
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
3D printed and stimulus responsive drug delivery systems based on synthetic polyelectrolyte hydrogels manufactured via digital light processing†
Sonja
Vaupel‡
 ab,
Robert
Mau‡
cd,
Selin
Kara
a,
Hermann
Seitz
cd,
Udo
Kragl
bc and
Johanna
Meyer
*abc
ab,
Robert
Mau‡
cd,
Selin
Kara
a,
Hermann
Seitz
cd,
Udo
Kragl
bc and
Johanna
Meyer
*abc
aInstitute of Technical Chemistry, Leibniz University Hannover, Callinstraße 5, 30167 Hannover, Germany. E-mail: johanna.meyer@iftc.uni-hannover.de
bInstitute of Chemistry, University of Rostock, Albert-Einstein-Str. 3a, 18059, Rostock, Germany
cDepartment Life, Light & Matter, Faculty for Interdisciplinary Research, University of Rostock, Albert-Einstein-Straße 25, 18059 Rostock, Germany
dMicrofluidics, University of Rostock, Justus-von-Liebig-Weg 6, 18059 Rostock, Germany
First published on 16th June 2023
Abstract
Hydrogels are three-dimensional hydrophilic polymeric networks absorbing up to and even more than 90 wt% of water. These superabsorbent polymers retain their shape during the swelling process while enlarging their volume and mass. In addition to their swelling behavior, hydrogels can possess other interesting properties, such as biocompatibility, good rheological behavior, or even antimicrobial activity. This versatility qualifies hydrogels for many medical applications, especially drug delivery systems. As recently shown, polyelectrolyte-based hydrogels offer beneficial properties for long-term and stimulus-responsive applications. However, the fabrication of complex structures and shapes can be difficult to achieve with common polymerization methods. This obstacle can be overcome by the use of additive manufacturing. 3D printing technology is gaining more and more attention as a method of producing materials for biomedical applications and medical devices. Photopolymerizing 3D printing methods offer superior resolution and high control of the photopolymerization process, allowing the fabrication of complex and customizable designs while being less wasteful. In this work, novel synthetic hydrogels, consisting of [2-(acryloyloxy) ethyl]trimethylammonium chloride (AETMA) as an electrolyte monomer and poly(ethylene glycol)-diacrylate (PEGDA) as a crosslinker, 3D printed via Digital Light Processing (DLP) using a layer height of 100 μm, are reported. The hydrogels obtained showed a high swelling degree q∞m,t ∼ 12 (24 h in PBS; pH 7; 37 °C) and adjustable mechanical properties with high stretchability (εmax ∼ 300%). Additionally, we embedded the model drug acetylsalicylic acid (ASA) and investigated its stimulus-responsive drug release behaviour in different release media. The stimulus responsiveness of the hydrogels is mirrored in their release behavior and could be exploited in triggered as well as sequential release studies, demonstrating a clear ion exchange behavior. The received 3D-printed drug depots could also be printed in complex hollow geometry, exemplarily demonstrated via an individualized frontal neo-ostium implant prototype. Consequently, a drug-releasing, flexible, and swellable material was obtained, combining the best of both worlds: the properties of hydrogels and the ability to print complex shapes.
Introduction
3D printing techniques have rapidly evolved in the last two decades, due to their ability to revolutionize various fields including health care and medicine. This has been accomplished by the relatively uncomplicated, fast, and cost-efficient way to fabricate prototypes and products with no need for high-volume manufacturing.1 The current research on 3D printing in the medical field has mostly been focused on (I) preoperative planning and surgical training,2–4 (II) bioactive and biodegradable scaffolds,5,6 (III) tissue and organ engineering,7 and (IV) customizable implants,8 and (V) pharmaceutical products,9 although many of these topics can also overlap. These customizable implants include biomedical materials, such as hard metals, ceramic materials, and polymers, ranging from thermoplastic and elastomeric polymers to soft hydrogels. Metallic-based implants are widely used in orthopedics and dentistry, mostly relying on stainless steel or cobalt-chromium-, titanium-, or tantalum alloys, as they are often described with good chemical, mechanical behaviour, and biocompatibility. Furthermore, hydrogels can also be used as 3D printed implants. These physically or chemically crosslinked hydrophilic polymer networks are able to absorb up to 90% of water or more, without dissolving or losing their three-dimensional structure.10 Their properties combine designable mechanical and swelling properties, biocompatibility, stimulus responsivity, and even antimicrobial behavior.11–13 At this point we would like to recommend previously published review articles that address the wide range and tunability of hydrogel properties and their synthesis.14–19 Due to these versatile features, hydrogels have a huge variety of applications in the medical field, ranging from tissue engineering to contact lenses and drug delivery systems (DDSs).20–22Controlled DDSs can overcome the limitations of conventional repeated drug administration, such as poor drug absorbance, rapid metabolization, and short half-life, which is why higher doses often need to be prescribed. Other important DDS factors to improve the effectiveness and safety of therapy include controlling the timing, rate, and location of the drug release and minimizing the impact on healthy cells in other parts of the body.23,24 Hydrogel drug delivery depots can have different concepts to incorporate active agents in the polymer matrix. On the one hand, it is possible to introduce an interaction between the polymer network and the active ingredient. These interactions can be created physically, such as electrostatic interactions, or chemically by cleavable covalent linkages.25,26 On the other hand, it is possible to physically impede the diffusion of the active agent.27 These different strategies can be implemented in an extensive diversity of hydrogel adjusting mechanisms. The composition of the hydrogel can be varied using diverse monomers and copolymers regarding their functional groups, which either physically interact with the active ingredient or chemically react with reversible linkers. The diffusion of the active ingredient can be fine-tuned via the pore and mesh size of the polymer matrix.28 An example of physical interactions between the polymer matrix and the drug is the hydrogel designed by Bhattacharyya et al. consisting of acrylated guar gum, acrylic acid, and 3-sulfopropyl acrylate potassium salt (AESO3) crosslinked with N,N′-methylenebisacrylamide (MBAA).29 The active agent gentamicin sulfate, a therapeutic agent for mild skin infections, was observed via Fourier-transform infrared spectroscopy (FTIR) within the polymer network, elucidating ionic interaction and hydrogen bonding between these composites. The authors monitored the drug release rates and their dependence on pH and temperature, with decreasing rates observed at higher temperatures. Furthermore, the drug release behaviour was dependent on the pH value, due to the pH responsiveness of the hydrogel, as the swelling degree affects the mesh size and thus also the drug release. In addition to these drug release properties, the mechanical quantities such as high elongation at break (∼90%) and tensile strength (∼35 MPa) were studied. Lau et al. reported a dextran-based drug delivery system, releasing fluorescein-tagged proteins from 10 days to 8 months.28 This could be achieved by (I) controlled distribution of the physically entrapped protein and (II) a dynamic network rearrangement via differently designed crosslinkers to incorporate various cleavage kinetics. Yue et al. documented a potential drug carrier consisting of poly (ethylene glycol) methyl ether methacrylate and acrylic acid.30 The thermo- and pH-sensitive drug delivery system was confirmed by FTIR and thermogravimetric analysis (TGA), as well as characterized by mechanical and swelling properties. Interestingly the Young's modulus of the obtained hydrogels decreased from 488.09 kPa to 461.08 kPa by modifying the pH value from 1.2 to 7.4. This correlation could be related to the weaker H-bonding interactions between the functional groups of the two hydrogel components. While the hydrogels required 10–20 h for equilibrium swelling, a drug release behaviour of 2–4 h of the model drug 5-fluorouracil was observed.
3D printing has significantly impacted the development of innovative drug delivery systems. In particular photopolymerizing 3D printing methods offer superior resolution and the possibility to tune material properties via adjusting the photopolymerizing parameters of the fluid feedstock.31 An interesting example was published by Martinez et al., featuring the photopolymerized hydrogel out of poly(ethylene glycol)diacrylate (PEGDA). The hydrogels, obtained by stereolithography (SLA) with the photoinitiator diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide (TPO), resulted in poor reproducibility in shape.32 These hydrogels, with a swelling ratio from 1.23 to 1.38, were investigated with regard to the drug release of ibuprofen dependent on the water content. As a result, a higher drug release with increased water content could be observed. Under the usage of PEGDA, we previously demonstrated a novel hybrid photopolymerizing 3D printing method combining SLA and inkjet printing for innovative DDSs with locally incorporated drugs for multimodal drug delivery.33 In addition to SLA printing, digital light processing (DLP) is also used for the 3D printing of hydrogels. DLP printers can fabricate samples efficiently and quickly because they convert 2D images into 3D objects. This is done by projecting a 2D image and polymerizing an entire layer at once. SLA or other laser-based printers print each layer line by line.34 Preobrazhenskiy et al. printed a gyroid structure in a cylindrical form from poly(ethylene glycol) methacrylate and PEGDA.35 These printed polymers could be degraded in citric acid by more than 40% mass loss. Larush et al. printed photopolymerizable hydrogels out of acrylic acid and PEGDA.36 The hydrogels, printed with the photoinitiator TPO, were reported with a swelling degree of 2 to 18 and with a drug release period of sulforhodamine B around 10 h. By studying these two characteristics, it was possible to establish a relationship between the swelling rate and release rate. This also demonstrated that a larger hydrogel surface led to quicker swelling and consequently to a faster release of the active ingredient.
We previously reported on a DDS consisting of polymerized electrolytes with the aforementioned crosslinker MBAA.37 Depending on the charge of the active agent used, either the positively charged monomer [2-(methacryloyloxy ethyl]trimethylammonium chloride (MAETMA) or the negatively charged monomer 3-sulfopropylmethacrylate potassium salt (MAESO3) could be utilized for the hydrogel. Through this technique, not only a purely diffusion-controlled release mechanism but also an ion-exchange behaviour could be established. This was examined with the two model drugs ibuprofen and timolol maleate in a sequential release study. These hydrogel DDSs could be very interesting for use in medical applications. In another previous study, we printed PEGDA with different amounts of water via DLP.38 It was possible to print complex and even hollow geometries, such as a prototype of a frontal sinus implant and a frontal neo—ostium implant, respectively. However, these materials lacked flexibility with low water contents and low printing accuracy with high water contents.
In this study, we present novel 3D printable biomaterials with the advantages of our previously published hydrogels such as stimulus responsivity, swellability, drug release behaviour, and flexibility, to combine them with the advantages of complex shapes in 3D printing, resulting in the first printable and synthetic polymer for biomedical application, to the best of our knowledge (Scheme 1).
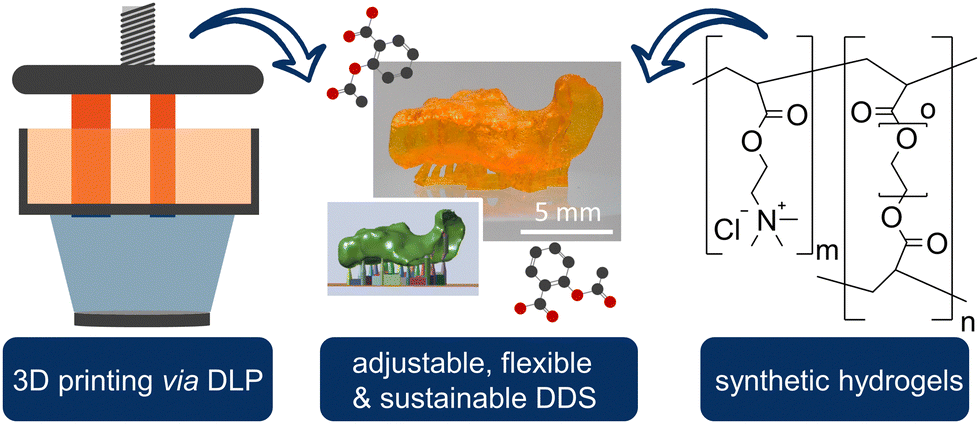 | ||
| Scheme 1 Combining the best of both worlds: the properties of synthetic hydrogels and the possibility of complex structures of 3D printing, obtaining an adjustable, flexible, and sustainable drug delivery system. | ||
In addition, the use of 3D printing reduces the amount of precursor polymer materials required due to the precision fit of the implant. Furthermore, the quantity of active ingredients can be significantly minimized through direct and local delivery, which contributes to the sustainability of the entire product. The obtained materials, consisting of AETMA and PEGDA, could be compressed and positioned to the target location due to their reversible swelling ability and elasticity. Furthermore, the conveniences of 3D printing could be exploited to produce complex structures that cannot be produced with simple hydrogel molds and to provide customization options for any patient specification.
Experimental
Chemicals
[2-(Acryloyloxy) ethyl]trimethylammonium chloride (AETMA; 80 wt%; Sigma-Aldrich; St. Louis, Missouri, USA), [2-(methacryloyloxy) ethyl]trimethylammonium chloride (MAETMA; 75 wt%; Aldrich), poly(ethyleneglycol)diacrylate (PEGDA; ≥99.0%; Mn = 700; Sigma-Aldrich), acetylsalicylic acid (ASA; ≥99.0%; Sigma-Aldrich), Orange G (80%; Sigma-Aldrich), lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP; ≥95%; Sigma-Aldrich), acetylsalicylic acid (ASA; ≥99%; Merck), salicylic acid (SA; ≥99%; Merck) and phosphoric acid (85 wt%; Sigma-Aldrich) were used as received. Solvents (water; acetonitrile) for HPLC analysis were of HPLC grade and used without further purification.3D printing of the hydrogels
The monomer MAETMA and respectively the monomer AETMA and the corresponding amounts of PEGDA, as well as the ultrapure water or ASA stock solution (0.244 g ASA in 100 mL ultrapure water; 13.54 mmol × L−1), were stirred at room temperature until homogenization. In the absence of light, the photoinitiator LAP and the dye Orange G were added and mixed again for 24 hours. The compositions, mentioned in Table 1, were chosen with respect to their miscibility and to create a composition variation as large as possible.| HG 1 (wt%) | HG 2 (wt%) | HG 3 (wt%) | Control (wt%) | |
|---|---|---|---|---|
| Monomer | 60 | 69.8 | 25.6 | — |
| PEGDA | 20 | 7.8 | 34.8 | 60.4 |
| Water | 20 | 22.4 | 39.6 | 39.6 |
For better comparability, a control with the same water content as HG 3 was selected. In addition, the supplemented water serves as a solubilizer for LAP and Orange G with the selected monomer and the PEGDA (Scheme 2). 3D printing was performed using a DLP 3D printer “VIDA” (EnvisionTEC GmbH, Gladbeck, Germany). The 3D printer features a high-resolution projector running at 1920 × 1080 pixels resolution with a wavelength of 405 nm. The build envelope dimensions are 140 mm × 79 mm × 100 mm (width × length × height). In this study, we use 3D printing parameters with a layer thickness of 100 μm and various exposure times of 5 s, 10 s, 15 s, and 30 s.
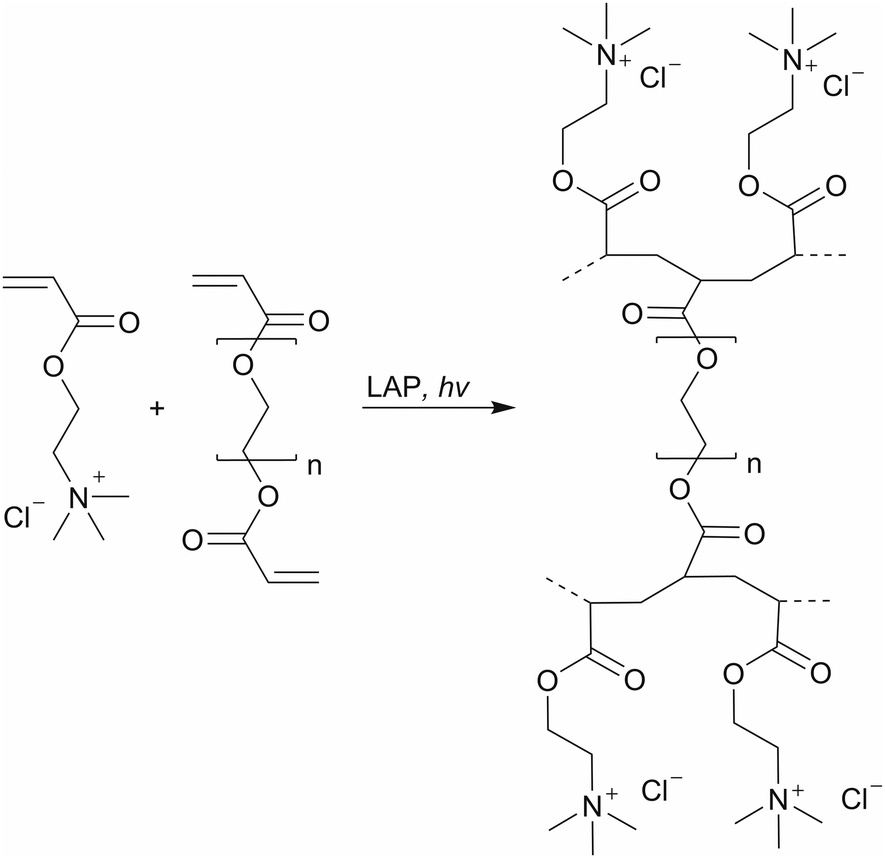 | ||
| Scheme 2 Synthesis of the synthetic polyelectrolyte hydrogels via photopolymerization with AETMA as the monomer and PEGDA as the crosslinker. | ||
The 3D printability of the hydrogels with these various exposure times was investigated by 3D printing of simple geometries of a shouldered test bar of 75 mm in length and a width of 10 mm. The specimen was checked optically for defects, for example delamination of the layers. Moreover, a tubular prototype of a frontal neo—ostium implant with a relatively complex shape, a length of ∼15 mm, and a wall thickness of ∼0.5 mm will be 3D printed exemplarily of HG 1, to test the 3D printing of complex structures (Fig. 1).
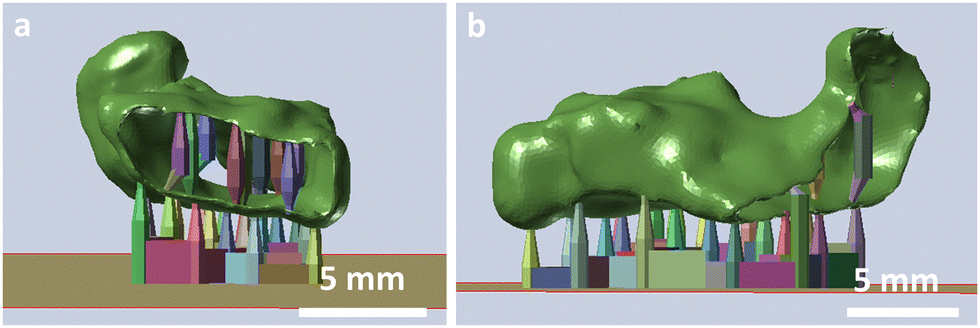 | ||
| Fig. 1 Front (A) and side view (B) of the CAD-model of the frontal neo-ostium implant prototype (green) with a supporting structure (coloured) for DLP 3D printing. | ||
Here, the focus lies on benchmarking our 3D printing process. Nevertheless, such patient-individualized implant prototypes might be helpful to develop novel concepts for the medical treatment of chronic rhinosinusitis via 3D printed implants made of soft, silicone-like materials.39,40 After 3D printing, the prototype will be checked for defects and will be stretched for exemplary handling tests.
Analytical characterization
To investigate the conversion, the hydrogels were washed in ultrapure water for a week and lyophilized. The resulting residue was weighed. IR spectra were recorded on a Nicolet 550 Fourier-transform infrared (FT-IR) spectrometer with an attenuated total reflection (ATR) sampling technique for solids and liquids. The differential scanning calorimetry (DSC) measurements were performed on a Mettler Toledo DSC 823e instrument (Mettler Toledo, Columbus, OH, USA) with a heating rate of 10 °C × min−1 under an argon flow of 100 mL × min−1 (DSC and ATR results see Fig. S1–S3, ESI†).Scanning electronic microscopy (SEM)
SEM was used to analyse the surface morphology of the 3D printed hydrogels. All hydrogel compositions were printed in shape 3 (Table 2), dried for 48 h at 70 °C, and cut in half. The hydrogel pieces were placed on the sample dish in order to use microscopy to look at the base and lateral surfaces of the printed cylinders. Before imaging the samples with the SEM (S3400N, Hitachi, Japan), they were sputter coated with Au/Pd for 45 s (SC7620 Sputter Coater). The images were taken with an acceleration voltage of 20 kV and a working distance of 10 mm, using ImageJ software (National Institutes of Health, Bethesda, MD, USA).| 1 | 2 | 3 | 4 | ||
|---|---|---|---|---|---|
| Diameter | mm | Outer: 15 | 14 | 10 | 6 |
| Inner: 11 | |||||
| Height | mm | 4.8 | 2.6 | 5 | 13.9 |
| Surface Area | mm2 | 556 | 420 | 314 | 319 |
Gravimetric sorption experiments
Solvent uptake kinetics of the 3D printed hydrogels independent of their composition and shape (Table 2) were studied gravimetrically in phosphate-buffered saline (PBS) at a pH of 7.4 and 37 ± 1 °C as a function of time.The weights of the swollen hydrogels were determined at different intervals. When reaching the equilibrium state of the swollen hydrogel, the mass remained constant and the measurement was finished. After determination of the dry mass, the samples were placed in a strainer and dipped in a beaker containing 250 mL of PBS buffer. The strainers with the hydrogels were taken out at different time intervals and the water was carefully removed before weighing and returning to the beaker. The swelling degree (qm,t) of the hydrogels was calculated according to the following equation:
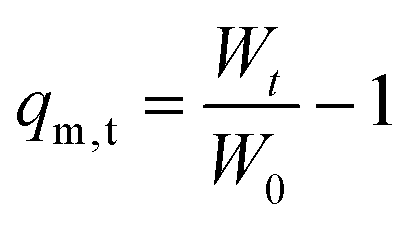 | (1) |
Mechanical characterization
Mechanical properties are investigated via compression and tensile tests. For the compression tests, each hydrogel specimen was characterized in fresh (directly after 3D printing), dried, as well as in equilibrated swollen status using rectangular sample dimensions of 10 mm × 10 mm × 8 mm (± 1 mm). The printed hydrogel samples were dried in a drying oven at 40 °C for 72 h. The hydrogels were printed oversized to compensate for the shrinking while drying. Fresh and dried specimens were tested using a zwickiLine Z5.0 testing machine (ZwickRoell AG, Ulm, Germany) equipped with a 5 kN load cell. The swelling of the dried specimens was carried out in a PBS bath (pH 7.4, ∼0.15 M, Merck KGaA, Darmstadt, Germany) at a temperature of T = 37 °C and a time of t = 48 h. The swollen specimen was cut with a scalpel to the desired sample dimensions and was compression tested using a MCR 702 (Anton Paar Group AG, Graz, Austria) equipped with a 50 N load cell. All specimens were compressed at a speed of 2 mm × min−1. For dried hydrogels, a preload of 2 N was used while a preload of 0.5 N was applied for fresh and swollen hydrogels. Tensile tests were performed for the hydrogel specimen in the fresh and dried state. Shouldered test bars in accordance with DIN EN ISO 527 with sample dimensions of a full length of 140 mm, a gauge length of 50 mm, a width of 10 mm, and a strength of 4 mm were used. The specimens were tested at a speed of 10 mm min−1. A preload of 0.1 N was used for dried samples and a preload of 0.1 N was used for fresh samples. Tensile tests were not performed with swollen hydrogels, because the measurement range of the tensile test device was not suitable.Drug release profiles
The drug-loaded and printed hydrogels were incubated in 10 mL PBS at 37 ± 1 °C under decent shaking. After a defined time t, media changes were performed and samples of the media were measured via HPLC. Each release study was performed with a minimum of three specimens. Sequential drug release studies were performed in different media and with media changes every 60 min. The HPLC analyses were performed on a Knauer HPLC system and the data was processed using Clarity software. Since ASA degrades in a humid environment into SA, the drug release of ASA and SA was investigated. The separation of ASA and SA was performed on a Kinetex C18 column (150 mm × 3 mm, 2.6 μm) at a temperature of 30 °C (for the calibration see Fig. S5, ESI†). The mobile phase consisted of a mixture of water/acetonitrile/phosphoric acid (680 mL/320 mL/2 mL) with a flow rate of 0.3 ml × min−1 and an injection volume of 20 μL. Samples were detected at a wavelength of 237 nm using a Knauer UV detector (2500). Each sample was measured in triplicate. Since SA is the active agent, we calculated the overall mass of the model drug released.Results and discussion
3D printing and characterization of the hydrogel specimens
In this study, we used a DLP 3D printer to produce DDS in the form of an implant using photoreactive polymers. Based on our earlier results on the DDS of hydrogels mentioned above, the monomer MAETMA was considered first. But this monomer was difficult to crosslink with PEGDA. The photopolymerization was slow, which is not desirable for application in DLP printing. This is caused by the additional methyl group, which stabilizes the resulting MAETMA radical. In addition to the positive inductive effect of the methyl group, steric hindrance occurs, which prevents the initiation of the monomer. In particular, the mobility of the radicals is strongly limited by the additional group, which significantly decreases the reaction rate. These effects and the effects of oxygen inhibition of methacrylates were already described in the literature.41,42 Therefore, AETMA was selected as a monomer and the photopolymerization proceeded smoothly and at a sufficient rate. Further parameters for the DLP printing of hydrogels were the exposure times of the individual layers, which were optimized with some test samples. The short exposure time of 5 s (tex1; Fig. 2a) resulted in insufficient curing of the layer over the exposed area and very poor contour sharpness. When the exposure time was increased to 10 s (tex2), the contour sharpness was significantly improved, but the surface and the contour still showed little defects. The result was even better with an exposure time of 15 s (tex1) as there is a smooth surface and sharp-edged contouring. At 30 s (tex4) exposure time, excessive over-curing leads to a loss of contour sharpness. Thus, a slightly too large area was cured, and the edges fray. As a result, an exposure time of 15 s was selected for the following hydrogel specimens for optimal time efficiency and shape reproducibility. These printing parameters were used to print different hydrogel compositions (Fig. 2b and Table 1). | ||
| Fig. 2 Overview of the 3D printed hydrogel samples. (a) Comparison of the different exposure times for the optimization of the contour sharpness (5 s; 10 s; 15 s; 30 s), (b) composition, and (c) surface areas. | ||
A conversion of ∼98% of the monomer was observed. The remaining monomers were eluted in water over several days and weighed after lyophilizing the aqueous solution. In addition, we printed a control specimen consisting entirely of PEGDA and water, to compare the influence of AETMA on the swelling behaviour and drug release. Furthermore, different shapes and surface areas of the hydrogel composition HG 1 were printed in order to observe and demonstrate the influence of the surface areas on swelling and active ingredient release (Table 2). These shapes could easily be printed with good shape reproducibility (Fig. 2c).
