
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and catalytic application of nanostructured metal oxides and phosphates
Keigo
Kamata
 *,
Takeshi
Aihara
and
Keiju
Wachi
*,
Takeshi
Aihara
and
Keiju
Wachi
Laboratory for Materials and Structures, Institute of Innovative Research, Tokyo Institute of Technology, Nagatsuta-cho 4259-R3-6, Midori-ku, Yokohama-city, Kanagawa 226-8501, Japan. E-mail: kamata.k.ac@m.titech.ac.jp
First published on 16th September 2024
Abstract
The design and development of new high-performance catalysts is one of the most important and challenging issues to achieve sustainable chemical and energy production. This Feature Article describes the synthesis of nanostructured metal oxides and phosphates mainly based on earth-abundant metals and their thermocatalytic application to selective oxidation and acid–base reactions. A simple and versatile methodology for the control of nanostructures based on crystalline complex oxides and phosphates with diverse structures and compositions is proposed as another approach to catalyst design. Herein, two unique and verstile methods for the synthesis of metal oxide and phosphate nanostructures are introduced; an amino acid-aided method for metal oxides and phosphates and a precursor crystallization method for porous manganese oxides. Nanomaterials based on perovskite oxides, manganese oxides, and metal phosphates can function as effective heterogeneous catalysts for selective aerobic oxidation, biomass conversion, direct methane conversion, one-pot synthesis, acid–base reactions, and water electrolysis. Furthermore, the structure–activity relationship is clarified based on experimental and computational approaches, and the influence of oxygen vacancy formation, concerted activation of molecules, and the redox/acid–base properties of the outermost surface are discussed. The proposed methodology for nanostructure control would be useful not only for the design and understanding of the complexity of metal oxide catalysts, but also for the development of innovative catalysts.
 Keigo Kamata | Keigo Kamata received his PhD degree from the Department of Applied Chemistry, University of Tokyo in 2006 under the supervision of Prof. Noritaka Mizuno. In 2007, he joined the University of Tokyo as a research associate in the Department of Applied Chemistry, and moved to the Materials and Structures Laboratory at Tokyo Institute of Technology as an assistant professor in 2014. Now, he is a full professor in the same institute. His research interests include the design and synthesis of advanced inorganic catalyst materials and the construction of highly atom-efficient catalytic systems. |
 Takeshi Aihara | Takeshi Aihara received his PhD in engineering from Tokyo Metropolitan University in Japan in 2021. In the same year, he joined the Laboratory for Materials and Structures at Tokyo Institute of Technology as an Assistant Professor. His research interests lie in the development of acid and base catalysis of nanosized-crystalline materials. |
 Keiju Wachi | Keiju Wachi is a Specially Appointed Assistant Professor in the Laboratory for Materials and Structures at Tokyo Institute of Technology. He received his PhD in engineering from the University of Tokyo in Japan in 2023. His current research interests are in multicomponent oxide catalysts for gas- and liquid-phase aerobic oxidation. |
Introduction
The situation surrounding the global environment is worsening year by year; therefore, it is necessary to reduce CO2 emissions through product manufacture and energy production with low environmental impact to build a sustainable society.1 Catalysis is a core technology that accounts for 90% of chemical processes and plays a crucial role in the sustainable production of useful chemicals and the conversion of energy. A shift from current processes based on petrochemical refining to the sustainable production of energy and chemicals from renewable resources has become a necessity. The development of nanostructured catalysts with active sites controlled at the atomic and/or molecular levels is one of the most promising approaches to solve these urgent problems, and much effort has been devoted to the synthesis, application, and understanding of nanostructured catalyst materials.1–10 Metal nanoparticles supported on high-surface-area oxide solids are common heterogeneous catalysts in industrial chemical reactions; therefore, the high functionalization of supported catalysts has mainly been investigated based on approaches such as reduction of the metal nanoparticle size (e.g., single atoms, subnano-/nano-clusters),2,3 shape control,4,5 metal–oxide interfaces,6 porosity,7 heterostructures (e.g., core/yolk–shell, hollow),8,9 and alloying of metal nanoparticles.10 Recent advances in computational methods,11operando studies under reaction conditions,12,13 and catalyst design using informatics14 also play an important role in the understanding of catalytic active sites at the atomic scale and in the development of new catalysts.Metal oxide-based catalysts have redox and/or acid–base active sites that promote various types of chemical reactions such as oxidation,15–18 hydrogenation,19,20 dehydrogenation,21 acid–base reactions,22–24 biomass conversion,25–27 environmental protection,28,29 photocatalysis,30,31 and electrocatalysis32,33 because their physicochemical properties can be controlled according to the crystal structure, morphology, composition, defects, and doping. In the field of condensed matter science, the unique structures and electronic states of crystalline complex oxides have been widely studied for application as novel functional materials with optoelectronic, conductive, magnetic, and superconductive properties;34–38 however, these complex oxides are typically synthesized by the conventional solid-state method to obtain highly pure materials from metal precursors without impurities, which limits the catalytic application and overall performance (especially, in liquid-phase reactions) of the bulk catalyst materials. Versatile, reproducible, scalable, and high-yield synthesis of nanostructure-controlled crystalline complex oxide materials with diverse compositions and crystal structures is essential to more effectively utilize the complexity of such materials as catalysts and realize efficient catalytic systems under mild reaction conditions.38 Although complexity has made it difficult to elucidate mechanistic aspects, recent advances in the synthesis of high-quality materials has gradually enabled the clarification of catalyst local structures and structure–activity correlations. For example, the roles of promoters in industrial multicomponent molybdenum oxide-based catalysts for the selective oxidation of propylene to acrylic acid can be discussed at the atomic scale.39,40 With this research background, we have developed simple but versatile methods for the synthesis of nanostructure controlled crystalline complex oxides with diverse compositions and structures,41–44 and these methods have contributed to the discovery of new catalytic properties (e.g., aerobic oxidation,42–50 direct methane conversion,51–54 concerted acid–base catalysis,55–58 one-pot synthesis,59,60 biomass conversion,43,57,61,62 water electrolysis63–67) of nanomaterials that have not received much attention until now. Herein, we first introduce methods used for nanostructural control of crystalline metal oxides and phosphates mainly based on earth-abundant metals, and then touch upon the development of catalytic materials and reactions with a focus on thermocatalysis (Fig. 1).
 | ||
| Fig. 1 Nanostructure control and catalytic application of complex oxide materials. | ||
Synthesis of nanostructured complex oxides
Nanostructured materials exhibit unique functionalities in various fields compared with their bulk counterparts.68,69 Precise tuning of the physicochemical properties of nanomaterials requires control of the structure, morphology, and texture, which requires the development of convenient synthesis methods.70–74 The solid-state synthesis method is typically used for optoelectronic applications; however, the specific surface areas of these materials are extremely low. Therefore, several other synthesis methods for nanostructured complex oxide catalysts have been reported. The methods employed have a strong influence on the purity, the size/shape of particles, the surface area, and the size/amount of pores.7,75–78 Representative synthesis methods such as co-precipitation, sol–gel, solution combustion, and soft/hard templating methods are briefly explained here along with the characteristics of the resultant complex oxide nanomaterials (Fig. 2).41,76 While the hydro(solvo)thermal method is also an effective technique for the synthesis of materials with defined crystal and surface structures,17,54,57 its effectiveness and applicability have already been summarized in many excellent review articles.79,80 | ||
| Fig. 2 Schematic representations of representative synthesis methods for nanostructured complex oxide catalysts. (Reproduced from ref. 41 with permission from The Chemical Society of Japan). | ||
The co-precipitation method is one of the most useful techniques for the facile synthesis of complex oxides. The addition of precipitants (e.g., NaOH, NH3, amine) to an aqueous solution containing two or more metal species components gives homogeneous precursors, and the precursors recovered after washing the collected precipitates with solvents are calcined at an appropriate temperature to give the product.81 Control of the chemical composition of multicomponent complex oxides is difficult because specific soluble metal species are formed during the precipitation and/or the washing steps. Another useful technique is the sol–gel method, whereby complex oxides with relatively high surface areas can be synthesized. The solution sol containing the metal sources and additives are gradually changed to a solid-phase gel by heating, pH control, and drying. Calcination of the dried gel precursors gives the desired and homogeneous complex oxides with controlled chemical compositions. Frequently used sol–gel methods include the hydroxyacid-aided method using citric acid (CA),82 the Pechini method using ethylenediaminetetraacetic acid–ethylene glycol (EG) complexation,83 and the polymerized complex (PC) method using CA–EG complexation.84 The solution combustion method involves heat treatment of a mixture at lower temperatures than those used for conventional synthesis routes because the heat required for phase formation is supplied by the self-sustaining exothermic chemical reactions of metal nitrates with suitable organic fuels.85 This method has been applied to the rapid synthesis of various multicomponent complex oxides, although consistent control of the process parameters and the final product quality is limited. Soft/hard templating methods are effective for the synthesis of porous complex oxides using polymeric materials and porous silicates as soft and hard templates, respectively, in contrast to non-porous bulk metal oxides.7 Although the templating methods offer high surface areas of more than 100 m2 g−1 and ordered pore structures, their applicability is limited due to the need for expensive templates and their subsequent removal, which involves multi-step complicated processes.
Synthesis of crystalline complex oxide nanoparticles using the amino acid-aided method
The sol–gel methods have attracted attention from the viewpoint of catalytic applications because of the controllable chemical composition and nanostructure. While common sol–gel methods have been utilized for the synthesis of complex oxide nanomaterials, they typically require a multi-step process, an excess amount of toxic organic reagents, and high-temperature calcination to remove carbonaceous precursors.82–84 Against this background, we have begun to develop a simple and versatile sol–gel method for the facile synthesis of crystalline complex oxides with desired chemical compositions and high surface areas (Fig. 3(a)). | ||
| Fig. 3 Schematic representations for (a) amino acid-aided method for the synthesis of crystalline complex oxide nanoparticles and (b) precursor crystallization method for the synthesis of mesoporous manganese oxide nanoparticles. | ||
The formation of an amorphous precursor in which various constituent metal species are highly dispersed would be required to synthesize highly pure crystalline complex oxide materials, and the ligand exchange between the metal sources and additive reagents affects the structure of the precursors.41,44–49,53,55,56,63–67 First, we investigated the effect of carboxylic acids and metal sources on the synthesis of hexagonal SrMnO3 as a benchmark material because of its unique catalysis properties for aerobic oxidation of various types of organic substrates.44 The precursor formation process influences the purity of SrMnO3 and the resultant surface areas. When using metal nitrates (i.e., Sr(NO3)2 and Mn(NO3)2) as starting materials in the presence of malic acid, only X-ray diffraction (XRD) peaks assignable to Sr(NO3)2 and complex mixed phases were observed, despite pH adjustment (pH 3.0–9.0) during precursor preparation and/or the calcination of the precursor at high temperatures (∼950 °C). On the other hand, the precursor prepared from metal acetates (i.e., Sr(OAc)2 and Mn(OAc)2) was completely amorphous and calcination at 550 °C gave single-phase SrMnO3 (Fig. 4). Formation of the amorphous precursor through ligand exchange reaction between the metal nitrates and malic acid is likely prevented by the presence of NO3− in the starting materials, and the weaker acidity of acetic acid (pKa = 4.76) than malic acid (pKa1 = 3.4) would accelerate the ligand exchange reactions. On the basis of infrared (IR) spectroscopy and electrospray ionization time-of-flight mass spectrometry (ESI-TOF-MS) measurements, the precursor is an amorphous malate salt with the metal cations linked by malate anions in a bridging bidentate manner.
 | ||
| Fig. 4 XRD patterns for the precursors and calcined samples prepared from (a), (e) metal nitrates/malic acid, (b), (f) metal acetates/aspartic acid, (c), (g) metal acetates/malic acid, (d) Sr(NO3)2 (JCPDS 01-087-0557), and (h) SrMnO3 (SMO, JCPDS 01-084-1612). (Reproduced from ref. 44 with permission from The American Chemical Society). | ||
The effect of various carboxylic acid additives on precursor formation was examined, and only aspartic acid and malic acid gave single phase SrMnO3. In particular, the specific surface area (SBET) for SrMnO3 prepared using aspartic acid was up to 47 m2 g−1 and much larger than those (2–25 m2 g−1) for SrMnO3 synthesized by conventional methods such as the polymerized complex, acetate, sol–gel combustion, cellulose templating, and citrate methods.50,86–88 The amorphous precursors with relatively low carbon contents are readily decomposed; therefore, the present method using aspartic acid (i.e., the amino acid-aided method) yields pure SrMnO3 at lower calcination temperatures (by 100–400 K). The high pH of the reaction solution containing metal acetates and aspartic acid containing amino groups (pH = 4.23) resulted in acceleration of the ligand exchange reaction without pH adjustment. Scanning electron microscopy (SEM) observation of SrMnO3 prepared using aspartic acid showed that the nanoparticles have a sphere-like morphology and the particle size was estimated to be 10–40 nm. On the other hand, sintering of SrMnO3 nanoparticles was observed in the case of the polymerized complex method because a higher calcination temperature is required.
Various crystalline complex oxides can be synthesized by calcination of precursors prepared with different metal sources and molar ratios at an appropriate temperature using the amino acid-aided method. In particular, this method has advantages in the synthesis of hexagonal perovskite nanoparticles that contain alkaline earth metals, which tend to segregate as carbonates with the conventional method including carbonaceous precursors (Fig. 3(a)). In contrast to the cubic perovskite structure (3C structure) in which typical BO6 octahedra are connected by corner-sharing, the hexagonal perovskite contains unique face-sharing octahedral units involving high-valency metal species (e.g., Mn4+, Ru4+, Fe4+) that are similar to the active sites of oxidases.89 We have found for the first time that hexagonal perovskite nanoparticles (4H-SrMnO3, 9R-BaRuO3, 6H-BaFeO3−δ) function as effective solid catalysts for the liquid-phase selective oxidation of various organic substrates using O2 as the sole oxidant.41,44,45,47–50 Although this sol–gel method is mainly limited to the synthesis of perovskite oxides with B-site metal cations of group 7–10 elements due to the requirement of metal acetates as starting materials, control of the solution states of d0-transition metals (Ti4+, Zr4+, and Nb5+) in the presence of dicarboxylic acids enables the synthesis of high-surface-area perovskite oxides containing d0-transition metals.55,56 In addition, the specific surface area can be increased by a simple procedure of changing the atmosphere from N2 to air during calcination of the amorphous precursor (e.g., SBET of SrTiO3: 46 m2 g−1 from 30 m2 g−1) because the decomposition processes of the precursor (i.e., combustion and pyrolysis under oxidative and inert atmospheres, respectively) likely affects the growth and aggregation of SrTiO3 nanoparticles (Fig. 5). This sol–gel method can be applied not only to the perovskite nanostructure of ABO3, but also to other crystalline metal oxides such as simple oxides,48 metal phosphates,51,53 and murdochite-type Mg6MnO8 nanostructures.46
 | ||
| Fig. 5 (a) SEM and (b) TEM images of SrTiO3 synthesized by a simple procedure of changing the atmosphere from N2 to air during calcination of an amorphous precursor. (Reproduced from ref. 56 with permission from The American Chemical Society). | ||
Synthesis of porous manganese oxide nanoparticles by the precursor crystallization method
V-,90,91 Fe-,92,93 Co-,94,95 Ni-,96,97 and Cu-based98,99 oxides have been extensively investigated as solid catalysts applicable to versatile reactions such as gas-phase oxidation, organic synthesis, environmental cleanup, water splitting, and electrochemical and photocatalytic CO2 reduction. Manganese dioxide (MnO2) has also attracted attention as an important functional metal oxide material with wide applications in catalysis, energy storage, magnetics, and sensors because of its structural diversity and the numerous Mn oxidation states available.100,101 MnO2 nanostructures with different crystal structures have been synthesized by several methods including the solid-state,102 reflux, hydrothermal,103–106 and sol–gel approaches.107,108 In particular, the effect of the reaction conditions on the morphology and crystal structure of MnO2 under reflux/hydrothermal conditions has been extensively studied.103–106 As described later, we have developed effective aerobic oxidation systems using only O2 based on β-MnO2 nanoparticles.42,43,59–61 The activation barrier for the transformation of other manganese oxides to β-MnO2 has been reported to be high, despite β-MnO2 being the most thermodynamically stable phase among the MnO2 polymorphs,105,109,110 and studies on the controlled synthesis of β-MnO2 with high surface area are still limited compared to those on other MnO2 polymorphs.100,101 Therefore, the hydrothermal synthesis of β-MnO2 typically requires high reaction temperatures or long reaction times, which lead to the formation of large β-MnO2 particles with low surface areas (3–35 m2 g−1).111,112 Although templating methods for the synthesis of mesoporous β-MnO2 materials with high surface areas have been reported, multi-step procedures including the utilization of organic and inorganic templates are required.107,108,113,114 Consequently, we have successfully synthesized mesoporous β-MnO2 nanoparticles without the use of template molecules by the solid-state transformation of a low-crystallinity layered manganese oxide precursor (i.e., the precursor crystallization method) (Fig. 3(b)).43,61Mn4+ precursors (low-crystallinity layered Mn4+ oxide (c-distorted H+-birnessite)) were prepared by the redox reaction of MnO4− and Mn2+ reagents with a molar ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, and the formation was confirmed by XRD and IR analyses.43 While non-porous β-MnO2 nanorods, hollow microrods, and/or nanowires are typically obtained under hydrothermal conditions from the solution-derived Mn4+ precursors,103–106,111,112 the present method involves the simple solid-state transformation (i.e., heat treatment at 400 °C) of isolated precursors. The structure, surface area, and morphology of manganese oxides are significantly dependent on the synthesis conditions. The amount of residual alkali metal cations in the precursors and the calcination temperature have a significant effect on the crystal structure of the manganese oxides formed because the content and type of interlayer metal cations play important roles as structure-directing templates in the transformation of layered birnessite to manganese oxides with different tunnel sizes (Fig. 3(b)). Heat treatment of precursors with a low content of Na+ cations promotes desorption of water and/or dehydrative condensation to give β-MnO2 nanoparticles at 400 °C. Calcination of precursors at 450 °C leads to the formation of Mn2O3 through deoxygenation of the MnO2 samples.
:3, and the formation was confirmed by XRD and IR analyses.43 While non-porous β-MnO2 nanorods, hollow microrods, and/or nanowires are typically obtained under hydrothermal conditions from the solution-derived Mn4+ precursors,103–106,111,112 the present method involves the simple solid-state transformation (i.e., heat treatment at 400 °C) of isolated precursors. The structure, surface area, and morphology of manganese oxides are significantly dependent on the synthesis conditions. The amount of residual alkali metal cations in the precursors and the calcination temperature have a significant effect on the crystal structure of the manganese oxides formed because the content and type of interlayer metal cations play important roles as structure-directing templates in the transformation of layered birnessite to manganese oxides with different tunnel sizes (Fig. 3(b)). Heat treatment of precursors with a low content of Na+ cations promotes desorption of water and/or dehydrative condensation to give β-MnO2 nanoparticles at 400 °C. Calcination of precursors at 450 °C leads to the formation of Mn2O3 through deoxygenation of the MnO2 samples.
The morphology and porous structure of the manganese oxides are dependent on the pH conditions during precipitation of the Mn4+ precursors, and plate-like and sphere-like nanoparticles are formed under acidic and weakly acidic conditions, which results in slit-like and ink-bottle shaped mesopores, respectively (Fig. 6). Non-rigid aggregates of plate-like particles with slit-like pores (β-MnO2-1 and -2) were obtained from combinations of NaMnO4/MnSO4 and NaMnO4/Mn(NO3)2, respectively. On the other hand, sphere-like particles with ink-bottle shaped pores (β-MnO2-3) were formed in NaMnO4/Mn(OAc)2 with pH adjustment (pH 0.8). The specific surface areas for β-MnO2-1, -2, and -3 were 106, 100, and 122 m2 g−1, respectively, which are comparable to those (68–195 m2 g−1) for the mesoporous β-MnO2 materials synthesized by the soft- and hard-template methods,107,108,113,114 and much higher than that (14 m2 g−1) for non-porous β-MnO2 nanorods synthesized using the typical hydrothermal method (β-MnO2-HT). In the case of β-MnO2-1, the number of pores with a size of ∼3 nm in the precursor gradually decreased (≤300 °C), while the number of pores with a size of ∼7 nm increased at 400 °C. Similarly, the number of pores with a size of ∼6 nm in the precursor of β-MnO2-3 did not change significantly at ≤300 °C, but the number of pores with a size of ∼9 nm increased at 400 °C.
 | ||
| Fig. 6 Nitrogen adsorption–desorption isotherms and BJH pore size distributions (inset) for (a) β-MnO2-1, (b) β-MnO2-2, and (c) β-MnO2-3. (Reproduced from ref. 43 with permission from The American Chemical Society). | ||
The oxidation state of Mn species in manganese oxides plays an important role in materials and catalytic science because the distortion due to the Jahn–Teller disproportionation effect of Mn3+ is related to various applications such as the synthesis of nanosheets by exfoliation,115 activity in oxidations of water and organic compounds,50,59–62,116 and improving the durability of battery materials.117 On the other hand, there was no significant difference among β-MnO2-1, -3, and -HT in the bulk and surface Mn valence states and the amount of adsorbed oxygen species (i.e., surface defects) determined using iodometry and Mn 2p and O 1s XPS, which indicates that the difference in synthesis conditions does not significantly affect the bulk and surface oxidation states of manganese and defects. Despite the similar electronic structures for β-MnO2-1, -3, and -HT as mentioned above, the catalytic activity of β-MnO2-1 and -3 for the aerobic oxidation of various substrates was much higher than that of β-MnO2-HT, which suggests that the increase in the catalytic reactivity of β-MnO2 nanoparticles would be mainly caused by their high surface area.
Focus on the ion exchange reaction of a K+-containing precursor with H3O+ under acidic conditions has realized the synthesis of β-MnO2 nanoparticles using inexpensive and readily available KMnO4. The K+-containing precursor obtained from KMnO4 and Mn(NO3)2 is isolated followed by treatment in 0.1 M aqueous HNO3 solution to give the proton-exchanged precursor. In contrast to the K+-containing precursor,43 a single phase of β-MnO2 (β-MnO2-4) is formed by calcination of the proton-exchanged precursor.60 The specific surface area of β-MnO2-4 was 124 m2 g−1 and comparable to those (100–122 m2 g−1) of β-MnO2-1, -2, -3. SEM and transmission electron microscopy (TEM) measurements also showed the presence of flower-like spherical particles composed of small nanoplates (∼10–80 nm).
This method was applied to synthesize the todorokite-type manganese oxide octahedral molecular sieve (OMS-1) nanoparticle catalyst with larger tunnel structures than α-MnO2.42 OMS-1 has 6.9 × 6.9 Å (3 × 3) edge-sharing MnO6 octahedra chains that are corner-connected to form a one-dimensional tunnel structure,100,101 and its related materials have been studied as electrodes and/or catalysts.116,117 The insertion/desertion of various metal cations or the sorption of organic compounds in the tunnel structure is possible, so that metal-modified OMS-1 can act as a catalyst for the oxidation of inert molecules, and for the decomposition of ozone.118–120 A multi-step process that includes ion exchange and crystallization under hydrothermal or reflux conditions has been typically utilized to synthesize OMS-1.100,118,121–124 However, this can sometimes cause a particle size increase and/or a decrease in the specific surface area, which are detrimental to improvement of the catalytic performance.125,126 Although ultrasmall nanocrystalline OMS-1 catalysts have been synthesized by the reduction of MnO4− in an organic solvent,127 the synthesis and catalytic application of OMS-1 nanoparticles are still limited in sharp contrast to the widely investigated cryptomelane-type manganese oxide (OMS-2) with a 2 × 2 tunnel structure.
Mesoporous OMS-1 nanoparticles with high specific surface areas were synthesized in one step by solid-state transformation of a precursor formed by redox reaction between Mg(MnO4)2 and Mn2+ reagents.42 Similar to the synthesis of β-MnO2, the conditions for preparation of the precursors and the calcination temperature of the precursors affect the formation of OMS-1. Low-crystallinity buserite precursors were prepared from Mg(MnO4)2, MnX (X = 2OAc−, 2NO3−, and SO42−), and MgX with various molar ratios (0.30, 0.40, 0.50, and 0.67) of MnO4−/Mn2+. The pH conditions during precipitation of the precursors have a strong effect on the morphology, particle size, and specific surface area of OMS-1 (Fig. 7). Aggregates of small nanoparticles were formed under weakly acidic conditions when using acetate salts, while the acidic conditions when using nitrates and sulfates led to the formation of flowerlike particles that contained relatively large nanoplates. The present solid-state transformation of a layered buserite to OMS-1 with 3 × 3 tunnel structures requires precursors of nanosized Mg-rich Mn3+-containing layered manganese oxides with orthogonal layer symmetry because the layer-to-tunnel transition would be initiated by the release of layer distortion and strain caused by locally concentrated Jahn–Teller [Mn3+O6] octahedra. The crystal structure of the final manganese oxides is dependent on the calcination temperature of the buserite precursors. The calcination of low-crystallinity buserite at 200 °C gives mesoporous OMS-1 nanoparticles, whereas the collapse of the OMS-1 structures to other phases such as Mn2O3 and MgMn2O4 gradually occurs at higher temperatures. From the viewpoints of both inorganic synthesis and catalytic application, ultrasmall OMS-1 nanoparticles synthesized by heat treatment of a precursor prepared using acetate salts (MnO4−/Mn2+ = 0.40) at 200 °C were the most effective catalyst.
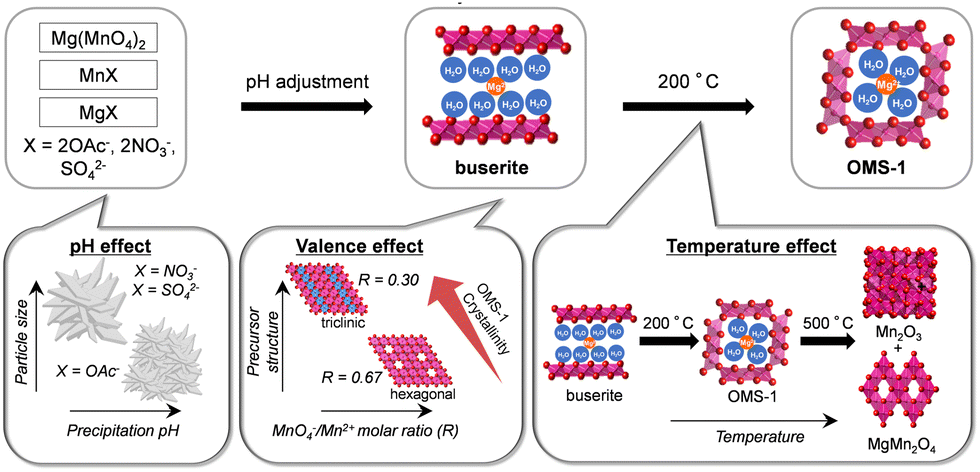 | ||
| Fig. 7 Schematic representation of the proposed mechanism for the formation of mesoporous OMS-1 nanoparticles. (Reproduced from ref. 42 with permission from The American Chemical Society). | ||
The specific surface area of the optimal OMS-1 ultrasmall nanoparticles was 249 m2 g−1, which was higher than those of OMS-1 synthesized by a hydrothermal method (35 m2 g−1) and previously reported Mg-OMS-1 prepared by the reduction of MnO4− in an organic solvent (155 m2 g−1),127 by reaction under hydrothermal or reflux conditions (13–185 m2 g−1),118,120,128–130 and by microwave irradiation (13–48 m2 g−1).130 N2 adsorption/desorption isotherms for the optimal OMS-1 ultrasmall nanoparticles showed a type IV pattern with hysteresis loops, which indicates a mesoporous structure. The shapes of the loops were similar to type H3, which is observed in non-rigid agglomerates of plate-like particles with slit-like pores. According to the pore size distribution, the optimal OMS-1 ultrasmall nanoparticles showed a broad peak around 7 nm, which corresponds to ink-bottle type pores, and the total pore volume was up to 0.46 cm3 g−1.
Catalysis of crystalline oxide nanomaterials
Crystalline complex oxide nanomaterials synthesized according to the methods discussed so far could be applied to a variety of chemical reactions in both the liquid and gas phases, and exhibit unique catalytic properties according to their compositions and structure. In this section, the catalytic performance and proposed mechanism, including the correlation between the bulk/surface catalyst structures and their activity, are considered with a focus on thermocatalysis including oxidation and acid–base reactions. In particular, oxygen vacancy formation and/or O2 activation of the metal oxide catalysts play important roles to efficiently promote oxidation reactions, and the synergistic action of acid–base sites is involved in improvement of the performance of the acid–base catalysts.Liquid-phase aerobic oxidation catalyzed by mesoporous manganese oxides
Although current chemical industries are heavily reliant on non-renewable fossil fuel-based petrochemicals, chemical processes that employ more sustainable feedstocks are desired.131,132 Cellulose, a main component of lignocellulosic biomass, could be depolymerized to glucose, which is in turn converted to platform biochemicals including 5-hydroxymethylfurfural (HMF). The catalytic conversion of HMF into value-added compounds has thus been extensively studied, and 2,5-furandicarboxylic acid (FDCA) is one of the most attractive raw materials for polyethylene furanoate, which can be a recyclable alternative to polyethylene terephthalate (Fig. 8). Various types of heterogeneous catalysts based on noble-metal catalysts (Au, Pt, Ru, and Pd), transition metals (Mn, Fe, Co, and Cu), and metal-free carbon materials have been developed to date.132 Mn oxide-based catalysts are potential heterogeneous catalysts for liquid-phase selective oxidation133–135 and gas-phase total oxidation of volatile organic compounds (VOCs), alcohols, NO, and CO;136,137 these catalysts are environmentally friendly and have specific chemical/physical properties that include diverse crystal structures and oxidation states. While several research groups have developed effective Mn-based composite oxide catalysts for the oxidation of HMF,132,138 there has been a lack of comprehensive studies on catalytic functions based on the crystal structure of MnO2 itself.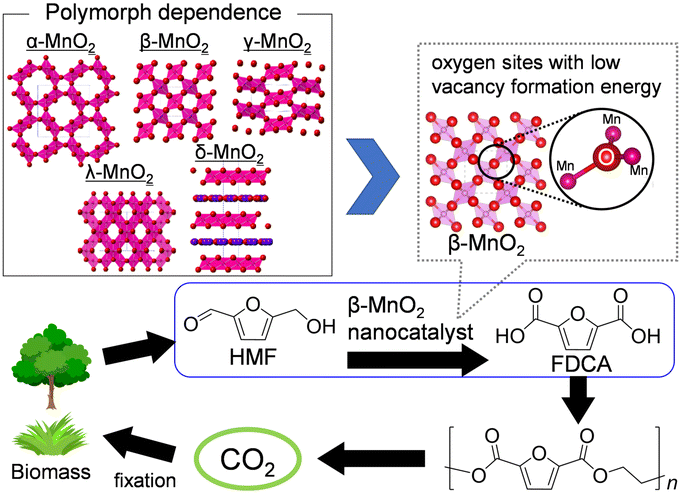 | ||
| Fig. 8 Polymorph dependence of MnO2 on the oxygen vacancy formation energy, and catalytic performance and high functionalization of β-MnO2 catalysts for the aerobic oxidation of HMF into FDCA. (Reproduced from ref. 43 with permission from The American Chemical Society). | ||
In our initial investigation on HMF oxidation using non-precious metal catalysts, a simple catalyst system based on commercially available activated MnO2 and NaHCO3 converted HMF to FDCA with O2 in 91% yield (eqn (1)).62 Other metal oxides such as Fe2O3, Fe3O4, FeO, Co3O4, CoO, NiO, CuO, and Cu2O were almost inactive. The MnO2 catalyst acted as a reusable heterogeneous catalyst with potential for application to large-scale oxidation, although the reaction mechanism for HMF oxidation, including the structure–activity relationship for MnO2, was ambiguous. Therefore, six types of crystalline MnO2 (α-, β-, γ-, δ-, ε- and λ-MnO2) were synthesized, and the dependence of HMF oxidation on the polymorphs was investigated.61 A kinetic simulation suggested that the pathway to oxidize HMF to FDCA via 2,5-diformylfuran (DFF) was dominant and that the oxidation of 5-formyl-2-furancarboxylic acid (FFCA) was the slowest step, which is in good agreement with the time course of each product during HMF oxidation. There was an obvious correlation between the FFCA oxidation rate per surface area and the reduction rate estimated from H2-temperature programmed reduction (H2-TPR) of various MnO2 polymorphs, and the β-MnO2 structure was determined as the most active. On the basis of detailed mechanistic studies, the present MnO2-catalyzed oxidation likely proceeds via a Mars–van Krevelen mechanism (i.e., substrate oxidation with oxygen supplied from the solid). The oxygen vacancy formation energy has been accepted as a good descriptor of the oxidizing power of a metal oxide. In the present case, density functional theory (DFT) calculations also suggested that β-MnO2 would be a good candidate as an oxidation catalyst because of the low vacancy formation energies at the bent oxygen sites of β-MnO2. The FDCA yield in HMF oxidation over mesoporous β-MnO2 nanoparticles (β-MnO2-1, -2, -3) was much higher than that for β-MnO2-HT.43 Furthermore, the narrow pore size distribution for mesoporous β-MnO2-3 imparts unique catalytic properties, especially for the oxidation of large organic molecules (e.g., 1-pyrenemethanol). These mesoporous β-MnO2 nanoparticles efficiently promoted the aerobic oxidation of various aromatic and heteroaromatic alcohols (14 examples) into the corresponding aldehydes and ketones under mild reaction conditions.60
 | (1) |
The dual nature of redox and acid sites on solids sometimes enhances the catalytic performance, and the oxidizability and Lewis acidity of the synthesized β-MnO2 nanoparticles were effective for the one-pot synthesis of nitriles and sulfonamides (eqn (2) and (3)).59,60 The β-MnO2-1-catalyzed system was applicable to the one-pot aerobic oxidative conversion of primary alcohols to the corresponding nitriles and amides in the presence of ammonia (NH3) as a nitrogen source. OMS-2 was an effective catalyst for the ammoxidation of various primary alcohols and aldehydes to the corresponding amides, and the Brønsted acidic sites on OMS-2 likely played a key role in the hydration of the nitrile.139 However, β-MnO2 was superior for the selective synthesis of nitriles from alcohols compared to OMS-2, which is considered to be due to the high alcohol oxidation activity and low nitrile hydration activity of β-MnO2. Detailed mechanistic studies have indicated that the surface lattice oxygen species of β-MnO2 with low oxygen vacancy formation energy contribute to the high activity for the oxidation of alcohols and aldimine intermediates to the corresponding carbonyl compounds and nitriles, respectively, without significant promotion of the hydration of nitriles.
 | (2) |
 | (3) |
In addition, β-MnO2-1 could efficiently catalyze the one-pot aerobic oxidative sulfonamidation of various aromatic and heteroaromatic thiols (12 examples) without the need for any additives.59 The present system was reusable and could be applied to the large-scale sulfonamidation of p-toluenethiol to yield the industrially important sulfonamide. The formation of disulfide and thiosulfonate as intermediates during sulfoamidation was proposed on the basis of mechanistic control experiments, and the reaction mechanism is shown in Fig. 9. DFT calculations confirmed that the reaction pathway is downhill and thermodynamically favourable. The Lewis acidic Mn sites on β-MnO2-1 promote the amination of a thiosulfonane intermediate through the interaction between sulfone and Mn; therefore, such dual-functionality of β-MnO2-1 results in highly efficient one-pot aerobic sulfonamidation.
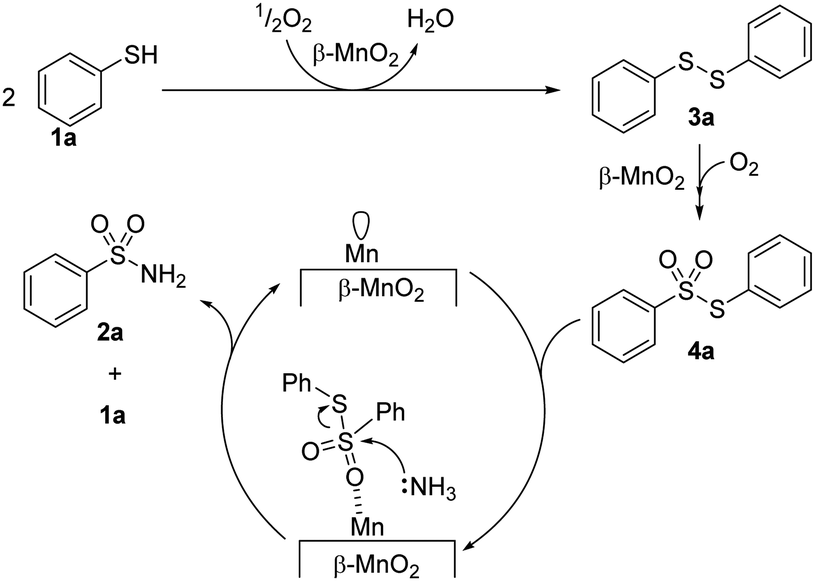 | ||
| Fig. 9 Proposed reaction mechanism for the one-pot aerobic sulfonamidation of benzenethiol (1a) to e benzenesulfonamide (2a) with β-MnO2-1. (Reproduced from ref. 59 with permission from Royal Society of Chemistry). | ||
The significantly high surface area (249 m2 g−1) of mesoporous OMS-1 ultrasmall nanoparticles42 facilitated the efficient aerobic oxidation of alcohols to the corresponding aldehydes over the OMS-1 catalyst (eqn (4)), and the activity was higher than those of other Mn4+-containing manganese oxide nanoparticles. OMS-1 was also applicable to the aerobic oxidation of sulfides to the corresponding sulfoxides and sulfones at 90 °C (eqn (5)). A trade-off relationship between particle size and catalytic performance has been observed in some cases because surface active sites increase with a decrease not only in particle size but also in the degree of crystallinity. However, the initial reaction rates for OMS-1 for the aerobic oxidation of benzyl alcohol and thioanisole increased linearly with the surface area without the trade-off relationship between particle size and catalytic performance, which suggests the formation of high-quality ultrasmall OMS-1 nanoparticles (Fig. 10).
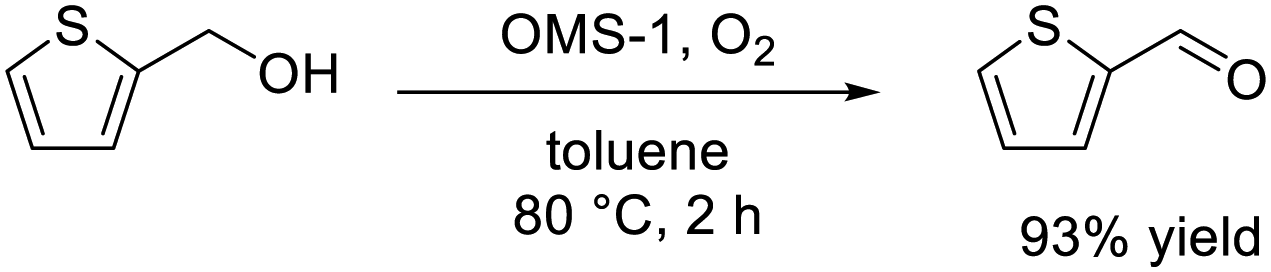 | (4) |
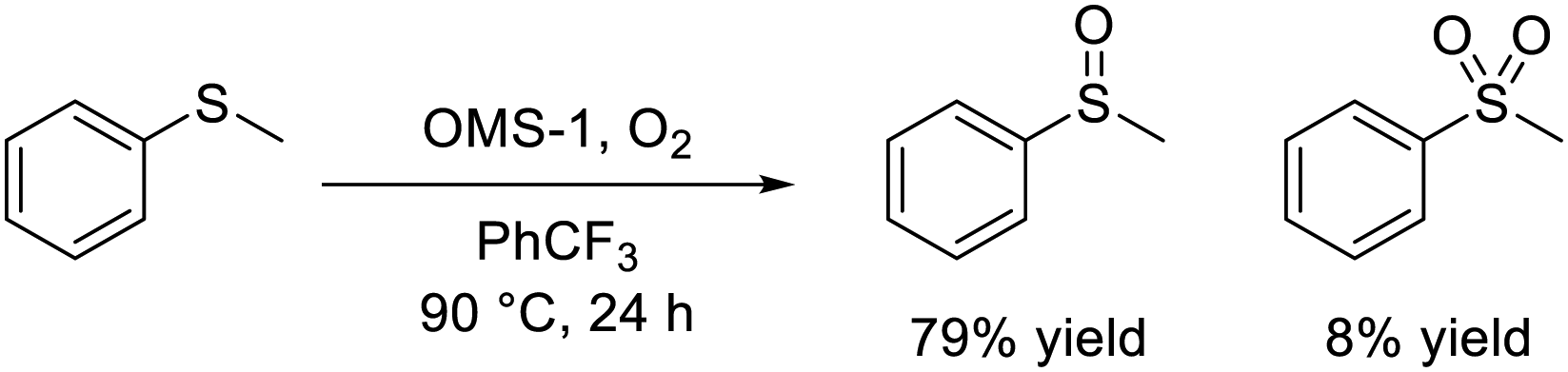 | (5) |
 | ||
| Fig. 10 Relationship between initial reaction rate (R0) and specific surface area of OMS-1 for the aerobic oxidation of (a) benzyl alcohol and (b) thioanisole. Reaction conditions were the same as those in eqn (4) and (5). (Reproduced from ref. 42 with permission from The American Chemical Society). | ||
Liquid-phase aerobic oxidation catalyzed by complex oxides
Oxygenated products are useful as platform commodities and specialty chemicals, and therefore the liquid-phase selective oxidation of organic substrates is an important reaction.140 The catalytic properties of perovskite oxides with the general formula ABO3 and related materials have been extensively researched.77 The ideal perovskite has a cubic structure and can be described as an A-site cation occupying the 12-fold coordination site between corner-sharing BO6 octahedra. On the other hand, a transformation from corner-sharing to face-sharing octahedra is induced when large alkaline-earth metals are employed as A-site cations, which results in hexagonal or rhombohedral structures. In these cases, the formation of high-valency B-site metal cations in close proximity and the resultant oxygen vacancies would be beneficial for the activation of organic molecules and O2, respectively. However, only electrochemical, photocatalytic, and high temperature gas-phase reactions over lanthanum-based perovskite oxides with corner-sharing BO6 octahedra have been mainly investigated.141 Given this context, our research group has applied various types of hexagonal perovskite oxides (e.g., SrMnO3, BaRuO3, and BaFeO3−δ), which were synthesized by the amino acid-aided method, to the liquid-phase selective oxidation of organic substrates and the relationships between crystal structure, valence state at the B site and catalytic activity were investigated.Hexagonal SrMnO3 synthesized by the polymerized complex method was an effective catalyst for the aerobic liquid-phase oxidation of various organic substrates.50 Differential IR spectra of 16O2- and 18O2-adsorved hexagonal SrMnO3 suggested that aerobic oxidation proceeded via Mn-superoxo species. The development of the amino-acid aided sol–gel method resulted in a high-surface-area catalyst with improved catalytic activity.44 The aerobic oxidation of fluorene to fluorenone proceeded at 60 °C (eqn (6)), and the reaction rate over hexagonal SrMnO3 synthesized by the sol–gel method using aspartic acid was five times higher than that of hexagonal SrMnO3 synthesized by the polymerized complex method.
 | (6) |
Not only crystal structure control of manganese oxides with Mn–O–Mn structures, but also the combination of isolated high-valency Mn species with base sites was effective for the aerobic oxidation of alkylarenes under mild reaction conditions. Murdochite-type oxide Mg6MnO8 nanoparticles with high surface area (104 m2 g−1) exhibited superior catalytic performance to those of other Mn- and Mg-based oxides for the oxidation of various alkylarenes under milder conditions than SrMnO3 (eqn (7)).46 Detailed kinetic and mechanistic studies suggested the present oxidation proceeds via a basicity-controlled C–H activation mechanism that involves O2 activation, which would be characteristic of Mg6MnO8 due to the crystal structure with isolated Mn4+ species located in a basic MgO matrix (Fig. 11). This study provided another catalyst design approach that is distinct from the incorporation of isolated Mn species into acidic matrices such as zeolites.142
 | (7) |
 | ||
| Fig. 11 Proposed reaction mechanism for the catalytic oxidation over Mg6MnO8-MA with O2. O*: schematic representation of surface oxygen species (O2−, O22−, O2−, OH−etc.). (Reproduced from ref. 46 with permission from The American Chemical Society). | ||
The approach based on hexagonal perovskite oxides was also applied to successfully accomplish the aerobic oxidation of alkylarenes and aryl sulfides under significantly milder conditions than those for previously reported systems. Rhombohedral BaRuO3 nanoparticles efficiently catalyzed the selective oxidation of various aryl and aliphatic sulfides into sulfoxides and/or sulfones at temperatures as low as 40 °C with O2 as the sole oxidant and without any additives.49 The present system was a reusable heterogeneous catalyst that has potential for application to the large-scale oxidation of thioanisole and quantitative conversion of dibenzothiophene, a model compound for the oxidative desulfurization of organic sulfur in fossil fuels, into the corresponding sulfone (eqn (8)). Measurements of the catalyst effect, and 18O-labeling, kinetic, mechanistic, and spectroscopic experiments indicated that sulfide oxidation most likely proceeds using lattice oxygen from the BaRuO3 oxide surface, which could be replenished by gaseous O2 (Mars–van Krevelen mechanism) (Fig. 12(a)). Therefore, the oxygen vacancy formation energy is useful information in a similar manner to that for MnO2. DFT calculations of BaRuO3, SrRuO3, CaRuO3, and RuO2 revealed that face-sharing oxygen in BaRuO3 exhibited a lower oxygen vacancy formation energy than any other corner- or edge-sharing oxygen in SrRuO3, CaRuO3 and RuO2, which may contribute to more feasible oxygen transfer from BaRuO3 to the sulfide (Fig. 12(b)).
 | (8) |
 | ||
| Fig. 12 (a) Proposed mechanism for sulfide oxidation over BaRuO3 catalyst. (b) Oxygen vacancy formation energies of BaRuO3, SrRuO3, CaRuO3 and RuO2. (Reproduced from ref. 49 with permission from The American Chemical Society). | ||
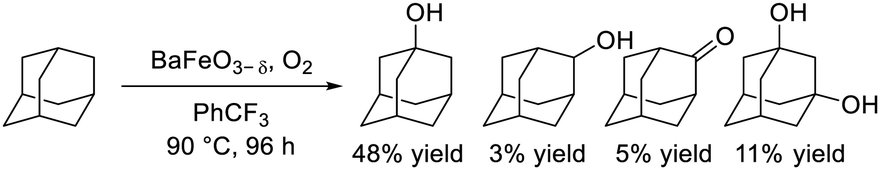
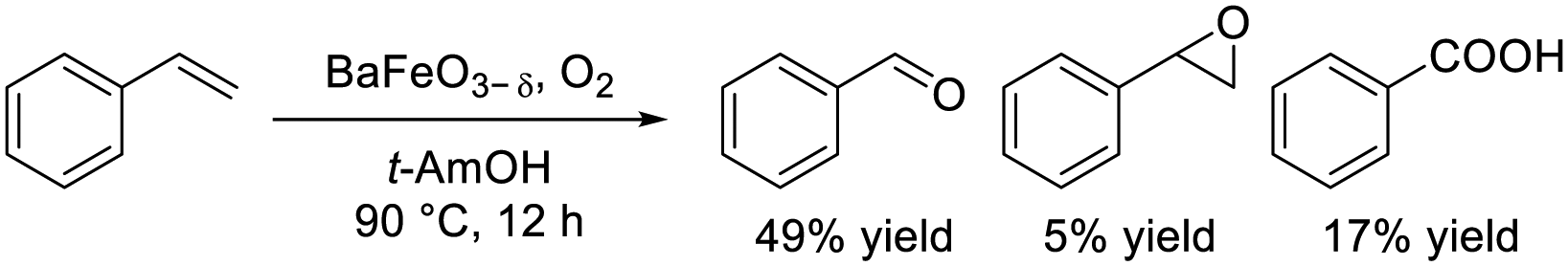
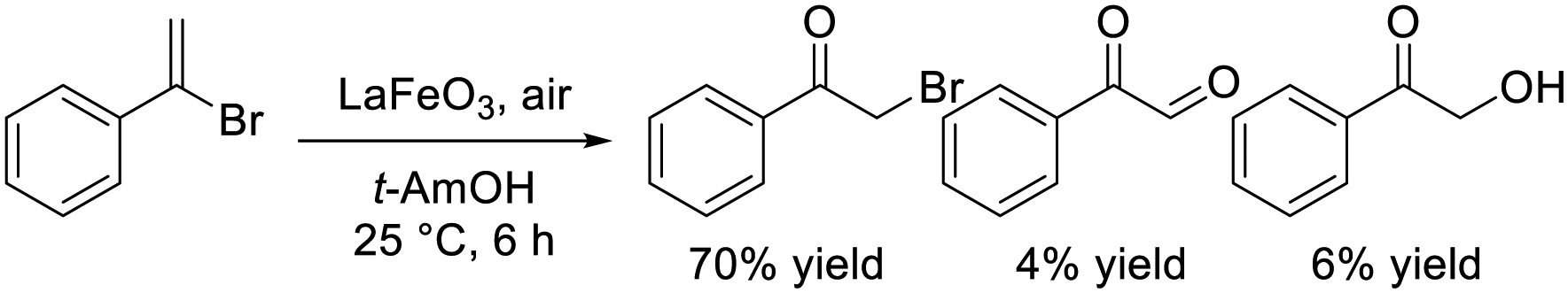
Dimeric high-valency iron-oxo species have been postulated to be strong active oxidants under mild conditions; therefore, we focused on hexagonal BaFeO3−δ containing face-sharing dimeric Fe2O9 units based on high-valency iron species (i.e., Fe4+).47,48 Hexagonal BaFeO3−δ acted as an efficient heterogeneous catalyst for the aerobic oxidation of alkanes to the corresponding alcohols and ketones, and for the oxidative C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond cleavage of various aromatic alkenes to the corresponding carbonyl compounds without the need for additives (eqn (9) and (10)). The activity of BaFeO3−δ was much higher than that of typical Fe3+/Fe2+-containing iron oxide-based catalysts including CaFeO2.5, LaFeO3 and Fe2O3, and H2-TPR measurement confirmed the high oxidizability of BaFeO3−δ The recovered BaFeO3−δ catalyst could be reused without significant loss of the catalytic performance, and both the oxidation of alkanes and alkenes likely involves a radical mechanism where BaFeO3−δ activates the substrate to form an active radical species. Despite the outstanding oxidation activity due to high-valency iron, BaFeO3−δ could not be reused for the oxidative transformation of α-bromostyrene to phenacyl bromide, because of a lack of structural stability.45 Alternatively, LaFeO3 exhibited high activity and reusability, although other Fe3+/Fe2+-containing simple oxides such as Fe2O3, Fe3O4, and FeO were much less active or were inactive. In this case, a similar radical-type mechanism that involves surface adsorbed O2 species over p-type LaFeO3 nanoparticles has been proposed (eqn (11)).
C bond cleavage of various aromatic alkenes to the corresponding carbonyl compounds without the need for additives (eqn (9) and (10)). The activity of BaFeO3−δ was much higher than that of typical Fe3+/Fe2+-containing iron oxide-based catalysts including CaFeO2.5, LaFeO3 and Fe2O3, and H2-TPR measurement confirmed the high oxidizability of BaFeO3−δ The recovered BaFeO3−δ catalyst could be reused without significant loss of the catalytic performance, and both the oxidation of alkanes and alkenes likely involves a radical mechanism where BaFeO3−δ activates the substrate to form an active radical species. Despite the outstanding oxidation activity due to high-valency iron, BaFeO3−δ could not be reused for the oxidative transformation of α-bromostyrene to phenacyl bromide, because of a lack of structural stability.45 Alternatively, LaFeO3 exhibited high activity and reusability, although other Fe3+/Fe2+-containing simple oxides such as Fe2O3, Fe3O4, and FeO were much less active or were inactive. In this case, a similar radical-type mechanism that involves surface adsorbed O2 species over p-type LaFeO3 nanoparticles has been proposed (eqn (11)).
 | (9) |
 | (10) |
 | (11) |
Methane oxidation catalyzed by metal phosphate
The conversion of methane (CH4), a main component of natural gas, has been viewed as a significant challenge for catalysis chemists for over a century. Every decade, a new approach has been proposed that seems to herald a new route to effective catalysis; however, as yet there has been no large-scale application of this research.143 The oxidative conversion of CH4 using O2 can be separated into oxidative coupling of CH4 (OCM) into C2 hydrocarbons and selective oxidation of CH4 into methanol and formaldehyde (HCHO). Since the pioneering studies on OCM by Keller and Bhasin in 1980s, numerous research efforts focused on oxide catalysts such as Li/MgO and Mn-Na2WO4/SiO2 have been published.144 In contrast to OCM systems, the yield to C1 oxygenates in the selective oxidation of CH4 was less than 10% in many reports, which indicates there is significant room for improvement. Recent advances in catalyst design for the selective oxidation of CH4 into C1 oxygenates have emphasized the approach of site isolation inspired by the active sites of enzymes. Single atoms or small clusters of Fe and Cu as active sites were constructed within zeolite145 and metal–organic framework (MOF) matrices146 or stabilized using polyoxometalate precursors147 for efficient activation of CH4 with O2. Crystalline complex oxides could also be potential candidates for selective oxidation. Li2CaSiO4 exhibited OCM activity comparable to Mn–Na2WO4/SiO2,148 and Cu-based mixed oxides, particularly α-Cu2V2O7 and Cu2Mo2O9 crystalline phases, were also effective for the selective oxidation of methane to formaldehyde.149Despite the fewer reports than those for supported catalysts, MOFs, and zeolites, several metal phosphates have also attracted much attention as catalysts for the selective oxidation of CH4. Low-crystallinity FePO4 with isolated tetrahedrally coordinated redox-active iron sites was an efficient catalyst for the selective oxidation of CH4 into CH3OH and HCHO with O2.150 Although Gomonaj and Toulhoat reported that the M–O bond strength and the acidity of various crystalline metal phosphates were correlated with the selectivity of CH4 oxidation toward HCHO using a flow circulation reactor with reaction mixture quenching,151 the effect of the metal phosphate crystal structures and the reaction mechanism on the direct oxidation of CH4 is still unclear. Therefore, various crystalline metal phosphate nanoparticles were systematically investigated for the oxidation of CH4 into HCHO with O2 as the sole oxidant.
Among four crystalline iron-based catalysts (trigonal FePO4, trigonal Fe3O3(PO4), monoclinic Fe4(P2O7)3 and trigonal Fe2O3), FePO4 exhibited the highest HCHO yield (0.19%) at 500 °C.53 CH4 conversion tended to increase with the Fe/P ratio, while the selectivity to HCHO decreased. FePO4 also exhibited durability without significant change in the HCHO, CO, and CO2 yields at 500 °C for 24 h time-on-stream. Kinetic studies and CH4 pulse reaction experiments suggested that the structure of bulk FePO4 was preserved during CH4 oxidation, and that lattice oxygen on the FePO4 surface was responsible for the selective oxidation of CH4 into HCHO with structural preservation of the bulk FePO4 (Fig. 13). IR spectroscopy measurements were conducted using adsorbed pyridine, chloroform, and methanol as probe molecules to investigate the detailed surface structures. Both FePO4 and Fe2O3 possess Lewis acid sites and base sites; however, the basicity of FePO4 was much lower than that of Fe2O3. Heterolytic cleavage of the C–H bond of CH4 could proceed using strong acidity via a proton-coupled electron transfer (PCET) mechanism;152 therefore, the redox-active Lewis acidic Fe3+ species likely play an important role in CH4 activation. Methanol-adsorbed IR spectroscopy measurements indicated that FePO4 without significant base sites had only a weak interaction with methanol, in sharp contrast to basic Fe2O3 that forms methoxide species. The temperature-programmed reaction of adsorbed methanol indicated that the molecularly adsorbed methanol on FePO4 easily desorbed as methanol without significant formation of CO2, but that methanol adsorbed by methoxy species caused only desorption of CO2 and CO. Therefore, this study suggests the importance of the effect of phosphate units as weak base sites to suppress the overoxidation of C1 products.
 | ||
| Fig. 13 Proposed reaction mechanism for oxidation of CH4 over FePO4 into HCHO with O2 using FePO4 and complete oxidation of CH4 over Fe2O3 into CO2 with O2 using Fe2O3. (Reproduced from ref. 53 with permission from Royal Society of Chemistry). | ||
BiPO4 nanoparticles synthesized in a mixed solvent of diethylene glycol and water exhibited a higher HCHO yield than that of FePO4 above 550 °C, while FePO4 showed a higher HCHO yield than BiPO4 below 550 °C.52 The lack of correlation between the catalytic activity and oxidizability estimated from H2-TPR measurements and the absence of any oxidation products in the CH4 pulse reaction experiments suggested that CH4 oxidation did not proceed with lattice oxygen supplied from the BiPO4 solid. Therefore, the reactive oxygen species for CH4 activation over BiPO4 is different from that over FePO4. Mechanistic studies including the catalyst effect, kinetics, and near-ambient pressure XPS (NAP-XPS) suggest that the oxidation of CH4 may proceed at surface adsorbed oxygen species generated on the surface Bi atoms of BiPO4 (Fig. 14(a)). Two pathways for H-abstraction from CH4 using surface O atoms on Bi or lattice O atoms on PO4 units were then compared by DFT calculations to further investigate the possible involvement of surface oxygen species on BiPO4 (Fig. 14(b) and (c)). H-abstraction using surface O atoms on Bi (ΔE = −1.65 eV) was more favourable than H-abstraction by lattice O atoms on PO4 units (ΔE = 0.94 eV). Such an O2 activation mechanism is similar to that proposed for BOx-based benchmark catalysts with relatively high HCHO yields,153 which suggests that such an O2 activation mode for BiPO4 would result in the highly selective formation of HCHO in sharp contrast to FePO4 nanoparticles with the redox mechanism.
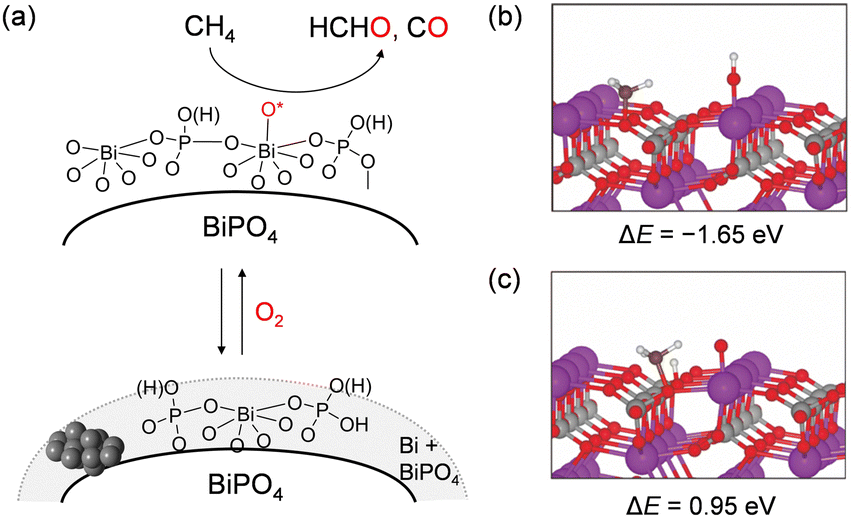 | ||
| Fig. 14 (a) Proposed mechanism for CH4 oxidation over BiPO4 catalyst. DFT calculation for H abstraction from CH4 by (b) the O atom adsorbed on surface Bi and (c) the lattice O atom. (Reproduced from ref. 52 with permission from Royal Society of Chemistry). | ||
Inspired by the different catalytic properties of FePO4 and BiPO4, various crystalline metal phosphates (37 examples including Cu, Fe, Bi, Ga, Ti, V, Cr, Mn, Co, Ni, Zn, In, K, Ba, B, La, Ce, Nb, Mo, Sb, and Sn) were surveyed, which revealed that Cu is the most active metal species for the direct oxidation of CH4 to HCHO.51 To further elucidate the relationship between structure and activity, four types of copper phosphates (monoclinic Cu2P2O7, monoclinic Cu2(P4O12), triclinic Cu3(PO4)2, and triclinic Cu4O(PO4)2) were investigated (Fig. 15). The CH4 conversion rate increased with Cu/P ratio, while the selectivity toward formaldehyde decreased. Based on H2-TPR and IR spectroscopy measurements of adsorbed probe molecules, the trends for conversion and selectivity could be explained by the changes in the base properties and oxidizability associated with variations in the Cu/P ratio as with that for FePO4. Furthermore, the HCHO yield at 550 °C was increased to 0.79% from 0.42% when the starting material was changed from Cu(OAc)2 to Cu(NO3)2. TEM measurements, IR spectral analysis with pyridine adsorption, and H2-TPR analysis revealed that the enhancement of surface crystallinity led to an increase in Lewis acidic Cu2+ sites, which facilitated CH4 activation and thereby enhanced oxidative ability. Therefore, the catalytic performance for the direct oxidation of CH4 is highly sensitive to the surface nanostructures as well as the bulk structure. Kinetic studies, isotope experiments using CD4 or 18O2 and the pulse reaction on Cu2P2O7 showed that CH4 was oxidized by surface lattice oxygen and that the C–H activation of CH4 is most likely the rate-determining step in a similar manner to that with FePO4. DFT calculations revealed that the oxygen vacancy formation energy for Cu2P2O7 decreases upon phase transition from α-Cu2P2O7 to β-Cu2P2O7 during the catalysis, which contributes to the high catalytic performance of Cu2P2O7.
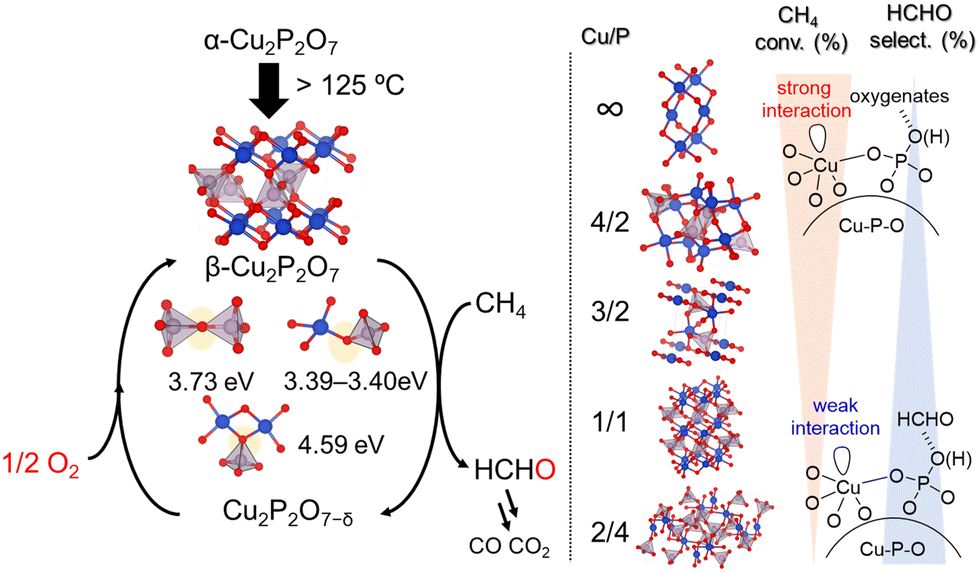 | ||
| Fig. 15 Proposed reaction mechanism for oxidation of CH4 over copper phosphates into HCHO with O2. (Reproduced from ref. 51 with permission from The American Chemical Society). | ||
The oxidation reactions discussed in this article can be classified into three main mechanism depending on the types of catalyst and reaction based on the experimental studies using spectroscopy, kinetics, and isotopes: (i) Mars–van Krevelen mechanism,42,43,49,51,53,60–62 (ii) mechanism involving O2 activation on surface redox-active Lewis acidic metals,44,46,50,52,54 and (iii) radical-mediated oxidation mechanism induced by high-valent metal oxygen species.45,47,48 In particular, the oxygen vacancy formation energy determined from DFT calculations is a good descriptor for both liquid- and gas-phase oxidations in which Mars–van Krevelen mechanism has been proposed. We have shown that structurally unique metal–oxygen sites, such as face-sharing octahedra in BaRuO3,49 planar oxygen sites in β-MnO2,61 and Cu–O–P sites in β-Cu2P2O7 formed by the phase transition of α-Cu2P2O7 under the catalytic conditions,51 are likely possible active sites.
Acid–base catalysis
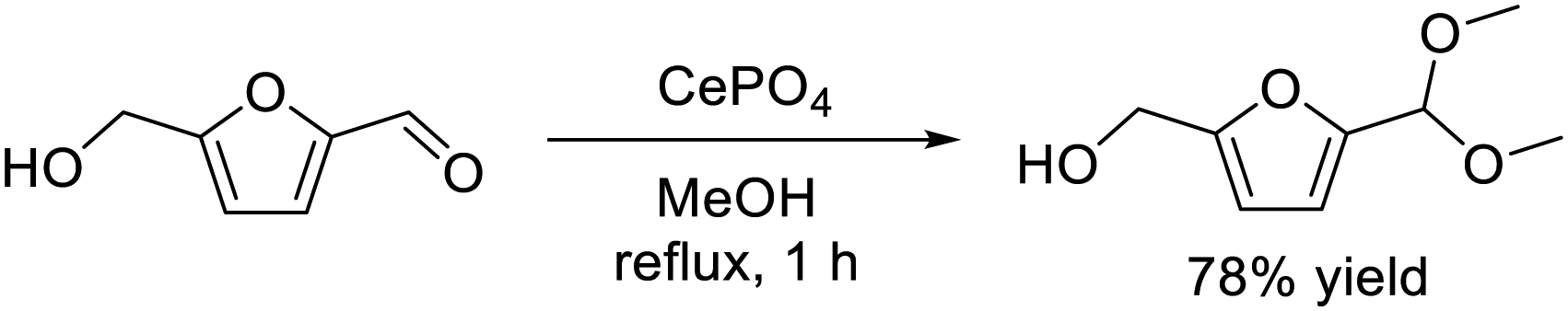
Acid and base catalysts play an important role because they are widely used in industrial chemical processes such as petroleum refining, biomass conversion, and the synthesis of fine chemicals.22,23,154 In contrast to traditional acids (e.g. HCl, H2SO4, AlCl3) and bases (e.g. NaOH, NH3, pyridine), solid acid and base catalysts are important materials because they can be easily separated, recovered, and reused. Acid–base bifunctional catalysts have recently attracted attention because nucleophiles and electrophiles are activated by acid and base sites, respectively, which results in higher catalytic activity and/or specific selectivity than that for catalysts with only acid or base sites.155–157 In the field of heterogeneous catalysis, difficulty in the construction of structurally controlled acid–base sites often leads to a problem where the fine-tuning of the catalyst structure and the reactivity are restrained. Therefore, we have focused on crystalline complex oxides to design effective solid acid–base bifunctional catalysts.Metal phosphates based on Ca, V, Ti, Zr, Fe, and Co are effective materials in the fields of catalysis,158 energy storage and conversion,159,160 biology,161 and biomass conversion.162 In addition, the acidic and basic characteristics of metal phosphate catalysts have been extensively investigated.51–53,163,164 Monoclinic CePO4 nanorods synthesized by the hydrothermal reaction of Ce(NO3)3 and (NH4)2HPO4 exhibited unique catalysis for the reaction of biomass-derived HMF with methanol.57 CePO4 nanorods selectively promoted the reaction to form the corresponding acetal derivative, 5-(dimethoxymethyl)-2-furanmethanol, in 78% yield (eqn (12)), while simple oxides (SiO2, ZrO2, CeO2, Al2O3, MgO, TiO2, and SnO2), Ce- and P-based materials, and a physical mixture of the precursors (Ce(NO3)3 and (NH4)2HPO4) were not effective for the acetalization. In addition, typical solid acid catalysts such as sulfated zirconia, sulfonated carbon, mordenite, and montmorillonite gave complex mixtures of acetal and ether derivatives. CePO4 could function as reusable solid catalyst for efficient acetalization of various carbonyl compounds with alcohols in good to excellent yields. The present catalytic system was also applicable to a gram-scale reaction of HMF (10.5 mmol scale) with methanol and 1.46 g of analytically pure product could be isolated.
 | (12) |
IR measurements of various probe molecules adsorbed on the catalyst surface were conducted to investigate the reaction mechanism of acetalization over CePO4 nanorods. The IR spectra of CePO4 nanorods with adsorbed pyridine and chloroform showed the presence of both Lewis acid sites and weak base sites, and the base sites were located in close proximity to the Lewis acid sites (Fig. 16(a) and (b)). IR spectra of acetone and methanol adsorbed on CePO4 were also measured to clarify the adsorption mode of the substrates (carbonyl compounds and alcohols). One band that corresponded to νCO (1699 cm−1) in acetone was shifted to a lower position than that of acetone adsorbed on CeO2 (1673 cm−1) or in the gas phase (1731 cm−1) (Fig. 16(c)), which indicated that CePO4 has moderate Lewis acid sites compared with the strength of the acid sites on CeO2. The new bands in the νCH3 region and negative broad band in the νO–H region were observed by the exposure of CePO4 to methanol (Fig. 16(d)), which indicated that methanol may be adsorbed on the surface molecularly via hydrogen bonds. Therefore, CePO4 nanorods most likely act as a bifunctional catalyst through interaction of the uniform Lewis acid sites and weak base sites with carbonyl compound and alcohols, respectively.
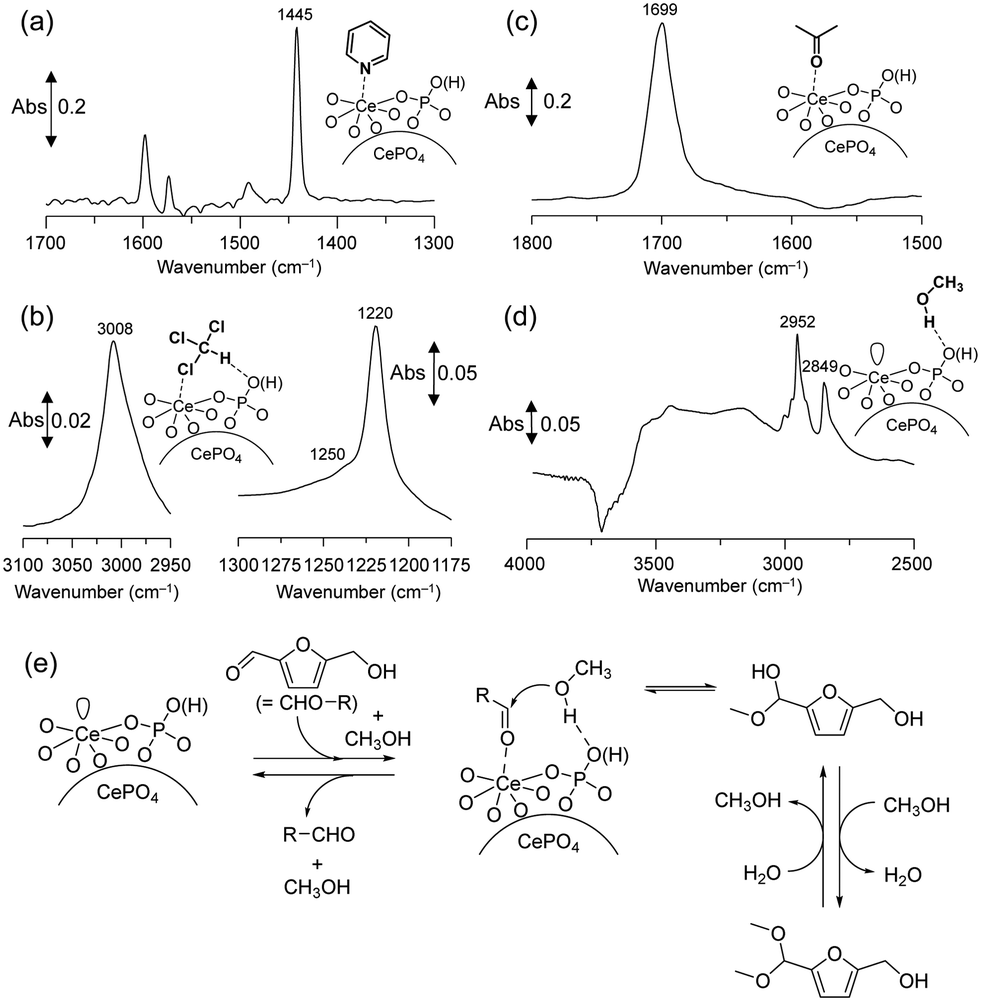 | ||
| Fig. 16 Difference IR spectra for (a) pyridine-, (b) chloroform-, (c) acetone-, and (d) methanol-adsorbed CePO4 at 25 °C. (e) Proposed reaction mechanism for the CePO4-catalyzed acetalization of HMF with methanol. (Reproduced from ref. 49 with permission from Royal Society of Chemistry). | ||
Based on the results of these investigations, Fig. 16(e) shows a proposed reaction mechanism for the CePO4-catalyzed acetalization of HMF with methanol. First, the activation of both HMF and methanol by adjacent acid–base pairs on CePO4 facilitates the nucleophilic attack of the OH group in methanol on the carbon atom of the carbonyl group in HMF to give the corresponding hemiacetal derivative. Further reaction of the hemiacetal with methanol then occurs, most likely with the assistance of the catalyst, to give the corresponding acetal derivative. Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) observations confirmed that the dominant facets exposed on the CePO4 nanorods are (110) and (100), with (110) being particularly dominant.54 The surface density (1.6 nm−2) of Lewis acidic Ce3+ cations at the (110) facet was in good agreement with the experimental value (1.6 nm−2) calculated from SBET and pyridine-IR results, which supports the presence of uniform surface Ce species on the CePO4 nanorods.
The acid–base catalysis of various perovskite oxides165–167 has been reported since the 1990s, and is typically estimated by titration using Hammett indicators, spectroscopy measurements such as IR and electron spin resonance (ESR) using probe molecules, and simple test reactions (e.g., 2-propanol conversion, esterification, and transfer hydrogenation).168,169 Metal cations generally act as Lewis acid sites and oxygen anions as base sites, and the catalytic performance is strongly dependent on the bulk and surface configurations. Foo and co-workers systematically investigated the acidity/basicity of Ti and Zr-based perovskite oxides such as SrTiO3, BaTiO3, SrZrO3, and BaZrO3 with a focus on the conversion of 2-propanol to propylene and acetone mainly promoted by acid and base sites, respectively.170 However, the catalytic application of perovskite oxides to liquid-phase organic reactions has been limited.
First, perovskite oxide nanoparticles with d0-transition metal cations at B-sites were investigated as catalysts for liquid-phase organic reaction such as cyanosilylation and Knoevenagel condensation, which are reactions known to be promoted by acid and/or base sites.55,56 From the catalyst effect on the cyanosilylation of acetophenone with trimethylsilyl cyanide (TMSCN) without a catalyst pretreatment, titanates showed higher activity than zirconates, LiNbO3, and KNbO3 and SrTiO3 nanoparticles were the most effective catalyst. In addition, the catalytic activity and specific surface area of SrTiO3 nanoparticles could be improved by a simple change of the atmosphere from N2 to air during calcination of the precursor. Although commercially available SrTiO3 (4 m2 g−1) was inactive for this cyanosilylation, the reaction occurred smoothly over SrTiO3 with a large specific surface area calcined in N2-air (denoted as SrTiO3_N2-air, 46 m2 g−1) and the reaction rate was 2.5 times greater than that for SrTiO3 (denoted as SrTiO3_air, 30 m2 g−1) calcined in air (eqn (13)). The SrTiO3_N2-air catalyst could be easily recovered from the reaction mixture by simple filtration and the recovered catalyst was reused five times without a substantial loss of catalytic performance. In addition, the reaction proceeded efficiently under solvent-free conditions with a high product formation rate comparable to that for benchmark catalysts such as Sn-Mont171 and Al-MCM-41,172 even though the SrTiO3_N2-air catalyst did not require thermal pretreatment.
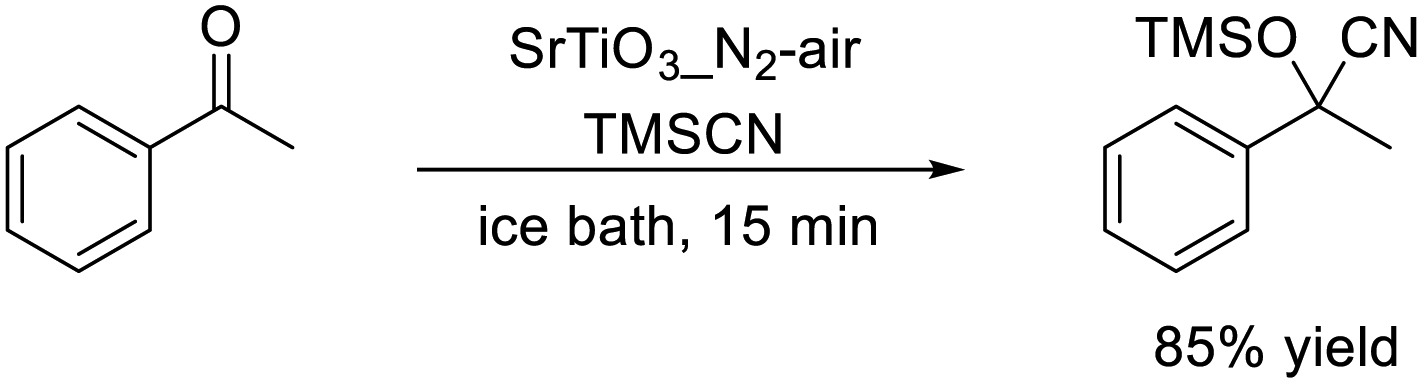 | (13) |
The acid–base properties of SrTiO3_N2-air were investigated using pyridine-, chloroform-, and acetophenone-adsorbed IR and CO2-TPD measurements, which showed the presence of both Lewis acid and base sites in the catalyst and that acetophenone was activated by Lewis acid Ti sites. Next, reactions were conducted by the addition of pyridine and acetic acid (AcOH) to poison the acidic and basic active sites on SrTiO3_N2-air, respectively (Fig. 17(a)). The reaction rate decreased with increasing amounts of both poisoning molecules, which suggested that both the acid and base sites on the catalyst surface contribute to the cyanosilylation, although the poisoning effect with AcOH was much stronger than that with pyridine. These results suggest that the reaction is mainly promoted by the base sites with possible cooperative action of the acid sites on the catalyst.
 | ||
| Fig. 17 (a) Poisoning effect of pyridine (black circle) and AcOH (white circle) and (b) possible reaction mechanism for cyanosilylation of carbonyl compounds with TMSCN over SrTiO3_N2-air catalyst. Adsorption configurations of TMSCN on SrTiO3 for (c) Sr–O terminated (100) and (d) Ti–O terminated (100) surfaces from DFT geometry optimization. (Reproduced from ref. 56 with permission from The American Chemical Society). | ||
Fig. 17(b) shows a possible mechanism for cyanosilylation over the SrTiO3_N2-air catalyst. First, TMSCN is activated by surface oxygens that act as base sites. Similarly, carbonyl compounds are adsorbed and activated by Lewis acid sites on the catalyst surface. Such cooperative activation facilitates the nucleophilic attack of a CN− anion at the carbon atom of the carbonyl groups, followed by desorption of the corresponding products. In this case, the distance between the acid and base sites may also be important for cooperative acid–base catalysis, and the presence of adjacent acid–base sites was confirmed by CHCl3-adsorbed IR spectroscopy measurements.
To investigate the effect of the calcination atmosphere, the surface atomic ratio of Sr to Ti (Sr/Ti) was estimated from the Sr 3d and Ti 2p peaks in the XPS spectra of the different catalysts. The Sr/Ti value for SrTiO3_N2-air was 1.51 and higher than that for SrTiO3_air (1.37), which suggested that the enrichment of Sr–O termination at the surface of SrTiO3 led to an increase in the amount of base sites. The effect of the outermost surface structure of SrTiO3(100) facets of SrTiO3 for the activation of TMSCN, was investigated using DFT calculations (Fig. 17(c) and (d)). The adsorption energy of chemisorbed TMSCN on the Sr–O terminated (100) facet was calculated to be −2.01 eV and lower than that (−1.01 eV) on the Ti-terminated (100) facet, which suggests that TMSCN is strongly activated on the SrO-rich surface. These results are consistent with the basicity and catalytic activity differences between the SrTiO3 samples treated in different atmospheres. Such a significant effect of the surface composition of perovskite oxides on the catalysis has been reported by Polo-Garzon and co-workers; thermal treatment of commercially available SrTiO3 in oxygen improved the selectivity toward acetone in 2-propanol conversion compared with that for bare SrTiO3.173 In contrast, SrTiO3 treated with HNO3 showed high selectivity toward propylene produced from 2-propanol by dehydration on acid sites. Various techniques, including calculation of the surface Sr/Ti ratio using XPS and low-energy ion scattering (LEIS) analysis, which is a more surface-sensitive measurement, direct observation of catalyst surfaces including HAADF-STEM analysis and DFT calculations revealed that the basicity of the Sr-rich surface was enriched by thermal treatment of SrTiO3 and the Ti-rich surface with acidic characteristics likely appeared due to chemical etching of SrTiO3 by HNO3. Other approaches based on the morphology/facets of perovskite oxides170 and the engineering of oxygen vacancies on the catalyst surface174–176 have been reported to be effective for controlling the surface composition.
We applied the Ti-based perovskite oxide nanoparticles to the Knoevenagel condensation of benzaldehyde with phenylacetonitrile, which is an inactive donor with a high pKa value (21.9).55 Among the tested Ti-based perovskite oxides, SrTiO3_N2-air showed the highest catalytic activity for the reaction and the yield of product (88%) was higher than that over SrTiO3_air (48%) (eqn (14)). SrTiO3_N2-air could be reused without a substantial loss of its catalytic performance. Only a few examples of reusable catalytic systems applicable to phenylacetonitrile as a donor have been reported, and this study provides the first example of recyclable heterogeneous catalysts applicable to phenylacetonitrile.177,178 The reactivity of diethyl malonate was lower than that of phenylacetonitrile, despite a lower pKa value (16.4) of diethyl malonate than phenylacetonitrile, which suggests a specific interaction of the active methylene compounds containing cyano groups with the SrTiO3 surface, as supported by IR measurements of phenylacetonitrile on SrTiO3. Lewis acid sites on the SrTiO3 surface can interact with active methylene compounds to promote the abstraction of an acidic α-proton by a base site adjacent to an acid site. This cooperative activation by acid–base pair sites promotes the coupling reaction of benzaldehyde with active methylene compounds having cyano groups to form α,β-unsaturated nitrile.
 | (14) |
Summary and outlook
Catalyst design methodology based on the nanostructural control of crystalline complex oxides is expected not only to lead to the discovery of new catalytic functions of materials that have not previously been investigated as catalysts, but also to lead to the development of new catalysts through the structural and compositional effects that emerge from combinations of versatile elements. The establishment of two unique and versatile synthetic methods, the amino acid-aided method and the precursor crystallization method, is key to the successful development of efficient catalytic systems for selective oxidation and acid–base reactions based on nanocatalyst materials such as perovskite oxides, manganese oxides, and metal phosphates. Furthermore, the combined approaches of precise experimental methods and highly accurate computational methods will provide new proposals and demonstrations of mechanistic aspects that include the catalytic roles of oxygen vacancies, bulk and surface structures, and the concerted effects of redox and acid–base properties in metal oxides.The unique structures and valences of hexagonal perovskite oxides and β-MnO2 contribute to promote the selective oxidation of alkanes, alcohols, and sulfides with O2 as the sole oxidant under milder reaction conditions than previously reported catalysts. In the case of the Mars–van Krevelen mechanism where surface lattice oxygen is involved in the reaction, the oxygen vacancy formation energy becomes an important descriptor that enables the design of catalysts with desirable oxidation properties. The concerted action of redox and acid–base properties is also an effective method for the activation of C–H bonds using Murdochite-type oxides. In addition, the combination of Lewis acidic metal species and base units makes it possible to develop acid–base catalysts based on a variety of metal phosphates and perovskites. The use of weakly basic phosphate units is effective for the conversion of unstable biomass-based substrates and the direct selective oxidation of CH4 through the inhibition effect of successive oxidation. The synergistic and cooperative activation of molecules on acid and base sites results in high catalytic performance when using more basic oxo units.
Further elemental complexity is expected to be an important factor in the development of high-performance catalysts in the future, as exemplified by the recent developments of multimetal-doped29,38,40,179 and/or high-entropy metal oxides180,181 as catalysts for selective oxidation, NO reduction, and electrochemical oxygen evolution reaction and energy storage materials. Control of electronic structures (e.g., spin-state, magnetic ordering)179,182 and design of catalytically active interfaces based on a synergy effect of binary oxide composites183,184 would be a promising approach to achieve highly active and durable catalysts. There is also an urgent need to design and develop catalysts in a shorter time through limited experiments. Therefore, in addition to the present approaches based on precise experiments and first-principle calculations, these fields will be developed by the incorporation of new methods such as high-throughput synthesis and machine learning. Such a continuous approach to catalyst design is likely to revolutionize the entire process of catalyst development to achieve potentially difficult reactions required to build a sustainable society.
Author contributions
Takeshi Aihara and Keiju Wachi: writing – original draft. Keigo Kamata: writing – original draft/review editing; conceptualization; supervision; funding acquisition.Data availability
No primary research results, software or code was included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was funded in part by JST CREST (JPMJCR22O1), JSPS KAKENHI grant number 24H00393, and “Design & Engineering by Joint Inverse Innovation for Materials Architecture” program of the Ministry of Education, Culture, Science, Sports and Technology (MEXT), Japan.References
- S. Mitchell, R. Qin, N. Zheng and J. Perez-Ramirez, Nat. Nanotechnol., 2021, 16, 129–139 CrossRef CAS PubMed.
- X. F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- F. X. Xiao, J. Miao, H. B. Tao, S. F. Hung, H. Y. Wang, H. B. Yang, J. Chen, R. Chen and B. Liu, Small, 2015, 11, 2115–2131 CrossRef CAS.
- F. Zaera, Chem. Rev., 2022, 122, 8594–8757 CrossRef CAS.
- Q. Fu, F. Yang and X. Bao, Acc. Chem. Res., 2013, 46, 1692–1701 CrossRef CAS.
- Y. Wang, H. Arandiyan, J. Scott, A. Bagheri, H. Dai and R. Amal, J. Mater. Chem. A, 2017, 5, 8825–8846 RSC.
- G. Prieto, H. Tüysüz, N. Duyckaerts, J. Knossalla, G. H. Wang and F. Schüth, Chem. Rev., 2016, 116, 14056–14119 CrossRef CAS.
- G. Li and Z. Tang, Nanoscale, 2014, 6, 3995–4011 RSC.
- Y. Nakaya and S. Furukawa, Chem. Rev., 2023, 123, 5859–5947 CrossRef CAS PubMed.
- B. W. J. Chen, L. Xu and M. Mavrikakis, Chem. Rev., 2021, 121, 1007–1048 CrossRef CAS PubMed.
- H. W. Cheng, S. Wang, G. Chen, Z. Liu, D. Caracciolo, M. Madiou, S. Shan, J. Zhang, H. He, R. Che and C. J. Zhong, Adv. Energy Mater., 2022, 12, 2202097 CrossRef CAS.
- S. W. Chee, T. Lunkenbein, R. Schlogl and B. Roldan Cuenya, Chem. Rev., 2023, 123, 13374–13418 CrossRef CAS PubMed.
- K. Takahashi, J. Ohyama, S. Nishimura, J. Fujima, L. Takahashi, T. Uno and T. Taniike, Chem. Commun., 2023, 59, 2222–2238 RSC.
- J. C. Vedrine, ChemSusChem, 2019, 12, 577–588 CrossRef CAS.
- J. T. Grant, J. M. Venegas, W. P. McDermott and I. Hermans, Chem. Rev., 2018, 118, 2769–2815 CrossRef CAS PubMed.
- S. Ishikawa, Z. Zhang and W. Ueda, ACS Catal., 2018, 8, 2935–2943 CrossRef CAS.
- N. Mizuno and K. Kamata, Coord. Chem. Rev., 2011, 255, 2358–2370 CrossRef CAS.
- S. Zhang, Z. Wu, X. Liu, K. Hua, Z. Shao, B. Wei, C. Huang, H. Wang and Y. Sun, Top. Catal., 2021, 64, 371–394 CrossRef CAS.
- G. Vilé, D. Albani, N. Almora-Barrios, N. López and J. Pérez-Ramírez, ChemCatChem, 2015, 8, 21–33 CrossRef.
- T. Otroshchenko, G. Jiang, V. A. Kondratenko, U. Rodemerck and E. V. Kondratenko, Chem. Soc. Rev., 2021, 50, 473–527 RSC.
- H. Hattori and Y. Ono, Solid Acid Catalysis: From Fundamentals to Applications, Taylor & Francis Group, New York, 2015 Search PubMed.
- Y. Ono and H. Hattori, Solid Base Catalysis, Springer, Berlin, Heidelberg, 2011 Search PubMed.
- J. C. Védrine, Appl. Catal., A, 2019, 575, 170–179 CrossRef.
- P. Sudarsanam, N. K. Gupta, B. Mallesham, N. Singh, P. N. Kalbande, B. M. Reddy and B. F. Sels, ACS Catal., 2021, 11, 13603–13648 CrossRef CAS.
- M. Hara, K. Nakajima and K. Kamata, Sci. Technol. Adv. Mater., 2015, 16, 034903 CrossRef.
- A. Shivhare, A. Kumar and R. Srivastava, Green Chem., 2021, 23, 3818–3841 RSC.
- F. Liu, Y. Yu and H. He, Chem. Commun., 2014, 50, 8445–8463 RSC.
- T. Hirakawa, Y. Shimokawa, W. Tokuzumi, T. Sato, M. Tsushida, H. Yoshida, S. Hinokuma, J. Ohyama and M. Machida, ACS Catal., 2019, 9, 11763–11773 CrossRef CAS.
- Y. Qu and X. Duan, Chem. Soc. Rev., 2013, 42, 2568–2580 RSC.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS.
- X. Li, H. Zhao, J. Liang, Y. Luo, G. Chen, X. Shi, S. Lu, S. Gao, J. Hu, Q. Liu and X. Sun, J. Mater. Chem. A, 2021, 9, 6650–6670 RSC.
- J. Hwang, R. R. Rao, L. Giordano, Y. Katayama, Y. Yu and Y. Shao-Horn, Science, 2017, 358, 751–756 CrossRef CAS PubMed.
- Y. Kobayashi, O. J. Hernandez, T. Sakaguchi, T. Yajima, T. Roisnel, Y. Tsujimoto, M. Morita, Y. Noda, Y. Mogami, A. Kitada, M. Ohkura, S. Hosokawa, Z. Li, K. Hayashi, Y. Kusano, J. Kim, N. Tsuji, A. Fujiwara, Y. Matsushita, K. Yoshimura, K. Takegoshi, M. Inoue, M. Takano and H. Kageyama, Nat. Mater., 2012, 11, 507–511 CrossRef CAS PubMed.
- X. He, S. Kimura, T. Katase, T. Tadano, S. Matsuishi, M. Minohara, H. Hiramatsu, H. Kumigashira, H. Hosono and T. Kamiya, Adv. Sci., 2024, 11, e2307058 CrossRef PubMed.
- Y. Chen, Z. Lun, X. Zhao, K. P. Koirala, L. Li, Y. Sun, C. A. O'Keefe, X. Yang, Z. Cai, C. Wang, H. Ji, C. P. Grey, B. Ouyang and G. Ceder, Nat. Mater., 2024, 23, 535–542 CrossRef CAS.
- N. J. Szymanski, P. Nevatia, C. J. Bartel, Y. Zeng and G. Ceder, Nat. Commun., 2023, 14, 6956 CrossRef CAS.
- W. Ueda, Crystalline Metal Oxide Catalysts, Springer, Singapore, 2022 Search PubMed.
- K. Amakawa, J. M. Mauss, P. Muller, B. Hinrichsen, S. Hirth, A. Bader, S. W. T. Price, S. D. M. Jacques and J. Macht, Sci. Adv., 2023, 9, eadh5331 CrossRef CAS PubMed.
- S. Ishikawa, Y. Yamada, N. Kashio, N. Noda, K. Shimoda, M. Hayashi, T. Murayama and W. Ueda, ACS Catal., 2021, 11, 10294–10307 CrossRef CAS.
- K. Kamata, Bull. Chem. Soc. Jpn., 2019, 92, 133–151 CrossRef CAS.
- M. Koutani, E. Hayashi, K. Kamata and M. Hara, J. Am. Chem. Soc., 2022, 144, 14090–14100 CrossRef CAS PubMed.
- Y. Yamaguchi, R. Aono, E. Hayashi, K. Kamata and M. Hara, ACS Appl. Mater. Interfaces, 2020, 12, 36004–36013 CrossRef CAS.
- K. Sugahara, K. Kamata, S. Muratsugu and M. Hara, ACS Omega, 2017, 2, 1608–1616 CrossRef CAS PubMed.
- S. Shibata, K. Kamata and M. Hara, ChemCatChem, 2022, 14, e202200395 CrossRef CAS.
- E. Hayashi, T. Tamura, T. Aihara, K. Kamata and M. Hara, ACS Appl. Mater. Interfaces, 2022, 14, 6528–6537 CrossRef CAS PubMed.
- S. Shibata, K. Kamata and M. Hara, Catal. Sci. Technol., 2021, 11, 2369–2373 RSC.
- S. Shibata, K. Sugahara, K. Kamata and M. Hara, Chem. Commun., 2018, 54, 6772–6775 RSC.
- K. Kamata, K. Sugahara, Y. Kato, S. Muratsugu, Y. Kumagai, F. Oba and M. Hara, ACS Appl. Mater. Interfaces, 2018, 10, 23792–23801 CrossRef CAS.
- S. Kawasaki, K. Kamata and M. Hara, ChemCatChem, 2016, 8, 3247–3253 CrossRef CAS.
- A. Matsuda, T. Aihara, S. Kiyohara, Y. Kumagai, M. Hara and K. Kamata, ACS Appl. Nano Mater., 2024, 7, 10155–10167 CrossRef CAS.
- A. Matsuda, K. Obara, A. Ishikawa, M.-H. Tsai, C.-H. Wang, Y.-C. Lin, M. Hara and K. Kamata, Catal. Sci. Technol., 2023, 13, 5180–5189 RSC.
- A. Matsuda, H. Tateno, K. Kamata and M. Hara, Catal. Sci. Technol., 2021, 11, 6987–6998 RSC.
- A. Sato, S. Ogo, K. Kamata, Y. Takeno, T. Yabe, T. Yamamoto, S. Matsumura, M. Hara and Y. Sekine, Chem. Commun., 2019, 55, 4019–4022 RSC.
- T. Aihara, W. Aoki, M. Hara and K. Kamata, Catal. Today, 2024, 428, 114448 CrossRef CAS.
- T. Aihara, W. Aoki, S. Kiyohara, Y. Kumagai, K. Kamata and M. Hara, ACS Appl. Mater. Interfaces, 2023, 15, 17957–17968 CrossRef CAS.
- S. Kanai, I. Nagahara, Y. Kita, K. Kamata and M. Hara, Chem. Sci., 2017, 8, 3146–3153 RSC.
- K. Kamata and K. Sugahara, Catalysts, 2017, 7, 345 CrossRef.
- E. Hayashi, Y. Yamaguchi, Y. Kita, K. Kamata and M. Hara, Chem. Commun., 2020, 56, 2095–2098 RSC.
- K. Kamata, N. Kinoshita, M. Koutani, R. Aono, E. Hayashi and M. Hara, Catal. Sci. Technol., 2022, 12, 6219–6230 RSC.
- E. Hayashi, Y. Yamaguchi, K. Kamata, N. Tsunoda, Y. Kumagai, F. Oba and M. Hara, J. Am. Chem. Soc., 2019, 141, 890–900 CrossRef CAS.
- E. Hayashi, T. Komanoya, K. Kamata and M. Hara, ChemSusChem, 2017, 10, 654–658 CrossRef CAS.
- Y. Sugawara, K. Kamata, A. Matsuda and T. Yamaguchi, ACS Appl. Energy Mater., 2023, 6, 7928–7934 CrossRef CAS.
- Y. Sugawara, S. Ueno, K. Kamata and T. Yamaguchi, ChemElectroChem, 2022, 9 Search PubMed.
- Y. Sugawara, T. Hihara, G. M. Anilkumar, K. Kamata and T. Yamaguchi, Sustainable Energy Fuels, 2021, 5, 1374–1378 RSC.
- Y. Sugawara, K. Kamata, A. Ishikawa, Y. Tateyama and T. Yamaguchi, ACS Appl. Energy Mater., 2021, 4, 3057–3066 CrossRef CAS.
- Y. Sugawara, K. Kamata, E. Hayashi, M. Itoh, Y. Hamasaki and T. Yamaguchi, ChemElectroChem, 2021, 8, 4466–4471 CrossRef CAS.
- K. Ariga and M. Ebara, Materials Nanoarchitectonics, Wiley-VCH, Weinheim, 2018 Search PubMed.
- E. Pomerantseva, F. Bonaccorso, X. Feng, Y. Cui and Y. Gogotsi, Science, 2019, 366, eaan8285 CrossRef CAS PubMed.
- D. Portehault, S. Delacroix, G. Gouget, R. Grosjean and T.-H.-C. Chan-Chang, Acc. Chem. Res., 2018, 51, 930–939 CrossRef CAS.
- M. B. Gawande, A. Goswami, F.-X. Felpin, T. Asefa, X. Huang, R. Silva, X. Zou, R. Zboril and R. S. Varma, Chem. Rev., 2016, 116, 3722–3811 CrossRef CAS PubMed.
- R. Ma and T. Sasaki, Acc. Chem. Res., 2015, 48, 136–143 CrossRef CAS.
- Q. Lu, Y. Yu, Q. Ma, B. Chen and H. Zhang, Adv. Mater., 2016, 28, 1917–1933 CrossRef CAS.
- Y. Li and W. Shen, Chem. Soc. Rev., 2014, 43, 1543–1574 RSC.
- P. Granger, V. I. Parvulescu, V. I. Parvulescu and W. Prellier, Perovskites and Related Mixed Oxides, Wiley-VCH, Weinheim, 2016 Search PubMed.
- D. D. Athayde, D. F. Souza, A. M. A. Silva, D. Vasconcelos, E. H. M. Nunes, J. C. Diniz da Costa and W. L. Vasconcelos, Ceram. Int., 2016, 42, 6555–6571 CrossRef CAS.
- S. Royer, D. Duprez, F. Can, X. Courtois, C. Batiot-Dupeyrat, S. Laassiri and H. Alamdari, Chem. Rev., 2014, 114, 10292–10368 CrossRef CAS.
- D. Chen, C. Chen, Z. M. Baiyee, Z. Shao and F. Ciucci, Chem. Rev., 2015, 115, 9869–9921 CrossRef CAS.
- R. I. Walton, Chem. Soc. Rev., 2002, 31, 230–238 RSC.
- J. A. Darr, J. Zhang, N. M. Makwana and X. Weng, Chem. Rev., 2017, 117, 11125–11238 CrossRef CAS PubMed.
- C. D. Evans, S. A. Kondrat, P. J. Smith, T. D. Manning, P. J. Miedziak, G. L. Brett, R. D. Armstrong, J. K. Bartley, S. H. Taylor, M. J. Rosseinsky and G. J. Hutchings, Faraday Discuss., 2016, 188, 427 RSC.
- Y. Teraoka, H. Kakebayashi, I. Moriguchi and S. Kagawa, Chem. Lett., 1991, 673 CrossRef CAS.
- L. Dimesso, in Handbook of Sol-Gel Science and Technology, ed. L. Klein, M. Aparicio and A. Jitianu, Springer, Cham, 2nd edn, 2018, pp. 1067–1088 Search PubMed.
- M. Kakihana, J. Sol-Gel Sci. Technol., 1996, 6, 7–55 CrossRef CAS.
- A. Varma, A. S. Mukasyan, A. S. Rogachev and K. V. Manukyan, Chem. Rev., 2016, 116, 14493–14586 CrossRef CAS.
- H. M. Zhang, Y. Teraoka and N. Yamazoe, Hyomen Kagaku, 1987, 8, 23–29 CrossRef CAS.
- N. Rezlescu, E. Rezlescu, P. D. Popa, C. Doroftei and M. Ignat, Composites, Part B, 2014, 60, 515–522 CrossRef CAS.
- K. Langfeld, R. Marschner, B. Frank and R. Schomaecker, ChemCatChem, 2011, 3, 1354–1358 CrossRef CAS.
- S. Fop, K. S. McCombie, E. J. Wildman, J. M. S. Skakle and A. C. McLaughlin, Chem. Commun., 2019, 55, 2127–2137 RSC.
- P. Hu, P. Hu, T. D. Vu, M. Li, S. Wang, Y. Ke, X. Zeng, L. Mai and Y. Long, Chem. Rev., 2023, 123, 4353–4415 CrossRef CAS PubMed.
- W. T. Broomhead, W. Tian, J. E. Herrera and Y.-H. C. Chin, ACS Catal., 2022, 12, 11801–11820 CrossRef CAS.
- P. Kumar, V. Tomar, D. Kumar, R. K. Joshi and M. Nemiwal, Tetrahedron, 2022, 106–107, 132641 CrossRef CAS.
- S. Rahim Pouran, A. A. Abdul Raman and W. M. A. Wan Daud, J. Cleaner Prod., 2014, 64, 24–35 CrossRef CAS.
- X. Deng and H. Tüysüz, ACS Catal., 2014, 4, 3701–3714 CrossRef CAS.
- M. Haneda, Y. Kintaichi, N. Bion and H. Hamada, Appl. Catal., B, 2003, 46, 473–482 CrossRef CAS.
- K. Suzuki, T. Yamaguchi, K. Matsushita, C. Iitsuka, J. Miura, T. Akaogi and H. Ishida, ACS Catal., 2013, 3, 1845–1849 CrossRef CAS.
- K. Fominykh, J. M. Feckl, J. Sicklinger, M. Doeblinger, S. Boecklein, J. Ziegler, L. Peter, J. Rathousky, E.-W. Scheidt, T. Bein and D. Fattakhova-Rohlfing, Adv. Funct. Mater., 2014, 24, 3123–3129 CrossRef CAS.
- D. Ren, Y. Deng, A. D. Handoko, C. S. Chen, S. Malkhandi and B. S. Yeo, ACS Catal., 2015, 5, 2814–2821 CrossRef CAS.
- W. Wang, C. Deng, S. Xie, Y. Li, W. Zhang, H. Sheng, C. Chen and J. Zhao, J. Am. Chem. Soc., 2021, 143, 2984–2993 CrossRef CAS PubMed.
- Q. Feng, H. Kanoh and K. Ooi, J. Mater. Chem., 1999, 9, 319–333 RSC.
- S. L. Suib, Acc. Chem. Res., 2008, 41, 479–487 CrossRef CAS.
- W. Si, Y. Wang, Y. Peng, X. Li, K. Li and J. Li, Chem. Commun., 2015, 51, 14977–14980 RSC.
- X. F. Shen, Y. S. Ding, J. Liu, J. Cai, K. Laubernds, R. P. Zerger, A. Vasiliev, M. Aindow and S. L. Suib, Adv. Mater., 2005, 17, 805–809 CrossRef CAS.
- X. Wang and Y. Li, J. Am. Chem. Soc., 2002, 124, 2880–2881 CrossRef CAS PubMed.
- W. N. Li, J. Yuan, X. F. Shen, S. Gomez-Mower, L. P. Xu, S. Sithambaram, M. Aindow and S. L. Suib, Adv. Funct. Mater., 2006, 16, 1247–1253 CrossRef CAS.
- G.-H. Qiu, H. Huang, S. Dharmarathna, E. Benbow, L. Stafford and S. L. Suib, Chem. Mater., 2011, 23, 3892–3901 CrossRef CAS.
- F. Jiao and P. G. Bruce, Adv. Mater., 2007, 19, 657–660 CrossRef CAS.
- Y. Ren, A. R. Armstrong, F. Jiao and P. G. Bruce, J. Am. Chem. Soc., 2010, 132, 996–1004 CrossRef CAS.
- D. A. Kitchaev, H. Peng, Y. Liu, J. Sun, J. P. Perdew and G. Ceder, Phys. Rev. B, 2016, 93, 045132 CrossRef.
- Y. F. Li, S. C. Zhu and Z. P. Liu, J. Am. Chem. Soc., 2016, 138, 5371–5379 CrossRef CAS.
- O. Ghodbane, J. L. Pascal and F. Favier, ACS Appl. Mater. Interfaces, 2009, 1, 1130–1139 CrossRef CAS.
- M. Sun, B. Lan, T. Lin, G. Cheng, F. Ye, L. Yu, X. Cheng and X. Zheng, CrystEngComm, 2013, 15, 7010 RSC.
- J.-J. Zhu, L.-L. Yu and J.-T. Zhao, J. Power Sources, 2014, 270, 411–417 CrossRef CAS.
- L.-L. Yu, J.-J. Zhu and J.-T. Zhao, J. Mater. Chem. A, 2014, 2, 9353 RSC.
- Y. Jiang, L. Yuan, X. Wang, W. Zhang, J. Liu, X. Wu, K. Huang, Y. Li, Z. Liu and S. Feng, Angew. Chem., Int. Ed., 2020, 59, 22659–22666 CrossRef CAS PubMed.
- L. Schwiedrzik, V. Brieskorn and L. Gonzalez, ACS Catal., 2021, 11, 13320–13329 CrossRef CAS PubMed.
- Y. Liu, C. Wang, S. Zhao, L. Zhang, K. Zhang, F. Li and J. Chen, Chem. Sci., 2020, 12, 1062–1067 RSC.
- Y. F. Shen, R. P. Zerger, R. N. Deguzman, S. L. Suib, L. McCurdy, D. I. Potter and C. L. O’Young, Science, 1993, 260, 511–515 CrossRef CAS.
- J.-Y. Wang, G.-G. Xia, Y.-G. Yin, S. L. Suib and C. L. O'Young, J. Catal., 1998, 176, 275–284 CrossRef CAS.
- Y. Liu and P. Zhang, Appl. Catal., A, 2017, 530, 102–110 CrossRef CAS.
- Q. Feng, H. Kanoh, Y. Miyai and K. Ooi, Chem. Mater., 1995, 7, 1722–1727 CrossRef CAS.
- B. Zhang, C. D. Quilty, L. Wang, X. Hu, A. Poyraz, D. C. Bock, Y. R. Li, L. Gerhardt, L. Wu, Y. Zhu, A. C. Marschilok, E. S. Takeuchi and K. J. Takeuchi, J. Electrochem. Soc., 2020, 167, 110528 CrossRef CAS.
- D. C. Golden, C. C. Chen and J. B. Dixon, Science, 1986, 231, 717–719 CrossRef CAS.
- X. H. Feng, W. F. Tan, F. Liu, J. B. Wang and H. D. Ruan, Chem. Mater., 2004, 16, 4330–4336 CrossRef CAS.
- S. Dharmarathna, C. K. King’ondu, W. Pedrick, L. Pahalagedara and S. L. Suib, Chem. Mater., 2012, 24, 705–712 CrossRef CAS.
- Y. Miyamoto, Y. Kuroda, T. Uematsu, H. Oshikawa, N. Shibata, Y. Ikuhara, K. Suzuki, M. Hibino, K. Yamaguchi and N. Mizuno, Sci. Rep., 2015, 5, 15011 CrossRef CAS PubMed.
- Y. Miyamoto, Y. Kuroda, T. Uematsu, H. Oshikawa, N. Shibata, Y. Ikuhara, K. Suzuki, M. Hibino, K. Yamaguchi and N. Mizuno, ChemNanoMat, 2016, 2, 297–306 CrossRef CAS.
- F. A. Al-Sagheer and M. I. Zaki, Microporous Mesoporous Mater., 2004, 67, 43–52 CrossRef CAS.
- S. Min and Y. Kim, Minerals, 2020, 10, 884 CrossRef CAS.
- K. A. Malinger, K. Laubernds, Y.-C. Son and S. L. Suib, Chem. Mater., 2004, 16, 4296–4303 CrossRef CAS.
- M. Sajid, X. Zhao and D. Liu, Green Chem., 2018, 20, 5427–5453 RSC.
- Y. Rang, W. Lv, Y. Wang, H. Chen, Y. Li, C. Wang and G. Xu, ChemCatChem, 2024, 16, e202301376 CrossRef.
- B. Sarmah, R. Srivastava, P. Manjunathan and G. V. Shanbhag, ACS Sustainable Chem. Eng., 2015, 3, 2933–2943 CrossRef CAS.
- J. R. Kona, C. K. King’ondu, A. R. Howell and S. L. Suib, ChemCatChem, 2014, 6, 749–752 CrossRef CAS.
- Y. Wang, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2012, 51, 7250–7253 CrossRef CAS PubMed.
- B. Chen, B. Wu, L. Yu, M. Crocker and C. Shi, ACS Catal., 2020, 10, 6176–6187 CrossRef CAS.
- H. Chen, Y. Wang and Y.-K. Lv, RSC Adv., 2016, 6, 54032–54040 RSC.
- S. Nakai, T. Uematsu, Y. Ogasawara, K. Suzuki, K. Yamaguchi and N. Mizuno, ChemCatChem, 2018, 10, 1096–1106 CrossRef CAS.
- K. Yamaguchi, H. Kobayashi, Y. Wang, T. Oishi, Y. Ogasawara and N. Mizuno, Catal. Sci. Technol., 2013, 3, 318–327 RSC.
- T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329–2363 CrossRef CAS.
- H. Chu, P. Zhang and S. Dai, ACS Catal., 2015, 5, 6370–6385 CrossRef.
- Y. Meng, H. C. Genuino, C.-H. Kuo, H. Huang, S.-Y. Chen, L. Zhang, A. Rossi and S. L. Suib, J. Am. Chem. Soc., 2013, 135, 8594–8605 CrossRef CAS PubMed.
- N. F. Dummer, D. J. Willock, Q. He, M. J. Howard, R. J. Lewis, G. Qi, S. H. Taylor, J. Xu, D. Bethell, C. J. Kiely and G. J. Hutchings, Chem. Rev., 2023, 123, 6359–6411 CrossRef CAS PubMed.
- E. V. Kondratenko, T. Peppel, D. Seeburg, V. A. Kondratenko, N. Kalevaru, A. Martin and S. Wohlrab, Catal. Sci. Technol., 2017, 7, 366–381 RSC.
- A. R. Kulkarni, Z.-J. Zhao, S. Siahrostami, J. K. Nørskov and F. Studt, Catal. Sci. Technol., 2018, 8, 114–123 RSC.
- L. S. Andrade, H. H. L. B. Lima, C. T. P. Silva, W. L. N. Amorim, J. G. R. Poço, A. López-Castillo, M. V. Kirillova, W. A. Carvalho, A. M. Kirillov and D. Mandelli, Coord. Chem. Rev., 2023, 481, 215042 CrossRef CAS.
- K. Wachi, T. Yabe, T. Suzuki, K. Yonesato, K. Suzuki and K. Yamaguchi, Appl. Catal., B, 2022, 314, 121420 CrossRef CAS.
- T. Matsumoto, M. Saito, S. Ichikawa, K. Fujii, M. Yashima, W. Ueda and T. Motohashi, ChemCatChem, 2020, 12, 1968–1972 CrossRef CAS.
- T. Akiyama, R. Sei and S. Takenaka, Catal. Sci. Technol., 2021, 11, 5273–5281 RSC.
- Y. Wang and K. Otsuka, J. Catal., 1995, 155, 256–267 CrossRef CAS.
- V. Gomonaj and H. Toulhoat, ACS Catal., 2018, 8, 8263–8272 CrossRef CAS.
- J. Li, S. Zhou, J. Zhang, M. Schlangen, D. Usharani, S. Shaik and H. Schwarz, J. Am. Chem. Soc., 2016, 138, 11368–11377 CrossRef CAS PubMed.
- P. Han, R. Yan, Y. Wei, L. Li, J. Luo, Y. Pan, B. Wang, J. Lin, S. Wan, H. Xiong, Y. Wang and S. Wang, J. Am. Chem. Soc., 2023, 145, 10564–10575 CrossRef CAS.
- G. Busca, Chem. Rev., 2007, 107, 5366–5410 CrossRef CAS.
- E. Iglesia, D. G. Barton, J. A. Biscardi, M. J. L. Gines and S. L. Soled, Catal. Today, 1997, 38, 339–360 CrossRef.
- M. J. Climent, A. Corma, S. Iborra and M. J. Sabater, ACS Catal., 2014, 4, 870–891 CrossRef CAS.
- J.-A. Ma and D. Cahard, Angew. Chem., Int. Ed., 2004, 43, 4566–4583 CrossRef CAS.
- A. Clearfield and D. S. Thakur, Appl. Catal., 1986, 26, 1–26 CrossRef CAS.
- H. Kim, J. Park, I. Park, K. Jin, S. E. Jerng, S. H. Kim, K. T. Nam and K. Kang, Nat. Commun., 2015, 6, 8253 CrossRef CAS PubMed.
- Q. Cheng, X. Zhao, G. Yang, L. Mao, F. Liao, L. Chen, P. He, D. Pan and S. Chen, Energy Storage Mater., 2021, 41, 842–882 CrossRef.
- S. V. Dorozhkin and M. Epple, Angew. Chem., Int. Ed., 2002, 41, 3130–3146 CrossRef CAS.
- A. Shivhare, A. Kumar and R. Srivastava, Green Chem., 2021, 23, 3818–3841 RSC.
- H. Yook, J. Hwang, W. Yeo, J. Bang, J. Kim, T. Y. Kim, J.-S. Choi and J. W. Han, Adv. Mater., 2023, 35, 2204938 CrossRef CAS.
- G. Innocenti, E. Papadopoulos, G. Fornasari, F. Cavani, A. J. Medford and C. Sievers, ACS Catal., 2020, 10, 11936–11950 CrossRef CAS.
- M. Miyazaki, H. Saito, K. Ogasawara, M. Kitano and H. Hosono, J. Am. Chem. Soc., 2023, 145, 25976–25982 CrossRef CAS.
- C. Srilakshmi, R. Saraf and C. Shivakumara, ACS Omega, 2018, 3, 10503–10512 CrossRef CAS PubMed.
- J. Ohyama, Y. Zhang, J. Ito and A. Satsuma, ChemCatChem, 2017, 9, 2864–2868 CrossRef CAS.
- M. Daturi, G. Busca and R. Willey, Chem. Mater., 1995, 7, 2115–2126 CrossRef.
- S. Sugunan and V. Meera, React. Kinet. Catal. Lett., 1997, 62, 327–332 CrossRef CAS.
- G. S. Foo, Z. D. Hood and Z. Wu, ACS Catal., 2018, 8, 555–565 CrossRef CAS.
- J. Wang, Y. Masui, K. Watanabe and M. Onaka, Adv. Synth. Catal., 2009, 351, 553–557 CrossRef CAS.
- K. Iwanami, J.-C. Choi, B. Lu, T. Sakakura and H. Yasuda, Chem. Commun., 2008, 1002–1004 RSC.
- F. Polo-Garzon, S.-Z. Yang, V. Fung, G. S. Foo, E. E. Bickel, M. F. Chisholm, D. Jiang and Z. Wu, Angew. Chem., Int. Ed., 2017, 56, 9820–9824 CrossRef CAS PubMed.
- R.-K. Chen, T.-F. Yu, M.-X. Wu, T.-W. Tzeng, P.-W. Chung and Y.-C. Lin, ACS Sustainable Chem. Eng., 2018, 6, 11949–11958 CrossRef CAS.
- Y. Zheng, R. Zhang, L. Zhang, Q. Gu and Z.-A. Qiao, Angew. Chem., Int. Ed., 2021, 60, 4774–4781 CrossRef CAS.
- P. Xiao, J. Zhu, D. Zhao, Z. Zhao, F. Zaera and Y. Zhu, ACS Appl. Mater. Interfaces, 2019, 11, 15517–15527 CrossRef CAS.
- T. Hara, S. Kanai, K. Mori, T. Mizugaki, K. Ebitani, K. Jitsukawa and K. Kaneda, J. Org. Chem., 2006, 71, 7455–7462 CrossRef CAS PubMed.
- K. Ebitani, K. Motokura, K. Mori, T. Mizugaki and K. Kaneda, J. Org. Chem., 2006, 71, 5440–5447 CrossRef CAS.
- J. S. Kim, B. Kim, H. Kim and K. Kang, Adv. Energy Mater., 2018, 8, 1702774 CrossRef.
- X. Zou, Y. R. Zhang, Z. P. Huang, K. Yue and Z. H. Guo, Chem. Commun., 2023, 59, 13535–13550 RSC.
- S. Hanabata, K. Kusada, T. Yamamoto, T. Toriyama, S. Matsumura, S. Kawaguchi, Y. Kubota, Y. Nishida, M. Haneda and H. Kitagawa, J. Am. Chem. Soc., 2024, 146, 181–186 CrossRef CAS PubMed.
- C. Biz, M. Fianchini and J. Gracia, ACS Catal., 2021, 11, 14249–14261 CrossRef CAS.
- S. Mourdikoudis, A. Kostopoulou and A. P. LaGrow, Adv. Sci., 2021, 8, 2004951 CrossRef CAS PubMed.
- Y. Dong, Y. Deng, J. Zeng, H. Song and S. Liao, J. Mater. Chem. A, 2017, 5, 5829–5837 RSC.
| This journal is © The Royal Society of Chemistry 2024 |