
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hypervalent zinc(I) complexes with an NNNN-macrocycle: C–H bond activation across the zinc(I)–zinc(I) bond†
Pritam
Mahawar
a,
Thayalan
Rajeshkumar
b,
Thomas P.
Spaniol
a,
Laurent
Maron
 b and
Jun
Okuda
*a
b and
Jun
Okuda
*a
aInstitute of Inorganic Chemistry, RWTH Aachen University, Landoltweg 1, 52056 Aachen, Germany. E-mail: jun.okuda@ac.rwth-aachen.de
bCNRS, INSA, UPS, UMR 5215, LPCNO, Université de Toulouse, 135 Avenue de Rangueil, 31077 Toulouse, France. E-mail: laurent.maron@irsamc.ups-tlse.fr
First published on 11th September 2024
Abstract
Hetero- and homoleptic dinuclear zinc(I) complexes containing the macrocycle Me4TACD (N,N′,N′′,N′′′-1,4,7,10-tetramethylcyclododecane) were prepared; the heteroleptic complex [(Me4TACD)Zn–ZnCp*]+ reacted with activated hydrocarbons R–H (R = CH2CN, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh) to give the corresponding hydrocarbyl zinc(II) complexes [(Me4TACD)ZnR]+.
CPh) to give the corresponding hydrocarbyl zinc(II) complexes [(Me4TACD)ZnR]+.
The discovery of decamethyldizincocene Cp*Zn–ZnCp* (Cp* = η5-C5Me5) by Carmona et al. in 20041 has prompted the isolation of other complexes featuring the remarkable zinc(I)–zinc(I) σ-bond. On the one hand, neutral zinc(I) analogs containing a LlX-type ligand (L = two-electron, X = one-electron ligand)2a such as bulky aryl (l = 0)3 and β-diketiminato (l = 1)4 became known, on the other hand protonolysis or oxidation of Cp*Zn–ZnCp* allowed the synthesis of mono(cations) of the type [((L3)Zn–ZnCp*)]+ (ref. 5) or dicationic complexes [(L3)Zn–Zn(L3)]2+(L = THF, DMAP).6 Regarding zinc as a main group element with filled 3d10 shell,2b the valence electron count of zinc in all these complexes does not exceed 8 electrons.2,7 Recently, we have reported that the heteroleptic zinc(I) cation [(TEEDA)(thf)Zn–ZnCp*]+[BAr4F]− (TEEDA = N,N,N′,N′-tetraethylethylenediamine; ArF = 3,5-((CF3)2C6H3)) can undergo a heterolytic dihydrogen cleavage.8 As recently suggested for the reactivity of diberyllocene,9 main group metal–metal bonds can be polarized, so for decamethyldizincocene a resonance structure [Cp*Zn(II)]+←[Zn(0)Cp*]− can be implied, accounting for some of the reactivity patterns (redox disproportionation) observed.10 We wondered whether introducing hypervalency7 at the zinc(I) center (with formal valence electron count higher than 8) would result in a higher reactivity of the zinc(I)–zinc(I) bond. Here we report on the preparation of both homo- and heteroleptic zinc(I) cations that contain the L4-type macrocycle Me4TACD (N,N′,N′′,N′′′-1,4,7,10-tetramethylcyclododecane).11 The heteroleptic zinc(I) cation was found to undergo a heterolytic C–H bond activation of acetonitrile and phenylacetylene.10,12
The versatile macrocyclic ligand Me4TACD is capable of coordinating s-,13 and p-block14 metal cations including low-valent triele cations Ga(I), In(I) and Tl(I). Thus, stoichiometric reaction of Cp*Zn–ZnCp* with the borate salt of the protonated Me4TACD [(Me4TACD)H][BAr4Me]15 (BAr4Me = [B{3,5-(CH3)2-C6H3}4]−) in THF at room temperature for one hour afforded the heteroleptic zinc(I) monocation [(Me4TACD)Zn–ZnCp*][BAr4Me] (1) in 90% yield with the elimination of one equivalent of Cp*H. Colorless compound 1 is stable under argon at room temperature and is soluble in THF, acetonitrile, and dichloromethane (Scheme 1).
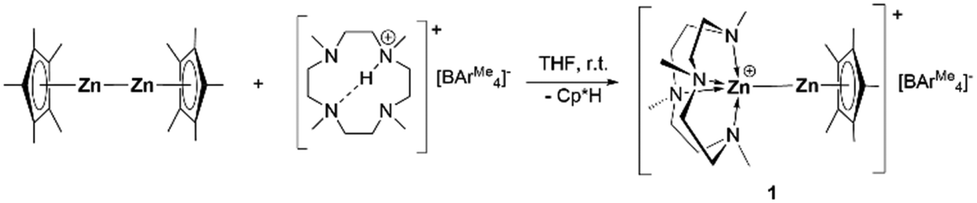 | ||
| Scheme 1 Synthesis of [(Me4TACD)Zn–ZnCp*][BAr4Me] (1). | ||
Compound 1 was characterized in solution using multinuclear NMR spectroscopy, including 1H, 13C, and 11B, and in the solid state using single crystal X-ray diffraction. The 1H NMR spectra indicate η5-Cp* coordination, displaying a characteristic single peak for all methyl groups of Cp* at δ 2.02 ppm and confirmed the ligand/borate ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The diastereotopic CH2CH2 protons of the Me4TACD ligand appear as multiplets of AA′BB′ spin system in the range of δ 2.14–2.31 ppm, as commonly observed for the coordinated Me4TACD ligand.14 The 13C{1H} NMR spectrum revealed two signals for the Cp* methyl and ring carbons at δ 10.5 and δ 108.4 ppm, respectively, along with the peaks for the Me4TACD ligand and borate counter-ion. Compound 1 crystallizes in the monoclinic space group P21/n with one ion pair per asymmetric unit. The structure of the molecular cation is depicted in Fig. 1 (see ESI† for details). The cationic penta-coordinate zinc center is positioned above the N4-basal plane of the Me4TACD ligand with the Zn–N(centroid) bond distance of 0.9416(6) Å, which is consistent with the Zn–N(centroid) bond distance (0.9371(15) Å) in the zinc(II) hydride cation [(Me4TACD)ZnH][HBPh3].16 The zinc–zinc distance of 2.3510(3) Å is marginally longer than the reported value for heteroleptic Zn(I) monocations [(Et2O)3Zn–ZnCp*][BAr4F] (2.324(2) Å)5 and [(TEEDA)Zn–ZnCp*][BAr4F] (2.3253(15) Å).8 The enhanced polarization effect caused by the increased coordination number and asymmetrical ligand environment causes the zinc–zinc bond distance to be longer than in Cp*Zn–ZnCp* with 2.302(1) Å.1
:1. The diastereotopic CH2CH2 protons of the Me4TACD ligand appear as multiplets of AA′BB′ spin system in the range of δ 2.14–2.31 ppm, as commonly observed for the coordinated Me4TACD ligand.14 The 13C{1H} NMR spectrum revealed two signals for the Cp* methyl and ring carbons at δ 10.5 and δ 108.4 ppm, respectively, along with the peaks for the Me4TACD ligand and borate counter-ion. Compound 1 crystallizes in the monoclinic space group P21/n with one ion pair per asymmetric unit. The structure of the molecular cation is depicted in Fig. 1 (see ESI† for details). The cationic penta-coordinate zinc center is positioned above the N4-basal plane of the Me4TACD ligand with the Zn–N(centroid) bond distance of 0.9416(6) Å, which is consistent with the Zn–N(centroid) bond distance (0.9371(15) Å) in the zinc(II) hydride cation [(Me4TACD)ZnH][HBPh3].16 The zinc–zinc distance of 2.3510(3) Å is marginally longer than the reported value for heteroleptic Zn(I) monocations [(Et2O)3Zn–ZnCp*][BAr4F] (2.324(2) Å)5 and [(TEEDA)Zn–ZnCp*][BAr4F] (2.3253(15) Å).8 The enhanced polarization effect caused by the increased coordination number and asymmetrical ligand environment causes the zinc–zinc bond distance to be longer than in Cp*Zn–ZnCp* with 2.302(1) Å.1
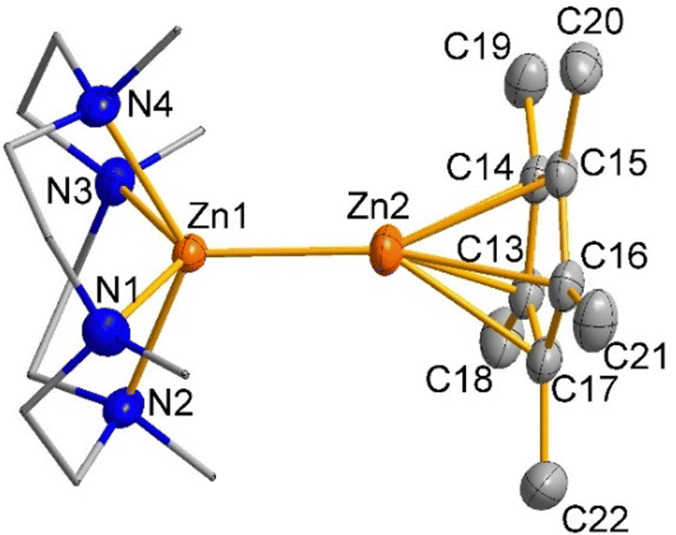 | ||
| Fig. 1 Cationic part of the molecular structure of compound 1. Selected interatomic distances [Å] and angles [°]: Zn1–Zn2 2.3510(3), Zn1–N1 2.2388(17), Zn1–N2 2.2177(15), Zn1–N3 2.2550(15), Zn1–N4 2.2181(15), Zn1–C13 2.3484(18), Zn1–C14 2.3168(18), Zn1–C15 2.2779(17), Zn1–C16 2.2889(18), Zn1–C17 2.3275(18), N1–Zn1–N2 80.40(6), N1–Zn1–N3 130.33(6), N1–Zn1–N4 79.98(6), N1–Zn1–Zn2 115.79(4), N2–Zn1–Zn2 112.12(4), N3–Zn1–Zn2 113.83(4), N4–Zn1–Zn2 117.99(4). | ||
Compound 1 shows a slight slipping of the Cp* ring from η5-coordination to Zn1, with the metal atom's projection displaced from the ring's centroid by 0.072 Å. The Zn1–Cp*(centroid) distance (1.971 Å) lies within the range of the Zn1–Cp*(centroid) bond distances in heteroleptic zinc(I) complexes [(TEEDA)Zn–ZnCp*][BAr4F] (1.954 Å)8 and [(HC{C(Me)NDipp}2)Zn–ZnCp*] (1.9215(3) Å).17 This slipped coordination results in the nonlinear alignment of the Zn2–Zn1–Cp*(centroid) bond angle of 164.75(1)° in 1.
Reactivity studies of zinc(I) complexes toward activated hydrocarbons are scarce,10 although reactions with phenylacetylene have been studied.10a–c While the zinc(I) cation 1 is kinetically robust in acetonitrile for at least 12 h, in the presence of 10 mol% of Me4TACD, the formation of a grey precipitate was observed within 5 min and zinc(II) cyanomethanide [(Me4TACD)Zn(CH2CN)][BAr4Me] (2) was isolated from the supernatant in 85% yield (Scheme 2). Formation of 2 can be interpreted as a product of oxidative C–H bond addition across the Zn–Zn bond of 1, presumably also forming unstable [Cp*ZnH],18 which is known to decompose via reductive elimination to form the observed byproducts Cp*H and metallic zinc. Compound 2 can also be synthesized in THF using 2 equivalents of acetonitrile in the presence of 10 mol% of Me4TACD. Likewise, in the presence of 10 mol% of Me4TACD, the reaction of compound 1 with phenylacetylene in THF at room temperature gave the zinc(II) acetylide complex [(Me4TACD)ZnCCPh][BAr4Me] (3) in 90% isolated yield (Scheme 2). The precise role of Me4TACD is unclear, it may act as a Brønsted base in these reactions. Compounds 2 and 3 are soluble in THF, acetonitrile, and dichloromethane and are stable at room temperature under argon. Compounds 2 and 3 were characterized using multinuclear NMR spectroscopy (1H, 13C, 11B) in the solution state. The solid-state characterization was performed using single-crystal XRD and IR spectroscopy. In the 1H and 13C{1H} NMR spectra of compound 2 the characteristic peaks for the CH2CN protons appear at δ 0.52 ppm and δ −14.3 ppm, respectively. For the acetylide compound 3 the 13C{1H} NMR spectrum shows the characteristic peaks for the acetylenic carbon atoms at δ 109.0 and 107.8 ppm. All peaks of the Me4TACD ligands in compounds 2 and 3 are downfield shifted compared to those of 1, due to the increase in oxidation number of zinc from +1 in compound 1 to +2 in compounds 2 and 3.
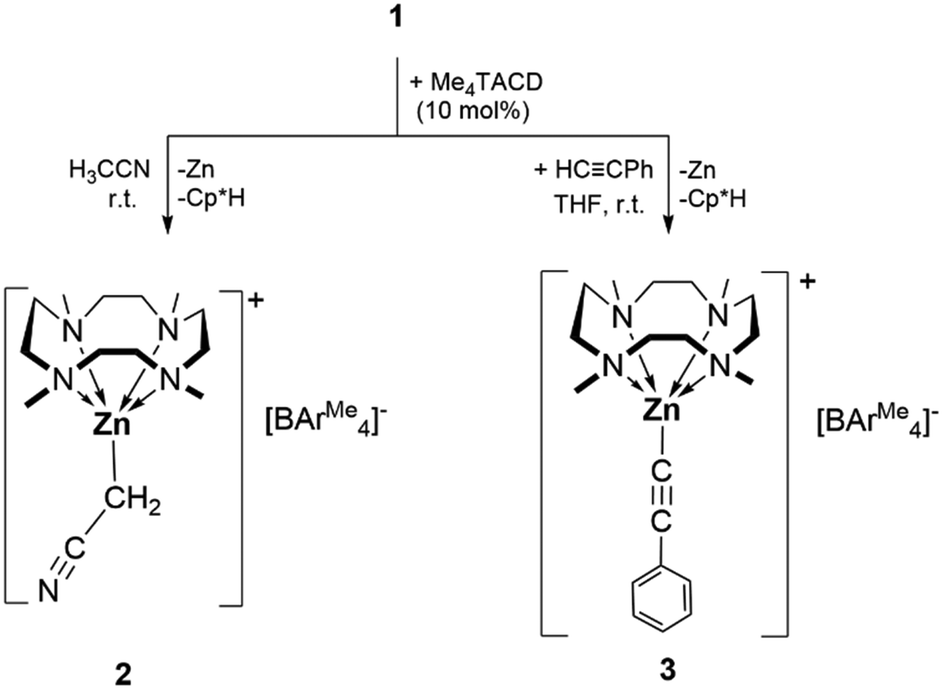 | ||
| Scheme 2 Reaction of [Me4TACDZn–ZnCp*][BAr4Me] (1) with activated hydrocarbons. | ||
Single crystal X-ray diffraction studies revealed the monomeric structure of compounds 2 and 3 (see ESI†). The coordination of the Me4TACD ligand at the cationic zinc(II) center in compounds 2 and 3 is comparatively stronger than in precursor 1 with zinc(I) cation which can be seen by the decrease in the Zn–N(centroid) bond distance (0.8856(16) Å for 2 and 0. 8776(9) Å for 3) from 0.9416(6) (for 1). The Zn–CH2 bond distance in compound 2 of 2.025(3) Å is comparable to the Zn–CH2 bond length in the pyrazolylborate- ([(TpPh,Me)Zn(CH2CN)]; TpPh,Me = hydrotris((5,3-methylphenyl-pyrazolyl)borate) 2.052(3) Å)19a and PMDTA-supported zinc cyanomethanide and (PMDTA = N,N,N′,N′′,N′′-pentamethyldiethylenetriamine, 1.991(6) Å) reported.19b The CC bond length in 3 (1.207(3) Å) is longer than that in free phenylacetylene (1.183(2) Å) due to the donation of π-electron to zinc vacant orbitals. The Zn–C bond length in compound 3 (1.9519(17) Å) lies within the range observed for the monomeric [{(dipp)NacNac}ZnCCPh] (1.906(2) Å) ((dipp)NacNac = 2-{(2,6-diisopropyl-phenyl)amino}-4-{(2,6-diisopropylphenyl)imino}pent-2-enyl).20 In the IR spectrum of compound 2 a stretching band at ν(CN) = 2193 cm−1 indicates the presence of a terminal CN group.
In analogy to the synthesis of 1 through protonation of one of the Cp* ligands in Cp*Zn–ZnCp*, we attempted to protonate both Cp* ligands by reacting with 2 equivalents of the acid [(Me4TACD)H][BAr4Me]. This only led to the formation of zinc(I) cation 1 along with unreacted acid. Recently, we reported that the zinc–zinc bond of zinc(I) cation [(TEEDA)Zn–ZnCp*][BAr4F] can cleave dihydrogen in a heterolysis, similar to a frustrated Lewis acid–base type activation.8 When the reaction of compound 1 was carried out with a large excess (5 equivalents) of HBpin in acetonitrile at 60 °C for 3 days, the zinc(I)–zinc(I) dication [(Me4TACD)Zn–Zn(Me4TACD)][BAr4Me]2 (4) was formed along with Cp*H, zinc metal, and B2pin2 (Scheme 3). Compound 4 was isolated in 30% yield (based on (Me4TACD)Zn) and characterized in the solution state using multinuclear NMR spectroscopy and in the solid state using single crystal XRD studies.
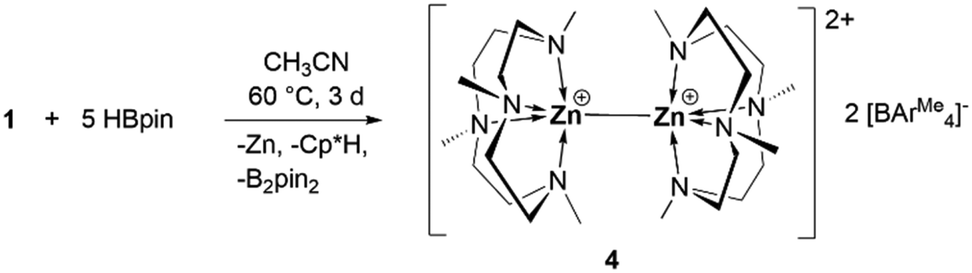 | ||
| Scheme 3 Formation of [(Me4TACD)Zn–Zn(Me4TACD)][BAr4Me]2 (4). | ||
In the 1H NMR spectrum of compound 4, all the Me4TACD protons (δ 2.33–2.51 ppm (CH2) and δ 2.20 ppm (CH3)) are deshielded compared to those in 1 (δ 2.14–2.31 ppm (CH2) and δ 2.11 ppm (CH3)) due to the increase in the cationic charge of the zinc centers. 13C NMR spectra show all the corresponding signals for the Me4TACD ligand and borate counterion. Compound 4 crystallizes in the orthorhombic space group Pbca with one ion pair in the asymmetric unit. The dinuclear structure of 4 with a zinc(I)–zinc(I) distance of 2.4860(6) Å was confirmed using single crystal XRD diffraction (Fig. 2a). Due to the higher coordination number in 4, the zinc–zinc bond distance is significantly longer than the zinc–zinc bond in the reported dications of the type [Zn2(L6)]2+ [Zn2(dmap)6][Al{OC(CF3)3}4]2 (2.419(2) Å)6a and [Zn2(thf)6][BAr4F]2 (2.363(2) Å).6b As can be seen from the space-filling model (Fig. 2b), the two 9-electron [Zn(Me4TACD)]+ units are closely meshed and the two Me4TACD ligands adopt a staggered conformation (Fig. 2c). Notably, both ligands show δδδδ or λλλλ conformation of the CH2CH2 units and the overall molecular symmetry of the homochiral dimer corresponds to the rare pointgroup D4.
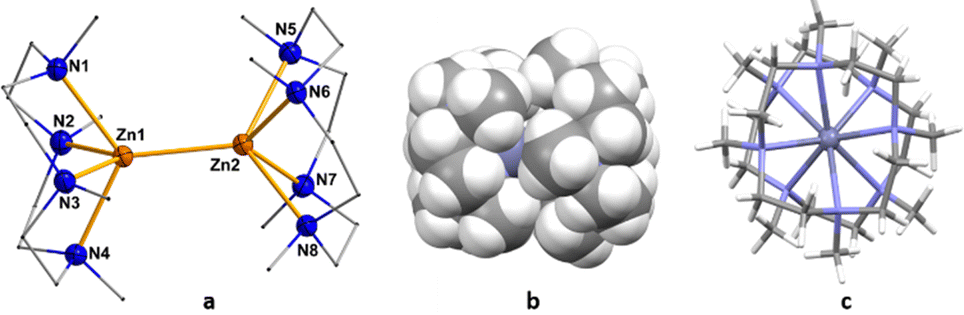 | ||
| Fig. 2 (a) Left: Cationic part of the molecular structure of compound 4. The anion part [BAr4Me] and all H atoms are omitted for clarity. Displacement parameters are shown at 30% probability; selected interatomic distances [Å] and angles [°]: Zn1–N1 2.378(3), Zn1–N2 2.337(3), Zn1–N3 2.390(3), Zn1–N4 2.326(3), Zn1–Zn2 2.4860(6), Zn2–N5 2.480(3), Zn2–N6 2.291(3), Zn2–N7 2.463(3), Zn2–N8 2.278(3); N1–Zn1–N2 76.31(11), N1–Zn1–N3 120.82(11), N1–Zn1–N4 75.92(11), N1–Zn1–Zn2 120.99(8), N2–Zn1–Zn2 119.19(8), N3–Zn1– Zn2 118.18(8), N4–Zn1–Zn2 119.86(8). (b) Middle: Space filling model of 4. (c) Right: View of 4 along the Zn–Zn axis, highlighting the D4 symmetry. | ||
To provide further insight into the bonding in compounds 1 and 4, DFT calculations were performed at the B3PW91 level of theory. Gas phase optimized structure agrees well with the experimentally determined structures of 1 and 4 from X-ray diffraction studies. The LUMO is mainly located on the Me4TACD ligand in both compounds. The zinc–zinc bond in compound 1 constitutes the HOMO−2, while the HOMO is localized on Zn–Cp* bond. In contrast, the HOMO is mainly localized on the zinc–zinc bond in compound 4 (Fig. 3). This is consistent with the apparent longer zinc–zinc bond in compound 4 (2.4860(6) Å) compared to 1 (2.3510(3) Å). Moreover, the presence of HOMO contribution in Zn–Cp* moiety goes in line with its reaction with the acidic proton of CH3CN and HCCPh for the formation of HZnCp*.
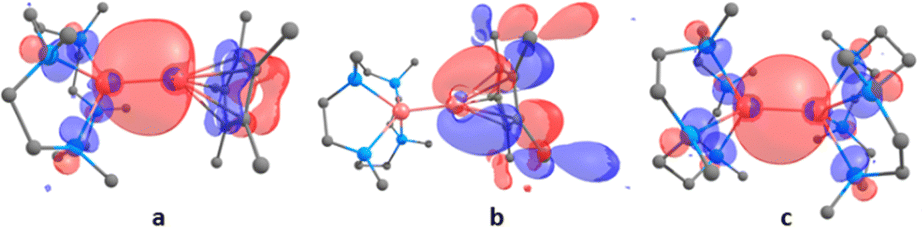 | ||
| Fig. 3 (a) HOMO−2 for compound 1. (b) HOMO for compound 1. (c) HOMO for compound 4. | ||
The mechanism for the formation of 4 remains obscure. It seems plausible that HBpin may act as a hydride transfer reagent to provide short-lived zinc hydride and boryl species as intermediates during the formation of 4 with elimination of Zn, Cp*H, and B2Pin2. We have previously reported somewhat unstable Zn(I) hydridoborate species [(TEEDA)Zn(HBPh3)–ZnCp*].16 The formation of zinc(II) hydrides from HBpin was reported by Ingleson et al.21 However, the zinc(II) hydride cation [(Me4TACD)ZnH]+, previously isolated as [(Me4TACD)ZnH][HBPh3]16 is stable with respect to dehydrocoupling. Xu et al. reported that the dehydrocoupling of zinc(II) hydride with a tridentate L2X-type ligand forms the zinc(I)–zinc(I) bonded complex, but the reaction requires the presence of catalytic [Ni(CO)2(PPh3)2] or stoichiometric [Pd(PPh3)4].22 At this point, however, we cannot exclude other mechanistic pathways for the formation of 4, including radical intermediates.22d,23
In conclusion, we have prepared hypervalent zinc(I) complexes that contain the L4-type macrocycle Me4TACD 1 and 4. While the heteroleptic complex 1 can be accessed by protonolysis of dizincocene Cp*Zn–ZnCp* using the conjugate acid of Me4TACD, the homoleptic complex 4 was only obtained by the treatment of 1 with the hydride reagent HBpin in a somewhat complicated reaction. The reaction of 1 with activated hydrocarbons acetonitrile (pKa = 25) and phenylacetylene (pKa = 29) suggests that C–H bond cleavage by the dinuclear zinc(I)–zinc(I) complexes can occur by a polarized zinc(I)–zinc(I) bond, possibly in the presence of a Brønsted base. While C–H bond activation has been reported for d-block transition metals,24 zinc(I) complexes appear to show similar reactivity with relevance to C–H bond functionalization.10
We thank Dr Louis J. Morris for helpful discussions.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) I. Resa, E. Carmona, E. Gutierrez-Puebla and A. Monge, Science, 2004, 305, 1136–1138 CrossRef PubMed; (b) A. Grirrane, I. Resa, A. Rodriguez, E. Carmona, E. Alvarez, E. Gutierrez-Puebla, A. Monge, A. Galindo, D. del Rio and R. A. Andersen, J. Am. Chem. Soc., 2006, 129, 693–703 Search PubMed.
- (a) M. L. H. Green and G. Parkin, J. Chem. Educ., 2014, 91, 807–816 Search PubMed; (b) J. B. Jensen, J. Chem. Educ., 2003, 80, 952–961 Search PubMed.
- Z. Zhu, M. Brynda, R. J. Wright, R. C. Fischer, W. A. Merrill, E. Rivard, R. Wolf, J. C. Fettinger, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 2007, 129, 10847–10857 CrossRef PubMed.
- Y. Wang, B. Quillian, P. Wei, H. Wang, X.-J. Yang, Y. Xie, R. B. King, P. V. R. Schleyer, H. F. Schaefer and G. H. Robinson, J. Am. Chem. Soc., 2005, 127, 11944–11945 CrossRef PubMed.
- K. Freitag, H. Banh, C. Ganesamoorthy, C. Gemel, R. W. Seidelb and R. A. Fischer, Dalton Trans., 2013, 42, 10540–10544 RSC.
- (a) S. Schulz, D. Schuchmann, I. Krossing, D. Himmel, D. Bläser and R. Boese, Angew. Chem., Int. Ed., 2009, 48, 5748–5751 CrossRef PubMed; (b) H. Banh, C. Gemel, R. W. Seidelb and R. A. Fischer, Chem. Commun., 2015, 51, 2170–2172 RSC.
- (a) M. L. H. Green and G. Parkin, Dalton Trans., 2016, 45, 18784–18795 RSC; (b) R. H. Crabtree, Chem. Soc. Rev., 2017, 46, 1720–1729 RSC.
- P. Mahawar, T. Rajeshkumar, T. P. Spaniol, L. Maron and J. Okuda, Inorg. Chem., 2024, 63, 8493–9501 CrossRef CAS PubMed.
- J. T. Boronski, A. E. Crumpton, A. F. Roper and S. Aldridge, Nat. Chem., 2024, 16, 1295–1300 CrossRef CAS PubMed.
- (a) I. L. Fedushkin, O. V. Eremenko, A. A. Skatova, A. V. Piskunov, G. K. Fukin, S. Y. Ketkov, E. Irran and H. Schumann, Organometallics, 2009, 28, 3863–3868 CrossRef CAS; (b) R. K. Sahoo, S. Rajput, A. G. Patro and S. Nembenna, Dalton Trans., 2022, 51, 16009–16016 RSC; (c) R. K. Sahoo, A. G. Patro, N. Sarkar and S. Nembenna, ACS Omega, 2023, 8, 3452–3460 CrossRef CAS PubMed; (d) T. Li, S. Schulz and P. W. Roesky, Chem. Soc. Rev., 2012, 41, 3759–3771 RSC.
- J. H. Coates, D. A. Hadi and S. F. Lincoln, Aust. J. Chem., 1982, 35, 903–909 CrossRef CAS.
- (a) F.-G. Fontaine and V. Desrosiers, Synthesis, 2021, 4599–4613 CrossRef CAS; (b) C. Jiang, O. Blacque and H. Berke, Organometallics, 2010, 29, 125–133 CrossRef CAS; (c) J. Guo, Ma Yan and D. W. Stephan, Org. Chem. Front., 2024, 11, 2375–2396 RSC.
- (a) J. Dyke, W. Levason, M. E. Light, D. Pugh, G. Reid, H. Bhakhoa, P. Ramasami and L. Rhyman, Dalton Trans., 2015, 44, 13853–13866 RSC; (b) H. Bhakhoa, L. Rhyman, E. P. Lee, D. K. W. Mok, P. Ramasami and J. M. Dyke, Dalton Trans., 2017, 46, 15301–15310 RSC; (c) P. Ghana and J. Okuda, Bull. Jpn. Soc. Coord. Chem., 2021, 77, 37–45 CrossRef.
- L. J. Morris, P. Ghana, T. Rajeshkumar, A. Carpentier, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2022, 61, e202114629 CrossRef CAS PubMed.
- D. Schuhknecht, T. P. Spaniol, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2020, 59, 310–314 CrossRef CAS PubMed.
- F. Ritter, L. J. Morris, K. N. McCabe, T. P. Spaniol, L. Maron and J. Okuda, Inorg. Chem., 2021, 60, 15583–15592 CrossRef CAS PubMed.
- B. Li, K. Huse, C. Wölper and S. Schulz, Chem. Commun., 2021, 57, 13692–13695 RSC.
- (a) P. Jochmann and D. W. Stephan, Angew. Chem., Int. Ed., 2013, 52, 9831–9835 CrossRef CAS PubMed; (b) P. Jochmann and D. W. Stephan, Chem. – Eur. J., 2014, 20, 8370–8378 CrossRef CAS PubMed.
- (a) H. Brombacher and H. Vahrenkamp, Inorg. Chem., 2004, 43, 6054–6060 CrossRef CAS PubMed; (b) P. Mahawar, T. Rajeshkumar, T. P. Spaniol, L. Maron and J. Okuda, Chem. – Eur. J., 2024, 30, e202401262 CrossRef CAS PubMed.
- J. Prust, H. Hohmeister, A. Stasch, H. W. Roesky, J. Magull, E. Alexopoulos, I. Usón, H.-G. Schmidt and M. Noltemeyer, Eur. J. Inorg. Chem., 2002, 2156–2162 CrossRef CAS.
- M. Uzelac, K. Yuan, G. S. Nichol and M. L. J. Ingleson, Dalton Trans., 2021, 50, 14018–14026 RSC.
- (a) S. Jiang, Y. Cai, A. Carpentier, I. del Rosal, L. Maron and X. Xu, Chem. Commun., 2021, 57, 13696–13699 RSC; (b) M. Chen, S. Jiang, L. Maron and X. Xu, Dalton Trans., 2019, 48, 1931–1935 RSC; (c) S. Jiang, M. Chen and X. Xu, Inorg. Chem., 2019, 58, 13213–13220 CrossRef CAS PubMed; (d) S. Xu, Q. Wang, T. Rajeshkumar, S. Jiang, L. Maron and X. Xu, J. Am. Chem. Soc., 2024, 146, 19590–19598 CrossRef CAS PubMed.
- Y. L. Phang, J.-K. Jin, F.-L. Zhang and Y.-F. Wang, Chem. Commun., 2024, 60, 4275–4289 RSC.
- (a) T. Tanabe, M. E. Evans, W. W. Brennessel and W. D. Jones, Organometallics, 2011, 30, 834–843 CrossRef CAS; (b) Y.-H. Chang, K. Takeuchi, M. Wakioka and F. Ozawa, Organometallics, 2015, 34, 1957–1962 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, analytical (NMR, IR spectra, elemental analysis) and crystallographic data of 1–4. CCDC 2378377 (1), 2378378 (2), 2378379 (3) and 2378380 (4). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc04266b |
| This journal is © The Royal Society of Chemistry 2024 |