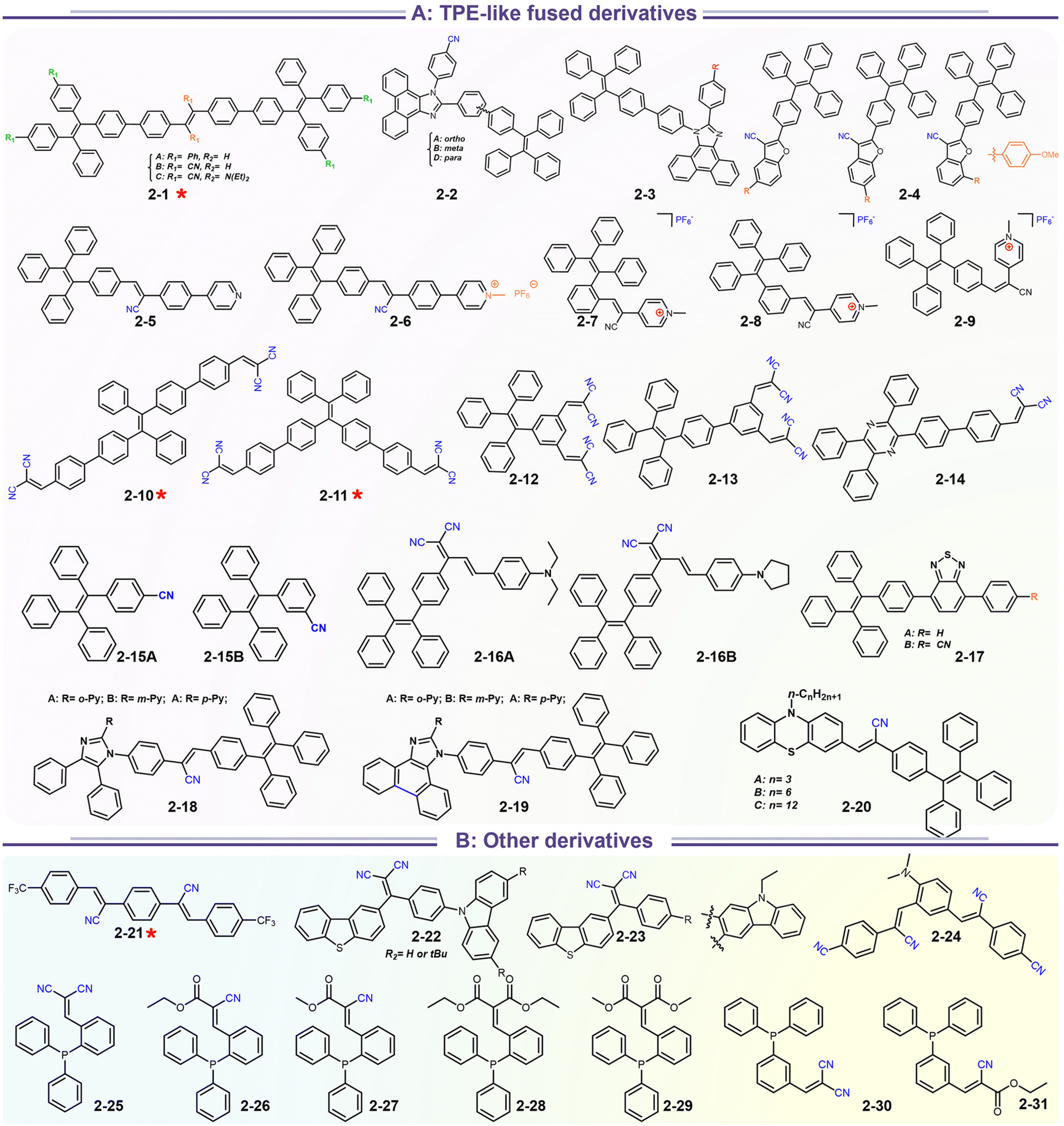
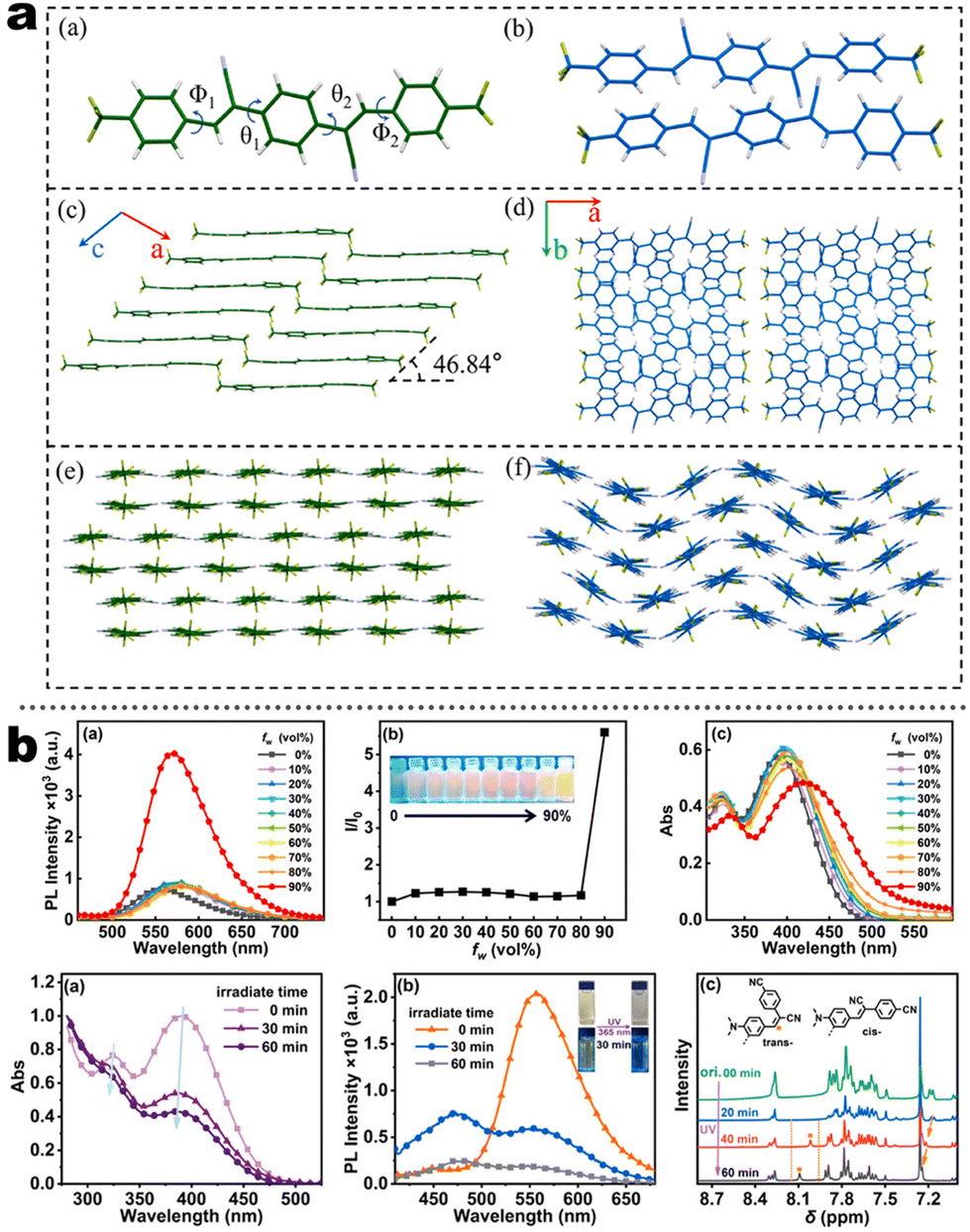
At the beginning of the 2000s, Tang's group and successor promoted the AIE applications.63 In 2013, Tang and co-workers pioneered the TPE-fused MCL materials based on cyano-incorporated TPE dimer (2-1). In these compounds, an efficient AIE characteristic (αAIE = 154), and reversible MCL have been realized by grinding-fuming (2-1A, Fig. 5a). In contrast, by replacing two phenyls with two cyano groups (2-1B, 2-1C) on the central TPE moiety, the resultant compounds showed efficient orange fluorescence (λem = 575 nm, ΦF = 100%) and evident AIE (αAIE = 13). The enhanced CT ability of donor and polar cyano endowed better self-assembled ability, red-shifted fluorescence, and solvatochromism.65 To explore MCL and electroluminescence (EL) capacities of TPE-fused phenanthroimidazoles, Misra et al. have reported three positional isomers (2-2, -ortho, -meta, and -para). The 2-2A and 2-2B isomers exhibited a large PL shift of 98 nm while the 2-2C exhibited a small spectral shift of 43 nm after grinding. Non-doped blue emitters of 2-2C provided the highest EQE of 4.0% in the blue OLEDs.66 Besides, the substituent effect (R = H, Me, F, CN) has been disclosed in their other works (2-3). It was found that the solid-state emission and MCL behavior of 2-3 were related to the terminal substituents and the CN substituted one showed the maximum MCL contrast (Δλem = 89 nm).67 In addition, the positional effect of distal groups in TPE-fused cyanobenzofurans (2-4B) displayed small red-shifted MCL but better OLED performance due to a higher degree of linear conjugation.68 It seemed that the MCL and OLED performance was contradictory because of the very contrary needs of molecular rigidity and close packing degree. Compared to the neutral emitters, the protonable pyridine unit could endow a narrow optical gap and multiple interionic NCIs. These pyridine-embedded TPEs have received much attention from Chi's (2-5, 2-6, Fig. 5b) and Niu's works (2-7 to 2-9). In Chi's report, one organic TPE-AIEgen and its cationic species were highly emissive in the solid state (PLQY up to 90% vs. 52%). Interestingly, neutral 2-5 only possessed insignificant MCL properties (Δλem = 14 nm), but the ionic pair species exhibited state-of-the-art MCL with the Δλem up to 90 nm.69 Similarly, Niu's works further revealed both protonation and positional effects in pyridine derivatives 2-7 (-ortho), 2-8 (-meta), and 2-9 (-para) as well as exploration of bioimaging. Compounds 2-7 and 2-8 showed similar yellow solid-state emission (∼526 nm). However, 2-9 (para-position) with stronger ICT instinct exhibited red-shifted solid-state emission of 616 nm as expected. Furthermore, compared with 2-7, both 2-8 and 2-9 displayed slight variations of MCL behavior, which may be caused by their less twisted configuration and rigid planar structures. Importantly, the wash-free bioimaging results indicated a hypotoxicity and vivo bioimaging ability for these ionic dyes.70 On the other hand, the protonation could also quench the fluorescence of precursors in Yang's work (2-18, 2-19), in which the detection of picric acid was realized via protonation and photo-induced electron transfer (PET).71 Polytropic structure-symmetry brought more complexity to molecular packing in the condensed state. In our previous progress, isomeric E/Z compounds contained two phenyl dicyanos at different positions on TPE (2-10, 2-11, Fig. 6a). These luminogens exhibited distinguishing MCL turn-on and morphology-dependent solid-state fluorescence, where the PL wavelength of the Z-isomer was much more sensitive to its morphology than the E-isomer. Moreover, the Z-isomer exhibited distinct blue-shifted MCL contrast to the red-shifted E-isomer. Based on the crystallography data, the absence and presence of π–π interactions between adjacent phenyl dicyanos were suggested to be responsible for the distinct solid MCL properties of these isomers.72 Further work revealed that two polymorphous meso-dicyanovinyl-substituted TPEs also followed the above rule (2-12, 2-13, Fig. 6b), where the compounds showed ultrahigh-contrast MCL (Δλem = 141 nm).73 To tailor molecular packing via the peripheral arms, Yang's group designed two TPE(D)–A–D type derivates with diethylamino or pyrrolidino donors as terminal substituents (2-16A, 2-16B). Compared with 2-16B, SCXRD analyses verified looser intermolecular stacking in 2-16A because of flexibility and nonplanarity in the diethylamino groups. Thus, 2-16A exhibited more red-shifted (25 nm) MCL behavior coupled with the loss of brightness.74 This TPE(D)–A–D integration strategy was fashionable for CT absorption enhancement, solvatochromism, and AIE. For instance, when benzothiadiazoles or thiodiphenylamines were integrated into TPE (2-17, 2-20), the cyano group in 2-17B facilitated stronger reversible MCL and solvatochromism than the H atom in 2-17A (Fig. 7b).75 It was interesting that the alkyl chain could affect the MCL behavior as evidenced by TPE and alkyl-substituted thiodiphenylamine-connected cyanoethylene (2-20). It was found that three compounds had the same monomer behavior but different aggregation behavior, where the shorter alkyl chain in 2-20A resulted in looser packing and higher crystallinity endowing the distinct MCL activity. In contrast, the longer alkyl chain will bring out more flexibility and poor crystallization.76 In 2019, Tang et al. reported a new AIE-active tetraphenylpyrazine backbone and its dicyano derivatives can serve as sensitive ratiometric fluorescent probes for detecting hydrogen sulfide with high specialty and low detection limit of 5 × 10−7 mol L−1 (2-14, Fig. 8). Research on the probing mechanism manifested a process involving the nucleophilic addition of H2S to the malonitrile site at first and subsequent oxidation and dimerization. This work demonstrated additional development prospects in chemical probes of cyano-derived luminophores.77
 |
| | Fig. 5 (a) Solvatochromism, AIE, and MCL features in one system of 2-1. Reproduced from ref. 65 with permission. Copyright (2013), American Chemical Society. (b) PL spectra and NCI analysis of compounds 2-5 and 2-6 at different states. Reproduced from ref. 69 with permission. Copyright (2020), The Royal Society of Chemistry and the Chinese Chemical Society. | |
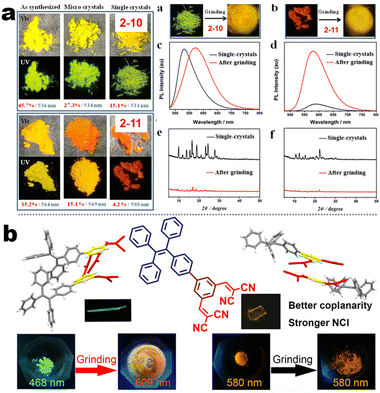 |
| | Fig. 6 (a) Crystal morphology, PL spectra, and PXRD diagrams of compounds 2-10 and 2-11. Reproduced from ref. 72 with permission. Copyright (2022), American Chemical Society. (b) Crystal structure of 2-13 as well as MCL pictures of 2-13 and 2-12. Reproduced from ref. 73 with permission. Copyright (2023), American Chemical Society. | |
 |
| | Fig. 7 (a) TL pictures and PL/TL spectra of compounds 2-15A and 2-15B. Reproduced from ref. 78 with permission. Copyright (2020), The Royal Society of Chemistry and the Chinese Chemical Society. (b) PL spectra and MCL analysis of compounds 2-17. Reproduced from ref. 75 with permission. Copyright (2015), The Royal Society of Chemistry. | |
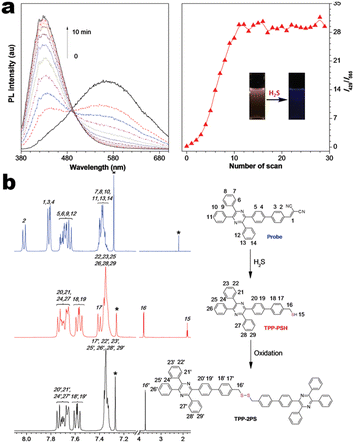 |
| | Fig. 8 (a) Time-dependent PL spectra of 2-14 treated with NaHS in DMSO/PBS buffer mixture. (b) 1H-NMR spectra of 2-14 and possible reaction intermediate. Reproduced from ref. 77 with permission. Copyright (2018), Wiley Online Library. | |
The emerging triboluminescence (TL) in pure organics and coordination compounds received great attention in recent years.7 The triboluminescence has also been found in cyanosubstituted TPE isomers (2-15, Fig. 7a). The cyano group was directly introduced at the para- and meta-positions on TPE. Two solid compounds exhibited bright blue TL after extrusion. SCXRD and powder-XRD analysis suggested that their TL nature was derived from the interlocked molecular packing and intact 3D supramolecular packing, which were stabilized by multiple strong NCIs from the cyanogroup. However, the TL state was irreversible in 2-15B but restorable in 2-15A, which could be ascribed to the polymorphous transition in 2-15B.78
In addition to popular TPE-like SRL AIE-emitters, some emissive D–A/D–π–A nonplanar backbones also endow strong AIE or AIEE effects. In fact, many above-discussed emitters in Scheme 4 have this common feature (i.e.1-5 to 1-8etc.), but most of them display AIEE because of the strongly intrinsic CT emission. For instance, taking recently reported compounds (2-21 to 2-31) as the discussion objects (Scheme 5), the symmetrical compound 2-21 has general styrene–cyanide with rotatable bonding specialty for terminal trifluoromethyl, which incubates the typical AIEE and remarkable photochromism (Fig. 9a).79 Importantly, polymorphic analysis from XRD suggests MCL derived from crystalline transformation, where the packing slip brings about a poor π-interaction. The weak π-interaction derived blue emitter is changed into a green one after grounding. The rare photodimerization results might be related to the slight steric hindrance of trifluoromethyl and the short π–π distance of alkene fragments. Nevertheless, the photoinduced cis–trans isomerism is more considerable based on many more demonstrations, which could also produce photochromism.72 In the report of Sun's group, a simple cyanostilbene derivative also displays a complex solvatochromism, acidichromism, mechanochromism, and photochromism (2-24). This multiple stimuli response benefits from an additional dimethylamino group to afford enhanced CT specialty and protonation site. NMR analysis points out that the photochromism is originated from the cis–trans isomerization (Fig. 9b).80 Thus, the intermolecular distance of alkene fragments is important for photodimerization. After introducing the stronger donors (carbazole, arylamine, and benzothiophene units), the photochromism and AIE/AIEE become more significant in SRL emitters (2-22, 2-23 to 2-31).81–83 However, the SRL of triphenylphosphine (TPP)-based emitters has not been disclosed in Zhu's report.83
 |
| | Scheme 5 Representative structures of SRL materials based on TPE-like fused cyano derivatives. | |
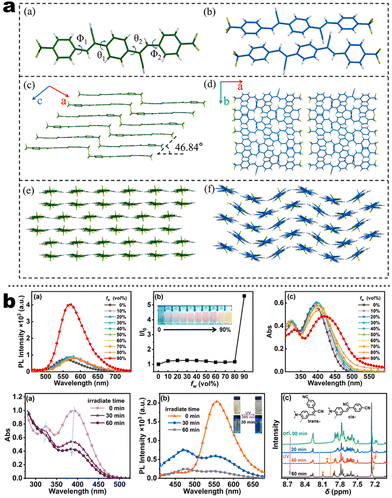 |
| | Fig. 9 (a) Single-crystal structures of two polymorphic 2-21. Reproduced from ref. 79 with permission. Copyright (2023), Elsevier Ltd. (b) PL emission spectra characterization of AIE behavior and photochromism of 2-24. Reproduced from ref. 80 with permission. Copyright (2023), Elsevier Ltd. | |
AIE-type molecule's inherent twisted non-planar structures are quite conducive to shaping different molecular conformation. And most cases of cyano-derived emitters are AIEE ascription instead of AIE, which could bring out additional photophysical analysis about SRL and potential applications in both solution and solid. Importantly, the effective charge transfer modulation entrusts a desired SRL with high contrast and switch. This strategy and emissive AIE skeleton need further exploration because of limited deep red or near-infrared (NIR) competence due to poor π-stacking.
2.3.3. Donor–acceptor involved CT system.
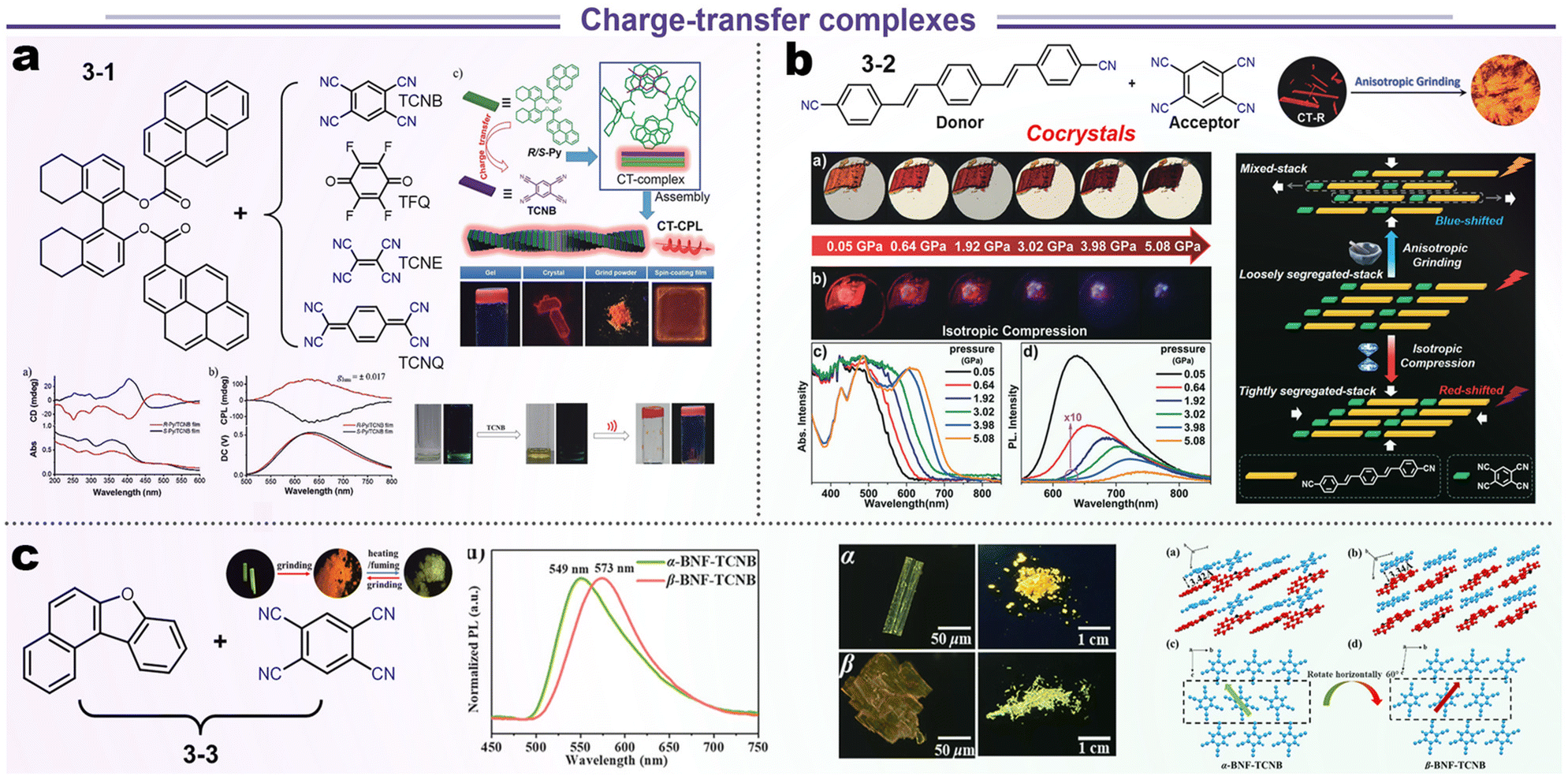
A standard charge transfer emission system involves the transfer of energy from the donor to acceptor (D–A) modules in a phase-compatible matrix (via Förster or Dexter process). Theoretically, efficient energy transfer needs highly ordered spatial organization as well as an appropriate energy gradient of the D–A system.84,85 Hence, the relative location of D–A modules could be influenced by external stimulation, leading to a variational energy transfer and luminescence. We reviewed some typical cases from recent work about stimuli-responsive materials (Scheme 6). Duan and Liu et al. fabricated four kinds of chiral CT complexes through various approaches such as grinding, crystallization, spin coating, and gelatinization, which were based on the chiral 8-H-binaphthyl (donor) and tetracyano derivatives (acceptor). Systematic research manifested that the optical gaps were decreased from TCNB, TFQ, TCNE to TCNQ due to the difference in falling energy barrier (3-1, Scheme 6a). Although the all-electron acceptors TFQ, TCNE, and TCNQ could also form CT complexes with the chiral electron donor, only the combination of TCNB with the electron donor was emissive because of the longest wavelength (at the NIR region) and non-radiative processes in TFQ, TCNE, and TCNQ systems. SCXRD illustrated a short contact of chiral D–A units about 3.499 and 3.484 Å, endowing this CT complex with higher CPL (glum = ±0.017).86 In other reports by Xu's group, TCNB and 1,4-bis-p-cyanostyrylbenzene (donor) could be assembled into two polymorphous cocrystals (CT-R and CT-O forms in 3-2, Scheme 6b). Interestingly, this CT-R form showed bathochromic-shifted emission after grinding but hypochromic-shifted emission under the hydrostatic pressure. Such distinct luminescence responses to anisotropic and isotropic compression could be ascribed to two factors. The anisotropic blue-shifted outcome might be ascribed to the structural reorganization from loosely segregated stacking to mixed packing for the former, and the red-shifted case for isotropic compression originated from the formation of a tighter packing structure.87 This common phenomenon of polymorphs has also been found in aphtho[2,1-b]benzofuran (BNF) and TCNB cocrystals (3-3, Scheme 6c), which displayed a reversible crystal transition between α- and β-forms and consistent red-shifted emission for anisotropic grinding and isotropic compression. Spectroscopic and structural analyses disclosed that the emission changes were mainly attributed to the packing transformation from crystalline to amorphous states.88
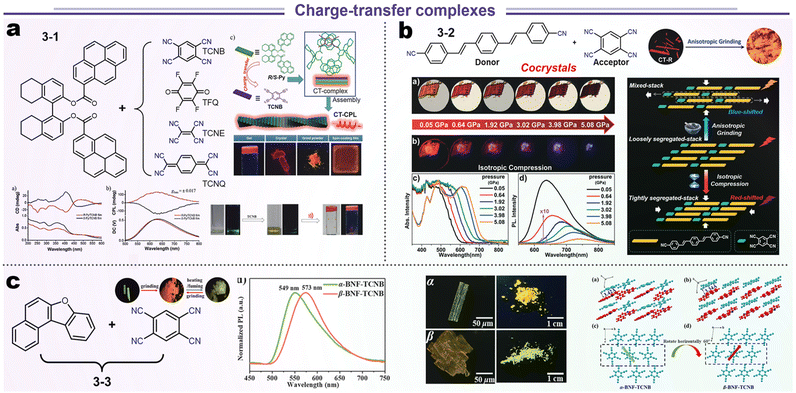 |
| | Scheme 6 Representative structures and data presentations of SRL based on charge-transfer complexes. (a) Chiral CT complexes system. Reproduced from ref. 86 with permission for 3-1. Copyright (2019), Wiley Online Library. (b) Pressure-dependent CT system. Reproduced from ref. 87 with permission for 3-2. (c) CT complexes with polymorphism behavior. Copyright (2018), Wiley Online Library. Reproduced from ref. 88 with permission for 3-3. Copyright (2023), The Royal Society of Chemistry. | |
D–A strategy involved CT complexes requiring precise crystallographic structures to understand their charger transfer channels. Generally, the energy gradient forces lower energy emission from yellow to red, which makes it easier to realize larger wavelength contrast after the external stimuli. Hence, precise energy level design of the D–A module via density functional theory (DFT) calculations should be considered. Besides, crystallographic engineering plays a pivotal role in obtaining polymorphism and SRL.
2.3.4. Cyano-derived luminescent soft matters and chiral emitters.
2.3.4.1. Multi-component assembly for LCs emitters.
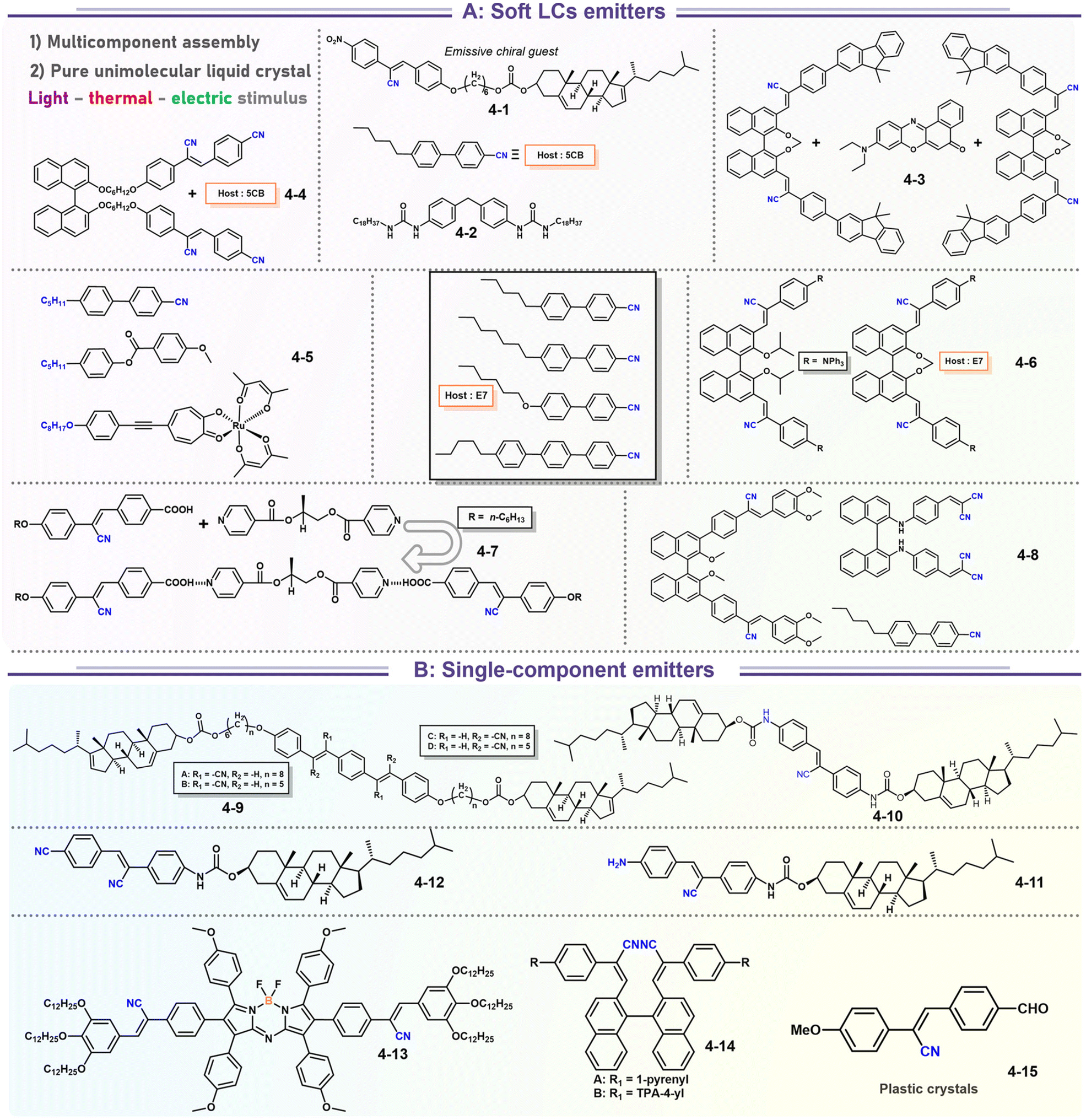
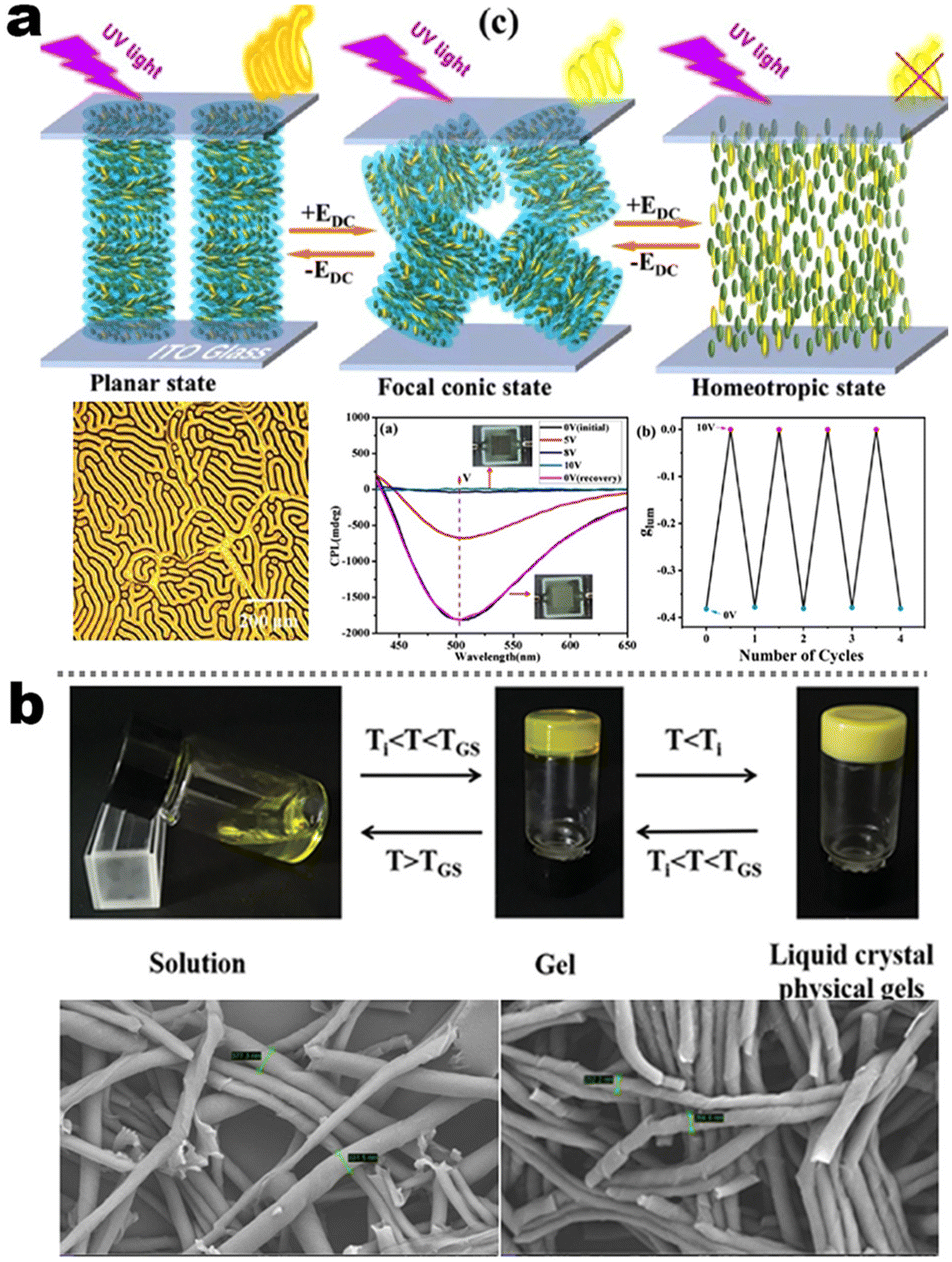
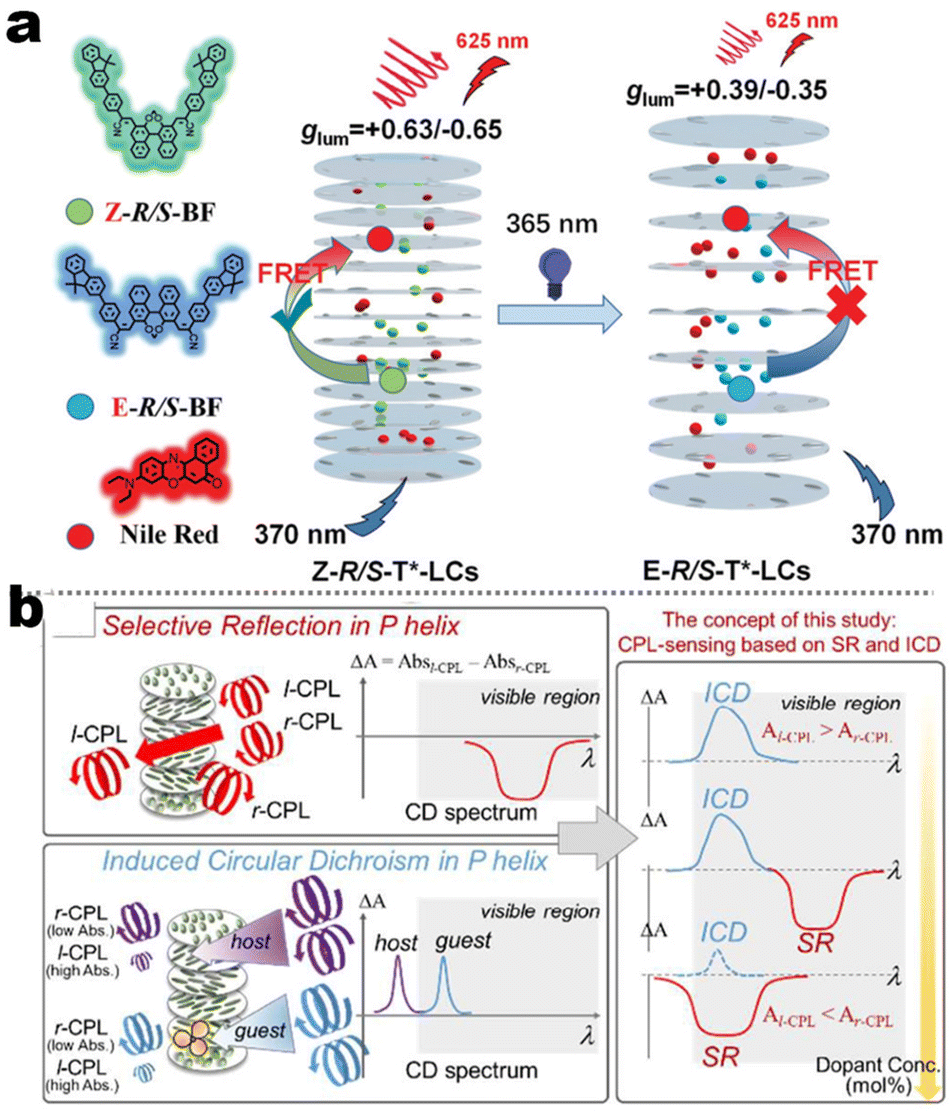
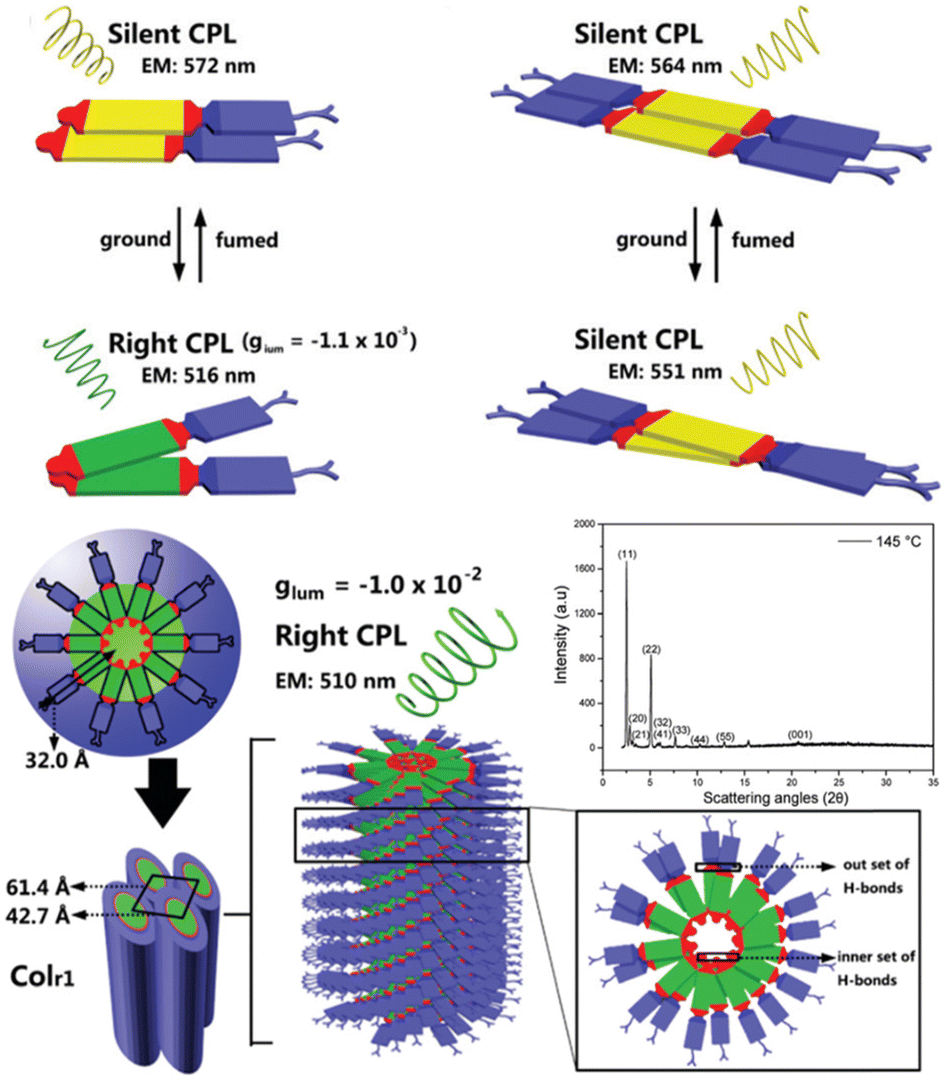
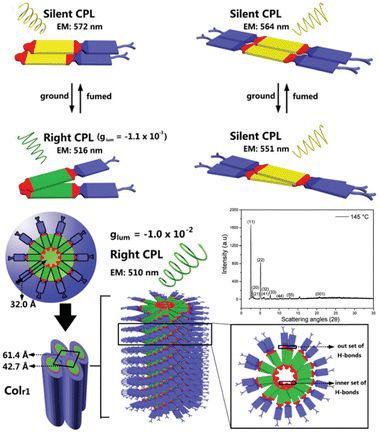
It's well-known that cyanobiphenyl was an important structural unit for building liquid crystals (LCs) because of its strong polar effect and versatile NCIs in the aggregation state.15,89,90 Hence, introducing both flexible units and cyanobiphenyl into an appropriate luminophore will generate a multistage phase sequence, where the molecular packing is widely different after thermal stimuli. This mesomorphic polymorphism was accompanied by different luminescence. In fact, traditional commercial liquid crystals in modern liquid crystal display technology were nonemissive, where the additional inorganic backlight layer was necessary. For example, the cyanobiphenyl derivatives 5CB and E7 LCs were nonluminous (Scheme 7). Theoretically, the superiority of self-luminous liquid crystal emitters can simplify the device construction in LCD or used in OLEDs. Hence, the construction of luminescent liquid crystal (LLC) molecular systems could promote the development of display technology as well as intelligent optical switches and lighting.91 So far, two main routes have been established to obtain LLC. The first approach was multi-component (including LC host and emitter guest) self-assembly technology and the second one was the design of pure LLC molecules. It seemed that the latter tactic was simpler and more promising because it could avoid the light loss and phase separation. In recent years, the above-mentioned methods have also been attempted to design stimuli-responsive soft matters and chiral emitters, in which multiple stimuli-responsive luminescence (light/thermal/electric stimulates) could be easily acquired because of rich mesophase states. Compared to solid stimuli-responsive emitters, the stimuli-responsive LC systems had high operability, consistency, integration capability, and potential application in flexible devices. For the first kind of multi-component self-assembly, Zhang et al. reported a chiral nematic LLC (N*-LLC) by doping a luminescent chiral cholesterol additive (4-1) into a commercial nematic liquid crystal (5CB). The self-assembled helical structures produced circularly polarized luminescence (CPL) with larger glum values in binary N*-LLC, where the glum value increases with a decrease in the helical pitch (P). Importantly, the CPL signal of N*-LLC can be repeatedly shuttled between the “On” and “Off” states by adjusting the applied voltage. On the other hand, the CPL signal can be switched between the “On” and “Off” states by regulating the temperature because of the switched phases between chiral N*-phase and isotropic liquid (Fig. 10a).92 This double-switching effect was extensive in many reports of this section (4-2, 4-6, Fig. 11). Actually, the subsequent study demonstrated that pure 4-1 was an LLC unit gestating two chiral mesophases (N* and SmC*), and their emission peaks were different. The isotropic melt showed a longer emission wavelength and silent CPL compared to LCs, and higher glum values in the N* phase (−0.17) were found than those of the SmC* phase (−0.021).93 Unfortunately, the reason for this disparity was unknown according to the current result. Next, this group prepared a ternary circularly polarized LLC gel using 4-1, 4-2, and 5CB (Fig. 10b). The ternary system can be assembled to form a gel network with nanofiber morphology, which limited the fluxion of the LLC and enhanced the stability of the CPL (glum = −0.31). In the mesophase structure and its associated CPL, transmittance properties could be adjusted by direct-current (DC) electric field and temperature as same as the above discussions.94 Another contactless stimulation was the light-induced structural transformation of molecules and mesophasic superstructures. In Cheng's design, the integrated chiral transfer and FRET advantages outputted a photosensitive CPL activity (glum = +0.28/−0.25) in a ternary LLC (4-3, Fig. 12a). The photo-isomeric binaphthyl derivates acted as the janus role of chiral inducer and energy donor, and their energy (E-isomer) could be delivered to an achiral dye Nile Red (energy acceptor), which benefited from the matched energy level and proper distance between donor and acceptor. Remarkably, this FRET-type CPL can be intercepted after 365 nm UV irradiation, leading to a separation of overlap between the absorption of the acceptor Nile Red and the emission of the Z-isomers of the donor.95 Very recently, this same group also realized the light-driven sign inversion of CPL by the dichroism modulation.96 These chiral binaphthyl partners were popular in the stimuli-responsive CPL-type LLC fields, which supported many similar works (4-4, 4-6, 4-8, 4-14).97–100 Among them, the maximal glum was found in 4-6 (−0.83 and +0.86 in Fig. 11).98 Multi-component self-assembly has also been attempted in coordination compounds emitters and we believe this topic will be promoted in the future. In Watanabeb's reports, a ternary LLC system, composed of achiral 5CB (host), PPMB (host), and chiral Δ/Λ-Ru(III) complexes (guest emitter), was fabricated to generate both right- and left-handed CPL (Fig. 12b), which depended on the guest concentrations (4-5). Their CPL signals can be reversibly switched between “On” and “Off” upon mechanical stress and heating. Based on the transmitted and CD spectral observation, this concentration-dependent CPL inversion phenomenon was attributed to different contributions between induced circular dichroism (CD) and selective Bragg reflection.101 Further, we abolish the exotic LC host employment to successfully fabricate exclusively Pt(II) metallomesogens emissive host and binary-mediated deep red CPL with stimuli-responsive switch with inherent color constancy and Pt⋯Pt/π–π interaction.102 In addition, intermolecular hydrogen-bonding interaction-induced binary LLC can emit strong CPL signals with smaller glum +0.076/−0.064 (4-7).103
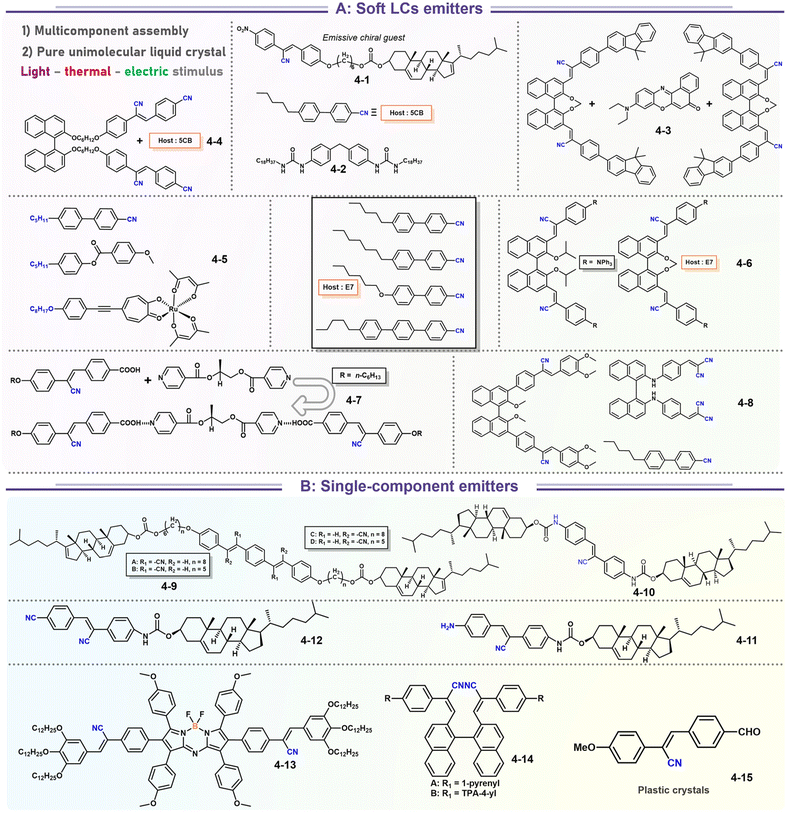 |
| | Scheme 7 Representative structures of stimuli-responsive luminescence materials based on soft matters and chiral emitters. | |
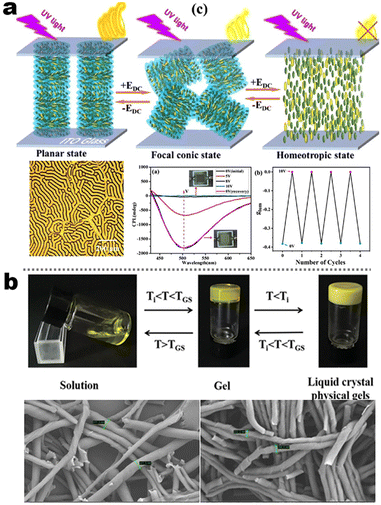 |
| | Fig. 10 (a) Schematic illustration of electric-induced CPL switching, LC assembly model, and POM image for system 4-1. Reproduced from ref. 92 with permission. Copyright (2020), American Chemical Society. (b) Gelation process and its SEM images of 4-2 system. Reproduced from ref. 93 with permission. Copyright (2022), The Royal Society of Chemistry. | |
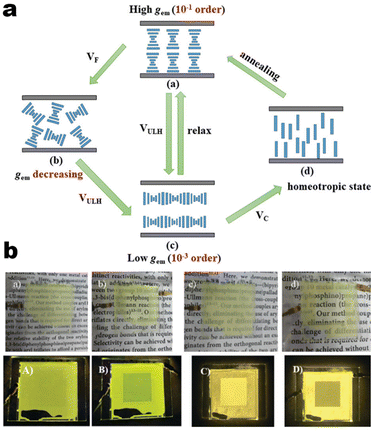 |
| | Fig. 11 (a) Schematic illustration of thermo-electric-induced CPL switching, LC assembly model for system 4-6. (b) Images of N*-LCs-4-6 system device under enhanced DC electric field. Reproduced from ref. 98 with permission. Copyright (2022), The Royal Society of Chemistry and the Chinese Chemical Society. | |
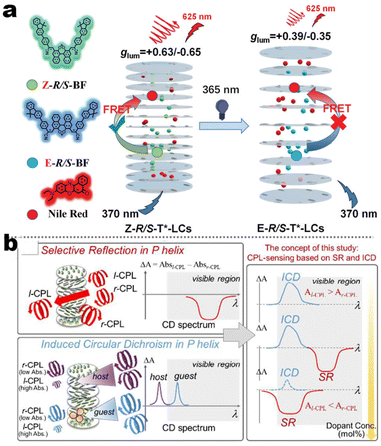 |
| | Fig. 12 (a) Schematic illustration of FRET-induced CPL switching, LC assembly model for system 4-3. Reproduced from ref. 95 with permission. Copyright (2021), The Royal Society of Chemistry. (b) Schematic illustration CD-inversion of 4-5 system under different doping concentrations. Reproduced from ref. 101 with permission. Copyright (2018), The Royal Society of Chemistry. | |
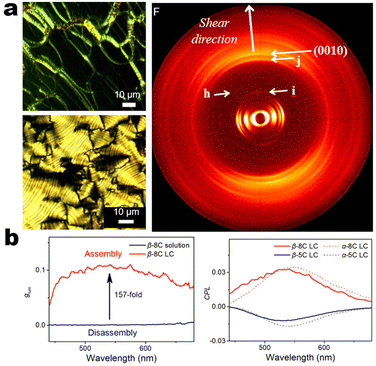 |
| | Fig. 13 (a) POM and SAXS images system 4-9. (b) CPL and glum spectra of 4-9 films under different states. Reproduced from ref. 104 with permission. Copyright (2020), American Chemical Society. | |
 |
| | Fig. 14 Schematic illustration of thermo-mechanic-induced CPL switching and LC assembly model for 4-10 and 4-11. Reproduced from ref. 105 with permission. Copyright (2020), The Royal Society of Chemistry. | |
 |
| | Fig. 15 (a) Fluorescence color changes under 365 nm UV light and fluorescence spectra of 4-14. Reproduced from ref. 100 with permission. Copyright (2023), Wiley Online Library. (b) Images of photosalience and photochromism of 4-15 crystals. Reproduced from ref. 108 with permission. Copyright (2023), The Royal Society of Chemistry. | |
Chiral LCs emitters with multiple SRL make an outstanding impression at CPL. Their stimuli-responsive CPL performance is desirable after the above-mentioned research breakthroughs. We believe that the emission wavelength contrast of SRL needs further optimization by photoinduced structure change or electric-induced structure redox. Furthermore, the higher sensitivity and desired polarization reversal of stimuli-responsive CPL should also be explored in the future.
2.4. Cyano and isocyano-based complexes
2.4.1. Cyano/isocyano-based MCL complexes with d10-metal configuration.
Emissive coordination compounds are promising phosphorescence species composed of coordinated metal ions and rigid organic ligands. Up to now, many phosphorescent coordination compounds have been synthesized and researched due to their attractive features at room-temperature phosphorescence and organic light-emitting diode devices (OLEDs) in the family of luminescent materials, because they can simultaneously harvest singlet and triplet excitons for better device efficiency.16,17 In addition, parts of the complexes can show multivariate luminescence phenomena during the stimulation of mechanical stresses owing to the deconstruction and reconstitution of molecular structures and supramolecular stacking structures. The relationship between mechano-responsive luminescence and supramolecular structures is being resolved. In this section, based on the recent studies on coordination compounds, we reviewed relevant advances with self-assembled coordination compounds with cyano and isocyano groups, mainly focused on emissive Cu(I), Au(I), Ag(I), and Pt(II) complexes (Scheme 8). The cyano group can be used to regulate the NCIs of molecular fragments, while the isocyano group acts as a coordination target. The isocyano group takes a coordination role but suppresses the activity of hydrogen-bonding receptors and ion-pair interactions. In addition, the metal complexes provide extra metallophilic interactions after packing compression via mechanical stimulation. Sometimes, this interaction is quite stronger than some π–π and H-bonding interactions. The emission specialty of complexes is highly sensitive to the intermetallic distance. Thus, the luminescence variation could be easily adjusted between monomer and aggregate.
 |
| | Scheme 8 Representative structures of stimulate-responsive luminescence materials based on coordination complexes emitters. | |
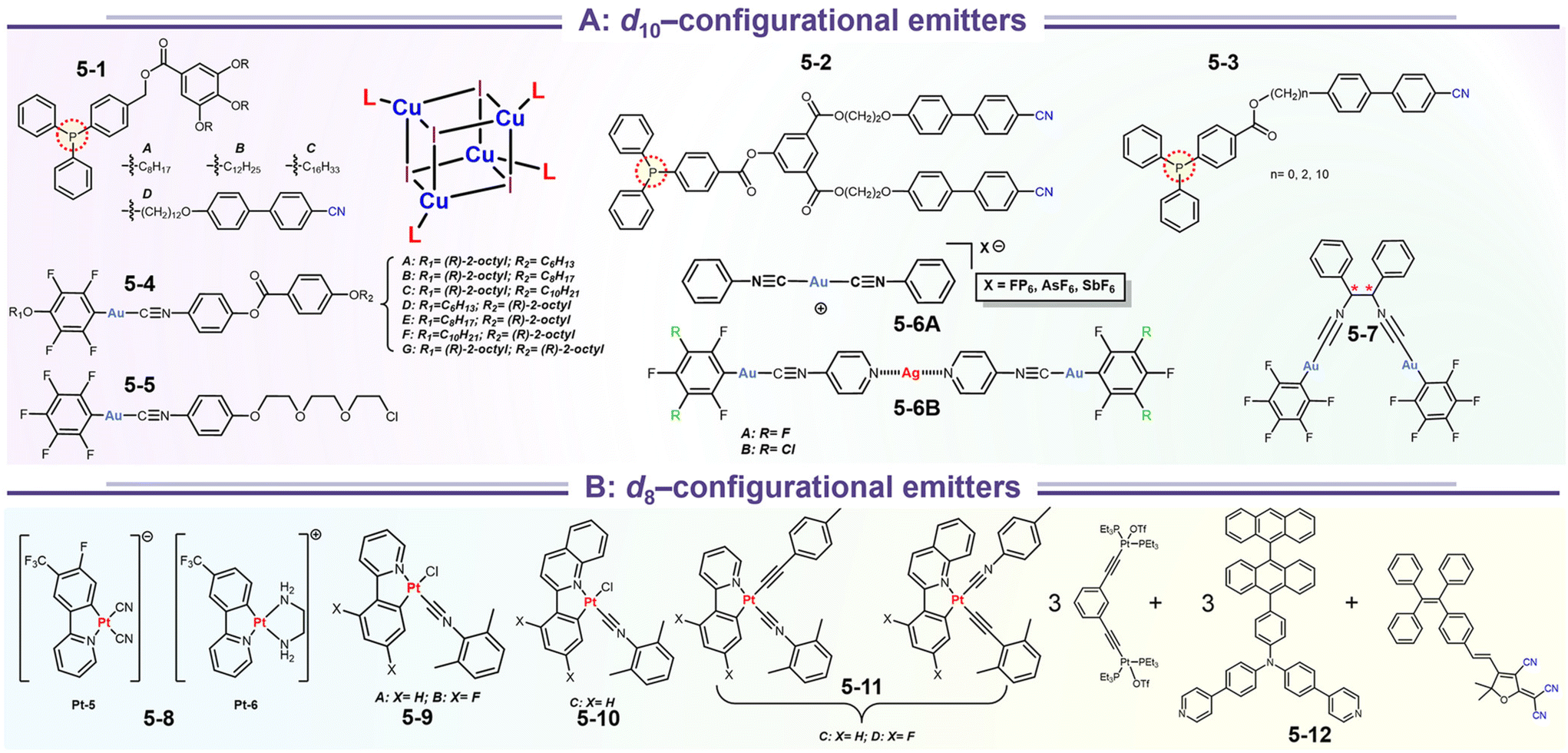
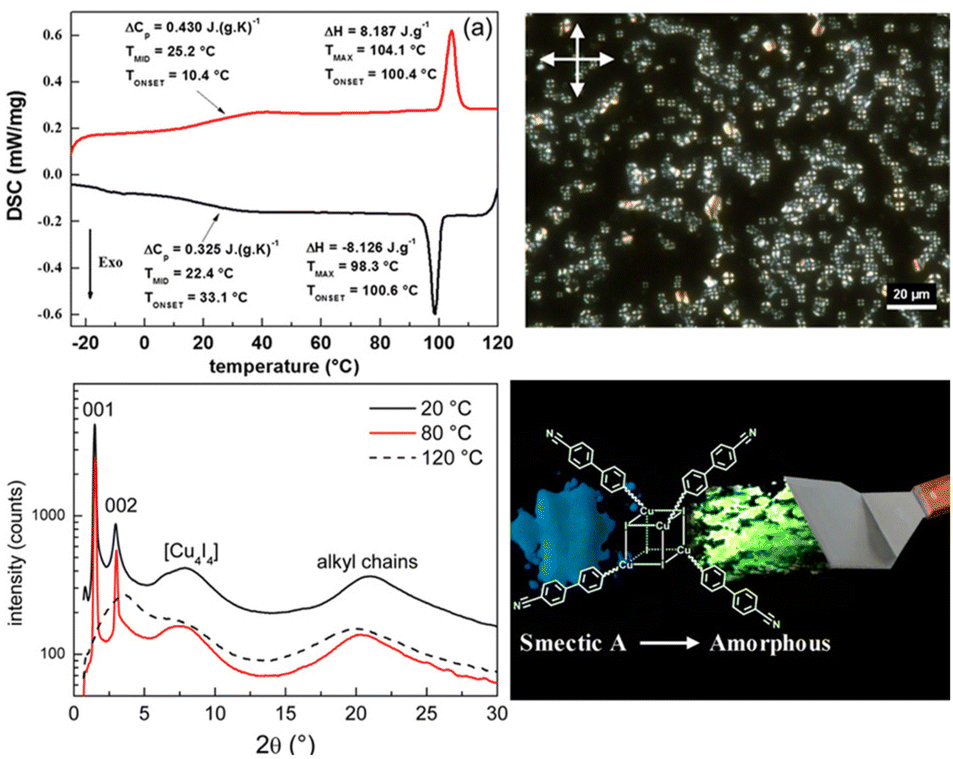
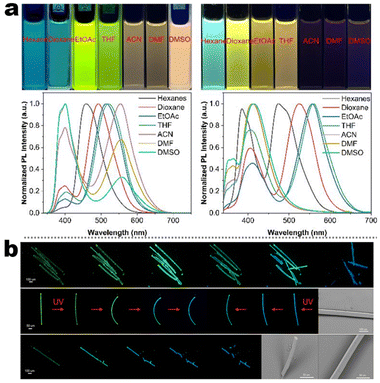
In systematic works from Camerel's group, a series of luminescent units of [Cu4I4] clusters have been functionalized by phosphine ligands carrying mesogenic gallate-based derivatives bearing either long alkoxy chains (–OC8H17 to –OC16H33) or polar cyanobiphenyl (5-1, Fig. 16).109 Importantly, these clusters functionalized solely with long alkyl chains presented amorphous or crystalline states (5-1A to 5-1C), but only the cluster 5-1D carrying cyanobiphenyl fragments displayed LC properties with the formation of a smectic A (SmA) mesophase from room temperature up to 100 °C. The placed cyanobiphenyl (CBP) fragments contributed to the formation of LCs mesophase via key dipole–dipole interactions. Furthermore, temperature-dependent photoluminescence features showed that the cyanobiphenyl-embedded cubane cluster displayed an unexpected thermochromism luminescence which might be caused by the energy transfer mechanism between the emissive [Cu4I4] acceptors and CBP donors. Moreover, the LCs properties imported on the bulky Cu4I4 core allowed for a facile deformation of its local environment, leading to double MCL and thermochromic luminescence (TCL) properties related to the modulation of intramolecular interactions.109 It should be noted that the MCL and TCL behavior of these particular compounds was induced by a modulation of the intramolecular interactions between luminophores instead of intermolecular interactions. This kind of molecular framework has been enriched so far in their research group (5-2, 5-3).110 Although the emissive character was maintained in 5-2 and 5-3, the LC property was lost. These results might relate to fewer cyanobiphenyl fragments and the modulation of ligands.
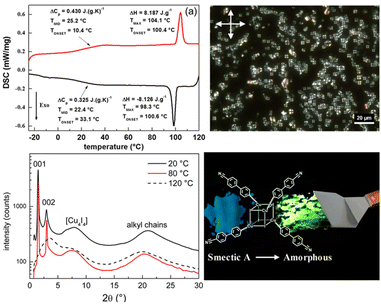 |
| | Fig. 16 DSC curve, POM image, and PXRD spectra of LC [Cu4I4] cluster 5-1. Schematic illustration of grinding-induced MCL activity. Reproduced from ref. 109 with permission. Copyright (2016), American Chemical Society. | |
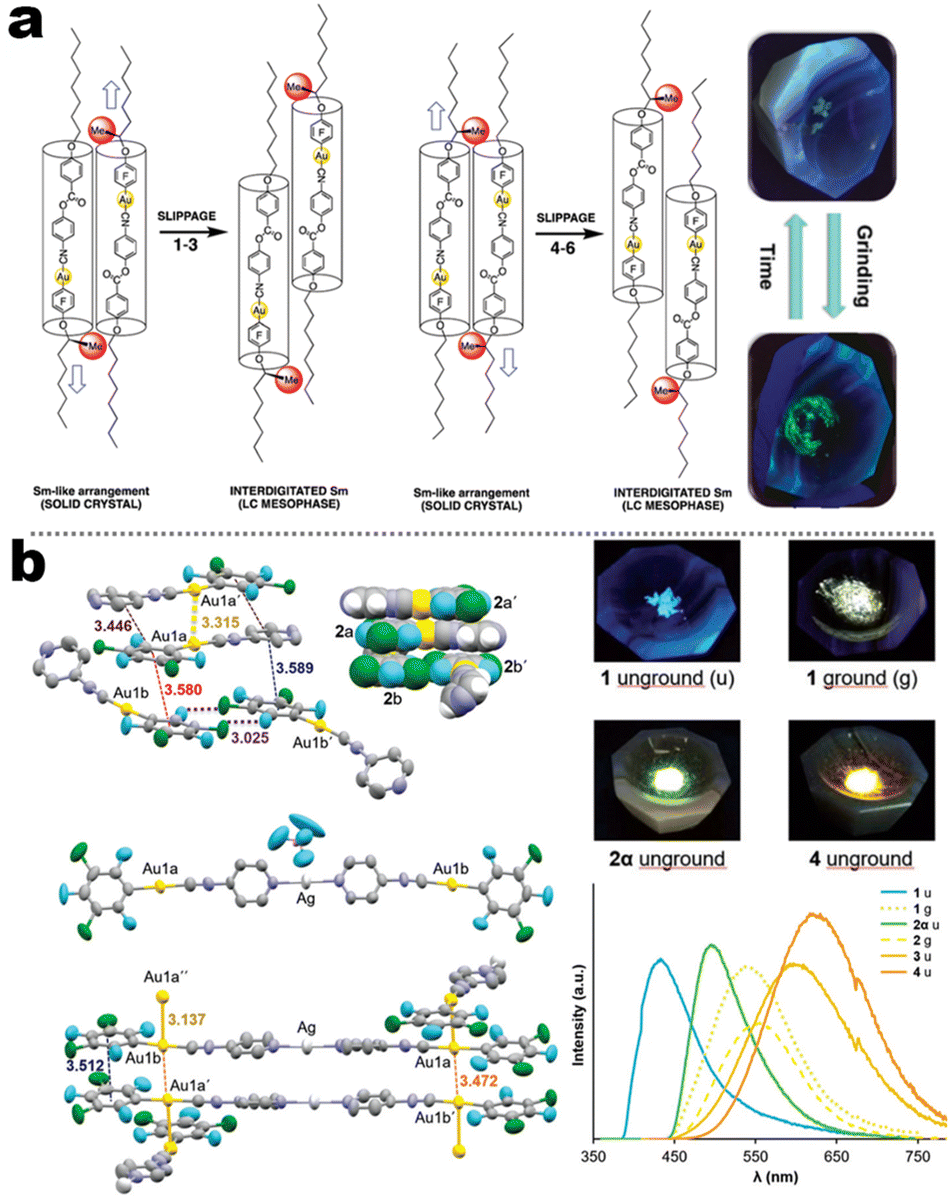
In contrast to the cyano group, the isocyano group possessed a strong bonding capacity with noble–metal cations. The groups of Espinet and Liu et al. reported many Au(I) complexes, such as 5-4, 5-5, 5-6, and early samples.111,112 For 5-4 and 5-6 in Espinet's work, two different strategies have been achieved, which contained LC or bimetallic chelation pathways. In the LC involving 5-4 derivates (Fig. 17a),113 the different oxy-alkyl chains were placed at two terminals of ligands, and these complexes displayed an unprecedented luminescence response to mechanical or thermal stimulation. The X-ray analysis disclosed that the phase transition of the crystals via mechanical or thermal treatment produced a reconstruction from a regular smectic (Sm) arrangement in the crystals to an interdigitated Sm packing in the LCs state. Importantly, this molecular slippage in the interdigitated Sm phase was conditioned by the position of the bulky tertiary butyl in the backbone, which allowed or restrained the generation of the Au⋯Au contacts that gave rise to enhanced emission. That was why only the tertiary carbon interrelated complexes displayed strong luminescence. In order to understand the bimetallism effect in emission, 5-6B represented a good case (Fig. 17b). Crystallography and DFT simulation of the Au(I) producers and Au(I)&Ag(I) bimetallic species pointed out that the marked luminescence red-shifts between Au(I) producers and Au(I)&Ag(I) bimetallic complexes were derived from structural changes originating form stronger π–π stacking and shorter Au⋯Au separations.114 In addition, the adjustable NCI also facilitated high-contrast MCL from 434 nm of pristine crystals to 540 nm of ground powder for Au(I) 5-6B precursor. However, the MCL disappeared in 5-6B because of the ultrashort Au⋯Au contact (3.137 Å) in pristine crystals, where the further approach in the Au⋯Au distance was impossible after grinding.114
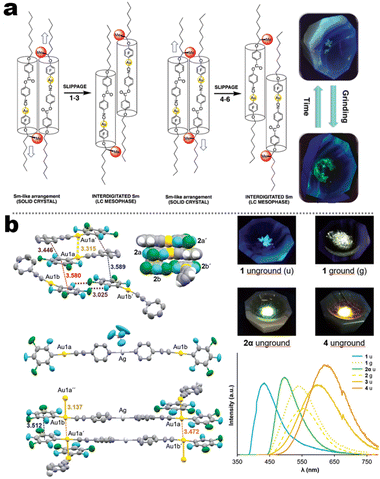 |
| | Fig. 17 (a) Schematic illustration of spacer-dependent solid-assembly model of complexes 5-4. Reproduced from ref. 113 with permission. Copyright (2022), The Royal Society of Chemistry. (b) Crystal illustration and Au⋯Au interaction of complex 5-6B. Reproduced from ref. 114 with permission. Copyright (2019), The Royal Society of Chemistry. | |
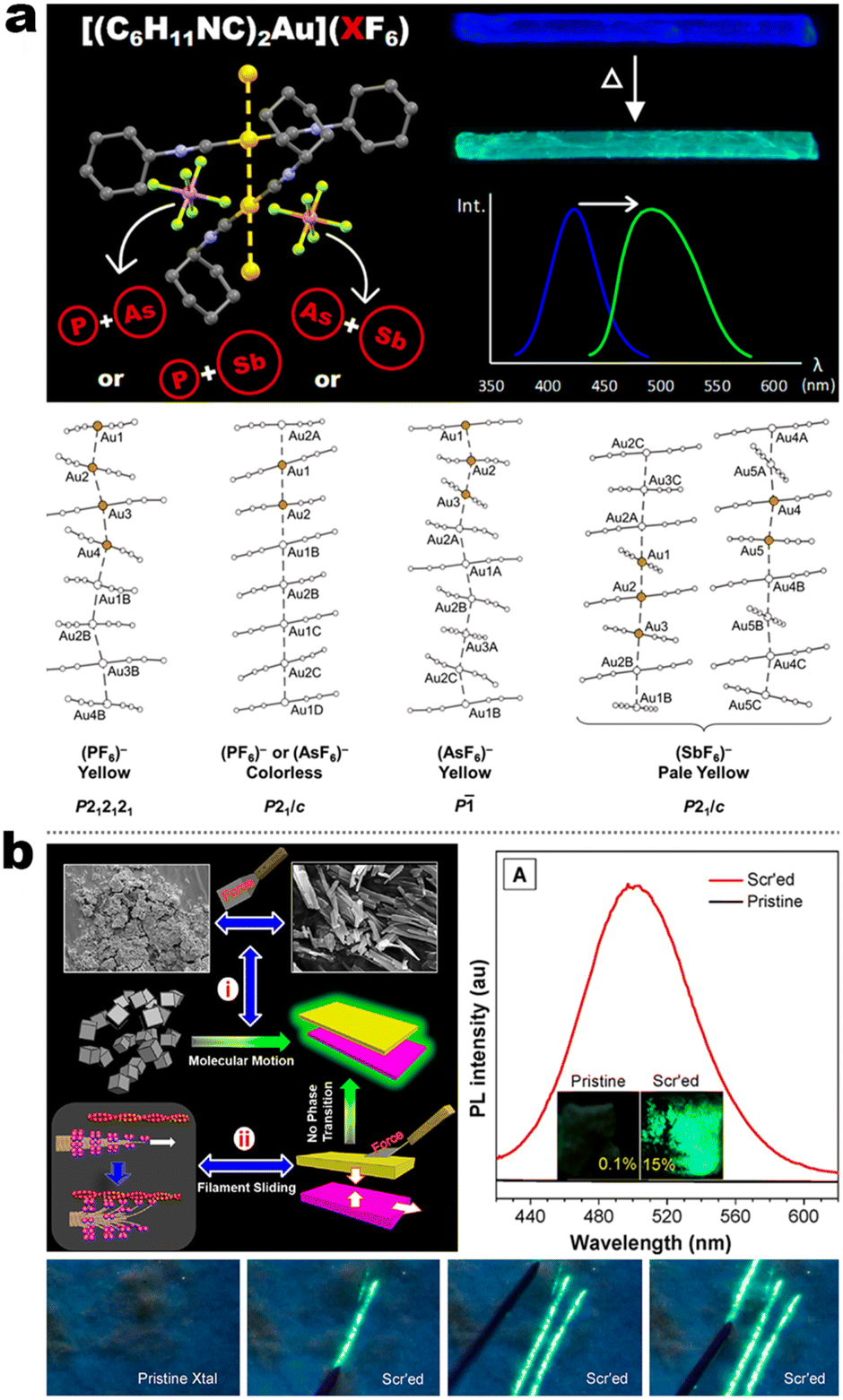
Integrating RTP into emissive coordination compounds has great significance in the field of chiral electronic devices. Liu and other groups also explored the potency of persistent room-temperature phosphorescence (RTP) or high-contrast phosphorescent MCL in Au(I) complexes. For example, 5-5 afforded two polymorphs (blue and yellow-green emission) in the same condition concomitantly, in which their structures were identified by SCXRD. The blue emissive crystal form exhibited high-contrast phosphorescent MCL behavior, while the yellow-green emission form showed a persistent RTP according to the time-gated luminescence spectra. Surprisingly, differing from a shorter decay (49.2 μs) of blue crystals, the room RTP decay of the yellow-green form at the PL peak of 492 nm was as high as 42.8 ms, which was about three orders of magnitude longer than that of blue form. Besides, the RTP at the longer wavelength region was changeless, while the shorter emission band was quenched. However, the persistent RTP feature was annihilated after grinding, resulting in an “On–Off” RTP switch. This enhanced persistent RTP in yellow-green form could be assigned to stronger NCIs and shorter Au⋯Au distance between adjacent molecules, which promoted intersystem crossing (ISC) and induced RTP propagation.115 Another interesting finding revealed the anion influence on thermochromism, vapochromism, and polymorph formation of luminescent crystals in the Au(I) cation (5-6A), in which the [5-6A]+[PF6]− and [5-6A]+[AsF6]− contained single chains of cations and were vapochromic (Fig. 18a).116 But the yellow form [5-6A]+[SbF6]− contained two distinct chains of cations connected through aurophilic interactions, giving inactive vapochromism. Further crystal fusion engineering revealed the initial kinetic products [5-6A]+[(AsF6)x(PF6)1−x]−, [5-6A]+[(SbF6)x(PF6)1−x]−, and [5-6A]+[(AsF6)x(PF6)1−x]− as precipitates of fine yellow needles with green emission, which could be gradually transformed into finally colorless crystals with blue emission and thermochromic behavior. Importantly, this thermochromic crystalline phase transform potency for [5-6A]+[(AsF6)x(PF6)1−x]− was anionic doping concentration-dependent, where the thermochromic temperature threshold increased as the increased fraction of [PF6]− ions in the crystal. In addition, this thermochromic behavior only existed in [5-6A]+[(AsF6)x(PF6)1−x]− framework nor [5-6A]+[(SbF6)x(PF6)1−x]−, [5-6A]+[(SbF6)x(AsF6)1−x]−, serial of SCXRD data showed that the Au⋯Au separations in different systems could be responsible for the thermochromic emission, where the Au⋯Au distances increased with the increased sizes of anions. In 2020, Tang et al. reported a pair of enantiomeric binuclear Au(I) complexes by introducing the chiral isocyanogen ligands (Fig. 18b). Unprecedently, 5-7 powders could realize a counterintuitive transformation from nonemissive isolated crystallites to emissive regular microcrystals under the scratch of mechanical force, which seemingly violated the entropy increase law. Their photoluminescence quantum yield was increased to 15% after scratch and accompanied by raised CPL signals with luminescence dissymmetry factors (|glum|) of 4 × 10−3. Such a crystallization morphological shaping and luminescence enhancement were presumed to be caused by molecular motions driven by the formation of strong aurophilic interactions as well as multiple C–H⋯F and π–π interactions. This inference can be derived from the crystal structures, pressure-dependent Raman, and PL spectral measurements.117
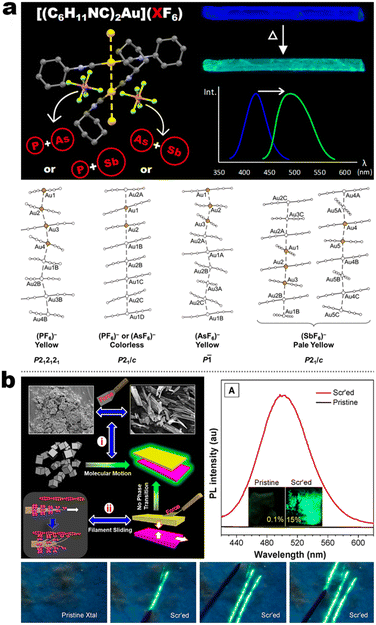 |
| | Fig. 18 (a) Schematic illustration of anion-dependent solid emission of complexes 5-6A and their crystal packing. Reproduced from ref. 116 with permission. Copyright (2020), American Chemical Society. (b) Schematic illustration of force-induced filament sliding of complex 5-7 and their spectra observations. Reproduced from ref. 117 with permission. Copyright (2020), American Chemical Society. | |
2.4.2. Cyano/isocyano-based MCL complexes with d8-metal configuration.
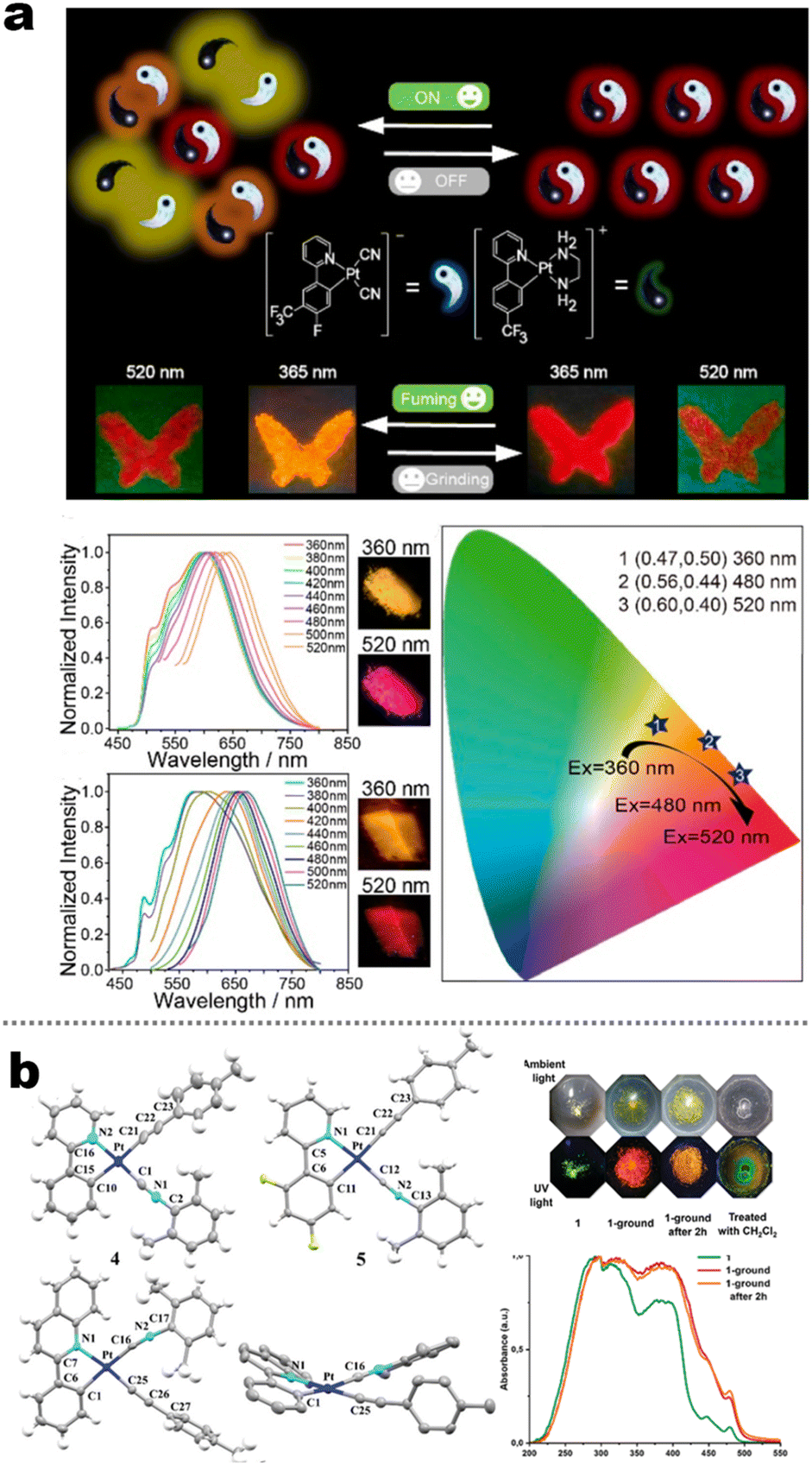
In the last few decades, the square coordination of d8 electronic Pt(II) coordination compounds has been extensively applied because they can simultaneously harvest singlet and triplet excitons in the phosphorescence OLED field. Indeed, the planar chelation chromophore could facilitate the molecular aggregation at solid state, the aggregation degree can be customized by tailoring the steric hindrance of ligands or strength of external stresses.37,118–120 In this field, some isocyano-coordinated Pt(II) complexes have been prepared and investigated (5-9 to 5-11, Fig. 19b).121,122 In these cases, their structural units contained basic aryl–pyridine ligands and modularized isocyano ligands, in which the photophysics involved 3IL/3LLCT//3MLCT characters. The solid photoluminescence resulting from the phenylpyridine-based complexes was mainly determined by intermolecular π–π aggregation, in which the bathochromic-shifted phosphorescence originated from 3π–π* (excimers or aggregates) or even Pt⋯Pt interactions (MMLCT, metal–metal-to-ligand charge transfer). Moreover, the MCL feature was found in these complexes because of enhanced π–π/Pt⋯Pt and planarity in the ground amorphous state.122 Wong et al. reported the Pt(II) soft salt based on fluorinated phenyl–pyridine derivates and CN− coordinating ions, and ethylenediamine coordinated cations (5-8, Fig. 19a). This pristine sample endowed excitation-wavelength-dependent emission but this nature was absent in uniform 5-8 nanostructures and ground powder. This outcome manifested that the variable Pt⋯Pt distances were the key factors for excitation-wavelength-dependent emission in pristine powder.123 Host–guest-involved coassembly systems received more attention due to the high-speed developments of supramolecular chemistry. Sun and his collaborators demonstrated a multi-component system (5-12), which involved [3 + 3] Pt(II) metallacycle and polycyano-fused TPE, leading to solid-state artificial light-harvesting and rare near-infrared MCL activities. The improved MCL was driven by the synergetic light harvesting effect and enhanced NCIs between host and guest. The MCL wavelength of ground mixtures was up to 747 nm.124
 |
| | Fig. 19 (a) Schematic illustration of excitation-wavelength-dependent emission of a Pt(II) complex 5-8 and their spectra observations. Reproduced from ref. 123 with permission. Copyright (2021), American Chemical Society. (b) Crystal structures of complex 5-9 to 5-11 and their MCL observations. Reproduced from ref. 122 with permission. Copyright (2020), The Royal Society of Chemistry. | |
Electronic configuration affects the coordination configuration, which directly decides the interactions between adjacent metal centers. Linear and planar coordination geometry have potential adjustability and large redundant space in loose packing compared to polyhedral geometry. That is why substantial SRL materials are derived from linear and near planar complexes in reports instead of a few polyhedral (tetrahedral or octahedral geometries) complexes.38
3. Conclusions and outlooks
The understanding of structure–function relationship between chemical structures and supramolecular structures or stimuli-responsive luminescence of polar cyano/isocyano-derived luminophores has made qualitative stimuli-responsive luminescence accessible. From the last few decades to the present, various tactics have been established to construct stimuli-responsive luminescence units containing D–A-like modules, AIE building blocks, chiral subassembly, multicomponent assembly, and phosphorescent coordination compounds. Among all possible chemical units, cyano derivatives were undoubtedly believed to be the most successful in organic compounds. However, cyano-containing, SRL coordination compounds still lagged certainly behind. Encouragingly, the developments of luminescent isocyano derivatives are advancing rapidly and the permission of phosphorescence or RTP emission capacity of unity where there is efficient spin–orbit coupling is an attractive prospect (i.e. large spin–orbit coupling parameter ζsoc in Pt 4481 cm−1 and Cu 857 cm−1).125 Nonetheless, there have been good attempts in isocyano complexes with a d8 or d10 electric configuration, while the design of cyano-substitutional complexes presents less attention because it just looks like a supplement to common organic partners. From our perspective, further supplements might be useful in the field of stimuli-responsive RTP.126,127
Stimuli-responsive CPL is a potentially useful feature, and the possibility of both molecular and supramolecular engineering in this chiral property requires careful consideration. The carried photon information of polarization state and color would bring out multiple response results after stimulation, which is quite useful for optical encryption and security. Similarly, polarised emission of emitters has significant values in polarized optical switches and 3D displays. However, while the concept of SRL cyano/isocyano derivatives is now quite well-established, challenges remain if the polymorphism–emission relevances could be predicted through the advanced database or theoretical calculation. We are happy to see some exciting efforts are being launched.128 At present, there needs to be precise control over the types of polymorphism as well as the emission behavior, which comes from a profound understanding of molecules and supermolecules, even a little contingency of unexpected outcomes in some cases. Furthermore, in light of the operability of the actual application, the soft matter and non-contacting stimuli mode have unique device processability, consistent reversibility, and flexibility.129 This superiority is demonstrated by the above discussion on the liquid-crystal self-assembly systems, where the light-electric-thermal stimulations could be realized by remote operations. Overall, SRL-active cyano/isocyano-derived luminophores have developed into a successful and worthy platform. We believe that integrating multiple functionalities and stimulations and even structure–property predictions is the direction of the future.
Abbreviations
| SRL | Stimuli-responsive luminescence |
| TADF | Thermally activated delayed fluorescence |
| AIE | Aggregation induced emission |
| AIEE | Aggregation induced emission enhancement |
| MCF | Mechanochromic luminescence |
| TL | Triboluminescence |
| TCL | Thermochromic luminescence |
| RTP | Room temperature phosphorescence |
| NCIs | Non-covalent interactions |
| OLEDs | Organic light-emitting diodes |
| SCXRD | Single crystal X-ray diffraction |
| PXRD | Powder X-ray diffraction |
| EL | Electroluminescence |
| PL | Photoluminescence |
| EQE | External quantum efficiency |
| PLQY | Photoluminescence quantum yield |
| NIR | Near Infra Red |
| DFT | Density functional theory |
| CZ | Carbazole |
| TPA | Triphenylamine |
| HOFs | Hydrogen-bonded organic frameworks |
| CIEE | Crystallization-induced emission enhancement |
| NMR | Nuclear magnetic resonance |
| CD | Circular dichroism |
| CPL | Circularly polarized luminescence |
| Colr | Columnar rectangular mesophase |
| Sm | Smectic phase |
| SmC* | Chiral smectic C phase |
| N* | Chiral nematic phase |
| FRET | Fluorescence resonance energy transfer |
| LCD | Liquid crystal display |
|
g
lum
| Asymmetry factor (for CPL) |
| LC(s) | Liquid crystal(s) |
| LLC | Luminescent liquid crystal |
| ICT | Intramolecular charge transfer |
| CT | Charge transfer |
| TPE | Tetraphenylethylene |
| ILCT | Intra-ligand charge transfer |
| LLCT | Ligand to ligand charge transfer |
| PICT | Planar intramolecular charge transfer |
| MLCT | Metal to ligand charge transfer |
| MMLCT | Metal–metal-to-ligand charge transfer |
| PET | Photo-induced electron transfer |
Author contributions
W. H. and B. Y. conceived the manuscript. B. Y. and W. H. wrote and revised the manuscript. All authors have approved the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21871133), the Natural Science Foundation of Jiangsu Province (No. BK20211146), and the Science, Technology, and Innovation Commission of Shenzhen Municipality (No. JCYJ20180307153251975).
References
- Y. Sun, Z. Lei and H. Ma, J. Mater. Chem. C, 2022, 10, 14834–14867 RSC.
- Y. Fan, Q. Li and Z. Li, Mater. Chem. Front., 2021, 5, 1525–1540 RSC.
- W. Sun, S. Guo, C. Hu, J. Fan and X. Peng, Chem. Rev., 2016, 116, 7768–7817 CrossRef CAS PubMed.
-
Z. Yang, J. Zhao, E. Ubba, Z. Yang, Y. Zhang and Z. Chi, in Handbook of Aggregation–Induced Emission, 2022, pp. 105–137 Search PubMed.
- T. Xu, J. Zhu, Y. Han and C. Chi, J. Mater. Chem. C, 2023, 7957–7969 RSC.
- Y. Chen, G. Mellot, D. Van Luijk, C. Creton and R. P. Sijbesma, Chem. Soc. Rev., 2021, 50, 4100–4140 RSC.
- Y. Xie and Z. Li, Mater. Chem. Front., 2020, 4, 317–331 RSC.
- S. K. Mellerup and S. Wang, Chem. Soc. Rev., 2019, 48, 3537–3549 RSC.
- Z. Yang, Z. Chi, Z. Mao, Y. Zhang, S. Liu, J. Zhao, M. P. Aldred and Z. Chi, Mater. Chem. Front., 2018, 2, 861–890 RSC.
- C. Wang and Z. Li, Mater. Chem. Front., 2017, 1, 2174–2194 RSC.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- H. Sun, S. Shen and L. Zhu, ACS Mater. Lett., 2022, 4, 1599–1615 CrossRef CAS.
- M. C. Tang, M. Y. Chan and V. W. W. Yam, Chem. Rev., 2021, 121, 7249–7279 CrossRef CAS PubMed.
- H. K. Bisoyi and Q. Li, Chem. Rev., 2022, 122, 4887–4926 CrossRef CAS PubMed.
- V. W. W. Yam, V. K. M. Au and S. Y. L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed.
- K. H. Kim and J. J. Kim, Adv. Mater., 2018, 30, 1705600 CrossRef PubMed.
- X. Yang, G. Zhou and W.-Y. Wong, Chem. Soc. Rev., 2015, 44, 8484–8575 RSC.
- B. Pashaei, S. Karimi, H. Shahroosvand, P. Abbasi, M. Pilkington, A. Bartolotta, E. Fresta, J. Fernandez-Cestau, R. D. Costa and F. Bonaccorso, Chem. Soc. Rev., 2019, 48, 5033–5139 RSC.
- S. J. Zou, Y. Shen, F. M. Xie, J. De Chen, Y. Q. Li and J. X. Tang, Mater. Chem. Front., 2020, 4, 788–820 RSC.
- Z. Xu, B. Z. Tang, Y. Wang and D. Ma, J. Mater. Chem. C, 2020, 8, 2614–2642 RSC.
- X. Xiong, F. Song, J. Wang, Y. Zhang, Y. Xue, L. Sun, N. Jiang, P. Gao, L. Tian and X. Peng, J. Am. Chem. Soc., 2014, 136, 9590–9597 CrossRef CAS PubMed.
- H. Zhu, J. Fan, J. Du and X. Peng, Acc. Chem. Res., 2016, 49, 2115–2126 CrossRef CAS PubMed.
- G. A. Zalmi, D. N. Nadimetla, P. Kotharkar, A. L. Puyad, M. Kowshik and S. V. Bhosale, ACS Omega, 2021, 6, 16704–16713 CrossRef CAS PubMed.
- E. Karakuş, M. Üçüncü and M. Emrullahoʇlu, Anal. Chem., 2016, 88, 1039–1043 CrossRef PubMed.
- J. Zhao, Z. Chi, Y. Zhang, Z. Mao, Z. Yang, E. Ubba and Z. Chi, J. Mater. Chem. C, 2018, 6, 6327–6353 RSC.
- R. Hojo, A. M. Polgar and Z. M. Hudson, ACS Sustainable Chem. Eng., 2022, 10, 9665–9678 CrossRef CAS.
- Y. Liu, X. L. Chen, X. Y. Li, S. S. Zhu, S. J. Li, Y. Song, L. B. Qu and B. Yu, J. Am. Chem. Soc., 2021, 143, 964–972 CrossRef CAS PubMed.
- P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed.
- P. A. S. Smith and N. W. Kalenda, J. Org. Chem., 1958, 23, 1599–1603 CrossRef CAS.
-
L. Kurti and B. Czakó, Strategic applications of named reactions in organic synthesis, Elsevier, 2005 Search PubMed.
- Z. Li, S. Shi and J. Yang, Synlett, 2006, 2495–2497 CrossRef CAS.
- N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 20, 3437–3440 CrossRef.
- S. Ito, CrystEngComm, 2022, 24, 1112–1126 RSC.
- P. Xue, J. Ding, P. Wang and R. Lu, J. Mater. Chem. C, 2016, 4, 6688–6706 RSC.
- X. Zhang, Z. Chi, Y. Zhang, S. Liu and J. Xu, J. Mater. Chem. C, 2013, 1, 3376–3390 RSC.
- A. Aliprandi, D. Genovese, M. Mauro and L. De Cola, Chem. Lett., 2015, 44, 1152–1169 CrossRef CAS.
- C. Cuerva, M. Cano and C. Lodeiro, Chem. Rev., 2021, 121, 12966–13010 CrossRef CAS PubMed.
- K. Li, Q. Chen and P. Xue, Tetrahedron, 2022, 121, 132924 CrossRef CAS.
- X. Xiang, Y. Zhan, W. Yang and F. Jin, Dyes Pigm., 2022, 206, 110670 CrossRef CAS.
- P. Chen, Y. Yin, Y. Ni, H. Zhu, J. Huang, J. Yang, J. Yang and L. Kong, CrystEngComm, 2021, 23, 2289–2296 RSC.
- J. Jayabharathi, V. Thanikachalam, U. Karunakaran, J. Anudeebhana and S. Thilagavathy, Ind. Eng. Chem. Res., 2022, 61, 6537–6550 CrossRef CAS.
- V. Thanikachalam, U. Karunakaran, J. Jayabharathi and S. Thilagavathy, J. Photochem. Photobiol., A, 2021, 420, 113489 CrossRef CAS.
- P. Gayathri, M. Pannipara, A. G. Al-Sehemi, D. Moon and S. P. Anthony, Mater. Adv., 2021, 2, 996–1005 RSC.
- Y. Liu, Z. Ma, X. Cheng, C. Qian, J. Liu, X. Zhang, M. Chen, X. Jia and Z. Ma, J. Mater. Chem. C, 2021, 9, 5227–5233 RSC.
- L. Yue, Y. Wang, J. Ma, S. Yuan, S. Xue, Q. Sun and W. Yang, Mater. Chem. Front., 2021, 5, 5497–5502 RSC.
- H. Zhu, J. Huang, L. Kong, Y. Tian and J. Yang, Dyes Pigm., 2018, 151, 140–148 CrossRef CAS.
- Y. Wang, D. Xu, H. Gao, Y. Wang, X. Liu, A. Han, C. Zhang and L. Zang, J. Phys. Chem. C, 2018, 122, 2297–2306 CrossRef CAS.
- J. Li, J. Xie, R. Zhang, S. Guo, F. Zhang, K. Guo and S. Dong, Chem. Commun., 2018, 54, 11455–11458 RSC.
- Y. X. Peng, Y. T. Gan, R. G. Shi, W. Huang and T. Tao, J. Phys. Chem. C, 2018, 122, 29488–29497 CrossRef CAS.
- C. Zhu, Q. Luo, Y. Shen, C. Lv, S. Zhao, X. Lv, F. Cao, K. Wang, Q. Song, C. Zhang and Y. Zhang, Angew. Chem., Int. Ed., 2021, 60, 8510–8514 CrossRef PubMed.
- Y. Xie, Z. Wang, X. Liu, M. Liu, Y. Lei, Y. Zhou, W. Gao, X. Huang and H. Wu, CrystEngComm, 2020, 22, 6529–6535 RSC.
- Y. Wang, J. Liu, W. Yuan, Y. Wang, H. Zhou, X. Liu, J. Cao and C. Zhang, Dyes Pigm., 2019, 167, 135–142 CrossRef CAS.
- T. Zhang, Y. Han, Q. Chen, X. Chen and P. Xue, Dyes Pigm., 2021, 190, 109296 CrossRef CAS.
- T. Zhang, Y. Han, K. Wang, M. Liang, W. Bian, Y. Zhang, X. Li, C. Zhang and P. Xue, Dyes Pigm., 2020, 172, 107835 CrossRef CAS.
- N. Hasan, Z. Ma, J. Liu, Z. Li, C. Qian, Y. Liu, M. Chen, H. Jiang, X. Jia and Z. Ma, ChemPhysChem, 2021, 22, 2093–2098 CrossRef CAS PubMed.
- Z. Wan, Y. Zan, B. Wang, W. Fang, S. Cui, Y. Teng, F. Zhang, Y. Li, L. Chen and G. Bai, J. Lumin., 2022, 242, 118555 CrossRef CAS.
- Y. Ooyama, Y. Kagawa, H. Fukuoka, G. Ito and Y. Harima, Eur. J. Org. Chem., 2009, 31, 5321–5326 CrossRef.
- C. Qian, Z. Ma, J. Liu, X. Zhang, S. Wang and Z. Ma, Chem. – Asian J., 2021, 16, 3713–3718 CrossRef CAS PubMed.
- K. Zheng, F. Ni, Z. Chen, C. Zhong and C. Yang, Angew. Chem., Int. Ed., 2020, 59, 9972–9976 CrossRef CAS PubMed.
- Y. Sang, J. Han, T. Zhao, P. Duan and M. Liu, Adv. Mater., 2019, 32, 1900110 CrossRef PubMed.
- D. W. Zhang, M. Li and C. F. Chen, Chem. Soc. Rev., 2020, 49, 1331–1343 RSC.
- J. Luo, Z. Xie, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 18, 1740–1741 RSC.
- L. Bian, Y. Liang and Z. Liu, ACS Appl. Nano Mater., 2022, 5, 13940–13958 CrossRef CAS.
- X. Y. Shen, Y. J. Wang, E. Zhao, W. Z. Yuan, Y. Liu, P. Lu, A. Qin, Y. Ma, J. Z. Sun and B. Z. Tang, J. Phys. Chem. C, 2013, 117, 7334–7347 CrossRef CAS.
- T. Jadhav, J. M. Choi, J. Shinde, J. Y. Lee and R. Misra, J. Mater. Chem. C, 2017, 5, 6014–6020 RSC.
- T. Jadhav, J. M. Choi, B. Dhokale, S. M. Mobin, J. Y. Lee and R. Misra, J. Phys. Chem. C, 2016, 120, 18487–18495 CrossRef CAS.
- Z. Peng, K. Huang, Y. Tao, X. Li, L. Zhang, P. Lu and Y. Wang, Mater. Chem. Front., 2017, 1, 1858–1865 RSC.
- X. Yang, Q. Wang, P. Hu, C. Xu, W. Guo, Z. Wang, Z. Mao, Z. Yang, C. Liu, G. Shi, L. Chen, B. Xu and Z. Chi, Mater. Chem. Front., 2020, 4, 941–949 RSC.
- X. Tian, H. Wang, S. Cao, Y. Liu, F. Meng, X. Zheng and G. Niu, J. Mater. Chem. C, 2023, 11, 5987–5994 RSC.
- S. Guo, G. Zhang, F. Chen, Y. Ni, J. Huang, L. Kong and J. Yang, New J. Chem., 2021, 45, 21327–21333 RSC.
- Y. X. Peng, H. Q. Liu, W. Huang and T. Tao, J. Phys. Chem. C, 2022, 126, 6491–6498 CrossRef CAS.
- F. Da Feng, B. Yang, Y. X. Peng, S. Q. Yan, T. Tao and W. Huang, J. Phys. Chem. C, 2023, 127, 8362–8372 CrossRef.
- H. Zhu, S. Zhang, J. Yang, M. Wu, Q. Wu, J. Liu, J. Zhang, L. Kong and J. Yang, J. Solid State Chem., 2022, 305, 122706 CrossRef CAS.
- T. Jadhav, B. Dhokale and R. Misra, J. Mater. Chem. C, 2015, 3, 9063–9068 RSC.
- C. Ma, X. Zhang, Y. Yang, Z. Ma, L. Yang, Y. Wu, H. Liu, X. Jia and Y. Wei, J. Mater. Chem. C, 2016, 4, 4786–4791 RSC.
- M. Chen, R. Chen, Y. Shi, J. Wang, Y. Cheng, Y. Li, X. Gao, Y. Yan, J. Z. Sun, A. Qin, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Adv. Funct. Mater., 2018, 28, 1704689 CrossRef.
- Y. Jiang, X. Chang, W. Xie, G. Huang and B. S. Li, Mater. Chem. Front., 2021, 5, 885–892 RSC.
- H. Yu, W. Wu, H. Zhao, K. Chen, S. Li, M. Tan, T. Wang, X. Huang, N. Wang and H. Hao, Dyes Pigm., 2023, 220, 111727 CrossRef CAS.
- C. Zeng, J. Dai, T. Yang, Z. Wang, Y. Gao, J. Xia, Y. Chen and M. Sun, Dyes Pigm., 2024, 222, 111906 CrossRef CAS.
- N. Zhou, Y. Lu, J. Zhou, D. Xu, Q. Jia and X. Liu, J. Lumin., 2024, 266, 120332 CrossRef CAS.
- N. Zhou, J. Zhou, Y. Lu, W. Zhang, X. Liu and D. Xu, J. Lumin., 2024, 267, 120385 CrossRef CAS.
- Q. Yu and Q. Zhu, Dyes Pigm., 2023, 223, 111903 CrossRef.
- H. Q. Peng, L. Y. Niu, Y. Z. Chen, L. Z. Wu, C. H. Tung and Q. Z. Yang, Chem. Rev., 2015, 115, 7502–7542 CrossRef CAS PubMed.
- C. Fan, L. Wei, T. Niu, M. Rao, G. Cheng, J. J. Chruma, W. Wu and C. Yang, J. Am. Chem. Soc., 2019, 141, 15070–15077 CrossRef CAS PubMed.
- J. Han, D. Yang, X. Jin, Y. Jiang, M. Liu and P. Duan, Angew. Chem., Int. Ed., 2019, 58, 7013–7019 CrossRef CAS PubMed.
- Y. Liu, Q. Zeng, B. Zou, Y. Liu, B. Xu and W. Tian, Angew. Chem., Int. Ed., 2018, 57, 15670–15674 CrossRef CAS PubMed.
- S. Wang, W. Xiang, C. Pan, J. Chen, W. Li, J. Zhang, J. Zhao and G. Liu, CrystEngComm, 2023, 25, 3861–3865 RSC.
- B. Yang, S. Yan, C. Li, H. Ma, F. Feng, Y. Zhang and W. Huang, Chem. Sci., 2023, 14, 10446–10457 RSC.
- S. Tokunaga, Y. Itoh, H. Tanaka, F. Araoka and T. Aida, J. Am. Chem. Soc., 2018, 140, 10946–10949 CrossRef CAS PubMed.
- G. Hong, X. Gan, C. Leonhardt, Z. Zhang, J. Seibert, J. M. Busch and S. Bräse, Adv. Mater., 2021, 33, 2005630 CrossRef CAS PubMed.
- Y. Chen, P. Lu, Z. Li, Y. Yuan, Q. Ye and H. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 56604–56614 CrossRef CAS PubMed.
- H. Wang, Z. Chen, Y. Yuan and H. Zhang, Soft Matter, 2022, 18, 5483–5491 RSC.
- Y. Chen, P. Lu, Q. Gui, Z. Li, Y. Yuan and H. Zhang, J. Mater. Chem. C, 2021, 9, 1279–1286 RSC.
- K. Yao, Y. Li, Y. Shen, Y. Quan and Y. Cheng, J. Mater. Chem. C, 2021, 9, 12590–12595 RSC.
- Y. Li, Y. Chen, J. Luo, Y. Quan and Y. Cheng, Adv. Mater., 2024, 2312331, 2312331 CrossRef PubMed.
- X. Gao, X. Qin, X. Yang, Y. Li and P. Duan, Chem. Commun., 2019, 55, 5914–5917 RSC.
- X. Li, Y. Shen, K. Liu, Y. Quan and Y. Cheng, Mater. Chem. Front., 2020, 4, 2954–2961 RSC.
- P. Lu, Y. Chen, Z. Chen, Y. Yuan and H. Zhang, J. Mater. Chem. C, 2021, 9, 6589–6596 RSC.
- Q. Zhang, X. Liao, C. Song, X. Meng, Y. He, Z. Li, X. Zhang and Z. Cao, Chem. – Eur. J., 2023, 29, e202301766 CrossRef PubMed.
- J. Yoshida, S. Tamura, H. Yuge and G. Watanabe, Soft Matter, 2017, 14, 27–30 RSC.
- B. Yang, S. Yan, Y. Zhang, H. Ma, F. Feng and W. Huang, Dyes Pigm., 2024, 221, 111813 CrossRef CAS.
- Y. Shen, K. Yao, H. Li, Z. Xu, Y. Quan and Y. Cheng, Soft Matter, 2022, 18, 477–481 RSC.
- Y. Wu, L. H. You, Z. Q. Yu, J. H. Wang, Z. Meng, Y. Liu, X. S. Li, K. Fu, X. K. Ren and B. Z. Tang, ACS Mater. Lett., 2020, 2, 505–510 CrossRef CAS.
- S. Chen, S. Jiang, J. Qiu, H. Guo and F. Yang, Chem. Commun., 2020, 56, 7745–7748 RSC.
- L. Wen, J. Sun, C. Li, C. Zhu, X. Zhang, Z. Wang, Q. Song, C. Lv and Y. Zhang, New J. Chem., 2021, 45, 11530–11535 RSC.
- W. Ding, S. Chen, X. Du and X. Cheng, J. Mol. Liq., 2022, 363, 119906 CrossRef CAS.
- Z. Jiang, H. Zhao, W. Wu, K. Chen, H. Yu, T. Wang, X. Huang, N. Wang, L. Zhou and H. Hao, J. Mater. Chem. C, 2023, 11, 4375–4383 RSC.
- B. Huitorel, Q. Benito, A. Fargues, A. Garcia, T. Gacoin, J. P. Boilot, S. Perruchas and F. Camerel, Chem. Mater., 2016, 28, 8190–8200 CrossRef CAS.
- R. Utrera-Melero, F. Massuyeau, C. Latouche, F. Camerel, S. Perruchas, C. Latouche and S. Perruchas, Inorg. Chem., 2022, 61, 4080–4091 CrossRef CAS PubMed.
- S. Cheng, Z. Chen, Y. Yin, Y. Sun and S. Liu, Chin. Chem. Lett., 2021, 32, 3718–3732 CrossRef CAS.
- M. Jin and H. Ito, J. Photochem. Photobiol., C, 2022, 51, 100478 CrossRef CAS.
- M. Barcenilla, C. L. Folcia, J. Ortega, J. Etxebarria, S. Coco and P. Espinet, J. Mater. Chem. C, 2022, 10, 941–946 RSC.
- V. Conejo-Rodríguez, M. N. Peñas-Defrutos and P. Espinet, Dalton Trans., 2019, 48, 10412–10416 RSC.
- X. Y. Wang, Y. Yin, J. Yin, Z. Chen and S. H. Liu, Dalton Trans., 2021, 50, 7744–7749 RSC.
- L. M. C. Luong, M. A. Malwitz, V. Moshayedi, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2020, 142, 5689–5701 CrossRef CAS PubMed.
- J. Zhang, B. He, W. Wu, P. Alam, H. Zhang, J. Gong, F. Song, Z. Wang, H. H. Y. Sung, I. D. Williams, Z. Wang, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2020, 142, 14608–14618 CrossRef CAS PubMed.
- X. Wu, M. Zhu, D. W. Bruce, W. Zhu and Y. Wang, J. Mater. Chem. C, 2018, 6, 9848–9860 RSC.
- R. Li, F. F. Xu, Z. L. Gong and Y. W. Zhong, Inorg. Chem. Front., 2020, 7, 3258–3281 RSC.
- I. Eryazici, C. N. Moorefield and G. R. Newkome, Chem. Rev., 2008, 108, 1834–1895 CrossRef CAS PubMed.
- S. A. Katkova, A. S. Mikherdov, E. V. Sokolova, A. S. Novikov, G. L. Starova and M. A. Kinzhalov, J. Mol. Struct., 2022, 1253, 132230 CrossRef CAS.
- M. Martínez-Junquera, R. Lara, E. Lalinde and M. T. Moreno, J. Mater. Chem. C, 2020, 8, 7221–7233 RSC.
- J. Li, K. Chen, J. Wei, Y. Ma, R. Zhou, S. Liu, Q. Zhao and W. Y. Wong, J. Am. Chem. Soc., 2021, 143, 18317–18324 CrossRef CAS PubMed.
- Y. Yin, Z. Chen, R. H. Li, C. Yuan, T. Y. Shao, K. Wang, H. Tan and Y. Sun, Inorg. Chem., 2021, 60, 9387–9393 CrossRef CAS PubMed.
-
S. Fraga, J. Karwowski and K. Saxena, Handbook of atomic data. Physical Science Data, Elsevier Scientific Publishing Company, New York, NY, 1976 Search PubMed.
- A. Lázaro, C. Cunha, R. Bosque, J. Pina, J. S. Ward, K. N. Truong, K. Rissanen, J. C. Lima, M. Crespo, J. S. Seixas De Melo and L. Rodríguez, Inorg. Chem., 2020, 59, 8220–8230 CrossRef PubMed.
- T. Theiss, S. Buss, I. Maisuls, R. López-Arteaga, D. Brünink, J. Kösters, A. Hepp, N. L. Doltsinis, E. A. Weiss and C. A. Strassert, J. Am. Chem. Soc., 2023, 145, 3937–3951 CrossRef CAS PubMed.
- W. Wang, R. Li, S. Xiao, Q. Xing, X. Yan, J. Zhang, X. Zhang, H. Lan and T. Yi, CCS Chem., 2022, 4, 899–909 CrossRef CAS.
- X. Yang, X. Jin, T. Zhao and P. Duan, Mater. Chem. Front., 2021, 5, 4821–4832 RSC.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab