
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile surface reconstructions of cobalt–copper phosphide heterostructures enable efficient electrocatalytic glycerol oxidation for energy-saving hydrogen evolution†
Zhengzhe
Xie‡
a,
Kang
Wang‡
 b,
Yu
Zou
a,
Guobing
Ying
*b and
Jiang
Jiang
*a
b,
Yu
Zou
a,
Guobing
Ying
*b and
Jiang
Jiang
*a
ai-Lab, CAS Key Laboratory of Nano-Bio Interface, Suzhou Institute of Nano-Tech and Nano-Bionics, Chinese Academy of Sciences, Suzhou 215123, China. E-mail: jjiang2010@sinano.ac.cn
bDepartment of Materials Science and Engineering, College of Mechanics and Materials, Hohai University, Nanjing 211100, China. E-mail: yinggb001@hhu.edu.cn
First published on 12th December 2022
Abstract
The glycerol oxidation reaction (GOR) affords an energetically more favorable route for assisted H2 production compared to the oxygen evolution reaction, with concurrent anodic value-added chemical generation. Herein, a self-supported CoP–Cu3P composite on carbon cloth (CoP–Cu3P/CC) has been fabricated, which exhibits excellent activity toward the GOR, requiring a low applied potential of 1.13 V (vs. reversible hydrogen electrode) to reach 10 mA cm−2. Through combined surface elemental analysis, cyclic voltammetry, and in situ Raman spectroscopy characterizations, we reveal that metal elements in the mixed phosphide phases interact synergistically, leading to facilitated surface reconstruction and improved electrochemical activity. We further develop an electrolytic cell where the GOR is paired with the hydrogen evolution reaction using CoP–Cu3P/CC as both the anode and cathode, which only needs the application of 1.21 V to reach a geometric current density of 10 mA cm−2. This work provides a facile strategy to couple glycerol upgrading in an energy-saving water electrolysis system.
Introduction
Due to its high energy density and ultimate zero carbon emission, hydrogen is being considered as the most promising energy carrier for the future.1 Renewable (green) hydrogen gas of high purity can be generated by electrochemical water splitting using intermittent energy supplies such as solar and wind power.2–5 However, its commercialization is severely hindered by the high energy demand required to drive the thermodynamically uphill and kinetically sluggish anodic oxygen evolution reaction (OER).6 Therefore, research endeavours have been focused on finding thermodynamically more favourable reactions to replace the anodic OER half-reaction in recent years, where electro-oxidation reactions of small organic molecules such as alcohol,7–11 glycerol,12–14 5-hydroxymethylfurfural (HMF),15–17 glucose,18,19 and amines20,21 have been applied as alternative oxidation half-reactions. By doing so, the overall voltage input for hydrogen evolution can be reduced significantly, the possible formation of an explosive O2/H2 gas mixture is avoided, and valuable chemicals are co-produced to further reduce the potential cost.22–28Among the various small organic molecules, glycerol stands out as an attractive economically viable feedstock to assist the hydrogen evolution reaction (HER) due to several reasons: (1) glycerol oxidation reaction (GOR) requires a significantly lower thermodynamic oxidation potential (0.003 V vs. reversible hydrogen electrode, RHE) compared to the OER (1.23 V vs. RHE);29 (2) a variety of high value-added products can be generated via selective glycerol oxidation;30 (3) glycerol is a cheap by-product in over-supply from biodiesel and the soap industry.31,32
The most effective electrocatalysts for the GOR reported so far are often based on noble metals33 such as Au, Pd, Pt and their alloys (PtBi and PdBi),34,35 which suffer from high cost and poor stability against poisoning, making them less competitive for economically feasible hydrogen production. In recent reports, transition metal-based catalysts have shown great promise as alternatives to noble metals for the GOR in alkaline media, including transition metal oxides,36–39 hydroxides,40–44 and nitrides.12,45,46 In particular, metal oxides and hydroxides containing both Co and Cu have demonstrated good activity toward the GOR.47–49 Han et al.47 conducted an investigation using cobalt-based spinel oxide (MCo2O4) as an electrocatalyst for the GOR, and discovered that CuCo2O4 was the best-performing catalyst with a high formate selectivity of ∼80%. Braun et al.48 synthesized Cu–Co hydroxycarbonates (Cu0.8Co0.2)2CO3(OH)2 for alcohol oxidation reactions, and found that Cu leaching could lead to increased catalytic activity. Unfortunately, transition metal oxides and hydroxides are generally poor conductors, which may limit their electrocatalytic activity. On the other hand, given the metallic nature of transition metal phosphides (TMPs) and their ability to form a thin layer of metal (oxy)hydroxide in an oxidative environment,50 TMPs have demonstrated superb activity toward water splitting applications.51 However, TMPs have been rarely explored for glycerol oxidation reactions.
Herein, we have prepared self-supporting CoP–Cu3P composite nanoarrays via transformation of mixed Co–Cu hydroxycarbonates. The self-supported structure can not only expose more active sites but also ensure good contact between the nanoarrays and the conductive substrate, accelerating the electron transport and mass transfer. We have found that the CoP–Cu3P heterostructures result in modulated electronic structures, which facilitates facile surface reconstruction to generate high-valent Co species during the electrocatalytic process. The as prepared CoP–Cu3P electrode only needs applied potentials of 1.13 and 1.22 V (vs. RHE) to drive geometric current densities of 10 and 20 mA cm−2 for the GOR in alkaline electrolyte (1 M KOH containing 0.1 M glycerol). Its practical feasibility is further demonstrated in an alkaline electrolyser using CoP–Cu3P/CC as both the anode and cathode. A geometric current density of 10 mA cm−2 has been achieved at a low cell voltage of 1.21 V, 210 mV lower than what is required for conventional electrolytic overall water splitting.
Experimental
Materials
Copper nitrate trihydrate (Cu(NO3)2·3H2O, >99.0%), cobalt nitrate hexahydrate (Co(NO3)2·6H2O, 98.5%), ammonium fluoride (NH4F, 96.0%), sodium hypophosphite monohydrate (NaH2PO2·H2O, >98.0%), urea (CO(NH2)2, 99.0%), acetone (99.5%), ethanol (99.7%), KOH (85.0%), HCl (36.0–38.0%) and glycerol (99.0%) were all obtained from Sinopharm Chemical Reagent Co., Ltd. All chemicals were used as received without purification. Carbon cloth (CC) was purchased from Shanghai Hesen Electric Co., Ltd. The deionized water was taken from a Millipore system.Synthesis of Co–Cu hydroxycarbonates
(Co,Cu)2CO3(OH)2/CC was synthesized via a hydrothermal reaction. To remove the organic impurities, carbon cloth was first cleaned sequentially using hydrochloric acid, deionized water, acetone, and ethanol. For a typical procedure, 1 mmol Cu(NO3)2·3H2O, 3 mmol Co(NO3)2·6H2O, 2 mmol NH4F and 5 mmol urea were dissolved in 50 mL deionized water under magnetic stirring. The obtained transparent solution was then transferred into a Teflon-lined stainless steel autoclave (100 mL). Subsequently, a piece of carbon cloth (2 × 5 cm2) was placed against the autoclave wall and immersed into the solution. The autoclave was then sealed and heated to 120 °C for a duration of 12 h. After that, the carbon cloth with sample grown on it was taken out of the autoclave, and rinsed thoroughly with deionized water. Mild ultrasonication was then applied in order to remove weakly surface bound hydroxycarbonates on the carbon cloth substrate. Finally, the (Co, Cu)2CO3(OH)2/CC was left to dry at 70 °C in a vacuum oven for 12 h.The synthetic procedures for Co(CO3)0.5(OH)·0.11H2O/CC and Cu2(CO3)(OH)2/CC were the same as that of (Co,Cu)2CO3(OH)2/CC, except using Co(NO3)2·6H2O or Cu(NO3)2·3H2O as the only precursor during the hydrothermal reaction.
Synthesis of Co–Cu phosphides
Prior to the synthesis of phosphides, carbon cloth was first cut into 1 × 2 cm2 pieces. In a typical phosphidation procedure, NaH2PO2·H2O (1 g) was placed in a ceramic boat inside a quartz tube at the upstream position of the gas flow relative to a piece of carbon cloth (with corresponding metal hydroxycarbonates). After purging with N2, the furnace was heated up to 300 °C at 5 °C min−1 ramping rate, and remained for 1 h.Material characterizations
The prepared electrode morphologies were acquired using scanning electron microscopy operated at 10 kV (SEM, JEOL JSM-7500F), and transmission electron microscopy at an accelerating voltage of 200 kV (TEM, FEI Tecnai G2 F20 S-Twin). For TEM characterizations, the samples were first detached from the carbon cloth, redispersed in deionized water under ultrasonication, and then deposited on a Cu or Ni grid. The crystalline structure of the electrodes was surveyed using X-ray diffraction (XRD) on a Bruker D8 advance (Cu Kα radiation, 40 kV and 40 mA, scan speed 0.1° s−1). The catalyst surface composition and valence states were investigated by X-ray photoelectron spectroscopy (XPS) on a Thermo Scientific K-Alpha (Al Kα X-ray source, 1486.6 eV). In situ Raman spectra were acquired on a Renishaw Raman InVia microscope with a home-designed electrochemical cell, using a 785 nm laser as the excitation source with a 50× objective lens.Electrochemical measurements
The electrocatalytic performance for the GOR was tested by applying a three-electrode system on an electrochemical workstation (CHI 760E, Shanghai Chenhua Instrument Co., Ltd) at room temperature. The working electrodes were fabricated directly using the as-synthesized phosphide nanostructures grown on carbon cloth. For all electrochemical measurements, Hg/HgO (1 M KOH) was used as the reference electrode, and Pt foil was set as the counter electrode. The GOR was performed in 1 M KOH solution (25 mL) containing 0.1 M glycerol. Prior to recording electrochemical tests, activation processes were first conducted by continuous cyclic voltammogram (CV). Linear sweep voltammetry (LSV) was obtained by positively scanning the potential at a rate of 2 mV s−1 under stirring. By performing CV in the non-faradaic potential windows with different scanning rates, electrochemical double layer capacitance (Cdl) of the electrodes was determined, which was then used to infer the electrochemical active surface area (ECSA). Electrochemical impedance spectroscopy (EIS) was performed with 5 mV amplitude in the frequency range from 0.1 Hz to 100 kHz. All the denoted potentials were referenced to the reversible hydrogen electrode (RHE) according to the following equation: ERHE = EHg/HgO + 0.059 × pH + 0.098. The iR-corrected LSV curves were plotted using solution resistance that has been determined from EIS measurements.Two-electrode glycerol electrolysis was conducted using CoP–Cu3P/CC as both the anode and cathode, with the LSV curves recorded at a sweep rate of 2 mV s−1.
Product analysis
The products from the GOR were separated and quantified by high-performance liquid chromatography (HPLC) using an UltiMate 3000 (Thermo Fisher Scientific). For the HPLC measurements, aliquots of electrolyte solution (500 μL) extracted from the H-type cell were diluted by H2SO4 (0.5 M, 1.5 mL), and 20 μL of this diluted solution was then injected directly into a ChromCore Sugar-10H column. The eluent was H2SO4 solution (5 mM) at a 0.5 mL min−1 flow rate. For the individual GOR product, standard calibration curves were first obtained using sample solutions of known concentrations, as demonstrated in our previous work.52Calculations of faradaic efficiency (FE) for the GOR are performed using the following half-reactions, which correspond to the oxidation of glycerol (C3H8O3) into various products (glycerate: C3H5O4−; glycolate: C2H3O3−; oxalate: C2O42−; formate: HCOO−) in alkaline solution:
| C3H8O3 + 5OH− → C3H5O4− + 4H2O + 4e− | (1) |
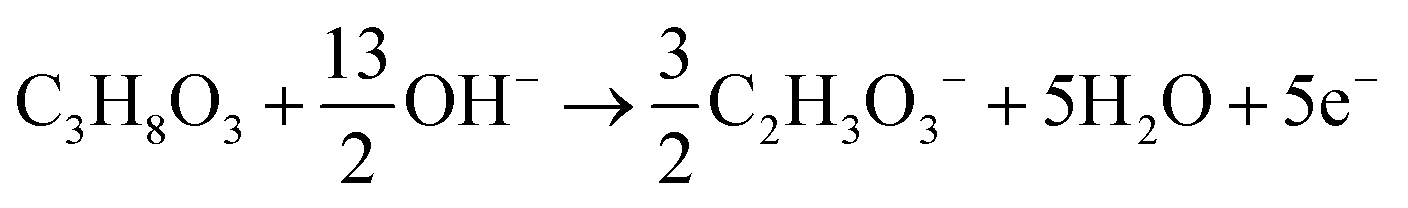 | (2) |
 | (3) |
| C3H8O3 + 11OH− → 3HCOO− + 8H2O + 8e− | (4) |
 | (5) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), and Qtotal represents the total passed charges (C).
485 C mol−1), and Qtotal represents the total passed charges (C).
Results and discussion
Cobalt–copper phosphide synthesis and characterization
Various methods for synthesizing TMPs have been developed, including electrodeposition,53 vapor-phase deposition,54 and solid phase reaction.55 In order to expose more active sites, we synthesized a self-supported mixed Co–Cu phosphide nanostructure electrode via a two-step procedure (schematically shown in Fig. 1a), where Co–Cu hydroxycarbonate precursors on carbon cloth were formed first through hydrothermal reaction, and then subjected to a phosphidation reaction at 300 °C for 1 h in a ceramic boat under a phosphine atmosphere. For comparative studies, self-supported single phase cobalt phosphides and copper phosphides from their corresponding hydroxycarbonates were also prepared in the same way.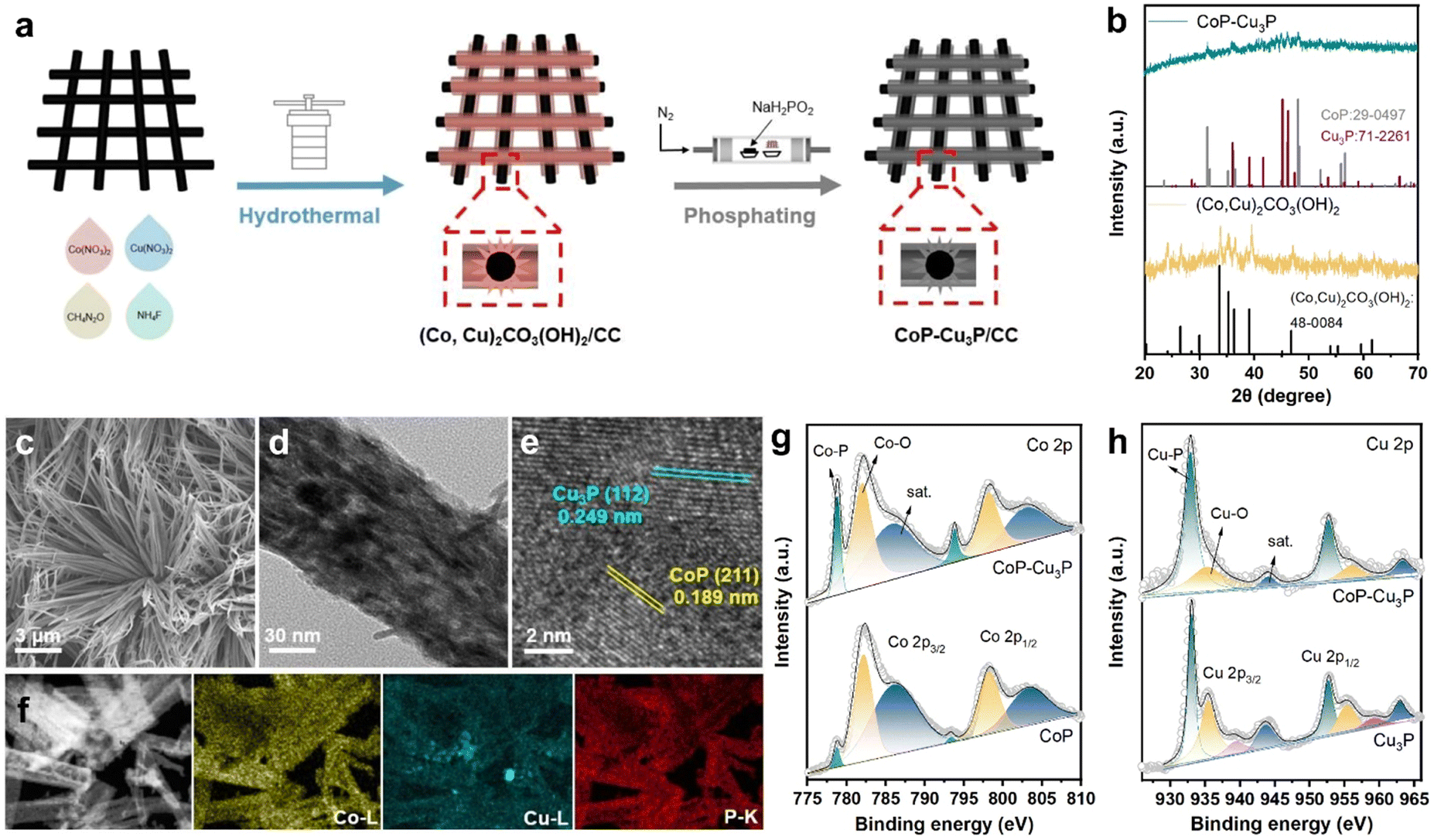 | ||
| Fig. 1 (a) Schematic illustration of the procedures to produce mixed metal phosphides CoP–Cu3P/CC. (b) XRD patterns of Co–Cu hydroxycarbonates (Co,Cu)2CO3(OH)2 and CoP–Cu3P. (c) SEM, (d) TEM, and (e) HRTEM images of CoP–Cu3P. (f) Scanning transmission electron microscopy (STEM) image and energy dispersive spectroscopy (EDS) elemental mapping of Co, Cu and P elements in CoP–Cu3P. (g) High resolution Co 2p XPS spectra of CoP and CoP–Cu3P. (h) High resolution Cu 2p XPS spectra of Cu3P and CoP–Cu3P. | ||
The structures of the as-formed Co–Cu hydroxycarbonate precursors and the corresponding metal phosphides scratched from the carbon cloth substrate were examined by powder XRD. The diffractogram of Co–Cu hydroxycarbonates showed peaks at 20.2, 24.1, 26.4, 33.6, and 39.0 degrees (as displayed in Fig. 1b), which can be indexed to the (001), (111), (220), (221), and (231) planes in the orthorhombic (Co,Cu)2CO3(OH)2 phase (JCPDS No. 48-0084), matching well with the literature reports on Co–Cu hydroxycarbonates.48,56 After annealing under a phosphine atmosphere, two new sets of XRD peaks appeared in the obtained sample. The diffraction peaks at 31.6° and 48.1° could be assigned to the (011) and (211) planes of CoP (JCPDS No. 29-0497), while those that appeared at 36.0, 39.1, 41.6, 45.1, and 46.2 degrees corresponded well to the diffractions from the (112), (202), (211), (300), and (113) planes of Cu3P (JCPDS No. 71-2261), respectively. Using the same synthetic protocol, pure CoP and Cu3P were also made by phosphiding the Co(CO3)0.5(OH)·0.11H2O and Cu2(CO3)(OH)2 precursors, respectively. The XRD patterns confirmed the successful synthesis of single-phase CoP and Cu3P (Fig. S1, ESI†), which served as control samples.
Under SEM, the (Co,Cu)2CO3(OH)2 precursor was in the form of bundles of nanowires on the carbon cloth substrate (Fig. S2, ESI†). After phosphidation conversion, the morphology of the initial self-supported mixed Co–Cu hydroxycarbonates has been preserved well, as indicated in the respective SEM (Fig. 1c) and TEM (Fig. 1d) images. Generally speaking, a self-supporting structure is beneficial for enhanced electrocatalysis, via exposing active sites and facilitating rapid mass transfer. Finer detailed structures of the CoP–Cu3P nanowires peeling off from the carbon cloth were revealed under high-resolution TEM (HRTEM). The distinct lattice fringes with 0.189 nm spacing can be ascribed to the (211) plane of orthorhombic CoP, and the ones with 0.249 nm spacing were in accordance with the (112) plane of hexagonal Cu3P (Fig. 1e), matching well with the XRD results. Furthermore, elemental mapping by energy dispersive spectroscopy (EDS) confirmed the presence of Co (yellow), Cu (blue), and P (red), with all the elements homogeneously and uniformly distributed across the selected area (Fig. 1f), indicating abundant heterojunctions between CoP and Cu3P. From the EDS spectrum (Fig. S3, ESI†), the Co/Cu molar ratio in CoP–Cu3P was determined to be ∼2.6, close to the initial feeding ratio of 3.
The surface elemental composition and valence states of the CoP–Cu3P nanostructures were further characterized by XPS. The Co 2p, Cu 2p, P 2p, and O 1s peaks were clearly revealed in the XPS survey spectrum of CoP–Cu3P, indicating the existence of the respective elements in the sample (Fig. S4a, ESI†). In the high-resolution Co 2p region of CoP and CoP–Cu3P (Fig. 1g), the broad peaks at 778.8 eV and 793.8 eV were attributed to metallic state (Co0) in CoP due to spin–orbit coupling effects, consistent with previous reports on the XPS of cobalt phosphides.50,57,58 Furthermore, the peaks that appeared at 782.1/797.1 eV and 786.3/802.7 eV can be ascribed to the surface oxidized Co species and the satellite peaks, respectively.59 For the high-resolution Cu 2p of Cu3P and CoP–Cu3P (Fig. 1h), the peaks at 933.1 eV and 952.8 eV are ascribed to Cu 2p3/2 and 2p1/2 in Cu3P. The peaks at 935.5, 939.4, and 943.7 eV (satellite) as well as 955.4, 959.3, and 963.6 eV corresponded to the surface oxidized copper species.60 Moreover, for the profile of the P 2p spectra of CoP, Cu3P, and CoP–Cu3P (Fig. S4b–d, ESI†), the characteristic peaks at around 129.5, 130.4, and 134.4 eV can be ascribed to P 2p3/2 and P 2p1/2 in the phosphides and the oxidized species such as PO43− or P2O5, respectively. In addition, in single-phase CoP, the Co 2p3/2 peak (778.7 eV) was shifted positively from metallic Co (778.3 eV), while the P 2p3/2 peak (129.3 eV) had a smaller binding energy compared to P (130.1 eV), suggesting that Co possessed positive charge while P possessed negative charge. Similarly, in single-phase Cu3P, the Cu 2p3/2 peak (933.1 eV) was also shifted positively from metallic Cu (932.6 eV), while the P 2p3/2 peak (129.0 eV) was shifted negatively from red P (130.1 eV), which also indicated that Cu and P possessed positive and negative charges, respectively. As shown in Fig. 1g and h, compared to the single-phase CoP and Cu3P, the Co 2p3/2 and Cu 2p3/2 binding energies in CoP–Cu3P were shifted positively to 778.8 and 932.9 eV, respectively. Furthermore, the P 2p3/2 and P 2p1/2 binding energies in CoP–Cu3P also displayed positive shifts relative to those in CoP and Cu3P (Fig. S4, ESI†). These results suggested that the electronic structures were modified for the CoP–Cu3P nanocomposites, due to interfacial charge transfer between CoP and Cu3P.
Electrocatalytic glycerol oxidation activity
After the successful synthesis of self-supporting CoP–Cu3P nanowire arrays, their electrocatalytic activities for the GOR under alkaline conditions were then investigated in a single-compartment cell with the three-electrode configuration at room temperature. Given that water oxidation is the main competing anodic reaction, the OER was evaluated for comparison purposes, by keeping all other experimental conditions the same except without adding glycerol.Prior to the actual electrochemical catalytic experiments, CoP–Cu3P/CC was first electrochemically activated by continuous cyclic voltammetry (reaching equilibrium after 4–5 cycles, Fig. S5, ESI†), and then LSV was recorded in 1 M KOH solution with 0.1 M glycerol. As shown in Fig. 2a (cyan solid line), the CoP–Cu3P/CC is highly active toward the glycerol oxidation reaction, requiring a potential of only 1.13 and 1.22 V (vs. RHE, unless otherwise noted) to achieve a geometric current density of 10 and 20 mA cm−2, respectively. In the absence of glycerol, CoP–Cu3P/CC can also catalyze electrochemical water oxidation (Fig. 2a, cyan dashed line), but at considerably more positive potentials, requiring a potential of 1.32 and 1.50 V to drive the same geometric current density of 10 and 20 mA cm−2. Moreover, comparative studies using CoP were also conducted under GOR and OER conditions (Fig. 2a, yellow solid and dashed lines), showing only moderate electrocatalytic activities. Cu3P displayed similar electrochemical activities toward the GOR compared to that of CoP. Under all the tested conditions, the GOR is electrochemically more favorable than the OER. In Fig. 2b, the anodic potentials to drive the glycerol oxidation reaction and water oxidation reaction at a geometric current density of 10 and 20 mA cm−2 were plotted together for comparison, showing 190 or 280 mV reduction in the driving potentials to reach a geometric current density of 10 or 20 mA cm−2 when switching the OER to the GOR using CoP–Cu3P/CC as the electrocatalyst. The measured GOR activity of CoP–Cu3P/CC in 1 M KOH solution containing 0.1 M glycerol is comparable or even slightly better than some recently reported state of the art precious-metal-free GOR electrocatalysts, such as CuCo2O4 (1.30 V),47 Ni–Mo–N (1.30 V),12 and NiOx/MWCNTs (1.31 V).38 A more complete comparison with recent literature reports is tabulated in Table S1 (ESI†). LSV curves on (Co,Cu)2CO3(OH)2 precursors were also recorded for comparison, which exhibited very poor activity toward the electrocatalytic GOR, requiring an applied potential of 1.92 V to reach a current density of 10 mA cm−2 (Fig. S6, ESI†). We also varied the feeding ratio of Co/Cu in preparing the mixed Co–Cu phosphides to 1:1 and 1:3, and the resulting Co–Cu phosphides had lost the original nanowire morphology, and also displayed worse performance in the GOR than CoP–Cu3P nanocomposites with 3:1 Co/Cu feeding ratio (Fig. S7, ESI†).
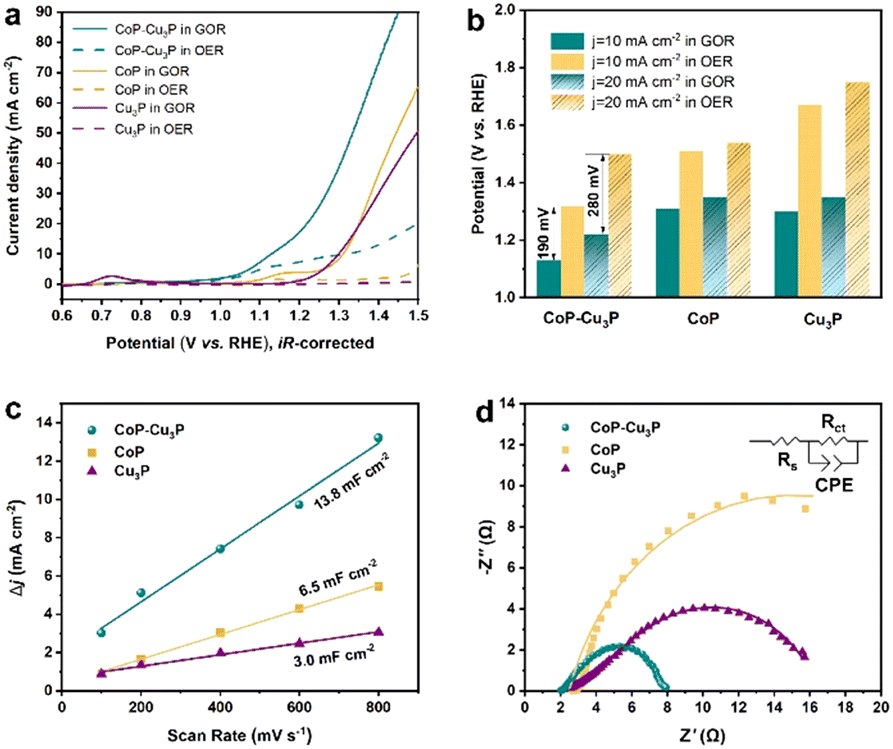 | ||
| Fig. 2 (a) LSV curves of CoP, Cu3P, and CoP–Cu3P grown on carbon cloth in 1 M KOH solution with (solid lines) and without (dashed lines) 0.1 M glycerol addition. (b) Comparison of the applied potential at 10 and 20 mA cm−2 for various electrodes under GOR and OER conditions. (c) Estimation of the ECSA of various electrodes based on Cdl values extracted from CV measurements in the non-faradaic regions. (d) Nyquist plots of various electrodes from EIS measurements performed at onset potentials. | ||
To understand the synergistically enhanced electrochemical GOR activity of CoP–Cu3P, we first compared their respective electrochemical surface area (ECSA). The ECSA was determined from the measured double-layer capacitance (Cdl) using cyclic voltammetry (Fig. S8, ESI†). As shown in Fig. 2c, the CoP–Cu3P nanocomposite possesses a considerably higher 2Cdl (13.8 mF cm−2) than CoP (6.5 mF cm−2) and Cu3P (3.0 mF cm−2), suggesting that the formation of nanocomposites resulted in more exposed active sites, which are beneficial to achieve high electrocatalytic GOR activity. Electrochemical impedance spectroscopy (EIS) measurement was then conducted to evaluate the charge transfer kinetics on CoP, Cu3P, and CoP–Cu3P during the GOR process (Fig. 2d). The EIS spectra show that these phosphide catalysts displayed similar solution resistances (Rs), but quite different charge transfer resistances (Rct). The CoP–Cu3P nanocomposite showed much smaller charge transfer resistance Rct of 5.70 Ω, compared to that of CoP (22.69 Ω) and Cu3P (17.78 Ω), indicating that the charge transfer kinetics are facilitated by forming a CoP–Cu3P heterostructure. Therefore, both increased active surface area and faster charge transfer kinetics contributed to the enhanced activity of the CoP–Cu3P nanocomposite toward the GOR.
Surface reconstructions of CoP–Cu3P during electrocatalysis
TMPs are often considered as the pre-catalyst in electrochemical oxidation reactions, as the surfaces of metal phosphides are converted to metal (oxy)hydroxides, which serve as the actual catalysts.50,61,62 To investigate the structural evolution and stability of the CoP–Cu3P catalyst, morphological examination under SEM and TEM was conducted after the electrochemical glycerol oxidation reactions. The SEM image (Fig. S9a, ESI†) demonstrates that the nanowire morphology of CoP–Cu3P was largely maintained after the GOR process. More detailed surface morphology was surveyed by carrying out TEM measurement. As shown in Fig. S9b (ESI†), a thin amorphous surface layer of 6–8 nm appeared to cover the inner crystalline nanowire, which not only served as the catalytic center but also protected the underlying metal phosphides from further oxidation. Moreover, the XRD pattern of CoP–Cu3P after the GOR confirmed the formation of cobalt oxyhydroxide. As shown in Fig. S10 (ESI†), the newly appeared diffraction peaks at 36.9°, 38.2°, and 65.7° could be assigned to the (021), (040), and (002) planes in CoOOH (JCPDS No. 26-0480), respectively. The surface composition and valence states of the CoP–Cu3P catalyst after the GOR were further examined by XPS. For both CoP–Cu3P and CoP, the peaks of Co0 species disappeared after the GOR, while Co3+ emerged in the high-resolution Co 2p spectra (Fig. S11a, ESI†). This indicates that near-surface Co0 species were oxidized to a higher valence state, forming a surface oxide-rich layer during the GOR process. In addition, the Co 2p3/2 spectra showed lower binding energy (0.6 eV) of Co3+ in CoP–Cu3P compared to that in CoP, suggesting possible electron transfer from Cu3P to CoP. Fig. S11b (ESI†) displays the high-resolution XPS spectrum of Cu 2p, with peaks for Cu–P bonds remaining. Moreover, the Cu 2p binding energy exhibited an upward shift (0.23 eV) for Cu–P in CoP–Cu3P, in comparison to that of Cu3P. The observed opposite shifts in Co and Cu binding energies suggested that interfacial charge transfer occurred between CoP and Cu3P in the mixed bimetallic phosphide heterostructures. Heterostructures with modulated interfacial electronic structures have been demonstrated to be active electrocatalysts toward various reactions.63–67CV and in situ Raman spectroscopy measurements provided more evidence and insight into the surface oxidation and reconstruction behaviors of TMPs. As shown in Fig. 3a, three anodic peaks at 0.80, 1.03, and 1.22 V, which correspond to the oxidation of Cu and Co (Co2+/Co3+ and Co3+/Co4+ redox pairs), were observed in the cyclic voltammogram of CoP–Cu3P. For pure Cu3P, the anodic oxidation peak appeared at 0.82 V, close to that in the CoP–Cu3P nanocomposites (Fig. S12a, ESI†). However, the cyclic voltammogram of pure CoP displayed a significant anodic shift of the Co oxidation peaks, with Co2+/Co3+ and Co3+/Co4+ oxidation occurring at 1.18 and 1.53 V, respectively (Fig. 3b). This is also consistent with the observed negative Co3+ binding energy shift. Given that the high-valency Co species (Co3+/Co4+) have been suggested to be involved actively in water oxidation, biomass and glycerol oxidation reactions,68–70 we postulate that the facile generation of high-valent Co species on the surface of CoP–Cu3P is likely the key to achieving efficient GOR. This is confirmed by in situ Raman spectroscopic characterizations. Fig. 3c shows the in situ Raman spectra that had been recorded on CoP–Cu3P in 1 M KOH, scanning at a rate of 1 mV s−1 from the open circuit potential (OCP) to 1.40 V. Two broad peaks located at 484 and 691 cm−1, corresponding to the Eg vibrational mode of Co–O and A1g vibrational mode of Co3O4, were observed at OCP. The appearance of CoOx on the catalyst surface is due to oxidation by the electrochemical activation treatment, which is common for metal phosphides.71–73 After the applied potential was raised above 1.05 V, new peaks started to emerge at 474 and 572 cm−1, which can be ascribed to the Eg and A1g vibrational modes of CoO2, accompanied by the disappearance of Eg and A1g vibrational modes of CoO and Co3O4. This indicated that a chemical conversion process on CoP–Cu3P started to occur from 1.05 V, generating new amorphous surface species with high-valent Co. As a control, in situ Raman spectroscopy was also conducted similarly on CoP and Cu3P as a function of the applied potential. As displayed in Fig. 3c, a weak peak located at 691 cm−1 was present at OCP, which can be assigned to CoOx on the CoP surface. Two new peaks at 484 and 608 cm−1 emerged from 1.2 V onward, together with the peak at 691 cm−1 showing increasing intensity, indicating more surface Co2+ and Co3+ generation. These results indicate that CoOx on the CoP surface is electrochemically rather stable, with very little Co3+ generation and accumulation after the voltage exceeds 1.2 V. Surprisingly, no Raman signals were detected on Cu3P, either at the OCP or upon raising the applied potential to 1.6 V (Fig. S12b, ESI†). A similar finding was reported by Zhou et al.,74 but the exact reason remains elusive. Both redox behaviors in CV and the in situ Raman spectra support the conclusion that the CoP–Cu3P surface is more readily reconstructed to produce high-valence surface Co3+ and Co4+ species in comparison to CoP, which is beneficial for the enhanced GOR activity.
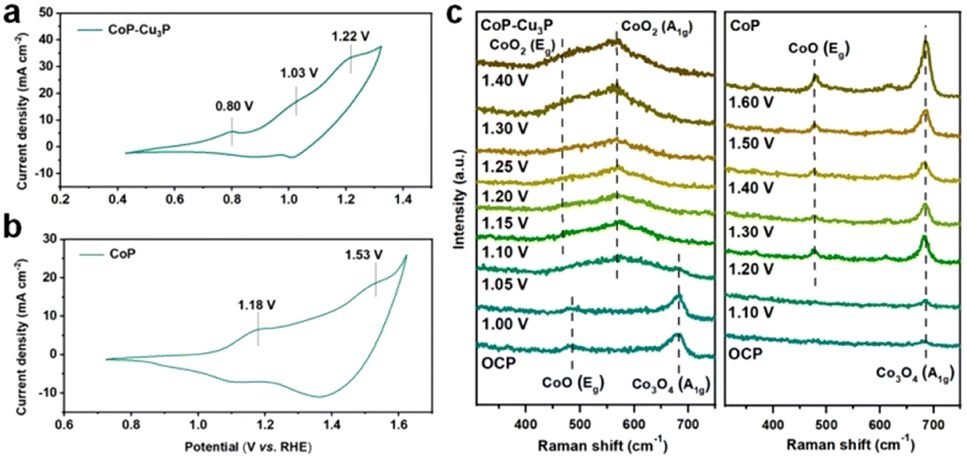 | ||
| Fig. 3 (a) CV curve in the potential range of 0.4–1.3 V vs. RHE of CoP–Cu3P, with a scan rate of 5 mV s−1. (b) CV curve in the potential range of 0.7–1.6 V vs. RHE of CoP, with a scan rate of 5 mV s−1. (c) In situ Raman spectroscopy conducted on the CoP–Cu3P electrode at increasing applied potentials from OCP to 1.4 V (left) and on the CoP electrode at increasing applied potentials from OCP to 1.6 V (right). | ||
Based on the above electrochemical, XPS, and in situ Raman characterization results, we therefore postulate that the excellent activity of the self-supporting CoP–Cu3P heterostructures toward electrocatalytic glycerol oxidation reactions likely originates from multiple factors: (1) abundant exposed active sites; (2) improved charge transfer kinetics on metallic metal phosphides; (3) formation of surface high-valent cobalt acting as a catalytic center, facilitated by electron transfer across the bimetallic phosphide interface.
Glycerol oxidation selectivity and coupled hydrogen evolution reaction
Electrochemical oxidation of glycerol can be a very complex process with various possible intermediate products involved. In the following section, the GOR was conducted on CoP–Cu3P/CC electrodes in an H-cell with a conventional three-electrode system, with the oxidation products quantitatively analyzed using HPLC. Typical HPLC chromatograms (Fig. S13, ESI†) display multiple elution peaks at retention times of 16.0, 14.5, 13.1, and 10.4 min, corresponding to formic acid, glycolic acid, glyceric acid, and oxalic acid, respectively. Calibration curves were obtained using pure chemicals with varying known concentrations, before the quantitative analysis of specific oxidation products (Fig. S14, ESI†). Holding the GOR at fixed applied potentials over a certain duration, the I–t data were acquired at 1.20, 1.30, 1.40 and 1.50 V (Fig. 4a), with oxidation products collected and analyzed to calculate the respective faradaic efficiencies. As shown in Fig. 4b, the total faradaic efficiency for all the detected solution products remained over 80% after 6 h of electrocatalysis at 1.2 V. When the applied potential was raised to 1.5 V, the overall faradaic efficiency dropped to ∼60%. In terms of specific oxidation products, the faradaic efficiencies of oxalic acid and glycolic acid decreased, with a concomitant increase in that of formic acid, upon raising the applied potentials. At 1.5 V, formic acid and oxalic acid remained as the only detectable oxidation products in the solution. This can be understood by the respective stability of various glycerol oxidation products. On CoP–Cu3P/CC electrodes, the electrochemical reactivity followed the order of glycerol > glycolic acid > formic acid > oxalic acid (Fig. S15, ESI†). Upon increasing the applied potential, glycolic acid is likely to be oxidized further to oxalic acid or to formic acid by breaking the C–C bond, while formic acid may be further mineralized completely to CO2 at sufficiently positive anodic potentials, thus resulting in reduced overall faradaic efficiency. We also analyzed the gaseous products using gas chromatography. However, no O2 or CO2 had been detected, indicating that the OER is not contributing to the lost faradaic efficiency, and the generated CO2 was dissolved in alkaline electrolyte forming CO32−. By assuming that the missing FE is solely due to the complete oxidation to CO2, the glycerol conversion, oxidation product selectivity, and carbon balance have been calculated and shown in Fig. S16 (ESI†). The calculated carbon balance values remained all above 90% at different applied potentials. After electrolysis at 1.5 V over 6 h, 65% of the original glycerol was oxidized mainly to formate (68% selectivity) and CO2, with only little oxalate and a trace amount of glycolic acid. Based on the above data, we propose a possible oxidation pathway for the electrocatalytic GOR on the CoP–Cu3P nanocomposites (Fig. 4c). The initial oxidation took place selectively on the terminal hydroxyl group of glycerol to generate glyceraldehyde, which was subsequently oxidized to glyceric acid. Glyceric acid can undergo a further oxidation step, forming glycolic acid and formic acid via C–C bond cleavage. Meanwhile, glycolic acid can be directly oxidized to oxalic acid or oxidized to formic acid by C–C bond cleavage.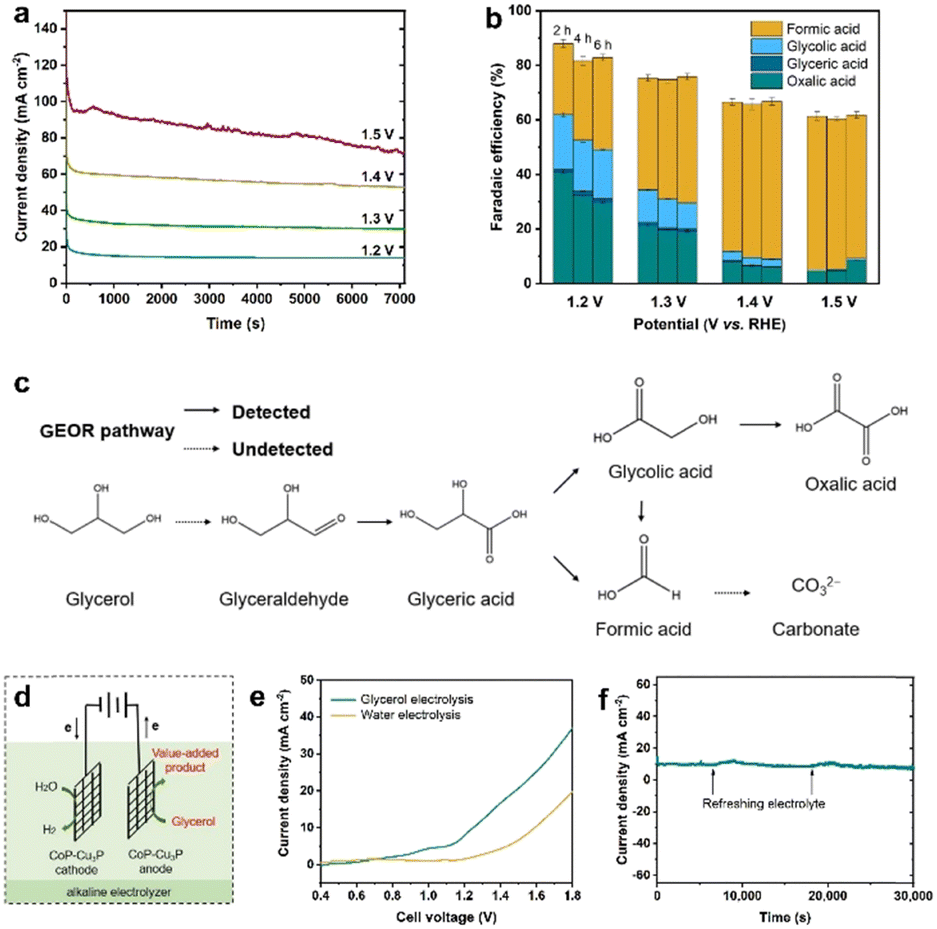 | ||
| Fig. 4 (a) I–t curves of GOR performed at different applied potentials in 1 M KOH containing 0.1 M glycerol. (b) The faradaic efficiencies toward various solution oxidized products in the GOR at different applied potentials in 1 M KOH that contained 0.1 M glycerol. (c) Proposed reaction pathways over CoP–Cu3P electrocatalysts for the GOR. (d) Schematic illustration of the two-electrode electrolysis system by integrating the GOR with the HER. (e) LSV curves for the CoP–Cu3P/CC||CoP–Cu3P/CC cell in 1.0 M KOH solution with and without 0.1 M glycerol addition. (f) I–t curve of the CoP–Cu3P/CC||CoP–Cu3P/CC cell with a constant applied potential of 1.21 V for 30000 s; the arrows indicate addition of extra glycerol to refresh the electrolyte. | ||
Finally, being inspired by the demonstrated high activity of metal phosphides toward hydrogen evolution,55,75–77 we went on to evaluate the performance of the as-prepared CoP–Cu3P/CC electrode for the HER in alkaline solution. As expected, the LSV profile of the CoP–Cu3P/CC electrode exhibited high activity for the catalytic HER, requiring an overpotential of ∼75 mV to reach a geometric current density of 10 mA cm−2 in 1 M KOH with or without glycerol (Fig. S17, ESI†). Given the demonstrated excellent activity of the CoP–Cu3P catalyst toward both the HER and GOR, a two-electrode alkaline electrolyzer was assembled using CoP–Cu3P/CC as both the cathode and anode for glycerol electrolysis (Fig. 4d). For comparison, the performance of the CoP–Cu3P/CC||CoP–Cu3P/CC electrolyzer for overall water splitting was also investigated in 1 M KOH electrolyte. As shown in Fig. 4e, a cell voltage of 1.42 V was required to drive 10 mA cm−2 in 1 M KOH solution. After the introduction of 0.1 M glycerol, the minimum required cell voltage to drive 10 mA cm−2 was reduced to 1.21 V, demonstrating that it is considerably more energy-efficient to produce hydrogen by replacing anodic oxygen evolution with glycerol oxidation. Actually, the current CoP–Cu3P/CC electrode system has surpassed many of the previously reported energy-saving hydrogen evolution systems using alternative anode oxidation reactions to replace the conventional OER (Table S2, ESI†). Moreover, the CoP–Cu3P/CC||CoP–Cu3P/CC electrolyzer has been demonstrated to maintain excellent durability for coupled HER/GOR reactions, as suggested by the I–t curve shown in Fig. 4f.
Conclusions
In conclusion, we have successfully synthesized 3D binary phosphide nanowire arrays on carbon cloth (CoP–Cu3P/CC) via transformation of mixed Co–Cu hydroxycarbonates. The as-synthesized self-supporting CoP–Cu3P shows outstanding activity of catalyzing glycerol oxidation in the alkaline electrolyte, requiring a low applied potential of 1.13 V vs. RHE to yield 10 mA cm−2 in 1 M KOH solution with 0.1 M glycerol. Their enhanced electrocatalytic performance originates from the abundantly exposed active sites and excellent charge transfer characteristics. Through systematic investigation using XPS, CV, and in situ Raman, we have unveiled that the electronic interaction between CoP and Cu3P leads to facile high-valent Co species generation during the electrocatalytic process. In addition, a two-electrode alkaline electrolyzer was assembled by employing CoP–Cu3P/CC as both the cathode and anode for the HER/GOR. This electrolyzer requires only an impressively low voltage of 1.21 V to drive a geometric current density of 10 mA cm−2. This work may provide guidance for designing advanced multiphase transition metal phosphide catalysts in the future, which can be used for glycerol as well as other organic electrooxidation-coupled electrolysis systems.Author contributions
Zhengzhe Xie: methodology, validation, investigation, visualization, writing original draft; Kang Wang: methodology, validation, investigation, visualization; Yu Zou: methodology, writing – review & editing; Guobing Ying: writing-review&editing, supervision; Jiang Jiang: conceptualization, writing – review & editing, supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Jiangsu Provincial Double-innovation Doctor Program (No. JSSCBS20211432) and the National Natural Science Foundation of China (No. 21873113). We thank Xin Huang for helping with the in situ Raman spectroscopy.Notes and references
- X. Tao, Y. Zhao, S. Wang, C. Li and R. Li, Chem. Soc. Rev., 2022, 51, 3561–3608 RSC.
- Q. Zhang, N. M. Bedford, J. Pan, X. Lu and R. Amal, Adv. Energy Mater., 2019, 9, 1901312 CrossRef.
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K.-Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS PubMed.
- Z. Xie, W. Wang, D. Ding, Y. Zou, Y. Cui, L. Xu and J. Jiang, J. Mater. Chem. A, 2020, 8, 12169–12176 RSC.
- F. W. S. Lucas, R. G. Grim, S. A. Tacey, C. A. Downes, J. Hasse, A. M. Roman, C. A. Farberow, J. A. Schaidle and A. Holewinski, ACS Energy Lett., 2021, 6, 1205–1270 CrossRef CAS.
- L. Zhang, W. Cai and N. Bao, Adv. Mater., 2021, 33, 2100745 CrossRef CAS PubMed.
- G.-F. Chen, Y. Luo, L.-X. Ding and H. Wang, ACS Catal., 2017, 8, 526–530 CrossRef.
- B. You, X. Liu, X. Liu and Y. Sun, ACS Catal., 2017, 7, 4564–4570 CrossRef CAS.
- X. Deng, M. Li, Y. Fan, L. Wang, X.-Z. Fu and J.-L. Luo, Appl. Catal., B, 2020, 278, 119339 CrossRef CAS.
- D. Wu, J. Hao, W. Wang, Y. Yu, X. Z. Fu and J. L. Luo, ChemSusChem, 2021, 14, 5450–5459 CrossRef CAS PubMed.
- Z. Li, Y. Yan, S. M. Xu, H. Zhou, M. Xu, L. Ma, M. Shao, X. Kong, B. Wang, L. Zheng and H. Duan, Nat. Commun., 2022, 13, 147 CrossRef CAS PubMed.
- Y. Li, X. Wei, L. Chen, J. Shi and M. He, Nat. Commun., 2019, 10, 5335 CrossRef PubMed.
- L. Fan, Y. Ji, G. Wang, J. Chen, K. Chen, X. Liu and Z. Wen, J. Am. Chem. Soc., 2022, 144, 7224–7235 CrossRef CAS PubMed.
- X. Yu, E. C. Dos Santos, J. White, G. Salazar-Alvarez, L. G. M. Pettersson, A. Cornell and M. Johnsson, Small, 2021, 17, 2104288 CrossRef CAS PubMed.
- B. You, X. Liu, N. Jiang and Y. Sun, J. Am. Chem. Soc., 2016, 138, 13639–13646 CrossRef CAS PubMed.
- G. Yang, Y. Jiao, H. Yan, Y. Xie, A. Wu, X. Dong, D. Guo, C. Tian and H. Fu, Adv. Mater., 2020, 32, 2000455 CrossRef CAS PubMed.
- T. Wang, Z. Huang, T. Liu, L. Tao, J. Tian, K. Gu, X. Wei, P. Zhou, L. Gan, S. Du, Y. Zou, R. Chen, Y. Li, X. Z. Fu and S. Wang, Angew. Chem., Int. Ed., 2021, 61, 202115636 Search PubMed.
- W. J. Liu, Z. Xu, D. Zhao, X. Q. Pan, H. C. Li, X. Hu, Z. Y. Fan, W. K. Wang, G. H. Zhao, S. Jin, G. W. Huber and H. Q. Yu, Nat. Commun., 2020, 11, 265 CrossRef CAS PubMed.
- Y. Zhang, B. Zhou, Z. Wei, W. Zhou, D. Wang, J. Tian, T. Wang, S. Zhao, J. Liu, L. Tao and S. Wang, Adv. Mater., 2021, 33, 2104791 CrossRef CAS PubMed.
- Y. Huang, X. Chong, C. Liu, Y. Liang and B. Zhang, Angew. Chem., Int. Ed., 2018, 57, 13163–13166 CrossRef CAS PubMed.
- X. Chong, C. Liu, C. Wang, R. Yang and B. Zhang, Angew. Chem., Int. Ed., 2021, 60, 22010–22016 CrossRef CAS PubMed.
- Y. Xu and B. Zhang, ChemElectroChem, 2019, 6, 3214–3226 CrossRef CAS.
- B. Garlyyev, S. Xue, J. Fichtner, A. S. Bandarenka and C. Andronescu, ChemSusChem, 2020, 13, 2513–2521 CrossRef CAS PubMed.
- Y. Holade, N. Tuleushova, S. Tingry, K. Servat, T. W. Napporn, H. Guesmi, D. Cornu and K. B. Kokoh, Catal. Sci. Technol., 2020, 10, 3071–3112 RSC.
- Y. Li, X. Wei, L. Chen and J. Shi, Angew. Chem., Int. Ed., 2021, 60, 19550–19571 CrossRef CAS PubMed.
- J. Jack, W. Zhu, J. L. Avalos, J. Gong and Z. J. Ren, Green Chem., 2021, 23, 7917–7936 RSC.
- C. Deng, C. Y. Toe, X. Li, J. Tan, H. Yang, Q. Hu and C. He, Adv. Energy Mater., 2022, 12, 2201047 CrossRef CAS.
- H. Luo, J. Barrio, N. Sunny, A. Li, L. Steier, N. Shah, I. E. L. Stephens and M. M. Titirici, Adv. Energy Mater., 2021, 11, 2101180 CrossRef CAS.
- M. Simoes, S. Baranton and C. Coutanceau, ChemSusChem, 2012, 5, 2106–2124 CrossRef CAS PubMed.
- A. Behr, J. Eilting, K. Irawadi, J. Leschinski and F. Lindner, Green Chem., 2008, 10, 13–30 RSC.
- G. Dodekatos, S. Schünemann and H. Tüysüz, ACS Catal., 2018, 8, 6301–6333 CrossRef CAS.
- L. Fan, B. Liu, X. Liu, N. Senthilkumar, G. Wang and Z. Wen, Energy Technol., 2020, 9, 2000804 CrossRef.
- T. Li and D. A. Harrington, ChemSusChem, 2021, 14, 1472–1495 CrossRef CAS PubMed.
- Y. Kwon, Y. Birdja, I. Spanos, P. Rodriguez and M. T. M. Koper, ACS Catal., 2012, 2, 759–764 CrossRef CAS.
- A. Zalineeva, A. Serov, M. Padilla, U. Martinez, K. Artyushkova, S. Baranton, C. Coutanceau and P. B. Atanassov, J. Am. Chem. Soc., 2014, 136, 3937–3945 CrossRef CAS PubMed.
- Y. Li, X. Wei, S. Han, L. Chen and J. Shi, Angew. Chem., Int. Ed., 2021, 60, 21464–21472 CrossRef CAS PubMed.
- P. V. B. Santiago, C. C. Lima, J. L. Bott-Neto, P. S. Fernández, C. A. Angelucci and J. Souza-Garcia, J. Electroanal. Chem., 2021, 896, 115198 CrossRef CAS.
- D. M. Morales, D. Jambrec, M. A. Kazakova, M. Braun, N. Sikdar, A. Koul, A. C. Brix, S. Seisel, C. Andronescu and W. Schuhmann, ACS Catal., 2022, 12, 982–992 CrossRef CAS.
- X. Yu, R. B. Araujo, Z. Qiu, E. Campos dos Santos, A. Anil, A. Cornell, L. G. M. Pettersson and M. Johnsson, Adv. Energy Mater., 2022, 12, 2103750 CrossRef CAS.
- J. Wu, J. Li, Y. Li, X. Y. Ma, W. Y. Zhang, Y. Hao, W. B. Cai, Z. P. Liu and M. Gong, Angew. Chem., Int. Ed., 2021, 61, 202113362 Search PubMed.
- L. Dong, G.-R. Chang, Y. Feng, X.-Z. Yao and X.-Y. Yu, Rare Met., 2022, 41, 1583–1594 CrossRef CAS.
- Z. He, J. Hwang, Z. Gong, M. Zhou, N. Zhang, X. Kang, J. W. Han and Y. Chen, Nat. Commun., 2022, 13, 3777 CrossRef CAS PubMed.
- N. Shilpa, A. Pandikassala, P. Krishnaraj, P. S. Walko, R. N. Devi and S. Kurungot, ACS Appl. Mater. Interfaces, 2022, 14, 16222–16232 CrossRef CAS PubMed.
- Y. Pei, Z. Pi, H. Zhong, J. Cheng and F. Jin, J. Mater. Chem. A, 2022, 10, 1309–1319 RSC.
- Y. Xu, M. Liu, S. Wang, K. Ren, M. Wang, Z. Wang, X. Li, L. Wang and H. Wang, Appl. Catal., B, 2021, 298, 120493 CrossRef CAS.
- X. Liu, Z. Fang, X. Teng, Y. Niu, S. Gong, W. Chen, T. J. Meyer and Z. Chen, J. Energy Chem., 2022, 72, 432–441 CrossRef CAS.
- X. Han, H. Sheng, C. Yu, T. W. Walker, G. W. Huber, J. Qiu and S. Jin, ACS Catal., 2020, 10, 6741–6752 CrossRef CAS.
- M. Braun, G. Behrendt, M. L. Krebs, P. Dimitri, P. Kumar, I. Sanjuán, S. Cychy, A. C. Brix, D. M. Morales, J. Hörlöck, B. Hartke, M. Muhler, W. Schuhmann, M. Behrens and C. Andronescu, ChemElectroChem, 2022, 9, e202200267 CAS.
- L. S. Oh, M. Park, Y. S. Park, Y. Kim, W. Yoon, J. Hwang, E. Lim, J. H. Park, S. M. Choi, M. H. Seo, W. B. Kim and H. J. Kim, Adv. Mater., 2022, 34, 2203285 CrossRef PubMed.
- J. Chang, Y. Xiao, M. Xiao, J. Ge, C. Liu and W. Xing, ACS Catal., 2015, 5, 6874–6878 CrossRef CAS.
- Z. Li, H. Feng, M. Song, C. He, W. Zhuang and L. Tian, Mater. Today Energy, 2021, 20, 100698 CrossRef CAS.
- X. Huang, Y. Zou and J. Jiang, ACS Sustainable Chem. Eng., 2021, 9, 14470–14479 CrossRef.
- F. Yu, H. Zhou, Y. Huang, J. Sun, F. Qin, J. Bao, W. A. Goddard, 3rd, S. Chen and Z. Ren, Nat. Commun., 2018, 9, 2551 CrossRef PubMed.
- Z. Zhao, D. E. Schipper, A. P. Leitner, H. Thirumalai, J.-H. Chen, L. Xie, F. Qin, M. K. Alam, L. C. Grabow, S. Chen, D. Wang, Z. Ren, Z. Wang, K. H. Whitmire and J. Bao, Nano Energy, 2017, 39, 444–453 CrossRef CAS.
- J. F. Callejas, C. G. Read, E. J. Popczun, J. M. McEnaney and R. E. Schaak, Chem. Mater., 2015, 27, 3769–3774 CrossRef CAS.
- H. Yu, S. Qu, P.-R. Chen, K.-Q. Ou, J.-Y. Lin, Z.-H. Guo, L. Zheng, J.-K. Li, S. Huang, Y. Teng, L. Zou and J.-L. Song, J. Hazard. Mater., 2022, 430, 128351 CrossRef CAS PubMed.
- A. P. Grosvenor, S. D. Wik, R. G. Cavell and A. Mar, Inorg. Chem., 2005, 44, 8988–8998 CrossRef CAS PubMed.
- Z. Zheng, M. Retana, X. Hu, R. Luna, Y. H. Ikuhara and W. Zhou, ACS Appl. Mater. Interfaces, 2017, 9, 16986–16994 CrossRef CAS PubMed.
- L. Ji, J. Wang, X. Teng, T. J. Meyer and Z. Chen, ACS Catal., 2020, 10, 412–419 CrossRef CAS.
- R. Shen, J. Xie, Y. Ding, S.-Y. Liu, A. Adamski, X. Chen and X. Li, ACS Sustainable Chem. Eng., 2019, 7, 3243–3250 CrossRef CAS.
- A. Saad, Z. Cheng, H. Shen, H. Guo, J. P. Attfield, T. Thomas and M. Yang, Sustainable Energy Fuels, 2021, 5, 1366–1373 RSC.
- S. Jin, ACS Energy Lett., 2017, 2, 1937–1938 CrossRef CAS.
- J. Hu, A. Al-Salihy, J. Wang, X. Li, Y. Fu, Z. Li, X. Han, B. Song and P. Xu, Adv. Sci., 2021, 8, 2103314 CrossRef CAS PubMed.
- H. Wang, Y. Zhou and S. Tao, Appl. Catal., B, 2022, 315, 121588 CrossRef CAS.
- L. Yan, B. Zhang, J. Zhu, Y. Li, P. Tsiakaras and P. K. Shen, Appl. Catal., B, 2020, 265, 118555 CrossRef CAS.
- B. Zhao, J. Liu, C. Xu, R. Feng, P. Sui, J.-X. Luo, L. Wang, J. Zhang, J.-L. Luo and X.-Z. Fu, Appl. Catal., B, 2021, 285, 119800 CrossRef CAS.
- X. Xu, A. Cao, W. You, Z. Tao, L. Kang and J. Liu, Small, 2021, 17, 2101725 CrossRef CAS PubMed.
- H. Y. Wang, S. F. Hung, H. Y. Chen, T. S. Chan, H. M. Chen and B. Liu, J. Am. Chem. Soc., 2016, 138, 36–39 CrossRef CAS PubMed.
- X. Deng, G. Y. Xu, Y. J. Zhang, L. Wang, J. Zhang, J. F. Li, X. Z. Fu and J. L. Luo, Angew. Chem., Int. Ed., 2021, 60, 20535–20542 CrossRef CAS PubMed.
- X. Huang, Y. Guo, Y. Zou and J. Jiang, Appl. Catal., B, 2022, 309, 121247 CrossRef CAS.
- J. Yang, H. Liu, W. N. Martens and R. L. Frost, J. Phys. Chem. C, 2010, 114, 111–119 CrossRef CAS.
- A. Moysiadou, S. Lee, C. S. Hsu, H. M. Chen and X. Hu, J. Am. Chem. Soc., 2020, 142, 11901–11914 CrossRef CAS PubMed.
- L. Reith, C. A. Triana, F. Pazoki, M. Amiri, M. Nyman and G. R. Patzke, J. Am. Chem. Soc., 2021, 143, 15022–15038 CrossRef CAS PubMed.
- M. Kong, H. Song and J. Zhou, Adv. Energy Mater., 2018, 8, 1801489 CrossRef.
- M.-Q. Wang, C. Ye, H. Liu, M. Xu and S.-J. Bao, Angew. Chem., Int. Ed., 2018, 57, 1963–1967 CrossRef CAS PubMed.
- L. Yan, B. Zhang, J. Zhu, S. Zhao, Y. Li, B. Zhang, J. Jiang, X. Ji, H. Zhang and P. K. Shen, J. Mater. Chem. A, 2019, 7, 14271–14279 RSC.
- L. Zhang, G. Wang, X. Hao, Z. Jin and Y. Wang, Chem. Eng. J., 2020, 395, 125113 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional figures and tables. See DOI: https://doi.org/10.1039/d2ya00311b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |