
A propanesultone-based polymer electrolyte for high-energy solid-state lithium batteries with lithium-rich layered oxides†
Xin
Yin‡
ab,
Shu
Zhao‡
ab,
Zhiyuan
Lin‡
ab,
Xianwei
Guo
*ab,
Chenjie
Lou
c,
Shiqi
Liu
ab,
Boya
Wang
ab,
Peipei
Ding
ab,
Mingxue
Tang
c,
Lingqiao
Wu
ab and
Haijun
Yu
 *ab
*ab
aInstitute of Advanced Battery Materials and Devices, Faculty of Materials and Manufacturing, Beijing University of Technology, Beijing 100124, PR China. E-mail: hj-yu@bjut.edu.cn; xwguo@bjut.edu.cn
bKey Laboratory of Advanced Functional Materials, Ministry of Education, Beijing University of Technology, Beijing 100124, PR China
cCenter for High Pressure Science & Technology Advanced Research, Beijing 100193, PR China
First published on 12th August 2023
Abstract
Polymer electrolyte-based solid-state lithium batteries (SSLBs) with lithium-rich layered oxide (LLO) cathode materials can provide high energy density and safety. However, the development of these batteries is hindered by the poor anti-oxidation ability of polymer electrolytes. Herein, a propanesultone-based polymer electrolyte (PPS-PE) is designed, and a wide electrochemical stability window (∼5.0 V vs. Li+/Li) and high ion transference number (∼0.78) at 25 °C can be achieved. The strong anti-oxidation ability of PPS-PE is contributed by the design of the chain-like molecular structure, and the hydrogen bond interactions are beneficial for inhibiting the anion movement of Li salt. The PPS-PE-based SSLBs with LLO cathode materials show characteristic charge/discharge profiles with a high initial discharge capacity of ∼270 mA h g−1 and good cycling stability at 25 °C. Therefore, this work not only reports a novel polymer electrolyte to couple with high-voltage cathodes but also promotes the application of LLO cathode materials in high-energy SSLBs.
 Xianwei Guo | Dr Xianwei Guo is an Associate Professor in the Faculty of Materials and Manufacturing, Beijing University of Technology. He received his PhD degree in Condensed Matter Physics from the Institute of Physics, Chinese Academy of Sciences under the supervision of Prof. Zhaoxiang Wang. He then worked as the Research Associate at the Advanced Institute for Materials Research (WPI-AIMR) of Tohoku University in Japan under the supervision of Prof. Mingwei Chen. In 2016, he joined the Faculty of Materials and Manufacturing, Beijing University of Technology. His current research interests include the design of solid electrolytes and interface construction of composite cathodes for solid-state batteries and the interface modification of high-energy cathode materials for lithium-ion batteries. |
1. Introduction
Lithium-ion batteries (LIBs) have been widely used in electric vehicles and large-scale energy storage, and more attention is paid to the safety issues caused by the utilization of liquid electrolytes, especially for improving the energy density by applying high-voltage and high-capacity intercalation cathode materials and a Li metal anode.1–6 Solid-state lithium batteries (SSLBs) are proposed to improve safety and stability, and research and technology development has been carried out rapidly.7–9 For SSLBs, solid electrolytes are the critical bottlenecks that determine the electrochemical performances.10,11 Compared with inorganic solid electrolytes,12–14 polymer electrolytes (PEs) have the advantages of light weight, high flexibility, and easy processing, as well as high compatibility with the production line of liquid electrolyte-based batteries and have been considered as the preferable choice for practical applications.15–18 However, the comprehensive properties of SSLBs are still restricted by the disadvantages of PEs, including the low ionic conductivity, narrow electrochemical stability window, and low ion transference number, and the improvement of energy density and rate performance is severely affected.3,19–22In recent years, a series of PEs with a wide electrochemical stability window have been developed to couple with high-voltage cathodes. For instance, a plastic-crystal-embedded elastomer electrolyte, a polyethylene oxide electrolyte with the –OCH3 group on the terminal of the molecular chain, and a fluorine-containing polycarbonate electrolyte can ensure the operation of SSLBs with a Ni-rich ternary cathode material at a charge voltage of 4.5 V.23–25 In addition, a polymer-in-salt electrolyte, a poly(vinyl ethylene carbonate) electrolyte, and a thiol-branched polymer electrolyte were designed for the stable operations of SSLBs with the LiCoO2 cathode when charged to 4.5 V.26–28 Furthermore, a polyglycol oxide-based electrolyte from deep eutectic solvents and a deep eutectic solvent-based composite polymer electrolyte were proposed for the 4.6 V SSLBs with the LiCoO2 cathode.29,30 Even more, a poly(fluoroethylene carbonate) electrolyte and a polysiloxane electrolyte were synthesized for the 4.9 V LiNi0.5Mn1.5O4 cathode material in SSLBs.31,32 Although the above SSLBs can be charged to relatively high voltages, the low discharge capacities of cathode materials still limit the increase of energy density.
Lithium-rich layered oxide (LLO) cathode materials display a high discharge capacity near 300 mA h g−1 and can greatly improve the energy density of SSLBs by using the Li metal anode.33–35 However, the electrochemical stability windows of PEs cannot be compatible with the high charge voltage of the LLO cathode, and thus, the progress of SSLBs is slow, although a complexed polymer electrolyte is prepared with strange charge/discharge curves,29 and a gel polymer electrolyte is designed for a quasi-solid-state battery.36 Different from the design of complex PEs, in our recent work, the anti-oxidation ability of poly(vinyl ethylene carbonate) electrolyte has been enhanced by adjusting the molecular structure.27 And the interface compatibility between the PE and high-voltage cathode is significantly improved by breaking the weak bonding during the polymerization process.
Herein, a propanesultone-based polymer electrolyte (PPS-PE) with a chain-like molecular structure is fabricated for coupling with the LLO cathode material, which possesses a wide electrochemical stability window and high ion transference number. Hydrogen bonding as the intermolecular interaction can inhibit the anion movement of the Li salt, and the ion migration mechanism in PPS-PE is revealed by combining theoretical calculations and experimental results. More importantly, steady charge/discharge processes of PPS-PE-based SSLBs with LLO cathode materials can be achieved when charged to 4.8 V, and thus, the practical application of high-energy SSLBs has been greatly promoted.
2. Experimental section
2.1. Fabrication of the polymer electrolyte and solid-state battery
The PPS-PE and SSLBs were prepared by an in situ polymerization process. First, the liquid PS monomer (10 g) and bistrifluoromethanesulfonimide lithium salt (LiTFSI, 5 g) were added to glass bottles to form a uniform solution. Then, tin(II) 2-ethylhexanoate (Sn(Oct)2) was added to the solution as a catalyst and stirred in a glove box for 3 h, and a uniform solution formed without other solvents. For the fabrication of PPS-PE, the above solution was injected into a Whatman® filter (100% borosilicate glass fiber) in a coin cell (CR2032). For the preparation of PEO-PE, 1 g PEO powder (Mw ∼400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was placed in the glass bottle, and 20 ml acetonitrile (AN) was added and evenly stirred for 24 h. Then, the LiTFSI salt (the molar ratio of EO:Li is 16:1) was also added and stirred again for 3 h. Then, the mixed solution was scraped on the surface of a Teflon template, and the AN evaporated naturally at room temperature. Finally, the Teflon template was transferred to an oven and dried at 60 °C for 6 h to obtain the PEO-PE. A battery with a symmetrical structure of stainless steel as electrodes was assembled and transferred to a vacuum oven at 80 °C for 12–24 h for polymerization. And the battery was disassembled for the characterization. For the fabrication of a solid-state battery, a similar process was adopted, where the LLO cathode and Li are used.
000) was placed in the glass bottle, and 20 ml acetonitrile (AN) was added and evenly stirred for 24 h. Then, the LiTFSI salt (the molar ratio of EO:Li is 16:1) was also added and stirred again for 3 h. Then, the mixed solution was scraped on the surface of a Teflon template, and the AN evaporated naturally at room temperature. Finally, the Teflon template was transferred to an oven and dried at 60 °C for 6 h to obtain the PEO-PE. A battery with a symmetrical structure of stainless steel as electrodes was assembled and transferred to a vacuum oven at 80 °C for 12–24 h for polymerization. And the battery was disassembled for the characterization. For the fabrication of a solid-state battery, a similar process was adopted, where the LLO cathode and Li are used.
2.2. Microstructural characterization
Fourier transform infrared (FT-IR) spectroscopy was carried out with a Nexus 870 FTIR spectrometer and ATR instrument to distinguish the structures of the liquid PS monomer and PPS-PE. The Raman spectra were recorded using a confocal Raman microscope (WITec, alpha300, German) with a laser wavelength of 532 nm. The morphologies of PPS-PE, Li metal and the composite cathode after cycling were characterized by using a field emission scanning electron microscope (SEM, Hitachi S-4800). The high-resolution solid-state magic angle spinning nuclear magnetic resonance (MAS-NMR) spectra were recorded on a Bruker 600 MHz spectrometer (AVANCE NEO) equipped with a Bruker 3.2 mm HXY MAS probe at the 1H, 6Li, and 7Li Larmor frequencies of 600.25, 88.33 and 233.28 MHz, respectively. All samples were packed into 3.2 mm zirconia rotors with Kel-F drive caps in an argon-filled glove box and were then spun at 14.0 kHz ± 3 Hz using a Bruker pneumatic MAS unit. The 90° pulse for 1H excitation was determined as 2.9 μs and a total of 4 scans were accumulated for one spectrum. The 1H chemical shifts were referenced to the proton and the secondary carbon atom in adamantane at 1.8 ppm and 38 ppm, respectively. Spectra were recorded after single-pulse excitation with repetition times of 5, 1 and 1 s for acquiring 6Li and 7Li signals respectively. GPC measurements were performed by dissolving the PPS-PE in trichloromethane and eluting the solutions in a Waters ambient temperature GPC equipped with triple detection capability for absolute polymer molecular weight determination. The crystal structure of PPS-PE was characterized via X-ray diffraction (XRD, Bruker D8 Advance) in the range of 10°–80°. For the X-ray photoelectron spectroscopy (XPS), the binding energies were calibrated according to the peak of 284.8 eV in the C1s spectrum.The ionic conductivity of the PE was obtained from the bulk resistance (Rb) of the electrolyte film via electrochemical impedance spectra at varied temperatures ranging from −15 to 55 °C. The Rb of the PE was determined from the impedance spectrum of the film, which was sandwiched between two stainless-steel (SS) plate electrodes. And the spectra were recorded in the frequency range of 0.1 to 105 Hz with an AC amplitude of 10 mV. The ionic conductivity was calculated from the equation: σ = L/(RbS), where L and S are the thickness and area of the PE film, respectively. The electrochemical stability window was investigated by linear sweep voltammetry (LSV) performed on a working electrode of SS, with Li metal as the counter and reference electrode of PE at a scan rate of 1.0 mV s−1 between 2.5 and 5.5 V. The cyclic voltammetry of the SS/PPS-PE/Li was performed at a scan rate of 0.2 mV s−1 between −0.5 and 3.0 V. The Li ion transference number (tLi+) was calculated by the chronoamperometry test on the Li/PPS-PE/Li symmetric battery with an applied voltage of 0.01 V and was determined from the equation of (Iss/I0) × (V − I0R0)/(V − IssRss), where I0 and Iss are the initial and steady-state currents, and R0 and Rss are the first and last resistances, respectively. The tests were completed by EIS measurements taken before and after the polarization scans over a frequency range of 0.1 to 105 Hz with 10 mV amplitude at 25 °C.
2.3. Density functional theory (DFT) calculations
The density functional theory (DFT) calculations were carried out by using the Gaussian 09 program. Geometric configurations were optimized at the B3LYP level with a 6-311G (d,p) basis set. The 6-311G (d,p) basis set is a split-valence triple-zeta basis with diffuse s and p functions for nonhydrogen atoms. The dispersion effect and thermal free energies are taken into account in the calculations. The atomic charge and bond order were calculated using Multifwn.37,382.4. Battery characterization
The composite cathode was composed of 80 wt% LLOs, 10 wt% carbon black and 10 wt% binder (PVDF:PS-LiTFSI-Sn(Oct)2 = 7:3). The mass loading of the cathode material was 2–5 mg cm−2. The C rates in all the electrochemical measurements are defined based on 1C = 200 mA g−1 for LLOs. The battery was assembled in an argon-filled glove box, with oxygen content and H2O content less than 0.1 ppm. The galvanostatic charge/discharge tests of CR2032 were conducted on a LAND testing system (Wuhan LAND Electronics Co., Ltd) at 25 °C.
3. Results and discussion
3.1. Preparation and structure of PPS-PE
The PPS-PE is fabricated by the solution casting method described in the experimental section. Typically, the PS liquid monomer, Li salt and Sn(Oct)2 catalyst are dispersed homogeneously in a glass bottle by intense stirring. After that the solution is poured into a glass filter paper with full permeation on the polytetrafluoroethylene plate, followed by thermal polymerization in a vacuum oven. Then, the self-supporting and flexible PPS-PE film is obtained. Fig. 1a shows the schematic diagram for the formation of PPS with a chain-like structure from the ring-opening polymerization of the PS monomer by Sn(Oct)2 catalyzation. And the polymerization mechanism is further confirmed by the theoretical calculation and structural analysis. As shown in Fig. 1b, the position for the breaking of chemical bonding during the polymerization is explored by comparing the electron localization function (ELF) and bond order of S–O, S–C, C–C and C–O bonds in the ring structure of the PS monomer. The bond order of the S–C bond is lower than that of other bonds, and it is weak and easy to break. Thus, the PPS with a chain-like structure can be obtained by breaking the S–C bond during the ring-opening polymerization of the PS monomer.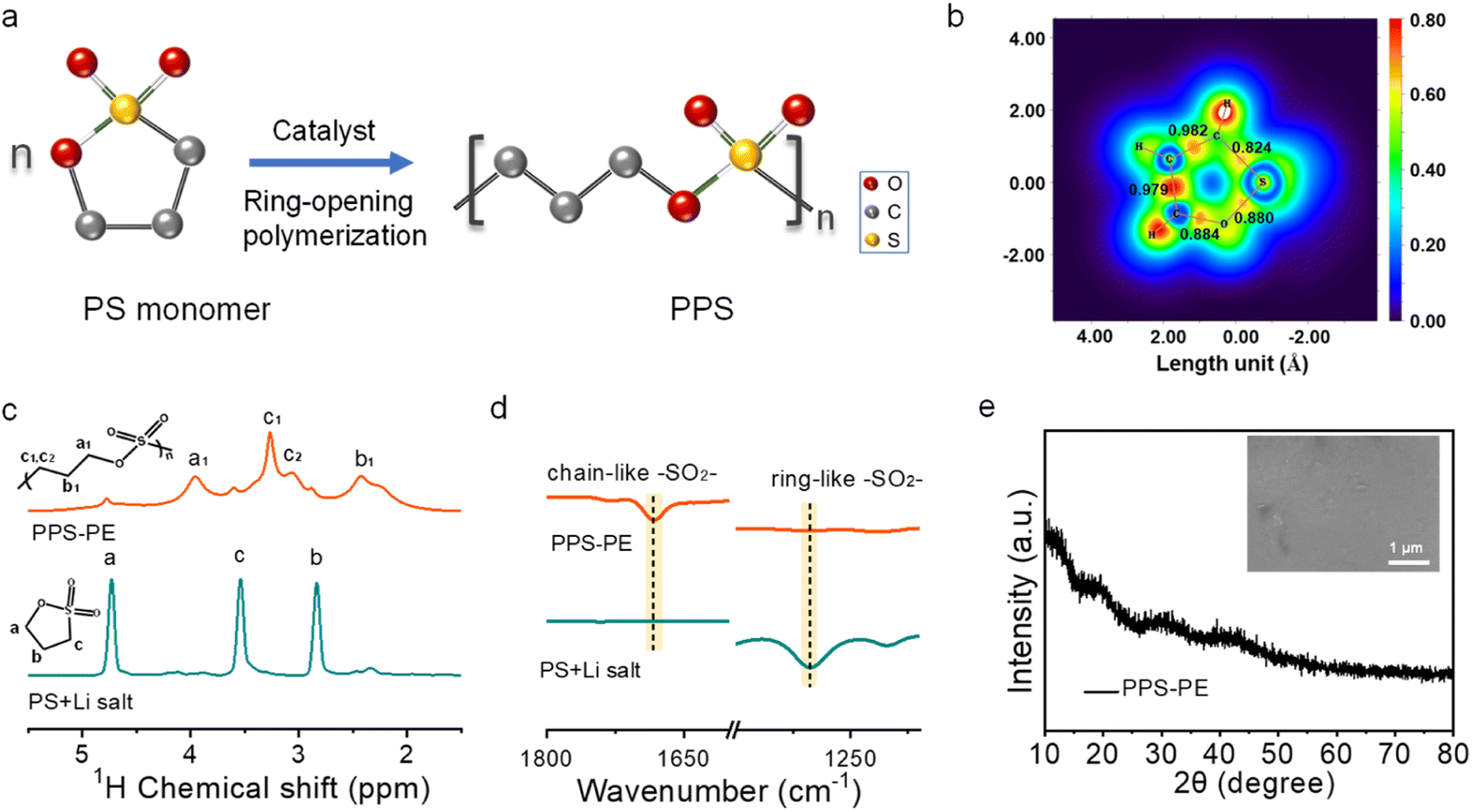 | ||
| Fig. 1 (a) The schematic diagram for the formation of PPS with a chain-like structure from the ring-opening polymerization of the PS monomer by Sn(Oct)2 catalyzation. (b) The electron localization function of the PS monomer. The number is the bond order of S–O, S–C, C–C and C–O bonds in the ring structure. The comparison of (c) 1H NMR spectra and (d) FT-IR spectra of PS + Li salt and PPS-PE. The H atoms at different positions of the PS monomer and PPS are marked respectively. (e) The XRD pattern of the PPS-PE film. The inset is the SEM image of the surface of the electrolyte film. | ||
Furthermore, the polymerization of PPS-PE with a chain-like structure is probed by solid-state 1H magic angle spinning nuclear magnetic resonance (MAS-NMR) spectroscopy. From Fig. 1c, the typical signals for H atoms at a, b and c positions (4.74, 2.84, and 3.55 ppm) on the ring structure of the PS monomer almost disappear after polymerization. Differently, there are some new peaks at a1, b1 and c1/c2 positions (at 3.95, 2.42 and 3.27/3.06 ppm) that can be found for the H atoms on the chain-like structure in PPS-PE. The shifts of peak positions of H atoms between the PS monomer and PPS-PE have confirmed the ring-opening polymerization, and are generated by the change of electron configuration from the five-membered ring structure to a chain-like structure.
The peak splitting at 3.27 ppm (c1) and 3.06 ppm (c2) for PPS-PE originated from the change of environment for the H atom at the c position after the breaking of the S–C bond during the ring-opening polymerization of the PS monomer.27 Besides, the peaks in PPS-PE have weaker intensity with broader linewidth than that of the PS monomer, which should be caused by the rigid chain-like structure after polymerization. Thus, the ring-opening polymerization process of PPS-PE from the monomer is clearly revealed by solid-state NMR spectra. The polymerization degree of PPS-PE from the monomer can be obtained by simulating the relative area of each peak, and the value can be up to 91% (Fig. S1 and Table S1†). The molecular weight of PPS-PE is obtained by the gel permeation chromatography (GPC) test, and the Mw is ∼1569524 g mol−1, indicating the formation of large molecules with a chain-like structure in PPS-PE after polymerization (Fig. S2a†).
Moreover, the change of the molecular structure between PPS-PE and the monomer is analyzed by using a Fourier transform infrared (FT-IR) spectrometer. The peak at ∼1268 cm−1 for the ring-like –SO2– bond in the PS monomer disappeared after polymerization (Fig. 1d).39 And the appearance of a new peak at ∼1683 cm−1 for the chain-like –SO2– bond can be found in PPS-PE.40 The changes in the above two peaks indicate the opening of the five-membered ring in the PS monomer and the formation of a chain-like structure of PPS-PE. And the peaks at ∼710 and ∼454 cm−1 for the S–C bond still can be found in PPS-PE (Fig. S2b†).41,42 However, the peak intensities reduce greatly, which is caused by the ring opening and formation of a chain-like structure before and after polymerization. Furthermore, the –OH bonds form after the reaction, corresponding to the terminal part of the molecular structure of PPS-PE (Fig. S2c and d†). Thus, the NMR and FT-IR spectra have revealed the ring-opening polymerization from the PS monomer and the formation of PPS-PE by breaking the weak bonding of the S–C bond.
The crystal structure of PPS-PE is detected by X-ray diffraction (XRD). From Fig. 1e, there are no obvious diffraction peaks in the XRD pattern. It demonstrates the low crystallinity of PPS-PE and is favorable for the Li ion migration. Besides, the formation of PPS-PE from the liquid monomer and the flexibility with good mechanical property can be found in Fig. S3,† which is helpful for assembling the SSLBs with tunable size and architecture and can effectively prevent the inner short-circuit and improve safety. Furthermore, from the scanning electron microscopy (SEM) image (inset in Fig. 1e), the surface of PPS-PE is smooth and homogeneous and is easy to make intimate interface contact with electrodes inside the solid-state battery.
3.2. Ion migration properties and mechanisms of PPS-PE
The ionic conductivity (σ) of PE plays a critical role in the electrochemical performances of SSLBs. Fig. S4† shows the bulk resistance (Rb) of the PPS-PE film from the electrochemical impedance spectra (EIS) of SS/PPS-PE/SS symmetrical batteries at different temperatures. And the σ of PPS-PE at various temperatures can be obtained. For comparison, the relative properties of polyethylene oxide electrolyte (PEO-PE) are also evaluated. As shown in Fig. 2a, with the increase in temperature, the σ also increases simultaneously. And the σ can be up to 1.5 × 10−4 S cm−1 at 25 °C, which is higher than that of PEO-PE. Besides, by the temperature-dependent behavior of σ, the activation energy of PPS-PE is ∼0.18 eV. It is estimated that the relatively low energy barrier is beneficial for Li ion migration at various temperatures and exhibits high ionic conductivity at 25 °C.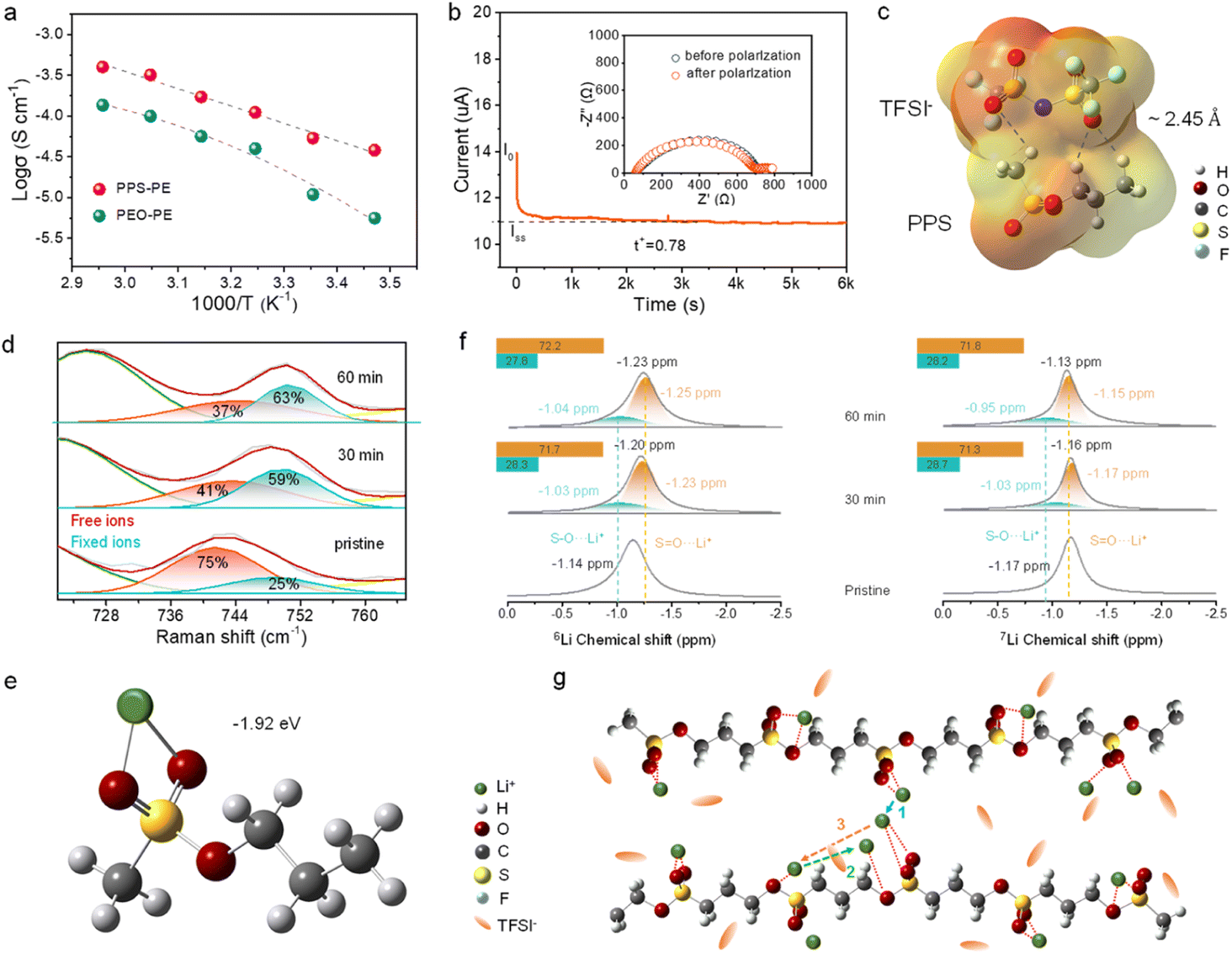 | ||
Fig. 2 (a) The temperature dependency of ionic conductivity of PPS-PE and PEO-PE. (b) Chronoamperometry profile of the Li/PPS-PE/Li symmetric battery at a polarization potential of 10 mV, and the EIS before and after the polarization (inset). (c) The molecular electrostatic potential energy mappings of PPS and TFSI− with the hydrogen bond interactions. (d) The Raman shift of ∼740 cm−1 for PPS-PE with the change of the area ratio of free and fixed ions. (e) The binding energy between the free Li ion and the two O atoms on the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group. (f) The 6Li and 7Li NMR spectra and the simulations of PPS-PE after different cycle times. (g) The schematic diagram of ion migration pathways in PPS-PE. O group. (f) The 6Li and 7Li NMR spectra and the simulations of PPS-PE after different cycle times. (g) The schematic diagram of ion migration pathways in PPS-PE. | ||
The Li ion transference number (tLi+) is the ratio between Li+ transport and the total ion (cation and anion) transport in the electrolyte. With low tLi+, the cation and anion accumulations will occur at the opposite electrodes and lead to the ion concentration gradients, side reactions, increase in interfacial impedance, and polarization of electrodes.43–45 Here, the tLi+ was tested by the Bruce–Vincent method with the Li/PPS-PE/Li symmetry battery, in which the initial and steady-state currents, and the first and last resistances before and after the EIS test were collected. The initial current was caused by the transport of both cations and anions, and the steady-state current was determined by only Li+ transport after 1 h (Fig. 2b). From the direct current polarization and EIS tests, the tLi+ of PPS-PE is calculated to be ∼0.78 at 25 °C. Besides, after the polarization test, there is no obvious change of Rb for the PPS-PE film and interfacial resistance between PPS-PE and the Li metal anode (inset in Fig. 2b), demonstrating the favorable interface compatibility with stable interface contact. Thus, the PPS-PE is beneficial for the fast ion transport, and then for the improvement of rate performances and enhancement of energy density for SSLBs.
The possible reason for the high tLi+ of PPS-PE is first explored by theoretical calculations. The interactions between anions (TFSI−) of Li salt and PPS in the electrolyte are explored by DFT calculations. In Fig. 2c, the electrostatic potentials (EPs) show the electric density maps of typical groups in TFSI− and PPS. As can be seen, the regions around O atoms in the S–O/SO group in PPS and the OSO group of TFSI− have higher electron density. By calculating the interaction between TFSI− and PPS, the hydrogen bonds form between O atoms in TFSI− and H atoms in PPS, while the bond length is ∼2.45 Å and binding energy is −0.59 eV. Accordingly, the hydrogen bonds between PPS and TFSI− are beneficial for anchoring of anions.
Besides, the bonding interactions between TFSI− of Li salt and H atoms of PPS are systematically explored by Raman spectroscopy and FT-IR, in which the ion pairing of TFSI− in Li salt is analyzed. The dissolution ability of Li salt in PPS-PE has been detected by FT-IR (Fig. S5†). The typical peak of Li salt has a shift to a lower wavenumber, indicating that the LiTFSI can completely dissociate in the PPS matrix. For the Raman test, the Li/PPS-PE/Li symmetric battery was cycled different times, and then the PPS-PE film was obtained after disassembling the battery. It is proved that the peak at ∼740 cm−1 is generated by the S–N–S stretching vibration of TFSI− anions, which is highly related to the ion pairing.46–48 As shown in Fig. 2d, there are two deconvoluted peaks at ∼741 and ∼749 cm−1 that can be found in the pristine state, corresponding to the free and fixed anions respectively.49 By the Gaussian fitting, the area ratio of free anions to fixed anions is ∼75%:25%. However, with the increase of cycling time, there is a shift of the S–N–S peak in TFSI− in the spectra and then the expansion of the area ratio for fixed anions consequently. Even more, after the battery was cycled for 60 min (corresponding to the steady-state current for 1 h in Fig. 2b), the area ratio of fixed anions has increased to ∼63%, which is much higher than that of the pristine state. The changes in the area ratios of fixed and free anions are systematically compared in Fig. S6.† Thus, from the change of area ratio for ion pairs by Raman shift of the S–N–S group in TFSI−, most of the free ion pairs are anchored by the interaction between anions and PPS, and then the high tLi+ can be obtained in PPS-PE. And the Raman tests and theoretical calculations are consistent with each other. These results confirm the responsibility of local hydrogen bond interactions for the increase of the area ratio for fixed anions, and the high tLi+ can be obtained.
The pathways for ion transport in the molecular structure of PPS-PE have been explored systematically in order to reveal the migration mechanism. Since the LiTFSI can be completely dissociated in PPS, the binding energies between free Li+ and oxygen (O) atoms of PPS are first calculated. As shown in Fig. S7,† the Li+ can interact with O atoms in both SO and S–O groups. The binding energy between Li+ and a single O atom in the SO group is −1.90 eV, while the combination of Li+ and O in the S–O group is unstable. By comparison, the binding energy of Li+ to both O atoms in SO and S–O groups is −1.87 eV, and the stable configuration is the interaction between Li+ and two O atoms in SO groups with a binding energy of −1.92 eV (Fig. 2e). So the Li+ can interact with O atoms in both SO and S–O groups, but the binding energy of the former is stronger, which plays an important role for the ion migration in PPS-PE. Besides, considering the surrounding environment of Li+ in LiTFSI, the binding energy between LiTFSI and the O atom in the SO group in PPS has been explored (Fig. S8†). Due to the interaction between Li+ and TFSI−, the bonding energy between Li+ and O atom in the SO group is −1.067 eV, demonstrating the relatively strong interaction. Thus, the interaction between Li+ and O atoms in the SO group plays an important role in the ion migration ability in PPS-PE.
Moreover, the interactions between conductive Li+ and functional units in PPS-PE have been probed by solid-state 6Li and 7Li NMR. Consistent with the Raman tests, the Li/PPS-PE/Li battery is disassembled after different cycles, and the electrolyte film is obtained for the characterization. In the spectra of Fig. 2f, there is only one 6Li NMR resonance centered at −1.14 ppm for the pristine PE, and the signal shifts to a high field (−1.23 ppm) after cycling for different time. The change of position for the polarized signal should be generated by the interactions between SO/S–O groups and Li+. In addition, the position for the interactions between Li+ and S–O/SO groups can be simulated by two peaks, and the area ratio of Li+ attached to the SO group is ∼71.7%, which is much larger than ∼28.3% of the Li+ interaction with the S–O group. Even more, there was no obvious change in the area ratio for these two characteristic peaks after more cycle time, indicating the stable interactions between SO/S–O groups and Li ions.
Similarly, for the 7Li NMR, the resonance centered at −1.17 ppm in the pristine state has shifted to −1.13 ppm after cycling. And the area ratios of interactions between Li+ and SO/S–O groups are the same as each other for the cycle time of 30 and 60 min. Thus, the interactions between Li+ and SO/S–O groups remain stable after cycles in PPS-PE, and the interactions between Li+ and SO groups play a more important role in the ion migration. According to the 6,7Li NMR results and theoretical simulations, the ion migration mechanism in PPS-PE is proposed and shown in Fig. 2g. The ion migration is mainly achieved through the coupling/decoupling between Li+ and oxygen atoms of SO groups (Path 1) during the process of segmental motion of molecular chains. And a portion of ions can migrate through the coupling/decoupling between Li+ and oxygen atoms of S–O groups (Path 2). Furthermore, the moving/exchange of Li+ between SO and S–O groups during the segmental motion of polymer chains may also exist (Path 3). The mixed ion migration pathways are beneficial for the fast ion transport in PPS-PE and for the enhancement of the electrochemical performances of SSLBs.
3.3. Electrochemical stability window of PPS-PE and interface contact with the Li metal anode
The anti-oxidation ability of PE is critical for enhancing the energy density of SSLBs with high-voltage cathodes. Here the electrochemical stability window of PPS-PE is explored by theoretical calculations and experimental analysis. It is proved that the electrochemical stability window of PE is relevant to the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels. So the LUMO and HOMO energy levels of PPS, PEO and Li salt are explored by DFT calculations. As shown in Fig. 3a, the PPS has a lower HOMO energy level than the PEO, and a strong anti-oxidation ability can be obtained. Besides, the HOMO energy level of Li salt is similar to that of PPS, indicating that LiTFSI is suitable for coupling with PPS to enhance the anti-oxidation ability of the electrolyte. The LUMO energy level of LiTFSI is lower than that of PPS, so the anti-reduction ability of PPS is better than that of Li salt and is favorable for the interface stability with the Li metal anode.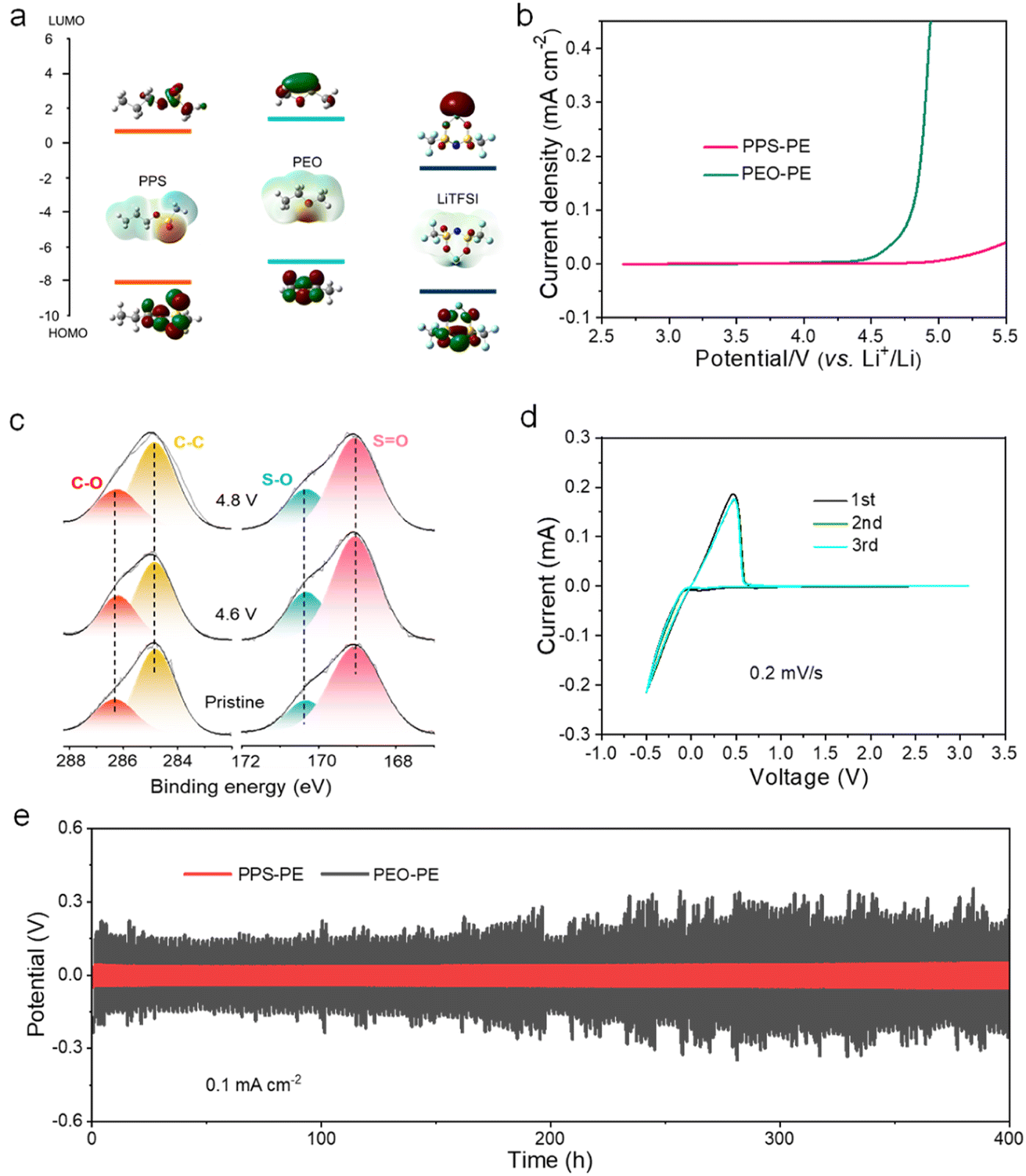 | ||
| Fig. 3 (a) The HOMO and LUMO energy levels of PPS, PEO, LiTFSI, and the interactions. (b) The electrochemical stability windows of PPS-PE and PEO-PE. (c) The XPS spectra of different chemical bonding energies in PPS-PE after different constant current–current voltage charging tests. (d) CV curve of the Li/PPS-PE/SS battery in the voltage range of −0.5 to 3.0 V. (e) The potential profiles of repeated Li plating/stripping in Li/PPS-PE/Li and Li/PEO-PE/Li symmetric batteries with a current density of 0.1 mA cm−2. | ||
The electrochemical stability window of PPS-PE is explored by the linear sweep voltammetry (LSV) test of an asymmetrical Li/PPS-PE/SS battery. As shown in Fig. 3b, the electrochemical stability window of PPS-PE can be up to ∼5.0 V (vs. Li+/Li), which is much higher than that of the reported polymer electrolytes.23,24,27 It has been confirmed that the anti-oxidation ability of polycarbonate polymer electrolytes with chain-like structures can be greatly enhanced than that with a five-membered ring structure.27 Following the same rule, the wide electrochemical stability window of PPS-PE with a chain-like structure can also be achieved and is beneficial for coupling with high-voltage and high-capacity intercalation cathode materials, and enabling the stable operation of high-energy SSLBs.
Furthermore, XPS is used to detect the changes of chemical components in PPS-PE before and after constant current-constant voltage charging at 4.6 V and 4.8 V for 50 h (Fig. 3c). The PE films are obtained by disassembling the Li/PPS-PE/SS batteries after the test. There are characteristic peaks in the spectra of PPS-PE with a ring structure in the pristine state, including the C–O bond (∼286.3 eV), C–C bond (∼284.8 eV), S–O bond (∼170.3 eV), and SO bond (∼169.1 eV). Obviously, there is no obvious change for the above peaks when the battery was charged to 4.6 V, and even to 4.8 V. Furthermore, there is no formation of new peaks for PPS-PE at different charge states.
Even more, the area ratio for each peak remains stable, indicating the strong anti-oxidation ability of PPS-PE at high charge voltages. Thus, from the theoretical calculations, LSV test and XPS spectra, the PPS-PE with a chain-like structure has a strong anti-oxidation ability and can couple with high-voltage cathode materials with a charge potential of up to 4.8 V.
Besides, the interface contact between PPS-PE and the Li metal anode is investigated by cyclic voltammetry (CV) and the polarization test. From the CV curves of the Li/PPS-PE/SS battery in a voltage range of −0.5 to 3.0 V (Fig. 3d), two peaks at −0.5 V and 0.5 V can be found, corresponding to the reversible lithium plating/stripping in each cycle, and the electrochemical stability of interface contact between PE and the Li anode. Furthermore, the polarization test of the Li/PPS-PE/Li battery at a current density of 0.1 mA cm−2 at 25 °C is shown in Fig. 3e. The battery shows low voltage polarization with an overpotential of ∼0.08 V, and works steadily for 400 h without short circuiting, which is much better than that of the Li/PEO-PE/Li battery at the elevated temperature. These results demonstrate the good reversibility of lithium plating/stripping in PPS-PE-based SSLBs. Even more, the uniform dendrite on the surface of the Li anode can form from the SSLBs after long cycles (Fig. S9†), and it is beneficial for the enhancement of electrochemical performances.
3.4. Electrochemical performances of PPS-PE based SSLBs with the LLO cathode material
As the electrochemical stability window of PPS-PE can be up to 5.0 V (vs. Li+/Li), high-capacity and high-voltage intercalation cathode materials can be applied for SSLBs to improve the energy density.Here, the LLO (Li1.13Mn0.517Ni0.256Co0.097O2) cathode material is adopted to assemble SSLBs by the in situ polymerization method.27,46Fig. 4a shows the initial galvanostatic charge/discharge curves of the LLOs/PPS-PE/Li battery at a current density of 0.1C (1C = 200 mA g−1) in the voltage range of 2.0-4.8 V at 25 °C. The battery can be charged to 4.8 V with the characteristic profiles of the LLO cathode material, and the initial discharge capacity of ∼270 mA h g−1 can be achieved, which is much similar to that of the liquid electrolyte-based batteries (Fig. S10†). The large discharge capacity with high initial coulombic efficiency of the LLOs/PPS-PE/Li battery should be contributed by the strong anti-oxidation ability and relatively high ion migration abilities of PPS-PE.
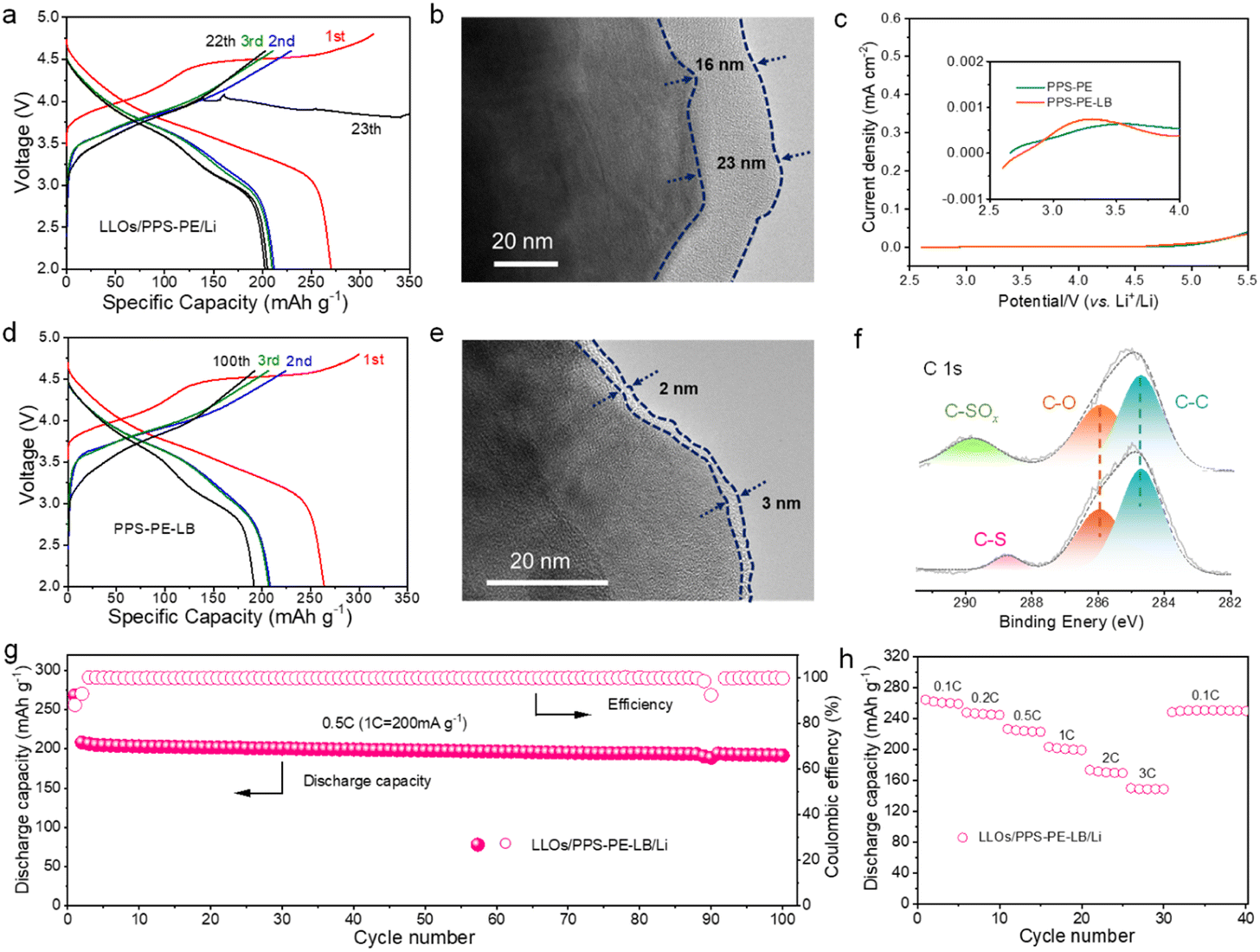 | ||
| Fig. 4 (a) The charge/discharge curves of the LLOs/PPS-PE/Li batteries with current densities of 0.1C and 0.5C (1C = 200 mA g−1). (b) The CEI on the surface of the cathode in LLOs/PPS-PE/Li batteries after cycling. (c) The linear sweep voltammetry of PPS-PE and PPS-PE-LB. (d) The charge/discharge curves of the LLOs/PPS-PE-LB/Li batteries with two current densities. (e) The CEI on the surface of the cathode in LLOs/PPS-PE-LB/Li batteries after cycling. (f) The XPS spectra of the PPS-PE film on the cathode side before and after the cycling of the battery. (g) The cycling stability of the LLOs/PPS-PE-LB/Li battery at 0.5C in the voltage range of 2.0–4.6 V. (h) The rate performances of the LLOs/PPS-PE-LB/Li battery. | ||
Furthermore, when the current density increases to 0.5C, the LLOs/PPS-PE/Li battery can work stably and show a discharge capacity over 200 mA h g−1 in the voltage range of 2.0–4.6 V, which is highly related to the high tLi+ of PPS-PE. However, the charge curve of the LLOs/PPS-PE/Li battery fluctuates after 22 cycles at 0.5C. The battery degradation should be caused by the uncontrollable interface reactions and mismatch between the LLO cathode and PPS-PE at high charge voltage after tens of cycles, which has been explored by the interface analysis. The cathode-electrolyte interphase (CEI) after battery cycling has been detected by transmission electron microscopy (TEM). As shown in Fig. 4b, the thick CEI layer with 16–23 nm can be found for the LLO cathode, which is generated by the uncontrollable side reactions, and possible oxidative decomposition of PPS-PE catalyzed by transition metal ions and oxygen redox reactions in the cathode material at high charge voltage.
It is proved that a small amount of LiBOB is helpful for forming a stable CEI and enhancing the cycling stability of batteries.50–52 Here, the LiBOB with a low amount of 2.0 wt% is adopted in PPS-PE to make the stable interface contact between the electrolyte and LLO cathode. As shown in Fig. 4c, the LiBOB in PPS-PE is pre-oxidized at the low charge voltage and is useful for interface compatibility, which can be confirmed by the theoretical calculations (Fig. S11†), and the reversible lithium plating/stripping in PPS-PE-LB-based SSLBs (Fig. S12†). For the PPS-PE with LiBOB (PPS-PE-LB), the corresponding SSLBs are assembled by the same procedure. As shown in Fig. 4d, the solid-state battery can also show the typical charge/discharge curves with high discharge capacities at 0.1C and 0.5C respectively. More importantly, the LLOs/PPS-PE-LB/Li battery has stable operation for 100 cycles at 0.5C, which is much better than that of the battery without LiBOB. Thus, with the help of LiBOB for the formation of the CEI, the lifetime of PPS-PE-LB-based SSLBs with high capacity has been greatly improved. The interface contact between the electrolyte and cathode in the LLOs/PPS-PE-LB/Li battery has been explored, and the CEI after cycles has been detected by TEM. As shown in Fig. 4e, the CEI on the cathode in the LLOs/PPS-PE-LB/Li battery is much thinner (2–3 nm) and uniform, indicating that the side reactions and oxidative decomposition of PPS-PE are greatly inhibited. Thus, the stable interface contact between PPS-PE and the LLO cathode material and the good cycling performance of SSLBs can be achieved from the strong anti-oxidation ability of PPS-PE, and the addition of LiBOB for forming a thinner and uniform CEI layer.
Moreover, the chemical components of the PPS-PE film on the cathode side before and after cycles in SSLBs have been detected by XPS (Fig. 4f). Compared to the pristine state in Fig. 3c, the characteristic peak of the C–S bond (∼286.3 eV) in the spectra of PPS-PE-LB remains stable after long cycles. However, in the spectra of PPS-PE without LiBOB, a new peak of the C–SOx bond (∼286.3 eV) formed, and the C–S bond cannot be distinguished. And the spectra of the B element in the CEI film of the cathode can be detected for the LLOs/PPS-PE-LB/Li battery, indicating that LiBOB is useful for CEI formation (Fig. S13†). Therefore, these results have further confirmed that the thin and uniform CEI is helpful for the favorable interface stability and compatibility between PPS-PE and the LLO cathode material at high charge voltage, and for enhancing the cycling performance of SSLBs. Besides, in the SEM images of the composite cathode after cycles (Fig. S14†), there are no obvious crack for the agglomerated spheres of the LLO cathode material, consistent with the good cycling stability of SSLBs.
Fig. 4g shows the cycling stability of SSLBs based on PPS-PE with LiBOB. The battery exhibits a high discharge capacity with good capacity retention of ∼92% over 100 cycles at 0.5C. As shown in Table S2,† compared to the other PE-based SSLBs with different cathode materials at a high charge voltage, the LLOs/PPS-PE-LB/Li battery exhibits a much higher discharge capacity at the initial cycle, and better cycling stability with high capacity retention. Moreover, good cycling stability of SSLBs with LiBOB at 1C and 2C can also be achieved (Fig. S15†). Besides, the LLOs/PPS-PE-LB/Li battery delivers a high discharge capacity of ∼210 mA h g−1 at 0.5C after 50 cycles at 50 °C, and ∼130 mA h g−1 at 0.5C after 50 cycles at −15 °C, which indicates that the PPS-PE based SSLBs can be applied in a wide temperature range (Fig. S16†). The good cycling stabilities of the LLOs/PPS-PE/Li battery are supported by the wide electrochemical stability window of chain-structured PPS-PE, and the excellent interface compatibility between PE and the LLO cathode with LiBOB.
The rate performances of LLOs/PPS-PE-LB/Li batteries with the increase in current densities from 0.1C to 3C are shown in Fig. 4h. The discharge capacity of ∼265 mA h g−1 at 0.1C, and ∼142 mA h g−1 at 3C can be obtained for the SSLBs. The excellent rate performances of SSLBs should be contributed by the high tLi+ of PPS-PE. Moreover, the soft-pack cell can light up a logo with 70 light-emitting diode (LED) lamps even at the cutting-off state (Fig. S17†). Thus, with the wide electrochemical stability window and high ion transference number, the PPS-PE can couple with the LLO cathode material for high-energy SSLBs with favorable electrochemical performances.
4. Conclusions
In summary, a propanesultone-based polymer electrolyte composed of monomer functional units is fabricated by ring-opening polymerization, and a high ion transference number of ∼0.78 and wide electrochemical stability window of ∼5.0 V (vs. Li+/Li) at 25 °C can be obtained. The strong anti-oxidation ability is highly related to the chain-like structure of PPS-PE, and a large amount of Li ion migration is provided by the local hydrogen bond interactions, which are revealed by both experimental results and theoretical calculations. The PPS-PE-based SSLBs with the LLO cathode material exhibit a high initial discharge capacity of ∼270 mA h g−1 with characteristic charge/discharge curves and a good capacity retention of ∼92% after 100 cycles at 25 °C. Therefore, this work has not only developed a novel polymer electrolyte for coupling with high-voltage cathodes but also promoted the practical applications of the LLO cathode material in high-energy SSLBs.Author contributions
Xin Yin, Zhiyuan Lin and Xianwei Guo: formal analysis, writing – original draft. Shu Zhao: computational simulation and analysis of data. Mingxue Tang and Chenjie Lou: solid-state NMR measurements and data analysis. Shiqi Liu, Peipei Ding and Boya Wang: Raman, FT-IR and SEM measurements. Lingqiao Wu: analyzing the experimental results. Haijun Yu: supervision, writing – original draft and formal analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Key Project of Science and Technology of Beijing Municipal Education Commission (KZ201910005002), the National Key R&D Program of China (Grant No. 2022YFB2404400), National Natural Science Foundation of China (92263206, 21975006 and 22075007, 22002004, 52002007), “The Youth Beijing Scholars program” (11000022T000000440694), and Beijing Natural Science Foundation (KZ202010005007, 2222001).Notes and references
- G. Cui, Matter, 2020, 2, 805–815 CrossRef.
- P. Fan, H. Liu, V. Marosz, N. T. Samuels, S. L. Suib, L. Sun and L. Liao, Adv. Funct. Mater., 2021, 31, 2103180 Search PubMed.
- P. Ding, Z. Lin, X. Guo, L. Wu, Y. Wang, H. Guo, L. Li and H. Yu, Mater. Today, 2021, 2021(51), 449–474 CrossRef.
- M. H. Braga, C. M. Subramaniyam, A. J. Murchison and J. B. Goodenough, J. Am. Chem. Soc., 2018, 140, 6343–6352 CrossRef CAS PubMed.
- T. Dong, J. Zhang, G. Xu, J. Chai, H. Du, L. Wang, H. Wen, X. Zang, A. Du, Q. Jia, X. Zhou and G. Cui, Energy Environ. Sci., 2018, 11, 1197–1203 RSC.
- Y.-G. Lee, S. Fujiki, C. Jung, N. Suzuki, N. Yashiro, R. Omoda, D.-S. Ko, T. Shiratsuchi, T. Sugimoto, S. Ryu, J. H. Ku, T. Watanabe, Y. Park, Y. Aihara, D. Im and I. T. Han, Nat. Energy, 2020, 5, 299–308 CrossRef CAS.
- A. Manthiram, X. Yu and S. Wang, Nat. Rev. Mater., 2017, 2, 16103 CrossRef CAS.
- D. Wu, L. Chen, H. Li and F. Wu, Appl. Phys. Lett., 2022, 121, 120502 CrossRef CAS.
- Y. Guo, S. Wu, Y.-B. He, F. Kang, L. Chen, H. Li and Q.-H. Yang, eScience, 2022, 2, 138–163 CrossRef.
- Z. Zou, Y. Li, Z. Lu, D. Wang, Y. Cui, B. Guo, Y. Li, X. Liang, J. Feng, H. Li, C. W. Nan, M. Armand, L. Chen, K. Xu and S. Shi, Chem. Rev., 2020, 120, 4169–4221 CrossRef CAS PubMed.
- K. Qin, K. Holguin, M. Mohammadiroudbari, J. Huang, E. Y. S. Kim, R. Hall and C. Luo, Adv. Funct. Mater., 2021, 31, 2009694 CrossRef CAS.
- T. Famprikis, P. Canepa, J. A. Dawson, M. S. Islam and C. Masquelier, Nat. Mater., 2019, 18, 1278–1291 CrossRef CAS PubMed.
- X. Wang, J. Chen, D. Wang and Z. Mao, Nat. Commun., 2021, 12, 7109 CrossRef CAS PubMed.
- L.-Z. Fan, H. He and C.-W. Nan, Nat. Rev. Mater., 2021, 6, 1003–1019 CrossRef CAS.
- J. Wan, J. Xie, X. Kong, Z. Liu, K. Liu, F. Shi, A. Pei, H. Chen, W. Chen, J. Chen, X. Zhang, L. Zong, J. Wang, L. Q. Chen, J. Qin and Y. Cui, Nat. Nanotechnol., 2019, 14, 705–711 CrossRef CAS PubMed.
- D. Zhang, Y. Shi, J. An, S. Yang and B. Li, J. Mater. Chem. A, 2022, 10, 23095–23102 RSC.
- W. Du, X. Du, M. Ma, S. Huang, X. Sun and L. Xiong, Adv. Funct. Mater., 2022, 32, 2110871 CrossRef CAS.
- J. Popovic, D. Brandell, S. Ohno, K. B. Hatzell, J. Zheng and Y.-Y. Hu, J. Mater. Chem. A, 2021, 9, 6050–6069 RSC.
- T. Xie, A. France-Lanord, Y. Wang, J. Lopez, M. A. Stolberg, M. Hill, G. M. Leverick, R. Gomez-Bombarelli, J. A. Johnson, Y. Shao-Horn and J. C. Grossman, Nat. Commun., 2022, 13, 3415 CrossRef CAS PubMed.
- X. Wang, R. Kerr, F. Chen, N. Goujon, J. M. Pringle, D. Mecerreyes, M. Forsyth and P. C. Howlett, Adv. Mater., 2020, 32, 1905219 CrossRef CAS PubMed.
- S. Lee, D. Sasaki, D. Kim, M. Mori, T. Yokota, H. Lee, S. Park, K. Fukuda, M. Sekino, K. Matsuura, T. Shimizu and T. Someya, Nat. Nanotechnol., 2019, 14, 156–160 CrossRef CAS PubMed.
- L. Zhang, T. Yang, C. Du, Q. Liu, Y. Tang, J. Zhao, B. Wang, T. Chen, Y. Sun, P. Jia, H. Li, L. Geng, J. Chen, H. Ye, Z. Wang, Y. Li, H. Sun, X. Li, Q. Dai, Y. Tang, Q. Peng, T. Shen, S. Zhang, T. Zhu and J. Huang, Nat. Nanotechnol., 2020, 15, 94–98 CrossRef CAS PubMed.
- M. J. Lee, J. Han, K. Lee, Y. J. Lee, B. G. Kim, K. N. Jung, B. J. Kim and S. W. Lee, Nature, 2022, 601, 217–222 CrossRef CAS PubMed.
- X. Yang, M. Jiang, X. Gao, D. Bao, Q. Sun, N. Holmes, H. Duan, S. Mukherjee, K. Adair, C. Zhao, J. Liang, W. Li, J. Li, Y. Liu, H. Huang, L. Zhang, S. Lu, Q. Lu, R. Li, C. V. Singh and X. Sun, Energy Environ. Sci., 2020, 13, 1318–1325 RSC.
- Y. Wang, S. Chen, Z. Li, C. Peng, Y. Li and W. Feng, Energy Stor. Mater., 2022, 45, 474–483 Search PubMed.
- H. Li, Y. Du, X. Wu, J. Xie and F. Lian, Adv. Funct. Mater., 2021, 31, 2103149 Search PubMed.
- Z. Lin, X. Guo, R. Zhang, M. Tang, P. Ding, Z. Zhang, L. Wu, Y. Wang, S. Zhao, Q. Zhang and H. Yu, Nano Energy, 2022, 98, 107330 CrossRef CAS.
- H. Wang, Q. Wang, X. Cao, Y. He, K. Wu, J. Yang, H. Zhou, W. Liu and X. Sun, Adv. Mater., 2020, 32, 2001259 CrossRef CAS PubMed.
- H. Wang, J. Song, K. Zhang, Q. Fang, Y. Zuo, T. Yang, Y. Yang, C. Gao, X. Wang, Q. Pang and D. Xia, Energy Environ. Sci., 2022, 15, 5149–5158 RSC.
- C. Wang, H. Zhang, S. Dong, Z. Hu, R. Hu, Z. Guo, T. Wang, G. Cui and L. Chen, Chem. Mater., 2020, 32, 9167–9175 CrossRef CAS.
- J. Liu, X. Shen, J. Zhou, M. Wang, C. Niu, T. Qian and C. Yan, ACS Appl. Mater. Interfaces, 2019, 11, 45048–45056 CrossRef CAS PubMed.
- M. Liu, B. Jin, Q. Zhang, X. Zhan and F. Chen, J. Alloys Compd., 2018, 742, 619–628 CrossRef CAS.
- H. Yu, Y.-G. So, A. Kuwabara, E. Tochigi, N. Shibata, T. Kudo, H. Zhou and Y. Ikuhara, Nano Lett., 2016, 16, 2907–2915 CrossRef CAS PubMed.
- H. Yu, Y. G. So, Y. Ren, T. Wu, G. Guo, R. Xiao, J. Lu, H. Li, Y. Yang, H. Zhou, R. Wang, K. Amine and Y. Ikuhara, J. Am. Chem. Soc., 2018, 140, 15279–15289 CrossRef CAS PubMed.
- Y. Yang, Z. Zhang, S. Liu, B. Wang, J. Liu, Y. Ren, X. Zhang, S. Zhao, D. Liu and H. Yu, Matter, 2022, 5, 3869–3882 CrossRef CAS.
- S. Cui, X. Wu, Y. Yang, M. Fei, S. Liu, G. Li and X.-P. Gao, ACS Energy Lett., 2021, 7, 42–52 CrossRef.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- S. Zhao, T. H. Wu, D. He, Z. h. Zhang, B. Y. Wang, L. H. Wang and H. J. Yu, Rare Met., 2022, 41, 3412–3420 CrossRef CAS.
- Y. Zhang, M. Irfan, Z. Yang, K. Liu, J. Su and W. Zhang, Chem. Eng. J., 2022, 435, 134775 CrossRef CAS.
- A. R. Kim, M. Vinothkannan and D. J. Yoo, Bull. Korean Chem. Soc., 2018, 39, 913–919 CrossRef CAS.
- N. M. Shishlov and S. L. Khursan, J. Mol. Struct., 2016, 1123, 360–366 CrossRef CAS.
- K. A. Uno Toyozo, Y. Saito and K. Machida, Bull. Chem. Soc. Jpn., 1975, 48, 2231–2235 CrossRef.
- Y. Zhai, W. Hou, M. Tao, Z. Wang, Z. Chen, Z. Zeng, X. Liang, P. Paoprasert, Y. Yang, N. Hu and S. Song, Adv. Mater., 2022, 34, 202205560 Search PubMed.
- F. Chen, X. Wang, M. Armand and M. Forsyth, Nat. Mater., 2022, 21, 1175–1182 CrossRef CAS PubMed.
- Y. Zhao, L. Wang, Y. Zhou, Z. Liang, N. Tavajohi, B. Li and T. Li, Adv. Sci., 2021, 8, 2003675 CrossRef CAS PubMed.
- Y. Wang, L. Wu, Z. Lin, M. Tang, P. Ding, X. Guo, Z. Zhang, S. Liu, B. Wang, X. Yin, Z. Chen, K. Amine and H. Yu, Nano Energy, 2022, 96, 107105 CrossRef CAS.
- B. Xu, X. Li, C. Yang, Y. Li, N. S. Grundish, P. H. Chien, K. Dong, I. Manke, R. Fang, N. Wu, H. Xu, A. Dolocan and J. B. Goodenough, J. Am. Chem. Soc., 2021, 143, 6542–6550 CrossRef CAS PubMed.
- S. G. Albert Bakker, L. Jan and K. Hermansson, Michael m. Probst, Polym. J., 1994, 36, 4371–4378 Search PubMed.
- N. Wu, P. H. Chien, Y. Li, A. Dolocan, H. Xu, B. Xu, N. S. Grundish, H. Jin, Y. Y. Hu and J. B. Goodenough, J. Am. Chem. Soc., 2020, 142, 2497–2505 CrossRef CAS PubMed.
- Q. Zhou, C. Fu, R. Li, X. Zhang, B. Xie, Y. Gao, G. Yin and P. Zuo, Chem. Eng. J., 2022, 437, 135419 CrossRef CAS.
- W. Liang, Y. Shao, Y.-M. Chen and Y. Zhu, ACS Appl. Energy Mater., 2018, 1, 6064–6071 CrossRef CAS.
- S. Li, Y.-M. Chen, W. Liang, Y. Shao, K. Liu, Z. Nikolov and Y. Zhu, Joule, 2018, 2, 1838–1856 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta01968c |
| ‡ These authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2023 |