
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFlow-through reductive catalytic fractionation of beech wood sawdust†
Francesco
Brandi‡
 ac,
Bruno
Pandalone‡
ac and
Majd
Al-Naji
*ab
ac,
Bruno
Pandalone‡
ac and
Majd
Al-Naji
*ab
aMax Planck Institute of Colloids and Interfaces, Colloid Chemistry Department, Am Mühlenberg 1, 14476 Potsdam, Germany
bBasCat – UniCat BASF JointLab, Technische Universität Berlin, Sekr. EW K-01, Hardenbergstraße 36, Berlin 10623, Germany. E-mail: majd.al-naji@tu-berlin.de; Fax: +49 (0)30 314-27331; Tel: +49 (0)30 314-73727
cCenter for Sustainable Catalysis and Engineering, KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium
First published on 26th January 2023
Abstract
The implementation of flow-through (FT) systems in the lignin-first approach can be a strategic tool for increasing the proficiency of biorefineries. Herein, the reductive catalytic fractionation (RCF) of waste beech wood sawdust (BWS) was conducted in an FT system using Ni on a nitrogen-doped carbon catalyst (35Ni/NDC) in pellet shape and MeOH and MeTHF as solvents. Lignin extraction was maximized in the first 4 h of time on stream (TOS), yielding maximum cumulative monomers of 247 mg gKL−1 using MeOH as solvent, extraction temperature of 235 °C, and reduction temperature of 225 °C. Importantly, the catalyst was used for two cycles and total time on stream (TOS) of 14 h without losing initial activity. These findings show that FT systems represent a promising solution for applications in lignin-first biorefineries.
Sustainability spotlight statementCommodity chemicals from fossil resources are directly linked to the linear increase of greenhouse gases emission. In line with goal 9 (industry, innovation, and infrastructure), goal 11 (sustainable cities and communities), goal 12 (responsible consumption and production), and goal 13 (climate action), establishing a sustainable wood biorefinery is greatly needed to promote a fast transition toward safer and less hazardous and CO2 neutral chemicals, materials and fuel production. In this work, a biorefinery of waste wood via reductive catalytic fractionation in a flow system was successfully implemented. The developed process resulted in extraordinary efficiency in the conversion of the complex native lignin biopolymer with respect to the existing methodology. |
Introduction
Lignocellulosic biomass is one of the most promising renewable and sustainable feedstocks for replacing fossil resources to produce fuels and fine chemicals in the so-called biorefinery process. Within the UN sustainability plan (17 goals), it is clear that the establishment of lignocellulosic biomass biorefineries is needed to promote a fast transition toward safe, less hazardous, and CO2 neutral chemicals.1–7 Moreover, non-edible lignocellulosic biomass is already produced as a waste product from agriculture, forestry, lumberjack, pulp, and paper industries.8–11 Lignocellulosic biomass is comprised of three major biopolymers, i.e., cellulose (30–40%), hemicellulose (10–20%), and lignin (20–30%).12,13 However, the complex structures of these major constituents and the variety of chemical linkages as well as the difficulty in selectively separating these constituents are still obstacles to their efficient usage as feedstock.12,14 Cellulose and hemicellulose are polysaccharides composed of C5 and C6 sugars, whereas lignin is formed by aromatic phenylpropanoid units condensed together. These units are connected via various linkages, including carbon–oxygen (e.g., β-O-4, α-O-4, γ-O-α, and 4-O-5) and carbon–carbon bonds (e.g., 5–5, and β–β).12,15–18 Owing to its aromatic nature, lignin is the major source of aromatics on our planet.15,17,19 Traditionally, the lignocellulosic biomass valorisation strategies have been focused on the sugar fraction, i.e., cellulose and hemicellulose, producing lignin as a side product.20–23In these strategies, lignin often undergoes condensation that irreversibly prevents efficient conversion toward aromatic units.17,22,24 Nevertheless, the aromatic value is high and lignin valorisation has been recognized as an essential process for the success of bio-based economy.25–28 Therefore, new holistic methodologies have been developed for biomass valorisation, which preserves both polysaccharides and lignin fractions.14,28–30 These methodologies are grouped under the umbrella term the ‘lignin-first approach’ and are generally based on the extraction and stabilization of lignin through solvothermal methods, catalysis, and protection-group chemistry.14,15,24,29
One of the most promising lignin-first technologies is reductive catalytic fractionation (RCF), which applies redox-active catalysts, such as Pt, Pd, Ru, or Ni.15,31–34 RCF comprises a heat-induced solvolytic extraction of lignin from biomass using water, alcohol, cyclic ethers, or mixtures as a solvent followed by catalytic hydrogenation and hydrogenolysis of the extracted lignin using molecular or pressurized hydrogen in batch systems.24,33,35–37 In the first step, lignin is extracted and partially depolymerized, producing unsaturated fragments that are prone to re-polymerize. Then, in the catalytic step, the extracted fragments are hydrogenated and further depolymerized via hydrogenolysis.33 The major advantage of RCF is the capability of producing high monomer yield without compromising the structure of cellulose. To date, RCF conducted in batch systems has shown lignin-derived monomers and oligomers yields close to the theoretical maximum, mostly reported by the Sels group.37–39
Nowadays, the vast majority of RCF studies have been conducted using batch systems. Besides the excellent performance obtained, batch systems present some intrinsic disadvantages when compared with flow-through (FT) systems, such as the non-continuous cycle of operations, difficult separation of products, catalysts, and polysaccharide pulp, mechanical disruption of pulp due to stirring, and slow heating profile.14,40 Additionally, batch systems require high reactor volumes for process intensification, which imply hazards and big space requirements. In contrast to batch, RCF in FT systems present physically separated feedstocks and catalyst beds with the feedstock located upstream from the catalyst bed. Accordingly, in the first bed, lignin is extracted from the lignocellulosic biomass and undergoes solvolytic fragmentation; afterwards, in the downstream reactor, lignin is catalytically depolymerized via hydrogenolysis and eventually, non-stable double-bond tails are stabilized via hydrogenation.33 Moreover, the de-coupled biomass and catalyst beds allow straightforward separation and different conditions apply for the two RCF steps.14 For these reasons, RCF in FT systems (FT-RCF) would become more favorable and cost-effective when a large production (>10 ktons per year) is targeted.40 Noteworthily, the FT-RCF is intrinsically a semi-continuous process owing to the necessity to replace the biomass bed once lignin is fully extracted. Nevertheless, it is possible to design the systems with multiple reactors and switch valves, which allow continuous operation.41 Additionally, FT systems have been indicated by IUPAC as a key technology that will make the chemical industry more sustainable, and they are intrinsically less hazardous.42,43
In a circular and green bio-economy, it is important to minimize waste and to design effective strategies to valorise the already existing waste.44,45 Beech is the most prevalent and economically relevant hardwood species in Europe, with an industrial usage of 160 ktons per year.46 As a side product of wood cutting, beech wood sawdust (BWS) represents a waste produced from the lumberjack industry with a low commercial value and it is commonly used as fuel, livestock bedding, adsorbent material, or particleboard panels.47,48 From a compositional perspective, hardwood lignin consists of both syringyl (S) and guaiacyl (G) units, with a typical S![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :G ratio of three.49,50 Consequently, hardwood has relatively abundant aryl-ether bonds (mostly β-O-4 linkages) and lower C–C linkages compared to softwood. Due to these properties, the RCF of hardwood achieved a monomer yield close to the maximum theoretical yield (∼40%). Therefore, the waste BWS has the potential to be upgraded in a biorefinery aimed at lignin valorization toward monomers.
:G ratio of three.49,50 Consequently, hardwood has relatively abundant aryl-ether bonds (mostly β-O-4 linkages) and lower C–C linkages compared to softwood. Due to these properties, the RCF of hardwood achieved a monomer yield close to the maximum theoretical yield (∼40%). Therefore, the waste BWS has the potential to be upgraded in a biorefinery aimed at lignin valorization toward monomers.
Nonetheless, only a few studies have been reported for RCF in FT systems. Firstly, Samec et al. reported a reductive lignocellulose fractionation in an FT reactor in which the extraction and the hydrogenolysis were separated by coupling two systems, achieving a maximum monomer yield of 31 wt% using MeOH/H2O as a solvent mixture and H-donor (7:3 wt% ratio, 0.1 mL min−1 educt flow), 5 wt% Pd/C as catalyst (150 mg) and H3PO4 as a co-catalyst (2.8 g L−1), birch wood (150 mg) as feedstock, and 200 °C and 180 °C as the extraction and reduction temperatures, respectively.51 Similarly, the Beckham and Roman Leshkov groups designed the FT process coupling a catalyst-packed bed reactor with a dual bed “switchable” packed bed reactor.41,52 In this study, the extraction bed was filled with 1 g of poplar wood while the catalyst bed was filled with 600 mg of pelletized Ni/C catalyst (15% Ni/C:SiO2, 1:1 wt ratio), yielding a maximum of 17 wt% monomer yield at a temperature of 190 °C, MeOH as solvent (flow rate 0.5 mL min−1) and a flow rate of 50 mL min−1 of H2 at 6 MPa.41 The same groups recently optimized the lignin extraction and minimized solvent consumption via recycling the lignin oil in a multi-pass FT-RCF.53 Tessonier and co-workers carried out the catalyst-free solvolysis of corn stover lignin with MeOH and EtOH in both subcritical and supercritical conditions. Notably, the maximum monomer yield was higher when using FT systems compared to batch, i.e., 4 wt%, 10 wt%, at 170 °C and 3.0 MPa of pressure. Interestingly, Li et al. combined a traditional batch solvolysis approach with catalytic flow hydrogenation of lignin.54 This study demonstrated the improved product selectivity (>80%) of RCF in flow as compared to a batch system using dioxane/water (9:1 vol. ratio) as a solvent, Pd/C catalyst (Pd/C:Al2O3 1:10 wt. ratio), reduction temperature of 140–190 °C, H2 flow of 20 cm3 min−1 at 35–60 MPa, educt flow of 0.2 cm3 min−1, and poplar wood as feedstock. In a complementary study from Brandner et al., an FT solvolytic extraction of lignin was combined with batch, and with in situ and ex situ FT-RCF, respectively, demonstrating that FT systems enable the production of native-like lignin.64 Noteworthily, the D'Angelo group reported a non-RCF hydrogen-free lignin-first approach using the FT system approach using birch and spruce wood as feedstock, ethanol:water 1:1 v/v as a solvent, and β-zeolite as a catalyst, obtaining 18 wt% monomer yield at 220 °C for extraction.56
Nevertheless, the application of FT systems in lignin valorisation is still in its infancy. The above-mentioned studies apply minimal quantities of feedstock and catalysts (mg-scale). Additionally, these works use catalysts pelletized with the addition of an inert phase (i.e. Pd/C with Al2O3,52 and Ni/C with SiO54), which presents intrinsic disadvantages for scaling up compared to catalysts already in pellet shape.56
Herein, we present the RCF of waste BWS in the FT system using two consecutive packed bed reactors for both BWS and catalyst beds, i.e., 10 g each, which is a larger amount than that used in literature, cf., Scheme 1. Moreover, the efficiencies of two different biomass-derivable solvents, MeTHF and MeOH, are investigated. Further, the temperature effect on lignin extractability as well as its catalytic hydrogenation is separately investigated. Noteworthily, these investigations were conducted using the waste product, i.e., beech wood sawdust (BWS) without further manipulation.
 | ||
| Scheme 1 Representation of the flow-through (FT) reductive catalytic (RCF) fractionation of beech wood sawdust (BWS); in the first reactor BWS is placed and lignin is extracted and solvolytically fragmented, while the 35 wt% Ni on nitrogen-doped carbon catalyst (35Ni/NDC) is placed in the second downstream reactor and catalytic depolymerization and hydrogenation occurs. | ||
Results and discussion
Catalyst synthesis and characterization
The used catalyst, i.e. pellet-shaped 35 wt% Ni on nitrogen-doped carbon (35Ni/NDC) was synthesized and characterized according to our “kitchen-lab” approach, as previously reported by our group.57 The prepared 35Ni/NDC was characterized using combustion elemental analysis (EA), inductively coupled plasma optical emission spectroscopy (ICP-OES), N2 physisorption, and powder X-ray diffraction spectroscopy (XRD). The catalyst synthetic procedure, as well as all the applied procedures for catalyst characterization, are described in detail in the ESI†.Compositional analysis of raw beech wood sawdust
To ensure an accurate product quantitative analysis the raw BWS feedstock was subjected to compositional investigations. The chemical composition was evaluated using elemental analysis for C, O and H, giving a respective content of 46 wt%, 44 wt% and 5.6 wt% (Table S1 in ESI†), which corresponds to a C/O ratio of 1:1. The Klason lignin (KL) was found to be 21 wt%, which is the expected value according to the literature.29 Moreover, the cellulose, and hemicellulose contents were 42 wt%, and 17 wt%, which are also in line with the literature (with arabinan, xylan, mannan, galactan and glucan, respectively, of 0.37, 16.12, 1.03, 0.63, and 41.69 wt%).29 All the values are listed in Tables S1 and S2 in ESI.† Additionally, the moisture and ash content in the BWS were calculated to be 4.7 wt% and 6.6 wt%, respectively (Fig. S7 in ESI†). The BWS crystallinity was investigated with XRD, showing the two typical cellulose peaks at 2θ of 15° and 22° (Fig. S8 in ESI†).63
Flow-through solvolytic extraction
The flow-through (FT) solvolytic extraction of lignin was conducted in a single BWS-packed reactor, cf. Fig. S4 in the ESI.† Accordingly, the effects of temperature and extraction time were studied using methanol (MeOH) and 2-methyltetrahydrofuran (MeTHF) as a solvent. MeOH is well known as an efficient solvent for lignin extraction.39,41,51,62 In contrast, MeTHF was scarcely investigated and can be alternatively used to simplify the separation of sugars from lignin via H2O–MeTHF liquid–liquid extraction.22 In addition, both solvents, i.e., MeOH and MeTHF, can be derived from lignocellulosic biomass in a biorefinery process.Initially, the effect of temperature on the efficiency of the lignin solvolytic extraction was evaluated in terms of residual BWS mass, residual wood crystallinity (XRD), and molecular weight distributions of the extracted products from size exclusion chromatography (SEC). Firstly, increasing the temperature from 175 °C to 200 °C and 225 °C corresponded, respectively, to a decrease in residual mass from 9.4 g to 8.6 g, and 6.5 g (Fig. 1), using MeTHF as the solvent. When MeOH was used as the solvent, the residual mass showed a decrease similar to MeTHF with 8.3 g, 7.6 g, and 6.9 g at 175 °C, 200 °C, and 225 °C, respectively (Fig. 1).
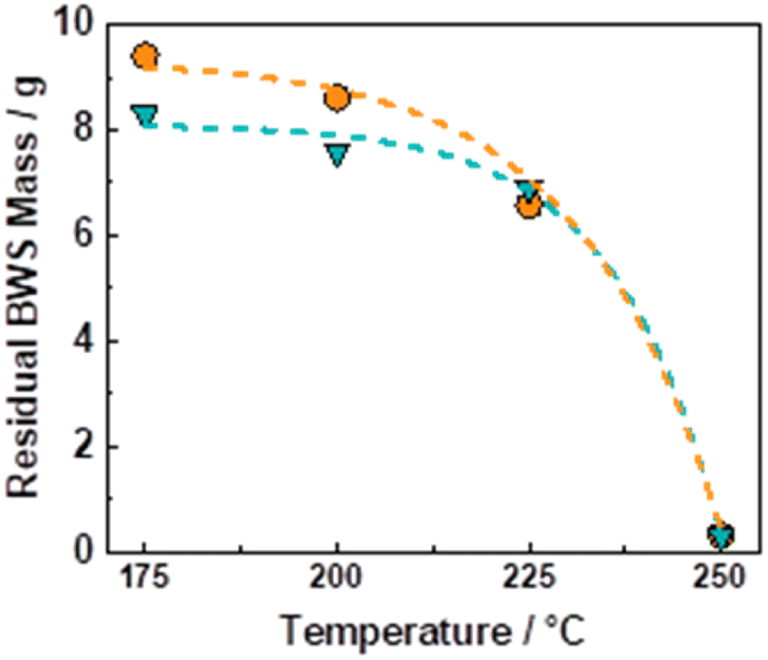 | ||
| Fig. 1 Residual BWS mass as a function of temperature in the FT-solvolytic lignin extraction process using MeTHF (blue triangle) and MeOH (orange circles) as solvents. Reaction conditions: mBWS = 10 g, Textraction = 175 °C, 200 °C, 225 °C, 250 °C, p = 7.0 MPa, Qeduct = 1.0 mL min−1, tresidence = 50 min, TOS = 4 h. | ||
In both cases, the low residual mass at 225 °C suggests the removal of lignin combined with the partial removal of the sugar fractions, i.e., hemicellulose. Accordingly, the XRD of the residual wood displayed cellulose peaks at 2θ of 15° and 22° (Fig. S9 in ESI†). This indicates the removal from the BWS of the amorphous components, i.e., lignin and hemicellulose, whereas the crystallinity of cellulose was preserved. A further increase in temperature from 225 °C to 250 °C corresponded to the almost complete BWS mass loss, yielding a residual mass of 0.3 g for both solvents (Fig. 1). This is combined with the complete loss of crystallinity of the residual wood displayed by XRD (Fig. S9 in ESI†) and is attributed to the complete degradation and liquefaction of cellulose. Therefore, the 6.9 g of residual BWS obtained using MeOH at 225 °C was the more suitable result and presented a KL content of 12 wt%, corresponding to a removal of 60 wt% of the KL from native BWS.
The sample at 225 °C was selected for further analysis and investigations. Firstly, the molecular weight distributions were analysed with SEC, resulting in a weighted average molecular weight distribution (Mw) of 1960 g mol−1 and a number average molecular weight distribution (Mn) of 896 g mol−1 for the experiment using MeTHF as a solvent. When MeOH was used, the Mw and Mn were slightly lower, being 1352, and 925 g mol−1, respectively (Fig. S10 in ESI†). The calculated Mw values are in good agreement with a study conducted by Sahayaraj et al. using FT systems with MeOH as a solvent.63 Moreover, the structural properties of the extracted lignin were investigated with the 2D-HSQC NMR. The sample using MeOH as a solvent was selected for further analysis due to its lower residual BWS weight after the extraction. As shown in Fig. 2, the extracted lignin presented intact native-like ether moieties, such as the β-O-4 aryl-ether (δC/δH = 70–75/4.5–5.5 and 80–82/4.0–4.5 ppm – in green), and β–β resinol (δC/δH = 84–87/4.3–4.7 in red). The presence of these bonds is a strong indication that the solvolysis conditions are not sufficiently strong to cleave all the native-like lignin. The typical hardwood signal of syringil (S) and guaiacyl (G) phenolic units are in the aromatic region (δC/δH = 100–115/6.0–7.5 ppm). Noteworthily, the FT solvolysis process produced a double bond tail (δC/δH = 120–130/6.0–6.5 ppm), which indicated that the solvothermal cleavage temperature was not followed by re-condensation at high temperatures. In this respect, the capacity of the FT system to isolate native-like lignin was recently reported by Roman-Leshkov and Beckham groups using poplar wood (i.e. hardwood).64 Herein, our findings confirm these results. The presence of ester signals (ROOCH3) is attributed to the condensation of MeOH with carboxylic functionalities, mostly from fatty acid. Additionally, the solvolysis produced signals that are commonly attributed to hemicellulosic xylans (δC/δH = 110–120/4.0–5.5 ppm).65 The presence of these signals, combined with the low residual BWS mass is a strong indication that hemicellulose is also removed together with lignin.
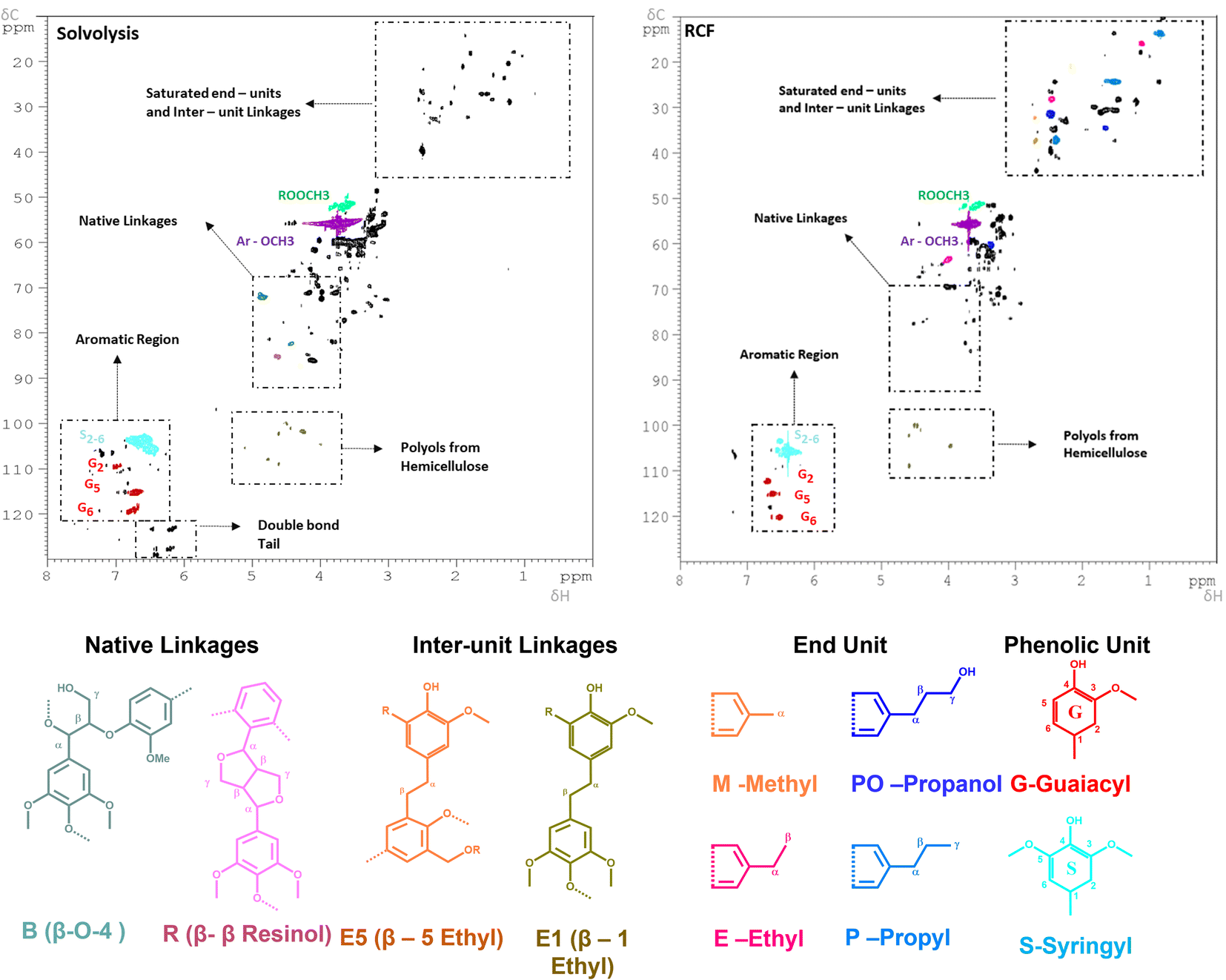 | ||
| Fig. 2 2D-HSQC NMR spectra of the (left) FT-solvolytic and (right) FT-RCF (MeOH) extraction of lignin. The detailed assignments of the saturated end-units/inter-unit linkages (δC/δH = 0–60/0–4 ppm) and native linkages regions (δC/δH = 0–60/0–4 ppm) are shown in Fig. S11 and S12 in the ESI.† Reaction conditions: m35Ni/NDC = 10 g, MBWS = 10 g, Textraction = 225 °C Treduction = 225 °C, p = 7.0 MPa, Qeduct = 1.0 mL min−1, QH2 = 48 mL min−1, tresidence1 = tresidence2 = 50 min. | ||
One of the advantages of operating in FT systems is the possibility of providing precise time-resolved investigations. Therefore, the UV-vis spectra of the extracted solutions were recorded once for every hour of time on stream. Accordingly, the first sample, at 1 h of TOS using MeOH showed the two typical lignin maxima at wavelengths of 208 nm and 250 nm, corresponding to the π → π* electronic transition in the aromatic ring and the conjugated methoxyphenyl group, respectively (Fig. 3).66,67 However, the presence of the UV inhibitor, BHT, as a stabilizer in MeTHF strongly interfered with the measurement, hampering a precise peak attribution (Fig. S13 in ESI†).
 | ||
| Fig. 3 UV-vis spectra of FT-solvolytic lignin extraction using MeOH as the solvent at different times. Reaction conditions: mBWS = 10 g, Textraction = 225 °C, p = 7.0 MPa, Qeduct = 1.0 mL min−1, tresidence = 50 min, time on stream = 1 h, 2 h, 3 h, 4 h. | ||
Nevertheless, in both cases, increasing the TOS from 1 h, to 2 h, 3 h and 4 h, corresponds to a progressive decrease in the absorbance intensity of the identified peaks. This is attributed to the progressive decrease in the aromatic chromophore concentration, i.e., lignin concentration in the extracted solution. Thus, the efficiency of lignin extraction is maximized at the beginning of the process and decreases with increasing TOS. Therefore, the lignin extraction is maximized within the first two hours of extraction, which is in complete agreement with the previous work by the Beckham and Roman Leshkov groups.41 Moreover, the presence of lignin even after 4 h is due to an incomplete lignin extraction process, which is attributed to lignin being non-accessible by the solvent.
Flow-through reductive catalytic fractionation
In this section, the solvolytic lignin extraction described previously was coupled with a catalytic step to perform the FT-RCF process using 35Ni/NDC as a catalyst. The parent carbonized support and the 35Ni/NDC showed a specific surface area (ABET) of 755 m2 g−1 and 578 m2 g−1, respectively as well as a type IV isotherm, which is typical of mesoporous materials (Fig. S14 and Table S3 in ESI†). The XRD pattern of 35Ni/NDC exhibited two typical Ni0 reflections at 44° and 51° (Fig. S15 in ESI†). Moreover, elemental analysis confirmed the Ni loading of the desired 35 wt% as well as the C/N ratio of 22, indicating relatively high N doping (Table S3 in ESI†).Based on the solvolysis experiments, a temperature of 225 °C, was selected. For this purpose, two reactors were coupled, one for lignin extraction and solvothermal fragmentation, and the other for catalytic reduction, cf. Fig. S3 in the ESI.† The efficiencies of MeTHF and MeOH as solvents for the RCF were investigated in terms of monomer yield and will be noted as RCF-MeTHF and RCF-MeOH. Initially, the temperature was set to 225 °C for both the fixed bed reactors, i.e., the BWS bed and the 35Ni/NDC bed. A time-resolved analysis was conducted by collecting samples every hour for 10 h for both solvents, MeOH and MeTHF (Fig. 4). The lignin-derived monomers were identified via GC-MS and quantified with GC-FID (Fig. 4 and Table S4 in ESI†). In the case of MeTHF, the cumulative monomer yield was found to progressively decrease with the increase of time on stream (TOS), from 11.0 mg gKL−1 after 1 h to 9.5 mg gKL−1 after 2 h and to 6.8 mg gKL−1 after 3 h. Moreover, extending the TOS to 4, 8, and 10 h corresponds to a decrease in the cumulative monomer yield from 4.7 mg gKL−1 to 2.8 mg gKL−1 and 2.7 mg gKL−1, respectively (Fig. 4). The same progressive decrease in cumulative monomer yield was found using MeOH as a solvent, namely from 22 mg gKL−1 at 1 h to 14 mg gKL−1 at 2 h of TOS, to 7.6 mg gKL−1 at 3 h and 5.0 mg gKL−1 at 4 h of TOS (Fig. 4). Extending the TOS to 8 and 10 h showed a cumulative monomer yield of 2.97 and 2.78 mg gKL−1, respectively. In the first 2 h of TOS, the cumulative monomer yield using MeOH as a solvent was found to be double in comparison to MeTHF, (22.2 and 11.0 mg gKL−1, respectively). Increasing the TOS from 2 to 10 h corresponded to a progressive decrease in cumulative monomer yield until 2.7 mg gKL−1 for both solvents (Fig. 4). These findings indicate that the monomers yield is maximized within the first 4 h of TOS, which is correlated to the decay in lignin extraction from BWS observed with UV-vis analysis (Fig. 3). This is in good agreement with the findings of the Beckham and Roman Leshkov groups who reported a similar decrease after the first 4 hours of TOS.41
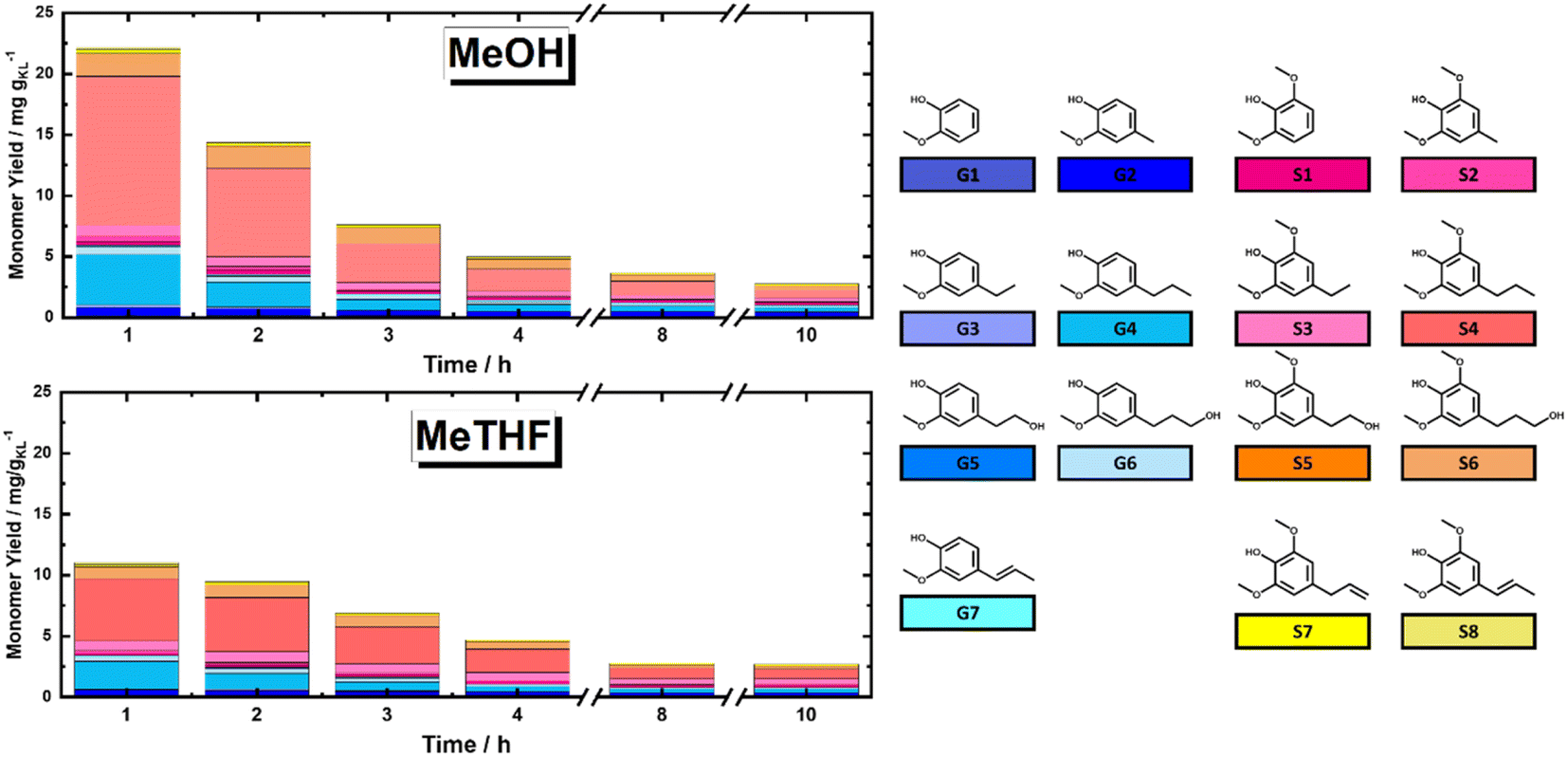 | ||
| Fig. 4 Monomer yield of FT-RCF process as a function of time on stream (TOS) using (top) MeOH and MeTHF (bottom) as solvent. The structures of quantified monomers are reported with the corresponding color. Reaction conditions: m35Ni/NDC = 10 g, MBWS = 10 g, Textraction = 225 °C Treduction = 225 °C, p = 7.0 MPa, Qeduct = 1.0 mL min−1, QH2 = 48 mL min−1, tresidence1 = tresidence2 = 50 min, TOS = 1 h, 2 h, 3 h, 4 h, 8 h, 10 h. | ||
To maximize the lignin extraction, the reaction products were collected from the beginning to the end of the reaction (after 10 of TOS, around 600 mL) using both MeOH and MeTHF as solvents (Fig. 5). As expected, the total monomer yield using MeOH as a solvent was doubled in comparison to MeTHF, i.e., 186 mg gKL−1, and 98.3 mg gKL−1, respectively. Moreover, the molecular weight distribution calculated from the SEC profile of RCF-MeOH was slightly smaller than in the case of RCF-MeTHF at 225 °C, with Mw of 638 g mol−1 and 943 g mol−1, respectively (Fig. S16 in ESI†). This finding, in combination with a high monomer yield, is evidence that MeOH is a more favorable solvent for obtaining a high cumulative monomer yield. This higher MeOH efficiency is attributed to a higher polarity (and Lewis acidity) with respect to MeTHF, which allows deeper penetration of the solvent inside the wood matrix and higher lignin accessibility.68 After 10 h of TOS, a residual wood mass of 6.3 and 6.6 g was found for MeOH and MeTHF, respectively. This residual mass is only slightly lower than that obtained after 4 h with the pure solvolytic experiment of 6.5 and 6.9 g, respectively, for MeOH and MeTHF, indicating poor lignin extractability after the 4th hour of TOS. Importantly, the 186 mg gKL−1 of the MeOH experiment corresponds to a yield of 30 wt% of the extracted KL from the BWS.
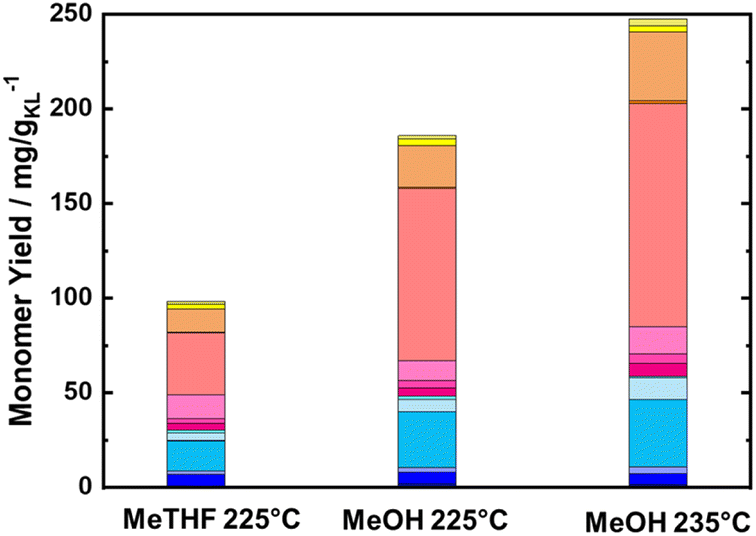 | ||
| Fig. 5 Monomer yield of the FT-RCF process after 10 h of TOS in a function of the used solvent, i.e., MeTHF and MeOH. Reaction conditions: m35Ni/NDC = 10 g, mBWS = 10 g, Textraction = 225 °C, and 235 °C Treduction = 225 °C, p = 7.0 MPa, Qeduct = 1.0 mL min−1, QH2 = 48 mL min−1, tresidence1 = tresidence2 = 50 min, TOS = 10 h. The colors refer to the compounds listed in Fig. 4. | ||
Nevertheless, despite its lower cumulative monomer yield, the results obtained with MeTHF are interesting for establishing a liquid–liquid extraction process with water to separate the saccharide-derived fraction (in the water phase) from the aromatics (in the oil phase), as our group previously reported.22
Nevertheless, both solvents displayed similar monomer distribution all over the investigated TOS, with 4-propyl syringol and 4-propyl guaiacol (G1 and S4) being the most abundant quantified monomers (Fig. 4). These monomers are typically obtained from the RCF of hardwood and present a fully reduced alkyl tail, indicating the successful reduction process.15 It is noteworthy that the selectivity for predominant monomers remains unchanged in reaction time (after 10 hours), which allows the exclusion of catalyst deactivation as a reason for lowering the rate of extraction. Moreover, for the investigated TOS, the syringyl to guacyl ratio (S/G) was ∼3, which was expected with respect to the initial BWS S/G ratio (3.36). The constant S/G ratio indicates a near-homogeneous extraction process of lignin from the LCB matrix. Additionally, the monomer yield shows a clear advantage of MeOH over MeTHF as a solvent. This is in agreement with the findings of the Sels group, which reported the efficiency of MeOH as a solvent in batch systems.37,62
The 2D HSQC NMR (Fig. 2) of the RCF-MeOH at 225 °C showed the complete absence of the native ether linkages, aryl-ether (δC/δH = 70–75/4.5–5.5 and 80–82/4.0–4.5 ppm), and β–β resinol (δC/δH = 84–87/4.3–4.7). Moreover, a reduced number of signals in the ether region (δC/δH = 50–90/2–6) was found when compared with the solvolytic experiment using MeOH at 225 °C (Fig. S11 in ESI†). These indicate that the hydrogenation and cleavage of ethers bond are more effective in the presence of the 35Ni/NDC catalyst. In addition, the signals in the aliphatic region δC/δH = 20–35/1–2.5 ppm were found to be intensified when compared to the solvolytic experiment, proving the presence of saturated end-units and the effect of the reduction step. In particular, the signal of the identified monomer's end-unit was found to appear with clear intensity in the spectra after FT-RCF, viz. Fig. S12 in the ESI.† Here, all the expected end-units quantified by FID were found, i.e. methyl-, ethyl-propyl-, and propanol-end units, viz.Fig. 2. These observations are a clear indication of the successful reduction obtained via the RCF process. Moreover, additional signals related to the inter-unit linkages were found (β-1 ethyl and β-5 ethyl), (δC/δH = 36–37/2.6–2.7 ppm, and δC/δH = 30–32/2.5–2.7 ppm), which proves the presence of lignin-derived dimers, trimers, and larger oligomers. Moreover, the RCF spectra showed a reduced number of xylans (δC/δH = 110–120/4.0–5.5 ppm) than in the solvolytic experiment, which is attributed to the degradation and hydrogenation of the sugars in the 35Ni/NDC catalyst reactor. GC-MS revealed the presence of furanic compounds obtained by sugar dehydration.
To maximize the cumulative monomer yield, a further experiment using MeOH as a solvent was performed, increasing the temperature for the first reactor (BWS bed) from 225 °C to 235 °C, while the second reactor (RCF) was kept constant at 225 °C. Similar to the experiment at 225 °C, a time-resolved study was conducted for the RCF with 235 °C as the extraction temperature. The cumulative monomer yield of this experiment for the first 4 h of TOS was found to be higher than that with 225 °C as the extraction temperature, yielding 34.1 mg gKL−1 at 1 h of TOS, 19.7 mg gKL−1 at 2 h of TOS, 10.4 mg gKL−1 at 3 h of TOS, and 6.4 mg gKL−1 at 4 h of TOS (Fig. 6). Furthermore, the complete quantification of lignin was made by collecting samples for 10 h (600 mL) and the cumulative yield was found to increase from 185 mg gKL−1 to 247 mg gKL−1 (Fig. 5). The higher total monomer yield at 235 °C is attributed to more accessible lignin in comparison with the experiment at 225 °C.
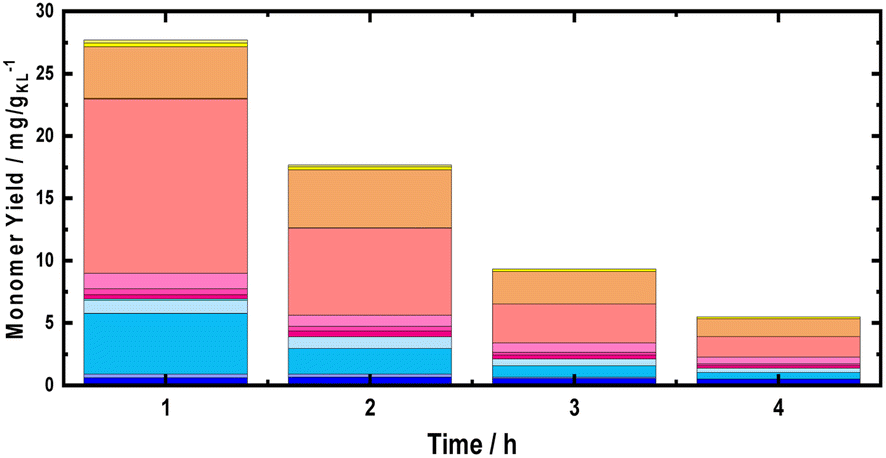 | ||
| Fig. 6 Monomer yield of the FT-RCF process as a function of time on stream using MeOH as the solvent. Reaction conditions: m35Ni/NDC = 10 g, mBWS = 10 g, Textraction = 235 °C Treduction = 225 °C, p = 7.0 MPa, Qeduct = 1.0 mL min−1, QH2 = 48 mL min−1, tresidence1 = tresidence2 = 50 min, TOS = 1 h, 2 h, 3 h, 4 h. The colors refer to the compounds listed in Fig. 4. | ||
Nonetheless, the total monomer yield of 247 mg gKL−1 is lower than the reported values for batch systems using hardwood (400–550 mg gKL−1), which is attributed to the lack of accessibility to lignin due to the large BWS size.36 The residual BWS mass was found to decrease from 6.4 g to 5.9 g, with the increased extraction temperature of 225 °C to 235 °C, which indicates that a greater amount of sugar was extracted. Moreover, the Klason lignin content was found to be 11 wt%, corresponding to 0.62 g of lignin (30% of the native BWS). Therefore, the cumulative monomer yield referring to the extracted lignin is 37 wt%, i.e., close to the theoretical maximum yield of 40 wt%.
However, the total monomer yield of 247 mg gKL−1 is slightly lower than the 310 mg gKL−1 reported by the Samec group51 and higher than the 172 mg gKL−1 from the Beckham and Roman Leshkov groups.41 These values correspond to a cumulative monomer yield of 25 wt%, 31 wt%, and 17 wt%, viz, Fig. S17 in the ESI† for comparison. However, these studies were conducted with different catalysts (Pd/C with H3PO4 as a cocatalyst51 and Ni/C mixed with SiO2 (ref. 41)) as well as with different feedstocks (birch51 and poplar41). Compared with the Samec group,51 herein, we did not include any co-catalyst, which could have contributed to the lower yield. When compared to the Leshkov group,41 this higher monomer yield is attributed to a long TOS of 10 h, as well as to the usage of a pellet-shaped 35Ni/NDC catalyst rather than mixed Ni/C with SiO2. Moreover, we were able to quantify 15 compounds compared to 8 from the Beckham and Roman Leshkov groups, which we attribute to the larger amount of biomass used.
Conducting the RCF in FT systems with two distinct tubular reactors allows for selectively tuning the reaction conditions of the single process. Therefore, to investigate the optimal reduction temperature, the extraction reactor was kept at a constant temperature of 235 °C, while the reduction temperature was varied (Fig. 7). The catalyst performances were evaluated in terms of cumulative monomer yield after 1 h of time on stream, i.e., when the lignin extraction is maximized. Initially, the temperature was set to 200 °C, and increased to 225 °C, 235 °C, and 250 °C. At 200 °C, the cumulative monomer yield was 20 mg gKL−1, lower than the 26 mg gKL−1 at 225 °C (Fig. 7). Moreover, a further increase in the temperature of 10 °C to reach 235 °C produced only a slight change in the cumulative monomer yield, i.e., from 26 to 27 mg gKL−1. We attribute the difference in cumulative monomer yield to the hydrogenolysis of aryl ether groups, which requires a high temperature (225 °C–235 °C) to be efficient. Finally, at a reduction temperature of 250 °C, the cumulative monomer yield drastically decreased to 4 mg gKL−1. This decrease is a clear indication of the lack of stability of the produced compounds, which undergo condensation at 250 °C.
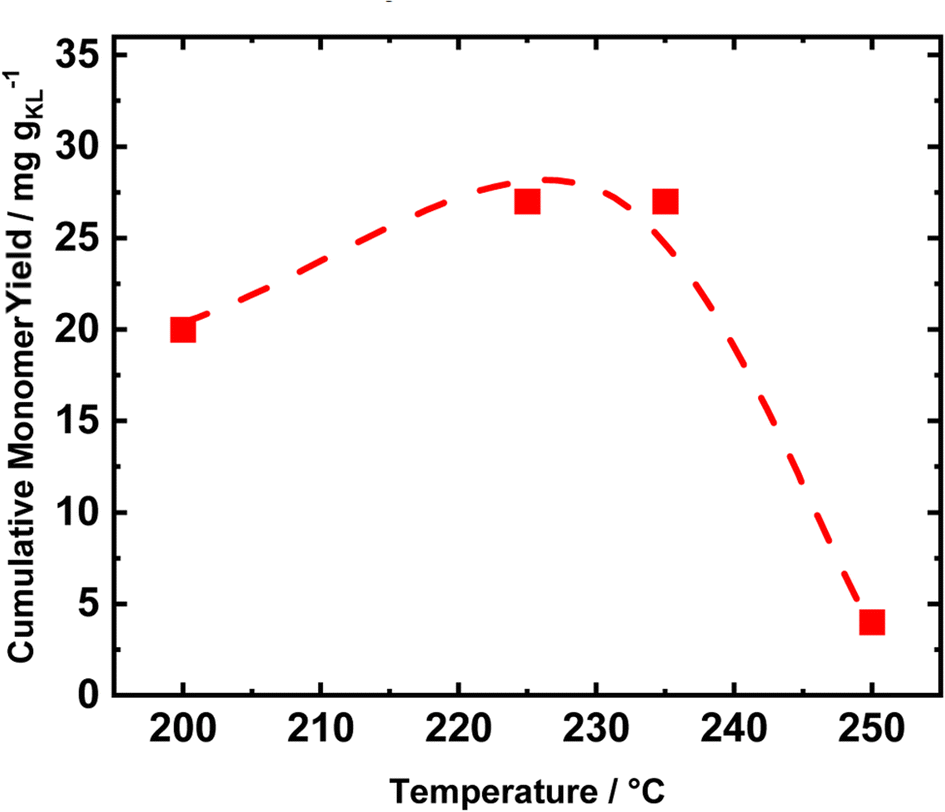 | ||
| Fig. 7 Cumulative monomer yield of the FT-RCF process as a function of temperature after 1 h of time on stream using MeOH as the solvent. Reaction conditions: m35Ni/NDC = 10 g, mBWS = 10 g, Textraction = 225 °C Treduction = 200°, 225 °C, 235 °C, and 250 °C p = 7.0 MPa, Qeduct = 1.0 mL min−1, QH2 = 48 mL min−1, tresidence1 = tresidence2 = 50 min, TOS = 1 h. | ||
The intensification of the RCF process using BWS feedstock requires the continuous replacement of spent BWS feedstock with fresh ones. One of the advantages of the FT tubular reactor is the possibility to replace spent beds with new ones by simply switching them, which can be ideally done by switchable valves.
Moreover, using multiple BWS beds without changing the catalyst bed allows the investigation of the catalytic performances of 35Ni/NDC over a longer TOS, assessing its recyclability over multiple RCF cycles. Herein, a second fresh BWS bed was placed to substitute for the spent one after 10 h of the above-discussed reaction. Previously, we demonstrated that the first 4 h of reactions were the most efficient for extracting lignin; therefore, the catalytic performance over the second bed was investigated for 4 h, which corresponds to a total of 14 h (Fig. 8).
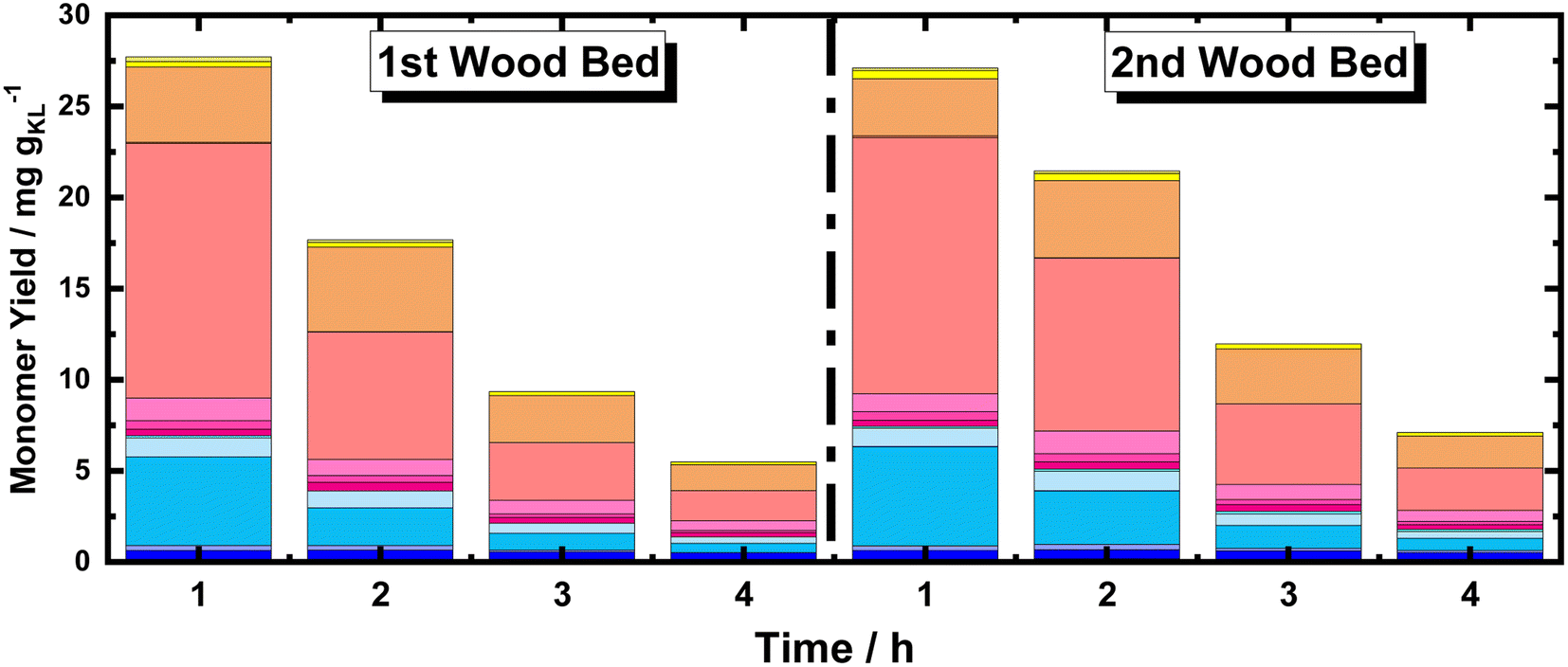 | ||
| Fig. 8 Monomer yield of the FT-RCF process as a function of time on stream (TOS) using MeOH as solvent. Reaction conditions: m35Ni/NDC = 10 g, mBWS = 10 g, Textraction = 235 °C Treduction = 225 °C, p = 7.0 MPa, Qeduct = 1.0 mL min−1, QH2 = 48 mL min−1, tresidence1 = tresidence2 = 50 min. The colors refer to the compounds listed in Fig. 4. | ||
Despite a long time on stream, the cumulative monomer yield of the second batch was found to be analogous to the fresh one. In both cases, around 26 mg gKL−1 were found in the first hour of extraction, which linearly decreased to around 4 mg gKL−1 after 4 h of TOS. Importantly, the product distribution was almost identical, indicating no substantial changes in the reductive catalytic fractionation after a total of 14 h of TOS and 2 cycles of RCF. Moreover, the spent catalyst textural properties were analysed with XRD and N2 physisorption. The N2 physisorption of the spent catalyst (35Ni/NDC spent) showed a slightly decreased specific surface area if compared to the fresh 35Ni/NDC, decreasing from ABET of 578 m2 g−1 to 525 m2 g−1 (Fig. S18 in ESI†). This decrease is attributed to the deposition of some unreacted lignin into the surface of the catalysts. The XRD of the spent catalyst exhibited the same typical Ni0 reflection of the fresh catalyst, indicating the stability of the catalyst in this high solvothermal condition (Fig. S19 in ESI†). These findings indicate that the 34Ni/NDC catalyst was stable and active for two RCF cycles and a total of 14 h. Moreover, the unchanged monomer yield, as well as the preserved catalytic textural properties of the second cycle confirmed that the decrease over the 4 h of TOS was due to lignin extractability rather than catalyst deactivation.
Conclusions
The implementation of a flow-through (FT) process into the lignin biorefinery has the potential to increase the overall efficiency of the system, and consequently, its sustainability. In this work, we successfully applied a cheap and scalable 35 wt% Ni on nitrogen-doped carbon catalyst (35NI/NDC) in pellet shape for the FT-RCF of beech wood sawdust (BWS). This was accomplished using two biomass-derivable solvents, i.e. MeOH and MeTHF. The best results in terms of the cumulative yield of 247 mg gKL−1 were obtained using MeOH as a solvent after 10 h of time on stream (TOS) at 235 °C and 225 °C as extraction and reduction temperatures, respectively. This yield corresponds to a 37 wt% yield of the extracted lignin, close to the maximum theoretical yield of 40 wt% and with more identified monomers when compared to other FT-RCFs.The solvent was found to play a key role in lignin extraction, and MeOH outperformed MeTHF due to its polarity. Nevertheless, MeTHF usage in such systems was pioneered, obtaining 98.3 mg gKL−1 as the highest cumulative monomer yield. Additionally, the lignin extraction from BWS was found to happen fast, with the maximized efficiency within the first 4 h of time on stream (TOS). To this end, we proved the catalyst stability over two cycles of RCF, replacing the spent BWS bed with a fresh one without observing any changes in the catalytic activity after a total of 14 h of TOS.
To our knowledge, this is the first example of FT-RCF that exceeds the mg-scale in both wood and catalyst beds. Therefore, the presented results of BWS-RCF in FT systems can provide a powerful tool that enables the implementation of flow processes into the lignin biorefinery, and potentially increases the efficiency of such systems. We are currently working on making this process continuous via the modification of the presented FT system in this study. In this approach, we are connecting parallel wood bed reactors with solvent switching valves to each of these reactors (every 4 hours the solvent is switched to a fresh wood bed). Furthermore, we are developing a twin-screw extruder reactor, in which the wood bed and solvent are fed continuously at the extruder (solvolytic extraction and fragmentation), and then the extracted and pulp-free fragmented lignin oil passes to a tubular reactor containing the fixed bed RCF catalyst.
Experimental
Materials
All the materials were utilized as received from the supplier without any further purification. A detailed list of the used chemicals, suppliers, and purities can be found in the ESI†.Catalyst synthesis and characterization
The used catalyst, i.e. pellet-shaped 35 wt% Ni on nitrogen-doped carbon (35Ni/NDC), was synthesized and characterized according to our “kitchen-lab” approach, as previously reported by our group.57 The prepared and spent 35Ni/NDC was characterized using combustion elemental analysis (EA), inductively coupled plasma optical emission spectroscopy (ICP-OES), N2 physisorption, and powder X-ray diffraction spectroscopy (XRD). The catalyst synthetic procedure, as well as all the applied procedures for catalyst characterization, are described in detail in the ESI†.Flow-through setups
The flow-through solvolysis and reductive catalytic fractionation (FT-RCF) of lignin extracted from beech wood sawdust experiments were conducted in a homemade FT fixed-bed system, similar to what was already reported by our group.22,57–60 In the case of solvolytic lignin extraction, the setup consisted of (A) an HPLC pump equipped with a pressure sensor, (B) two-side-opened independent heating units equipped with a heat controller, and (C) a sampling unit equipped with proportional relief valves as a pressure regulator (Fig. S2 in ESI†). The setup for RCF was similar to that described above and reported with its flowchart in Fig. S3 in the ESI.† After pumping (A), (B) the H2 was supplied via a mass flow controller (Brooks Instruments, Model SLA5850SC1AF1B2A1) connected through (C) a “T” union for H2-reactant mixing (Swagelok SS-400-30) before reaching the pre-heating unit and then (D1) and (D2) the two-side-opened independent heating units and (E) the sampling unit. To ensure efficient heating, an aluminum cylinder with three holes was placed inside the heating unit, i.e., a preheating unit for the reactant before it comes in contact with the catalyst, a hole for the thermocouple (Model # A472 × 105 Parr Instrument Company), and a hole for the reactor (Fig. S4 in ESI†). The investigations were conducted using a stainless steel tubular reactor with an inner diameter of 21 mm, an outer diameter of 25 mm, and a length of 280 mm (Fig. S5 in ESI†).Catalytic experiment
In a typical RCF experiment, two reactors were coupled in series, filled with 10 g of BWS, followed by 10 g on 35Ni/NDC, and named the BWS-bed and 35Ni/NDC-bed, respectively. In both the 35Ni/NDC and BWS beds, 1 g of glass wool was located at the entrance and exit of the reactor to ensure efficient heating and to prevent clogging of the system.The solvent solution (MeOH or MeTHF) was fed via HPLC pump at 1.0 mL min−1 with the addition of 40 mL min−1 of H2 at 7.0 MPa passed through the preheating unit and tubular reactor. The temperature and pressure were kept constant under ambient conditions for 30 minutes. Afterward, the system was pressurized to 7.0 MPa to ensure the presence of the solvent system in a liquid state. Later, the system was heated to the desired reaction temperature (175 °C, 200 °C, 225 °C, 235 °C, and 250 °C). Samples (20 mL) were collected once the steady state was reached (ca. 120 min). In the case of the continuous solvolytic extraction, an analogous procedure was followed, with the exception of only using the BWS reactor, as reported in Fig. S2 of the ESI†.
The collected sample was injected in the SEC without further processing, while a product analysis procedure was established to analyze the samples in GC-MS, GC-FID, and 2D HSQC NMR, viz Scheme 1 in the ESI.† The used product analysis procedure is described in detail in the ESI†.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors are grateful to the Max Planck Society for the financial support. The authors are grateful to Irina Shekova for her support in catalyst synthesis. Jessica Brandt, Antje Völkel, and Marlies Gräwert from Max Planck Institute of Colloids and Interfaces are acknowledged for ICP, EA, and SEC analysis respectively. Thanks are also extended to Klaus Bienert, Michael Born, Marco Bott, Tobias Schmidt, and Paul Meiβner from the electrical and mechanical workshop and IT at the Max Planck Institute of Colloids and Interfaces. The author are thankful to Matteo Faggioli for the idea and design of the cover artwork.References
- J. E. Holladay, J. F. White, J. J. Bozell and D. Johnson, Top value-added chemicals from biomass-Volume II—Results of screening for potential candidates from biorefinery lignin, Pacific Northwest National Lab (PNNL), Richland, WA (United States), 2007 Search PubMed.
- T. Werpy and G. Petersen, Top Value Added Chemicals from Biomass: Volume I – Results of Screening for Potential Candidates from Sugars and Synthesis Gas, Office of Scientific and Technical Information, Oak Ridge, TN, USA, 2004, https://www.osti.gov/servlets/purl/15008859 Search PubMed.
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2014, 16, 516–547 RSC.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS PubMed.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- D. Esposito and M. Antonietti, Chem. Soc. Rev., 2015, 44, 5821–5835 RSC.
- R. A. Sheldon, Philos. Trans. R. Soc., A, 2020, 378, 20190274 CrossRef CAS PubMed.
- J. Martin and M. Haggith, Enviromental Paper Network: The State of Global Paper Industry, http://www.environmentalpaper.org Search PubMed.
- E. Calcio Gaudino, G. Cravotto, M. Manzoli and S. Tabasso, Green Chem., 2019, 21, 1202–1235 RSC.
- F. H. Isikgor and C. R. Becer, Polym. Chem., 2015, 6, 4497–4559 RSC.
- L. Taiz, E. Zeiger, I. M. Moller and A. Murphy, Plant Physiology and Development, Oxford University Press Inc., United States (US), 6th edn, 2018 Search PubMed.
- M. M. Abu-Omar, K. Barta, G. T. Beckham, J. Luterbacher, J. Ralph, R. Rinaldi, Y. Roman-Leshkov, J. Samec, B. Sels and F. Wang, Energy Environ. Sci., 2021, 14, 262–292 RSC.
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- R. Vanholme, B. Demedts, K. Morreel, J. Ralph and W. Boerjan, Plant Physiol., 2010, 153, 895–905 CrossRef CAS PubMed.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- C. H. Zhou, X. Xia, C. X. Lin, D. S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588–5617 RSC.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- X. L. Tong, Y. Ma and Y. D. Li, Appl. Catal., A, 2010, 385, 1–13 CrossRef CAS.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- F. Brandi, M. Antonietti and M. Al-Naji, Green Chem., 2021, 23, 9894–9905 RSC.
- B. Kamm, P. R. Gruber and M. Kamm, Ullman's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KgaA, 2016, pp. 1–38 Search PubMed.
- T. Renders, S. Van den Bosch, S. F. Koelewijn, W. Schutyser and B. F. Sels, Energy Environ. Sci., 2017, 10, 1551–1557 RSC.
- M. Y. Balakshin, E. A. Capanema, I. Sulaeva, P. Schlee, Z. Huang, M. Feng, M. Borghei, O. J. Rojas, A. Potthast and T. Rosenau, ChemSusChem, 2021, 14, 1016–1036 CrossRef CAS PubMed.
- A. Corona, M. J. Biddy, D. R. Vardon, M. Birkved, M. Z. Hauschild and G. T. Beckham, Green Chem., 2018, 20, 3857–3866 RSC.
- R. Davis, N. J. Grundl, L. Tao, M. J. Biddy, E. C. Tan, G. T. Beckham, D. Humbird and M. S. Roni, Process Design and Economics for the Conversion of Lignocellulosic Biomass to Hydrocarbon Fuels and Coproducts: 2018 Biochemical Design Case Update; Biochemical Deconstruction and Conversion of Biomass to Fuels and Products via Integrated Biorefinery Path, 2018, https://www.nrel.gov/docs/fy19osti/71949.pdf Search PubMed.
- Y. Liao, S.-F. Koelewijn, G. Van den Bossche, J. Van Aelst, S. Van den Bosch, T. Renders, K. Navare, T. Nicolaï, K. Van Aelst, M. Maesen and B. Sels, Science, 2020, 367, 1385–1390 CrossRef CAS PubMed.
- Y. M. Questell-Santiago, M. V Galkin, K. Barta and J. S. Luterbacher, Nat. Rev. Chem., 2020, 4, 311–330 CrossRef CAS.
- Z. Sun, G. Bottari, A. Afanasenko, M. C. A. Stuart, P. J. Deuss, B. Fridrich and K. Barta, Nat. Catal., 2018, 1, 82–92 CrossRef CAS.
- H. P. Godard, J. L. McCarthy and H. Hibbert, J. Am. Chem. Soc., 1940, 62, 988 CrossRef CAS.
- J. M. Pepper, W. F. Steck, R. Swoboda and J. C. Karapally, in Lignin Structure and Reactions, ed. A. C. Society, 1966, pp. 238–248 Search PubMed.
- T. Renders, G. Van den Bossche, T. Vangeel, K. Van Aelst and B. Sels, Curr. Opin. Biotechnol., 2019, 56, 193–201 CrossRef CAS PubMed.
- M. Al-Naji, F. Brandi, M. Driess and F. Rosowski, Chem. Ing. Tech., 2022, 94, 1611–1627 CrossRef CAS.
- T. Renders, E. Cooreman, S. Van den Bosch, W. Schutyser, S. F. Koelewijn, T. Vangeel, A. Deneyer, G. Van den Bossche, C. M. Courtin and B. F. Sels, Green Chem., 2018, 20, 4607–4619 RSC.
- S. Van den Bosch, S. F. Koelewijn, T. Renders, G. Van den Bossche, T. Vangeel, W. Schutyser and B. F. Sels, in Topics in Current Chemistry, 2018, vol. 376 Search PubMed.
- S. Van den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S. F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Energy Environ. Sci., 2015, 8, 1748–1763 RSC.
- W. Schutyser, S. Van den Bosch, T. Renders, T. De Boe, S. F. Koelewijn, A. Dewaele, T. Ennaert, O. Verkinderen, B. Goderis, C. M. Courtin and B. F. Sels, Green Chem., 2015, 17, 5035–5045 RSC.
- K. Van Aelst, E. Van Sinay, T. Vangeel, E. Cooreman, G. Van den Bossche, T. Renders, J. Van Aelst, S. Van den Bosch and B. F. Sels, Chem. Sci., 2020, 11, 11498–11508 RSC.
- E. Cooreman, T. Vangeel, K. Van Aelst, J. Van Aelst, J. Lauwaert, J. W. Thybaut, S. Van den Bosch and B. F. Sels, Ind. Eng. Chem. Res., 2020, 59, 17035–17045 CrossRef CAS.
- E. M. Anderson, M. L. Stone, R. Katahira, M. Reed, G. T. Beckham and Y. Román-Leshkov, Joule, 2017, 1, 613–622 CrossRef CAS.
- F. Gomollon-Bel, Chem. Int., 2019, 41, 12–17 CAS.
- R. Gerardy, D. P. Debecker, J. Estager, P. Luis and J. M. Monbaliu, Chem. Rev., 2020, 120(15), 7219–7347 CrossRef CAS PubMed.
- J. H. Clark, T. J. Farmer, L. Herrero-Davila and J. Sherwood, Green Chem., 2016, 18, 3914–3934 RSC.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- A. Kozuch and J. Banaś, Forests, 2020, 11, 902 CrossRef.
- N. M. Clauser, S. Gutiérrez, M. C. Area, F. E. Felissia and M. E. Vallejos, Biofuels, Bioprod. Biorefin., 2018, 12, 997–1012 CrossRef CAS.
- J. M. Harkin, Uses for Sawdust, Shavings, and Waste Chips, U.S. Department of Agriculture – Forest Service – Forest Products Laboratory, 1969, vol. 48 Search PubMed.
- T. Kishimoto, W. Chiba, K. Saito, K. Fukushima, Y. Uraki and M. Ubukata, J. Agric. Food Chem., 2010, 58, 895–901 CrossRef CAS PubMed.
- M. Graglia, N. Kanna and D. Esposito, ChemBioEng Rev., 2015, 2, 377–392 CrossRef.
- I. Kumaniaev, E. Subbotina, J. Sävmarker, M. Larhed, M. V Galkin and J. S. M. Samec, Green Chem., 2017, 19, 5767–5771 RSC.
- E. M. Anderson, M. L. Stone, M. J. Hülsey, G. T. Beckham and Y. Román-Leshkov, ACS Sustainable Chem. Eng., 2018, 6, 7951–7959 CrossRef CAS.
- J. H. Jang, G. David, R. J. Dreiling, J. R. Bielenberg, T. Gregg, J. H. Jang, D. G. Brandner, R. J. Dreiling, A. J. Ringsby, J. R. Bussard, L. M. Stanley, R. M. Happs, A. S. Kovvali, J. I. Cutler, T. Renders, J. R. Bielenberg and Y. Roma, Joule, 2022, 6(8), 1859–1875 CrossRef CAS.
- Y. Li, B. Demir, L. M. Vázquez Ramos, M. Chen, J. A. Dumesic and J. Ralph, Green Chem., 2019, 21, 3561–3572 RSC.
- A. Kramarenko, D. Etit, G. Laudadio and M. F. N. D’angelo, ChemSusChem, 2021, 14, 3838–3849 CrossRef CAS PubMed.
- S. Mitchell, N. L. Michels and J. Perez-Ramirez, Chem. Soc. Rev., 2013, 42, 6094–6112 RSC.
- F. Brandi, M. Bäumel, V. Molinari, I. Shekova, I. Lauermann, T. Heil, M. Antonietti and M. Al-Naji, Green Chem., 2020, 22, 2755–2766 RSC.
- F. Brandi, I. Khalil, M. Antonietti and M. Al-Naji, ACS Sustainable Chem. Eng., 2021, 9, 927–935 CrossRef CAS.
- F. Brandi, M. Bäumel, I. Shekova, V. Molinari and M. Al-Naji, Sustainable Chem., 2020, 1, 106–115 CrossRef.
- J. A. Mendoza Mesa, F. Brandi, I. Shekova, M. Antonietti and M. Al-Naji, Green Chem., 2020, 22, 7398–7405 RSC.
- J. Gong, J. Li, J. Xu, Z. Xiang and L. Mo, RSC Adv., 2017, 7, 33486–33493 RSC.
- T. Renders, S. Van den Bosch, T. Vangeel, T. Ennaert, S.-F. Koelewijn, G. Van den Bossche, C. M. Courtin, W. Schutyser and B. F. Sels, ACS Sustainable Chem. Eng., 2016, 4, 6894–6904 CrossRef CAS.
- D. Vincent Sahayaraj, L. A., E. M. Mitchell, X. Bai and J.-P. Tessonnier, Green Chem., 2021, 23, 7731–7742 RSC.
- D. G. Brandner, J. S. Kruger, N. E. Thornburg, G. G. Facas, J. K. Kenny, R. J. Dreiling, A. R. C. Morais, T. Renders, N. S. Cleveland, R. M. Happs, R. Katahira, T. B. Vinzant, D. G. Wilcox, Y. Román-Leshkov and G. T. Beckham, Green Chem., 2021, 23, 5437–5441 RSC.
- N. Giummarella and M. Lawoko, ACS Sustainable Chem. Eng., 2017, 5, 5156–5165 CrossRef CAS.
- R. El Hage, N. Brosse, L. Chrusciel, C. Sanchez, P. Sannigrahi and A. Ragauskas, Polym. Degrad. Stab., 2009, 94, 1632–1638 CrossRef CAS.
- A. M. L. Seca, J. A. S. Cavaleiro, F. M. J. Domingues, A. J. D. Silvestre, D. Evtuguin and C. Pascoal Neto, J. Agric. Food Chem., 2000, 48, 817–824 CrossRef CAS PubMed.
- Y. Gu and F. Jerome, Chem. Soc. Rev., 2013, 42, 9550–9570 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2su00076h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |