
A case study on the thermal-stability of polymerized small molecular acceptor-based polymer solar cells†
Hongmei
Qin
a,
Shijie
Ju
a,
Wenyan
Su
*a,
Baofeng
Zhao
 b,
Qunping
Fan
c,
Zhaozhao
Bi
c,
Sen
Zhang
c,
Jifa
Yu
c,
Guanghao
Lu
c,
Ji-Ting
Hou
d,
Wei
Ma
c,
Chao
Gao
*b and
Yuxiang
Li
*a
b,
Qunping
Fan
c,
Zhaozhao
Bi
c,
Sen
Zhang
c,
Jifa
Yu
c,
Guanghao
Lu
c,
Ji-Ting
Hou
d,
Wei
Ma
c,
Chao
Gao
*b and
Yuxiang
Li
*a
aSchool of Materials Science and Engineering, Xi’an University of Science and Technology, Xi’an 710054, China. E-mail: suwy1027@xust.edu.cn; liyuxianggj687@xust.edu.cn
bXi’an Key Laboratory of Liquid Crystal and Organic Photovoltaic Materials, State Key Laboratory of Fluorine & Nitrogen Chemicals, Xi’an Modern Chemistry Research Institute, Xi’an 710065, China. E-mail: chaogao1974@hotmail.com
cState Key Laboratory for Mechanical Behavior of Materials & Frontier Institute of Science and Technology, Xi’an Jiaotong University, Xi’an 710049, China
dSchool of Ophthalmology & Optometry, School of Biomedical Engineering, Wenzhou Medical University, Wenzhou, 325000, China
First published on 8th December 2022
Abstract
Despite the relatively unique advantages of morphological stability in PSMA-based polymer solar cells (PSCs), the microstructure evolution of the active layer over the thermal stress is highly sensitive to the degraded efficiency still remains a challenge. To reveal the specific roles of polymer donors (PDs) and polymerized small molecular acceptors (PSMAs) in the microstructure evolution of active layers over thermal stress, we developed a new PSMA named P50-γ using near-infrared absorption via a random ternary copolymerization strategy of combining two different small molecule precursors with BPT-core and BPTz-core. The resultant PSMA-based PSCs have achieved a high-power conversion efficiency (PCE) of 15.1% when paired with the commonly used PD (PBDB-T). Furthermore, we reveal the respective influences of PD and PSMA in the active layer on the thermal stability of devices. Specifically, after the thermal annealing treatment at 80 °C for 150 hours, PSC maintained ∼80% of the initial PCE, in which the photocurrent was slightly decreased while the fill factor (FF) significantly reduced. Systematical studies indicate that the decrease of PCE over thermal stress is mainly attributed to the burn-in disaggregation of PD, which is supported by the absorption spectra and crystalline characteristics of the annealed active layer. Our work not only presents a new PSMA to achieve efficient and relatively stable PSCs but also reveals for the first time that PD dominates the thermal-disaggregation process of PSMA-based PSCs.
1. Introduction
Polymer solar cells (PSCs) with bulk-heterojunction active layers have shown multiple advantages of low-cost processability and versatile potential applications in transparent, lightweight, and flexible electronic devices.1–4 By virtue of the breakthrough of photovoltaic materials, especially the recently developed non-fullerene small-molecule acceptors (SMAs),5–11 SMA-based PSCs have achieved the state-of-the-art power conversion efficiencies (PCEs) of over 19%.12–14 Comparatively, all-polymer solar cells (all-PSCs) based on a tightly entangled active layer of the polymer donor (PD) and polymer acceptor (PA) are considered to be the most promising photovoltaic technologies with more application prospects in wearable electronics due to the high mechanical strength and superior morphological stabilities,15–19 yet they still suffer from the relatively low PCEs, which is primarily due to the lack of high-performance of PAs compared to the diverse SMAs. Therefore, further improving the PCEs of all-PSCs is critical for advancing commercial organic photovoltaics.20–22 From this perspective, developing novel PAs with favorable properties to match well with high-performance PDs is the key to further advancing all-PSCs.Recently, as the most recent and important milestone in the development of PAs, Li et al. have developed an ingenious strategy to construct high-performance PAs, namely polymerized SMAs (PSMAs), by polymerizing SMAs with desirable optoelectronic properties.23 For example, as the first PSMA, PZ1 originated from an SMA precursor, namely IDIC-C16, with mixed 5(or 6)-brominated end-groups has achieved an impressively high PCE of over 9% in all PSCs due to its narrow bandgap and high extinction coefficient,23 attesting the effectiveness of this strategy. More recently, encouraged by the great success of Y6 and its derivatives with near-infrared (NIR) absorption and low energy loss,24,25 a series of efficient PSMAs originating from Y6 derivatives as precursors have been developed and have achieved significantly increased PCEs.26–35 Although those PSMA-based all-PSCs have achieved the highest PCE of up to 18%,36 it is imperative to adopt novel strategies and develop diversified polymer acceptors to improve their PCEs further.
Additionally, excellent thermal stability is one of the prerequisites for the practical applications of efficient PSMA-based all-PSCs,37–42 however, it is still not satisfactory for the lab-scale device due to the slightly degraded efficiency over thermal stress. For example, Fu et al. recently reported regio-regular PSMA, namely PZT-γ,43 providing a higher PCE of 15.8% when paired with a typical polymer donor PBDB-T, while its all-PSCs maintained ∼81% of the initial PCE upon the thermal stability test under high-temperature of 70 °C for 120 h. Specifically, a significantly degraded fill factor of ∼20% after prolonged heating at 70 °C was observed in three PSMA-based all-PSCs, which is generally regarded as due to the film morphological evolution over thermal stress.44 However, the in-depth roles of PDs and PAs in the microstructure evolution of active layers over thermal stress and related device performance are poorly understood so far. Furthermore, very few efforts on long-term thermal stability have been directed toward defining the rational molecule design that overcomes the above challenges for further advancing PSMA-based PSCs.
Herein, a new PSMA named P50-γ was developed via a random ternary copolymerization strategy of combining two different SMA precursors with the BPT-core and BPTz-core to realize the expective optoelectronic properties.43 The fundamental studies show that P50-γ has strong NIR absorption, suitable energy levels, high electron mobility, strong crystallinity, and excellent thermal stability. When paired with a classical PD named PBDB-T, the PSMA-based PSCs achieved a high PCE of 15.1%. After the thermal annealing treatment at 80 °C for 150 hours, PSMA-based PSCs maintained ∼80% of the initial PCE, in which the short-circuit current density (Jsc) was slightly decreased and the fill factor (FF) obviously decreased. Further studies revealed that the decrease in PCE is mainly attributed to the burn-in disaggregation of PD, which is supported by the absorption and morphological properties of the annealed active layers. As a result, our study not only developed a new PSMA named P50-γ to achieve efficient and relatively stable PSMA-based PSCs, but also revealed for the first time that PD dominates the thermal-disaggregation process of PSMA-based PSCs.
2. Results and discussion
Herein, we designed and synthesized a new PSMA named P50-γ with near-infrared absorption via a random ternary copolymerization strategy of combining two small molecular accepting precursors (BPT-core and BPTz-core) with a thiophene monomer. We expected this random ternary copolymerization strategy to enable the P50-γ with the desired optoelectronic properties as demonstrated in recent studies.45,46 The molecular structure and synthetic route of P50-γ are displayed in Fig. 1a and Scheme S1 (ESI†). BPT-2HO and BPTz-2CHO were synthesized according to previous reports.24,43 YIC-Br-γ, ZIC-Br-γ, and P50-γ were characterized by 1H NMR (Fig. S1–S3, ESI†). P50-γ was obtained by random ternary copolymerization using Pd2(dba)3 and P(o-tol)3 as catalytic systems. The detailed synthetic information is described in the ESI.† As shown in Fig. S4 (ESI†) of high-temperature gel permeation chromatography (GPC) measurement using 1,2,4-trichlorobenzene as the eluent at 150 °C, P50-γ has a number average molecular weight (Mn) of 8.7 kDa with a polydispersity index (PDI) of 1.72. Note that P50-γ has a low molecular weight with 2-3 monomers, which indicates a low degree of polymerization. We believe that the further advanced synthesized method will assist PSMAs with a higher molecular weight. P50-γ can be readily dissolved in common organic solvents, such as chloroform, toluene, and chlorobenzene. As shown in Fig. S6 (ESI†), thermogravimetric analysis (TGA), P50-γ has a high thermal decomposition temperature (Td) of ∼350 °C under a weight loss of 5%, which is sufficient for device manufacturing.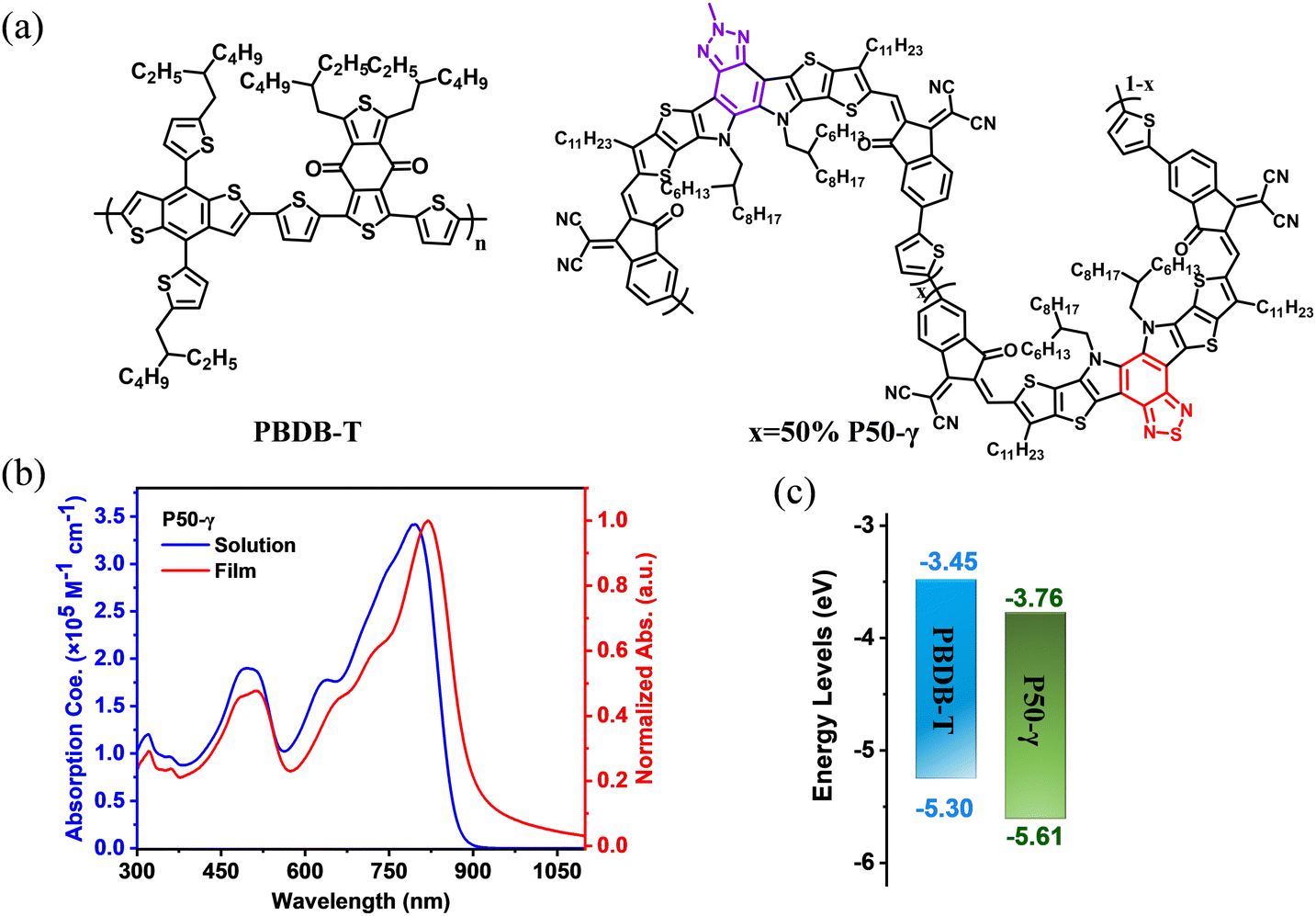 | ||
| Fig. 1 (a) Molecular structures of PBDB-T and P50-γ. (b) Absorption spectra of P50-γ in dilute chloroform (<10−6 mol L−1) and thin film. (c) Energy levels of PBDB-T and P50-γ. | ||
The UV-vis absorption spectra of P50-γ in the solution and thin films are shown in Fig. 1b and corresponding data are summarized in Table 1. In dilute chloroform, P50-γ shows a high maximum extinction coefficient (ε) of 3.42 × 105 M−1 cm−1 at the absorption peak (λmax) of 797 nm. Moreover, P50-γ film exhibits an obviously red-shifted λmax at 820 nm compared to the solution one, indicating a solid intramolecular interaction.47 Furthermore, the P50-γ film has a NIR-absorption onset (λonset) located at 931 nm that corresponds to a small optical bandgap (Eoptg) of 1.33 eV, which can well complement a wide bandgap PBDB-T for harvesting more photons. In addition, significant π–π* transition peaks from P50-γ were observed at ∼500 nm in both the solution and thin film, indicating efficient charge delocalization.48
| Polymer | M n [kDa] | PDI | ε solmax [M−1 cm−1] | λ solmax [nm] | λ filmmax [nm] | λ filmonset [nm] | E optg [eV]a | E LUMO [eV]b | E HOMO [eV]b | T d [°C] |
|---|---|---|---|---|---|---|---|---|---|---|
| a E optg = 1240/λonset. b From CV curve: EHOMO = −[Eox + (4.8 − Fc/Fc+)], ELUMO = −[Ered + (4.8 − Fc/Fc+)]. | ||||||||||
| PBDB-T | 21.3 | 6.58 | 0.48 × 105 | 612 | 630 | 695 | 1.78 | −3.45 | −5.30 | 392 |
| P50-γ | 8.69 | 1.72 | 3.42 × 105 | 797 | 820 | 931 | 1.33 | −3.75 | −5.54 | 356 |
Cyclic voltammetry (CV) measurements were carried out to study the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) levels of active layer materials (Fig. S8, ESI†). As shown in Fig. 1c and Table 1, the HOMO/LUMO levels were determined to be −5.30 eV/−3.45 eV for PBDB-T and −5.61 eV/−3.76 eV for P50-γ, respectively.
As shown in Fig. S9 and Table S1 (ESI†), grazing incidence wide-angle X-ray scattering (GIWAXS) measurements were performed to investigate the molecular orientation, crystallinity, and packing properties of PBDB-T and P50-γ neat films. As evidenced by the strong (010) π–π stacking diffraction peaks in the out-of-plane (OOP) direction, two neat films exhibit a face-on dominant orientation. On the other hand, in the in-plane (IP) direction, P50-γ shows a strong (100) diffraction peak at 0.392 Å−1 with a small d-spacing of 1.60 nm and a high crystal coherence length (CCL) of 9.75 nm, while PBDB-T has a sharp (100) diffraction peak at 0.318 Å−1 with a d-spacing of 1.97 nm and a CCL of 6.75 nm. Notably, PBDB-T displays an additional diffraction peak at 0.692 Å−1 compared to P50-γ, which offers an excellent channel to investigate the thermal stability of each component in the PBDB-T:P50-γ active layer.
To probe the photovoltaic performance of P50-γ, PSMA-based PSCs with a device structure of ITO/PEDOT:PSS/PBDB-T:P50-γ/PFN-Br/Ag were fabricated. The detailed device optimization processes and relevant performance parameters are summarized in Fig. S11, S12 and Tables S2, S3 (ESI†). As shown in Fig. 2a, the current density–voltage (J–V) curves and Table 2 of photovoltaic parameters, the PBDB-T:P50-γ-based PSCs achieved an ideal PCE of 15.10% with both high Jsc (24.85 mA cm−2) and FF (71.65%). Since good stability is a prerequisite for practical applications of OSCs devices, herein, the storage and thermal stabilities in the N2-filled glovebox of PSMA-based PSCs were probed. Under continuous annealing at 80 °C for 150 hours, the PSMA-based PSCs maintained ∼80% of the initial PCE, indicating good thermal stability. The external quantum efficiency (EQE) spectrum is shown in Fig. 2b, and the integral values are shown in Table 2. All devices exhibit broad and strong EQE response at 300–900 nm, while the EQE spectrum in the 700–900 nm region gradually decreased with increasing annealing time, which may be due to the minimal morphological evolution in the mixed phase, inferred by the slightly decreased Jsc. As shown in Fig. 2c, the PCE decay of PSMA-based PSCs is mainly due to the apparent decrease in FF from 71.65% to 59.08%, while the open-circuit voltage (Voc, from 0.85 to 0.81 V) and Jsc (from 24.85 to 24.58 mA cm−2) well retained 95.3% and 98.9% of the initial values. A similar phenomenon was also observed for the storage stability in PSMA-based PSCs, as shown in Fig. S13 and Table S4 (ESI†).
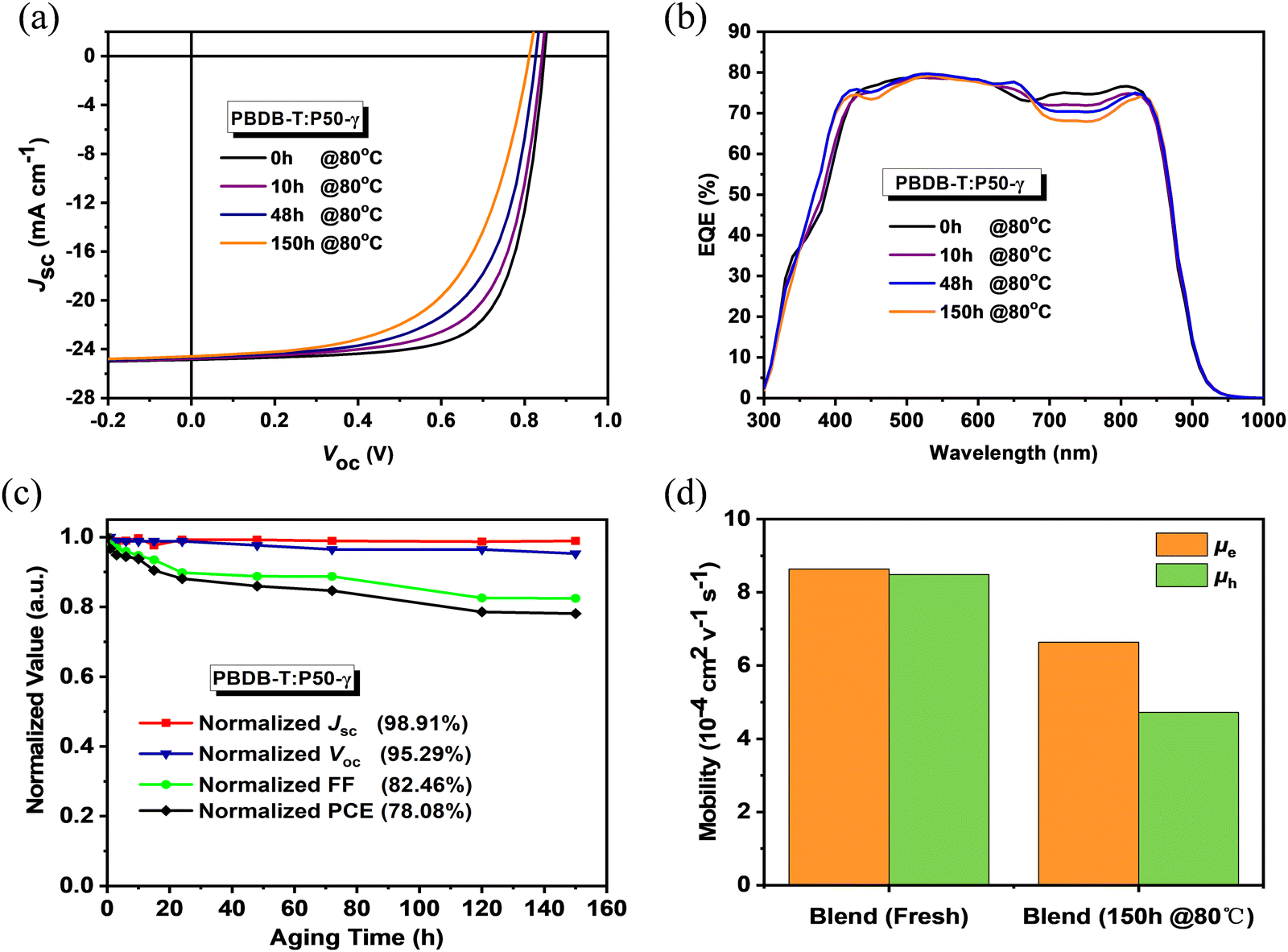 | ||
| Fig. 2 (a) J–V curves, (b) EQE spectra, and (c) normalized photovoltaic parameters of the PBDB-T:P50-γ-based PSCs with different storage times under 80 °C in the N2-filled glovebox. (d) The mobilities of the PBDB-T:P50-γ-based devices with fresh active layer and annealed active layer at 80 °C for 150 hours. | ||
| Annealing Time [h] | V oc [V] | J sc [mA cm−2] | J cal [mA cm−2] | FF [%] | PCEb [%] |
|---|---|---|---|---|---|
| a Integrated values from EQE spectra. b The average parameters were calculated from over 10 devices. | |||||
| 0 | 0.85 (0.83 ± 0.01) | 24.85 (24.59 ± 0.26) | 24.12 | 71.65 (71.10 ± 0.72) | 15.10 (15.00 ± 0.10) |
| 10 | 0.84 (0.82 ± 0.01) | 24.77 (24.40 ± 0.31) | 23.94 | 67.88 (67.14 ± 0.68) | 14.16 (14.03 ± 0.13) |
| 48 | 0.83 (0.81 ± 0.02) | 24.66 (24.44 ± 0.22) | 23.77 | 63.60 (63.00 ± 0.81) | 12.98 (12.81 ± 0.17) |
| 150 | 0.81 (0.80 ± 0.02) | 24.58 (24.40 ± 0.18) | 23.65 | 59.08 (59.08 ± 0.71) | 11.79 (11.64 ± 0.15) |
Since the PCE decay of PSMA-based PSCs is mainly due to the apparent decrease of FF, the charge-transporting properties of PBDB-T and P50-γ neat films and related PBDB-T:P50-γ blend were studied using the space-charge-limited current (SCLC) method. As shown in Fig. S14 and Table S5 (ESI†), both fresh neat films achieved high electron-mobility (μe) of 9.91 × 10−4 cm2 V−1 s−1 for P50-γ and high hole-mobility (μh) of 29.5 × 10−4 cm2 V−1 s−1 for PBDB-T. Notably, after continuous annealing at 80 °C for 150 hours, P50-γ film had a well-kept initial μe (8.03 × 10−4 cm2 V−1 s−1), while PBDB-T film obtained a 38% significantly reduced μh (18.4 × 10−4 cm2 V−1 s−1). As exhibited in Fig. 2d, the fresh PBDB-T:P50-γ blend achieved both high μe (8.64 × 10−4 cm2 V−1 s−1) and μh (8.49 × 10−4 cm2 V−1 s−1) with a balanced μe/μh ratio close to 1. Similar to the annealed neat films, the PBDB-T:P50-γ blend annealed at 80 °C for 150 hours also achieved well-kept μe of 6.64 × 10−4 cm2 V−1 s−1 and much-decreased μh of 4.72 × 10−4 cm2 V−1 s−1. The decay of μh results in a relatively unbalanced charge transport property with an increased μe/μh ratio of 1.41, thus resulting in a reduced FF in PSMA-based PSCs.
To further probe the in-depth roles of PD and PSMA in the microstructure evolution of active layers under thermal stress and related device performance, the absorption spectra of PBDB-T, P50-γ, and PBDB-T:P50-γ films with different annealing times at 80 °C were measured. With continuous annealing from 0 to 150 hours, the PBDB-T film showed obviously and gradually lower absorption intensity (Fig. 3a), implying that the intermolecular stacking was disrupted to some extent. Conversely, the P50-γ film showed much weaker temperature dependence in absorption intensity with a barely perceptible decay rate (Fig. 3b), suggesting that thermal annealing has less effect on intermolecular aggregation and stacking behavior. Similarly, the PBDB-T:P50-γ blend also exhibited a much lower absorption intensity belonging to PBDB-T, and a slightly reduced absorption peak belonging to P50-γ (Fig. 3c). Moreover, to better understand the absorption decay of these pure and blended films under thermal annealing, the absorption intensities of 0–0 peaks were normalized. As shown in Fig. 3d–f, the 0–0 absorption peaks belonging to PBDB-T showed a sharp decrease to ∼58% after 80 °C annealing for 150 hours in both pure and blend films, indicating that the PBDB-T film is thermodynamically unstable. On the other hand, the 0–0 absorption peaks of P50-γ displayed very small decays of 16.7% in pure film and 10.9% in blend film, suggesting the molecular stacking of P50-γ is more robust in thermodynamics with better molecular ordering. The above results imply that PD dominates the thermal-disaggregation processes of PSMA-based efficient PSCs in this work.
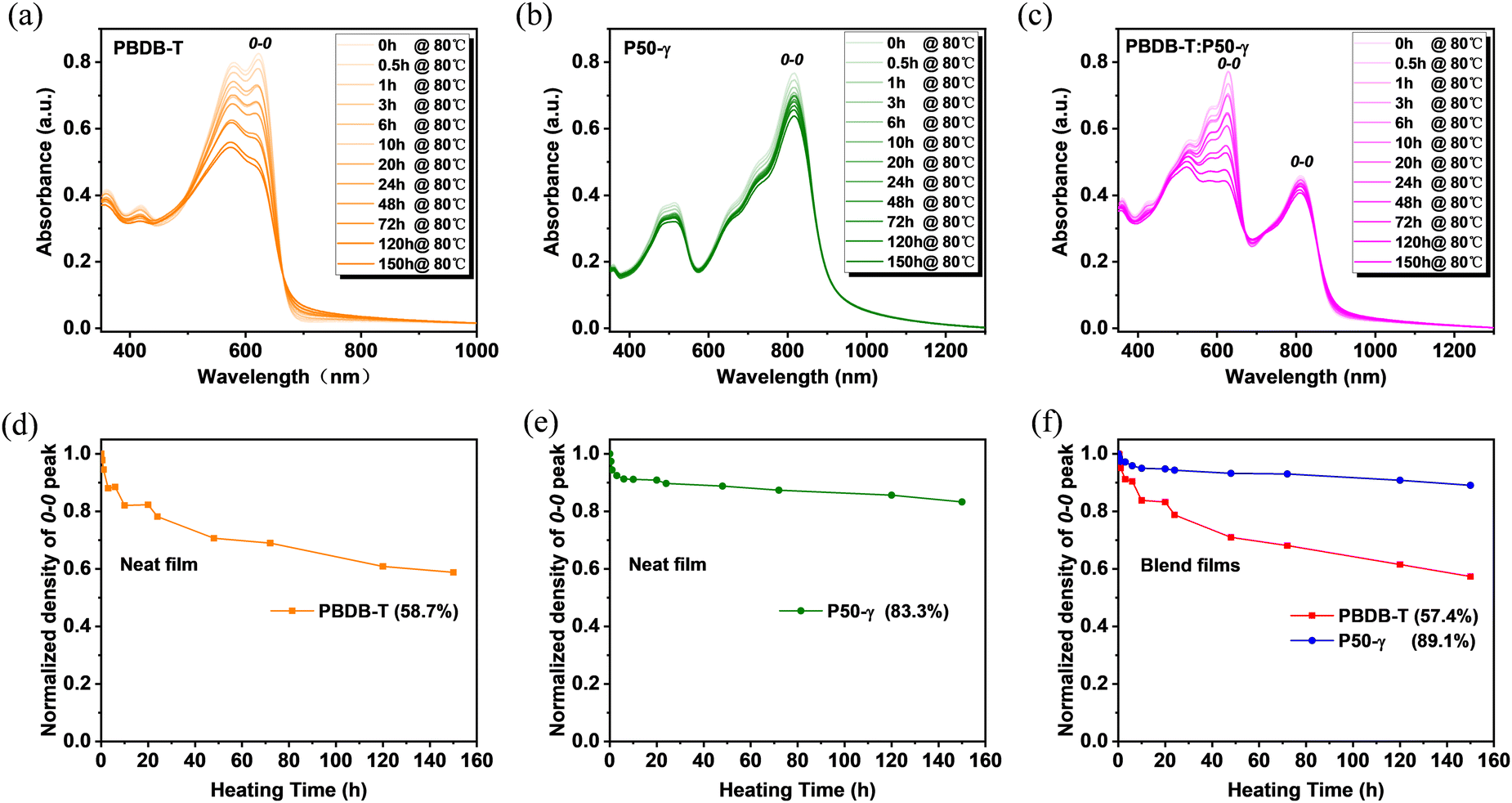 | ||
| Fig. 3 (a–c) Absorption spectra and (d–f) Normalized absorption intensity from 0–0 peaks of PBDB-T, P50-γ, and PBDB-T:P50-γ films with prolonged annealing times under 80 °C. | ||
To explore the effect of the active layer morphology with different annealing times at 80 °C on device performance, atomic force microscopy (AFM) images were measured (Fig. 4). The PBDB-T neat film and annealed film at 80 °C for 150 hours showed a relatively smooth and homogeneous surface, while the annealed film had a slightly decreased root-mean-square (RMS) roughness from 0.71 to 0.69 nm (Fig. 4a–d). Compared to PBDB-T, the P50-γ fresh and annealed neat films showed rough surface morphologies (Fig. 4e–h) with higher RMS values of 3.40–3.68 nm, implying a solid aggregation behavior. On the other hand, unlike the neat films, the PBDB-T:P50-γ blends with an RMS of 1.20–1.45 nm formed a network-like structure of protofibrils (Fig. 4i–l). Moreover, the suitable phase separation of active layers indicates good miscibility between PBDB-T and P50-γ, which facilitates the exciton dissociation and charge transport. Overall, both neat and blend films show decreased RMS values after thermal annealing, implying diminished aggregation or microstructure evolution, which is consistent with the absorption of fresh/annealed active layers (Fig. 3). In addition, the minor change in the phase separation after thermal annealing can partly explain the slightly decreased high Jsc in the devices with different storage times at 80 °C.
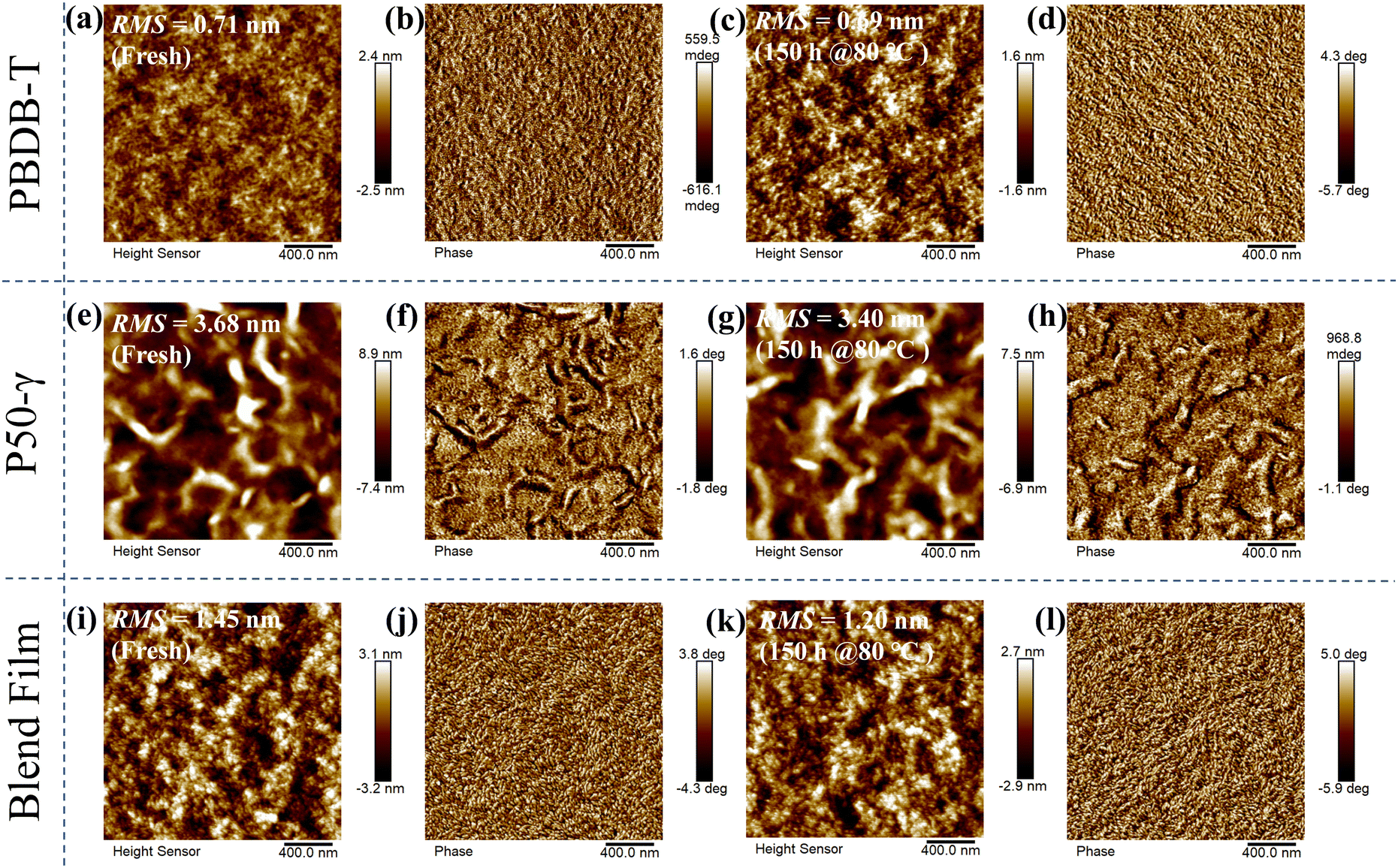 | ||
| Fig. 4 AFM height and phase images of fresh/annealed neat and blend films: (a–d) for PBDB-T; (e–h) for P50-γ; and (i–l) for PBDB-T:P50-γ, respectively. | ||
GIWAXS measurements were carried out to understand in-depth the effect of the molecular stacking and crystallization characteristics of blend films on device performance to reveal further the reasons for the PCE degradation of PSMA-based PSCs under thermal stress shown in Fig. 5. Similar to the PBDB-T and P50-γ pure films, PBDB-T:P50-γ blend films showed a predominant face-on orientation. Specifically, for the (100) diffraction peaks in the IP direction, four blend films with different annealing times had a similar peak position located at 0.318 Å−1 (corresponds to the same d-spacing of 1.97 nm) with an almost identical CCL of ∼9.5 nm. For the (010) diffraction peaks in the OOP direction, four blend films also showed a similar peak position of 1.769 Å−1 (corresponding to the same π–π stacking spacing of 0.36 nm), while the CCLs decreased from 1.52 to 1.44 nm with continuous annealing. The similar molecular orientation, packing, and crystallinity of blend films with different annealing times are consistent with the AFM results, which can further explain why the Jsc of PSMA-based PSCs remains at a high level under continuous thermal annealing.
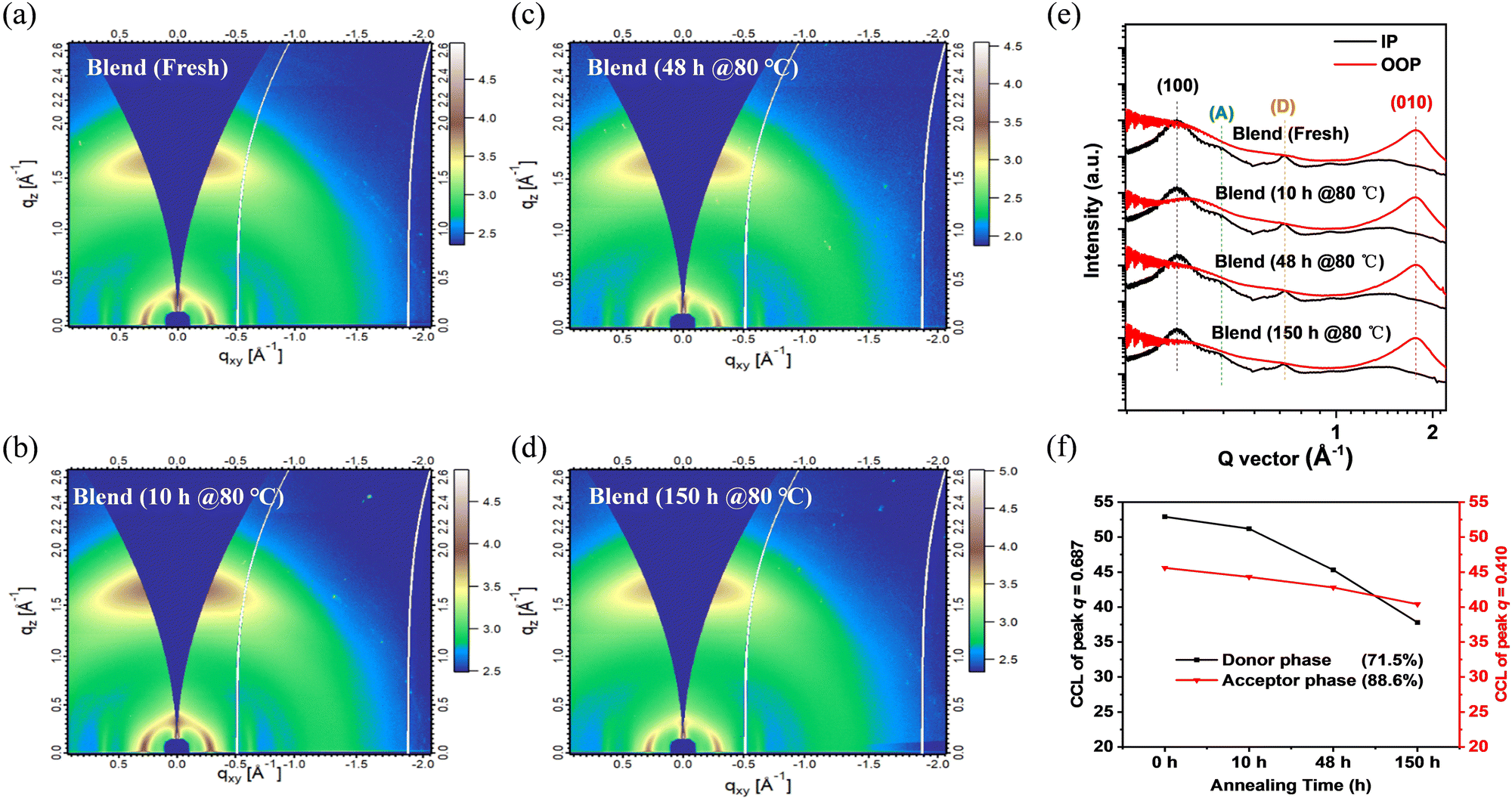 | ||
| Fig. 5 (a–d) 2D GIWAXS patterns of blend films with different annealing times of 0, 10, 48, and 150 hours. (e) Line-cut profiles of IP and OOP directions. (f) CCL values from (100) diffraction peaks in the IP direction of the donor (PBDB-T) and acceptor (P50-γ) phases of blend films with different annealing times. | ||
Notably, PBDB-T (0.687 Å−1) and P50-γ (0.410 Å−1) in blend films have their distinctive characteristic diffraction peaks in the IP direction (Fig. S10 (ESI†) and Fig. 5e). Therefore, the corresponding CCL values of the two peaks from blend films with different annealing times could be calculated to study further the specific roles of PBDB-T and P50-γ in the microstructure evolution of the active layers under thermal stress. As shown in Fig. 5f, with the increase in annealing time from 0 to 150 hours, the blend film showed a lower acceptor phase which dropped to 88.6% of its original CCL value of the diffraction peak at 0.410 Å−1. However, the donor phase featured by the diffraction peak at 0.687 Å−1 significantly decreased to 71.5%, directly affecting the hole mobility, and resulting in an unbalanced charge-transporting property. The above results are consistent with the SCLC measurements and can explain the sharp decrease of FF in the annealed PSMA-based PSCs.
To investigate the morphology evolution of the PBDB-T:P50-γ film in the vertical direction after thermal annealing, film-depth-dependent light absorption spectrometry (FLAS) was performed to understand the composition distribution and optical/electronic properties along the film-depth direction.49,50Fig. 6a and e show the absorption spectra at different depths of the fresh and annealed film, respectively. In the fresh film (Fig. 6a), the maximum absorption peaks of PD and PSMA showed a red-shift absorption spectrum of ∼6 nm and ∼10 nm from top to bottom, respectively, indicating the well-ordered structures of PD and PSMA in the vertical direction. After thermal annealing treatment for 150 h, the PSMA showed a red-shift absorption spectrum of ∼7 nm. The absorption intensity of PD decreased significantly, indicating a significant disaggregation process in the donor phase, consistent with the film absorption and GIWAXS results. The composition distribution in the fresh and annealed film is shown in Fig. 6b and f, where the depths of 0 and 100 nm represent the PEDOT:PSS/active-layer and active-layer/PFN-Br interfaces, respectively. In the fresh film (Fig. 6b), we observed a well-balanced mixture phase along the vertical direction, which benefits the efficient charge dissociation and transport.19 Although the mixed phase was observed at 10-75 nm in the annealed film (Fig. 6f), a higher composition ratio of PSMA of over 50% was observed at 10–75 nm relative to PD, indicating that the disaggregation process occurred in the PD phase. Besides, an unbalanced distribution of PSMA and PD was observed near the depths of 0 nm and 100 nm, which probably prevents the charge transport and collection.
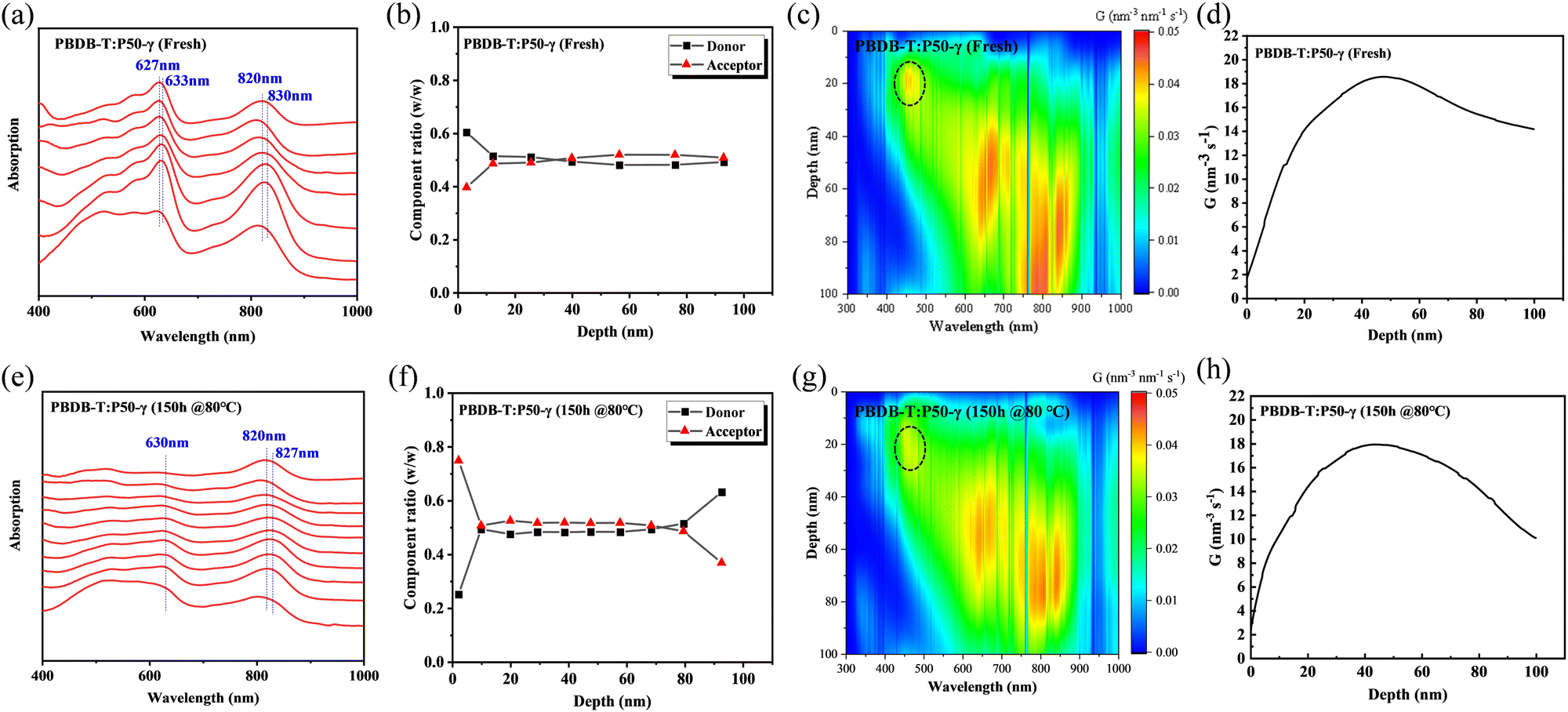 | ||
| Fig. 6 Film-depth-dependent absorption spectra of (a) fresh and (e) annealed blend films. The compositional distribution as a function of film depth for (b) fresh (f) annealed blend films. (c and g) Simulated exciton generation contours as a function of film depth. (d and h) Exciton generation rate G profiles simulated from film-depth-dependent absorption spectra with an optical transfer matrix model. | ||
In addition, we simulated the exciton generation as a function of film depth for both blend films by combining the depth-dependent spectra with an optical transfer matrix model.51Fig. 6c and g show the exciton generation contour of the fresh and annealed film derived from the harvested solar energy within the ITO/PEDOT: PSS/active layer/PFN-Br/Al device, where the incident light comes from the ITO/PEDOT: PSS side. The exciton generation in the fresh and annealed films was mainly distributed in the bottom half of the active layer. Meanwhile, a weaker density of excitons was observed in the annealed film relative to the fresh film at 400–900 nm, indicating a significant attenuation in the PD phase. Moreover, the curves of the exciton generation rate (G) for both devices are shown in Fig. 6d and h, as simulated from the film-depth-dependent absorption spectra of the active layer upon 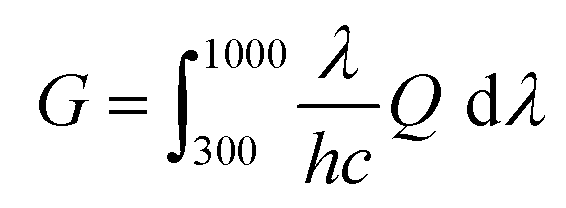 , where λ is the wavelength, h is Planck's constant, and c is the speed of light. The Gmax in both blend films is about 18 nm−3 s−1, and the G value of the fresh active layer drops slowly to 14 nm−3 s−1 at 70–100 nm. Comparatively, the annealed film presents a more significant drop of G value to ∼10 nm−3 s−1, indicating that the disaggregated PD caused the change of the partially mixed phase. Furthermore, the reduced G value in the annealed film may facilitate an imbalance in charge transport, which echoes the SCLC and FF results.
, where λ is the wavelength, h is Planck's constant, and c is the speed of light. The Gmax in both blend films is about 18 nm−3 s−1, and the G value of the fresh active layer drops slowly to 14 nm−3 s−1 at 70–100 nm. Comparatively, the annealed film presents a more significant drop of G value to ∼10 nm−3 s−1, indicating that the disaggregated PD caused the change of the partially mixed phase. Furthermore, the reduced G value in the annealed film may facilitate an imbalance in charge transport, which echoes the SCLC and FF results.
3. Conclusion
A near-infrared absorbing PSMA named P50-γ was developed via a random ternary copolymerization strategy of combining two different SMA precursors with the BPT-core and BPTz-core. The fundamental studies showed that P50-γ has strong NIR absorption, suitable energy levels, high electron mobility, strong crystallinity, and excellent thermal stability. When paired with PBDB-T, PSMA-based PSCs achieved a high PCE of 15.1%, and after storage at 80 °C for 150 hours, PSMA-based PSCs maintained ∼80% of the initial PCE, in which the Jsc was slightly decreased and FF was obviously reduced. Systematic studies indicate that the PCE decay is mainly attributed to the burn-in disaggregation of PBDB-T, which is supported by absorption spectra, charge transports, and crystalline morphology of annealed active layer. As a result, in this study, we not only developed a high-performance PSMA called P50-γ to achieve efficient and stable PSMA-based PSCs but also revealed for the first time that PD dominates the thermal-disaggregation process of PSMA-based PSCs.Author contributions
H. Qin and W. Su conceived the idea and designed the experiments; S. Ju synthesized the monomers and characterization; W. Su, Q. Fan, and B. Zhao performed the device fabrication and characterization, and data collection and analysis; Z. Bi, S. Zhang, and W. Ma performed the GIWAXS characterization and analysis; J. Yu and G. Lu performed the FLAS characterization and analysis; H. Qin and Y. Li participated in the material synthesis and characterization; S. Ju, W. Su, Q. Fan, and Y. Li wrote the original draft; J. T. Hou, C. Gao and Y. Li supervised and directed the project.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 52203241, 22005121, 22209131, 21905225), the Science and Technology Program of Shanxi Province (2022JM-229). X-ray data were acquired at beamlines 7.3.3 at the Advanced Light Source, which is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. The authors thank Dr Eric Schaible and Dr Chenhui Zhu at beamline 7.3.3 for assistance with data acquisition. The authors also thank Dr Zicheng Ding from Shaanxi Normal University for help with AFM measurement.References
- J. Hou, O. Inganäs, R. H. Friend and F. Gao, Nat. Mater., 2018, 17, 119–128 CrossRef CAS PubMed.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CrossRef CAS.
- A. J. Heeger, Adv. Mater., 2014, 26, 10–28 CrossRef CAS PubMed.
- P. Cheng, G. Li, X. Zhan and Y. Yang, Nat. Photonics, 2018, 12, 131–142 CrossRef CAS.
- L. Meng, Y. Zhang, X. Wan, C. Li, X. Zhang, Y. Wang, X. Ke, Z. Xiao, L. Ding, R. Xia, H.-L. Yip, Y. Cao and Y. Chen, Science, 2018, 361, 1094–1098 CrossRef CAS PubMed.
- K. Weng, L. Ye, L. Zhu, J. Xu, J. Zhou, X. Feng, G. Lu, S. Tan, F. Liu and Y. Sun, Nat. Commun., 2020, 11, 2855 CrossRef CAS PubMed.
- F. Lin, K. Jiang, W. Kaminsky, Z. Zhu and A. K. Y. Jen, J. Am. Chem. Soc., 2020, 142, 15246–15251 CrossRef CAS PubMed.
- C. He, Y. Pan, Y. Ouyang, Q. Shen, Y. Gao, K. Yan, J. Fang, Y. Chen, C.-Q. Ma, J. Min, C. Zhang, L. Zuo and H. Chen, Energy Environ. Sci., 2022, 15, 2537–2544 RSC.
- Z. Yao, X. Liao, K. Gao, F. Lin, X. Xu, X. Shi, L. Zuo, F. Liu, Y. Chen and A. K. Y. Jen, J. Am. Chem. Soc., 2018, 140, 2054–2057 CrossRef CAS PubMed.
- Y. Lin, Q. He, F. Zhao, L. Huo, J. Mai, X. Lu, C.-J. Su, T. Li, J. Wang, J. Zhu, Y. Sun, C. Wang and X. Zhan, J. Am. Chem. Soc., 2016, 138, 2973–2976 CrossRef CAS PubMed.
- Y. Lin, J. Wang, Z.-G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- K. Chong, X. Xu, H. Meng, J. Xue, L. Yu, W. Ma and Q. Peng, Adv. Mater., 2022, 34, 2109516 CrossRef CAS PubMed.
- Y. Cui, Y. Xu, H. Yao, P. Bi, L. Hong, J. Zhang, Y. Zu, T. Zhang, J. Qin, J. Ren, Z. Chen, C. He, X. Hao, Z. Wei and J. Hou, Adv. Mater., 2021, 33, 2102420 CrossRef CAS PubMed.
- L. Zhu, M. Zhang, J. Xu, C. Li, J. Yan, G. Zhou, W. Zhong, T. Hao, J. Song, X. Xue, Z. Zhou, R. Zeng, H. Zhu, C.-C. Chen, R. C. I. MacKenzie, Y. Zou, J. Nelson, Y. Zhang, Y. Sun and F. Liu, Nat. Mater., 2022, 21, 656–663 CrossRef CAS PubMed.
- G. Wang, F. S. Melkonyan, A. Facchetti and T. J. Marks, Angew. Chem., Int. Ed., 2019, 58, 4129–4142 CrossRef CAS PubMed.
- Y. Xu, J. Yuan, S. Liang, J.-D. Chen, Y. Xia, B. W. Larson, Y. Wang, G. M. Su, Y. Zhang, C. Cui, M. Wang, H. Zhao and W. Ma, ACS Energy Lett., 2019, 4, 2277–2286 CrossRef CAS.
- T. Kim, J.-H. Kim, T. E. Kang, C. Lee, H. Kang, M. Shin, C. Wang, B. Ma, U. Jeong, T.-S. Kim and B. J. Kim, Nat. Commun., 2015, 6, 8547 CrossRef CAS.
- H. Benten, D. Mori, H. Ohkita and S. Ito, J. Mater. Chem. A, 2016, 4, 5340–5365 RSC.
- C. Lee, S. Lee, G.-U. Kim, W. Lee and B. J. Kim, Chem. Rev., 2019, 119, 8028–8086 CrossRef CAS PubMed.
- T. Liu, T. Yang, R. Ma, L. Zhan, Z. Luo, G. Zhang, Y. Li, K. Gao, Y. Xiao, J. Yu, X. Zou, H. Sun, M. Zhang, T. A. Dela Peña, Z. Xing, H. Liu, X. Li, G. Li, J. Huang, C. Duan, K. S. Wong, X. Lu, X. Guo, F. Gao, H. Chen, F. Huang, Y. Li, Y. Li, Y. Cao, B. Tang and H. Yan, Joule, 2021, 5, 914–930 CrossRef CAS.
- Q. Fan, W. Su, S. Chen, T. Liu, W. Zhuang, R. Ma, X. Wen, Z. Yin, Z. Luo, X. Guo, L. Hou, K. Moth-Poulsen, Y. Li, Z. Zhang, C. Yang, D. Yu, H. Yan, M. Zhang and E. Wang, Angew. Chem., Int. Ed., 2020, 59, 19835–19840 CrossRef CAS PubMed.
- T. Yang, R. Ma, H. Cheng, Y. Xiao, Z. Luo, Y. Chen, S. Luo, T. Liu, X. Lu and H. Yan, J. Mater. Chem. A, 2020, 8, 17706–17712 RSC.
- Z.-G. Zhang, Y. Yang, J. Yao, L. Xue, S. Chen, X. Li, W. Morrison, C. Yang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 13503–13507 CrossRef CAS PubMed.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- W. Zhu, A. P. Spencer, S. Mukherjee, J. M. Alzola, V. K. Sangwan, S. H. Amsterdam, S. M. Swick, L. O. Jones, M. C. Heiber, A. A. Herzing, G. Li, C. L. Stern, D. M. DeLongchamp, K. L. Kohlstedt, M. C. Hersam, G. C. Schatz, M. R. Wasielewski, L. X. Chen, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2020, 142, 14532–14547 CrossRef CAS PubMed.
- Q. Fan, H. Fu, M. Liu, J. Oh, X. Ma, F. R. Lin, C. Yang, F. Zhang and A. K. Y. Jen, ACS Appl. Mater. Interfaces, 2022, 14, 26970–26977 CrossRef CAS PubMed.
- Q. Fan, H. Fu, Z. Luo, J. Oh, B. Fan, F. Lin, C. Yang and A. K. Y. Jen, Nano Energy, 2022, 92, 106718 CrossRef CAS.
- Q. Fan, R. Ma, W. Su, Q. Zhu, Z. Luo, K. Chen, Y. Tang, F. R. Lin, Y. Li, H. Yan, C. Yang, A. K. Y. Jen and W. Ma, Carbon Energy, 2022 DOI:10.1002/cey2.267.
- Y. Li, M. Wang, Q. Zhang, Z. Wu, H. Lim, Y. Wang, H. Qin, J. Yang, C. Gao, H. Young Woo and J. Yuan, Chem. Eng. J., 2022, 428, 131232 CrossRef CAS.
- Y. Kong, Y. Li, J. Yuan and L. Ding, InfoMat, 2022, 4, e12271 CrossRef CAS.
- Q. Fan, H. Fu, Q. Wu, Z. Wu, F. Lin, Z. Zhu, J. Min, H. Y. Woo and A. K. Y. Jen, Angew. Chem., Int. Ed., 2021, 60, 15935–15943 CrossRef CAS.
- Z. Luo, T. Liu, R. Ma, Y. Xiao, L. Zhan, G. Zhang, H. Sun, F. Ni, G. Chai, J. Wang, C. Zhong, Y. Zou, X. Guo, X. Lu, H. Chen, H. Yan and C. Yang, Adv. Mater., 2020, 32, 2005942 CrossRef CAS PubMed.
- H. Yu, Y. Wang, H. K. Kim, X. Wu, Y. Li, Z. Yao, M. Pan, X. Zou, J. Zhang, S. Chen, D. Zhao, F. Huang, X. Lu, Z. Zhu and H. Yan, Adv. Mater., 2022, 34, 2200361 CrossRef CAS PubMed.
- R. Sun, W. Wang, H. Yu, Z. Chen, X. Xia, H. Shen, J. Guo, M. Shi, Y. Zheng, Y. Wu, W. Yang, T. Wang, Q. Wu, Y. Yang, X. Lu, J. Xia, C. J. Brabec, H. Yan, Y. Li and J. Min, Joule, 2021, 5, 1548–1565 CrossRef CAS.
- R. Ma, C. Yan, P. W.-K. Fong, J. Yu, H. Liu, J. Yin, J. Huang, X. Lu, H. Yan and G. Li, Energy Environ. Sci., 2022, 15, 2479–2488 RSC.
- J. Wang, Y. Cui, Y. Xu, K. Xian, P. Bi, Z. Chen, K. Zhou, L. Ma, T. Zhang, Y. Yang, Y. Zu, H. Yao, X. Hao, L. Ye and J. Hou, Adv. Mater., 2022, 34, 2205009 CrossRef CAS PubMed.
- Z. Li, X. Xu, W. Zhang, Z. Genene, W. Mammo, A. Yartsev, M. R. Andersson, R. A. J. Janssen and E. Wang, J. Mater. Chem. A, 2017, 5, 11693–11700 RSC.
- S. Holliday, R. S. Ashraf, C. B. Nielsen, M. Kirkus, J. A. Röhr, C.-H. Tan, E. Collado-Fregoso, A.-C. Knall, J. R. Durrant, J. Nelson and I. McCulloch, J. Am. Chem. Soc., 2015, 137, 898–904 CrossRef CAS PubMed.
- M. Jørgensen, K. Norrman, S. A. Gevorgyan, T. Tromholt, B. Andreasen and F. C. Krebs, Adv. Mater., 2012, 24, 580–612 CrossRef PubMed.
- R. Ma, C. Yan, J. Yu, T. Liu, H. Liu, Y. Li, J. Chen, Z. Luo, B. Tang, X. Lu, G. Li and H. Yan, ACS Energy Lett., 2022, 7, 2547–2556 CrossRef CAS.
- R. Ma, K. Zhou, Y. Sun, T. Liu, Y. Kan, Y. Xiao, T. A. Dela Peña, Y. Li, X. Zou, Z. Xing, Z. Luo, K. S. Wong, X. Lu, L. Ye, H. Yan and K. Gao, Matter, 2022, 5, 725–734 CrossRef CAS.
- S. Yao, T. Yang, X. Shen, T. Li, B. Huang, H. Liu, X. Lu, T. Liu and B. Zou, J. Mater. Chem. C, 2022, 10, 9723–9729 RSC.
- H. Fu, Y. Li, J. Yu, Z. Wu, Q. Fan, F. Lin, H. Y. Woo, F. Gao, Z. Zhu and A. K. Y. Jen, J. Am. Chem. Soc., 2021, 143, 2665–2670 CrossRef CAS PubMed.
- Y. Zhang, Y. Xu, M. J. Ford, F. Li, J. Sun, X. Ling, Y. Wang, J. Gu, J. Yuan and W. Ma, Adv. Energy Mater., 2018, 8, 1800029 CrossRef.
- J. Du, K. Hu, L. Meng, I. Angunawela, J. Zhang, S. Qin, A. Liebman-Pelaez, C. Zhu, Z. Zhang, H. Ade and Y. Li, Angew. Chem., Int. Ed., 2020, 59, 15181–15185 CrossRef CAS PubMed.
- J. Du, K. Hu, C. Zhu, J. Zhang, Z. Zhang, Z. Wei, L. Meng and Y. Li, Macromolecules, 2022, 55, 7481–7487 CrossRef CAS.
- L. Yan, H. Zhang, Q. An, M. Jiang, A. Mahmood, M. H. Jee, H.-R. Bai, H.-F. Zhi, S. Zhang, H. Y. Woo and J.-L. Wang, Angew. Chem., Int. Ed., 2022, 61, e202209454 CAS.
- Y. Li, D. H. Lee, J. Lee, T. L. Nguyen, S. Hwang, M. J. Park, D. H. Choi and H. Y. Woo, Adv. Funct. Mater., 2017, 27, 1701942 CrossRef.
- L. Bu, S. Gao, W. Wang, L. Zhou, S. Feng, X. Chen, D. Yu, S. Li and G. Lu, Adv. Energy Mater., 2016, 2, 1600359 Search PubMed.
- J. Wang, J. Zhang, Y. Xiao, T. Xiao, R. Zhu, C. Yan, Y. Fu, G. Lu, X. Lu, S. R. Marder and X. Zhan, J. Am. Chem. Soc., 2018, 140, 9140–9147 CrossRef CAS PubMed.
- Y. Huo, X.-T. Gong, T.-K. Lau, T. Xiao, C. Yan, X. Lu, G. Lu, X. Zhan and H.-L. Zhang, Chem. Mater., 2018, 30, 8661–8668 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc04362a |
| This journal is © The Royal Society of Chemistry 2023 |