With these compositions, the printing of tube-like structures with complex geometry was possible. We successfully printed an exemplary prototype of a frontal neo-ostium implant with a wall thickness of ∼0.5 mm without any defects (Fig. 3). This indicated an adequate proportion of the photoinitiator (LAP) and light absorbing agent (Orange G) for an appropriate light penetration depth at DLP 3D printing. There was no notable delamination of the layers. The contour is characterized by the single layers as it is common for 3D printing (staircase effect). Even small details of the CAD model were 3D printed successfully. The prototype showed high stretchability and elasticity, which can be beneficial for the minimally invasive implantation process of many implant applications, such as a patient individualized frontal neo-ostium implant (Fig. 3d–f). Such an implant is promising for treating chronic rhinosinusitis as described by Gao et al.40 At this point, concrete mechanical needs, such as the required elongation at the break of such an implant, are not known. Nevertheless, a soft and deformable material behaviour is helpful to ensure smooth insertion and the prevention of traumatizing sensitive anatomical structures.39
 | ||
| Fig. 3 Front (a and b) and side view (c) CAD-model of the frontal neo-ostium implant prototype with a supporting structure, exemplarily printed with HG 1. It was 3D printed successfully without defects, indicating an adequate proportion of the photoinitiator (LAP) and the light absorbing agent (Orange G) for appropriate light penetration depth for the DLP 3D printing. There is no notable delamination of the layers. The contour is characterized by the single layers, as is common for 3D printing (staircase effect). Even small details of the CAD model are 3D printed successfully (white circle). The prototype shows high stretchability and elasticity, which can be beneficial for the minimally invasive implantation process of many implant applications (d–f). | ||
As shown in Fig. 4, the surface morphology of the dried hydrogels correlates strongly with their properties, which are investigated in the following chapters. The lateral surface of the printed cylinders (Fig. 4a) was especially interesting because we could observe the horizontal layers formed during the printing process. These are mainly found in the hydrogel HG 3 and the control. In addition, the layer height of 100 μm in the 3D printing process can be detected via smooth vertical lines. A peculiarity of the control was the slightly slanted area, where the vertical lines were interrupted. These may indicate small defects respectively small delamination, which may have occurred during the printing process or the drying process. HG 1 and HG 2 showed small pores. Because of the higher amount of the short-chained PEGDA, HG 3 and the control from a harder and consequently more homogeneous surface. The bases (Fig. 4b) of all of the hydrogels showed an even more inhomogeneous surface than the lateral surfaces. This is a consequence of the 3D printing process itself, as the samples stick with this side to the building platform and need to be detached via scalpel because of relatively high platform adhesion, especially in the case of HG 1 and HG 2. Moreover, the drying process leads to a shrivelling of the surfaces, especially of HG 1 and HG 2 as these hydrogels are softer than HG 3 and the control, because of the long polymer strands of the AETMA backbone. As a result, the contour sharpness in the microscopic scale is best for HG 3 and the control as their surfaces are smoother than the surfaces of HG 1 and HG 2 (extended SEM photographs in Fig. S4, ESI†).
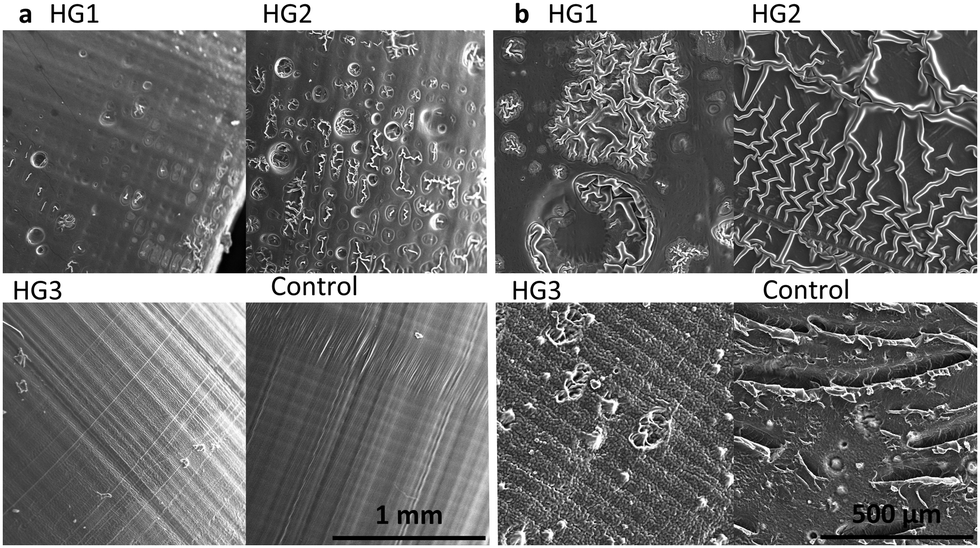 | ||
| Fig. 4 SEM photographs of (a) the lateral surface and (b) the base of the printed cylinders, indicating a higher contour sharpness in the microscopic scale for HG 3 and the control than for HG 1 and HG 2. | ||
Gravimetric sorption experiments
The swelling degree is a crucial characteristic of drug delivery systems. Herein, the sorption experiments of the dried hydrogels were performed in PBS buffer at 37 °C. As shown in Fig. 5a, the trends of the gravimetric swelling experiments are dependent on the composition of the gel. The hydrogels HG 1 and HG 2 swell much more than HG 3 and the control, due to a lower amount of the crosslinker PEGDA (20 wt% and 7.8 wt%) and a high ratio of the AETMA monomer (60 wt% and 69.8 wt%). This leads to a lower crosslinking density and therefore greater water absorption. The statement can be confirmed by the fact that our control sample, printed without a monomer and with a high amount of crosslinker (60.4 wt%), showed a much lower degree of swelling (1.0 ± 0.13). This trend can be explained by the crosslinking density which takes the number average molecular weight between crosslinks into account. Hereby, a theoretical exponential fit between the molar crosslinking ratio and the molecular weight between the crosslinks is typical.43 The swelling degree is 1.7 ± 0.16 even though the amount of PEGDA is reduced to half compared to the control sample. The 3D printed hydrogels published by Kadry et al., consisting of PEGDA 400 and polyethylene glycol dimethacrylate (PEGDMA 1000), had swelling degrees around 1. This low swelling degree was mainly caused by the high crosslinker amount, which is comparable to HG 3 and thecontrol.34 Comparable effects were reported by Preobazhenski et al. who used different concentrations of polyethylene glycol methacrylate (PEGMA) and PEGDA 350 as well as 575 to modify the swelling properties of the polymers.35 The printed different shapes of the hydrogels should have approximately the same volume, but a different surface area. The swelling of the hydrogels in equilibrium, therefore, differs only marginally, but the speed and thus also the increase in the swelling curves depends on the surface (Fig. 5b). The higher the surface area, the faster the swelling, the steeper the slope, and the higher the resulting swelling rate. Another interesting property of hydrogels is their stimulus-responsive behavior.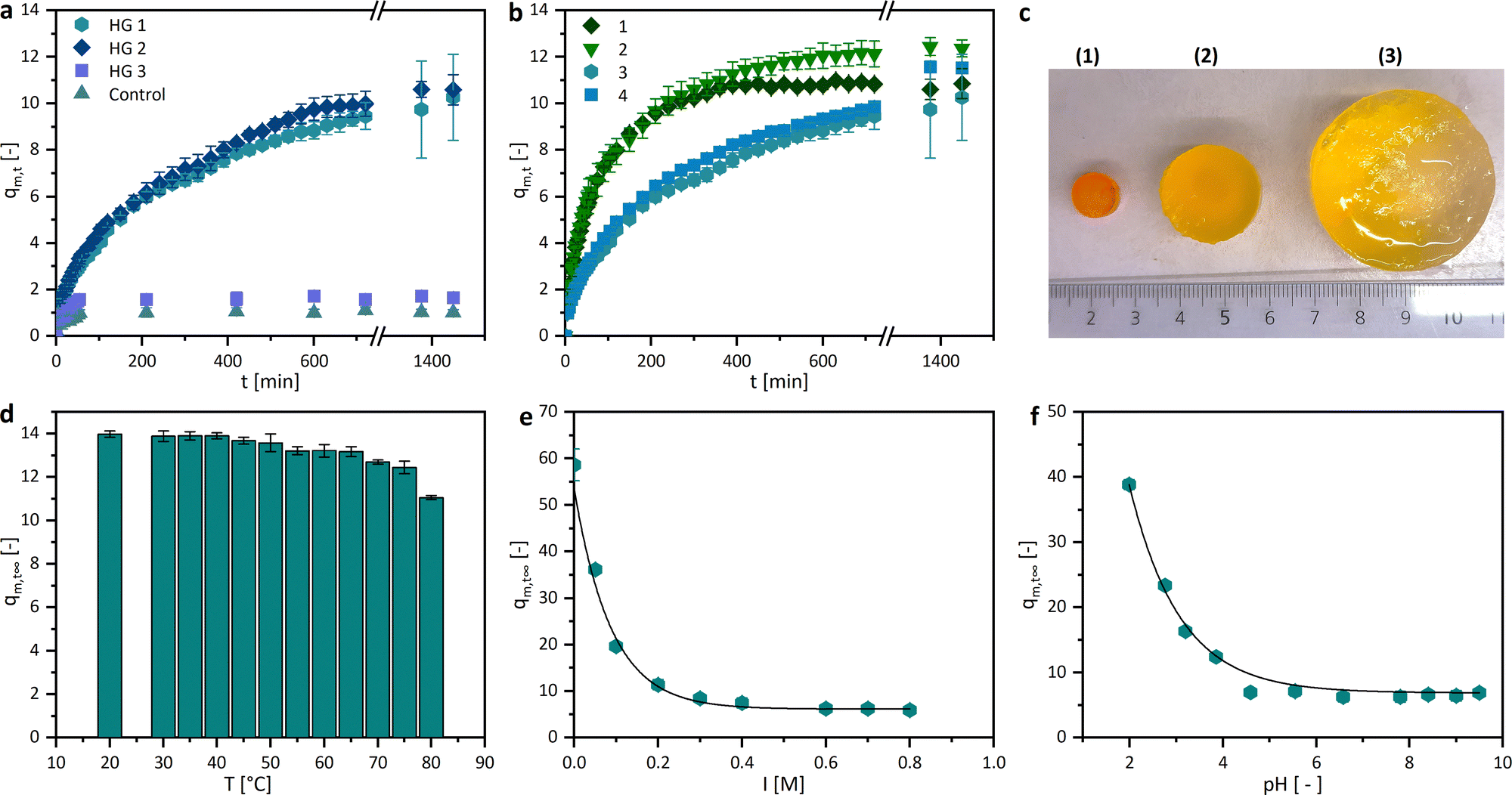 | ||
| Fig. 5 Overview of the sorption experiments. (a) Gravimetric swelling kinetics of the different compositions of polyAETMA hydrogels (PBS; pH 7.4; 37 °C; n = 3), (b) gravimetric swelling kinetics of the HG 1 composition with different surface values (PBS; pH 7.4; 37 °C; n = 3), (c) comparison of the (1) dried and swollen HG 1 in an aqueous KCl solution with (2) I = 0.6 M and (3) I = 0.0 M, (d) q∞m,t of HG 1 as a function of T (PBS; pH 7.4; n = 3), (e) q∞m,t of HG 1 as a function of the ionic strength (37 °C; n = 3; ultrapure Water) and (f) q∞m,t of HG 1 as a function of the pH-value (37 °C; n = 3), showing a stimulus-responsive swelling behaviour regarding the pH-value and the ionic strength I. | ||
We investigated their swelling behavior in relation to the temperature, the pH value, and the ionic strength of the surrounding media. The temperature dependency, illustrated with the equilibrium swelling degrees in Fig. 5d, shows a slightly decreasing tendency. However, this could be due to the very rapid swelling of the hydrogels at high temperatures, rather than a responsive behavior. If diffusion is so much higher than the chain relaxation, this not only indicates a “Case II transport”, it can also lead to macroscopic fractures in the hydrogel.44,45 This can also cause the mass loss shown above due to the breaking off of small polymer parts, which could be observed at temperatures higher than 70 °C. The pH dependence, in turn, is clearer and more recognizable, the more basic the surrounding milieu of the hydrogel, the smaller the swelling degree (Fig. 5e). This may be due to the different functional groups in the side chains of the hydrogel. Quaternary ammonium compounds can form stable quaternary ammonium hydroxides in the neutral and alkaline milieu. This stronger interaction between the hydroxide ions and the quaternary ammonium ions could lead to a more shielded charge of the side chains. The electrostatic repulsion between the side chains gets smaller and the swelling degree decreases. However, in a more neutral environment, the swelling degree approaches an equilibrium of 6.2 ± 0.7. Since the ionic strength strongly affects the swelling behaviour of hydrogels containing dissociative or other charged functional groups, the equilibrium swelling degree dependency of the hydrogels was investigated and showed, as expected, an increase in medium at a lower KCl concentration (Fig. 5c and f).
Mechanical characterization
Unconfined uniaxial compression and tensile tests were performed to quantify the mechanical behaviour of the 3D printed hydrogel samples on the stress–strain relationship. The compression tests were performed with fresh, dried, and swollen specimens of HG 1, HG 2, and HG 3. No results of the mechanical characterization of the control hydrogel are shown because the specimen was damaged (cracks and fractures) during both the drying and swelling process and was therefore discarded. We observed significant differences in the compression and fracture behaviour between the tested hydrogels. In the dried state, HG 1 and HG 2 showed relatively stiff material behaviour until yielding starts at a compressive strain between 10% and 15%. HG 1 and HG 2 showed typical yielding in their stress–strain curves, whereas HG 3 indicated no evidence for the yielding of the material (Fig. 6a). The intensive tangling of the AETMA polymer strands leads to the strengthening and stiffening of the polymer network. However, the applied mechanical stress causes the entanglement of the polymer strands, resulting in the typical yielding of the material. HG 2 showed the highest compressive stress at yield (from ∼20 MPa to ∼25 MPa) and the highest compressive strength (from ∼40 MPa to a maximum of ∼60 MPa). HG 2 was the hydrogel with the highest amount of the monomer AETMA and the lowest amount of PEGDA (Table 1). All hydrogels showed similar values for compressive strain at a compressive strength with values of about ∼60% to ∼70%. HG 1 and HG 2 show yielding, which was not the case for HG 3. The fracture behaviour of HG 3 was more brittle compared to the fracture behaviour of HG 1 or HG 2. This was indicated by the multiple abrupt breakdowns of the curve, due to macroscopic splintering and breakage of the specimen. The specimens of HG 3 splintered in multiple parts (photo series of HG 3 in Fig. 6a), while the specimens of HG 1 and HG 2 showed a ductile fracture behaviour, indicated by yielding. The specimens exhibited single ruptures but were mostly intact (photo series of HG 1 in Fig. 6a). The reason is also the described differences in strand lengths in the polymeric network. In comparison to the dried state, the fresh and the swollen hydrogels showed different behaviour in the compression test (Fig. 6b and c). The compressive strength decreases significantly for all hydrogels, the more water they hold. Especially for HG 1 and HG 2, the dominant drop of compressive strength demonstrated softer hydrogels. In contrast to the dried state, the curves no longer showed any yielding, indicated by the moderate slope of the compression curves. Additionally, in contrast to the dried state, at fresh and swollen state HG 3 was the hydrogel with the most mechanical strength and stiffness. This was a result of the diverse swelling behaviour. Due to the relatively long polymer strands of the AETMA backbone, the hydrogels HG 1 and HG 2 showed very intensive swelling, so the packing density and entanglement of the polymeric network decreased significantly, as described by Richbourg et al.46 As a result, the hydrogels become soft and easily compressible. The slopes of the hydrogels were comparable to the results of previous investigations by Claus et al.37 In this former study, the hydrogels were not polymerized via photopolymerizing 3D printing but via radical polymerization, using the often—described redox initiating system, ammonium peroxodisulfate, and tetramethylethylenediamine.47 This was different for HG 3. Because of a higher ratio of shorter PEGDA polymer strands (Mn = 700 Da), the polymer network was relatively short-chained and the polymer network was fully tensed at relatively low swelling ratios. As a result, HG 3 was not capable of high strain in both the fresh and the swollen states. Similar to the dried state of HG 3, there was a brittle-like fracture behaviour, indicated by the abrupt decrease and increase of the curves. At the breakage point, HG 3 intensively ruptured into multiple fragments (Fig. 6b and c, photo series of HG 3), which was not the case for HG 1 and HG 2, as these exhibited high strain capacity as the polymer network was entangled but not fully tensed from swelling. In the fresh state, with a moderate amount of water included, the specimens of HG 1 and HG 2 showed single ruptures but were mostly intact, what was shown exemplarily for HG 1 in the photo series of Fig. 6b. In the swollen state, the specimen burst at the end of the test, because the polymeric network is tensed because of the high swelling (Fig. 6c, photo series of HG 1). Fig. 6d and e showed the tensile stress–strain curves for HG 1 in a dried and fresh state. The mechanical behaviour of the hydrogels in the tensile test was analogous to the results of the compression test. The dried samples of HG 1 and HG 2 were relatively stiff and the material demonstrated yielding from the point of a relatively low strain of ∼10%. The curves continued without an increase in tension stress, because of detangling of the polymer network, respectively the detangling of the single polymer strands, similar to findings shown before by Chen et al. for other PEG-based hydrogels with different lengths of polymeric chains in the network.48 When there is a moderate amount of water included in the polymer network, as in the case of the samples in the fresh state after 3D printing, HG 1 and HG 2 displayed different stress–strain behaviour than in the dried state. The material did not show any yielding and was significantly softer, as it had a visible drop in stiffness and tensile strength but a higher maximum strain. Our materials show a maximum of elongation at break of about 300% in the fresh 3D printed state. This is respectful in comparison to many other hydrogels, as shown by Zhang et al. Nevertheless, the state of the art holds 3D printed hydrogels with values for elongation at break of more than 1000%.49 Because of the short polymer strands of PEGDA the HG 3 hydrogels showed relatively high stiffness and low strain both in the dried and fresh state. Tensile strength was quite low because of the brittle material behaviour as it is common for PEGDA with fairly low Mn as already shown by Rekowska et al. and Zhang et al.49,50 Generally, the soft mechanical behaviour and the relatively high stretchability of our hydrogels might be promising for many implant applications, especially when there is no need for intensive load-bearing capacity. e.g. the key role of the described frontal neo-ostium implant might be drug release for the reduction of the risk of inflammation and restenosis to preserve frontal sinus drainage. Moreover, a soft and stretchable mechanical behaviour benefits minimal invasive and smooth insertion of the implant.39 Another example of a novel concept of an implant, which could profit from our hydrogels might be a drug-releasing implant for the medical treatment of the inner ear.51 Following this concept, a patient-individualized implant (dimensions of about 3 × 2 × 1 mm) will be placed into the round window niche in the middle ear and enable a long-term diffusion of the drug through the round window membrane in the inner ear.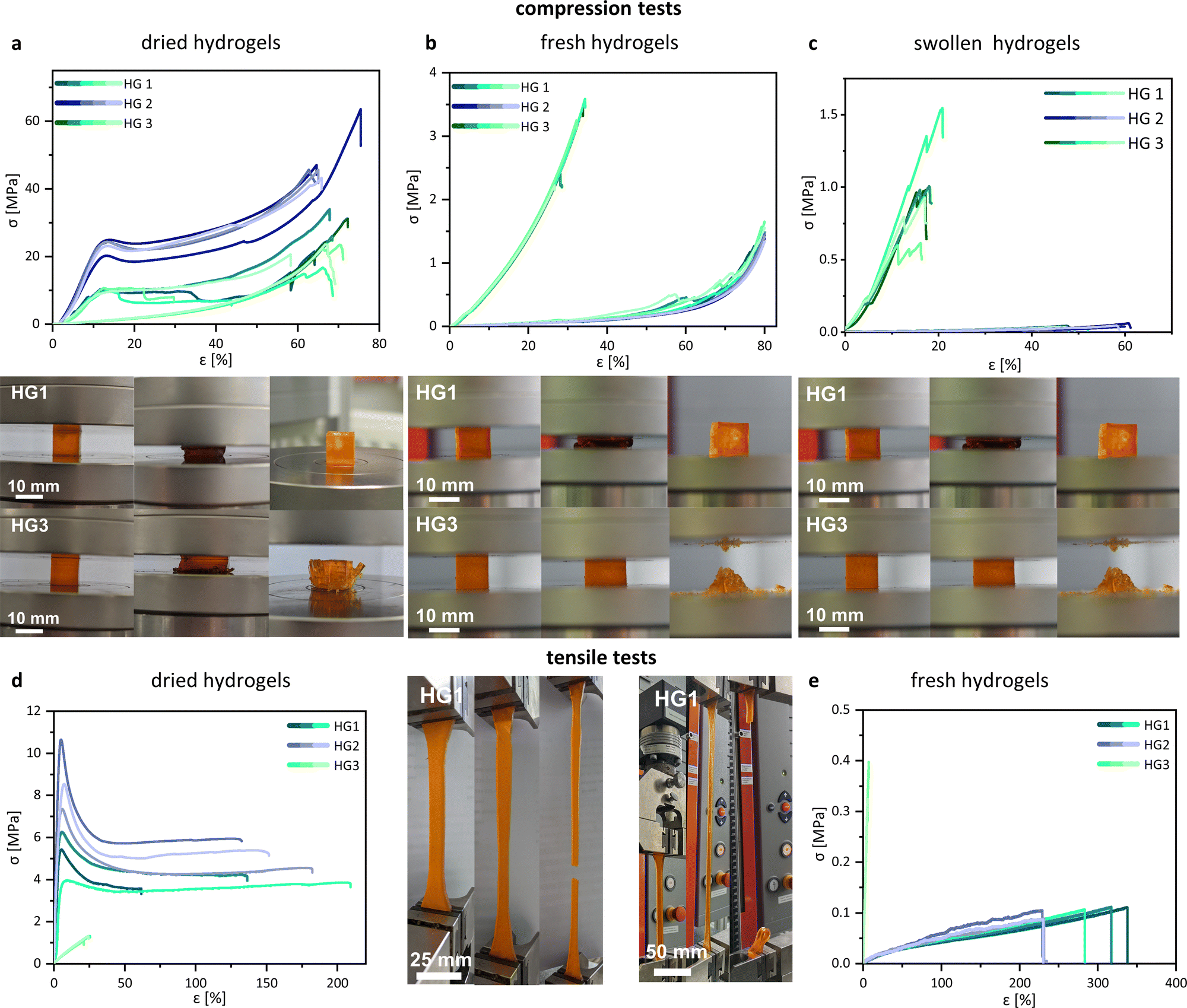 | ||
| Fig. 6 Mechanical properties of 3D printed hydrogels. Compression stress–strain curves of the (a) dried, (b) fresh and (c) swollen (in PBS) hydrogels with exemplary photos of HG 1 and HG 3, as well as tensile stress–strain curves of the (d) dried and (e) fresh hydrogels with exemplary photos of HG 1 (20 °C; n ≤ 3). HG 1 and HG 2 showed relatively high strains, coming with yielding at the dried state. Fracture behaviour for HG 1 and HG 2 was mostly ductile. The greater the amount of water included, the stronger the softening of the materials. These effects resulted due to the dominance of long-chained AETMA backbone in the polymeric network. HG 3 did not show yielding and relatively low strains. The fracture behaviour for HG 3 was brittle in the dried, fresh and swollen state, due to the dominance of short-chained PEGDA (Mn = 700 Da). | ||
We reported in a previous study about the preferable mechanical properties of such implants.52 A soft and stretchable material (tensile strength ∼4.5 MPa, elongation at break ∼60%) was much more suitable for the implant insertion than stiff and brittle materials (tensile strength ∼25 MPa, elongation at break ∼9%). Stiff materials made it challenging to handle the relatively small implant prototypes by forceps because they slipped off the forceps. In contrast, the tested soft material was easy to grab. Moreover, the use of soft materials minimizes the risk of tissue trauma during insertion.
The mechanical properties of the investigated hydrogels strongly depend on the material composition as well as on the content of water. This might be advantageous for the tuning of the mechanical properties for specific needs. The swollen state might be most relevant for the intended use as implant materials because the materials may swell at the location of implantation. Consequently, the investigated hydrogels are strongly limited for implant applications, which require an intensive load-bearing capacity.
Drug release profiles
By investigating the drug release profiles of the printed hydrogels, we determined the influence of the polymer composition (the monomer and crosslinker amount), the sample design by association with the surface area, and the media change interval. The drug release profiles of ASA from the different hydrogel compositions, shown in Fig. 7a, were relatively similar in terms of growth rate. However, different total percentages of the overall drug were released. An influence of the different hydrogel surfaces was shown in Fig. 7b. It was found that growth rates in the release studies increased with the surface area. This is primarily attributed to the increased swelling rate with enlarged surface area, whereby the polymer network becomes wider and the active ingredient can be released more quickly. In addition, the percentage of drug mass released increases from 63.4 ± 0.2% to 97.3 ± 0.1%. For all further experiments, the printed hydrogel samples HG 1 with surface area 3 were chosen. The release study in Fig. 7e showed the ASA release with a media change every 24 h. In this case, the release of the active agent is considerably prolonged from approximately 10 h to one week. This could be of interest to drug delivery systems in locations with a low fluid exchange. To have a better understanding of the mechanism of drug release from the printed hydrogels, drug release experiments were performed with medium changes at different time intervals. However, it is noticeable that the release profiles are very similar when plotted against time (Fig. 7d), while they differ as a function of the media change (Fig. 7c), which indicates at first a release mechanism independent of the interval of media change. However, in these studies, the ionic strength I was kept constant at 0.6 M. Nevertheless when the results from the pH and I dependencies were considered, it can be concluded that the salinity of the surrounding medium of the hydrogel influenced the release. Consequently, it can be assumed that the release was mainly ion-exchange controlled by different I, rather than diffusion-controlled. Based on these results, we extended the drug release studies with sequential release profiles (Fig. 8). Thus, we first incubated the hydrogel samples in ultrapure water (I = 0.0 M) for six media changes and switched subsequently to a KCl solution at I = 0.6 M (Fig. 8a). The samples were then treated twice with salt solutions. If the hydrogel was first treated with ultrapure water (I = 0.0 M), only a little ASA was released very slowly.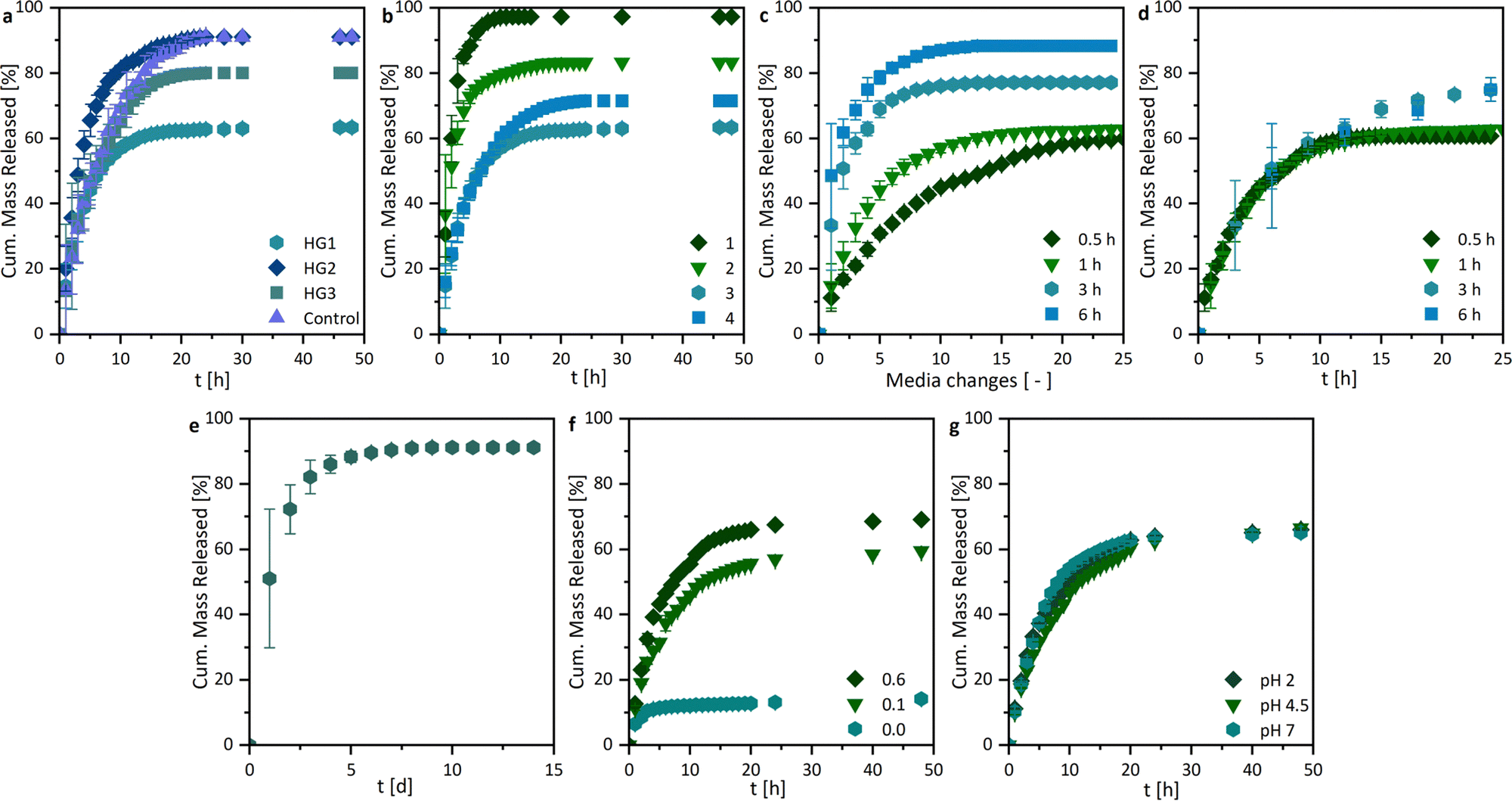 | ||
| Fig. 7 Drug release profiles of 3D printed hydrogels: (a) samples with different compositions and (b) samples with different surface areas (PBS; pH 7.4; 37 °C; n = 3). Drug release profiles showing the (c) cumulative mass released in terms of the dependency on the media change interval and (d) in terms of dependency on the time, as well as (e) the drug release profiles dependent on the media change interval of 24 h showing the cumulative mass released (HG 1; shape 3; PBS; pH 7.4; 37 °C; n = 3), the dependency on (f) the ionic strength I and (g) the pH-value (HG 1; shape 3; 37 °C; n = 3). | ||
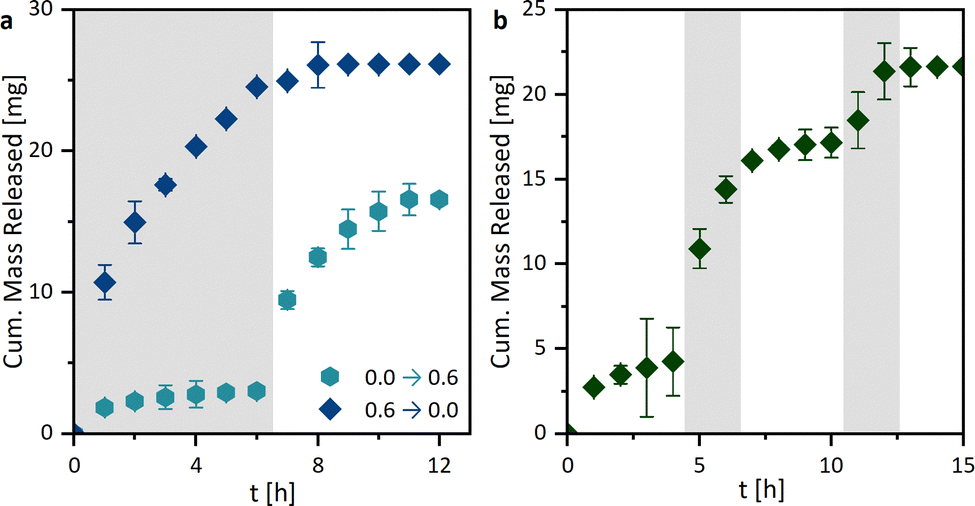 | ||
| Fig. 8 Triggered drug release profiles of 3D printed hydrogels: (a) One media change after 6 h from I = 0.0 M to 0.6 M and vice versa and (b) switch back and forth between I = 0.0 M (white) and I = 0.6 M (grey) (KCl solution in ultrapure water; 37 °C; n = 3). | ||
If after 6 h the surrounding salt solution has changed to I = 0.6 M, ASA is released abruptly. On the one hand, this is due to the ion-exchange behavior shown earlier (Fig. 8g), but also to the rapidly changing swelling degree of the hydrogel from 58.6 ± 3.4 to 6.3 ± 0.8. Vice versa, a lot of ASA was released at the beginning of the drug release profile, which was stopped by the change to ultrapure water.
An additional experiment was performed to test the switchability of the drug delivery system. For this purpose, the drug-loaded hydrogel samples were first incubated in ultrapure water for four media changes and then switched to the KCl solution (I = 0.6 M) for two media changes. This was followed by a change back to ultrapure water (Fig. 8b). Just as in the previous triggered experiments, an immediate release could be induced by changing to the KCl solution. This could also be repeated several times. In addition to the ion-exchange mechanism, a diffusion-controlled release was also a possibility. When the surrounding medium triggers the shrinkage of the hydrogel, the distance that the ASA molecules have to diffuse out of the hydrogel decreases. Additionally, the ASA molecules could be extruded, during the shrinkage. If this was the case, similar release profiles should be observed in the pH-dependent release systems (Fig. 7h). Since, however, this was not the case, we can assume that mainly an ion-exchange behavior is observed.
Conclusions
Previous work by our group has already described synthetic hydrogels, which are easy and cost-effective to synthesize and showed interesting properties such as antibacterial properties, mechanical and swelling tunability, and biocompatibility. Especially the biocompatibility of the eluate and direct contact showed the great cell viability of the AETMA hydrogels towards L929 mouse fibroblasts.13,53 Ion exchange behaviour has already been observed in drug release experiments.37 However, the synthesis of complex hydrogel shapes, beyond simple cylindrical structures, is difficult. To achieve this, additive manufacturing was used in this study. It was possible to print hydrogels into complex, hollow, and fine structures using DLP. The obtained hydrogels had a significant variation in their swelling properties, which we found to vary greatly depending on their composition.We found hydrogels with respectful ductility and stretchability. The maximum elongation at break was about 300% fresh after 3D printing. The release studies of the hydrogels could be investigated depending on a wide variety of factors. Release durations of a few hours up to one week could be observed. To our delight, we were able to determine a strongly pronounced ion exchange behaviour in a sequential release study, enabling a stimulus-responsive drug release.
However, this 3D printed DDS is not without limitations. Regardless of which method was used to load the hydrogels with the active agent, whether the drug was printed in water or swollen in an aqueous drug solution, the drug loading is restricted by the solubility of the used active agent in water. Additionally, the stability of the active agent under UV light is important, if the former mentioned method of hydrogel loading is chosen. Thus, the drug used for this system should be carefully chosen.34 Further investigations of these polymers will aim to characterize and optimize their biocompatibility and antimicrobial behaviour. Beyond medical applications, these 3D printable hydrogels could also be suitable as enzyme carriers in biocatalysis, as they provide a suitable aqueous microenvironment for the enzyme and the surface is designable via 3D printing.19,54 Furthermore, these hydrogels could be useful as ion exchangers or absorbers for downstream processing, wastewater treatment, or chromatography.55–57
Author contributions
Sonja Vaupel: methodology, formal analysis, and writing – review and editing. Robert Mau: conceptualization, methodology, formal analysis, and writing – original draft. Selin Kara: funding acquisition and writing – review and editing. Udo Kragl: funding acquisition and writing – review and editing. Hermann Seitz: funding acquisition, writing – review and editing. Johanna Meyer: conceptualization, methodology, formal analysis, writing – original draft, writing – review and editing, visualization, and supervision. All authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SV, RM, SH, UK, and JM thank the Bundesministerium für Bildung und Forschung (BMBF, Federal Ministry of Education and Research) within “Zwanzig20” (Response), grant number: 03ZZ0933L. SK and JM thank the Ministry for Science and Culture for Lower Saxony for the Holen & Halten starting grant (grant no 12.5-76251-17-9/20). We want to thank Sandra Diederich, Tom Kunde, Martina Weiß, Caroline Mueller, and Laura Schmitz, for their support in the daily laboratory routine and their help with the HPLC analytics. Furthermore, we thank Sven Barker for his help with the SEM images and David Knebel for his skillful assistance with the mechanical characterization. And most of all, we would like to thank the people who worked in systemically important jobs throughout all the lockdowns and got us through the last years of the pandemic.Notes and references
- G. Palmara, F. Frascella, I. Roppolo, A. Chiappone and A. Chiadò, Biosens. Bioelectron., 2021, 175, 112849, DOI:10.1016/j.bios.2020.112849.
- L. Piles, M. J. Reig, V. J. Segui, R. Pla, F. Martines and J. M. Segui, Procedia Manuf., 2019, 41, 739–746, DOI:10.1016/j.promfg.2019.09.065.
- P. Aseni, T. Santaniello, F. Rizzetto, L. Gentili, F. Pezzotta, F. Cavaliere, M. Vertemati and P. Milani, Diagnostics, 2021, 11, 1734, DOI:10.3390/diagnostics11091734.
- A. Ganguli, G. J. Pagan-Diaz, L. Grant, C. Cvetkovic, M. Bramlet, J. Vozenilek, T. Kesavadas and R. Bashir, Biomed. Microdevices, 2018, 20, 65, DOI:10.1007/s10544-018-0301-9.
- F. Ghorbani, D. Li, S. Ni, Y. Zhou and B. Yu, Mater. Today Commun., 2020, 22, 2352–4928, DOI:10.1016/j.mtcomm.2020.100979.
- Y. Hu, J. Wang, X. Li, X. Hu, W. Zhou, X. Dong, C. Wang, Z. Yang and B. P. Binks, J. Colloid Interface Sci., 2019, 545, 104–115, DOI:10.1016/j.jcis.2019.03.024.
- I. Matai, G. Kaur, A. Seyedsalehi, A. McClinton and C. T. Laurencin, Biomaterials, 2020, 226, 119536, DOI:10.1016/j.biomaterials.2019.119536.
- A. Manmadhachary, A. Aditya Mohan and M. Haranadha Reddy, Bioprinting, 2021, 21, e00118, DOI:10.1016/j.bprint.2020.e00118.
- V. M. Vaz and L. Kumar, AAPS PharmSciTech, 2021, 22, 2–20, DOI:10.1208/s12249-020-01905-8.
- J. Claus, F. O. Sommer and U. Kragl, Solid State Ion, 2018, 314, 119–128, DOI:10.1016/j.ssi.2017.11.012.
- A. Jastram, J. Claus, P. A. Janmey and U. Kragl, Polym. Test., 2021, 93, 106943, DOI:10.1016/j.polymertesting.2020.106943.
- J. Claus, A. Jastram, E. Piktel, R. Bucki, P. A. Janmey and U. Kragl, J. Appl. Polym. Sci., 2021, 138, e50222, DOI:10.1002/app.50222.
- J. Claus, A. Brietzke, C. Lehnert, S. Oschatz, N. Grabow and U. Kragl, PLoS One, 2020, 15, e0231421, DOI:10.1371/journal.pone.0231421.
- E. S. Dragan, Chem. Eng. J., 2014, 243, 572–590, DOI:10.1016/j.cej.2014.01.065.
- E. M. Ahmed, J. Adv. Res., 2015, 6, 105–121, DOI:10.1016/j.jare.2013.07.006.
- Y. Meng, J. Lu, Y. Cheng, Q. Li and H. Wang, Int. J. Biol. Macromol., 2019, 135, 1006–1019, DOI:10.1016/j.ijbiomac.2019.05.198.
- M. A. Haq, Y. Su and D. Wang, Mater. Sci. Eng., C, 2017, 70, 842–855, DOI:10.1016/j.msec.2016.09.081.
- W. A. Laftah, S. Hashim and A. N. Ibrahim, Polym. Plast Technol. Eng., 2011, 50, 1475–1486, DOI:10.1080/03602559.2011.593082.
- J. Meyer, L.-E. Meyer and S. Kara, Eng. Life Sci., 2022, 22, 165–177, DOI:10.1002/elsc.202100087.
- S. van Vlierberghe, P. Dubruel and E. Schacht, Biomacromolecules, 2011, 12, 1387–1408, DOI:10.1021/bm200083n.
- E. Caló and V. v Khutoryanskiy, Eur. Polym. J., 2015, 65, 252–267, DOI:10.1016/j.eurpolymj.2014.11.024.
- R. Dimatteo, N. J. Darling and T. Segura, Adv. Drug Deliv. Rev., 2018, 127, 167–184, DOI:10.1016/j.addr.2018.03.007.
- D. Rosenblum, N. Joshi, W. Tao, J. M. Karp and D. Peer, Nat. Commun., 2018, 9, 1410, DOI:10.1038/s41467-018-03705-y.
- E. Waleka, M. Mackiewicz, J. Romanski, A. Dybko, Z. Stojek and M. Karbarz, Int. J. Pharm., 2020, 579, 119188, DOI:10.1016/j.ijpharm.2020.119188.
- X. Xu, A. K. Jha, R. L. Duncan and X. Jia, Acta Biomater., 2011, 7, 3050–3059, DOI:10.1016/j.actbio.2011.04.018.
- N. Hammer, F. P. Brandl, S. Kirchhof and A. M. Goepferich, J. Controlled Release, 2014, 183, 67–76, DOI:10.1016/j.jconrel.2014.03.031.
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 1–17, DOI:10.1038/natrevmats.2016.71.
- C. M. L. Lau, G. Jahanmir, Y. Yu and Y. Chau, J. Controlled Release, 2021, 335, 75–85, DOI:10.1016/j.jconrel.2021.05.006.
- R. Bhattacharyya and P. Chowdhury, J. Polym. Res., 2021, 28, 286, DOI:10.1007/s10965-021-02633-8.
- Z. Yue, Y. J. Che, Z. Jin, S. Wang, Q. Ma, Q. Zhang, Y. Tan and F. Meng, J. Biomater. Sci., Polym. Ed., 2019, 30, 1375–1398, DOI:10.1080/09205063.2019.1634859.
- X. Xu, A. Awad, P. Robles-Martinez, S. Gaisford, A. Goyanes and A. W. Basit, J. Controlled Release, 2021, 329, 743–757, DOI:10.1016/j.jconrel.2020.10.008.
- P. R. Martinez, A. Goyanes, A. W. Basit and S. Gaisford, Int. J. Pharm., 2017, 532, 313–317, DOI:10.1016/j.ijpharm.2017.09.003.
- J. Konasch, A. Riess, R. Mau, M. Teske, N. Rekowska, T. Eickner, N. Grabow and H. Seitz, Pharmaceutics, 2019, 11, 1–14, DOI:10.3390/pharmaceutics11120661.
- H. Kadry, S. Wadnap, C. Xu and F. Ahsan, Eur. J. Pharm. Sci., 2019, 135, 60–67, DOI:10.1016/j.ejps.2019.05.008.
- I. I. Preobrazhenskiy, A. A. Tikhonov, P. V. Evdokimov, A. V. Shibaev and V. I. Putlyaev, Open Ceramics, 2021, 6, 100115, DOI:10.1016/j.oceram.2021.100115.
- L. Larush, I. Kaner, A. Fluksman, A. Tamsut, A. A. Pawar, P. Lesnovski, O. Benny and S. Magdassi, 3D Print. Med., 2017, 1, 219–229, DOI:10.2217/3dp-2017-0009.
- J. Claus, T. Eickner, N. Grabow, U. Kragl and S. Oschatz, Macromol. Biosci., 2020, 20, 2000152, DOI:10.1002/mabi.202000152.
- R. Mau, J. Nazir, S. John and H. Seitz, Curr. Dir. Biomed. Eng., 2019, 5, 249–252, DOI:10.1515/cdbme-2019-0063.
- Z. Gao, F. Matin, C. Wei, T. Lenarz, C. Weber, S. John and V. Scheper, Curr. Dir. Biomed. Eng., 2021, 7, 407–410, DOI:10.1515/cdbme-2021-2103.
- Z. Gao, F. Matin, C. Weber, S. John, T. Lenarz and V. Scheper, Life, 2020, 10, 1–10, DOI:10.3390/life10120353.
- A. M. van Herk, Macromol. Theory Simul., 2000, 9, 433–441, DOI:10.1002/1521-3919(20001101)9:8 < 433::AID-MATS433 > 3.0.CO;2-I.
- T. Y. Lee, C. A. Guymon, E. S. Jönsson and C. E. Hoyle, Polymer, 2004, 45, 6155–6162, DOI:10.1016/j.polymer.2004.06.060.
- N. A. Peppas, H. J. Moynihan and L. M. Lucht, J. Biomed. Mater. Res., 1985, 19, 397–411, DOI:10.1002/jbm.820190405.
- F. Ganji, S. Vasheghani-Farahani and E. Vasheghani-Farahani, Polym. J., 2010, 19, 375–398 CAS.
- A. Jastram, T. Lindner, C. Luebbert, G. Sadowski and U. Kragl, Polymers, 2021, 13, 1834, DOI:10.3390/polym13111834.
- N. R. Richbourg and N. A. Peppas, Prog. Polym. Sci., 2020, 105, 101243, DOI:10.1016/j.progpolymsci.2020.101243.
- B. Strachota, L. Matějka, A. Zhigunov, R. Konefał, J. Spěváček, J. Dybal and R. Puffr, Soft Matter, 2015, 11, 9291–9306, 10.1039/c5sm01996f.
- K. Chen, Y. Feng, Y. Zhang, L. Yu, X. Hao, F. Shao, Z. Dou, C. An, Z. Zhuang, Y. Luo, Y. Wang, J. Wu, P. Ji, T. Chen and H. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 36458–36468, DOI:10.1021/acsami.9b14348.
- B. Zhang, S. Li, H. Hingorani, A. Serjouei, L. Larush, A. A. Pawar, W. H. Goh, A. H. Sakhaei, M. Hashimoto, K. Kowsari, S. Magdassi and Q. Ge, J. Mater. Chem. B, 2018, 6, 3246–3253, 10.1039/c8tb00673c.
- N. Rekowska, J. Huling, A. Brietzke, D. Arbeiter, T. Eickner, J. Konasch, A. Riess, R. Mau, H. Seitz, N. Grabow and M. Teske, Pharmaceutics, 2022, 14, 628, DOI:10.3390/pharmaceutics14030628.
- F. Matin, Z. Gao, F. Repp, S. John, T. Lenarz and V. Scheper, J. Imaging, 2021, 7(5), 79, DOI:10.3390/jimaging7050079.
- R. Mau, J. Nazir, Z. Gao, D. A. Labrador, F. Repp, S. John, T. Lenarz, V. Scheper, H. Seitz and F. Matin-Mann, Curr. Dir. Biomed. Eng., 2022, 8, 157–160, DOI:10.1515/cdbme-2022-1041.
- A. Brietzke, C. Von Der Ehe, S. Illner, C. Matschegewski, N. Grabow and U. Kragl, Curr. Dir. Biomed. Eng., 2017, 3, 695–698, DOI:10.1515/cdbme-2017-0147.
- L.-E. Meyer, D. Horváth, S. Vaupel, J. Meyer, M. Alcalde and S. Kara, React. Chem. Eng., 2023, 8, 384–388, 10.1039/D3RE00058C.
- J. Seo, D. I. Kushner and M. A. Hickner, ACS Appl. Mater. Interfaces, 2016, 8, 16656–16663, DOI:10.1021/acsami.6b03455.
- O. Halevi, T. Y. Chen, P. S. Lee, S. Magdassi and J. A. Hriljac, RSC Adv., 2020, 10, 5766–5776, 10.1039/c9ra09967k.
- U. Simon and S. Dimartino, J. Chromatogr. A, 2019, 1587, 119–128, DOI:10.1016/j.chroma.2018.12.017.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tb00285c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |