
Renal-clearable nanoprobes for optical imaging and early diagnosis of diseases
Wei
An
ac,
Weiping
Xu
b,
Ya
Zhou
b,
Changwen
Huang
d,
Weiguo
Huang
 *ac and
Jiaguo
Huang
*b
*ac and
Jiaguo
Huang
*b
aState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, China
bDepartment School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou, 510006, China. E-mail: huangjg36@mail.sysu.edu.cn
cUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
dGeneral surgery department, the Sixth Affiliated Hospital of Guangzhou Medical University, Qingyuan People's Hospital, Qingyuan, Guangdong 511518, China
First published on 8th February 2024
Abstract
Optical imaging has played an indispensable role in clinical diagnostics and fundamental biomedical research due to its high sensitivity, high spatiotemporal resolution, cost-effectiveness, and easy accessibility. However, the issues of light scattering and low tissue penetration make them effective only for superficial imaging. To overcome these issues, renal-clearable optical nanoprobes have recently emerged, which are activated by abnormal disease-associated biomarkers and initiate a pharmacokinetic switch by undergoing degradation and eventually releasing signal reporters into urine, for simple imaging and sensitive optical in vitro urinalysis. In this review, we focus on the advancements of renal-clearable organic nanoprobes for optical imaging and remote urinalysis. The versatile design strategies of these nanoprobes are discussed along with their sensing mechanisms toward biomolecules of interest as well as their unique biological applications. Finally, challenges and perspectives are discussed to further advance the next-generation renal-clearable nanoprobes for in vivo imaging and in vitro urinalysis.
 Wei An | Wei An graduated in 2021 with a bachelor's degree in Polymer Materials and Engineering from North University of China. Currently, she is pursuing a master's degree under the supervision of Professor Weiguo Huang at the University of Chinese Academy of Sciences. Her primary research focus lies in the design of stimuli-responsive polymer materials and the synthesis of platinum-based drugs, with a particular emphasis on their applications in biomedicine. |
 Weiping Xu | Weiping Xu received his Ph.D. from Guangdong University of Technology in 2022. Then he joined Prof. Jiaguo Huang's group at the School of Pharmaceutical Sciences, Sun Yat-Sen University and worked as a postdoctoral fellow. His current research interests are focused on novel small molecular probes for in vivo imaging and disease diagnosis. |
 Ya Zhou | Ya Zhou is a PhD candidate in Sun Yat-sen University from 2021, under the supervision of Professor Jiaguo Huang. She received her Master of Medicine degree in pharmacology in 2020 and Bachelor of Science degree in pharmaceutical analysis in 2017, currently focuses on molecular probe imaging-related research, particularly on molecular pharmacology and urinalysis. |
 Changwen Huang | Changwen Huang received his PhD degree from Nanchang University in 2009. Since 2022, he has joined into general surgery department, the Sixth Affiliated Hospital of Guangzhou Medical University. His research interests include the pathology of hepatobiliary and pancreatic neoplasms and the basis of hepatobiliary and pancreatic surgery. |
 Weiguo Huang | Weiguo Huang received his PhD degree from the Institute of Chemistry, Chinese Academy of Sciences in 2011. After obtaining his PhD degree, he started his postdoctoral research programs in the Johns Hopkins University, and then in Umass-Amherst. Since 2018, he has been a full professor at Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences. His research interests include domino-effect triggered emission (DETE), biochemical sensors, organic electro-optics and photonics, and soft smart matter. |
 Jiaguo Huang | Jiaguo Huang received his Ph.D. from the University of Heidelberg (Germany) in 2016. Then he worked as a Postdoctoral Research Fellow in the University of Heidelberg and the School of Chemical and Biomedical Engineering (SCBE), Nanyang Technological University (NTU). Since 2021, he has been a full professor at Sun Yet-Sen University. His current research focuses on the development of activatable optical probes for real-time in vivo imaging and detection of life-threatening diseases. |
1. Introduction
Molecular imaging enables non-invasive and real-time visualization of physiological or pathological processes in living organisms.1–3 With the combination of specialized instruments and imaging probes that reflect imaging signals, molecular imaging has played significant roles in early diagnosis, proper disease management, and drug development and discovery.4–7 Several imaging modalities such as X-ray computed tomography (CT) imaging, positron emission tomography (PET), single-photon emission computerized tomography (SPECT), magnetic resonance imaging (MRI) and ultrasound (US) imaging are employed for deep tissue imaging in clinical practice.8–11 Unlike those conventional imaging modalities, optical imaging as a nonionizing radiation method offers unique opportunities to real-time visualize and quantify multiple dynamic biological events at the molecular level with high spatiotemporal resolution and high sensitivity.12–17 To correlate the signal with biomolecules or biological events in vivo, optical probes are of high significance. In clinical settings, imaging-guided surgery with clinically approved indocyanine green (ICG) and methylene blue (MB) have been widely investigated.18,19 Moreover, renal-clearable small molecular probes have been explored to detect versatile pathological parameters in kidney disease, bladder cancer, cancer immunotherapy, etc.20–22 However, these small molecular fluorophores typically experienced short retention time because of their rapid clearance.23,24 In contrast, optical nanoparticulate probes with prolonged circulation time and biomarker-triggered signals offer measurable and quantifiable real-time information on pathological status at the molecular level.25,26 Nevertheless, nanoprobes combined with optical imaging often encounter the shallow tissue penetration depth due to high photon absorption and scattering, which hampers imaging invisible or deep tissue diseases.27–29Nanoparticles could disassemble or degrade into small molecules with desired properties such as fluorescence turn on and renal clearance in the presence of biomarkers of interest, which not only offer measurable real-time information on pathological sites but also leverage the level of biomarkers to amplify the detection signals in urine, allowing highly sensitive detection of diseases.30 Thus far, only few nanoprobes built on metal materials such as gold nanoclusters have been reported for this purpose.31 However, the design of those inorganic nanoprobes simply relies on the surface anchor via noncovalent bonds and the use of heavy metals failed to thoroughly be degraded.32 Moreover, the in vivo stability and long-term biosafety remain to be optimized. By contrast, organic nanoprobes are more biocompatible, show faster clearance and have more tunable optical properties relative to inorganic nanomaterials. As most organic dye-based nanoparticles have been constructed through intraparticle doping, they are often unable to disassemble upon exposure to biological events, or their disassembled fragments failed to clear into urine. Therefore, organic nanoprobes that simultaneously possess activatable fluorogenic signals and undergo degradation into small molecules with high renal clearance efficiency are promising for both real-time optical imaging in vivo and urinalysis in vitro.
The publications and citations about optical nanoprobes are increasing in number, and there are some reviews summarizing their designs, optical properties, and general biosensing applications. However, the existing literature has showcased the design strategies and therapeutic prospects of optical nanoprobes, which are mostly excreted through hepatobiliary clearance pathway. In this review, we focus on the recent progress in the development of renal-clearable organic nanoprobes for optical imaging and urinalysis and highlight the nanoprobe design and sensing mechanisms toward biomolecules or biological events of interest (Table 1). In the following sections, we first discuss the design principles of renal clearable nanoprobes along with their biological applications towards various conditions including cancers, allograft rejection, immunotherapy, infection, and thrombosis (Fig. 1). Furthermore, we discuss the challenges that exist in this growing field in the setting of preclinical studies and strategies for clinical translation.
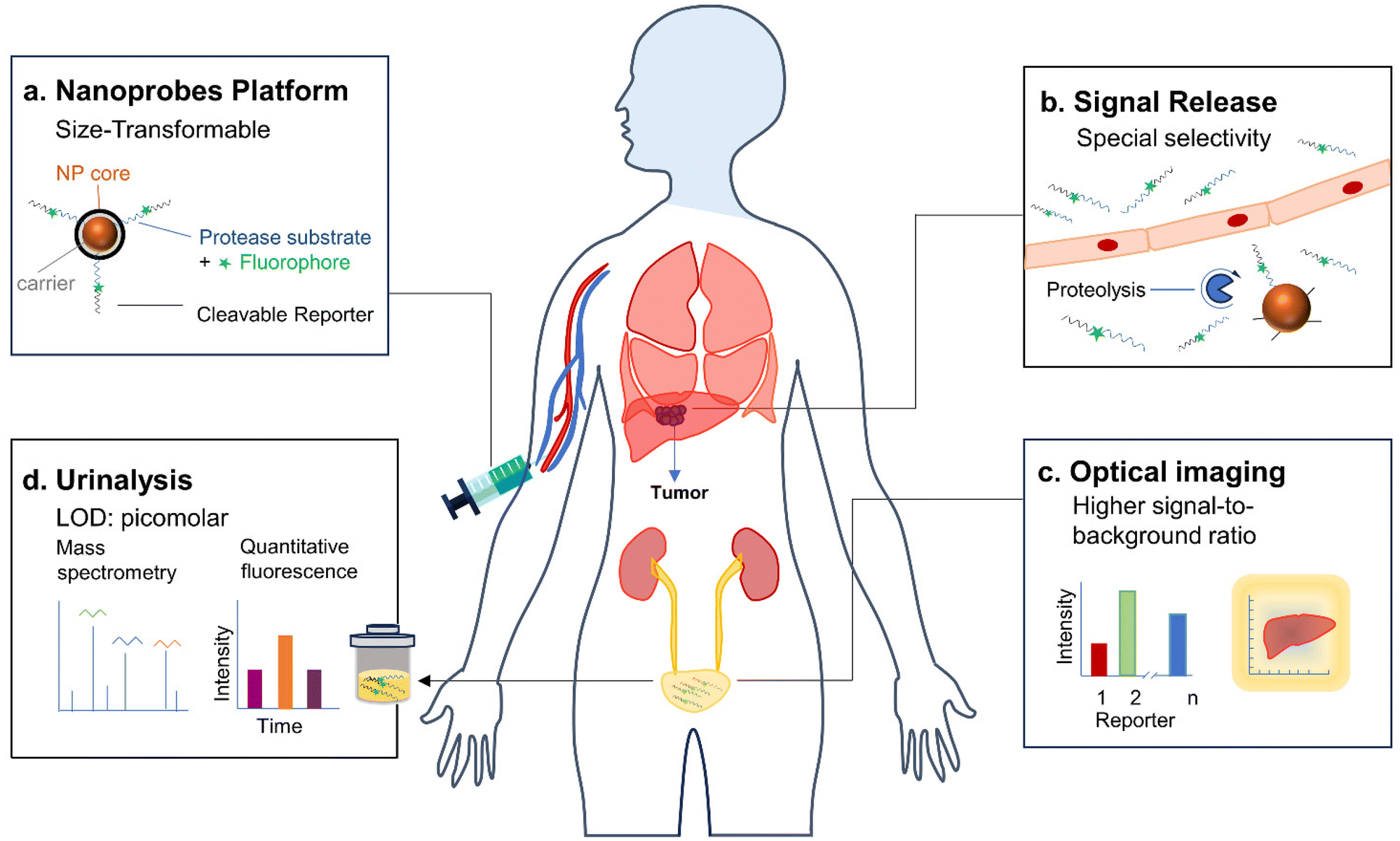 | ||
| Fig. 1 Schematic illustration of the applications of renal-clearable nanoprobes for imaging and urinalysis of diseases. | ||
| Disease type | Probe | Biomarkers | Application | Ref. |
|---|---|---|---|---|
| MMP9: matrix metalloproteinase-9, Hepatic IRI: hepatic ischemia-reperfusion injury, DILI: drug-induced liver injury, O2˙−: superoxide anion. | ||||
| Cancer | APNC | Cathepsin B | NIRF imaging in orthotopic liver tumour model and urinalysis in vitro | 33 |
| ABNS | MMP9 | Imaging in tumour nodules and urinalysis in vitro | 36 | |
| Allograft rejection | APNG | Granzyme B | Urinalysis in orthotopic liver transplantation of rat model | 33 |
| Immunotherapy | αPD1-GS | Granzyme B | Urinalysis in MC38 colon adenocarcinoma syngeneic tumour model and Imaging in tumour organs | 44 |
| Thrombosis | PEG-T1E | Thrombin | ELISA and paper ELISA | 60 |
| Inflammation | PEG-M1E | MMP9 | ELISA and paper ELISA | 60 |
| Hepatic IRI | APNSO | O2˙− | Detecting O2˙−, NIRF imaging and urinalysis | 55 |
| DILI | ADN2 | O2˙− | Detecting O2˙−, NIRF imaging and urinalysis | 63 |
2. Early detection of cancer and acute allograft rejection
Cancer is a leading cause of disease-related death worldwide.4 Early diagnosis plays a crucial role in the timely identification of cancers or precancerous lesions, enabling interventions that can enhance survival rates and reduce incidence. Conventional clinical radiological imaging modalities have limitations in detecting cancers at an early stage.4 Moreover, solid organ transplantation is the only option for patients with end-stage cancer. However, acute allograft rejection manifests as a sudden decline in graft function after transplantation and poses a serious medical complication with high mortality risk in clinical settings. Therefore, early detection of allograft rejection is critical for long-term graft function and survival in transplant patients. Although invasive biopsy is the gold standard routinely used to diagnose allograft rejection, this procedure is invasive and offers only a static and focal pathological state.33To overcome these issues, Pu and his colleagues have reported activatable polyfluorescent nanoprobes (APNS) for real-time near-infrared fluorescence (NIRF) imaging and urinalysis of tumours and allograft rejection.33 These APNS consist of fluorophores with azide groups incorporated into alkyl chains (NH2CyOH), along with an aniline group linked to a self-immolative linker (4-aminophenyl conjugated benzoyldimethanol) (Fig. 2b). This self-immolative linker is employed in conjunction with dipeptide acetyl-Phe-Lys (Ac-FK) or tetrapeptide acetyl-Ile-Glu-Phe-Asp (Ac-IEFD), resulting in the formation of polymer C and polymer G (Fig. 2a). To enhance their hydrophilicity and targeting ability, the azide groups on the side chains of polymer C/G are further coupled with cRGDfK/HPβCD or HPβCD, affording APNC and APNG, respectively.34,35 It should be noted that Ac-FK and Ac-IEFD peptide moieties can be specifically cleaved by Cathepsin B (Cat B) and Granzyme B (GzmB) proteases, respectively, which are highly expressed in tumour cells and lymphocyte cells during allograft rejection, respectively.
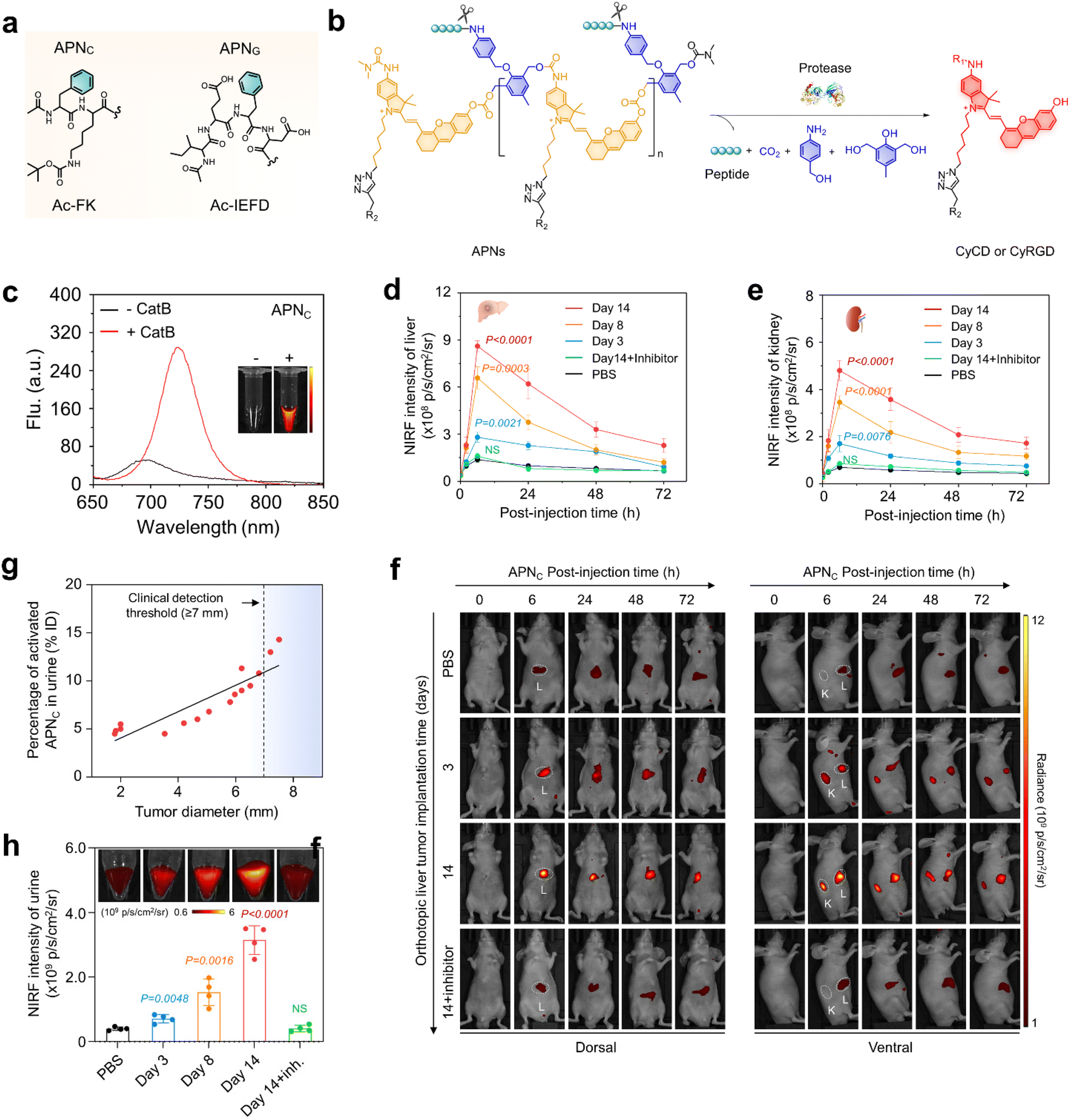 | ||
| Fig. 2 NIRF imaging and urinalysis of APNC for early cancer diagnosis. (a). Chemical structures of protease-responsive peptide brushes (Ac-FK and Ac-IEFD) for APNS design. (b). Chemical structures of APNS and the corresponding fluorogenic fragments in response to the respective proteases (CyCD or CyRGD). (R1 = H or (CH3)2NCO, R2 = HPβCD or cRGD, R3 = H or CH2CHOHCH3). (c). NIR-fluorescence spectra of APNC (10 μM) in the absence or presence of Cat B (0.5 μg) in a PBS solution at 37 °C. (d and e). NIRF intensity versus time curves of the liver (d) and kidney (e) after injection of APNC in live mice (n = 4). (f). NIRF imaging of live mice with implanted tumours after APNC injection at different time points (0, 3, and 14 d). White circles display the tumour (T), liver (L), and kidney (K). (g). The correlation between the percentage of activated APNC in urine and tumour size was studied by linear regression analysis. (h). APNC was injected at different time points and the fluorescence intensity of the activated probe was collected in urine from each group of mice (n = 4). PBS versus tumour-implantation groups. Reproduced with permission from ref. 33. Copyright 2022, Springer Nature. | ||
The amphiphilicity of APNC and APNG enabled spontaneous assembly into spherical nanoparticles with average dynamic sizes of ∼170 nm. APNC and APNG are naturally non-fluorescent in PBS solutions. After incubation with cathepsin B and granzyme B, the fluorescence at 720 nm increased by 11-fold and 10-fold (Fig. 2c and 3b) for APNC and APNG, respectively. This was attributed to the enzymatic cleavage of the amide linkage between the peptide substrate and the self-immolative moiety, followed by 1,6-elimination and 1,4-elimination, eventually leading to depolymerization to form fluorescent CyCD or CyRGD. Moreover, their sizes decreased dramatically upon incubation with respective proteases, as they underwent backbone degradation and thus stayed in a single-molecule state rather than nanoparticle state in aqueous solutions.
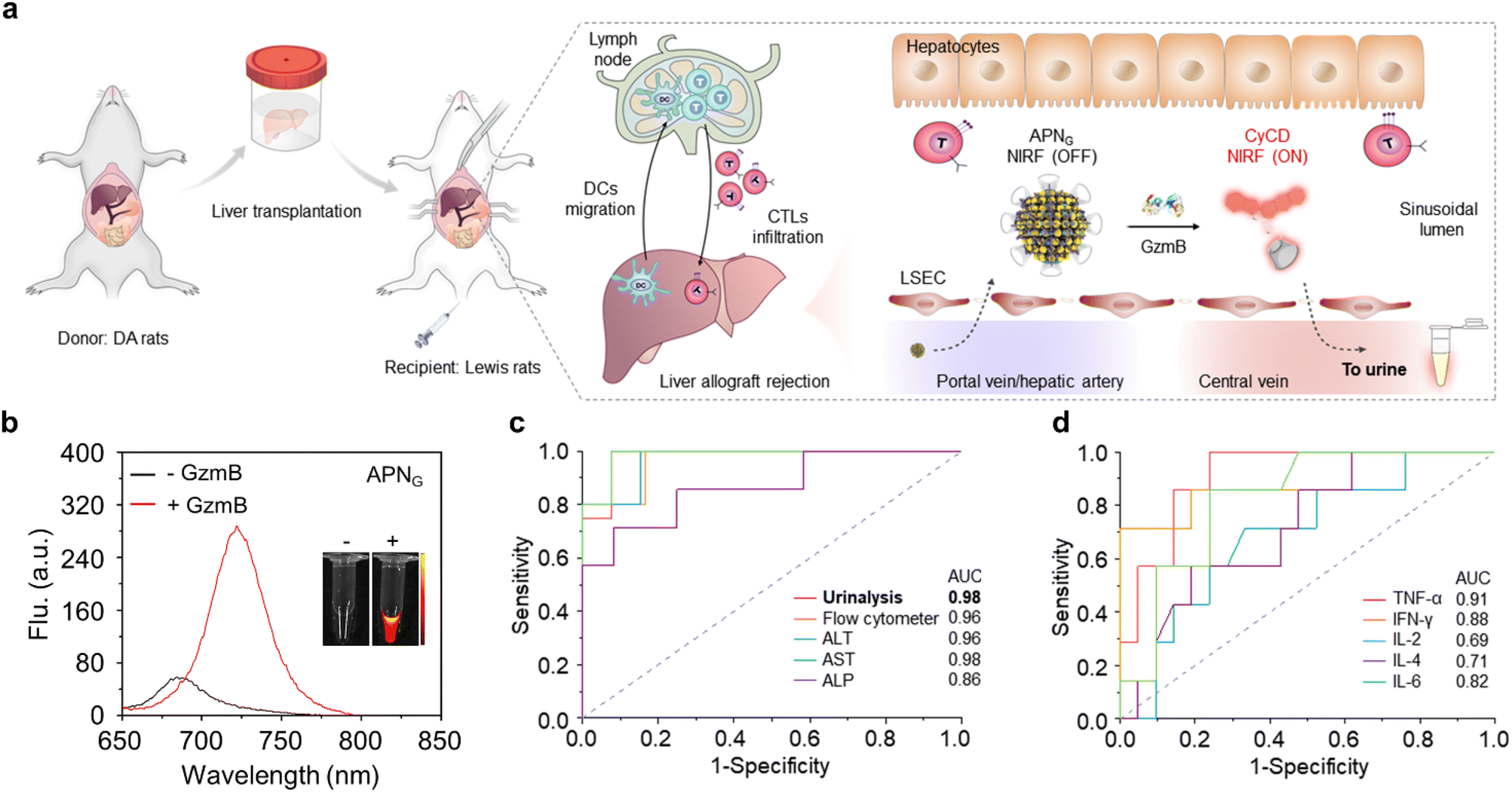 | ||
| Fig. 3 Urinalysis of acute liver allograft rejection in live rats. (a). Illustration of the liver transplantation surgical procedure, liver allograft rejection mechanisms, and APNG activation in the grafted liver for optical urinalysis in live mice. LSEC, liver sinusoidal endothelial cells. (b). NIR-fluorescence spectra of APNG in the absence or presence of granzyme B in a PBS solution at 37 °C. (c). Diagnostic specificity and sensitivity for urinalysis, flow cytometry, ALT, AST and ALP in differentiating between healthy rats (n = 12) and allografts (n = 8) 6 d after transplantation. (d). Diagnostic specificity and sensitivity for cytokines including TNF-α, IFN-γ, IL-2, IL-4 and IL-6 in differentiating between healthy rats (n = 21) and allografts (n = 14) 6 d after transplantation. Reproduced with permission from ref. 33. Copyright 2022, Springer Nature. | ||
Owing to the size of APNS (∼170 nm) being far over the renal filtration threshold (∼5 nm) and intrinsic non-fluorescent feature, APNS accumulated predominately in the liver and showed weak fluorescence. After systematic injection into live mice carrying an orthotopic liver tumour in a mouse model, the tumour in the liver was easily delineated through NIRF imaging. Meanwhile, the fluorescence signal was detected in the kidneys (Fig. 2f), because the overexpressed cathepsin B in the tumour effectively activated APNC, leading to depolymerization to produce fluorescent artificial urinary biomarker CyCD with a strong fluorescence signal. The maximum liver-to-background ratio (LBR) and kidney-to-background ratio (KBR) were 5.6-fold and 6.9-fold higher than those of the control mice, respectively (Fig. 2d and e). A gradual decline in these ratios was observed at later time points due to the renal clearance of CyCD. Furthermore, a separate cohort of tumour-bearing mice were treated with a cathepsin B inhibitor (CA-074) prior to APNC administration. Pharmacological inhibition of cathepsin B resulted in a remarkable decrease in signal for both liver and kidneys.
The intra-tumour Cathepsin B levels was detected by urinalysis of the CyCD signal following APNC injection. On days 3, 8, and 14 after tumour implantation, the intra-tumour Cathepsin B levels gradually increased, corresponding to CyCD signal increases of 1.8-fold, 3.8-fold, and 7.8-fold, respectively, in comparison to control mice (Fig. 2h). Notably, the percentage of activated APNC in urine closely correlated with the tumour size. APNC-based urinalysis demonstrated its capability to in situ detect liver tumours as small as approximately 1.9 mm in diameter, outperforming the conventional clinical liver function tests including glutamate transaminase (ALT) and aspartate transaminase (AST) (Fig. 2g). Hence, it is anticipated that APNC are superior to conventional clinical assays for the early diagnosis of liver cancer.
In an orthotopic liver transplantation of rat model (Fig. 3a), APNG was i.v. injected into live rats at different post-operation time points (2, 3, 4 and 6 days). Urinalysis was conducted and their signals were normalized by the fold of urinary protein in order to exclude the effect of urine volume between different rats. The first statistically significant NIRF enhancement for excreted CyCD was observed at 3 days (3.2-fold) post operation, which continued to increase to 4.9-fold at 4 days and 3.9-fold at 6 days post operation. Moreover, there was no significant elevation in urine signals from high-dose tacrolimus-treated grafted rats, iso-grafted rats and sham-operated rats with only trauma on the belly. The signal evolution behaviors of urinalysis coincided well with the level of granzyme B in CD8+ cytotoxic T cells from liver, spleen, lymph nodes and blood by flow cytometry. Moreover, the sensitivity and specificity for all tested assays including APNG-based urinalysis, flow cytometry, cytokines and liver function were evaluated (Fig. 3c and d). Different from the limited predictive power of cytokines for early detection (0.69 < AUC < 0.91), APNG-based urinalysis and flow cytometry assays were highly discriminatory, producing a higher AUC (0.94 < AUC < 0.98). Although ALT and AST had a comparable predictive power (AUC = 0.96 and 0.98), their detection time was delayed to 4 days post operation time. Therefore, APNG-based urinalysis not only enabled the detection of allograft rejection early, but also showed the highest sensitivity to in the prediction and prognosis of acute liver allograft rejection.
Besides, Kwon et al.36 developed a novel exogenous administered activity-based tumour-penetrating nanosensor (ABNS) that can effectively detect tumour lesions <5 mm as well as micro nodes based on abnormal protease expression in the tumour microenvironment (Fig. 4a). In the ABNS nanoplatform, Kwon et al. have chosen MMP9 as a protease target, modified with a tumour-penetrating peptide (LyP-1) to mediate the active internalization of ABNS and their transport through the tumour stroma into the tumour tissue, which significantly improved the sensitivity, as evidenced by detecting tumour sizes from 150 mm3 to 30 mm3 in volume. Furthermore, they selected another tumour penetrating peptide, iRGD, which is involved in the same active internalization pathway as LyP-1, but with an αvβ3 or αvβ5 integrin heterodimer as the central receptor. After intravenous (i.v.) administration, excellent tumour enrichment effects were observed in a mouse tumour model (Fig. 4b and c). Finally, in mice with tumours implanted for two weeks, the total tumour burden measured 36 mm3, with a median nodule diameter less than 2 mm. At this stage, urine test results show a notable increase in signals compared to the blood biomarker HE4 (Fig. 4b and e). When compared to clinically accepted imaging modalities capable of detecting only individual tumour nodules larger than 5 mm, the ABNS platform demonstrated greater sensitivity in the mouse model.
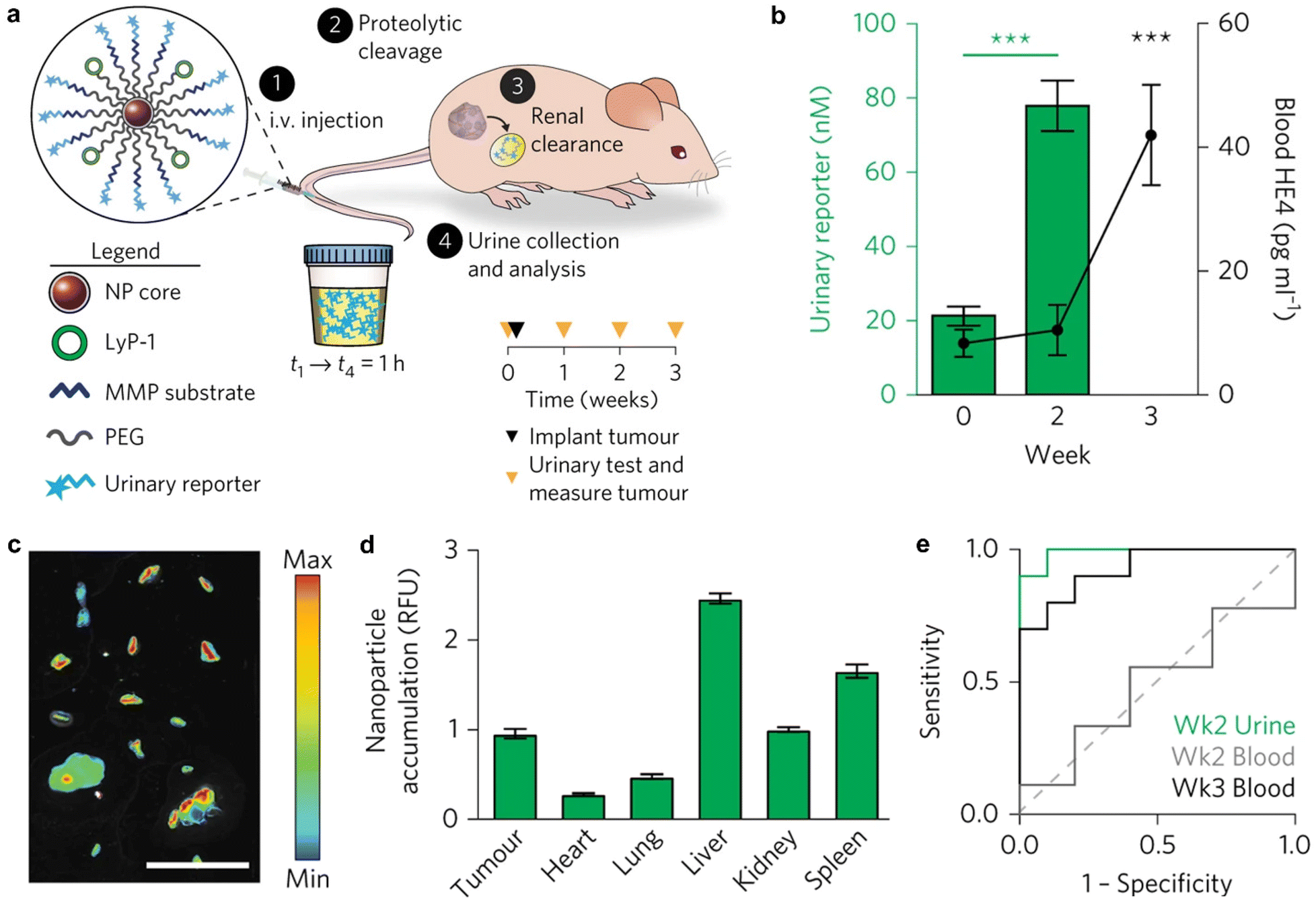 | ||
| Fig. 4 Tumour enrichment and urine detection mechanism of ABNS. (a). Illustration of the ABNS assembly and urine test pathway. (b). Urine and blood measurement of ABNS and HE4. (c). ABNS accumulation in tumour nodules in a representative animal. (d). Normalization accumulation of ABNS in tumours and major organs. (e). ROC curve of urinary diagnostic and blood biomarker. Reproduced with permission from ref. 36. Copyright 2017, Springer Nature. | ||
3. Detection of immune checkpoint blockade (ICB) responses
The advent of Immune Checkpoint Blockade (ICB) therapy has marked a significant progress in cancer treatment.37–39 Nevertheless, it is important to note that objective response rates still fall below approximately 25% across several cancer types.38 Currently, a combination of radiological assessments, tumour evaluations, and serum biomarkers is employed to gauge patient responses to ICB therapy.40 However, there is an evident need to enhance radiological evaluation following the initial cycle of ICB therapy, considering the occurrence of “pseudoprogression” during immunotherapy.41–45 Furthermore, tumour biomarkers typically require invasive biopsy procedures during treatment, underscoring the critical importance of developing non-invasive assays for monitoring the immune response to ICB therapy.44Kwong et al. have reported a category of active sensors as urinary biomarkers for assessing the ICB response.44 These sensors comprise anti-programmed cell death protein 1 antibodies (αPD1) in conjunction with fluorescently labelled, mass-barcoded protease substrates. Such substrates can be cleaved by proteases during ICB treatment, leading to the release of signal reporters that are subsequently filtered into the urine. Through urine sampling, both tumour and immuno-proteases can be quantified using fluorescence and mass spectrometry, allowing for the monitoring of ICB responses based on their distinctive mass barcodes. As a representative example, a fluorescently labelled peptide substrate that selectively targets murine granzyme B (GzmB; substrate IEFDSG46,47) was conjugated to an αPD1 (clone 8H3) antibody via cross-linking surface amines on the antibody, resulting in the formation of an αPD1-GzmB sensor conjugate, referred to as αPD1-GS (Fig. 5a).
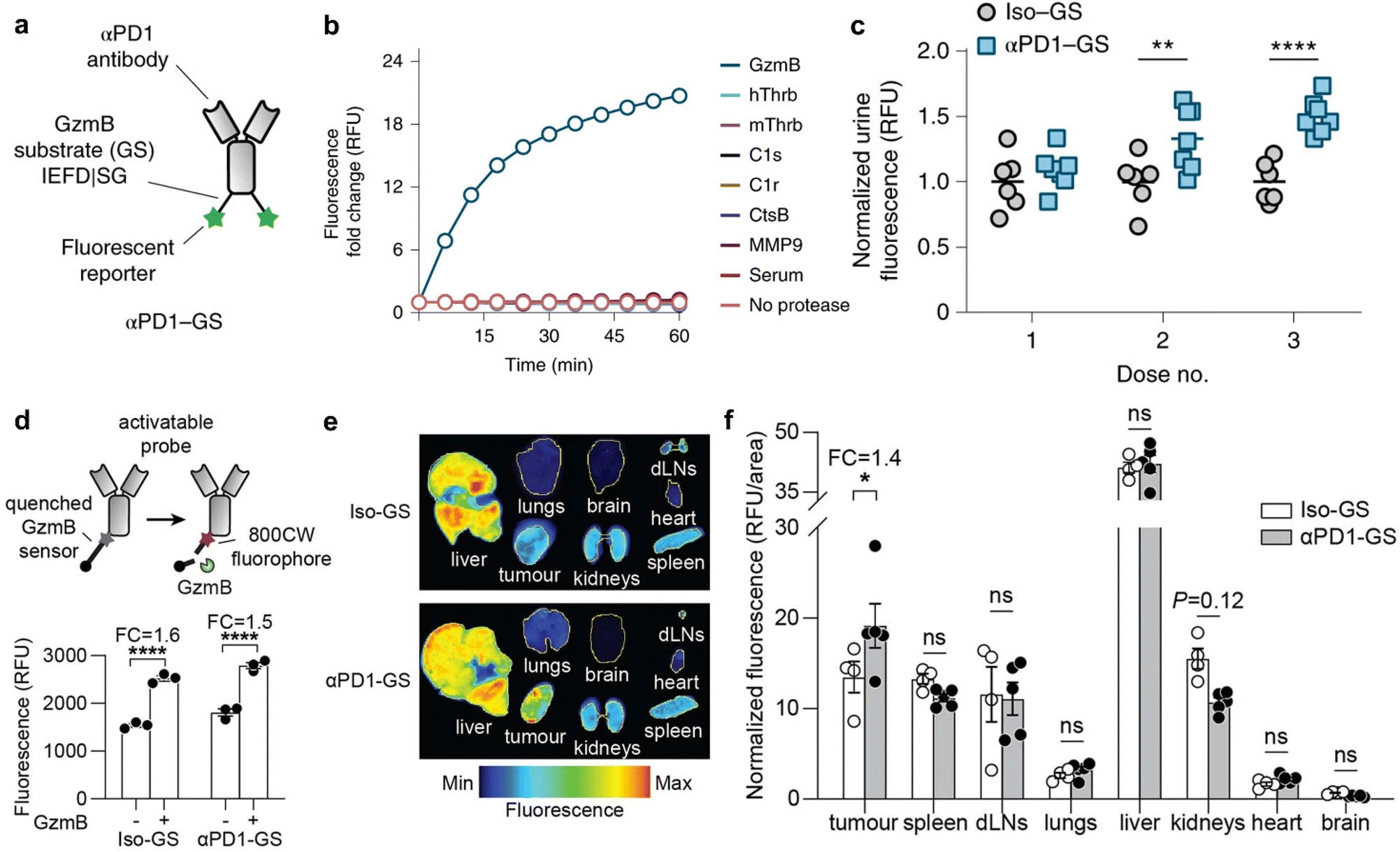 | ||
| Fig. 5 Assessment of ICB therapeutic response using antibody-GzmB sensor conjugates. (a). Schematic structure of the αPD1-GzmB sensor conjugate (αPD1-GS). (b). Normalized fluorescence (relative fluorescence unit, RFU) of recombinant GzmB (blue), mouse serum (red) and other proteases incubated with αPD1-GS (n = 3). (c). Normalized urinary fluorescence of MC38 tumour-bearing mice after treatment with αPD1-GS or Iso-GS (n = 6–7). (d). Schematic of the αPD1-GzmB probe labelled with a quencher and a near-infrared fluorophore 800CW and fluorescence obtained by cleavage in vitro (FC, fold change; n = 3) (e and f). Fluorescence images and quantitative analyses of tumour/non-tumour organs after treatment with Iso-GS or αPD1-GS (n = 4–5). Reproduced with permission from ref. 44. Copyright 2022, Springer Nature. | ||
The capacity of αPD1-GzmB sensor conjugates (αPD1-GS) to detect the T cell-mediated killing of tumour cells was assessed in the MC38 colon adenocarcinoma syngeneic tumour model.48 Following the administration of four doses of antibodies on days 7, 10, 14, and 17 to C57BL/6 mice bearing MC38 tumours, no statistically significant differences in tumour volume were observed between mice receiving αPD1-GS and those administered with unmodified αPD1. This corroborates that the coupling of the GzmB peptide does not affect the efficacy of ICB treatment. Subsequently, the capability of αPD1-GS in monitoring the GzmB activity within a T cell killing assay was evaluated. The co-incubation of this conjugate with Pmel T cells and B16 tumour cells resulted in the separation of fluorescence reporter signals from internal bursters and a subsequent increase in fluorescence levels in the samples. Conversely, no significant fluorescence increase was observed after incubating αPD1-GS with fresh mouse serum, tumour-associated proteases (e.g., histone B and MMP9), or coagulation and complement proteases (e.g., C1s and thrombin) (Fig. 5b). This observation suggests that αPD1-GS exhibits substrate-specificity during T cell killing and is particularly sensitive to GzmB activity. Finally, the non-invasive detection of αPD1-GS response in C57BL/6 mice carrying homozygous MC38 tumours was evaluated. To ascertain whether αPD1-GS undergoes cleavage within tumours, a designed activatable probe containing a near-infrared fluorophore (800CW) and a quenching agent (QC1) was employed (Fig. 5d). When activated αPD1-GS and Iso-GS were incubated with recombinant GzmB, the fluorescence intensity increased by ∼1.5 and ∼1.6 times, respectively (Fig. 5d). Mice bearing with MC38 tumours were injected with 2 doses of αPD1 or isotype control antibodies, followed by the activation of αPD1-GS or Iso-GS. There was no statistically significant difference in NIRF imaging of non-tumour organs (liver, spleen, dLNs (tumour draining lymph nodes), lung, kidney, heart, and brain), while NIR fluorescence in tumours was increased by 1.4 times (Fig. 5e and f), which suggested that αPD1-GS accumulates in MC38 tumours and is cleaved to release enhanced fluorescence during ICB treatment. Following the treatment of MC38 tumours with αPD1-GS or IgG1 isotype antibody conjugated to the GzmB peptide substrate (Iso-GS), representative flow cytometry plots revealed a significant increase in both the number and percentage of GzmB+ tumour-infiltrating lymphocytes (TILs) within the CD3+CD8+ T cell subsets. However, there was no observable response of CD3+ CD8- subsets to αPD1-GS compared to control mice receiving the same dose of Iso-GS, indicating that CD8+ TILs are the responsive cells to ICB treatment in this model.
To gauge the potential for continuous monitoring of the response to ICB treatment, two groups of MC38 tumour-bearing mice received treatment with either αPD1-GS or Iso-GS. The concentration of cleaved fluorescent reporter signals in urine samples was quantified within 3 hours of administration on days 7, 10, and 14 (Fig. 5c). Compared to the control mice treated with Iso-GS, there were no statistically significant differences in urinary fluorescence signals between the two groups after the first dose. However, at the initiation of the second dose on day 10, the αPD1-GS-treated mice exhibited significantly higher urinary fluorescence signals (P = 0.0093, n = 6–7) while maintaining statistically comparable tumour volumes (255 mm3vs. 441 mm3, P = 0.68, n = 6–7). Furthermore, the disparity in urinary fluorescence signals was further accentuated at the outset of the third dose on day 14 (P < 0.0001, n = 6–7), and tumour volume also showed a significant difference (P < 0.0001, n = 6–7). Receiver operating characteristic (ROC) analysis of urine samples at the reporting level indicated an area under the curve (AUC) of 0.86 for dose 2 and 1.00 for dose 3. This demonstrates the ability to discern ICB responses with a high degree of sensitivity and specificity. Furthermore, the effectively therapeutic response at the outset of the second dose within the first cycle of ICB treatment was manifested in a preclinical animal model.
4. Early detection of hepatic ischemia-reperfusion injury
Hepatic ischemia-reperfusion injury (IRI) represents a significant complication associated with various clinical procedures including hepatectomy, transplantation, and trauma.49 Oxidative stress serves as a key pathophysiological mechanism in the course of hepatic IRI, ultimately leading to cell death.50 Recently, clinical diagnostic approaches for hepatic IRI involve plasma markers (serum ALT/AST) and histological assays. In addition, non-invasive imaging techniques such as CT, MRI, and PET are employed.51–53 Nevertheless, these methods are primarily designed to identify late-stage liver function changes and prove ineffective in the early detection of molecular-level alterations associated with hepatic IRI.51–54Huang et al. have developed an activatable polymeric nanoprobe, APNSO, for real-time NIRF imaging of hepatic IRI and remote urinalysis in the early stage.55 APNSO is composed of four functional blocks: a superoxide anion (O2˙−) reactive moiety, a self-immolative linker, a caged fluorophore moiety and a renal clearance moiety. The fluorophore moieties are convergently connected with the self-immolative linkers to form the polymer backbone, which is conjugated by an O2˙−-cleavable trifluoromethanesulfonate group and grafted by a renal-clearance moiety HPβCD (Fig. 6a). The amphiphilic properties of these polymers enable them to spontaneously assemble into nanoparticles in aqueous solutions, with an average dynamic size of 160 nm. APNSO is naturally non-fluorescent. In the presence of O2˙−, the trifluoromethanesulfonate groups are cleaved, triggering self-depolymerization of the APNSO backbone. This process releases a renal-excretable fluorescent fragment referred to as the “fluorescent artificial urine biomarker” (FAUB) (Fig. 6a), leading to a new peak at 700 nm in the absorption spectrum (Fig. 6c) and an 11-fold fluorescence enhancement at 720 nm (Fig. 6d). The nanoprobe also exhibits a low limit of detection of about 10 × 10−9 M towards O2˙−. APNSO exhibited superior liver-oriented accumulation, which is probably due to its large particle size of ∼160 nm that exceeded the renal filtration threshold of ∼5 nm (Fig. 6b). Despite high accumulation in the liver, APNSO exhibited only a weak background signal in healthy mice as it was intrinsically non-fluorescent.
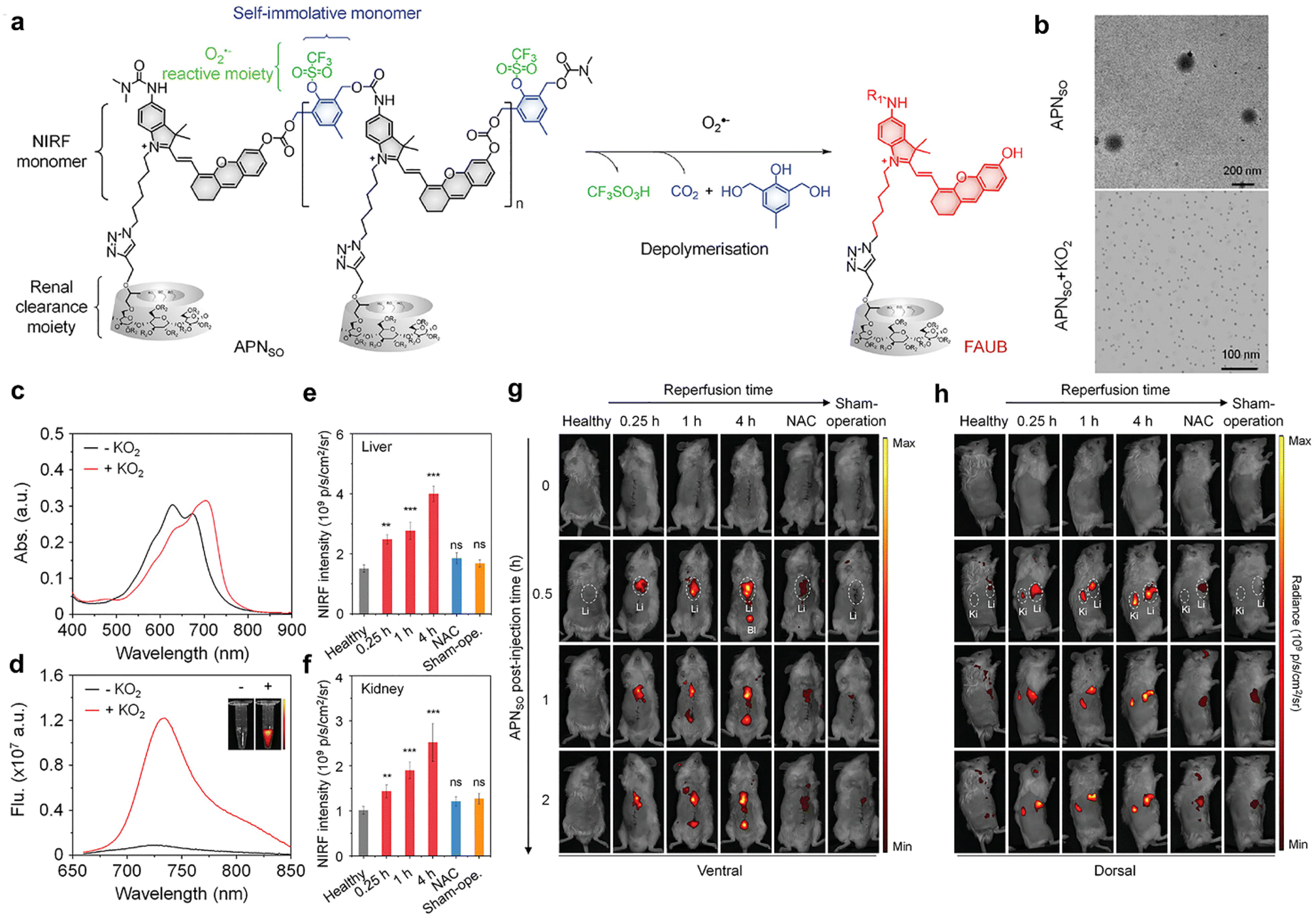 | ||
| Fig. 6 APNSO-based NIRF imaging and urinalysis of hepatic IRI. (a). Chemical structures of APNSO and its fluorescent fragment FAUB in the presence of O2˙−. (R1 = H or (CH3)2NCO, R2 = H or CH2CHOHCH3). (b). TME images of APNSO in a PBS solution before and after exposure to KO2. (c and d). Absorption and fluorescence spectra of APNSO in a PBS solution before and after exposure to KO2 at 37 °C. Inset: Fluorescence images at 720 nm obtained by the IVIS fluorescence system excited at 675 nm. (e and f). NIRF intensities of the liver and kidney of live mice with injection of APNSO at 0.5 h after different postreperfusion time points (n = 4). (g and h). NIRF images of live mice with injection of APNSO at different time points after reperfusion for 0.25, 1, or 4 h. Reproduced with permission from ref. 55. Copyright 2022, John Wiley and Sons. | ||
The ability of APNSO to monitor hepatic IRI was tested in a mouse model of hepatic warm IRI.55,56 APNSO was i.v. injected into live mice at various time points (0.25, 1, and 4 hours) after reperfusion. At 0.25 hours post reperfusion timepoint, the activated APNSO signal depicted both the liver and kidney in mice (Fig. 6g and h). In comparison, the maximal NIRF signals in the liver (and kidneys) of mice at 1 and 4 hours of reperfusion were 1.8 times (1.9 times) and 2.56 times (2.52 times), respectively (Fig. 6e and f). No significant differences in NIRF signals were observed in the NAC-pretreated group57 when compared to the control group (Fig. 6e and f). In comparison of clinical diagnostic methods, it was observed that serum ALT and AST did not exhibit statistical elevation until 8 hours after reperfusion. This suggests that ALT and AST serve as insensitive later indicators for monitoring the liver function in the mouse model of hepatic IRI. Moreover, hepatic IRI was remotely detected by urinalysis. Following i.v. injection of APNSO, the fluorescence signal of FAUB in urine was measured at 1 hour, 4 hours, and 8 hours post reperfusion, respectively. Statistically significant enhancements in NIRF signals of FAUB were observed at 1 hour (1.93-fold) post reperfusion. Furthermore, the contrasting NIRF signals of FAUB continued to exhibit enhancement at 4 hours (3.02-fold) and 8 hours (6.11-fold) post reperfusion. Histological studies revealed normal liver tissue morphology at 4 hours post reperfusion, while significant necrosis was observed at 8 hours post reperfusion. Therefore, APNSO-based imaging and urinalysis can detect hepatic IRI up to 7 hours earlier than clinical serological biomarkers (ALT/AST) and histological analyses.
5. Diagnosis of thrombosis and inflammation
Postoperative infection and thromboembolism are life-threatening complications in clinical settings and are responsible for high mortality annually worldwide.58 Currently, the levels of prothrombin fragment 1.2 and D-dimer that serve as indicators of thrombosis or genetic tests have been used for monitoring pathogenic infections for patients in hospitals.58 However, these indicators are often rapidly depleted in the bloodstream,59 which makes them unable to capture signals associated with long-lasting latent diseases after the patient's discharge.Bhatia et al.60 have constructed synthetic biomarkers for diagnosis of thrombosis and infection. These synthetic biomarkers comprised three components: poly(ethylene glycol) (PEG) with a molecular weight of 40 kDa, thrombin or matrix metalloproteinase (MMP9) cleavable peptide linkers, and a urinary reporter, which acted as the core scaffold, surface linker and signal moiety, respectively (Fig. 7a).
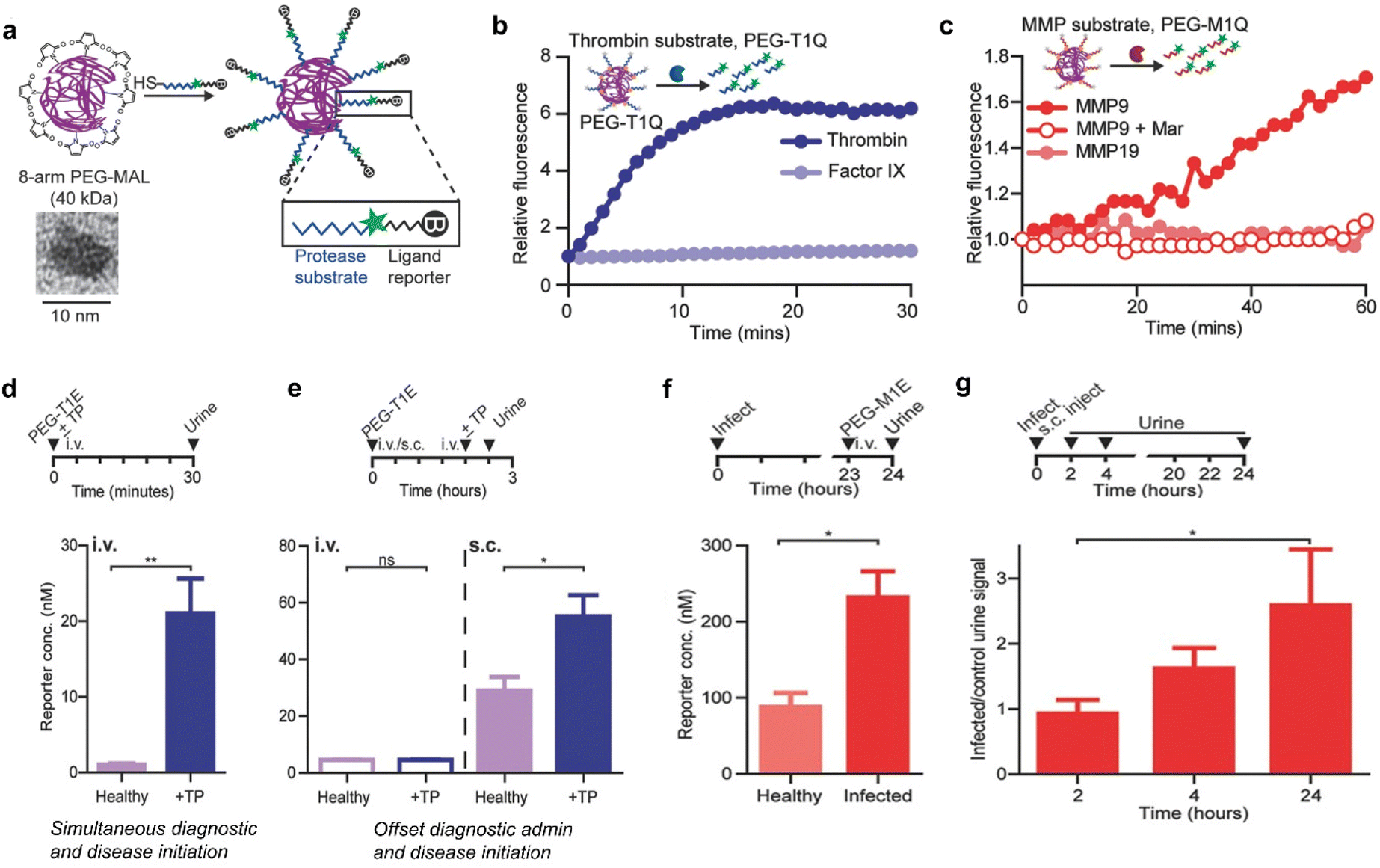 | ||
| Fig. 7 Diagnosis of thrombosis and inflammation using synthetic biomarkers. (a). Eight-arm PEG maleimide (PEG-MAL) reacts with peptides terminated with cysteine, which include urinary reporters and protease substrates. The inset shows a TEM image of the 40 kDa PEG molecule. (b). Representative dequenching measurements of PEG-T1Q (thrombin substrate) in the presence of recombinant thrombin, Factor IX. (c). Representative dequenching measurements of PEG-M1Q (MMP9 substrate) in the presence of MMPs; MMP9 inhibit Marimastat. (d). Diagnostically administered at the same time as the onset of disease in mice, PEG-T1E was effective in differentiating healthy mice from thrombosed mice. (e). There was no significant difference between diseased and healthy mice when administered intravenously prior to the onset of disease, whereas subcutaneous injection allowed diagnosis of pre-diseased mice. (f). P. aeruginosa was injected into the mouse lungs to establish an inflammation model, and 24 h later, PEG-M1E was administered intravenously, showing sensitivity to infection detection. (g). Subcutaneous injection of PEG-M1E detects later inflammation within 24 h. (I.D. = 4000 pmoles, n = 4–5; ±SEM). Reproduced with permission from ref. 60. Copyright 2016, John Wiley and Sons. | ||
After incubation with recombinant thrombin, significant fluorescence enhancement was observed within a short period of time, whereas PEG-conjugated peptide (PEG-T1Q) was negligibly cleaved by clotting cascade protease factor IX (Fig. 7b). In a parallel characterization study, remarkable fluorescence enhancement was observed after coincubation of PEG-M1Q with MMP9. However, negligible enhancements in fluorescence signals were determined for the distinct enzyme MMP19 and pretreated with an MMP9 inhibitor Marimastat (Fig. 7c).
The ability of PEG-T1E to detect ongoing thrombosis was tested using a mouse model of thrombosis. After i.v. injection of thromboplastin and PEG-T1E, blood clots, primarily in the lung vasculature, were formed via the extrinsic clotting cascade. In addition, the urinary signal was significantly elevated related to healthy controls (Fig. 7d). However, such model did not mimic the kinetics of the onset of a postoperative complication in clinical settings, because the realistic postoperative complication is more likely to be delayed relative to the time of diagnostic administration. As such, subcutaneous administration of PEG-T1E was employed to read out a response to thromboplastin. The results indicated that the same dose of PEG-T1E administered subcutaneously 2 hours prior to disease onset allowed us to clearly distinguish between diseased and healthy animals (Fig. 7e). Furthermore, the capability of the MMP-responsive synthetic biomarker (PEG-M1E) to detect inflammation associated with pneumonia was evaluated. In the course of infection, MMP9 is highly expressed by neutrophils and other immune cells as key mediators in innate immunity and inflammation. Followed by 24 hours post-intratracheal inoculation of P. aeruginosa, the urine reporter levels measured were significantly higher in infected mice compared to healthy controls after 1 hour i.v. injection of PEG-M1E (Fig. 7f). Finally, the PEG-M1E nanoprobe was subcutaneously injected to cohorts of mice that were either coinfected or not infected via pulmonary delivery of bacteria. The levels of urine reporter were measured periodically in both groups over a 24-hour period. In the initial period of 2 hours post injection, no difference between healthy and infected mice was observed. However, the urine signal in infected mice was significantly elevated at later time points from 2 to 24 hours due to the inflammatory response strengthened (Fig. 7g). Therefore, those synthetic biomarkers not only enable long-term monitoring of diseases with facile urinary readouts, but also have high potentials for further advancement in monitoring and diagnostic applications in human.
6. Early detection of drug-induced liver injury
Drug-induced liver injury (DILI) poses a significant global public health concern.61 Excessive exposure to hepatotoxic drugs can deplete glutathione, leading to the formation of toxic protein adducts, thereby increasing oxidative stress and ultimately resulting in liver injury.62 Conventional methods relying on the measurement of ALT/AST levels and histological analyses have limitations in terms of their low sensitivity and specificity.To tackle those issues, Huang et al. have recently reported an activatable nanoreporter, ADN2, with superoxide anion (O2˙−)-triggered size transformation and chemo-fluoro-luminescence response for crosstalk-free duplex imaging of DILI and in vitro urinalysis (Fig. 8a).63 ADN2 was prepared via co-nanoprecipitation of NIR naphthalocyanine NCBS with ADN1, which comprised four functional components: a cascade self-eliminating moiety responsive to superoxide anions, a chemiluminescent signaling moiety (adamantyl-1,2-dioxane), a fluorescent signaling moiety, and a renal-clearable moiety (HPβCD). These synthesized polymers could spontaneously form nanoparticles in an aqueous solution with an average diameter of ∼130 nm (Fig. 8c). ADN2 was initially non-fluorescent and non-chemiluminescent as the phenol group was caged. In the presence of O2˙−, the chemiluminescence signals gradually elevated with the increase in the doping weight percentage of NCBS, eventually saturating at 3.5% and decreasing at higher doping ratios (Fig. 8b). This was ascribed to a cascade self-elimination reaction and depolymerization of the polymer backbone occurred, leading to the release of renal-cleared fluorescent fragments (IPCDs) (Fig. 8a). This activation switched on both chemo-fluorescence channels, enabling crosstalk-free imaging and urinalysis.
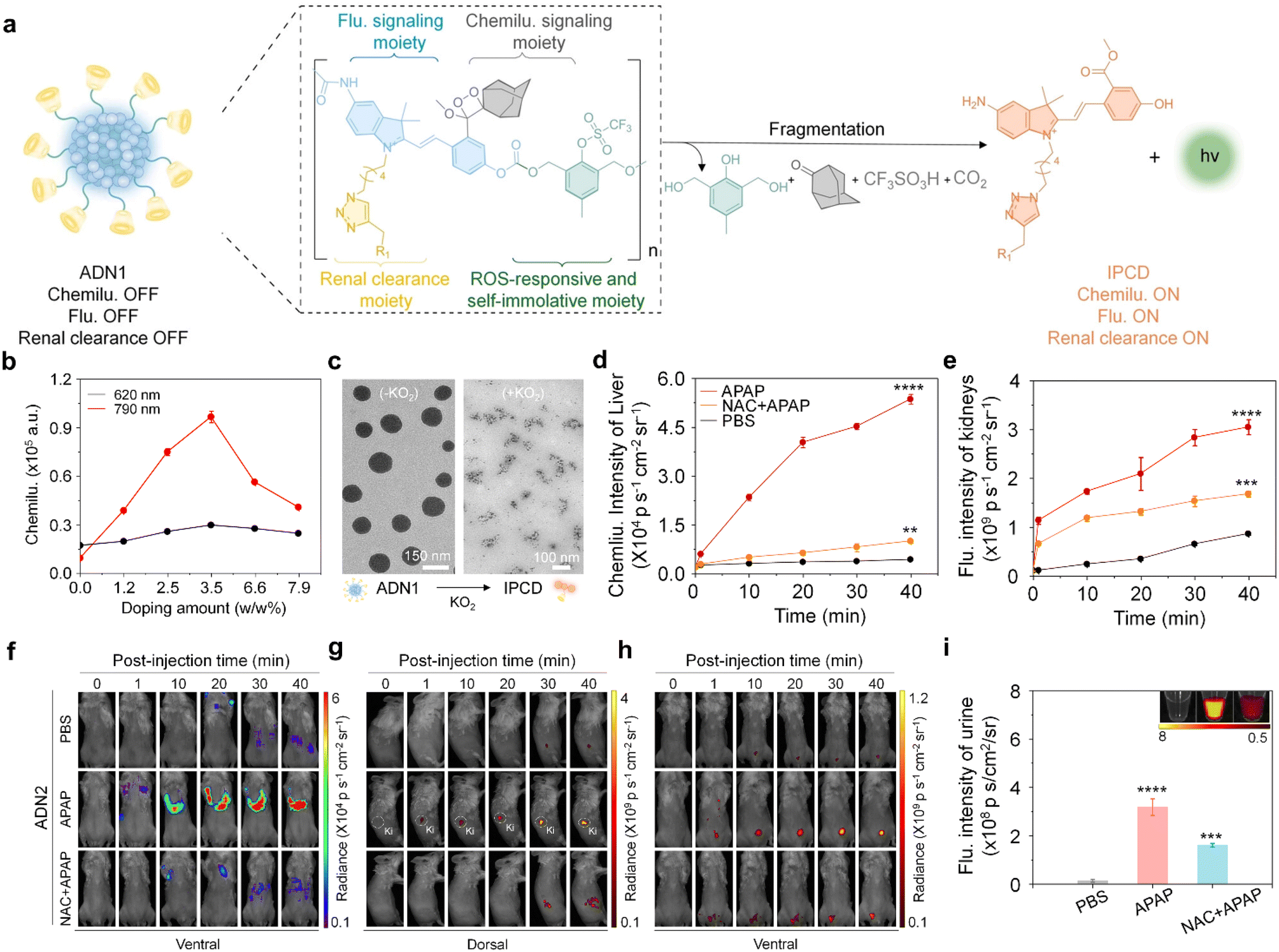 | ||
| Fig. 8 Duplex imaging and urinalysis of DILI using ADN2. (a). Chemical structure of ADN1 and its O2˙− activated fragments (R1 = HPβCD). (b). Doping content of the NCBS dye as a function of the chemiluminescence intensity of ADN2 in the presence of KO2 (80 μM) at 620 nm and 790 nm. (c). Representative TEM images of ADN1 in the absence or presence of KO2 in a PBS solution (similar size to ADN2). (d and e). Chemiluminescence intensities versus time curves of the liver (d) and kidney (e) after injection of ADN2 in live mice. (f–h). Chemiluminescence and fluorescence imaging of live mice after the injection of ADN2 at different time points. (i). The fluorescence intensities of excreted IPCD in the urine from live mice after intravenous injection of APN2 for 3 hours. Reproduced with permission from ref. 63. Copyright 2023, John Wiley and Sons. | ||
The feasibility of ADN2 for real-time duplex imaging and urinalysis of DILI was conducted in a mouse model of Acetaminophen (APAP)-induced hepatotoxicity.62,64 After injection of ADN2, the earliest time-point of the first statistically significant chemiluminescence signal increase in the liver was observed at 10 min, and gradually increased at later imaging time points for APAP-treated mice, but not for the control mice (Fig. 8d and e). The signal was 15.3-fold higher than that of the control mice at 40 min post-injection of ADN2, indicating the upregulation of O2˙− during the progression of hepatotoxicity. In addition, kidney and bladder were delineated at 10 min post-injection of ADN2. Moreover, similarly gradual increased fluorescence signals in the kidney and bladder were observed (Fig. 8e–h). These results validated the sequential depolymerization of ADN2 and excretion of IPCD from the liver to kidneys. However, both chemiluminescence and fluorescence signals were attenuated in the liver, kidney and bladder for NAC-treated mice. The SBR of chemiluminescence imaging was 5.2-fold higher than that of fluorescence imaging at 40 min post-injection of ADN2 in the liver of APAP-treated mice, implying that chemiluminescence had a higher sensitivity than that of fluorescence for liver imaging.
Moreover, the signals of urinary IPCD were measured after 3 hours of ADN2 injection. The fluorescence signals increased 16-fold and 1.3-fold for APAP-treated mice relative to the control mice and NAC-pretreated mice (Fig. 8i), respectively, validating ADN2-based urinalysis-enabled detection of hepatotoxicity. H&E staining showed normal hepatic morphology at 0.5 hours post treatment of APAP, but tissue necrosis at 6 hours post treatment. Through the comparison of ADN2-based duplex imaging/urinalysis and plasma/histological assays, ADN2-based duplex imaging/urinalysis possesses the superiority for early and sensitive detection of hepatotoxicity.
7. Conclusions
A substantial barrier facing early diagnosis of diseases using optical nanoprobes is the shallow tissue penetration depth due to high photon absorption and light scattering. To realize the diagnosis of deep tissue diseases, optical probes with degradable parameter, renal clearance and fluorescence activation are of high demand. Although various small molecular probes are reported for real-time optical imaging of various biomarkers for disease detection, their hepatobiliary clearance features impeded the feasibility of in vitro urinary detection.65–67 In contrast, activatable nanoprobes are activated by abnormal disease-associated biomarkers, and initiate a pharmacokinetic switch by undergoing degradation and eventually releasing signal reporters into urine, for simple imaging and sensitive optical urinalysis. These intrinsic advantages coupled with in vitro detection have enabled real-time ultrasensitive detection of pathological biomarkers (e.g., ROS and enzymes), physiological indexes (e.g., pH and hypoxia), etc. Although various renal-clearable nanoprobes have been constructed recently, this field is still in the early stage. Challenges and opportunities coexist, and several issues are to be tackled. First, owing to the challenges in nanoprobe design, very few renal clearable nanoprobes for optical imaging have been reported, making a major roadblock to explore their further applications. Although some inorganic renal-clearable nanoprobes have been designed, they simply rely on the surface anchor via noncovalent bonds, and the used heavy metals failed to be completely degraded. Furthermore, their instability in physiological environments and long-term safety remain yet to be addressed. Therefore, more efforts should be devoted to design novel and highly biocompatible and biodegradable materials for constructing renal-clearable nanoprobes. Second, existing renal-clearable nanoprobes suffer from low targeting ability. The resulting severe off-target signals significantly compromise the accuracy of diagnosis. As such, novel nanoprobes with high targeting capability are highly needed. Clinical approved antibodies can be selected and conjugated onto nanoprobes. Third, the luminescence of current renal-clearable optical nanoprobes is limited in the NIR-I window. Therefore, great efforts should be devoted to the construction of nanoprobes with the emission above 900 nm. Such improvements allow us to apply those nanoprobes in deeper seated diseases. Fourth, as the field is in its infancy, future clinical application has yet to proceed to pivotal trials to determine their use for disease diagnosis. Alternatively, as biomarkers are released from disease sites to circulation, efforts can be made to detect biomarkers via analysis of blood and other biofluids using nanoprobes. Through discussion, we hope to encourage the development of different optical probe platforms and hardware systems, obtaining high sensitivity and high selectivity in targeting tissues, thereby achieving early diagnosis and treatment of diseases.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. H. thanks the National Key R&D Program of China (2022YFA0912700), National Natural Science Foundation of China (22207130), the Key Research and Development Plan of Guangzhou City (202206080007), the Sun Yat-sen University Funded Program (2022_76220_B21127), the Fundamental Research Funds for the Central Universities (22lgqb35), the Introduced Innovative R&D Team Project of “The Pearl River Talent Recruitment Program” (2021ZT09Y544), Guangdong-H0.5ongkong-Macao Research Team Project of the Guangdong Basic and Applied Basic Research Foundation (2022b1515130008) for the financial support. J. H. acknowledges the award of “The Recruitment Program of Global Youth Experts” and the support from the start-up funding of Sun Yat-Sen University. W. X. thanks the Fundamental Research Funds for the Central Universities, Sun Yat-sen University (No. 23qnpy119). W. G. H. thanks the National Natural Science Foundation of China (22275193), the Natural Science Foundation of Fujian Province (2021J06034), Self-Deployment Project Research Program of Haixi Institutes, Chinese Academy of Science, CXZX-2022-GH09 (E255KF0101), Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences (E055AJ01).References
- V. Ntziachristos, J. Ripoll, L. H. V. Wang and R. Weissleder, Nat. Biotechnol., 2005, 23, 313–320 CrossRef CAS PubMed.
- A. V. Naumova, M. Modo, A. Moore, C. E. Murry and J. A. Frank, Nat. Biotechnol., 2014, 32, 804–818 CrossRef CAS PubMed.
- P. Cheng and K. Pu, Nat. Rev. Mater., 2021, 6, 1095–1113 CrossRef CAS.
- G. Hong, A. L. Antaris and H. Dai, Nat. Biomed. Eng., 2017, 1, 0010 CrossRef CAS.
- D. Yu, M. A. Baird, J. R. Allen, E. S. Howe, M. P. Klassen, A. Reade, K. Makhijani, Y. Song, S. Liu, Z. Murthy, S.-Q. Zhang, O. D. Weiner, T. B. Kornberg, Y.-N. Jan, M. W. Davidson and X. Shul, Nat. Methods, 2015, 12, 763–765 CrossRef CAS PubMed.
- S. Roy, N. Bag, S. Bardhan, I. Hasan and B. Guo, Adv. Drug Delivery Rev., 2023, 190, 114821 CrossRef PubMed.
- L. Zhang, Y. Liu, H. Huang, H. Xie, B. Zhang, W. Xia and B. Guo, Adv. Drug Delivery Rev., 2022, 190, 114536 CrossRef CAS PubMed.
- C. L. Walsh, P. Tafforeau, W. L. Wagner, D. J. Jafree, A. Bellier, C. Werlein, M. P. Kuhnel, E. Boller, S. Walker-Samuel, J. L. Robertus, D. A. Long, J. Jacob, S. Marussi, E. Brown, N. Holroyd, D. D. Jonigk, M. Ackermann and P. D. Lee, Nat. Methods, 2021, 18, 1532–1541 CrossRef CAS PubMed.
- J. Schwenck, D. Sonanini, J. M. Cotton, H.-G. Rammensee, C. la Fougere, L. Zender and B. J. Pichler, Nat. Rev. Cancer, 2023, 23, 474–490 CrossRef CAS PubMed.
- P. J. Keall, C. Brighi, C. Glide-Hurst, G. Liney, P. Z. Y. Liu, S. Lydiard, C. Paganelli, P. Trang, S. Shan, A. C. Tree, U. A. van der Heide, D. E. J. Waddington and B. Whelan, Nat. Rev. Clin. Oncol., 2022, 19, 458–470 CrossRef PubMed.
- S. Vilov, B. Arnal, E. Hojman, Y. C. Eldar, O. Katz and E. Bossy, Sci. Rep., 2020, 10, 4637 CrossRef CAS PubMed.
- M. J. Schnermann, Nature, 2017, 551, 176–177 CrossRef CAS PubMed.
- L. Wu, J. Huang, K. Pu and T. D. James, Nat. Rev. Chem., 2021, 5, 406–421 CrossRef CAS PubMed.
- J. Huang, C. Xie, X. Zhang, Y. Jiang, J. Li, Q. Fan and K. Pu, Angew. Chem., Int. Ed., 2019, 58, 15120–15127 CrossRef CAS PubMed.
- J. Huang and K. Pu, Chem. Sci., 2021, 12, 3379–3392 RSC.
- J. Huang, J. Li, Y. Lyu, Q. Miao and K. Pu, Nat. Mater., 2019, 18, 1133–1143 CrossRef CAS PubMed.
- B. You, Y. Zhang, Y. Jiao, K. Davey and S. Z. Qiao, Angew. Chem., Int. Ed., 2019, 58, 11796–11800 CrossRef CAS PubMed.
- A. L. Vahrmeijer, M. Hutteman, J. R. van der Vorst, C. J. H. van de Velde and J. V. Frangioni, Nat. Rev. Clin. Oncol., 2013, 10, 507–518 CrossRef CAS PubMed.
- N. Nishio, N. S. van den Berg, S. van Keulen, B. A. Martin, S. Fakurnejad, N. Teraphongphom, S. U. Chirita, N. J. Oberhelman, G. Lu, C. E. Horton, M. J. Kaplan, V. Divi, A. D. Colevas and E. L. Rosenthal, Nat. Commun., 2019, 10, 5044 CrossRef PubMed.
- J. G. Huang, Y. Y. Jiang, J. C. Li, S. S. He, J. S. Huang and K. Y. Pu, Angew. Chem., Int. Ed., 2020, 59, 4415–4420 CrossRef CAS PubMed.
- S. S. He, J. C. Li, Y. Lyu, J. G. Huang and K. Y. Pu, J. Am. Chem. Soc., 2020, 142, 7075–7082 CrossRef CAS PubMed.
- J. G. Huang, J. C. Li, Y. Lyu, Q. Q. Miao and K. Pu, Nat. Mater., 2019, 18, 1133–1143 CrossRef CAS PubMed.
- K. S. de Valk, H. J. Handgraaf, M. M. Deken, B. G. S. Mulder, A. R. Valentijn, A. G. T. van Scheltinga, J. Kuil, M. J. van Esdonk, J. Vuijk, R. F. Bevers, K. C. Peeters, F. A. Holman, J. V. Frangioni, J. Burggraaf and A. L. Vahrmeijer, Nat. Commun., 2019, 10, 3118 CrossRef PubMed.
- R. Tian, Q. Zeng, S. Zhu, J. Lau, S. Chandra, R. Ertsey, K. S. Hettie, T. Teraphongphom, Z. Hu, G. Niu, D. O. Kiesewetter, H. Sun, X. Zhang, A. L. Antaris, B. R. Brooks and X. Chen, Sci. Adv., 2019, 5, eaaw0672 CrossRef CAS PubMed.
- J. F. Lovell, C. S. Jin, E. Huynh, H. Jin, C. Kim, J. L. Rubinstein, W. C. W. Chan, W. Cao, L. V. Wang and G. Zheng, Nat. Mater., 2011, 10, 324–332 CrossRef CAS PubMed.
- Q. Miao, C. Xie, X. Zhen, Y. Lyu, H. Duan, X. Liu, J. V. Jokerst and K. Pu, Nat. Biotechnol., 2017, 35, 1102–1110 CrossRef CAS PubMed.
- H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. I. Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165–1170 CrossRef CAS PubMed.
- B. Du, X. Jiang, A. Das, Q. Zhou, M. Yu, R. Jin and J. Zheng, Nat. Nanotechnol., 2017, 12, 1096–1102 CrossRef CAS PubMed.
- W. Poon, Y.-N. Zhang, B. Ouyang, B. R. Kingston, J. L. Y. Wu, S. Wilhelm and W. C. W. Chan, ACS Nano, 2019, 13, 5785–5798 CrossRef CAS PubMed.
- C. N. Loynachan, A. P. Soleimany, J. S. Dudani, Y. Lin, A. Najer, A. Bekdemir, Q. Chen, S. N. Bhatia and M. M. Stevens, Nat. Nanotechnol., 2019, 14, 883–890 CrossRef CAS PubMed.
- X. Jiang, B. Du and J. Zheng, Nat. Nanotechnol., 2019, 14, 874–882 CrossRef CAS PubMed.
- S. D. Perrault and W. C. W. Chan, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11194–11199 CrossRef CAS PubMed.
- J. Huang, X. Chen, Y. Jiang, C. Zhang, S. He, H. Wang and K. Pu, Nat. Mater., 2022, 21, 598–607 CrossRef CAS PubMed.
- J. Huang, S. Weinfurter, P. C. Pinto, M. Pretze, B. Kraenzlin, J. Pill, R. Federica, R. Perciaccante, L. Della Ciana, R. Masereeuw and N. Gretz, Bioconjugate Chem., 2016, 27, 2513–2526 CrossRef CAS PubMed.
- J. Huang, S. Weinfurter, C. Daniele, R. Perciaccante, R. Federica, L. Della Ciana, J. Pill and N. Gretz, Chem. Sci., 2017, 8, 2652–2660 RSC.
- E. J. Kwon, J. S. Dudani and S. N. Bhatia, Nat. Biomed. Eng., 2017, 1, 0054 CrossRef CAS PubMed.
- G. Morad, B. A. Helmink, P. Sharma and J. A. Wargo, Cell, 2021, 184, 5309–5337 CrossRef CAS PubMed.
- A. Ribas and J. D. Wolchok, Science, 2018, 359, 1350–1355 CrossRef CAS PubMed.
- P. Sharma and J. P. Allison, Science, 2015, 348, 56–61 CrossRef CAS PubMed.
- M. Nishino, N. H. Ramaiya, H. Hatabu and F. S. Hodi, Nat. Rev. Clin. Oncol., 2017, 14, 655–668 CrossRef CAS PubMed.
- F. S. Hodi, W.-J. Hwu, R. Kefford, J. S. Weber, A. Daud, O. Hamid, A. Patnaik, A. Ribas, C. Robert, T. C. Gangadhar, A. M. Joshua, P. Hersey, R. Dronca, R. Joseph, D. Hille, D. Xue, X. N. Li, S. P. Kang, S. Ebbinghaus, A. Perrone and J. D. Wolchok, J. Clin. Oncol., 2016, 34, 1510–1517 CrossRef CAS PubMed.
- A. Vozy and G. Zalcman, Oncologie, 2015, 17, 407–408 Search PubMed.
- M. Nishino, A. Giobbie-Hurder, M. P. Manos, N. Bailey, E. I. Buchbinder, P. A. Ott, N. H. Ramaiya and F. S. Hodi, Clin. Cancer Res., 2017, 23, 4671–4679 CrossRef CAS PubMed.
- Q. D. Mac, A. Sivakumar, H. Phuengkham, C. Xu, J. R. Bowen, F.-Y. Su, S. Z. Stentz, H. Sim, A. M. Harris, T. T. Li, P. Qiu and G. A. Kwong, Nat. Biomed. Eng., 2022, 6, 310–324 CrossRef CAS PubMed.
- M. Gerwing, K. Herrmann, A. Helfen, C. Schliemann, W. E. Berdel, M. Eisenblaetter and M. Wildgruber, Nat. Rev. Clin. Oncol., 2019, 16, 442–458 CrossRef CAS PubMed.
- Q. D. Mac, D. V. Mathews, J. A. Kahla, C. M. Stoffers, O. M. Delmas, B. A. Holt, A. B. Adams and G. A. Kwong, Nat. Biomed. Eng., 2019, 3, 281–291 CrossRef CAS PubMed.
- L. Martinez-Lostao, A. Anel and J. Pardo, Clin. Cancer Res., 2015, 21, 5047–5056 CrossRef CAS PubMed.
- M. Efremova, D. Rieder, V. Klepsch, P. Charoentong, F. Finotello, H. Hackl, N. Hermann-Kleiter, M. Loewer, G. Baier, A. Krogsdam and Z. Trajanoski, Nat. Commun., 2018, 9, 32 CrossRef PubMed.
- H. K. Eltzschig and T. Eckle, Nat. Med., 2011, 17, 1391–1401 CrossRef CAS PubMed.
- M. Cannistra, M. Ruggiero, A. Zullo, G. Gallelli, S. Serafini, M. Maria, A. Naso, R. Grande, R. Serra and B. Nardo, Int. J. Surg., 2016, 33, S57–S70 CrossRef PubMed.
- M. Shoup, M. Gonen, M. D'Angelica, W. R. Jarnagin, R. P. DeMatteo, L. H. Schwartz, S. Tuorto, L. H. Blumgart and Y. M. Fong, J. Gastrointest. Surg., 2003, 7, 325–330 CrossRef PubMed.
- W. de Graaf, R. J. Bennink, R. Vetelainen and T. M. van Gulik, J. Nucl. Med., 2010, 51, 742–752 CrossRef PubMed.
- B. E. Van Beers, J.-L. Daire and P. Garteiser, J. Hepatol., 2015, 62, 690–700 CrossRef PubMed.
- D. N. Granger and P. R. Kvietys, Redox Biol., 2015, 6, 524–551 CrossRef CAS PubMed.
- J. Huang, S. Xian, Y. Liu, X. Chen, K. Pu and H. Wang, Adv. Mater., 2022, 34, 2201357 CrossRef CAS PubMed.
- Y. Abe, I. N. Hines, G. Zibari, K. Pavlick, L. Gray, Y. Kitagawa and M. B. Grisham, Free Radicals Biol. Med., 2009, 46, 1–7 CrossRef CAS PubMed.
- H. Nakano, K. Boudjema, E. Alexandre, P. Imbs, M. P. Chenard, P. Wolf, J. Cinqualbre and D. Jaeck, Hepatology, 1995, 22, 539–545 CrossRef CAS PubMed.
- M. F. Monn, X. Hui, B. D. Lau, M. Streiff, E. R. Haut, E. C. Wick, J. E. Efron and S. L. Gearhart, Dis. Colon Rectum, 2014, 57, 497–505 CrossRef PubMed.
- G. A. Kwong, S. Ghosh, L. Gamboa, C. Patriotis, S. Srivastava and S. N. Bhatia, Nat. Rev. Cancer, 2021, 21, 655–668 CrossRef CAS PubMed.
- J. S. Dudani, C. G. Buss, R. T. K. Akana, G. A. Kwong and S. N. Bhatia, Adv. Funct. Mater., 2016, 26, 2919–2928 CrossRef CAS PubMed.
- W. Bernal, G. Auzinger, A. Dhawan and J. Wendon, Lancet, 2010, 376, 190–201 CrossRef PubMed.
- M. Yan, Y. Huo, S. Yin and H. Hu, Redox Biol., 2018, 17, 274–283 CrossRef CAS PubMed.
- B. Ruan, M. Yu, Y. Zhou, W. Xu, Y. Liu, B. Liu, L. Zhu, S. Yi, Y. Jiang and J. Huang, Angew. Chem., 2023, 135, e202305812 CrossRef.
- C. Sheng, J. Zhao, Z. Di, Y. Huang, Y. Zhao and L. Li, Nat. Biomed. Eng., 2022, 6, 1074–1084 CrossRef CAS PubMed.
- J. J. Chen, L. Q. Chen, Y. L. Wu, Y. C. Fang, F. Zeng, S. Z. Wu and Y. L. Zhao, Nat. Commun., 2021, 12, 6870 CrossRef CAS PubMed.
- L. Y. Wu, Y. Ishigaki, Y. X. Hu, K. Sugimoto, W. H. Zeng, T. Harimoto, Y. D. Sun, J. He, T. Suzuki, X. Q. Jiang, H. Y. Chen and D. J. Ye, Nat. Commun., 2020, 11, 446 CrossRef CAS PubMed.
- J. H. Liu, W. Zhang, C. M. Zhou, M. M. Li, X. Wang, W. Zhang, Z. Z. Liu, L. L. Wu, T. D. James, P. Li and B. Tang, J. Am. Chem. Soc., 2022, 144, 13586–13599 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |