
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransesterification reaction of tristearin (TS) & glycerol mono stearate (GMS) over surface basified PAN fibrous solid catalyst
Rawaz A.
Ahmed
 ,
Sanaa
Rashid
,
Ketan
Ruparelia
and
Katherine
Huddersman
*
,
Sanaa
Rashid
,
Ketan
Ruparelia
and
Katherine
Huddersman
*
Faculty of Health and Life Sciences, De Montfort University, The Gateway, Leicester, LE1 9BH, UK. E-mail: huddzeo1@dmu.ac.uk
First published on 9th August 2023
Abstract
A promising solution for the near future is the substitution of non-renewable fossil fuels with sustainable liquid feedstock such as biofuel (biodiesel). The cost of conventional biodiesel production is higher than that of petroleum-based diesel production since it is produced mostly from expensive high-quality virgin oil. Conventionally, commercial biodiesel is produced via liquid phase base-catalyzed transesterification of the triglyceride components of the oil with short-chain alcohols. This study demonstrates the first effective conversion of the triglyceride, tristearin (TS) and the monoglyceride, glycerol monostearate (GMS) to biodiesel using novel protonated and then basified crosslinked modified polyacrylonitrile ion exchange fibres (PANF) in the form of a mesh and an investigation of their lifetime in batch recycling. 3 g of basified PANF in 26 mL of methanol with a molar ratio of methanol to tristearin (TS) of 274![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 at 65 °C could achieve 95% conversion of tristearin. The catalyst was re-used for 9 cycles (18 hours) before being effectively regenerated back to 70% conversion. Response surface methodology (RSM) with central composite design (CCD) gave an optimum biodiesel conversion of tristearin of 87.62% with 2.5 g of catalyst, methanol to TS molar ratio of 143:1 at 65 °C for 1 h (0.1936% error). Using glycerol monostearate at (a molar ratio of methanol to GMS of (115:1)) conversion to the methyl ester was above 97.63% at 65 °C in 60 min. These basified PANF ion exchange fibres showed comparable activity to conventional homogeneous base catalysts namely, NaOH; as well as exhibiting high stability and ease of use. The FT-IR spectra suggested that after use, active sites were blocked with most probably unreacted reactants which can be removed with a more extensive DCM washing regime. As PANF is produced in the form of a self-supporting mesh it is easy to use, regenerate in situ, maintain and replace in a continuous flow reactor. The study is promising as a basified catalyst for sustainable use in converting triglyceride (TGs) in fats, oils and greases (FOGs) and fatbergs in wastewater to biodiesel.
:1 at 65 °C could achieve 95% conversion of tristearin. The catalyst was re-used for 9 cycles (18 hours) before being effectively regenerated back to 70% conversion. Response surface methodology (RSM) with central composite design (CCD) gave an optimum biodiesel conversion of tristearin of 87.62% with 2.5 g of catalyst, methanol to TS molar ratio of 143:1 at 65 °C for 1 h (0.1936% error). Using glycerol monostearate at (a molar ratio of methanol to GMS of (115:1)) conversion to the methyl ester was above 97.63% at 65 °C in 60 min. These basified PANF ion exchange fibres showed comparable activity to conventional homogeneous base catalysts namely, NaOH; as well as exhibiting high stability and ease of use. The FT-IR spectra suggested that after use, active sites were blocked with most probably unreacted reactants which can be removed with a more extensive DCM washing regime. As PANF is produced in the form of a self-supporting mesh it is easy to use, regenerate in situ, maintain and replace in a continuous flow reactor. The study is promising as a basified catalyst for sustainable use in converting triglyceride (TGs) in fats, oils and greases (FOGs) and fatbergs in wastewater to biodiesel.
1. Introduction
As the availability of fossil fuel gradually declines, sustainable and renewable energy production is increasingly required with biodiesel currently being explored as an environmentally friendly substitute for diesel fuel.1 Biodiesel can be produced via the following processes: (1) alkaline-catalysed transesterification (suitable for feedstocks with low free fatty acid content); (2) enzyme (biocatalyst) catalysed transesterification (3) acid-catalysed transesterification/esterification (good for feedstocks with high free fatty acid (FFA) content); (4) transesterification via a two-step process (again good for feedstocks with high FFA content).2–4The commercial production of biodiesel from virgin oil via homogenous transesterification of the triglycerides uses base catalysts, most commonly, sodium hydroxide (NaOH) and potassium hydroxide (KOH). For instance, Borges et al.,5 studied the homogeneous catalysis of soybean oil transesterification via methylic and ethylic routes in a multivariate comparison. The highest yield of biodiesel (above 90%) for the ethylic route arose from the optimum reaction parameters; reaction time 60 min, stirring 100 rpm, ethanol/oil molar ratio = 12:1, 0.2 wt% potassium ethoxide and temperature 35 °C. However, the biodiesel yield was 93% for the methylic route using optimum reaction parameters such as reaction time 30 min, stirring 100 rpm, methanol/oil molar ratio of 6:1, 0.2 wt% KOH catalyst, reaction temperature 55 °C.5 Hariprasath et al.,6 reported biodiesel production via the homogeneous base (NaOH) catalysis in the transesterification of cashew and canola oil. The maximum biodiesel from canola oil was 85% which was 30% higher than the biodiesel from cashew oil. The optimized transesterification parameters used to yield biodiesel from canola oil were a temperature of 70 °C for 120 min, an alcohol/fatty oil molar ratio 6.5:1 mixing at 550 rpm.6 Dias et al.7 investigated different alkali catalysts for the transesterification of virgin and waste cooking oils. It was found that the virgin oils and waste oils produced biodiesel yields of 97 and 92% respectively.7 The authors found that process parameters such as catalyst concentration, feedstock-to-alcohol ratio, stock purity and temperature all affect the purity and yield of the resulting biodiesel.7 An increase in alkali concentration is also detrimental to the process as it leads to soap formation as well as excess glycerol production which dilutes the biodiesel and ultimately increases reaction time.8 Transesterification of oil which was extracted from Citrullus vulgaris (watermelon) seeds has been reported as a potential feedstock in biodiesel production. Initial catalyst loading of a 0.13 g NaOH yielded 70% biodiesel; further increasing the catalyst to 0.18 g gradually reduced the yield to 49%.9 The most recent literature has reported the transesterification of canola oil in a continuous flow reactor using homogeneous base as catalyst. The maximum yield (95.13%) was obtained using: a 9:1 molar ratio (methanol to oil), 0.5% (w/w) KOH, 300 rpm stirring speed, at 60 °C for 60 minutes under reflux of methanol.10
Unfortunately, free fatty acids in low-quality feedstocks have a negative effect on homogenous base catalysts and acid and neutralisation pre-treatment processes are required prior to use. These pre-treatments are undesirable as they add to operation costs and are not environmentally friendly. Solid catalysts are thus a favourable green and economical alternative for the conversion of waste cooking oils to biodiesel.11 Heterogenous base solid catalysts have been developed and successfully applied in the transesterification process of triglycerides (TGs). For instance, the solid base catalyst (K2O/CaO–ZnO) was tested in the transesterification of soybean oil, at a reaction temperature of 60 °C, with catalyst loading of 2 wt%, methanol to oil molar ratio = 15:1, time 4 h. The incorporation of K2O on the CaO–ZnO catalyst enhanced catalytic activity to yield a maximum conversion of around 81.08 w/w%, due to increased basicity and surface area.12 While 97 w/w% of biodiesel production was observed over 6 wt% of CaO/Fe3O4@SiO2 catalyst, at a molar ratio of oil to methanol of 1:15, 65 °C; mechanical stirring 500 rpm; time 5 h.13 CaO powder in the catalysis of crude jatropha oil gave around 95.8.% of fatty acid methyl ester (FAME) using an oil to methanol ratio of 1:5.15, temperature 65 °C; stirring rate 500 rpm, over 133 min.14 The optimum FAME product from soybean oil has been reported as about 90% over the novel Mg/Al/Zn hydrotalcite/SBA-15 catalyst, at a reaction temperature 180–300 °C, reaction time of 2 h, oil to methanol molar ratio in the range of 1:5 to 1:30.15
It should be noted that the empirical relationships based on process modelling are key to optimising biodiesel production parameters. An empirical modelling method that has been used to establish the relationship between experimental variables and observed results is response surface methodology (RSM).16 This method has been used by many researchers to optimise transesterification process parameters. For instance, Abubakar A. et al.,17 optimized biodiesel reaction conditions using RSM and central composite design (CCD) for Jatropha seed oil with 0.30 g of catalyst and ethanol to oil molar ratio of 12:1 at 65 °C for 2 h, FAME yield was 98.32%. Their experimental yield was in good agreement with the predicted yield, with a relatively small percentage error (0.58%).17
Also, Zabaruddin N. H. et al.18 applied response surface methodology (RSM) for biodiesel synthesis catalysed by radiation-induced kenaf catalyst in a packed-bed reactor. The radiation-treated kenaf (Hibiscus cannabinus L.) was functionalized by trimethylamine and then ion-exchanged into the base form with NaOH. The optimum conditions were 9.81 cm packed bed height, a molar ratio of refined palm oil to ethanol of 1:50, and a volumetric flow rate of 0.38 mL min−1. Good agreement between the predicted and actual conversions to fatty acid ethyl ester (FAEE) of 97.29% and 96.87%, were achieved respectively.18 Garg and Jain used both RSM and artificial neural networks (ANN) for the modelling of yield and process parameters. They reported a significant quadratic regression model with values of R2 of 0.99 and 0.96 for ANN (92% conversion) and RSM with Box–Behnken experimental design (94% conversion), respectively. Their optimum reaction conditions were methanol to algal oil (20–60% (v/v)), catalyst concentration (0–2 wt%) and reaction time (60–180 min) at a constant temperature of 50 °C.19
An extensive literature review was conducted20 on the advantages and disadvantages of the different methodologies in biodiesel production via catalytic transesterification. Catalyst structure, morphology, texture, optimization, and reaction parameters such as temperature, catalyst concentration, reaction time, alcohol to substrate molar ratio, and type of alcohol have a significant influence on catalytic activity in biodiesel production.20 Despite many studies carried out on heterogeneous solid base catalytic transesterification, there are still a number of drawbacks that hinder industrial application. The drive to decarbonisation means there is a need to develop more economically viable solid base catalysts that require less energy in terms of their process conditions yet still maintain optimal efficacy and lifetime.
In this work, the surface functionalized fibrous polyacrylonitrile (PANF) ion-exchange fibres in the form of a mesh, are adapted to produce a strong base ion-exchanger and explored for the first time for efficacy in transesterification reactions. The surface functionalised PANF ion-exchange mesh was obtained by modification of the cyano-group of PANF with a mixture of hydrazine sulphate and hydroxylamine sulphate to produce a crosslinked polymer containing amidoxime, hydrazine, amide and carboxylate groups.21 This modified PAN was then treated with acid followed by alklali to obtain the strong base catalyst. The main novelty of the present work is the production of protonated and crosslinked hydrazine groups acting as a strong base PANF catalyst and its subsequent use in the transesterification process together with optimization of the experimental parameters, such as temperature, the molar ratio between tristearin (TS) or glycerol mono stearate (GMS) and alcohol, catalyst amount, reaction time, and reusability of catalyst.
2 Experimental
2.1 Chemical materials
The PAN mesh was modified with dihydrazine sulphate (Aldrich with purity > 98%), and hydroxylamine sulphate (99%, Aldrich). The functionalised PANF mesh was acidified using hydrochloric acid (37%, Aldrich) and basified with potassium hydroxide (KOH) (97% Aldrich).Triglyceride (TG): tristearin-TS, composed of 56.4% stearic acid and 41.3% palmitic acid (Technical grade, Aldrich), glycerol monostearate-GMS, composed of 95.4% monoglycerides (mainly monostearate and monopalmitate), 0.4% free glycerin, 0.5% free fatty acid (Purified, Fisher Scientific). Methyl ester of fatty acid: methyl palmitate (95%, Fisher Scientific), methyl stearate (99%, Fischer Scientific). Toluene (≥99.7% GC), methanol (99.8% GC), dichloromethane (≥99.9%, GC), hexane (95% n-Hexane, Fisher Scientific), chloroform-D (99.8% + 0.05% TMS, Goss Scientific Instruments Ltd).
2.2 Preparation of the surface functionalized PANF mesh
The surface functionalized PANF mesh is made on an industrial scale, though it is not currently commercially available and is prepared by modification of the cyano-group of the PANF.21 Modification solutions were prepared from alkaline hydrazine sulphate and hydroxylamine sulphate at pH 9.5 and heated at 95 °C the PAN mesh for two hours. It was then treated with alkali at pH 12 at 60 °C for 15 minutes followed by washing with water and then drying. The surface functionalized PANF mesh contains approximately 50% PAN yarn and 50% polypropylene. Initially the PANF mesh was used as this was produced in a strongly alkaline environment, however it was found not to work in the transesterification reaction. This surface functionalised PANF mesh was then acidified at ambient temperature for 24 h with 2 M HCl to protonate NH groups such as the substituted hydrazine groups and then dried. This was followed by ion-exchange of Cl− with OH− by contact with 2 M NaOH at ambient temperature for 24 h under stirring and dried at ambient temperature for 24 h.2.3 Transesterification reaction
Transesterification of the model triglyceride (TG) tristearin (TS) and the model mono glyceride (MG) glycol monostearate (GMS) with methanol was carried out in a Radley carousel (see Chart 1) fitted with water reflux condenser. TG or GMS (1 g) and methanol were preheated and then mixed and magnetically stirred under reflux, at a temperature of 65 °C. Then the desired amount of basified catalytic PANF mesh was added to the reactor. To determine the effect of different process parameters, the reaction time, amount of catalyst, and temperature were varied. The catalyst lifetime was also studied by its re-use in fresh feed after each reaction. Most of the transesterification reactions were repeated twice with 0.2–10% error in the % conversion, whilst the average of the reactions was used to plot the results in the Figures below. The total volume of the reaction mixture was in the range of 107.6–109.52 mL to reduce error percentage below 10% on the removal of 1 mL of sample for analysis at each time point resulting in a total removal of 7 mL over the 180 min of the transesterification reaction. | ||
| Chart 1 Transesterification reaction under water-cooled reflux in Radley Carousel reactor. | ||
To determine the yield of the methyl ester product the methanol was evaporated from the final transesterification reaction and the FAME product was collected and redissolved in 5 up to 10 mL toluene and injected into the GC-FID. The products of the transesterification of tristearin (TS) and glycerol monostearate (GMS) are methyl stearate (MS) and methyl palmitate (MP), and both MP and MS conversion percentages have been calculated via GC-FID areas and with the total conversion labelled as FAME for all samples.
To confirm the production of the esters 1H-NMR was used. After analysis by GC-FID 1 mL of sample product from the transesterification reaction, was taken and dried in an oven at 80–100 °C to remove the toluene solvent used for redissolution and the final product methyl ester was collected and redissolved in chloroform (chloroform-D 99.8% + 0.05%) for 1H-NMR measurements.
2.4 Experimental design of the transesterification reaction
Response surface methodology (RSM) with central composite design (CCD) was used to model the optimum biodiesel production from tristearin (TS). Three independent parameters were evaluated (reaction time, catalyst loading and methanol to TS molar ratio), whilst the dependent variable was the conversion to the fatty acid methyl ester (FAME). The range and levels of the independent variables for the transesterification process are shown in Table 6.20 sets of experiments were carried out including the 23 factorial experiments, 6 axial points and 6 replicates of centre points as suggested by RSM, (see Table 7 and Section 3.4). The centre points are all variables at level zero which are vital in determining the level of experimental error and data reproducibility.45 The transesterification reactions for these twenty experiments comprised different amounts of tristearin in 13 mL of methanol. After the reaction was completed, the PANF catalyst was removed from the solution phase. The reaction mixture was slowly evaporated for 1–2 h at 80–100 °C to evaporate the excess methanol and water. After cooling to room temperature, the methyl ester product and unreacted TG were measured by GC-FID.
2.5 Model fitting and statical analysis
Design Expert software version 13 (STAT-EASE Inc., statistic made easy) was utilised for regression analysis of the experimental data (20 sets of experimental data as described in Section 3.4). The accuracy of the fitted model was determined from the value of correlation (R2), while the evaluation of the statistical significance of the equations developed was determined using an analysis of variance (ANOVA).452.6 Methyl stearate (MS)/methyl palmitate (MP) standard solution preparation
Quantitative analysis of the fatty acid methyl esters was performed by GC-FID using calibration curves. The concentrations of the standard fatty acid methyl esters were chosen as between 78 and 2500 ppm with the calibration curves consisting of six concentrations. Triplicate injections were performed for each standard solution for reproducibility. The correlation coefficient was no less than r2 = 0.999, thus confirming the linearity of the method.2.7 Characterization techniques
GC-FID analysis was conducted in a Thermofisher GC (TRACE1310), equipped with a flame ionization detector (FID) and manual sampler. Sample aliquots of 1 μl were injected using a spilt mode of (40:1) with both the injector and detector temperatures held at 250 °C. Hydrogen was used as carrier gas at constant flow (2.4 mL min−1). Chromatographic separation was performed using a nitro-terephthalic acid-modified polyethene glycol capillary column (Zebron ZB-FFAP, GC Cap. Column (60 m × 0.25 mm × 0.25 μm). The oven temperature was set at 200 °C and increased at a rate of 4 °C min−1 up to 260 °C. Standards and samples were measured with three triplicates.
1H NMR analyses were conducted using a JEOL ECZ 600 MHz spectrometer operating at 200–300 MHz. The solvent used was deuterated chloroform CDCl3 (chloroform-D 99.8% + 0.05% TMS). Chemical shifts (δ) were expressed in parts per million (ppm), and the values of the coupling constant (J) were expressed in Hertz (Hz). Conversion percentage (C, %), by 1H NMR, was calculated according to eqn (1) given in the literature.22
FTIR characterisation of PANF and its basified and regenerated analogues was performed using an ATR-FT-IR (Bruker Alpha Platinum ATR-FTIR) in the range 400–4000 cm−1. Spectra were produced in triplicate using 160 scans and a resolution of 4 cm−1. The following method of data manipulation was also used: Baseline correction, spectrum scaling and smoothing (2 × 25 smoothing points) and a final baseline correction and scaling.23
3. Results and discussion
The work in this paper is presented in two parts. Part one is to find values of the process parameters which will be reasonably close enough to the optimum values so as to produce meaningful answers in the DoE simulation.Thus part one, describes the batch transesterifications of tristearin (TS) and glycerol monostearate (GMS) with methanol by the basified PANF mesh catalyst. The reaction parameters (reaction temperature, amount of catalyst, reactant molar ratio, and reusability and regeneration of the catalyst) were optimized in the single variation method in laboratory-based experiments and the results presented in Section 3.1 up to Section 3.3 and Tables 2 and 3. In part two, a design of experiment (DoE) was performed to determine optimized reaction parameters for transesterification tristearin (TS) only. Here, the reaction temperature was kept constant at 65 °C as this was found to be optimum from the single variation method experiments given in Tables 2 and 3. 20 experiments were carried out using different values of the process parameters from those used in Tables 2 and 3, including 23 factorial experiments, 6 axial points, and 6 replicates of centre points. Central composite design (CCD) was utilized and the three process parameters considered were methanol to TS molar ratio (143–250), catalyst amount (1–2.5 g), and reaction time (60–150 min) at a constant temperature of 65 °C (see Table 6). The results are presented in Sections 3.4.1, 3.4.2 and 3.4.3.
3.1 Qualitative analysis of FAME product by NMR
The 1H NMR spectra of the model compounds tristearin (TS) and methyl ester (FAME) used in this work are shown in Fig. 1a and b. The 1H NMR spectra for the transesterification products of samples FAME-MR 571.1:1 and FAME-MR 285.5:1 (see Table 1) are shown in Fig. 2a and b. The products of the transesterification reaction of tristearin are methyl stearate (MS) and methyl palmitate (MP) and whilst the yields of MP and MS have been calculated individually via GC-FID with the total conversion labelled as FAME, the 1H NMR was found to be unable to easily differentiate between MS and MP. Table 1 summarizes the groups and their chemical shifts. The high intensity of the singlet signal (A) of the –CH3 group of the methyl ester occurs at 3.50 and 3.70 ppm and is clearly present at 3.60–3.7 ppm for the model methyl ester (Fig. 1b) and for the two transesterification products (see Fig. 2a and b),22 but is clearly absent for tristearin (TS) (see Fig. 1a). This signal will increase with the extent of conversion of tristearin to ester and was found to increase on decreasing the molar ratio of methanol to TS from 571.1:1 (FAME-MR 571.1:1) to 285.5:1 (FAME-MR 285.5:1). The triplet signal (B) at 2.24–2.29 ppm of the CH2 group adjacent to the carbonyl group in tristearin (TS) (see Fig. 1a) occurs at a slightly higher chemical shift (δ) value than that of methyl stearate because of greater deshielding by the carbonyl group of tristearin (TS) as compared to the ester group (Fig. 1b). The intensity and overall area of the triplet peak (B) of tristearin (TS) decreases which clearly indicates higher conversion of tristearin to methyl ester (see Fig. 2a and b).
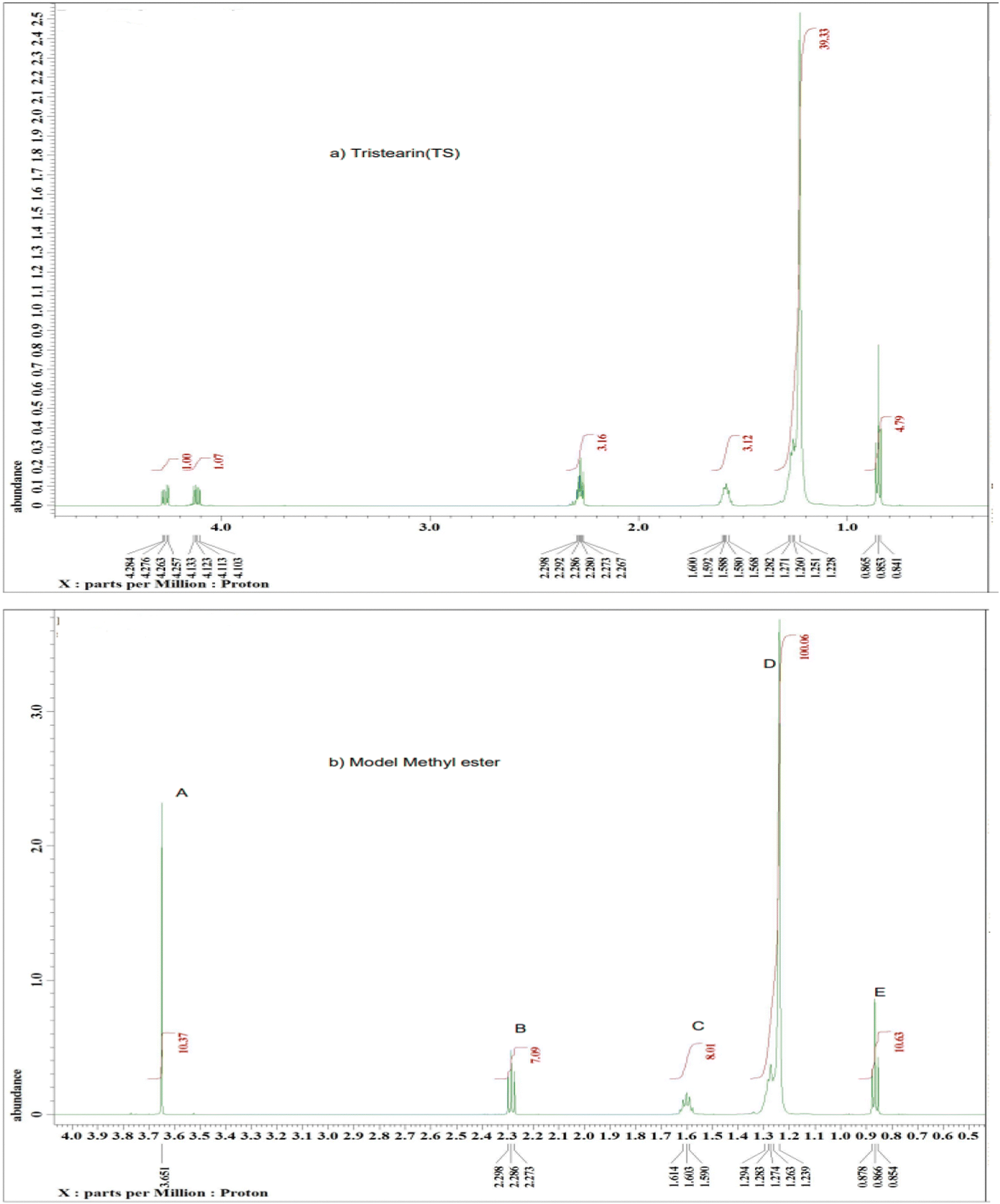 | ||
| Fig. 1 A typical 1H NMR spectrum of the model compounds (a) tristearin and (b) methyl stearate with labelling of the major peaks. | ||
| Signal | Moieties | Chemical shifts (ppm) |
|---|---|---|
| A | Methyl ester –CH3 | 3.50–3.70 |
| B | –CH2-adjacent to the carbonyl group | 2.24–2.29 |
| C | The aliphatic –CH2–s: CH2 group is one group away from the carbonyl group. | 1.61–1.28 |
| D | –CH2 – in CH2R: CH2 groups between the end CH3 group and the CH2 group | 1.23–1.29 |
| E | End of chain aliphatic –CH3 | 0.85–0.87 |
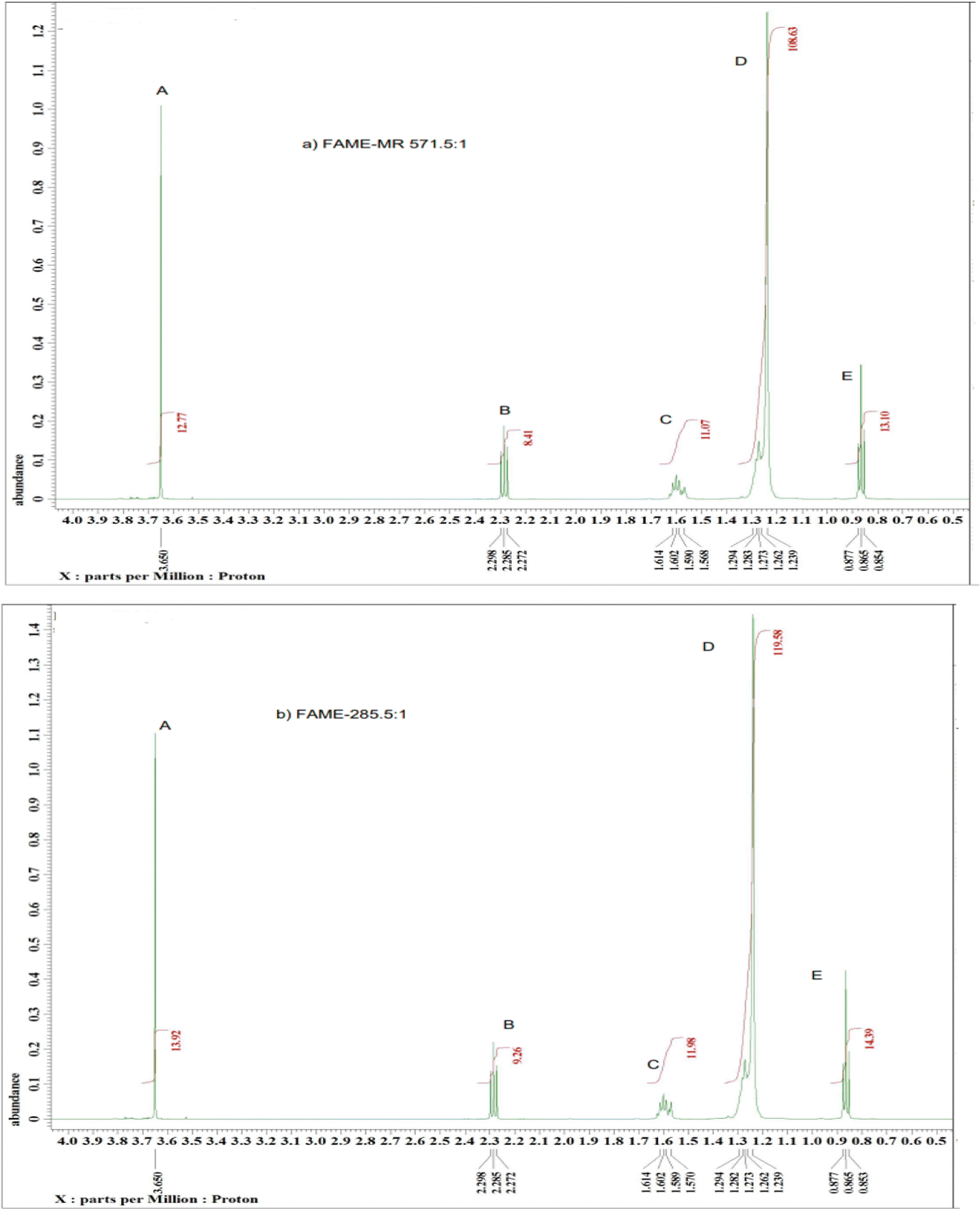 | ||
| Fig. 2 A typical 1H NMR spectrum of sample (a) FAME-MR 571.5:1 and (b) FAME-MR 285.5:1 with labelling of the major peaks, reaction conditions: reaction temperature 65 °C, 2 h and over 1.5 g of basified PANF fibrous solid catalyst. | ||
3.2 Effect of individual process parameters over basified PANF solid catalyst
As triglycerides and alcohol are immiscible, this limits the surface area available for transesterification and thus slows the reaction rate.24 Thus, a solid catalyst can increase the contact between reactants and improve the reaction rate.24 Thus, the transesterification process is limited by low conversion and a need for long reaction times with several approaches developed to avoid equilibrium establishment and to improve overall conversion and rate of reaction, with significant differences existing between current industrial practices and optimum transesterification processes/conditions. In the following section, tristearin (TS) glycerol monostearate (GMS) has been transesterified via solid base PANF fibrous catalyst mesh at moderate reaction temperatures.Methanol to TS molar ratios were varied from 142.9:1 to 571.5:1, keeping the remaining parameters constant, as illustrated in Fig. 3a and Table 2. At a lower methanol to TS molar ratio of 142.1:1, the conversion to FAME was 47.64% based on GC-FID analysis, which increased to 74.05% and 73.17% on increasing the methanol to tristearin molar ratios to 190.5:1 and 285.5:1, respectively, as the excess methanol helps to move the transesterification reaction in the forward direction. By further increasing the methanol to TS ratio to 571.5:1, the conversion to FAME decreased to 61.48%, due to a dilution effect. This can be explained as follows: the OH− anion and produced methoxy anion (CH3O−) are electrostatically held towards the protonated hydrazine groups on the mesh. Thus, it is likely that the dissolved TS and GMS need to approach closely to the mesh in order to undergo the transesterification reaction. As the solution becomes more dilute the probability of these molecules being found in the vicinity of the mesh decreases. Too much methanol could also hinder the separation of products, thus affecting the final yield of biodiesel. This was probably because the solubility of glycerol in the media increased as the amount of alcohol increased, making it difficult to separate glycerol out from the methyl ester mixture. Therefore, some glycerol remained in the solution and it reduced the conversion of the triglyceride by increasing the back reaction.30–32
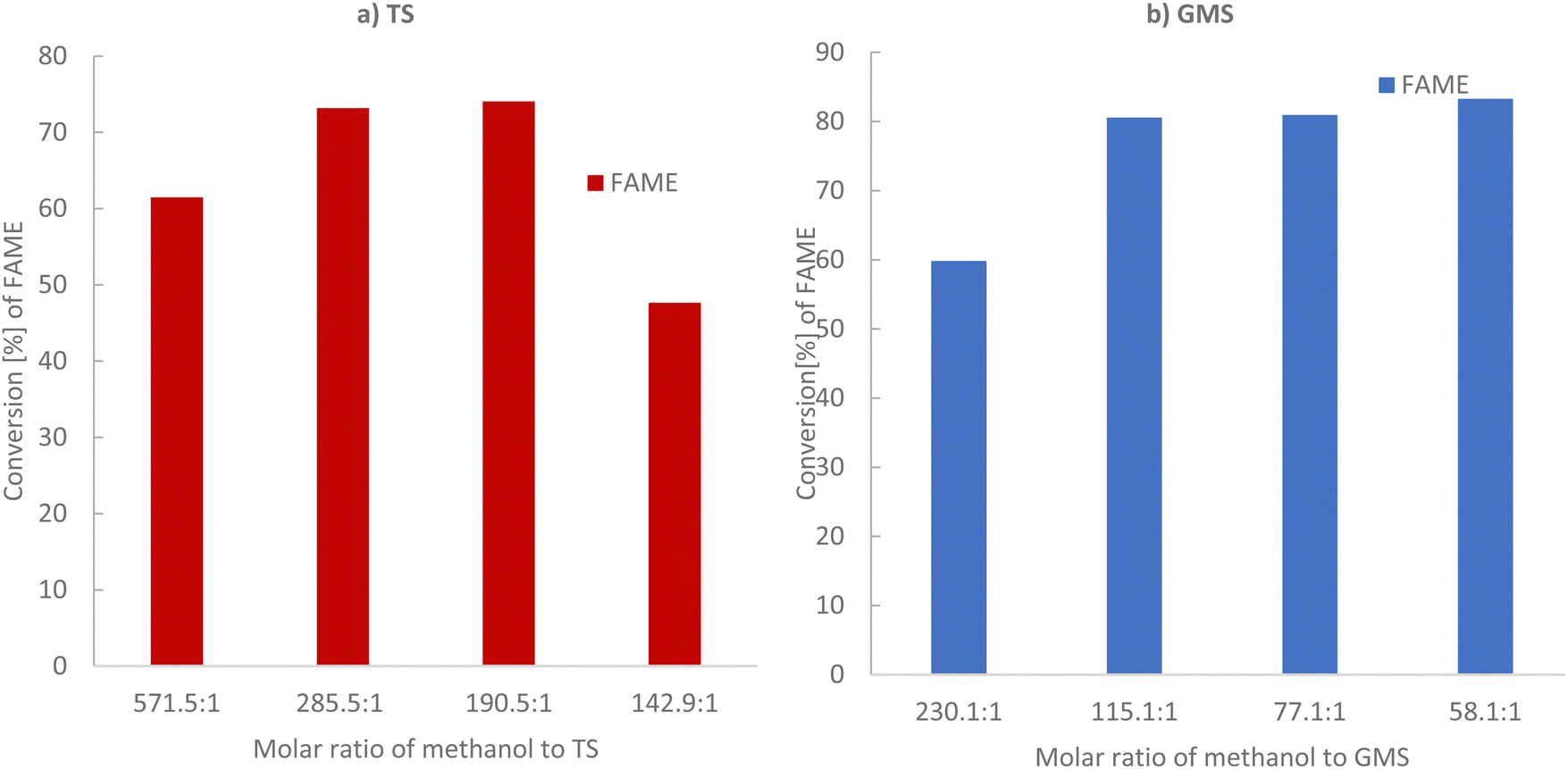 | ||
| Fig. 3 GC-FID analysis for conversion of triglyceride tristearin (TS) and monoglyceride glycerol monostearate (GMS) to (FAME) as a function of their molar ratio. (a) Tristearin (TS) conversion and (b) GMS conversion. Reaction conditions: 65 °C, 2 h, 1.5 g of solid base PANF catalyst. | ||
| Sample | MR | % conversion to MS ± SD (GC error%) | % conversion to MP ± SD (GC error%) | Total % conversion to FAME ± SD (Batch error%) |
|---|---|---|---|---|
| Note: The products of transesterification of tristearin are methyl stearate (MS) and methyl palmitate (MP). The SD is for triplicate injections of the reaction products on the GC. Peak areas of both MS and MP are used to obtain the total % conversion to FAME. | ||||
| Effect of molar ratio of methanol to TS (MR), 65 °C, 1.5 g cat,2 h, volume = 108.95 mL | ||||
| FAME-MR 571.5:1 |
571.5:1 |
26.77 ± 0.018 | 34.71 ± 0.019 | 61.48 ± 2.30 |
| FAME-MR 285.6:1 |
285:6:1 |
30.61 ± 0.03 | 42.56 ± 0.01 | 73.17 ± 1.43 |
| FAME-MR 190.5:1 |
190.5:1 |
30.98 ± 0.042 | 43.07 ± 0.06 | 74.05 ± 3.56 |
| FAME-MR 142.9:1 |
142.9:1 |
19.87 ± 0.003 | 27.77 ± 0.007 | 47.64 ± 4.76 |
| Samples | Time (min) | % conversion to MS ± SD (GC error%) | % conversion to MP ± SD (GC error%) | Total % conversion to FAME ± SD (bach error %) |
|---|---|---|---|---|
| Effect of catalyst amount, constant MR = 285.5:1, 65 °C, 2.5 g of catalyst volume = 108.95 mL |
||||
| FAME-15min | 15 | 5.64 ± 0.035 | 6.003 ± 0.018 | 11.643 ± 4.30 |
| FAME-30min | 30 | 8.68 ± 0.017 | 9.83 ± 0.016 | 18.51 ± 6.01 |
| FAME-60min | 60 | 13.53 ± 0.008 | 18.09 ± 0.006 | 31.62 ± 2.56 |
| FAME-90min | 90 | 15.8 ± 0.123 | 26.88 ± 0.041 | 42.68 ± 7.056 |
| FAME-120min | 120 | 29.32 ± 0.0052 | 40.89 ± 0.012 | 70.21 ± 3.33 |
| FAME-150min | 150 | 30.38 ± 0.058 | 45.95 ± 0.005 | 76.33 ± 4.24 |
| FAME-180min | 180 | 37.96 ± 0.012 | 51.54 ± 0.021 | 89.5 ± 5.01 |
| Effect of catalyst amount, constant MR = 285.5:1, 65 °C, 4 g of catalyst volume = 108.95 mL |
||||
| FAME-15min | 15 | 9.1 ± 0.021 | 14.54 ± 0.018 | 23.64 ± 5.30 |
| FAME-30min | 30 | 13.5 ± 0.008 | 17.98 ± 0.0060 | 31.48 ± 3.456 |
| FAME-60min | 60 | 17.66 ± 0.047 | 27.03 ± 0.0052 | 44.69 ± 2.30 |
| FAME-90min | 90 | 32.87 ± 0.11 | 46.47 ± 0.0033 | 79.34 ± 4.56 |
| FAME-120min | 120 | 41.54 ± 0.006 | 54.08 ± 0.016 | 95.62 ± 3.67 |
| FAME-150min | 150 | 34.34 ± 0.05 | 56.03 ± 0.12 | 90.37 ± 2.23 |
| FAME-180min | 180 | 33.88 ± 0.1 | 56.56 ± 0.012 | 90.44 ± 3.89 |
| Effect of catalyst amount, constant MR = 285.5:1, 65 °C, 6 g of catalyst volume = 108.95 mL |
||||
| FAME-15min | 15 | 25.4 ± 0.012 | 38.5 ± 0.013 | 63.9 ± 6.34 |
| FAME-30min | 30 | 25.87 ± 0.003 | 40.75 ± 0.015 | 66.62 ± 2.87 |
| FAME-60min | 60 | 32.53 ± 0.05 | 48.85 ± 0.08 | 81.38 ± 3.30 |
| FAME-90min | 90 | 41.94 ± 0.008 | 54.38 ± 0.13 | 96.32 ± 3.67 |
| FAME-120min | 120 | 37.74 ± 0.006 | 57.03 ± 0.003 | 94.77 ± 4.45 |
| FAME-150min | 150 | 34.59 ± 0.006 | 60.46 ± 0.004 | 95.05 ± 2.34 |
| FAME-180min | 180 | 35.48 ± 0.12 | 54.92 ± 0.008 | 90.4 ± 4.45 |
| Effect of catalyst amount, constant MR = 285.5:1, 65 °C, 8 g of catalyst volume = 108.95 mL |
||||
| FAME-15min | 15 | 24.97 ± 0.056 | 41.43 ± 0.014 | 66.4 ± 2.345 |
| FAME-30min | 30 | 31.43 ± 0.044 | 44 ± 0.024 | 75.43 ± 2.67 |
| FAME-60min | 60 | 39.2 ± 0.11 | 52.26 ± 0.11 | 91.46 ± 3.567 |
| FAME-90min | 90 | 37.78 ± 0.052 | 52.97 ± 0.008 | 90.75 ± 4.30 |
| FAME-120min | 120 | 37 ± 0.019 | 49.82 ± 0.0034 | 86.82 ± 4.56 |
| FAME-150min | 150 | 37.58 ± 0.018 | 49.38 ± 0.13 | 86.96 ± 1.89 |
| FAME-180min | 180 | 37.56 ± 0.023 | 51 ± 0.044 | 88.56 ± 2.30 |
| Effect of reaction temperature, constant MR = 285.5:1, 6 g of catalyst, 55 °C volume = 108.95 mL |
||||
| FAME-15min | 15 | 15.52 ± 0.12 | 28.27 ± 0.033 | 43.79 ± 5.60 |
| FAME-30min | 30 | 23.52 ± 0.022 | 32.94 ± 0.107 | 56.46 ± 4.30 |
| FAME-60min | 60 | 28.53 ± 0.031 | 40.8 ± 0.071 | 69.33 ± 7.30 |
| FAME-90min | 90 | 32.24 ± 0.017 | 42.96 ± 0.12 | 75.2 ± 2.67 |
| FAME-120min | 120 | 34.5 ± 0.11 | 47.36 ± 0.14 | 81.86 ± 2.98 |
| FAME-150min | 150 | 35.4 ± 0.056 | 49.19 ± 0.013 | 84.59 ± 4.05 |
| FAME-180min | 180 | 35.35 ± 0.068 | 50.78 ± 0.008 | 86.13 ± 2.78 |
| Effect of reaction temperature, constant MR = 285.5:1, 6 of catalyst, 45 °C volume = 108.95 mL |
||||
| FAME-15min | 15 | 4.3 ± 0.02 | 4.76 ± 0.076 | 9.06 ± 3.30 |
| FAME-30min | 30 | 6.35 ± 0.05 | 8.35 ± 0.002 | 14.7 ± 5.670 |
| FAME-60min | 60 | 13.35 ± 0.04 | 19.97 ± 0.07 | 33.32 ± 4.00 |
| FAME-90min | 90 | 22.28 ± 0.05 | 29.11 ± 0.03 | 51.39 ± 3.80 |
| FAME-120min | 120 | 23.89 ± 0.07 | 36.82 ± 0.019 | 60.71 ± 2.45 |
| FAME-150min | 150 | 33.79 ± 0.038 | 37.2 ± 0.21 | 70.99 ± 6.56 |
| FAME-180min | 180 | 37.26 ± 0.17 | 38.41 ± 0.23 | 75.67 ± 3.45 |
| FAME-210min | 210 | 36.27 ± 0.003 | 44.96 ± 0.066 | 81.23 ± 8.30 |
| FAME-240min | 240 | 43.27 ± 0.16 | 37.026 ± 0.026 | 80.296 ± 5.078 |
| Samples | Run no | % conversion to MS ± SD (GC error%) | % conversion to MP ± SD (GC error%) | Total % conversion to FAME ± SD (Batch error%) |
|---|---|---|---|---|
| Reusability of basified PAN solid catalyst (3 g), 65 °C, MR = 274.8:1, 2 h, total vol. 27.33 mL |
||||
| FAME-R1 | 1 | 45.06 ± 0.063 | 51.25 ± 0.029 | 96.31 ± 2.05 |
| FAME-R2 | 2 | 44.42 ± 0.056 | 50.91 ± 0.0072 | 95.33 ± 3.078 |
| FAME-R3 | 3 | 45.78 ± 0.0058 | 50.64 ± 0.0078 | 95.42 ± 4.65 |
| FAME-R4 | 4 | 39.05 ± 0.011 | 49.67 ± 0.0076 | 88.72 ± 3.34 |
| FAME-R5 | 5 | 33.16 ± 0.058 | 49.16 ± 0.0058 | 82.32 ± 5.08 |
| FAME-R6 | 6 | 27.17 ± 0.093 | 43.64 ± 0.0082 | 70.81 ± 5.66 |
| FAME-R7 | 7 | 29.39 ± 0.017 | 40.08 ± 0.082 | 69.47 ± 3.48 |
| FAME-R8 | 8 | 19.89 ± 0.026 | 26.84 ± 0.011 | 46.73 ± 5.98 |
| FAME-R9 | 9 | 14.56 ± 0.005 | 20.94 ± 0.047 | 35.5 ± 3.34 |
| Samples | Run no | % conversion to MS ± SD (GC error%) | % conversion to MP ± SD (GC error%) | Total % conversion to FAME |
|---|---|---|---|---|
| Reusability of basified PAN solid catalyst (3 g) after regeneration, 65 °C, MR = 274.8:1, 2 h, total vol. 27.33 mL |
||||
| FAME-R1 | 1 | 29.17 ± 0.005 | 41.63 ± 0.011 | 70.8 |
| FAME-R2 | 2 | 25.2 ± 0.0015 | 33.23 ± 0.067 | 58.43 |
| FAME-R3 | 3 | 13.73 ± 0.0056 | 18.45 ± 0.047 | 32.18 |
| FAME-R4 | 4 | 7.082 ± 0.0012 | 10.5 ± 0.0045 | 17.582 |
Fig. 3b and Table 3 show the lowest conversion, 59.83%, of glycerol monostearate (GMS) to FAME at high methanol to GMS molar ratio of 230:1. Conversion sharply increased to 80% with decreasing molar ratio of methanol to GMS of 115:1, with negligible further increase in conversion from 80% to 83% as the ratio decreased further (see Table 3 and Fig. 3b). Thus, regardless of the mechanism (Rideal or Langmuir–Hinshelwood), both imply that excess methanol adsorption on the catalyst surface results in poorer conversion. Considering these results, going forward the methanol/tristearin ratio of 285.5:1, and methanol/GMS molar ratio of 115:1 was selected for further study.
| Samples | MR | % conversion to MS ± SD (GC error%) | % conversion to MP ± SD (GC error%) | Total % conversion to FAME ± SD (batch error%) |
|---|---|---|---|---|
| Effect of molar ratio methanol to GMS (MR), 65 °C, 1.5 g cat, 2 h | ||||
| The products of transesterification of glycerol monostearate are methyl stearate (MS) and methyl palmitate (MP). The SD is for triplicate injections of the reaction products on the GC. Peak areas of both MS and MP are used to obtain the total % conversion to FAME. | ||||
| FAME-MR230:1 |
230:1 |
33.21 ± 0.05 | 26.62 ± 0.012 | 59.83 ± 3.45 |
| FAME-MR115:1 |
115:1 |
45 ± 0.035 | 35.58 ± 0.059 | 80.58 ± 2.078 |
| FAME-MR 77:1 |
77:1 |
45.13 ± 0.043 | 35.8 ± 0.025 | 80.93 ± 6.68 |
| FAME-MR 58:1 |
58:1 |
42.1 ± 0.077 | 41.28 ± 0.10 | 83.58 ± 3.38 |
| Samples | Time (min) | % conversion to MS ± SD (GC error%) | % conversion to MP ± SD (GC error%) | Total % conversion to FAME ± SD (batch error%) |
|---|---|---|---|---|
| Effect of catalyst amount, constant MR = 115, 65 °C, 1.5 g of catalyst, volume = 107.74 mL | ||||
| FAME-5min | 5 | 7.078 ± 0.063 | 6.8 ± 0.046 | 13.878 ± 5.56 |
| FAME-15min | 15 | 18.234 ± 0.07 | 12.97 ± 0.078 | 31.204 ± 3.078 |
| FAME-30min | 30 | 21.6 ± 0.015 | 18.81 ± 0.026 | 40.41 ± 5.03 |
| FAME-40min | 40 | 30.53 ± 0.036 | 28.11 ± 0.004 | 58.64 ± 4.45 |
| FAME-60min | 60 | 31.28 ± 0.08 | 37.63 ± 0.04 | 68.91 ± 2.78 |
| FAME-90min | 90 | 36.21 ± 0.007 | 34.89 ± 0.0062 | 71.1 ± 3.98 |
| FAME-120min | 120 | 43.82 ± 0.0095 | 36.14 ± 0.058 | 79.96 ± 5.00 |
| Effect of catalyst amount, constant MR = 115, 65 °C, 2.5 g of catalyst, volume = 107.74 mL | ||||
| FAME-5min | 5 | 26.16 ± 0.05 | 21.64 ± 0.004 | 47.8 ± 2.78 |
| FAME-15min | 15 | 36.51 ± 0.09 | 29.3 ± 0.0014 | 65.81 ± 4.38 |
| FAME-30min | 30 | 45.08 ± 0.008 | 35.08 ± 0.036 | 80.16 ± 5.34 |
| FAME-40min | 40 | 48.4 ± 0.08 | 39.92 ± 0.006 | 88.32 ± 3.88 |
| FAME-60min | 60 | 50.1 ± 0.09 | 47.4 ± 0.056 | 96.63 ± 2.34 |
| FAME-90min | 90 | 51.08 ± 0.05 | 44.5 ± 0.008 | 95.58 ± 3.45 |
| FAME-120min | 120 | 50.09 ± 0.007 | 40.9 ± 0.06 | 90.99 ± 56.45 |
| Effect of catalyst amount, constant MR = 115, 65 °C, 4 g of catalyst, volume = 107.74 mL | ||||
| FAME-5min | 5 | 31.73 ± 0.07 | 25.93 ± 0.02 | 57.66 ± 4.56 |
| FAME-15min | 15 | 40.55 ± 0.021 | 29.93 ± 0.05 | 70.01 ± 2.078 |
| FAME-30min | 30 | 40.38 ± 0.01 | 40.43 ± 0.03 | 80.81 ± 3.34 |
| FAME-40min | 40 | 42.48 ± 0.04 | 43.23 ± 0.026 | 85.71 ± 5.26 |
| FAME-60min | 60 | 47.93 ± 0.06 | 44.26 ± 0.0028 | 92.19 ± 4.50 |
| FAME-90min | 90 | 51.03 ± 0.004 | 39.64 ± 0.025 | 90.67 ± 2.056 |
| FAME-120min | 120 | 44.33 ± 0.02 | 45.08 ± 0.008 | 89.41 ± 4.98 |
| Effect of catalyst amount, constant MR = 115, 65 °C, 5.5 g of catalyst, volume = 107.74 mL | ||||
| FAME-5min | 5 | 36.65 ± 0.018 | 27.9 ± 0.022 | 64.55 ± 6.54 |
| FAME-15min | 15 | 39.64 ± 0.033 | 32.13 ± 0.0026 | 71.77 ± 5.23 |
| FAME-30min | 30 | 41.35 ± 0.089 | 34.6 ± 0.089 | 75.95 ± 3.45 |
| FAME-40min | 40 | 46.99 ± 0.035 | 32.85 ± 0.002 | 79.84 ± 2.56 |
| FAME-60min | 60 | 45.73 ± 0.044 | 36.59 ± 0.0026 | 82.32 ± 4.078 |
| FAME-90min | 90 | 34.22 ± 0.0025 | 50.68 ± 0.0018 | 84.9 ± 7.07 |
| FAME-120min | 120 | 55.035 ± 0.094 | 28.68 ± 0.13 | 83.72 ± 3.67 |
| Effect of reaction temperature, constant MR = 115, 2.5 of catalyst, 55 °C, volume = 107.74 mL | ||||
| FAME-5min | 5 | 21.44 ± 0.097 | 22.25 ± 0.075 | 43.69 ± 4.078 |
| FAME-15min | 15 | 26.55 ± 0.044 | 32.69 ± 0.032 | 59.24 ± 3.99 |
| FAME-30min | 30 | 43.3 ± 0.023 | 31.19 ± 0.098 | 74.69 ± 1.73 |
| FAME-40min | 40 | 47.83 ± 0.049 | 37.73 ± 0.036 | 85.56 ± 4.65 |
| FAME-60min | 60 | 50.47 ± 0.050 | 39.58 ± 0.086 | 90.05 ± 2.55 |
| FAME-90min | 90 | 50.58 ± 0.15 | 37.16 ± 0.011 | 87.74 ± 6.05 |
| FAME-120min | 120 | 47.85 ± 0.089 | 38.72 ± 0.078 | 88.57 ± 3.66 |
| Effect of reaction temperature, constant MR = 115, 2.5 of catalyst, 45 °C, volume = 107.74 mL | ||||
| FAME-5min | 5 | 21.56 ± 0.09 | 15.69 ± 0.04 | 37.25 ± 7.87 |
| FAME-15min | 15 | 32.32 ± 0.011 | 20.071 ± 0.031 | 53.391 ± 3.98 |
| FAME-30min | 30 | 35.78 ± 0.12 | 35.5 ± 0.02 | 71.28 ± 4.768 |
| FAME-40min | 40 | 40.65 ± 0.071 | 37.19 ± 0.03 | 77.84 ± 2.56 |
| FAME-60min | 60 | 44.23 ± 0.05 | 37.22 ± 0.014 | 81.45 ± 3.95 |
| FAME-90min | 90 | 43.69 ± 0.03 | 40.37 ± 0.11 | 84.06 ± 2.47 |
| FAME-120min | 120 | 42.66 ± 0.025 | 40.9 ± 0.06 | 83.56 ± 5.67 |
Whilst conversion to FAME (biodiesel) was in the range of 80% to 83% for glycerol monostearate (GMS) as compared with 73.17 and 74.47% for tristearin (TS), the optimum methanol molar ratios were different. Not unexpectedly the three backbones of TS needed at least double the amount of methanol than is required for GMS to obtain good conversion to the methyl ester (biodiesel) under the same reaction conditions.
Effect of catalyst amount. In theory, the more catalyst that is added the more products are produced. Our experiments gave results consistent with theory. The effect of the amount of basified PANF solid base catalyst on the transesterification reaction with a molar ratio of methanol to tristearin 285.5
:1 and methanol to GMS 115:1 at 65 °C as a function of the duration of treatment is shown in Fig. 4a and b, (see Tables 2 and 3). For tristearin there was a significant increase in FAME production on increasing the basified PANF solid catalyst from 2.5 g to 6 g, which did not increase further for 8 g of catalyst.33 4 g and 6 g of PANF base solid catalyst, gave high conversions of 95.32 and 96.32% w/w respectively, with 6 g of catalyst reaching optimum conversion at 80 min which was earlier than 4 g of catalyst which took 110 min. The results suggest that a catalyst dosage of 6 g provided enough catalytic sites for the reactants. When the amount of catalyst was in excess, mass transfer between the catalyst and reactants decreased, therefore reducing interactions between them and, ultimately, the FAME content.34 This effect can also be contributed to by the production of soap via an unwanted side reaction which could arise from any trapped NaOH.
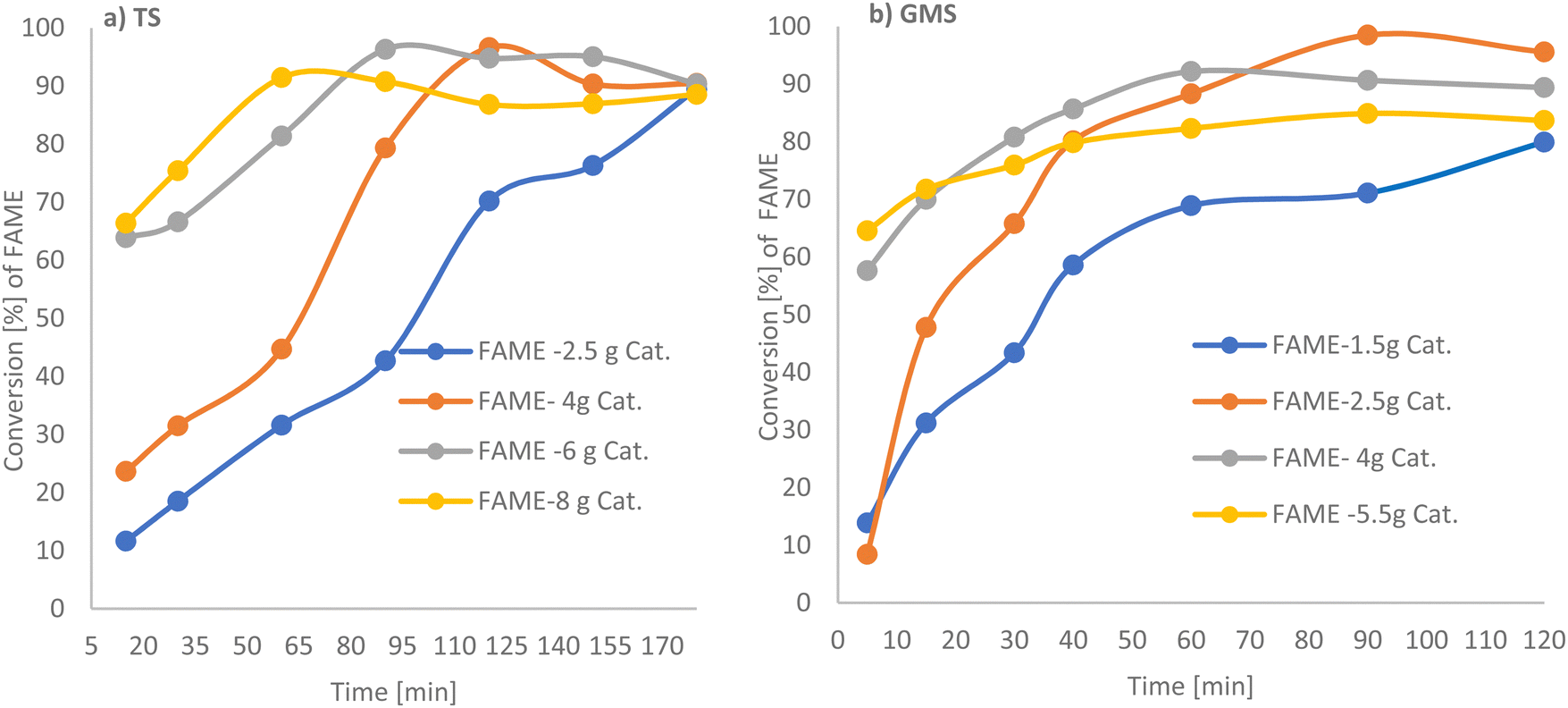 | ||
| Fig. 4 GC-FID analysis for the conversion of TS and GMS to FAME as a function of solid base PANF catalyst loading. (a) TS to methyl ester for a molar ratio of methanol to TS 285.5:1, total vol. 108.95; and (b) GMS to FAME for a molar ratio of methanol to GMS 115.1:1, total vol. 107.74 mL, at 65 °C. | ||
A similar trend in FAME production was obtained for glycerol monostearate (GMS) with a significant increase in methyl ester (FAME) production from 1.5 g to 4 g of basified PAN solid catalyst, with high conversions of 92.19% and 97.5%, respectively. FAME production was reduced to 84.9% by increasing the catalyst amount to 5.5 g. This has also been observed in homogeneous transesterification catalysis.35,36 Thus 6 g and 2.5 g basified PANF solid catalysts were selected for tristearin (TS) and glycerol monostearate (GMS) feedstock, respectively, going forward as higher amounts of catalyst did not significantly increase the reaction rate nor FAME production.
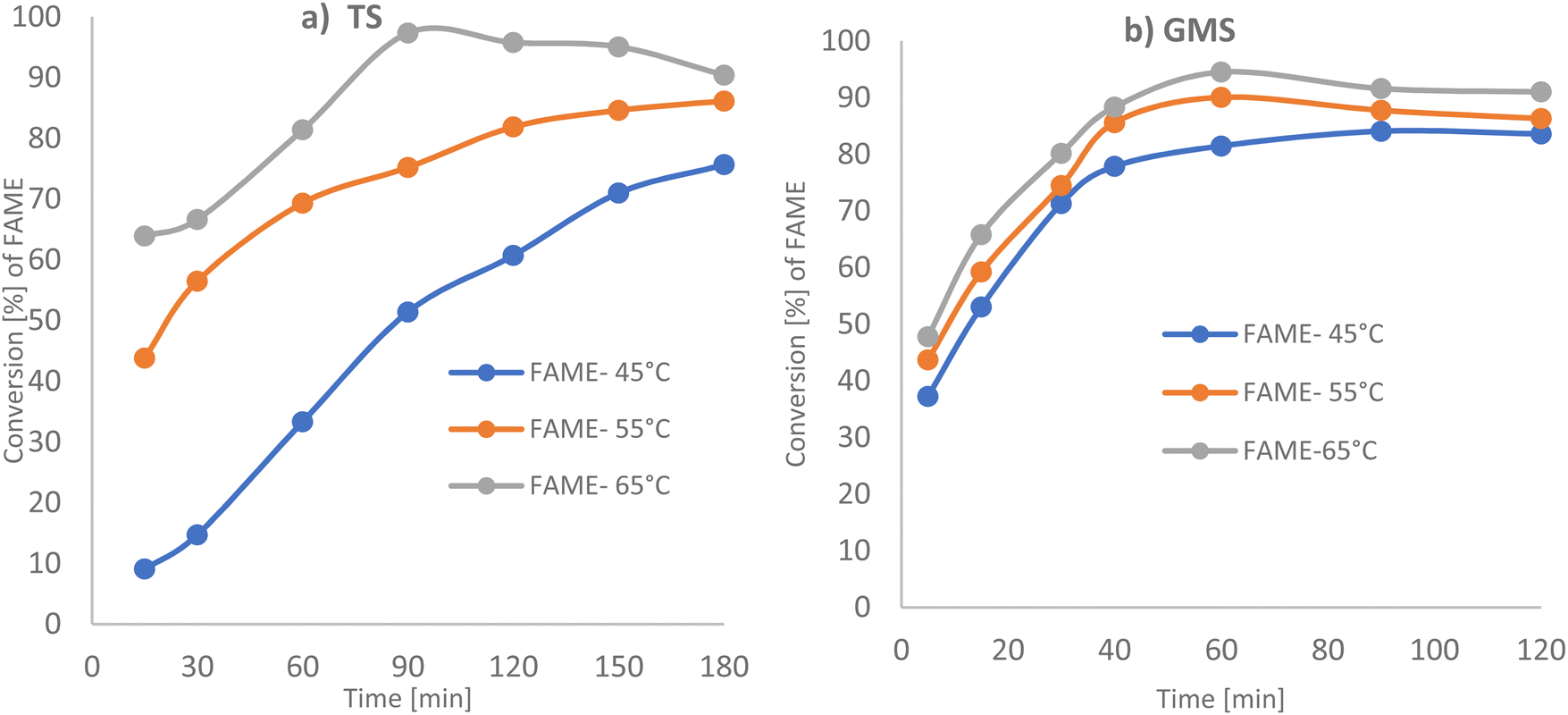 | ||
| Fig. 5 GC-FID analysis for conversion of TS and GMS to FAME as a function of temperature (a) TS to methyl stearate for the molar ratio of methanol to TS 285.5:1, total vol. 108.95 mL, 6 g of PANF base catalyst; (b) GMS to methyl stearate for the molar ratio of methanol to GMS 115.1:1, total vol. 107.74 mL, 2.5 g of PANF base catalyst. | ||
GMS requires a smaller amount of catalyst (2.5 g) than tristearin and in contrast to tristearin, high FAME production is observed for all reaction temperatures in the range 45 °C up to 65 °C. Glycerol monostearate is easier to transform to FAME product as it needs milder reaction conditions, as compared with the need to break three C–C bonds in the glycerol backbone of tristearin.
:1, total vol. 27.34 mL. as shown in Fig. 6. It was found that the catalyst maintained high catalytic activity for the first 6 cycles with conversion to the esters ranging from 95–82%, showing good stability of the catalyst similar to that obtained by Chumuang et al. using the heterogeneous catalyst calcium methoxide.37 Conversion decreased gradually to 35% for the ninth cycle (see Table 2 and Fig. 6). The reduction in activity after each cycle using the recycled catalyst indicated the deactivation of active sites.38,39
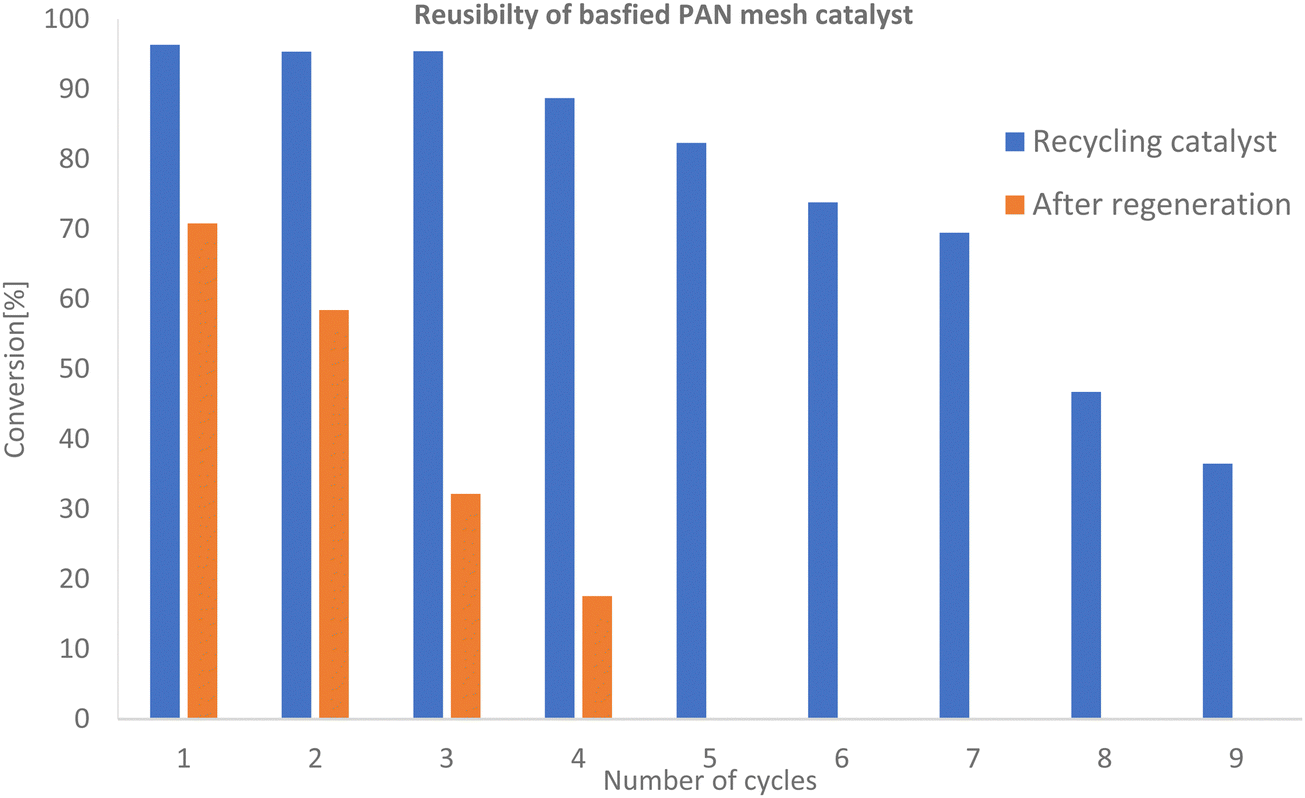 | ||
| Fig. 6 GC-FID analysis for conversion of tristearin (TS) to FAME as a function of the number of cycles. Reaction conditions: 65 °C, 3 h, 3 g of catalyst, Molar ratio of methanol to TS was 274:1, volume = 27.34 mL. | ||
To investigate regeneration of the basified PANF solid catalyst for repeated use, the PANF mesh was recovered after the 9th cycle of reaction and washed briefly with dichloromethane (DCM) to dissolve off any adsorbed organic reaction products/reactants. The PANF catalyst was regenerated with 30 mL of 2 M HCl solution for 24 h after which it was immersed in 2 M NaOH for 24 h under stirring and dried for 24 h. Subsequently, the regenerated PANF base catalyst was reused in the transesterification process. The catalytic activity of the regenerated PANF base solid catalyst was regained to give 70–58% conversion of TS to FAME for two more cycles (see Fig. 6 and Table 2). However catalytic activity significantly decreased after the third cycle with conversion reduced to about 17% for the fourth cycle. This suggests that the brief wash with dichloromethane was not sufficient to remove adsorbed compounds (Fig. 6). This is corroborated by the FTIR spectrum of the initial basified PANF catalyst in Fig. 7 and the deactivated and reused catalysts in Fig. 8.
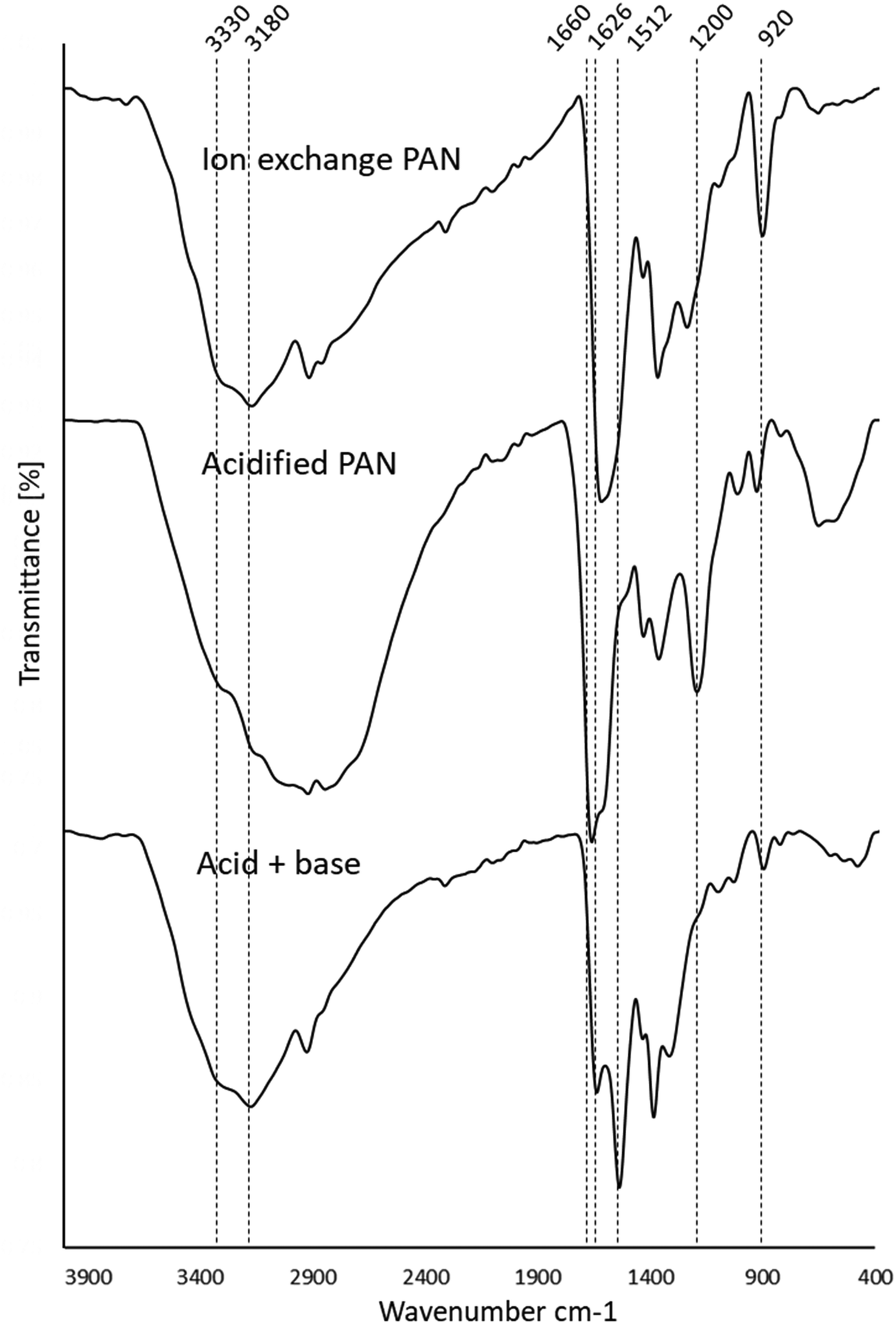 | ||
| Fig. 7 Stacked FTIR spectra of the ion exchange PANF, the ion exchange PAN in its acidified and basified forms. | ||
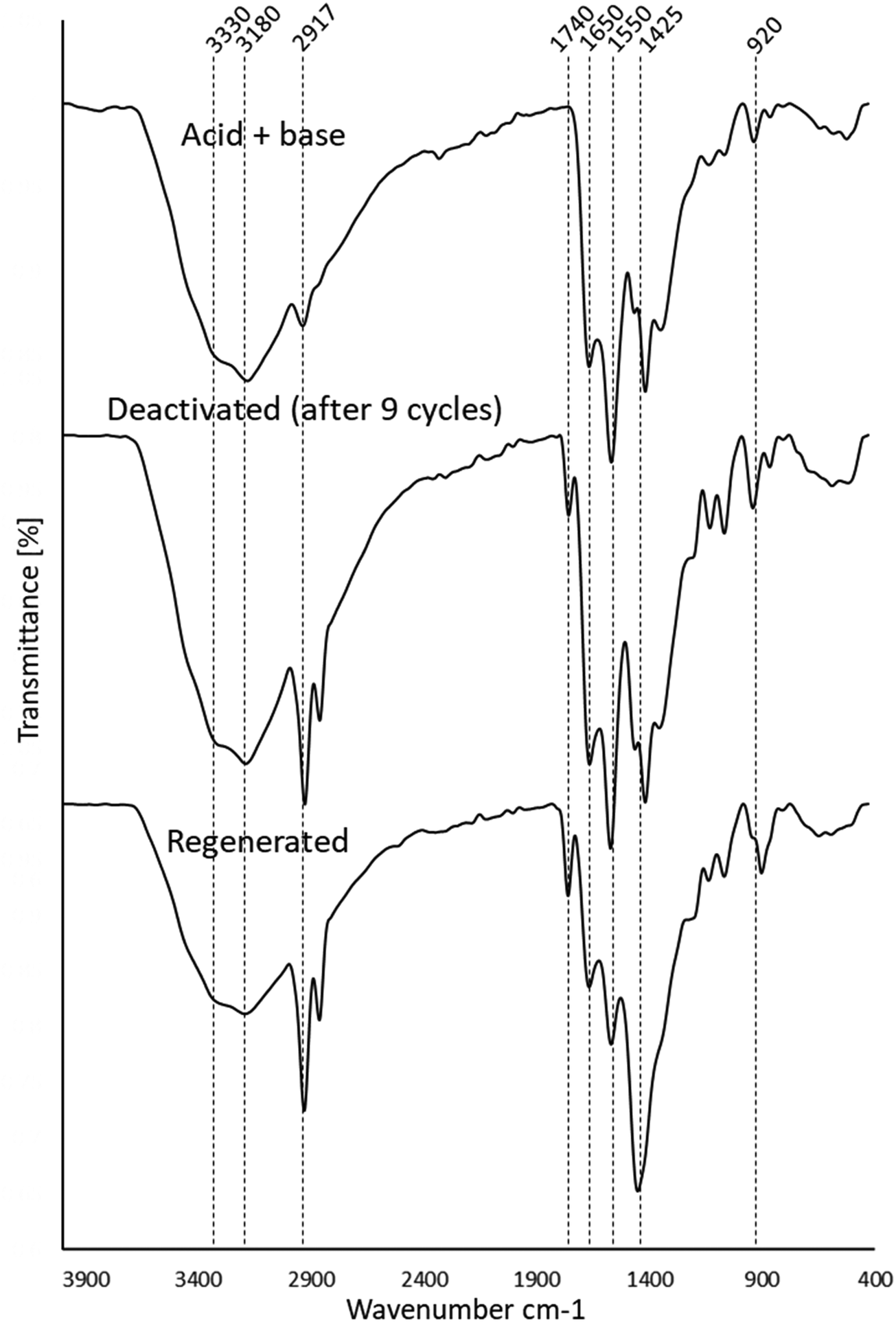 | ||
| Fig. 8 Stacked FTIR spectra of the basified catalyst, the deactivated and regenerated catalysts. | ||
Thus, one of the reasons for the drop in the catalytic activity was the leaching of the hydroxyl ion from the PANF-basified solid catalyst. It could also be due to the blocking of the basic sites of the catalyst. The first cycles after regeneration resulted in a significantly lower conversion to the ester than those of the first cycles before regeneration and unlike the initial cycles, there was a steep drop-in activity. It is possible that in each cycle before regeneration, a significant amount of organic product and byproduct such as the methyl ester product, unreacted tristearin and glycerol was gradually building up and deposited on the fibres of PANF mesh causing deactivation by blocking access to the active sites. It is also possible that all these by-products glycerol, unreacted TGs and the methyl ester could prevent the hydrochloric acid and sodium hydroxide treatment from hydroxylation of the amine groups on the amidoxime functionalized PANF. The regeneration of the base catalyst was then due solely to sodium hydroxide absorbed between the fibrils of the fibrous catalyst, which quickly leached out resulting in its poor performance. However, a more extensive regeneration protocol of the basified PANF catalyst of our work would likely show promise in improving efficacy after regeneration. Thus, further studies about the causes of this deactivation are required to clarify whether blocking of active sites or loss of basicity, among other factors, is responsible.
Nonetheless, our heterogenous basified PANF solid catalyst showed a very high catalytic activity after 9th recycles and for four cycles after regeneration as compared to other works.40–43
3.3 FTIR analysis of the basified catalyst and regenerated catalyst
The FTIR assignments of the ion exchange PANF can be found in Table 4 and as indicated in previous papers.23,44 In short, the groups present on the ion exchange PAN consist of amidoximes (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–OH) and hydrazines (C–NH–NH2) Scheme 1.
N–OH) and hydrazines (C–NH–NH2) Scheme 1.
| Wavenumber cm−1 | Ion exchange PAN fibres | Acidified ion exchange PAN fibres | Acidified and then basified PAN fibres | Assignment23,44 |
|---|---|---|---|---|
| 3330 | X | X | X | OH, amidoxime |
| OH, carboxylic acid | ||||
| 3180 | X | X | X | NH, amidoxime, hydrazine and amide |
| 3000–2800 | X | NH2+ and NH3+ amidoxime, hydrazine and amide | ||
| 1660 | X | X | X | CO amide/imide, carboxylate/carboxylic acid |
| 1626 | X | X | X | CN, amidoxime |
| 1550–1512 | X | X | NH2, amidoxime and hydrazine | |
| COO−, carboxylate | ||||
| 1200 | X | X | C–O carboxylic acid | |
| 920 | X | X | X | N–OH, amidoxime |
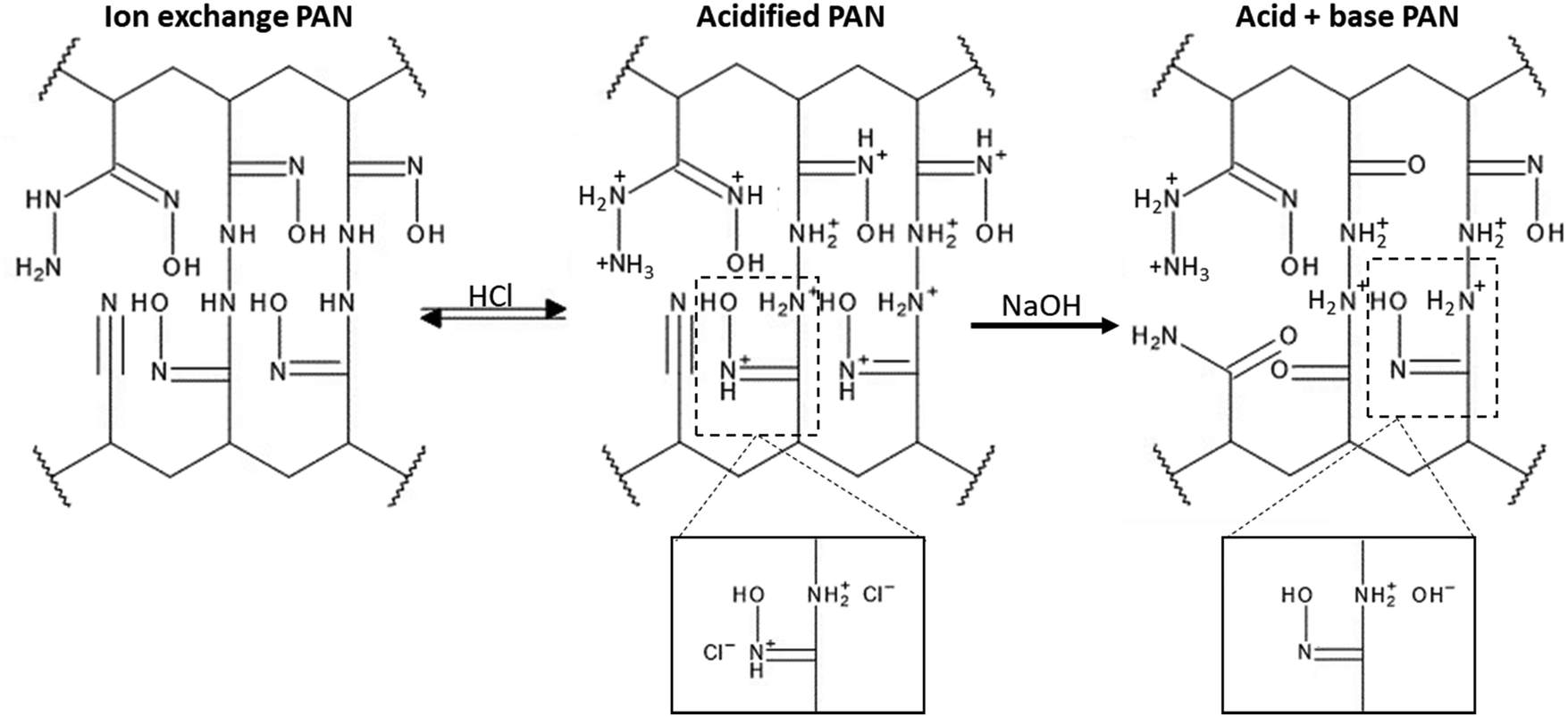 | ||
| Scheme 1 Structures of the ion exchange PAN upon acidification and subsequent basification (Partially reproduced from Ahmed R.)23 | ||
Upon acidification with 2 M HCl, there is a significant reduction in the 1512 cm−1 peak assigned to NH2 in amidoximes and hydrazines owing to protonation. There is a reduction in the 920 cm−1 N–OH amidoxime peak which is possibly due to a shift to lower wavenumbers upon protonation. There is also evidence in the FT-IR of some acid hydrolysis of the amidoxime group to amides with further hydrolysis to carboxylic acids, which could contribute to the growth of the CO 1660 cm−1 peak and the C–O peak at 1200 cm−1. The presence of carboxylic acid groups will also add OH groups which is demonstrated by the intense and broad peak at 3400–2800 cm−1 which centres ∼3000 cm−1. Amine salts also adsorb in the 2800–3000 cm−1 region which explains the growth of this peak on acidification.
Thus, acidification of PANF resulted in the protonation of the hydrazine groups in both crosslinked and uncrosslinked forms. Hydrazine is a strong base and once protonated it is difficult to deprotonate. These positively charged groups will form electrostatic bonds with the negatively charged chloride ions on acidification which ion-exchange with OH− during basification with NaOH to form a strong base. It is likely that the OH− then reacts with the methanol to form the methoxide ion which catalysis the transesterification in the usual manner.
From the stacked spectra in Fig. 7, it is likely that the basification process further converted some amide groups formed during acid hydrolysis to carboxylate groups via alkaline hydrolysis. The spectrum of basified PAN in Fig. 7 also shows further reduction of the 920 cm−1 N–OH amidoxime group, and the reduction in intensity of the 1626 cm−1 CN group of amidoxime. The re-basification of the ion exchange polymer also reforms the NH2 groups in amidoxime via deprotonation as indicated by the regrowth of the peak at 1512 cm−1. This peak at 1512 cm−1 is also assigned to the COO− peak which is also reformed on de-protonation.
In Fig. 8, the FT-IR spectrum of the deactivated catalyst is stacked alongside the fresh acidified and then basified catalyst and the regenerated catalyst. The deactivated catalyst was very similar to the fresh catalyst except for a few differences which heavily suggest active site blockage. The deactivated catalyst was visibly seen to have a white substance trapped within its fibres which was identified as an ester by FTIR analysis (Fig. 8 and Table 5) with a new low-intensity peak at 1740 cm−1 which can be assigned to CO of an ester. The band at 3000–3400 cm−1 has also increased significantly in intensity due to the deposition of the long chain esters adding to the C–H peaks at 2900 cm−1. Interestingly, both the CN (1626/1650 cm−1) and NH2/COO− (1550 cm−1) peaks do not change in either intensity or position. This indicates that these groups are not affected by deactivation and remain in their basified forms.
| Wavenumber cm−1 | Acidified and then basified PAN fibres | Deactivated PAN fibres | Regenerated PAN fibres | Assignment23,44 |
|---|---|---|---|---|
| 3330 | X | X | X | OH, amidoxime |
| OH, carboxylic acid | ||||
| 3180 | X | X | X | NH, amidoxime and hydrazine |
| 2917 | X | X | X | C–H, PAN fibres and ester (tristearin) |
| 1740 | X | X | CO, ester (tristearin) |
|
| 1650 | X | X | CO, amide/imide, carboxylate |
|
| 1626 | X | X | X | CN, amidoxime |
| 1550 | X | X | X | NH2, amidoxime and amide |
| COO−, carboxylate | ||||
| 1425 | X | X | X | CH2, PAN fibres and ester (tristearin) |
| 920 | X | X | X | N–OH, amidoxime |
The white substance was scraped off the deactivated catalyst, analysed on the FTIR and compared to the spectra of both tristearin and FAME product. As can be seen from Fig. 9, the spectrum of the white substance highly resembles that of tristearin with the positions and intensities of all the peaks identical. The spectrum of the white substance was also compared to the spectra of both methyl stearate and methyl palmitate and whilst the spectra are very similar there are noticeable differences especially in the C–O peaks at 1169 and 1100 cm−1 which are very much smaller for methyl stearate and methyl palmitate than for tristearin. The FT-IR of the FAME product also has a low intensity band at 3345 cm−1 ascribed to OH, most likely due to the presence of small amounts of mono and/or diglycerides or possibly fatty acid, however the white substance does not have any intensity in this region suggesting that it was unreacted tristearin.
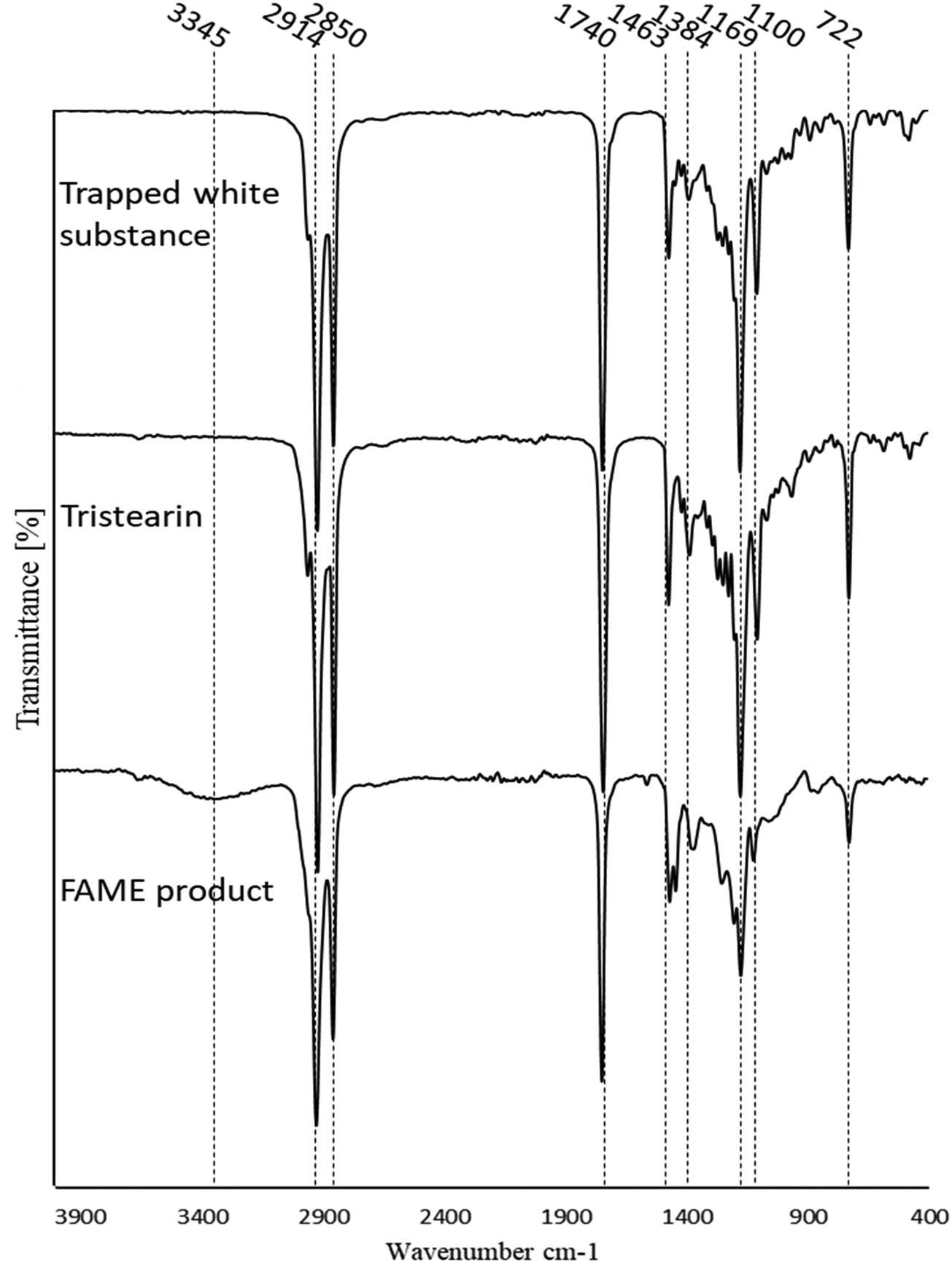 | ||
| Fig. 9 FTIR spectra of the white substance (esters) in deactivated fibres. | ||
This suggests that the PANF catalyst experienced a continuous loss of OH− ions over the 9 consecutive transesterification cycles, whereby conversion also decreased leading to a build-up of tristearin on the fibres.
Upon regeneration in Fig. 8, the broad, intense band at 3400–2000 cm−1 reduces in intensity, suggesting the loss of NH groups by hydrolysis to OH groups which are generally broader and less intense. The presence of the high-intensity C–H peak at 2917 cm−1 (also present in the white substance in Fig. 9) suggests that the washing regime with DCM was not sufficient to remove the tristearin (TS) blocking the catalyst active sites. This is further supported by the unchanged intensity of the tristearin CO peak at 1740 cm−1 and the high-intensity CH2 peak at 1425 cm−1. The CN (1626 cm−1) and NH2/COO− (1550 cm−1) groups have decreased in intensity in comparison to those of the initial acid and then basified catalyst suggesting that there has been a loss of these groups due to hydrolysis on regeneration. There is also the possibility that under the heated basic conditions of the transesterification reaction that the reactants and/or products reacted with the amine and/or carboxylate groups present on the catalyst to form fatty acid amides or fatty acid esters chemically bound to the catalyst, which could then not be washed off. It is difficult to interpret the FT-IR spectrum of the regenerated catalyst to definitively ascribe the mechanism of deactivation on regeneration. However, it seems that regeneration was not effective owing to both loss of sites on regeneration, chemical binding of active sites with reactant/products and blocking of the sites by the tristearin which were not removed by the DCM wash.
3.4 Design of experiment transesterification reaction
In this study, the design of experiment [DoE] was performed as follows: 20 experiments were carried out, including 23 factorial experiments, 6 axial points, and 6 replicates of centre points. Central composite design (CCD) was utilised and the three process parameters considered were methanol to TS molar ratio (143:1 to 250:1), catalyst amount (1 g to 2.5 g) and reaction time (60 to 150 min) at a constant temperature of 65 °C (see Table 6). These parameters were calculated from the regression formula (eqn (1)).
| Variables | Levels | ||||||
|---|---|---|---|---|---|---|---|
| Coding | Unit | −α | −1 | 0 | 1 | +α | |
| Molar ratio | A | Molar | 106.524 | 143 | 196.5 | 250 | 286.476 |
| Catalyst | B | g | 0.488655 | 1 | 1.75 | 2.5 | 3.01134 |
| Time | C | min | 29.3293 | 60 | 105 | 150 | 180.681 |
The batch transesterification reaction experiments were performed for all 20 reaction conditions as given in Table 7 for factors 1, 2 and 3 which are the outcomes of the design of the experiment [DoE] and calculated from the regression formula (eqn (1)). The products of these reactions were measured via GC-FID to yield the percentage FAME conversion which was then input (see Table 6) in the design of the experiment to run the DoE. The results from the experimental transesterification data were fitted to a mathematical model that correlates the FAME conversion with the independent reaction variables via a second-order polynomial equation as given below.45
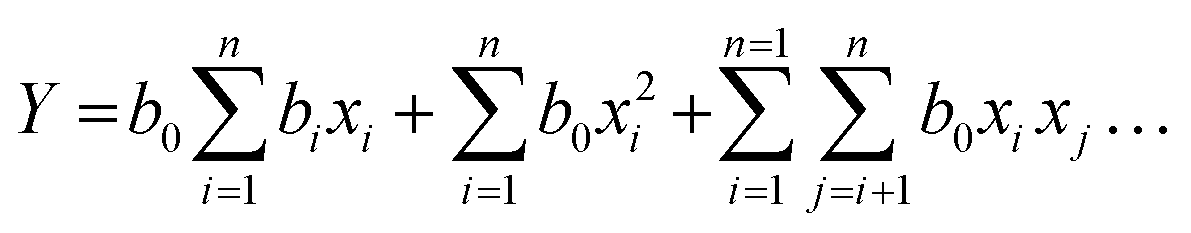 | (1) |
| Runs | Factor 1 | Factor 2 | Factor 3 | Response 1 | |
|---|---|---|---|---|---|
| A: molar ratio | B: catalyst amount (g) | C: reaction time (min) | Experimental value conversion (%) | Predicted value conversion (%) | |
| 1 | 250 | 1 | 60 | 50.95 | 51.18 |
| 2 | 143 | 1 | 150 | 40.64 | 40.58 |
| 3 | 196.5 | 0.50 | 105 | 55.85 | 55.83 |
| 4 | 250 | 1 | 150 | 59.23 | 59.08 |
| 5 | 106.52 | 1.75 | 105 | 49.28 | 49.33 |
| 6 | 196.5 | 1.75 | 105 | 57.44 | 58.41 |
| 7 | 286.5 | 1.75 | 105 | 44.75 | 44.68 |
| 8 | 196.5 | 1.75 | 105 | 58.84 | 58.41 |
| 9 | 196.5 | 1.75 | 105 | 58.15 | 58.41 |
| 10 | 250 | 2.5 | 150 | 48.17 | 48.15 |
| 11 | 196.5 | 1.75 | 105 | 58.37 | 58.41 |
| 12 | 196.5 | 1.75 | 105 | 59.13 | 58.41 |
| 13 | 196.5 | 1.75 | 105 | 58.51 | 58.41 |
| 14 | 196.5 | 1.75 | 30 | 83.46 | 83.17 |
| 15 | 196.5 | 3.00 | 105 | 62.66 | 62.66 |
| 16 | 250 | 2.5 | 60 | 63.69 | 63.76 |
| 17 | 143 | 2.5 | 150 | 36.35 | 36.14 |
| 18 | 143 | 2.5 | 60 | 87.62 | 87.79 |
| 19 | 143 | 1 | 60 | 68.69 | 68.72 |
| 20 | 196.5 | 1.75 | 181 | 46.12 | 46.39 |
| FAME [%] = +58.41 − 1.38A + 2.03B − 10.94C − 1.6AC − 88BC − 4.03A2 + 0.2965B2 + 2.25C2 | (2) |
| Source | Sum of squares | Df | Mean square | F-value | p-Value | |
|---|---|---|---|---|---|---|
| “Sequential model sum of square”: Select the highest order polynomial where the additional terms are significant, and the model is not aliased. | ||||||
| Mean vs. Total | 65883.72 | 1 | 65883.72 | |||
| Linear vs. Mean | 1716.03 | 3 | 572.01 | 7.10 | 0.0030 | |
| 2FI vs. Linear | 946.80 | 3 | 315.60 | 12.00 | 0.0005 | |
| Quadratic vs. 2FI | 339.74 | 3 | 113.25 | 554.67 | <0.0001 | Suggested |
| Cubic vs. Quadratic | 0.3172 | 4 | 0.0793 | 0.2759 | 0.8834 | Aliased |
| Residual | 1.72 | 6 | 0.2874 | |||
| Total | 68888.33 | 20 | 3444.42 | |||
| Summary Fit statistic | ||||||
| Std. Dev. | 0.4519 | R 2 | 0.9993 | |||
| Mean | 57.40 | Adjusted R2 | 0.9987 | |||
| C.V.% | 0.7873 | Predicted R2 | 0.9983 | |||
| Std. Dev. | 0.4519 | Adeq Precision | 161.6487 | |||
Positive signs in front of the terms indicate a synergic effect, while negative sign indicates an antagonistic effect.46 The terms B, AC, B2 and C2 therefore, play an important role in increasing the FAME conversion, whilst the other terms A, C, AB, BC and A2, play an important role in decreasing the biodiesel concentration.
| Source | Sum of squares | df | Mean square | F-Value | p-Value | |
|---|---|---|---|---|---|---|
| df = is the degree of freedom [the number of values of a system that varies independently is called degrees of freedom (DF)]. | ||||||
| Model | 3002.57 | 9 | 333.62 | 1634.02 | <0.0001 | Significant |
| A: molar ratio | 26.10 | 1 | 26.10 | 127.82 | <0.0001 | |
| B: catalyst amount | 56.48 | 1 | 56.48 | 276.63 | <0.0001 | |
| C: reaction time | 1633.45 | 1 | 1633.45 | 8000.43 | <0.0001 | |
| AB | 21.00 | 1 | 21.00 | 102.83 | <0.0001 | |
| AC | 649.44 | 1 | 649.44 | 3180.87 | <0.0001 | |
| BC | 276.36 | 1 | 276.36 | 1353.57 | <0.0001 | |
| A2 | 234.26 | 1 | 234.26 | 1147.36 | <0.0001 | |
| B2 | 1.26 | 1 | 1.26 | 6.18 | 0.0322 | |
| C2 | 73.14 | 1 | 73.14 | 358.24 | <0.0001 | |
| Residual | 2.04 | 10 | 0.2042 | |||
| Lack of fit | 0.3184 | 5 | 0.0637 | 0.1847 | 0.9563 | Not significant |
| Pure error | 1.72 | 5 | 0.3447 | |||
| Cor total | 3004.61 | 19 | ||||
R 2, adjusted R2, predicted R2, C.V., F value and P values were examined in the model produced by RSM. The significance of the equation was checked by the F-test technique. In addition, P-values are utilized to verify the importance of each coefficient, which indicates the power of each by-product to interact. The smaller P values lead the associated coefficients to be of great importance. An extremely low P-value (<0.0001) of the F model at a 95 percent confidence level indicates that the regression model is statistically significant. The determination coefficient (R2) that indicates connections between the actual FAME product and the anticipated product is estimated as 0.9993. Moreover, 0.9987 and 0.9983 respectively are adjusted and projected R2, with a low standard deviation value, was 0.4519 (see Table 8). These values reveal the fitted model to be outstanding. C.V., which must be less than 10%, is another key element for the evaluation of the model. This model is acceptable for the projected biodiesel produced by tristearin as the C.V. fitting model values are 0.7873%.49
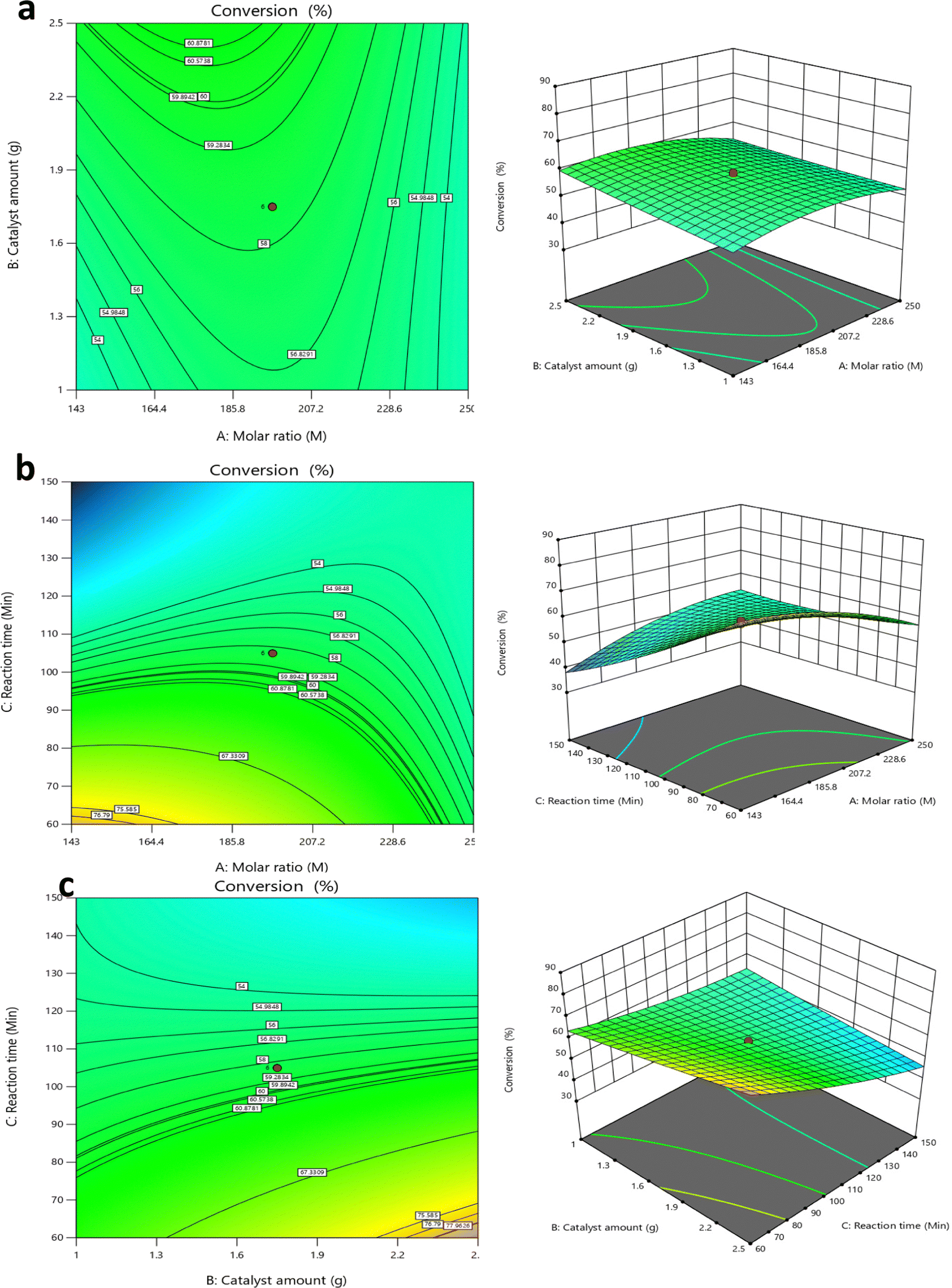 | ||
| Fig. 10 Contour plot and 3D response curve for conversion of TS to FAME (biodiesel), (a) interaction of catalyst amount and molar ratio, (b) interaction between reaction time and molar ratio and (c) interaction between reaction time and catalyst amount. | ||
The contour plot and the three-dimensional (3D) plot for the interaction effect between A (molar ratio) and C (time) are shown in Fig. 10b. The reaction temperature and catalyst amount were kept constant at 65 °C and 1.75 g respectively. Fig. 10b shows the contour plot that has higher biodiesel conversion (> 75%) was obtained at a minimum range molar ratio (143 up to 164.4 molar ratios) and reaction time in the range of 60 min up to 70 min. The 3D response surface shows that the biodiesel product increased with both decreasing reaction time and decreasing molar ratio, which again clearly shows that excess methanol has a significant negative impact on biodiesel conversion as shown by Lee et al.50 The contour plot and the three-dimensional (3D) plot for the interaction effect between B (catalyst) and C (time) are depicted in Fig. 10c. The reaction temperature and molar ratio were kept constant at 65 °C and 196.5 molar ratio respectively. The contour plot Fig. 10c shows that higher biodiesel conversion (>77%) was obtained between an intermediate and high amount of catalyst (2.2 to 2.5) and at low values of time (60 min to 30 min). The 3D response surface supports this. For example, at 60 min and 2.4 g catalyst, the biodiesel conversion was 77.96%, but when the time was increased to 180 min, a major decline in conversion was observed (less than 54%). This indicates that excess reaction time resulted in decreased biodiesel product.
In the present experiment, biodiesel production was set to the maximum value, while the other reaction parameters were set at a minimum value (see Table 10). The experimental conditions with the highest predicted biodiesel value were selected for further validation. The result of model validation is shown in Table 11. An optimum biodiesel conversion of 87.62% was obtained by transesterifying tristearin with 2.5 g of catalyst and a methanol to TS molar ratio of 143:1 at 65 °C for 60 min. The experimental product is in good agreement with the predicted value (87.79%) with a relatively small percentage error (0.1936) see Table 11. This shows that the proposed statistical model is suitable for the prediction of optimized biodiesel products and for the optimization of the transesterification process.
| Factors | Goal | Lower limit | Upper limit |
|---|---|---|---|
| A: molar ratio | Minimize | 143 | 250 |
| B: catalyst amount | Minimize | 1 | 2.5 |
| C: reaction time | Minimize | 60 | 150 |
| Conversion | Maximize | 36.35 | 87.62 |
| Catalyst (g) | The molar ratio (M) | Time (min) | Experimental Con. (%) | Predicted Con. value (%) | Percentage errors |
|---|---|---|---|---|---|
| 2.5 | 143 | 60 | 87.62 | 87.79 | 0.1936 |
In conclusion, the best values of the parameters for the transesterification reaction based on the design of the experiment [DoE] which was performed in the laboratory was 2.5 g of catalyst, molar ratio 143 and 60 min of reaction time [see the result from Table 11]. Table 11 gives the predicted conversion of 87.79% from the DoE simulation in close agreement with the experimental conversion of 87.62%. Thus, compared to our single variable strategy, the DoE resulted in a good optimisation of the transesterification reaction but with milder reaction conditions.
4. Conclusion
The surface is functionalised basified PANF mesh was shown to be an efficient solid base catalyst in the transesterification process with potential application for biodiesel production. The presence of strongly basic hydrazine groups on the PANF, once protonated, were able to electrostatically bind with Cl− and ultimately the Cl− anion could ion exchange with OH− ions. The OH− ions were thus able to react with methanol to produce the methoxide in transesterification. The transesterification reaction parameters of the molar ratio of TS or GMS to alcohol, reaction time, catalyst concentration and temperature were chosen to optimize the synthesis of the methyl ester. This work suggests that methyl esters can be synthesized at high percentage conversion (above 97 w/w%) at 65 °C at a molar ratio of methanol to GMS of 115:1, 1 h and 2.5 g basified PANF mesh catalyst in a total volume of 108.95 mL, greater than 95 w/w% conversions was observed at 65 °C with a molar ratio of methanol to tristearin of 285.5:1, 2 h and 6 g basified PANF mesh catalyst, total volume 107.48 mL.
RSM was successfully applied to assess the effects of multiple variables, including alcohol/TS molar ratio, catalyst mass and reaction time to produce biodiesel. The Predicted R2 of 0.9983 is in reasonable agreement with the Adjusted R2 of 0.9987; i.e., the difference is less than 0.2. In conclusion, the best values of the parameters for the transesterification reaction based on the design of the experiment [DoE] was 2.5 g of catalyst, molar ratio of methanol to TS of 143:1 and 60 min of reaction time. The predicted conversion of 87.79% from DoE simulation is in close agreement with the experimental conversion of 87.62%.
Thus, the novel basified PANF catalyst requires a relatively low temperature i.e. 65 °C and only a comparatively short reaction time, that is 2 hours to achieve maximum conversion albeit at a higher methanol to TGs molar ratio. However, the methanol is normally recovered in industry and reused back within the transesterification process. This together with the advantages of the surface functionalized PANF solid base catalyst such as its production at industrial quantities, recyclability, ease of use, economic viability, effectiveness, as well as its eco-friendly aspects suggests that it has the potential for transesterification of TGs and hence would be useful in biodiesel production.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by funding from the co-sponsors Daphne Jackson Trust, Society of Chemical Industry (SCI) and Royal Society of Chemistry (RSC), and hosted at De Montfort University, Leicester. I also thank the technical team (Nazmin and Unmesh) for their help with instrumentation, especially Dr Ketan Ruparelia to run the NMR technique.References
- W. Nabgan, A. A. Jalil, B. Nabgan, A. H. Jadhav, M. Ikram and A. Ul-Hamid, et al., Sustainable biodiesel generation through catalytic transesterification of waste sources: a literature review and bibliometric survey, RSC Adv., 2022, 12(3), 164–1627 RSC.
- D. Samios, F. Pedrotti, A. Nicolau, Q. B. Reiznautt, D. D. Martini and F. M. Dalcin, A Transesterification Double Step Process—TDSP for biodiesel preparation from fatty acids triglycerides, Fuel Process. Technol., 2009, 90(4), 599–605 CrossRef CAS.
- C. Urrutia, N. Sangaletti-Gerhard, M. Cea, A. Suazo, A. Aliberti and R. Navia, Two step esterification–transesterification process of wet greasy sewage sludge for biodiesel production, Bioresour. Technol., 2016, 200, 1044–1049 CrossRef CAS PubMed.
- M. K. Lam, K. T. Lee and A. R. Mohamed, Homogeneous, heterogeneous and enzymatic catalysis for transesterification of high free fatty acid oil (waste cooking oil) to biodiesel: A review, Biotechnol. Adv., 2010, 28(4), 500–518 CrossRef CAS PubMed.
- K. A. Borges, A. L. Squissato, D. Q. Santos, W. B. Neto, A. C. F. Batista and T. A. Silva, et al., Homogeneous catalysis of soybean oil transesterification via methylic and ethylic routes: Multivariate comparison, Energy, 2014, 67, 569–574 CrossRef CAS.
- P. Hariprasath, S. T. Selvamani, M. Vigneshwar, K. Palanikumar and D. Jayaperumal, Comparative analysis of cashew and canola oil biodiesel with homogeneous catalyst by transesterification method, Mater. Today: Proc., 2019, 16, 1357–1362 CrossRef CAS.
- J. M. Dias, M. C. M. Alvim-Ferraz and M. F. Almeida, Comparison of the performance of different homogeneous alkali catalysts during transesterification of waste and virgin oils and evaluation of biodiesel quality, Fuel, 2008, 87(17), 3572–3578 CrossRef CAS.
- G. Vicente, M. Martínez and J. Aracil, Optimisation of integrated biodiesel production. Part I. A study of the biodiesel purity and yield, Bioresour. Technol., 2007, 98(9), 1724–1733 CrossRef CAS PubMed.
- J. K. Efavi, D. Kanbogtah, V. Apalangya, E. Nyankson, E. K. Tiburu and D. Dodoo-Arhin, et al., The effect of NaOH catalyst concentration and extraction time on the yield and properties of Citrullus vulgaris seed oil as a potential biodiesel feed stock, S. Afr. J. Chem. Eng., 2018, 25(1), 98–102 Search PubMed.
- N. F. Hasanah, Process Parameter Optimisation for Transesterification of Canola Oil using Continuous Reactor with Homogeneous Base Catalyst, Adv. Sustainable Eng., 2021, 01, 1 Search PubMed.
- A. Munyentwali, H. Li and Q. Yang, Review of advances in bifunctional solid acid/base catalysts for sustainable biodiesel production, Appl. Catal., A, 2022, 118525 CrossRef CAS.
- I. Istadi, S. A. Prasetyo and T. S. Nugroho, Characterization of K2O/CaO-ZnO catalyst for transesterification of soybean oil to biodiesel, Procedia Environ. Sci., 2015, 23, 394–399 CrossRef CAS.
- M. Feyzi and L. Norouzi, Preparation, and kinetic study of magnetic Ca/Fe3O4@ SiO2 nanocatalysts for biodiesel production, Renewable Energy, 2016, 94, 579–586 CrossRef CAS.
- R. Anr, A. A. Saleh, M. S. Islam, S. Hamdan and M. A. Maleque, Biodiesel production from crude Jatropha oil using a highly active heterogeneous nanocatalyst by optimizing transesterification reaction parameters, Energy Fuels, 2016, 30(1), 334–343 CrossRef.
- M. Prabu, M. Manikandan, P. Kandasamy, P. R. Kalaivani, N. Rajendiran and T. Raja, Synthesis of biodiesel using the Mg/Al/Zn hydrotalcite/SBA-15 nanocomposite catalyst, ACS Omega, 2019, 4(2), 3500–3507 CrossRef CAS.
- R. Chamola, M. F. Khan, A. Raj, M. Verma and S. Jain, Response surface methodology based optimization of in situ transesterification of dry algae with methanol, H2SO4 and NaOH, Fuel, 2019, 239, 511–520 CrossRef CAS.
- A. Abubakar, A. A. Mahmoud, D. A. Ajiya, U. F. Hassan, K. Y. Muhammad and S. Aban Sallau, et al., Optimization Process of Parameters for the Transesterification of Jatropha curcas Seed Oil using Response Surface Methodology (RSM), Int. J. Res. Sci. Innov. Appl. Sci., Volume, 2022, VII(II), 2454 Search PubMed.
- N. H. Zabaruddin, L. C. Abdullah, N. H. Mohamed and T. S. Y. Choong, Optimization using response surface methodology (RSM) for biodiesel synthesis catalyzed by radiation-induced Kenaf catalyst in packed-bed reactor, Processes, 2020, 8(10), 1289 CrossRef CAS.
- A. Garg and S. Jain, Process parameter optimization of biodiesel production from algal oil by response surface methodology and artificial neural networks, Fuel, 2020, 277, 118254 CrossRef CAS.
- R. Ahmed and K. Huddersman, Review of biodiesel production by the esterification of wastewater containing fats oils and grease (FOGs), J. Ind. Eng. Chem., 2022, 110, 1–14, DOI:10.1016/j.jiec.2022.02.045.
- V. V. Ishtchenko, K. D. Huddersman and R. F. Vitkovskaya, Part 1. Production of a modified PAN fibrous catalyst and its optimisation towards the decomposition of hydrogen peroxide, Appl. Catal., A, 2003, 242(1), 123–137 CrossRef CAS.
- K. I. Doudin, Quantitative and qualitative analysis of biodiesel by NMR spectroscopic methods, Fuel, 2021, 284, 119114 CrossRef CAS.
- R. A. Ahmed, S. Rashid and K. Huddersman, Esterification of stearic acid using novel protonated and crosslinked amidoximated polyacrylonitrile ion exchange fibres, J. Ind. Eng. Chem., 2023, 119, 550–573, DOI:10.1016/j.jiec.2022.12.001.
- V. Mandari and S. K. Devarai, Biodiesel production using homogeneous, heterogeneous, and enzyme catalysts via transesterification and esterification reactions: A critical review, BioEnergy Res., 2022, 15(2), 935–961 CrossRef CAS PubMed.
- J. Otera, Transesterification, Chem. Rev., 1993, 93(4), 1449–1470 CrossRef CAS.
- M. V. Rodionova, R. S. Poudyal, I. Tiwari, R. A. Voloshin, S. K. Zharmukhamedov and H. G. Nam, et al., Biofuel production: challenges and opportunities, Int. J. Hydrogen Energy, 2017, 42(12), 8450–8461 CrossRef CAS.
- E. F. Aransiola, T. V. Ojumu, O. O. Oyekola, T. F. Madzimbamuto and D. Ikhu-Omoregbe, A review of current technology for biodiesel production: State of the art, Biomass Bioenergy, 2014, 61, 276–297 CrossRef CAS.
- I. M. Rizwanul Fattah, H. C. Ong, T. Mahlia, M. Mofijur, A. S. Silitonga and S. A. Rahman, et al., State of the art of catalysts for biodiesel production, Front. Energy Res., 2020, 8, 101 CrossRef.
- L. C. Meher, D. V. Sagar and S. N. Naik, Technical aspects of biodiesel production by transesterification—a review, Renewable Sustainable Energy Rev., 2006, 10(3), 248–268 CrossRef CAS.
- N. Rasimoglu and H. Temur, Cold flow properties of biodiesel obtained from corn oil, Energy, 2014, 68, 57–60 CrossRef CAS.
- F. Ezebor, M. Khairuddean, A. Z. Abdullah and P. L. Boey, Oil palm trunk and sugarcane bagasse derived heterogeneous acid catalysts for production of fatty acid methyl esters, Energy, 2014, 70, 493–503 CrossRef CAS.
- M. Satyanarayana and C. Muraleedharan, A comparative study of vegetable oil methyl esters (biodiesels), Energy, 2011, 36(4), 2129–2137 CrossRef CAS.
- J. M. Encinar, A. Pardal and G. Martínez, Transesterification of rapeseed oil in subcritical methanol conditions, Fuel Process. Technol., 2012, 94(1), 40–46 CrossRef CAS.
- M. A. Olutoye, S. W. Wong, L. H. Chin, H. Amani, M. Asif and B. H. Hameed, Synthesis of fatty acid methyl esters via the transesterification of waste cooking oil by methanol with a barium-modified montmorillonite K10 catalyst, Renewable Energy, 2016, 86, 392–398 CrossRef CAS.
- J. M. Encinar, J. F. González, A. Pardal and G. Martínez, Rape oil transesterification over heterogeneous catalysts, Fuel Process. Technol., 2010, 91(11), 1530–1536 CrossRef CAS.
- A. A. Koutsouki, E. Tegou, A. Badeka, S. Kontakos, P. J. Pomonis and M. G. Kontominas, In situ and conventional transesterification of rapeseeds for biodiesel production: The effect of direct sonication, Ind. Crops Prod., 2016, 84, 399–407 CrossRef CAS.
- N. Chumuang and V. Punsuvon, Response surface methodology for biodiesel production using calcium methoxide catalyst assisted with tetrahydrofuran as cosolvent, J. Chem., 2017, 2017, 1–9, DOI:10.1155/2017/4190818.
- Y. H. Tan, M. O. Abdullah, C. Nolasco-Hipolito and Y. H. Taufiq-Yap, Waste ostrich-and chicken-eggshells as heterogeneous base catalyst for biodiesel production from used cooking oil: Catalyst characterization and biodiesel yield performance, Appl. Energy, 2015, 160, 58–70 CrossRef CAS.
- A. S. Yusuff, O. D. Adeniyi, S. O. Azeez, M. A. Olutoye, U. G. Akpan, A. S. Yusuff, O. D. Adeniyi, S. O. Azeez, M. A. Olutoye and U. G. Akpan, Synthesis and characterization of anthill-eggshell-Ni-Co mixed oxides composite catalyst for biodiesel production from waste frying oil, Biofuels, Bioproducts and Biorefining, UK, 13: 37-47, 2019, Online ISSN: 1932-1031 DOI:10.1002/bbb.1914.
- N. Boz, N. Degirmenbasi and D. M. Kalyon, Esterification and transesterification of waste cooking oil over Amberlyst 15 and modified Amberlyst 15 catalysts, Appl. Catal., B, 2015, 165, 723–730 CrossRef CAS.
- T. Dong, J. Wang, C. Miao, Y. Zheng and S. Chen, Two-step in situ biodiesel production from microalgae with high free fatty acid content, Bioresour. Technol., 2013, 136, 8–15 CrossRef CAS PubMed.
- J. M. Encinar, J. F. González, G. Martínez and S. Nogales-Delgado, Use of NaNO3/SiAl as Heterogeneous Catalyst for Fatty Acid Methyl Ester Production from Rapeseed Oil, Catalysts, 2021, 11(11), 1405 CrossRef CAS.
- L. Hsieh, U. Kumar and J. C. Wu, Continuous production of biodiesel in a packed-bed reactor using shell–core structural Ca (C3H7O3) 2/CaCO3 catalyst, Chem. Eng. J., 2010, 158(2), 250–256 CrossRef CAS.
- C. A. Akinremi, S. Rashid, P. D. Upreti, G. T. Chi and K. Huddersman, Regeneration of a deactivated surface functionalised polyacrylonitrile supported Fenton catalyst for use in wastewater treatment, RSC Adv., 2020, 10(22), 12941–12952 RSC.
- Y. C. Wong, Y. P. Tan, Y. H. Taufiq-Yap and I. Ramli, An optimization study for transesterification of palm oil using response surface methodology (RSM), Sains Malays., 2015, 44(2), 281–290 CrossRef.
- S. H. Shuit, K. T. Lee, A. H. Kamaruddin and S. Yusup, Reactive extraction of Jatropha curcas L. seed for production of biodiesel: process optimization study, Environ. Sci. Technol., 2010, 44(11), 4361–4367 CrossRef CAS PubMed.
- N. G. Zanjani, A. Kamran-Pirzaman and M. Khalajzadeh, Synthesis of modified layered double hydroxide of MgAl catalyst with Ba and Li for the biodiesel production, Clean Technol. Environ. Policy, 2020, 22, 1173–1185 CrossRef CAS.
- P. Goyal, M. P. Sharma and S. Jain, Optimization of transesterification of Jatropha curcas oil to biodiesel using response surface methodology and its adulteration with kerosene, J. Mater. Environ. Sci., 2013, 4(2), 277–284 CAS.
- M. K. Yesilyurt, M. Arslan and T. Eryilmaz, Application of response surface methodology for the optimization of biodiesel production from yellow mustard (Sinapis alba L.) seed oil, Int. J. Green Energy, 2019, 16(1), 60–71 CrossRef CAS.
- H. V. Lee, R. Yunus, J. C. Juan and Y. H. Taufiq-Yap, Process optimization design for jatropha-based biodiesel production using response surface methodology, Fuel Process. Technol., 2011, 92(12), 2420–2428 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |