
Prediction of superhard C1+xN1−x compounds with metal-free magnetism and narrow band gaps
Haiping
Wu†
 *ab,
Yunhao
Zheng†
a,
Erjun
Kan
ab and
Yan
Qian
*ab
*ab,
Yunhao
Zheng†
a,
Erjun
Kan
ab and
Yan
Qian
*ab
aSchool of Physics, Nanjing University of Science and Technology, Nanjing 210094, China. E-mail: mrhpwu@njust.edu.cn; qianyan@njust.edu.cn
bMIIT Key Laboratory of Semiconductor Microstructure and Quantum Sensing, Nanjing University of Science and Technology, Nanjing 210094, China
First published on 10th April 2024
Abstract
The scarcity of superhard materials with magnetism or a narrow band gap, despite their potential applications in various fields, makes it desirable to design such materials. Here, a series of C1+xN1−x compounds are theoretically designed by replacing different numbers of nitrogen atoms with carbon atoms in the synthesized C1N1 compound. The results indicate that the compounds C5N3 and C7N1 possess both superhardness and antiferromagnetic ordering due to the introduction of low-coordinated carbon atoms. The hardness of the two compounds is about 40.3 and 54.5 GPa, respectively. The magnetism in both compounds is attributed to the unpaired electrons in low-coordinated carbon atoms, and the magnetic moments are 0.42 and 0.39 μB, respectively. Interestingly, the magnetism in C5N3 remains unaffected by the external pressure used in this study, whereas C7N1 becomes nonmagnetic when the pressure exceeds ∼80 GPa. Electronic calculations reveal that both compounds behave as indirect band gap semiconductors, with narrow energy gaps of about 0.30 and 0.20 eV, respectively. Additionally, the other two compounds, C6N2-I and C6N2-III, exhibit nonmagnetic ordering and possess hardness values of 52.6 and 35.0 GPa, respectively. C6N2-I behaves as a semiconductor with an energy gap of 0.79 eV, and C6N2-III shows metallic behavior. Notably, the energy gaps of C5N3 and C6N2-I remain nearly constant under arbitrary pressure due to their porous and superhard structure. These compounds fill the gap in magnetic or narrow band gap superhard materials, and they can be used in the spintronic or optoelectronic fields where conventional superhard materials are not suitable.
1. Introduction
Superhard materials, which possess high Vickers hardness (HV) (>40 GPa),1 play a crucial role in contemporary industries. Consequently, they have been widely used in deverse fields,2,3 such as cutting and polishing tools, wear-resistant coatings, etc. However, superhard materials that have been commercially applied are scarce yet. Carbon diamond and cubic boron nitride (c-BN) are the only two of them, and they have HV of ∼100 and 33–45 GPa,4,5 respectively. The two materials, however, have their limitations. For example, diamond exhibits inferior thermal stability and chemically reacts with ferrous metals when subjected to extremely high pressure, and c-BN shows relatively low hardness and is prohibitively expensive due to the extreme conditions required for synthesis.The high hardness of materials is primarily attributed to the presence of strong covalent bonds. Moreover, the strong covalent bonds usually result in the semiconducting or insulating properties of superhard materials. For example, both carbon diamond and c-BN exhibit insulating properties, with band gaps of 5.47 and 6.40 eV, respectively. However, the unique electronic properties hinder the applications of superhard materials to many other fields, such as hard coatings in electromechanical systems, special wear-resistant parts, electronic applications under high stress, etc. Additionally, magnetism also plays an important role in technological applications, for example, data storage, gap sensors utilizing the change of magnetism,6,7 and so on. Unfortunately, superhard materials with magnetism are rarely reported, especially those consisting lightweight elements (e.g., B, C, O, and N). This case hinders their applications in some special fields, such as magnetic bearings, magnetic rotors, etc.
Compounds composed of lightweight elements typically exhibit superhard properties due to the formation of strong and short bonds. For example, several superhard C3N4 compounds, with hardness ranging from 62.3 to 92.0 GPa, have been reported,8–14 although the structures of the synthesized C3N4 allotropes are still being debated due to the limited quantity and heterogeneity of samples. Notably, the hardness of certain C3N4 compounds closely approaches that of diamond. It was also suggested that there are thermodynamically favorable C–N compounds (with different C/N ratios) in comparison to C3N4. Inspired by this case, the C–N binary compounds gained great attention as potential rivals to diamond in terms of hardness. Subsequently, a series of superhard C–N compounds with superhard properties were explored by many researchers, for instance, semiconducting Pnnm-CN with a Hv of 62.3 GPa,15 semiconducting α-C3N2 and β-C3N2 having a Hv of 86 GPa,16 a simple cubic structure C4N with a Hv of 40.1 GPa and several C17N4 systems with a Hv of 64.0 GPa,17 a direct band gap semiconducting C5N2 (Hv = 74.9 GPa) and antiferromagnetic C4N3 (Hv = 54.4 GPa) predicted recently,18,19 and so on. Notably, the reported superhard C–N compounds are mainly limited to carbon rich systems (i.e., the nitrogen content lower than 50%), in that the synthesis of nitrogen-rich compounds is not beneficial due to the strong repulsions between nitrogen atoms caused by short nonbonded distances. Moreover, the aforementioned compounds are primarily reported through theoretical calculations.
Recently, the Pnnm C1N1 compound, which has been theoretically predicted by Wang,15 was synthesized using laser-heating technology under high external pressures. Tetracyanoethylene mixed with N2 or graphite mixed with N2 was chosen as the precursor by two research groups, respectively.8,20 Interestingly, one of the experimental studies, which utilized graphite and N2 as precursors, shows that diamond would be the sole product when quenched to room temperature at pressures above 70 GPa.20 This result suggests the possibility of synthesizing other C–N compounds between C1N1 and diamond (i.e., the C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N ratio from 1 to ∝) under different experimental conditions. Additionally, the C and N atoms in the synthesized C1N1 compound are coordinated in a four-fold and three-fold manner, respectively. Therefore, substituting some N atoms with C atoms may result in the presence of unpaired electrons within the system, potentially leading to the emergence of magnetism.
:N ratio from 1 to ∝) under different experimental conditions. Additionally, the C and N atoms in the synthesized C1N1 compound are coordinated in a four-fold and three-fold manner, respectively. Therefore, substituting some N atoms with C atoms may result in the presence of unpaired electrons within the system, potentially leading to the emergence of magnetism.
Based on the above fact and using first-principles calculations, we studied a series of C1+xN1−x by replacing different numbers of N atoms with C atoms in the C1N1 system. As expected, several superhard C1+xN1−x compounds with unique magnetic and electronic properties are predicted.
2. Computational methods
First-principles calculations employed in this work are executed using the VASP code.21 The calculations are performed using generalized-gradient approximations with the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional and projector augmented wave (PAW) potentials.22,23 The structures are relaxed until the Hellmann–Feynman force on each atom falls below 0.001 eV Å−1. The cutoff energy for the plane wave is set to be 500 eV, the total energy change is less than 10−6 eV, and Monkhorst–Pack k-point grids with a size of 9 × 11 × 21 are utilized for integrating the Brillouin-zone (BZ) in the first BZ. Since C1N1 and diamond were synthesized at different pressures, the structural relaxations are performed at the initial step under pressures of 0 and 70 GPa, respectively. The phonon dispersion is calculated by employing the supercell approach and the force-constant method. The force constants in real space for the supercells are determined using density functional perturbation theory (DFPT),24 and then the PHONOPY code25,26 is utilized to calculate the phonon dispersions.3. Results and discussion
It was reported experimentally that diamond was the only product synthesized at a pressure of 70 GPa.20 Therefore, the structure of C1+xN1−x with all N atoms replaced by C atoms is explored first. During the structural relaxations, two hydrostatic pressures of 0 and 70 GPa are used to simulate the synthesis conditions in the experiment. The results indicate that under pressures of 0 and 70 GPa, the system undergoes a transformation into graphite and diamond, respectively. The result is consistent with the experimental results,20 and Fig. 1 illustrates the transformation process under two distinct pressures. In detail, Pnnm C1N1 could be regarded as the stacked 2D C1N1 nanosheets that are covalently bonded to each other through C–C bonds (as drawn in Fig. 1(c) and (d)). As the pressure increases, carbon atoms may prefer to replace nitrogen atoms. After replacing all N atoms with C atoms, the structure of C8N0 (named as C8) could be regarded as the stacked graphene layers with buckling characteristic. As long as the pressure is high enough to overcome a high activation barrier, interlayer covalent bonds will be formed between the initial three-fold coordinated C atoms (the corresponding atoms forming bonds are indicated by red and blue arrows in Fig. 1(b), and the formed bonds are represented by red and blue ellipses in Fig. 1(h)), resulting in the transformation from the stacked graphene sheets to diamond. The transformation pressure is closely related to the stacking sequence of graphene layers,27,28 in that the height of the energy barrier deeply depends on the starting structure. Moreover, different experimental technologies are another significant factor that profoundly affects the transformation pressure. For example, the pressure is above 22 GPa for the highly oriented pyrolytic graphite to transform into hexagonal diamond, and a value higher than 46 GPa is required to transform the as-deposited pyrolytic graphite into cubic diamond.27 On the other hand, graphite is more energetically stable than diamond, which leads to the tendency of carbon allotropes with an initial hexagonal-like configuration to adopt a planar structure rather than a buckled one. This results in the transformation of the initial C8 into a graphite-like structure when it undergoes relaxation under ambient pressure.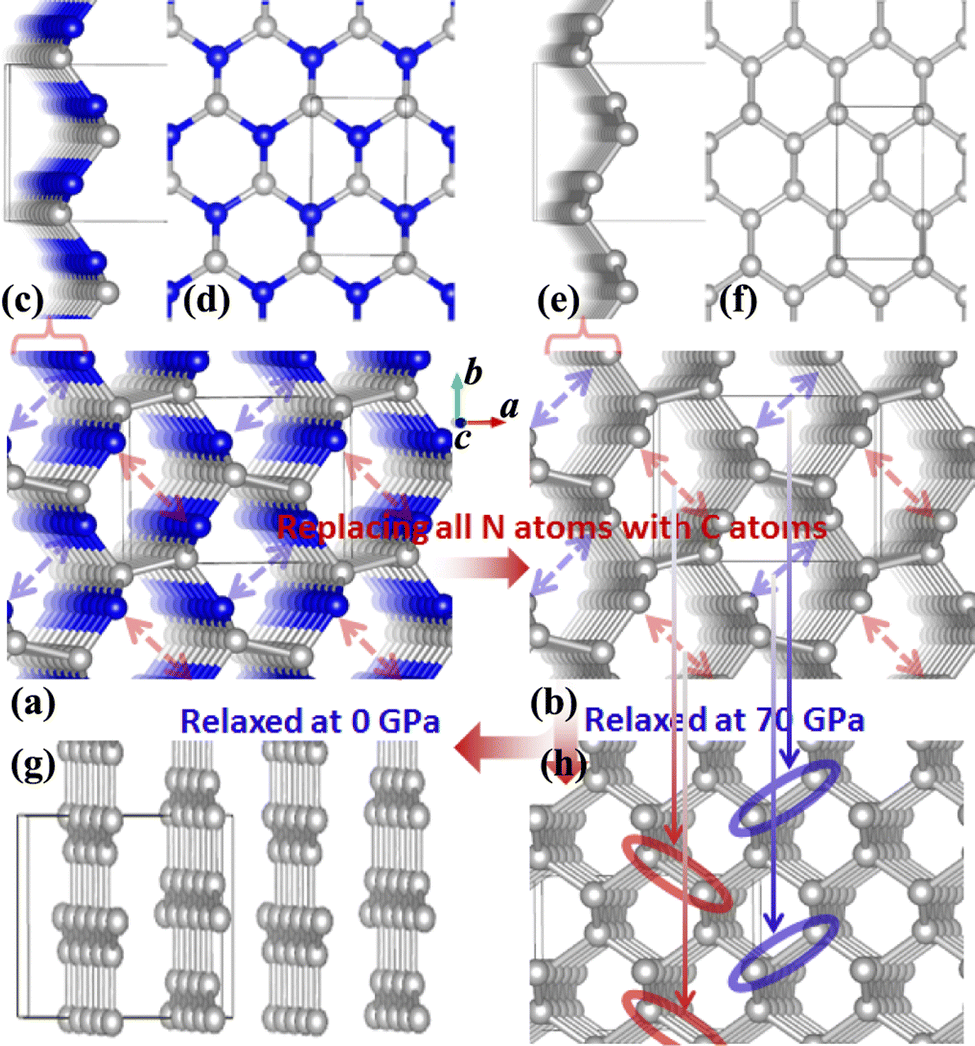 | ||
| Fig. 1 (a) The structure of Pnnm C1N1. (b) The initial structure of Pnnm C1N1 after replacing all N atoms with C atoms (named as C8). (c) and (d) The side and top views of the monolayer C1N1 exfoliated from the bulk Pnnm C1N1. (e) and (f) The side and top views of the monolayer C8 exfoliated from the initial bulk Pnnm C8. (g) and (h) The optimized structures of the bulk Pnnm C8 under the hydrostatic pressures of 0 and 70 GPa, respectively. The blue and brown spheres represent N and C atoms, respectively. | ||
To examine the possibility of synthesizing diamond through a denitriding reaction in C1N1, the thermodynamic properties are investigated by calculating the dependence of formation enthalpy on pressure. In an experiment,20 a platinum (Pt) or an iridium (Ir) ring spacer is employed and exposed to N2. Additionally, PtN, PtN2 and IrN2 were synthesized under high temperature and pressure conditions,29,30 and carbon nitride was successfully denitrified in the presence of magnesium (Mg) by Yuan et al.31 Therefore, the formation enthalpy is defined as:
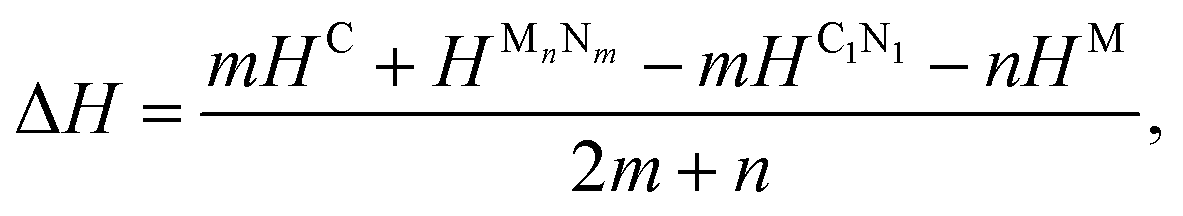 | (1) |
 | ||
| Fig. 2 The relationship between formation enthalpy and pressure in various denitriding reactions. | ||
Subsequently, a series of C1+xN1−x systems are studied through replacing different numbers of N atoms with C atoms. The initial structures are plotted in the left side of Fig. 3, and named as C5N3, C6N2-I, C6N2-II, C6N2-III, and C7N1, respectively. Then, the structures are relaxed at 0 and 70 GPa, respectively, and the optimized ones are drawn in the middle of Fig. 3. After relaxation under both pressures, the 3D-bonded framework of C1N1 remains intact in C5N3, i.e., the C–C bonds between adjacent layers remain unbroken, and no new bonds are formed to link them. This performance can be elucidated by the mechanism below. The three-fold coordinated N and C atoms in the adjacent layers, indicated by the red arrows in Fig. 3, are unable to form N–C bonds due to the preference of N atoms to be three-fold coordinated. Half of the carbon atoms in the C–C bonds that connect the two adjacent layers are three-fold coordinated with nitrogen atoms, resulting in sp3 hybridization of these carbon atoms (i.e., forming bonds with carbon atoms in the adjacent layer in order to achieve four-fold coordination). However, breaking these C–C bonds will result in the formation of numerous dangling bonds, which are energetically unfavorable. The structure of the C6N2-I phase transforms into a two-dimensional (2D) nanosheet layered crystal when relaxed at 0 GPa, but it becomes 3D-bonded at 70 GPa. It is clear that the strength of C–C bonds connecting the adjacent nanosheets varies. One type of C–C bonds is located in a carbon hexatomic ring. The other type of C–C bonds is situated on hexatomic rings consisting of both C and N atoms, which makes them much weaker due to the relatively larger electronegativity of N atoms. At 0 GPa, the weaker C–C bonds break and form double C–C bonds in nanosheets, while the stronger C–C bonds remain intact. This results in a layered configuration consisting of two interconnected C6N2 layers. Under a high external pressure of 70 GPa, the three-fold coordinated (sp2 hybridized) carbon atoms (indicated by the blue arrows in Fig. 3) undergo a transition to four-fold coordination (sp3 hybridization), leading to the formation of C–C bonds that connect the adjacent layers. This transformation is analogous to the conversion from graphite to cubic diamond. With the release of pressure, the 3D-bonded structure remains. For the C6N2-II phase, the C–C bonds are weak due to coordination of C atoms with N atoms. Thus, the C 2p electrons prefer to form C–C double bonds in the nanosheet plane, similar to the C6N2-I phase at ambient pressure. According to the fact that the N atoms are three-fold coordinated, no new bonds could be formed between the corresponding atoms (represented by the blue and red arrows in Fig. 3) even at high pressure. Consequently, the relaxed structure undergoes a transformation into a layered configuration upon the release of pressure into the ambient environment. For C6N2-III, the initial structure could be regarded as the stacking of monolayered C1N1 and graphene. In the monolayered C1N1, all carbon atoms are coordinated to three nitrogen atoms, resulting in the presence of the unpaired C 2p electrons. Thus, the system is energetically favorable by preserving the initial C–C bonds, regardless of pressure. For C7N1, the structure would turn into a 2D configuration at 0 GPa, similar to C6N2-II. Under high pressure, the C atoms that are three-fold coordinated (indicated by the blue arrows) prefer to form bonds, and this 3D-bonded structure persists even when the pressure is released to 0 GPa, similar to C6N2-I. The formation of bonds in the four 3D-bonded systems (in an ambient environment) can be confirmed by the electron localization functions (ELFs) as depicted in Fig. 4(a) and (b). It obviously shows that the neighboring atoms are bonded mainly with the σ state. The above result can also be proved by the charge density difference as shown in Fig. 4c. The formation of C–C bonds between the adjacent sublayers is evident from the accumulation of electrons. To further investigate the covalency of the bonds, Bader charges were calculated to assess the charge transfer, and the data are listed in Table 1. It is evident that there is minimal electron transfer between the atoms, attributed to the similar electronegativity of C and N atoms. This performance indicates a high level of bond covalency.
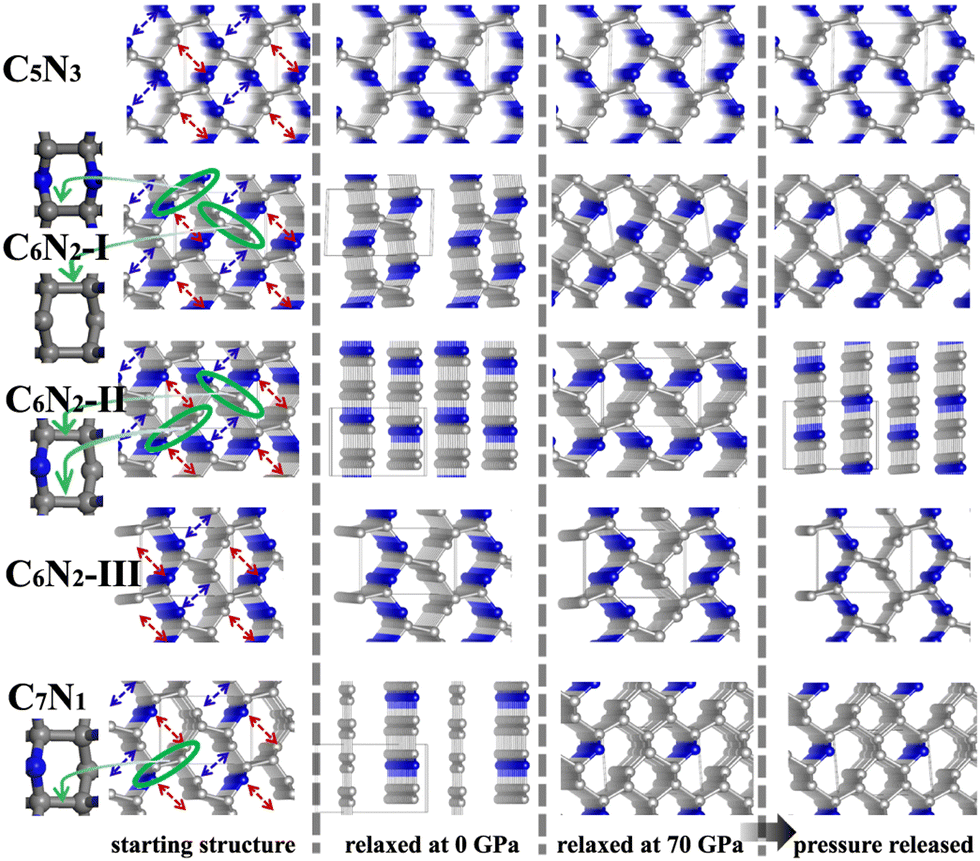 | ||
| Fig. 3 The initial and optimized structures of C1+xN1−x systems. The inset on the left side is the views of hexatomic rings in the systems. | ||
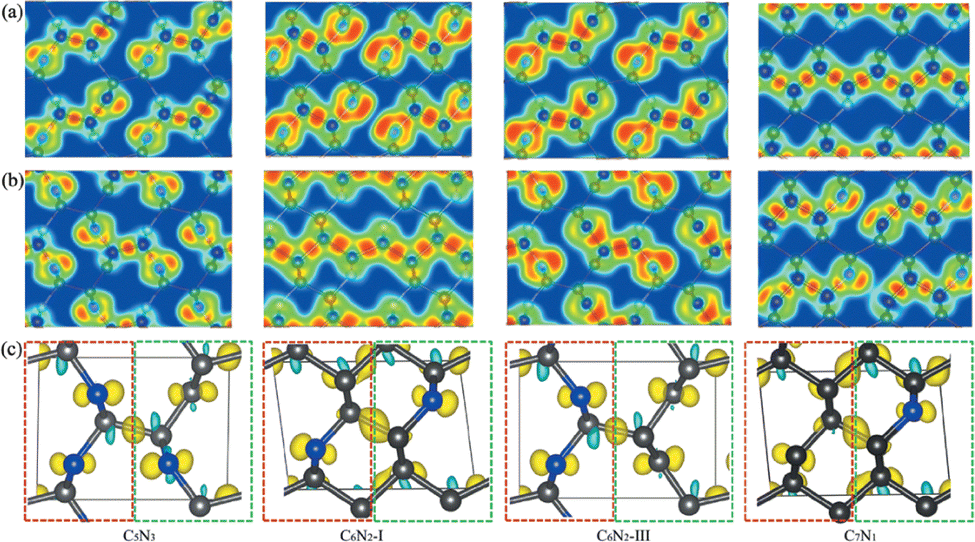 | ||
| Fig. 4 (a) and (b) The electron localization functions (ELF) for the C5N3, C6N2-I, C6N2-III, and C7N1 systems of the two different (001) planes, respectively. (c) The charge density difference for the four C5N3, C6N2-I, C6N2-III, and C7N1 systems. The isosurface value is set to be 0.001 e Å−1. The electron accumulation and depletion regions are indicated by yellow and cyan, respectively. The charge density difference is expressed as Δρ = ρCnNm − ρsublayer1 − ρsublayer2. Sublayer1 and sublayer2 are displayed by rectangles with dashed red and green lines, respectively. | ||
| Phase | C1 | C2 | C3 | C4 | C5 | C6 | C7 | N1 | N2 | N3 |
|---|---|---|---|---|---|---|---|---|---|---|
| C5N3 | 0.05 | 0.05 | 0.06 | 0.04 | −0.03 | — | — | −0.03 | −0.11 | −0.03 |
| C6N2-I | 0.05 | 0.05 | 0.00 | 0.00 | 0.04 | 0.04 | — | −0.09 | −0.09 | — |
| C6N2-III | 0.06 | 0.07 | 0.06 | 0.06 | −0.06 | −0.06 | — | −0.06 | −0.06 | — |
| C7N1 | 0.02 | 0.04 | 0.03 | 0.10 | −0.05 | 0.17 | −0.09 | — | −0.03 | — |
Some structural parameters for the C5N3, C6N2-I, C6N2-III, and C7N1 systems are given in Table 2. C5N3, C6N2-I, and C7N1 exhibit monoclinic crystals with Pm, P2/m, and Pm symmetries, respectively. C6N2-III shows orthorhombic crystals with Pmn21 symmetry. This demonstrates that substituting N atoms with C atoms alters the initial crystal symmetry of Pnnm C1N1. The lattice parameters a, b and c change significantly as well, for instance, both a and c increase after substitution. The relatively shorter C–C bonds are formed in the systems with values of ∼1.47 Å, much shorter than ∼1.58 Å in Pnnm C1N1. This is attributed to the formation of C–C double bonds resulting from the substitution of N atoms with C atoms. In contrast, Pnnm C1N1 has only C–C single bonds due to the four-fold coordination of all C atoms. Besides, these lengths are longer than ∼1.42 Å of the bonds in graphene, but shorter than ∼1.55 Å of the C–C single bonds in diamond. This is because nitrogen atoms have a higher electronegativity, causing electrons to transfer to nitrogen atoms and weakening the strength of C–C double bonds. The C–C bonds connecting the adjacent layers have a length of ∼1.61 Å, larger than ∼1.58 Å in C1N1. The three-fold coordinated C atoms prefer to construct a flat 2D layered structure, which results in an elongation of the interlayer distance.
| Bond | C5N3 | C6N2-I | C6N2-III | C7N1 | C1N1 | Gra | Dia |
|---|---|---|---|---|---|---|---|
| C–C | 1.46–1.61 | 1.52–1.62 | 1.47–1.60 | 1.47–1.62 | 1.58 | 1.42 | 1.55 |
| C–N | 1.45–1.50 | 1.43–1.45 | 1.46–1.47 | 1.46/1.50 | 1.42/1.47 | — | — |
| Symm | Pm (no. 6) | P2/m (no. 10) | Pmn21 (no. 31) | Pm (no. 6) | Pnnm (no. 58) | — | — |
| a | 5.37 | 5.29 | 5.56 | 5.27 | 5.26 | — | — |
| b | 3.97 | 3.65 | 3.86 | 3.65 | 3.93 | — | — |
| c | 2.41 | 2.43 | 2.45 | 2.49 | 2.37 | — | — |
The presence of three-fold coordination of carbon atoms in the C1+xN1−x systems potentially induces unpaired electrons, which may contribute to the excitation of magnetism. The structures of the C5N3, C6N2-III, and C7N1 systems reveal the presence of three-fold coordinated carbon atoms that are not bonded to one another. This suggests that there are unpaired electrons in these systems. In the C6N2-I system, all the C atoms are four-fold coordinated, implying that all of their electrons are bonded. This suggests that the C5N3, C6N2-III, and C7N1 systems may exhibit magnetic ordering, while the C6N2-I system is in a nonmagnetic phase. Consequently, the ferromagnetic (FM), antiferromagnetic (AFM) and nonmagnetic (NM) phases are investigated to determine the ground-state phases of these C1+xN1−x systems. The total energies of these phases are calculated and presented in Table 3. As assumed, the antiferromagnetic phases of both C5N3 and C7N1 compounds have the lowest total energies, at least 0.10 and 0.40 eV per cell lower than the other magnetic phases. This demonstrates that the ground states of both systems are antiferromagnetically coupled. The magnetic moments are located on the C atoms with three-fold coordinated, as evidenced by the net magnetic charge density (i.e., the electronic charge density difference between the spin-up and spin-down channels) in Fig. 5. The magnetic moment values on the corresponding atoms for the C5N3 and C7N1 systems are about 0.39 and 0.34 μB, respectively. For C6N2-I and C6N2-III systems, both the spin-polarized and spin-unpolarized calculations yield a nonmagnetic characteristic, indicating that their ground-states are nonmagnetic. This suggests the unpaired electrons in C6N2-III are delocalized.
| Phase | C5N3 | C6N2-I | C6N2-III | C7N1 |
|---|---|---|---|---|
| FM | −1.24 | 0 | 0 | −0.03 |
| AFM | −1.34 | 0 | 0 | −0.43 |
| NM | 0.00 | 0 | 0 | 0.00 |
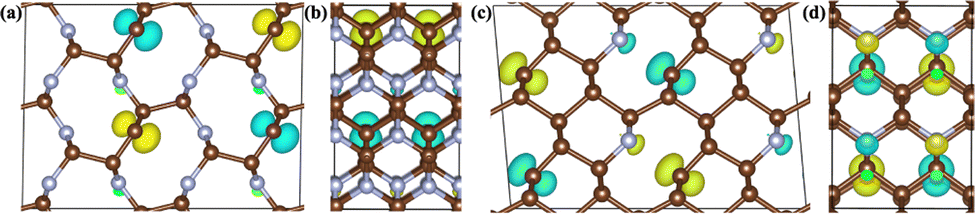 | ||
| Fig. 5 The 3D isosurface plots of magnetic charge density for the two AFM C5N3 (a) and (b) and C7N1 (c) and (d) phases, respectively. (a)–(d) The views from a and c directions, respectively. | ||
The magnetic transition temperature significantly influences the applications of materials. Therefore, the Néel temperature (Tc) of the two magnetic phases, C5N3 and C7N1, is approximately estimated by the mean-field approximation through the following expression,32
| 3/2kBTN = (EAFM − EFM)/N | (2) |
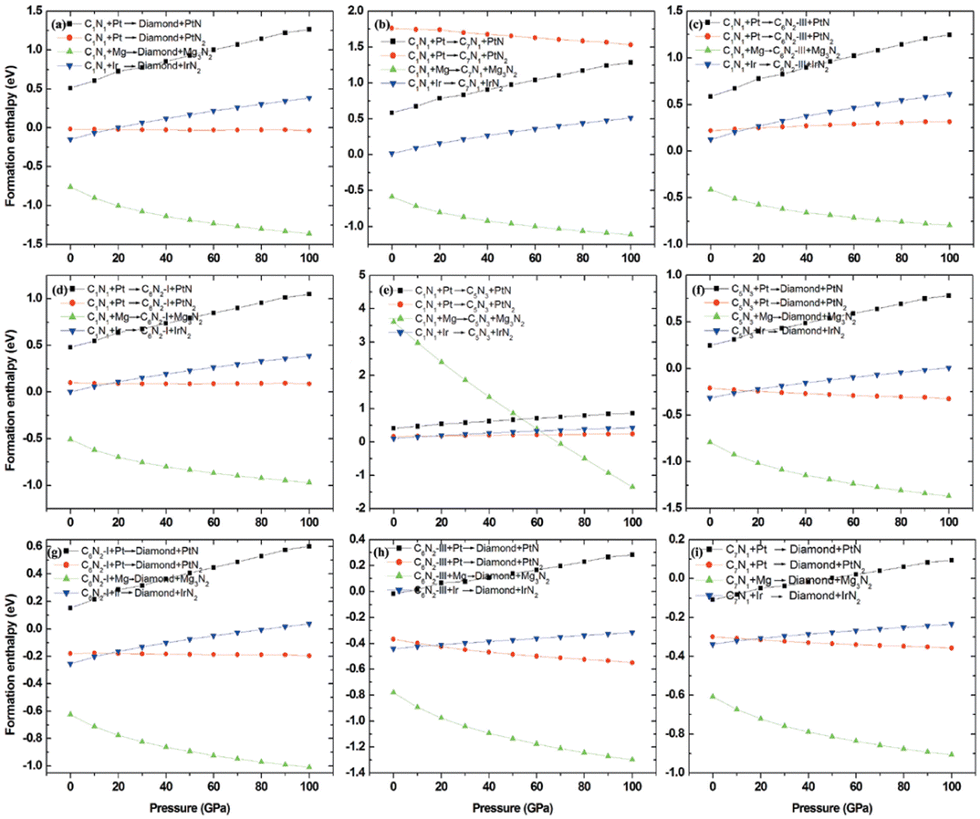
To investigate the feasibility of synthesizing C1+xN1−x systems via a denitridng reaction between C1N1 and Pt/Ir/Mg metals, the thermodynamic stability of C1+xN1−x is explored through formation enthalpy as given below
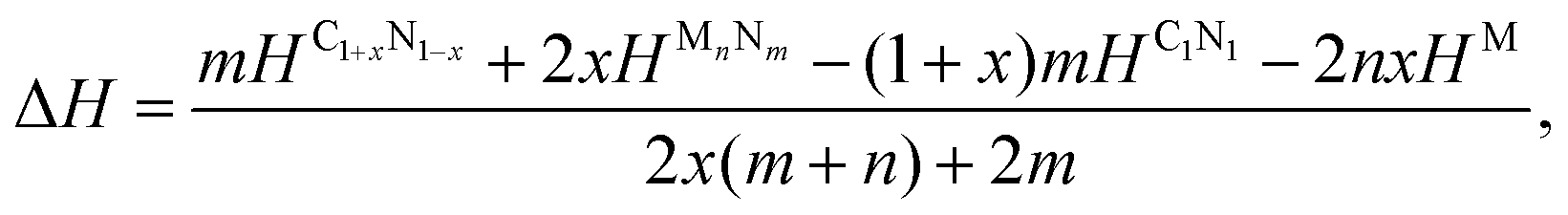 | (3) |
Additionally, the formation enthalpy is calculated by utilizing graphite and N2 as precursors, and ΔH is expressed as:
| ΔH = HC1+xN1−x − (1 + x)HC − (1 − x)HN, | (4) |
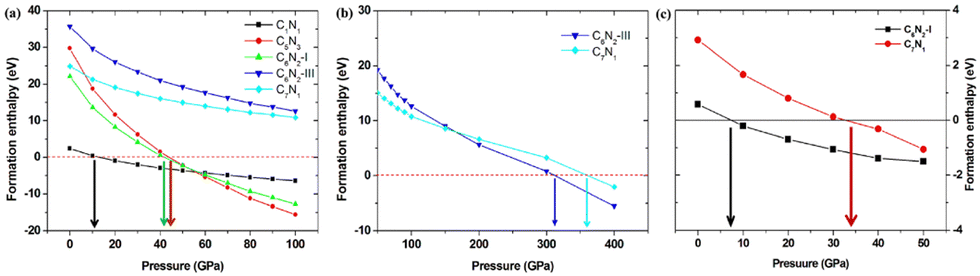 | ||
| Fig. 6 (a) The pressure-dependent formation enthalpies of the C1N1, C5N3, C6N2-I, C6N2-III, and C7N1 systems with pressure from 0 to 100 GPa, respectively. (b) The pressure-dependent formation enthalpies of C6N2-III and C7N1 systems with the pressure ranging from 100 to 400 GPa. (c) The pressure-dependent formation enthalpies of C6N2-I and C7N1 using the 2D layered nanosheets as precursors. | ||
In many applications, it is essential for materials to maintain stability in an ambient environment. Therefore, the dynamic stability of these C1+xN1−x systems under ambient pressure is studied, and the corresponding phonon dispersion curves are presented in Fig. 7. It obviously shows that there are no imaginary phonon frequencies for all the systems. The phonon density of states is consistent with the phonon dispersion curves, i.e., there are no states below zero. The absence of imaginary phonon frequencies demonstrates that the systems are dynamically stable under ambient pressure. Usually, a higher frequency indicates the presence of stronger bonds in materials. The highest frequencies, as shown in Fig. 7(g), are 1382, 1402, 1400, and 1368 cm−1 for C5N3, C6N2-I, C6N2-III, and C7N1, respectively. All these values are smaller than 1415 cm−1 of C1N1, due to certain shorter or stronger C–N bonds in C1N1 compared to those of the other C1+xN1−x systems. However, these frequencies are much higher than 1346 cm−1 of the carbon diamond, because some C–C bonds in these C1+xN1−x systems are much shorter or stronger than those in carbon diamond.
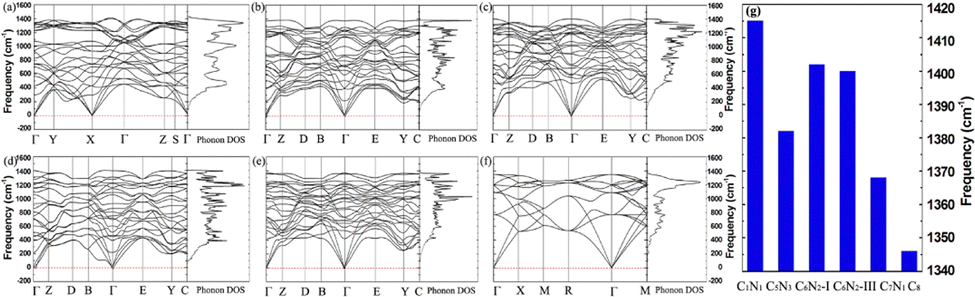 | ||
| Fig. 7 (a) The phonon-dispersion curves and phonon density of states for the Pnnm C1N1 phase. (b)–(e) The phonon-dispersion curves and phonon density of states for the C5N3, C6N2-I, C6N2-III, and C7N1 systems at ambient pressure. (f) The phonon–dispersion curves and phonon density of states for the diamond. (g) The highest phonon frequencies for C1N1, C5N3, C6N2-I, C6N2-III, C7N1, and carbon diamond (C8). | ||
Mechanical stability is another crucial factor in the application of materials and is investigated for all C1+xN1−x systems. The mechanical stability criterion is given by the following expression for all crystals, i.e., each order determinant of the elastic modulus matrix should be positive:
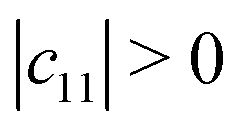
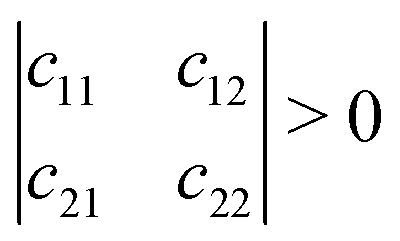
……
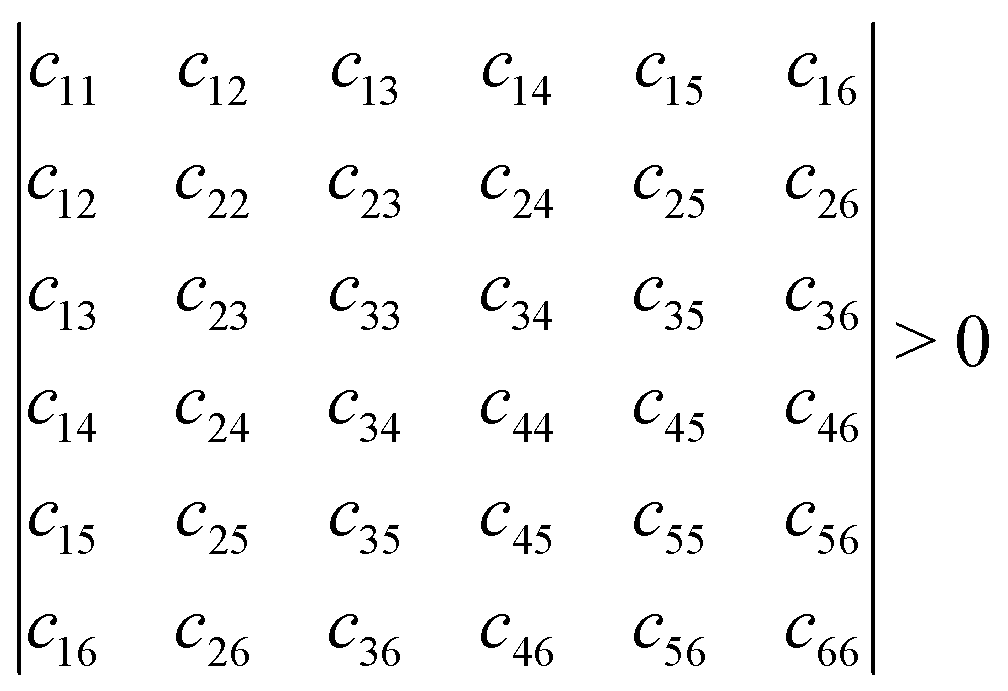
In order to evaluate the accuracy and validity of the method used, the mechanical properties of diamond are initially calculated first, and the results are listed in Table 3. The calculated elastic stiffness constants are in good agreement with the experimental data.33,34 Next, the elastic stiffness constants of the C1+xN1−x systems are calculated and given in Table 3. Consequently, the elastic stiffness constants of all systems satisfy the mechanical criteria, indicating their mechanical stability.
Elastic deformation is important for the applications of materials. Furthermore, various moduli that reflect a material's intrinsic response to different types of deformations, including bulk modulus (B), shear modulus (G), Young's modulus (E), Poisson's ratio (v), Pugh's ratio (p), etc., are explored. The Voigt–Reuss–Hill approximations35 are used to estimate these values. The calculated data are given in Table 4. The values of B, G, E, and v of diamond are in good agreement with the experimental values, confirming the reliability of our results. The moduli B, G, and E of the C1+xN1−x systems are much smaller than those of diamond, primarily because of their porous structure. Poisson's ratios (v) of all the C1+xN1−x systems are about 0.15, much larger than 0.07 of diamond. Usually, Poisson's ratio v reflects the strength of covalent bonding in a material, and it is typically less than 0.10 for covalently bonded materials. This suggests that the covalent bonds in the C1+xN1−x systems are weaker than those in diamond, primarily because of the electronegativity difference between carbon and nitrogen atoms. Additionally, the presence of relatively weak covalent bonds results in a smaller shear modulus (Table 5).
| Phases | C11 | C12 | C13 | C23 | C22 | C33 | C44 | C55 | C66 |
|---|---|---|---|---|---|---|---|---|---|
| Dia-Exp | 1076 | 125 | 577 | ||||||
| Diamond | 1052 | 125 | 560 | ||||||
| C1N1 | 546 | 218 | 83 | 144 | 697 | 1227 | 451 | 286 | 394 |
| C5N3 | 458 | 212 | 46 | 93 | 377 | 1076 | 376 | 243 | 289 |
| C6N2-I | 701 | 288 | 77 | 124 | 859 | 1217 | 441 | 315 | 441 |
| C6N2-III | 507 | 204 | 83 | 84 | 272 | 1100 | 216 | 251 | 305 |
| C7N1 | 698 | 213 | 54 | 112 | 824 | 1122 | 416 | 294 | 378 |
| Phases | B | G | E | v | p | v l | v t |
|---|---|---|---|---|---|---|---|
| Diamond | 434 | 519 | 1114 | 0.07 | 0.83 | 17910 |
12175 |
| Dia-Exp | 422 | 534 | 0.07 | ||||
| C1N1 | 360 | 341 | 778 | 0.14 | 1.05 | 15184 |
9825 |
| C5N3 | 275 | 245 | 567 | 0.15 | 1.12 | 13511 |
8624 |
| C6N2-I | 411 | 370 | 853 | 0.15 | 1.11 | 15930 |
10190 |
| C6N2-III | 262 | 220 | 515 | 0.17 | 1.20 | 13241 |
8327 |
| C7N1 | 368 | 351 | 799 | 0.14 | 1.05 | 15657 |
10141 |
Pugh's ratio (B/G) is used to assess the brittleness (B/G < 1.75) or ductility (B/G > 1.75) of materials.36 The estimated values of Pugh's ratio for the C1+xN1−x systems are listed in Table 4. The Pugh's ratio of the systems ranges from 1.05 to 1.20, all of which are much higher than 0.83 of diamond. This implies that the C1+xN1−x systems might exhibit higher ductility in comparison to diamond. To validate this result, the hardness of the C1+xN1−x systems is investigated. Here, a common metrics HV is estimated using both the Chen (HCV)37 and Mazhnik–Oganov (HMOV)38 models, as expressed in eqn (4) and (5):
| HCV = (2((1/p)2G)0.585 − (3)) | (5) |
| HMOV = 0.096E(1 − 8.5ν + 1.95)ν2/(1 − 7.5ν + 12.2ν2 + 19.6ν3), | (6) |
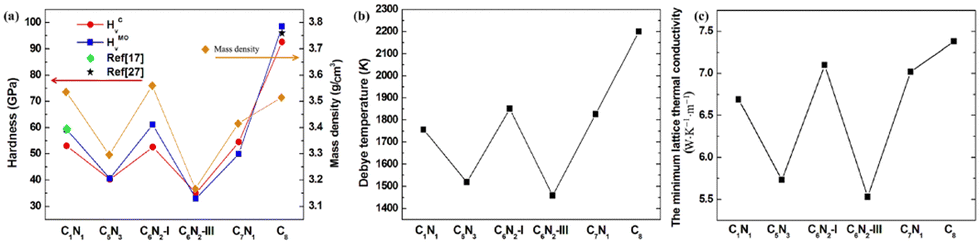 | ||
| Fig. 8 (a) The hardness and mass density for the C1+xN1−x systems and diamond. (b) and (c) The Debye temperature and the minimum lattice thermal conductivity for the C1+xN1−x systems and diamond, respectively. | ||
Clearly, the Debye temperature (ΘD) is closely associated with various physical properties of materials, such as specific heat, elasticity, melting point, and hardness, etc. It is defined as
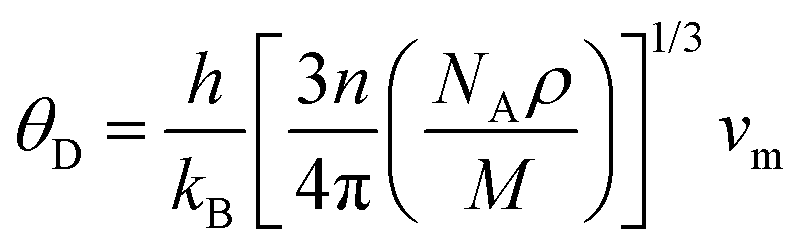 | (7) |
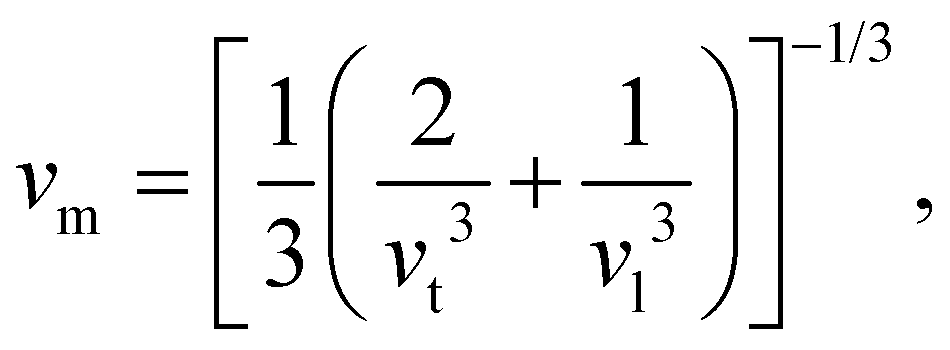 where vt and vl are the transverse and longitudinal elastic wave velocities of solids and calculated by using Navier's equations of
where vt and vl are the transverse and longitudinal elastic wave velocities of solids and calculated by using Navier's equations of 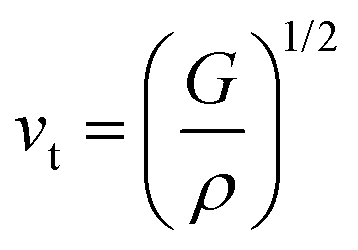 and
and 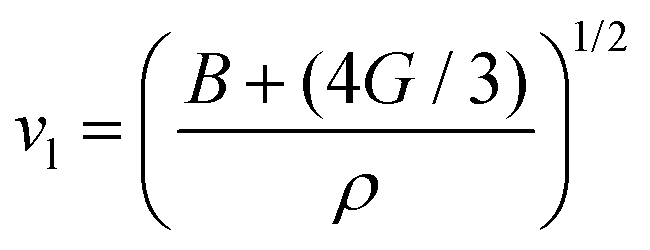 . The calculated vl and vt values are given in Table 4, and the ΘD values are plotted in Fig. 8(b). The ΘD value of the C1+xN1−x systems ranges from 1458 to 1826 K, greatly lower than 2200 K of diamond. Moreover, the Debye temperatures of these systems follow the same tendency as their hardness.
. The calculated vl and vt values are given in Table 4, and the ΘD values are plotted in Fig. 8(b). The ΘD value of the C1+xN1−x systems ranges from 1458 to 1826 K, greatly lower than 2200 K of diamond. Moreover, the Debye temperatures of these systems follow the same tendency as their hardness.
The thermal conductivity of these systems is also investigated, as it is another crucial factor for materials working at high temperatures. Therefore, the minimum lattice thermal conductivity is estimated using the Cahill's formula:41
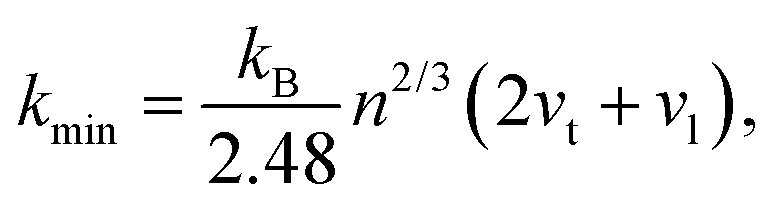 | (8) |
It is well known that Poisson's ratio v generally reflect the conductivity of materials, i.e., materials with higher v typically exhibit greater conductivity. To further explore the electronic properties of the C1+xN1−x systems, the band structures, density of electronic states (DOS), and partial density of electronic states (PDOS) of the systems are investigated and shown in Fig. 9. As predicted, the three superhard phases C5N3, C6N2-I, and C7N1 behave as semiconductors. The hard phase C6N2-III exhibits metallic properties. This is consistent with the existence of the delocalized unpaired electron on three-fold coordinated carbon atoms. The band gaps are about 0.3, 0.79, and 0.20 eV for the C5N3, C6N2-I, and C7N1 systems, respectively. Considering the fact that the PBE functional would generally underestimate the band gap, the screened hybrid functional of Heyd, Scuseria, and Ernzerhof (HSE),42 which typically provides more accurate band gaps compared to the PBE functional, is employed additionally. The results show that the band gaps are about 1.80, 2.00, and 1.10 eV for the C5N3, C6N2-I, and C7N1 systems, respectively. The C6N2-III phase still exhibits metallic behavior. These gaps are much smaller than those of traditional superhard materials (e.g., ∼5.60 eV of diamond, ∼6.40 eV of c-BN), and they are also smaller than ∼3.70 eV of C1N1.15 Therefore, these systems could address the gap in superhard materials with small band gaps or metallic features. Moreover, all of the systems behave as indirect band gap semiconductors, since the valence band maximum (VBM) and the conduction band minimum (CBM) are located at the different reciprocal lattice points. This case is the same as those of diamond, c-BN, and C1N1.
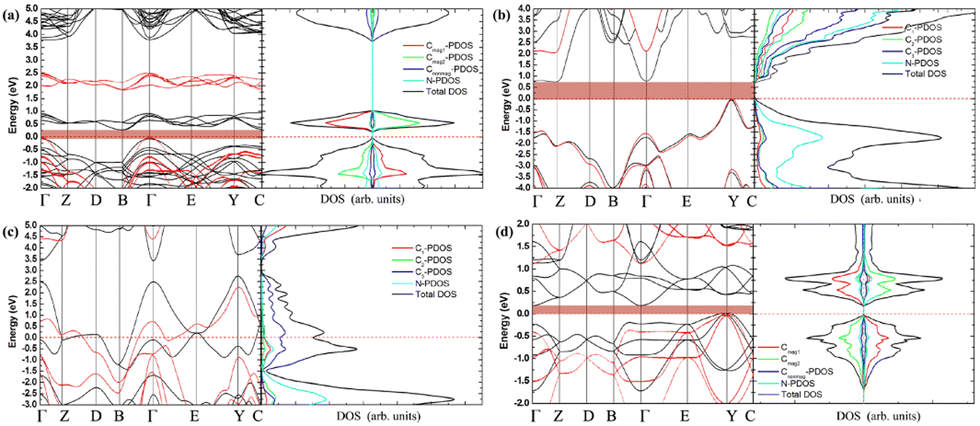 | ||
| Fig. 9 (a)–(d) The band structures, total density of electronic states, and partial density of electronic states (PDOS) for C5N3, C6N2-I, C6N2-III, and C7N1, respectively. For the C6N2-I system in (b), C1 is the C atoms coordinated by four C atoms, C2 is the C atoms coordinated by three C atoms and one N atom, C3 is the C atoms bonded to two N and two C atoms, as indicated by a green elliptic solid line in Fig. 2. For the C6N2-III system in (c), C1 represents the C atoms coordinated by four C atoms, C2 represents the C atoms coordinated by one C atom and three N atoms, C3 represents the three-fold coordinated C atoms. The black and red lines in the band structures present results given by the PBE and HSE functionals, respectively. | ||
The total DOS of C1+xN1−x systems are consistent with the band structures. There are no electronic states located at the Fermi energy level (EF) for the C5N3, C6N2-I, and C7N1 systems, and the energy gap corresponds to the values provided by the band structures. Some electronic states are situated at EF for the C6N2-III system, indicating the metallic nature as revealed by the band structure. Furthermore, the PDOS reveals that, in the same spin channel, the valence and conduction bands of both the antiferromagnetic C5N3 and C7N1 systems are mainly attributed to 2p states of the three-fold coordinated C atoms with opposite magnetic moments. For the nonmagnetic C6N2-I system, the valence and conduction bands are mainly composed of both C3 and N 2p orbitals. For the nonmagnetic C6N2-III system, the electronic states at EF are mainly composed of C3 2p orbitals.
Based on the fact that devices usually work under external pressure, therefore, it is essential to explore the effect of external pressure on the electronic structures and magnetic properties of these C1+xN1−x systems. Fig. 10(a) shows the dependence of enthalpy on the external pressure (ranging from 0 to 100 GPa) of the C1+xN1−x systems. It is clear that the AFM phase of C5N3 remains energetically stable in the whole pressure range, despite the enthalpy difference between the AFM and NM phases decreases with increasing external pressure. The AFM phase of C7N1 is energetically favored when the pressure is below ∼80 GPa, and then the NM phase becomes energetically stable. In addition, the AFM phases for both C5N3 and C7N1 systems are energetically stable than the FM ordering phases, except for the FM phase of C7N1, which becomes energetically stable when the pressure exceeds ∼85 GPa. Fig. 10(b) illustrates the relationship between the magnetic moment and the external pressure. For the AFM C5N3 system, the magnetic moment on C atoms decreases slightly from 0.42 to 0.38 μB, while the magnetic moment on the N atoms increases slightly from 0.03 to 0.09 μB. In the AFM C7N1 system, the magnetic moment on C atoms decreases significantly from 0.39 to 0.15 μB, and that on the N atoms decreases from 0.09 to 0.07 μB. The difference in the trend of the magnetic moment between the two AFM systems can be primarily attributed to their distinct structural properties. The pore in C5N3 is much larger than that in C7N1. Consequently, the overlap of the orbitals in C5N3 would be much weaker than that in C7N1 when subjected to external pressure. The greater overlap of orbitals would significantly weaken the magnetic moments, resulting in a faster decrease in magnetic moments in C7N1 compared to C5N3.
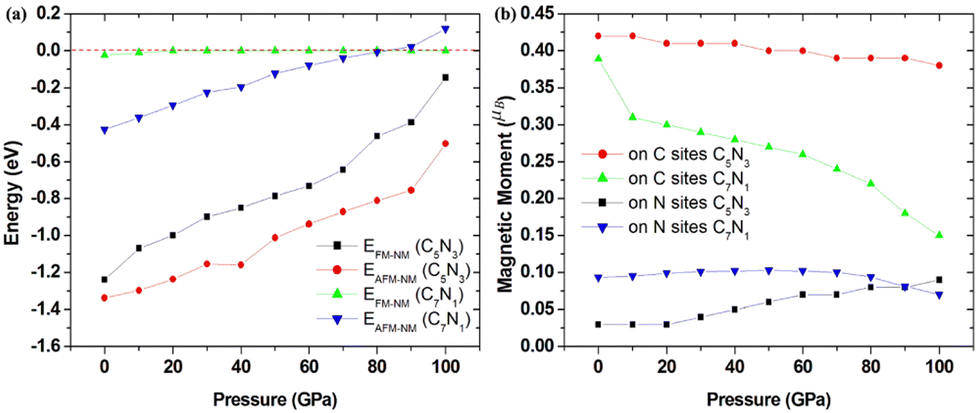 | ||
| Fig. 10 (a) The pressure-dependent enthalpy difference between the AFM/FM and NM phases for the C5N3 and C7N1 systems. (b) The pressure-dependent magnetic moment on the corresponding atomic sites for the C5N3 and C7N1 systems. | ||
Finally, the effect of external pressure on the band gaps of the three semiconducting systems is studied, and the corresponding results are shown in Fig. 11. Interestingly, the band gap of the C5N3 system increases from 0.30 to 0.50 eV (from 1.80 to 1.98 eV in the HSE level) as the pressure increases up to 50 GPa, and then the gap decreases to 0.35 eV (∼1.91 eV in HSE level) as the pressure continues to increase up to 100 GPa. For the C6N2-I system, the band gap changes slightly and remains about 0.79 eV (∼2.20 eV in HSE level), regardless of the external pressure. This changing behavior of the band gap under external pressure is scarce. It can be explained by the porous structure and superhardness of C5N3 and C6N2-I systems, which lead to weak overlap of orbitals when external pressure is applied. For the C7N1 system, the energy gap decreases from 0.20 eV to 0 eV as the pressure increases up to 50 GPa. A continuous increase in external pressure would cause the C7N1 system to transform into a metal. The results from HSE show a similar trend. This performance is attributed to the significant orbital overlap resulting from external pressure, which is facilitated by its dense structure in comparison to the C5N3 and C6N2-I systems.
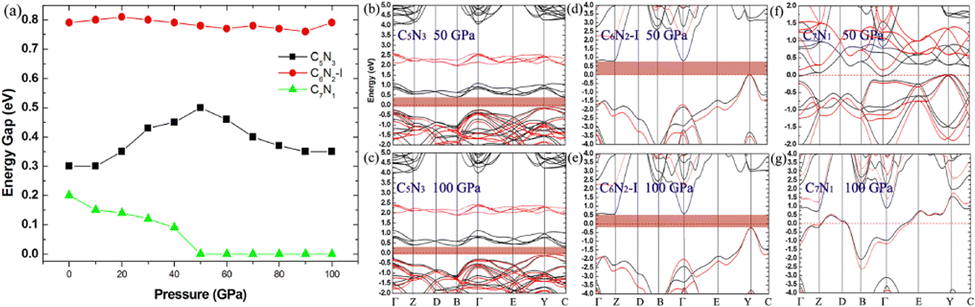 | ||
| Fig. 11 (a) The pressure-dependent energy gap of C5N3, C6N2-I, and C7N1 systems. (b)–(g) The band structures for the three systems under 50 and 100 GPa external pressures, respectively. The black and red lines in the band structures represent the results obtained from the PBE and HSE functionals, respectively. | ||
4 Conclusions
In summary, with the help of first-principles calculations, various C1+xN1−x systems have been designed by replacing different numbers of N atoms with C atoms in the synthesized Pnnm C1N1 system. Depending on the different replacement situations, they exhibit diverse physical properties, including magnetism, semiconductivity, superhardness, metallic behavior, etc. The presence of magnetism in C5N3 and C7N1 systems enriches the physical properties of superhard materials, which can expand their application into spintronic fields, etc. This case further confirms that the induction of three-fold coordinated C atoms is an effective method for synthesizing magnetic carbon-based superhard materials. For the C5N3 and C6N2-I systems, their electronic structures are slightly affected by external pressure, making them highly advantageous for applications that require resisting the adverse effects caused by external pressure. This suggests that porous materials with superhardness can retain their physical properties when subjected to external pressure, in that the strong covalent bonds can effectively resist the structural deformation caused by stress. This provides a potential approach to designing materials whose physical properties are insensitive to external pressure. The narrow band gaps of C5N3 and C7N1 systems make them suitable for unique photoelectric applications that cannot be accomplished using conventional superhard materials, particularly in the mid-far-infrared fields. This work also demonstrates that reducing the coordination number of certain constituent atoms in light element systems is feasible to design magnetic superhard materials. We hope that experimental researchers will be able to synthesize these materials in the near future.Data availability
Data are provided within the article.Author contributions
Haiping Wu: conceptualization, formal analysis, methodology, writing – review, and editing. Yunhao Zheng: investigation, computation, writing, and formal analysis. Erjun Kan: conceptualization, review, and formal analysis. Yan Qian: conceptualization, methodology, formal analysis, and review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 12274226).Notes and references
- J. Haines, J. M. Leger and G. Bocquillon, Annu. Rev. Mater. Res., 2001, 31, 1–23 CrossRef CAS.
- B. Xu and Y. Tian, Sci. China: Mater., 2015, 58, 132–142 CAS.
- R. B. Kaner, J. J. Gilman and S. H. Tolbert, Science, 2005, 308, 1268–1269 CrossRef CAS PubMed.
- A. Krauss, O. Auciello, D. Gruena, A. Jayatissa, A. Sumant, J. Tucek, D. Mancini, N. Moldovan, A. Erdemir, D. Ersoy, M. Gardos, H. Busmann, E. Meyer and M. Ding, Diamond Relat. Mater., 2001, 10, 1952 CrossRef CAS.
- R. Wentorf, R. DeVries and F. Bundy, Science, 1980, 208, 873 CrossRef CAS PubMed.
- Z. Cui, H. Wu, K. Yang, X. Wang and Y. Lv, Sens. Actuators, A, 2024, 366, 114954 CrossRef CAS.
- Z. Cui, K. Yang, Y. Shen, Z. Yuan, Y. Dong, P. Yuan and E. Li, Appl. Surf. Sci., 2023, 613, 155978 CrossRef CAS.
- D. Laniel, F. Trybel, A. Aslandukov, S. Khandarkhaeva, T. Fedotenko, Y. Yin, N. Miyajima, F. Tasnadi, A. V. Ponomareva, N. Jena, F. I. Akbar, B. Winkler, A. Neri, S. Chariton, V. Prakapenka, V. Milman, W. Schnick, A. N. Rudenko, M. I. Katsnelson, I. A. Abrikosov, L. Dubrovinsky and N. Dubrovinskaia, Adv. Mater., 2023, 2308030 Search PubMed.
- A. Y. Liu and M. L. Cohen, Science, 1989, 245, 841–842 CrossRef CAS PubMed.
- D. M. Teter and R. J. Hemley, Science, 1996, 271, 53–55 CrossRef CAS.
- A. Y. Liu and M. L. Cohen, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 10727 CrossRef CAS PubMed.
- A. Y. Liu and R. M. Wentzcovitch, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 10362 CrossRef CAS PubMed.
- C. M. Niu, Y. Z. Lu and C. M. Lieber, Science, 1993, 261, 334 CrossRef CAS PubMed.
- J. He, L. Guo, X. Guo, R. Liu, Y. Tian, H. Wang and C. Gao, Appl. Phys. Lett., 2006, 88, 101906 CrossRef.
- X. Wang, J. Chem. Phys., 2012, 138, 184506 CrossRef PubMed.
- F. Tian, J. Wang, Z. He, Y. Ma, L. Wang, T. Cui, C. Chen, B. Liu and G. Zou, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 235431 CrossRef.
- Z. Li, K. Luo, B. Liu, L. Sun, P. Ying, C. Liu, W. Hu and J. He, Carbon, 2021, 184, 846–854 CrossRef CAS.
- J. Shao, Y. Qian, E. Kan and H. Wu, J. Phys. D: Appl. Phys., 2023, 56, 205303 CrossRef.
- H. Wu, Y. Li, Y. Qian and E. Kan, Phys. Chem. Chem. Phys., 2023, 25, 21408 RSC.
- E. Stavrou, S. Lobanov, H. Dong, A. R. Oganov, V. B. Prakapenka, Z. Konopkova and A. F. Goncharov, Chem. Mater., 2016, 28, 6925–6933 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- S. Baroni, S. Gironcoli, A. Corso and P. Giannozzi, Rev. Mod. Phys., 2001, 73, 515 CrossRef CAS.
- K. Parlinski, Z. Li and Y. Kawazoe, Phys. Rev. Lett., 1997, 78, 4063 CrossRef CAS.
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef.
- T. J. Volz and Y. M. Gupta, Phys. Rev. B, 2021, 103, L100101 CrossRef CAS.
- J.-T. Wang, C. Chen and Y. Kawazoe, Phys. Rev. Lett., 2011, 106, 075501 CrossRef PubMed.
- E. Gregoryanz, C. Sanloup, M. Somayazulu, J. Badro, G. Fiquet, H.-K. Mao and R. J. Hemley, Nat. Mater., 2004, 3, 294–297 CrossRef CAS PubMed.
- J. C. Crowhurst, A. Goncharov, B. Sadigh, C. Evans, P. Morrall, J. Ferreira and A. Nelson, Science, 2006, 311, 1275–1278 CrossRef CAS PubMed.
- Y. Yuan, T. Wang, H. Chen, S. M. Mahurin, H. Luo, G. Veith, Z. Yang and S. Dai, Angew. Chem., Int. Ed., 2020, 59, 21935–21939 CrossRef CAS PubMed.
- P. Kurz, G. Bihlmayer and S. Blugel, J. Phys: Condens. Matter, 2002, 14, 6353 CrossRef CAS.
- M. Grimsditch and A. Ramdas, Phys. Rev. B: Condens. Matter Mater. Phys., 1975, 11, 3139 CrossRef CAS.
- R. A. Andrievski, Int. J. Refract. Met. Hard Mater., 2001, 19, 447 CrossRef CAS.
- H. Richard, Proc. Phys. Soc. A, 1952, 65, 349 CrossRef.
- S. Pugh, Philos. Mag. A, 1954, 45, 823 CrossRef CAS.
- X. Chen, H. Niu, D. Li and Y. Li, Intermetallics, 2011, 19, 1275 CrossRef CAS.
- E. Mazhnik and A. Oganov, J. Appl. Phys., 2019, 126, 125109 CrossRef.
- X. Wang, K. Bao, F. Tian, X. Meng, C. Chen, B. Dong, D. Li, B. Liu and T. Cui, J. Chem. Phys., 2010, 133, 044512 CrossRef PubMed.
- J. Du and X. Li, J. Alloys Compd., 2020, 815, 152324 CrossRef CAS.
- D. Cahill, S. Watson and R. Pohl, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6131–6140 CrossRef CAS PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207 CrossRef CAS.
Footnote |
| † Haiping Wu and Yunhao Zheng contributed equally to this work. |
| This journal is © the Owner Societies 2024 |