
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of substituted benzylboronates by light promoted homologation of boronic acids with N-sulfonylhydrazones†
Álvaro
Valdés-Maqueda
 ,
Lucía
López
,
Manuel
Plaza
* and
Carlos
Valdés
*
,
Lucía
López
,
Manuel
Plaza
* and
Carlos
Valdés
*
Departamento de Química Orgánica e Inorgánica, Instituto Universitario de Química Organometálica “Enrique Moles” and Centro de Innovación en Química Avanzada (ORFEO-CINQA), Universidad de Oviedo, C/Julián Clavería 8, 33006 Oviedo, Spain. E-mail: plazamanuel@uniovi.es; acvg@uniovi.es
First published on 21st November 2023
Abstract
The synthesis of benzylboronates by photochemical homologation of boronic acids with N-tosylhydrazones under basic conditions is described. The reaction involves the photolysis of the N-tosylhydrazone salt to give a diazoalkane followed by the geminal carboborylation of the diazoalkane. Under the mild reaction conditions, the protodeboronation of the unstable benzylboronic acid is circumvented and the pinacolboronates can be isolated after reaction of the benzylboronic acid with pinacol. The metholodogy has been applied to the reactions of alkylboronic acids with N-tosylhydrazones of aromatic aldehydes and ketones, and to the reactions of arylboronic acids with N-tosylhydrazones of aliphatic ketones. Moreover, the employment of the DBU/DIPEA bases combination allows for homogeneous reactions which have been adapted to photochemical continuous flow conditions. Additionally, the synthetic versatility of boronates enables their further transformation via Csp3–C or Csp3–X bond forming reactions converting this methodology into a novel method for the geminal difunctionalization of carbonyls via N-tosylhydrazones.
Introduction
Boronic acids and boronates are a very significant class of compounds due to their vast applications as synthetic intermediates as well as for their interesting properties in areas that span from materials to medicinal chemistry.1 Of particular interest is a specific class of boronic acid derivatives known as saturated boronic acids, distinguished by the presence of a Csp3–B bond.2 In recent years, a wide array of reactions have been devised to transform these systems through both C–C and C-heteroatom bond forming reactions significantly expanding their synthetic versatility.3–5On the other hand, the transition-metal free coupling of boronic acids with N-sulfonylhydrazones is a very powerful method for the modification of carbonyl compounds.6 In these reactions, a carbon–carbon bond and a carbon–boron bond are formed on the same carbon atom in a process that involves the decomposition of the N-sulfonylhydrazone to give a diazo compound followed by the carboborylation of the diazo compound with concomitant loss of nitrogen. However, under the relative harsh reaction conditions required for the generation of the diazo compound, the intermediate boronic acid formed may undergo protodeboronation to yield the product derived from the reductive coupling of the boronic acid and the N-sulfonylhydrazone (Scheme 1a). This methodology has proven to be a very powerful C–C bond forming reaction that have found wide applications in organic synthesis.7,8 Nevertheless, the possibility of circumventing the protodeboronation step, and achieving the geminal difunctionalization of the carbonyl group through the N-sulfonylhydrazone is a very attractive synthetic transformation.9 Our research group has successfully demonstrated the feasibility of this approach by capturing the boronic acid moiety via intramolecular cyclizations. Thus, we have developed cascade geminal C–C/C–C bond forming processes10 as well as C–C/C–N cyclizations11 (Scheme 1b) that have led to the development of new methods to synthesize quite complex Csp3-rich bicyclic and spirocyclic structures. Moreover, very recently, Merchant and Qin et al. showed in a remarkable study, that the secondary and tertiary boronic acids which are obtained by reaction of N-sulfonylhydrazones of alkyl ketones and aldehydes with alkylboronic acids are not prone to undergo protodeboronation, and therefore can be efficiently trapped as pinacol boronates, providing a general method for the construction of these important synthetic intermediates with wide structural diversity (Scheme 1c).12 This type of homologation has been also explored employing stable diazo compounds13 as well as other sources of unstabilized diazoalkanes.14 In particular, the photochemical decomposition of oxadiazolines under continuous flow has been employed by Ley et al. as a mild method to generate diazoalkanes, that upon treatment with aryl or alkenylboronic acids lead to the corresponding homologated boronic acids, that can be trapped as the corresponding boronates15 or upon a subsequent reaction.16 However, the synthesis of benzyl and allyl boronates from N-sulfonylhydrazones has remained elusive, due to their unstability under the thermal and basic reaction conditions required for the generation of the diazoalkane from the hydrazone.6,12a Nevertheless, considering the widespread availability and simplicity of obtaining N-sulfonylhydrazones from carbonyl compounds, the capability to synthesize benzylboronates under transition-metal-free conditions using such readily accessible starting materials would represent a highly valuable synthetic transformation. In this context, we became inspired by some recent publications that reported that the decomposition of N-tosylhydrazones can be promoted photochemically.17–20 In particular, König et al. discovered that the formation of diazo compounds from N-tosylhydrazones can be achieved by irradiation of the N-tosylhydrazone salts with visible light at rt, and applied this reaction in the homologation of carbonyl compounds (Scheme 1e).17 Taking these results into consideration we envisioned that under mild photochemical conditions the preparation of benzylboronates by homologation of boronic acids by reaction with N-tosylhydrazones might be possible.21,22 We expected that at room temperature the protodeboronation of the homologated boronic acid might not occur, enabling their capture as pinacolboronates. In this manuscript we wish to report our results, which have led to the development of a new method for the preparation of secondary and tertiary benzyl boronates by homologation of boronic acids with N-tosylhydrazones under mild photochemical conditions.
 | ||
| Scheme 1 (A) Reductive coupling of boronic acids and N-sulfonylhydrazones by the carboborylation/protodeboronation sequence. (B) Synthesis of pyrrolidines by a C–C/C–N bond forming cascade involving carboxylation of a N-tosylhydrazone and intramolecular interception by an azide. (C) Synthesis of pinacol boronates form alkyl N-tosylhydrazones and alkylboronic acids. (D) Synthesis of benzyl boronates by photochemically promoted homologation of arylboronic acids with oxadiazolines. (E) Photoinduced generation of diazo compounds from N-tosylhydrazones and their use in the homologation of aldehydes. (F) This work: synthesis of benzyl boronates by photochemically promoted homologation of alkyl and arylboronic acids with N-sulfonylhydrazones. | ||
Results and discussion
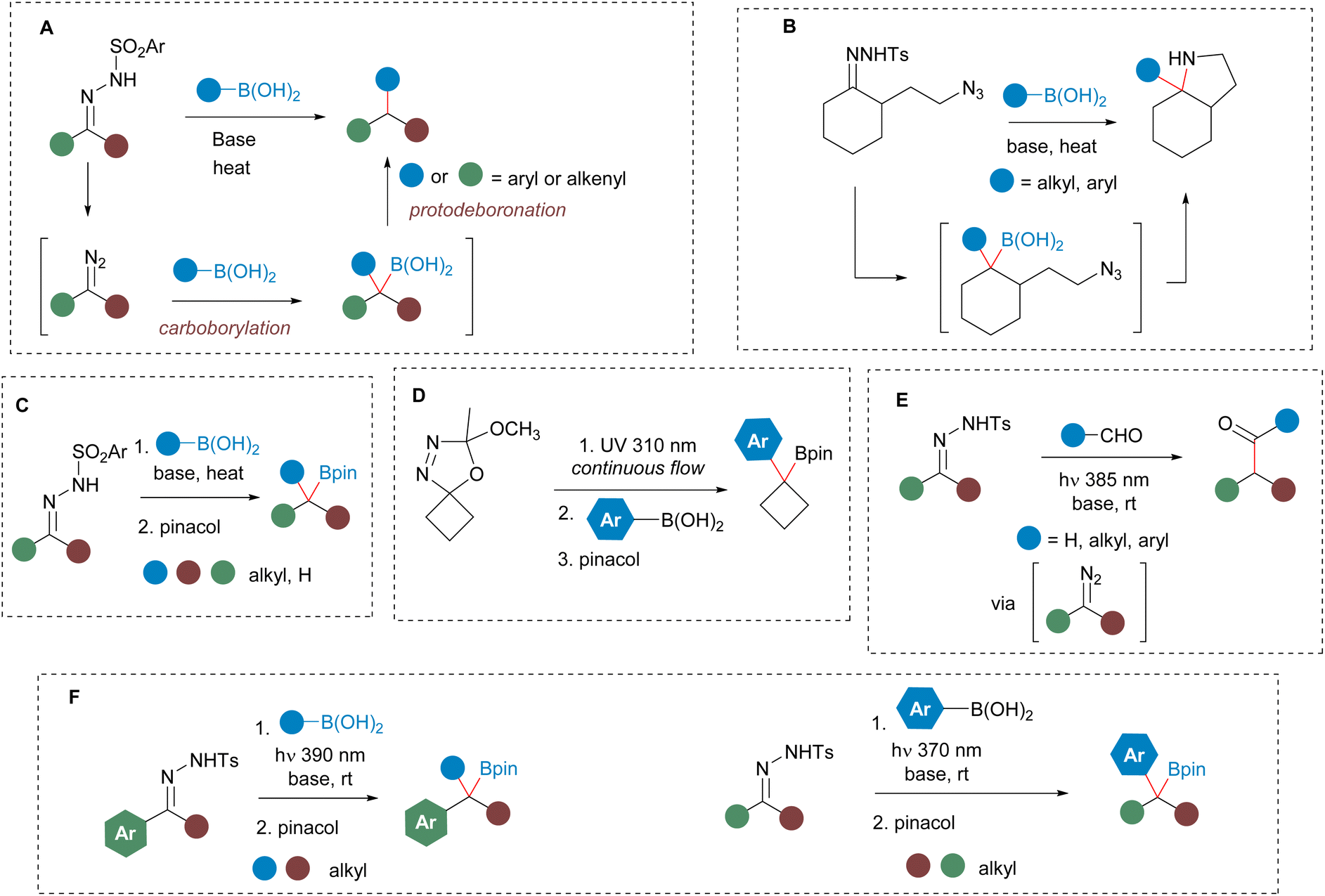
To check our hypothesis, we initiated our study by considering the reaction between propylboronic acid and the N-tosylhydrazone 1a derived from 1-tetralone. To select the proper irradiation wavelength, a UV spectrum of the solution of the N-sulfonylhydrazone in the presence of a base was registered. As described by König for N-tosylhydrazones of alkyl ketones,17 a bathochromic shift of the absorption was observed for the mixture in the presence of base when compared with the pure N-tosylhydrazone. Based on these measurements, violet light (390 nm) was selected as the excitation wavelength (see ESI† for details). Thus, we irradiated a mixture of the N-tosylhydrazone 1a, 3 equiv. of n-propylboronic acid and 3 equiv. of cesium carbonate in CH2Cl2 with a 390 nm LED lamp for 2 h at rt. Then, the mixture was treated with H2O2, to oxidize the expected homologated boronic acid (Scheme 2). Delightfully, the expected alcohol 2 was obtained with high yield, indicating that the homologation had taken place through the photochemically induced reaction, and importantly that no protodeboronation had occurred under the reaction conditions. This promising result pointed that the isolation of the benzyl boronate might be possible. However, when we attempted to trap the boronic acid as the pinacol boronate 3 by addition of pinacol instead of the oxidation with H2O2 upon completion of the reaction, the tertiary alcohol was still the main product, even in the absence of any oxidation agent and in dry and oxygen free conditions. Nevertheless, when the same conditions were applied to the reaction of the N-tosylhydrazone of 4-methoxybenzaldehyde 1b with butylboronic acid, this time the pinacolboronate 4a could be obtained in good yield, with no alcohol or protodeboronation byproduct being detected (Table 1, entry 1).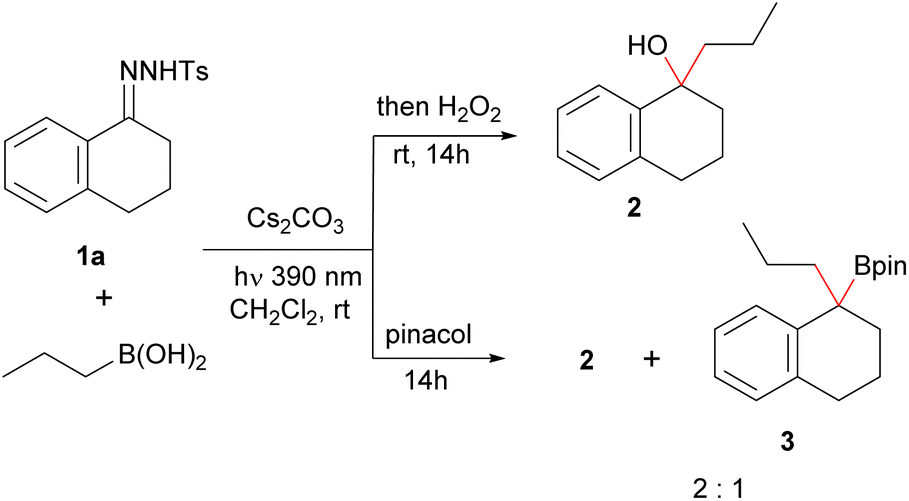 | ||
| Scheme 2 Initial experiments on the photochemical homologation of alkylboronic acids with aromatic N-tosylhydrazones. | ||
|
|
|||
|---|---|---|---|
| Entry | Deviation from standard conditionsa | Conversionb (%) | Yieldc (%) |
| a Standard conditions: hydrazone 1b 0.2 mmol, n-Bu-B(OH)2 (3 equiv.), solvent 2 mL, Kessil PR160L lamp 390 nm (52 W). b Determined by the disappearance of the N-tosylhydrazone signals by 1H NMR. c Isolated yield after column chromatography. DIPEA: diisopropylethylamine. | |||
| 1 | None | 100 | 78 |
| 2 | 2 equiv. of DIPEA added | 100 | 95 |
| 3 | With K2CO3 instead of Cs2CO3 and 2 equiv. of DIPEA | 100 | 88 |
| 4 | With LiOtBu instead of Cs2CO3 and 2 equiv. of DIPEA | 100 | 46 |
| 5 | No light | 0 | 0 |
| 6 | No base | 0 | 0 |
| 7 | 1,4-Dioxane instead of DCM | 100 | 0 |
| 8 | THF instead of DCM | 100 | 0 |
| 9 | Acetonitrile instead of DCM | 100 | 0 |
| 10 | 427 nm lamp, 4 h | 17 | — |
| 11 | 1 equiv. of boronic acid | 100 | 62 |
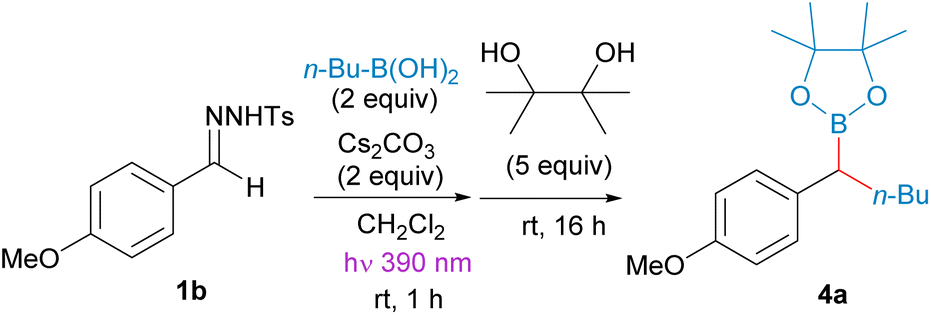
Thus, we selected the reaction between the N-tosylhydrazone of 4-methoxybenzaldehyde 1b and n-butylboronic acid as starting point to develop proper reaction conditions (Table 1). The reactions were carried out at room temperature, and after the reaction time indicated, the lamp was turned off and pinacol was added to trap the boronic acid as the benzyl boronate 4a. A selection of the conditions analyzed are presented in Table 1. We found that the addition of diisopropylethylamine (DIPEA) together with the inorganic base provided higher yields than the Cs2CO3 alone (Table 1, entry 2). Moreover, other bases such as K2CO3 and LiOtBu, which are widely employed for the thermal decomposition of N-sulfonylhydrazones at high temperatures also promoted the reaction, although with lower yields (Table 1, entries 3 and 4). The influence of the solvent is remarkable, in coordinating solvents such as 1,4-dioxane, THF and acetonitrile the N-tosylhydrazone undergoes decomposition, but the carboborylation reaction does not proceed at all (Table 1, entries 7–9).
Then, the scope of the reaction was studied under the reaction conditions developed (Scheme 3). Regarding the structure of the boronic acid, the reaction is compatible with the employment of methylboronic acid, primary alkylboronic acids, and cyclic secondary alkylboronic acids (4d–4f). Importantly, the reaction is also compatible with a variety of sensitive functional groups in the boronic acid, such as bromide 4i, nitrile 4j and an enolizable ketone 4k. Unfortunately, N-boc protected heterocyclic boronic acids did not react with the diazoalkane under this mild reaction conditions.
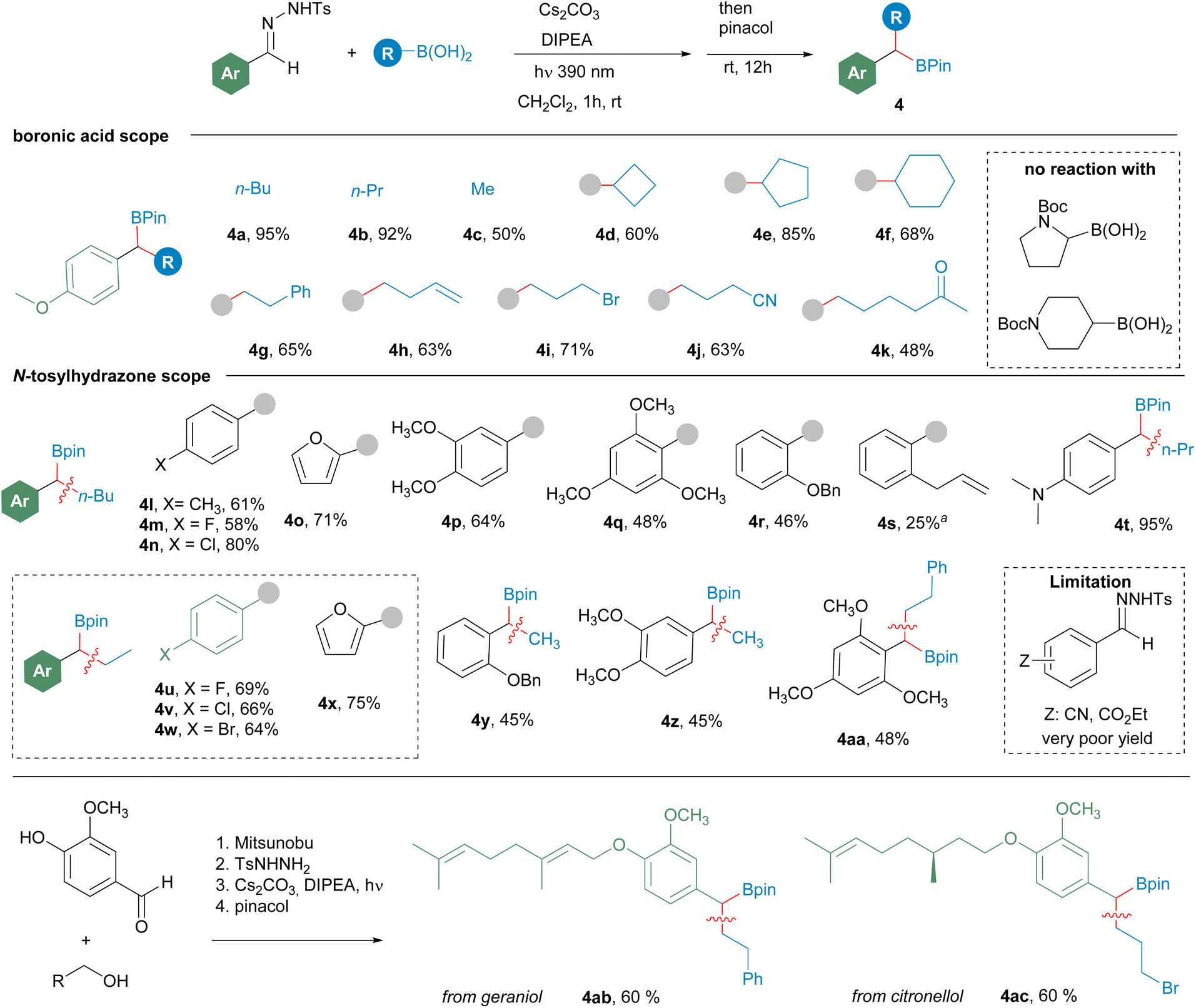 | ||
Scheme 3 Formation of benzylboronates 4 by reaction of N-tosylhydrazones of aromatic aldehydes with alkylboronic acids. Reaction conditions: N-tosylhydrazone (0.2 mmol), alkylboronic acid (0.6 mmol), Cs2CO3 (3 equiv.), DIPEA (3 equiv.), CH2Cl2 2 mL, irradiation with a 390 nm lamp (52 W), 1–3 h, rt; then pinacol (5 equiv.), 12 h, rt. Isolated yields after column chromatography are given. aA 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of the boronic ester and the pyrazoline derived from a 1,3-dipolar cycloaddition was detected in the reaction crude (see Scheme 8b for details). :1 mixture of the boronic ester and the pyrazoline derived from a 1,3-dipolar cycloaddition was detected in the reaction crude (see Scheme 8b for details). | ||
Regarding the structure of the aldehyde, the reaction proceeds successfully with electron-rich (4p, 4q, 4t), neutral (4l) and moderately electron-withdrawing aromatic systems (4m, 4n, 4v, 4w), but with N-tosylhydrazones with strong electron-withdrawing substituents the yields of the reactions are substantially reduced. Additionally, the reaction tolerates ortho mono (4r) and disubstitution (4q, 4aa). However, low yield was obtained for the o-allyl substituted N-tosylhydrazone 4s, since in this case a 1:1 mixture of 4s and the pyrazoline derived from the intramolecular 1,3-dipolar cycloaddition between the diazo compound and the double bond is obtained (see Scheme 8 below). To further illustrate the potential of this transformation to access to complex boronates, vanillin was used as a linker to attach hydroxylated natural products to the N-tosylhydrazone moiety. Then the homologation reaction provided the boronates with good yields, as shown by the synthesis of geraniol and (−)-citronellol containing boronates 4ab and 4ac respectively.
Once we had developed a useful method for the synthesis of secondary benzyl boronates from alkylboronic acids and aromatic aldehydes via their N-sulfonylhydrazones, we turned our attention to the reverse combination of partners, N-tosylhydrazones of alkyl ketones and arylboronic acids. We selected 4-methoxyphenylboronic acid and the N-tosylhydrazone 1c derived from benzyl methyl ketone as substrates for the model reaction (Table 2). When the conditions developed before for aromatic N-tosylhydrazones were applied no coupling product was obtained, recovering the N-tosylhydrazone untouched (Table 2, entry 2). Interestingly, upon irradiation for 16 h, 50% conversion of the hydrazone was achieved (Table 2, entry 3). These results indicated that the alkyl substituted hydrazone underwent very slow photochemical decomposition under these conditions. Indeed, a new UV-vis analysis revealed that the N-tosylhydrazone salts from alkyl ketones should be excited with low-energy UV light rather than violet light, due to their lower absorbance (see ESI† for details). Thus, when the irradiation wavelength was changed to 370 nm total conversion was achieved after 6 h to provide, after reaction with pinacol overnight, a mixture of the boronate 5a and the alcohol 6 derived from the oxidation of the homologated boronic acid (Table 2, entry 1). The attempts to eliminate the formation of the alcohol keeping Cs2CO3 as base were unsuccessful: reduction of the amount of Cs2CO3 led to a dramatic reduction in the conversion (entry 4) shorter reaction times increased the 5a:6 ratio, but also at the expenses of much lower conversion (entries 5 and 6). The employment of toluene as solvent (entry 7) and the exclusion of water by addition of molecular sieves (entry 8) did not prevent the formation of the alcohol either. The reaction employing DIPEA as only base (entry 9), and reactions employing a preformed hydrazone salt by deprotonation with NaH led to the recovery of the starting material (entries 11–13). Moreover, very low isolated yield of the boronate 5a was obtained when the reaction was conducted with Cs2CO3 in the absence of DIPEA (entry 10). Delightfully, when DBU was used as base, total conversion was achieved, and importantly the boronate 5a was isolated with no formation of the alcohol (entry 14). After some more experimentation it was found that the combination of DBU and DIPEA provided the best results leading to the boronate in a 70% yield (entry 15).
|
|
|||
|---|---|---|---|
| Entry | Deviation from standard conditionsa | Conversionb (%) | Ratioc5a:6 (Yieldd (%)) |
| a Standard conditions: N-tosylhydrazone (0.2 mmol), arylboronic acid (0.3 mmol), Cs2CO3 (3 equiv.), DIPEA (3 equiv.), CH2Cl2 2 mL, irradiation with a 390 nm lamp (52 W), 1–3 h, rt; then pinacol (5 equiv.), 12 h, rt. b Determined by the disappearance of the N-tosylhydrazone. c Determined by 1H NMR on the reaction crude. d Isolated yield after column chromatography. PMP: 4-methoxyphenyl; DBU: 1,8-diazabicyclo(5.4.0)undec-7-ene. | |||
| 1 | None | 100 | 1:1.6 65% (6) |
| 2 | 390 nm, 1 h | 0 | — |
| 3 | 390 nm, 16 h | 50 | — |
| 4 | 1 eq. Cs2CO3 | 25 | 1:2 |
| 5 | 1 h | 7 | 3:1 |
| 6 | 2 h | 31 | 3:1 |
| 7 | Toluene instead of CH2Cl2 | 30 | 5:1 |
| 8 | Addition of 4A MS | 100 | 1:1 |
| 9 | Only DIPEA (2 eq.) as base, 16 h | 0 | — |
| 10 | Overnight (w/o DIPEA) | 100 | 16% (5a) |
| 11 | NaH (1 eq.), 10 min, then boronic acid + DIPEA | 0 | — |
| 12 | NaH (1 eq.), 10 min, then boronic acid (w/o DIPEA) | 0 | — |
| 13 | NaH (1 eq.), 10 min, then boronic acid + DIPEA, 16 h | 0 | — |
| 14 | DBU (2 eq.) instead of Cs2CO3 | 100 | 1:0 |
| 15 | DBU (2 eq.) instead of Cs 2 CO 3 , 2 h | 100 | 1:0 70% (5a) |
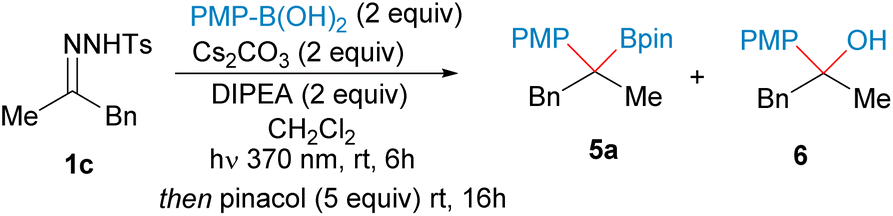
The scope of the reaction under these new conditions was studied for a set of N-tosylhydrazones leading to the tertiary boronic esters with moderate to good yields (Scheme 4). The reaction can be applied to linear N-tosylhydrazones (5a, 5b), carbocyclic (5c–5f) and heterocyclic systems (5g, 5h), and again tolerates the presence of sensitive functional groups, such as carboxylate (5h) and nitrile (5i). The reaction proceeded nicely for an α-substituted cyclohexanone leading to the tertiary boronate as a single diastereoisomer (5i).‡
 | ||
| Scheme 4 Synthesis of benzylboronates 5 by reaction of N-tosylhydrazones of aliphatic ketones with arylboronic acids. aReaction conditions: N-tosylhydrazone (0.2 mmol, 1 equiv.), arylboronic acid (0.4 mmol, 2 equiv.), DBU (2 equiv.), DIPEA (2 equiv.), CH2Cl2 2 mL, irradiation with a 370 nm lamp (43 W), 1–3 h, rt; then pinacol (5 equiv.), 12 h, rt. Isolated yields after column chromatography are given. bReaction conditions: N-tosylhydrazone (0.4 mmol, 2 equiv.), arylboronic acid (0.2 mmol, 1 equiv.), DBU (4 equiv.), DIPEA (4 equiv.), CH2Cl2 2 mL, irradiation with a 370 nm lamp (45 W), 1–3 h, rt; then pinacol (5 equiv.), 12 h, rt. Isolated yields after column chromatography are given. | ||
Very high diastereoselectivity was also achieved for the boronate derived from 4-phenylcyclohexanone N-tosylhydrazone 5j, highlighting the importance of the mild conditions to enhance the stereoselectivity of the reactions. However, lower stereoselectivity was obtained for boronate 5l, as in this case the hydrazone does not belong to the ring and the stereochemical control is marginal. The photochemical carboborylation could also be applied to cholestanone, as an example late-stage functionalization leading to the tertiary boronate 5m with moderate yields and stereoselectivities.
Quite unexpectedly, when the reaction was applied to other boronic acids such as 4-chlorophenyl boronic acid, under these specific reaction conditions poor conversion was detected. We found that in order to achieve good conversion it was necessary to perform the reactions in excess of the N-tosylhydrazone. Moreover, some of the benzylboronic esters are quite unstable under silica gel chromatography, and unactivated silica gel or neutral alumina is necessary in the purifications. Upon these modifications, the reaction can be carried out with a variety of arylboronic acids featuring electron donating (5p, 5q), neutral (5r) and electronwithdrawing substituents (5n).
Interestingly, these reaction conditions could be applied also to the homologation of an alkylboronic acid with alkylic N-tosylhydrazones towards the synthesis of tertiary alkyl boronates as represented by the synthesis of boronate 5v. These alkylboronates can be prepared efficiently under thermal conditions as reported by Qin et al.,12a but now their synthesis might be also accomplished under photochemical conditions at room temperature.
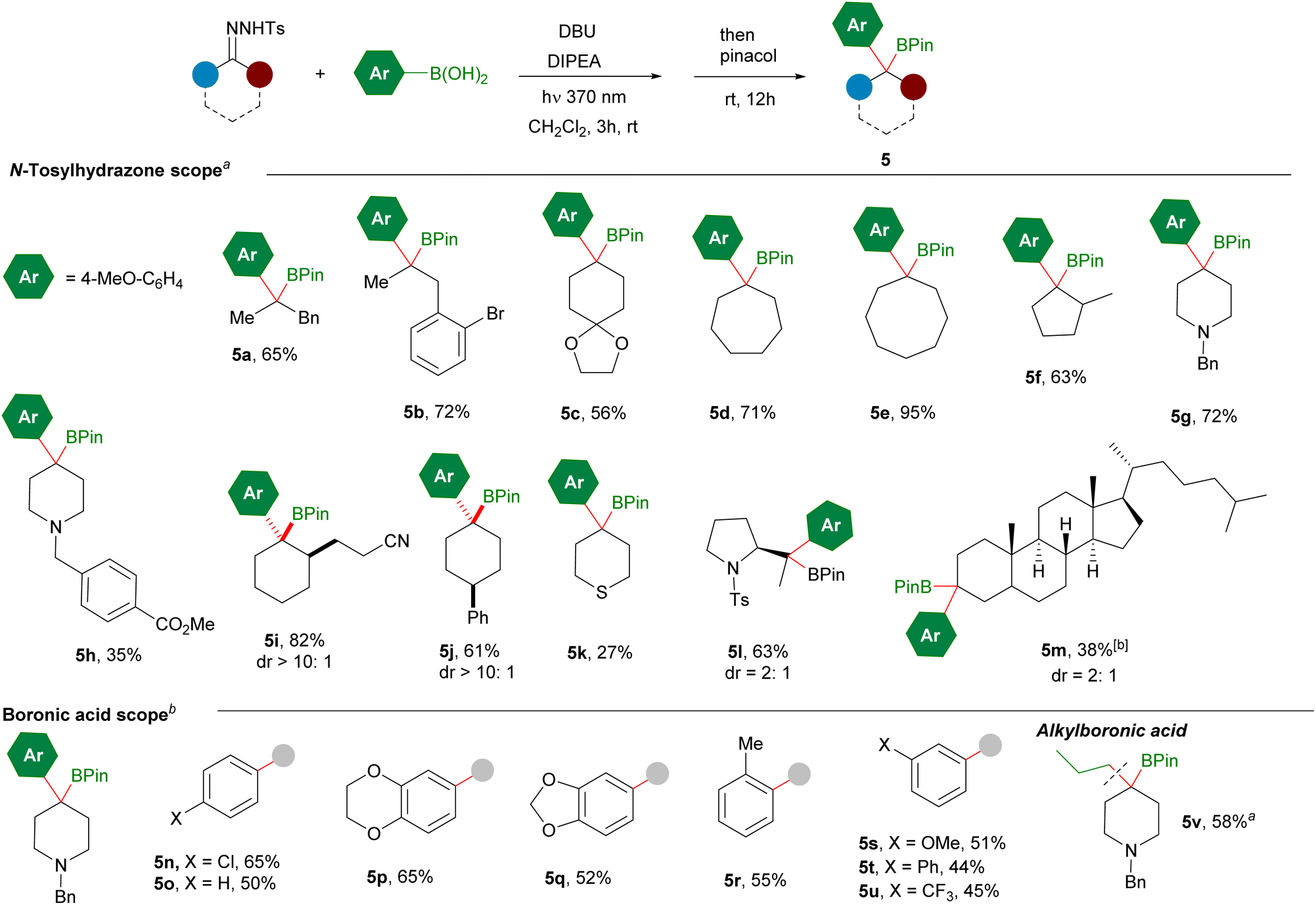
To continue with the study of the scope of this transformation, we then turned our attention to N-tosylhydrazones derived from aryl ketones, which would lead to tertiary benzyl boronates 7 upon reaction with alkylboronic acids (Scheme 5). Both protocols were examined. The reactions with the Cs2CO3/DIPEA combination seemed to be appropriate in several examples, although the DBU/DIPEA bases system gave consistently better yields avoiding the autooxidation of the homologated boronic acid. The reactions with hydrazones derived from acetophenones led to the tertiary benzylic boronates in yields comparable with the reactions with hydrazones from aldehydes (7a–7f). The presence of longer and functionalized alkyl substituents is also tolerated as represented by 7g and 7h, which feature an azide and an ester functionality at the γ-position respectively. It must be noted that in some examples very poor yields were achieved upon irradiation with the 390 nm light giving rise to degradation products derived from the N-sulfonylhydrazone. However, the employment of 370 nm light provided the benzyl boronate with acceptable yields. It is likely that in those cases the higher wavelength light may promote not only the decomposition of the N-tosylhydrazone but also the photolysis of the diazo compound into a carbene which then may evolve through different pathways.23
 | ||
| Scheme 5 Synthesis of benzylboronates 7 by reaction of N-tosylhydrazones of aromatic ketones with alkylboronic acids. aReaction conditions: N-tosylhydrazone (0.2 mmol, 1 equiv.), alkylboronic acid (0.6 mmol, 3 equiv.), Cs2CO3 (2 equiv.), DIPEA (2 equiv.), CH2Cl2 2 mL, irradiation with a 390 nm lamp (52 W), 1–3 h, rt; then pinacol (5 equiv.), 12 h, rt. Isolated yields after column chromatography are given. bReaction conditions: N-tosylhydrazone (0.2 mmol, 1 equiv.), alkylboronic acid (0.6 mmol, 3 equiv.), DBU (2 equiv.), DIPEA (2 equiv.), CH2Cl2 2 mL, irradiation with a 390 nm lamp (52 W), 1–3 h, rt; then pinacol (5 equiv.), 12 h, rt. Isolated yields after column chromatography are given. cReaction conditions: N-tosylhydrazone (0.2 mmol, 1 equiv.), alkylboronic acid (0.6 mmol, 3 equiv.), Cs2CO3 (2 equiv.), DIPEA (2 equiv.), CH2Cl2 2 mL, irradiation with a 370 nm lamp (43 W), 1–3 h, rt; then pinacol (5 equiv.), 12 h, rt. Isolated yields after column chromatography are given. PMP: 4-methoxyphenyl. | ||
In the case of the reactions with tetralones we had observed that the oxidation product could not be avoided employing Cs2CO3 as unique base (Scheme 2). Now, under the optimized conditions employing either the Cs2CO3/DIPEA or the DBU/DIPEA combinations, the autooxidation of the homologated boronic acid is prevented and the boronates could be isolated successfully (7i–7l). The scope of the reaction includes also the hydrazones derived from indanones (7m–7s) which provide the tertiary boronic esters with moderate to good yields. The reaction includes linear, functionalized and cyclic boronic acids. In contrast, poor yield was obtained under these conditions for the chromane derived benzyl ester 7t and no boronic ester 7u was obtained with the flavanone derivative.
The reaction of aryl ketone N-tosylhydrazones was also explored with arylboronic acids. However, in these cases the homologated boronic ester could not be trapped, as the diarylalkylboronic acids turned out to be too unstable and underwent very fast spontaneous protodeboronation even at rt to give the 1,1-diarylalkanes 8. These reductive coupling reactions are well developed under thermal conditions,6,7 but now we show that they might be also performed under photochemical conditions at room temperature.
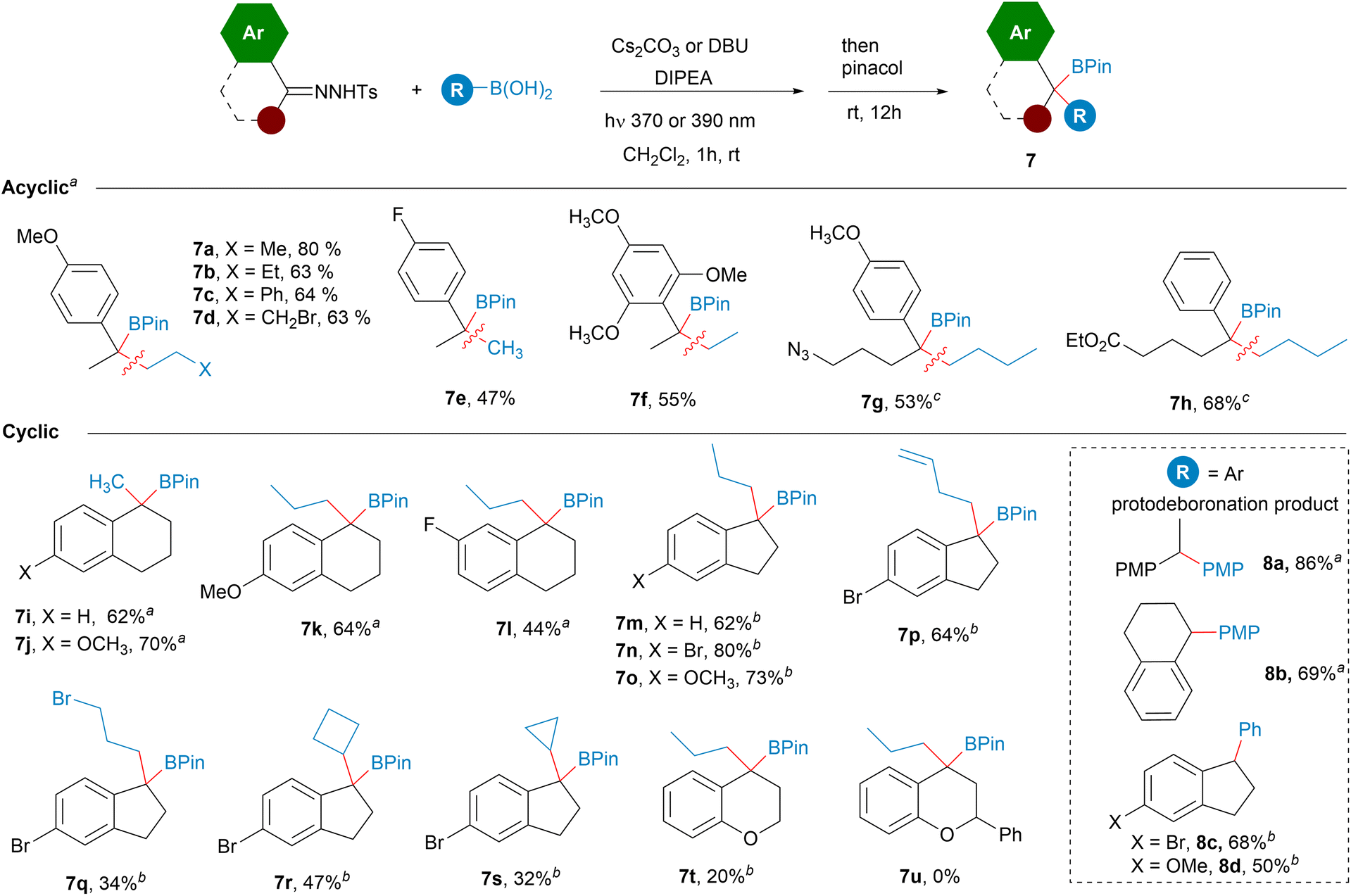
Importantly, the reactions employing the DBU/DIPEA bases system, unlike the reactions that use an inorganic base, are homogeneous. Therefore, we thought that it might be possible to adapt the reaction conditions to continuous flow, since it represents a very convenient option to scale-up photochemical reactions.24 Moreover, the in situ generation of the potentially hazardous non-stabilized diazoalkanes under continuous flow is also attractive for safety reasons.25,26 Taking all of this into consideration, the ability to perform the photochemically triggered homologations under continuous flow would be very appealing. Thus, with this idea we carried out an initial study to evaluate this possibility employing a PTFE tubing-made flow reactor. Delightfully, after some optimization, we were able to develop reaction conditions to achieve the homologation reactions under flow conditions for a set of hydrazones and boronic acids. In this manner, a solution containing the N-sulfonylhydrazone, the boronic acid, DBU and DIPEA was passed through the PTFE flow reactor illuminated by LED lamps of the proper wavelength to provide, after treatment with pinacol, the homologated boronic esters in yields comparable with the batch reaction conditions (Scheme 6). It must be pointed out that specific optimization is required for each particular substrate that included stoichiometry, radiation intensity and wavelength. Moreover, deoxygenated solvent as well as argon atmosphere over the complete process is recommended to avoid oxidation of some of the intermediate boronic acids. Specific details and description of the flow setup are given in the ESI.† Preliminary examples of the application of the flow methodology were illustrated by the synthesis of boronates derived from alkyl boronic acids and aryl N-tosylhydrazones and aryl boronic acids with alkyl N-tosylhydrazones (Scheme 6). The reactions with cyclic aromatic N-tosylhydrazones turned out to be particularly well suited for the continuous flow protocol. We used as prototype the reactions with indanone N-tosylhydrazones, which led to the expected boronates derived from primary boronic acids (7n, 7o, 7p, 7q, 7v), cyclic boronic acids (7t, 7u, 7w) and also functionalized boronic acids (7p, 7q). The flow methodology was also compatible with tetralone N-tosylhydrazones (7k). Moreover, even the reactions with chromanone and flavanone, which performed very poorly under batch conditions provided the boronates 7t and 7u with synthetically useful yields under continuous flow. This protocol could be also applied to the preparation of secondary benzyl boronates upon the employment of aldehyde N-tosylhydrazones, as shown by the synthesis of 4l, 4ad, although at this point of development with quite poor yields. The synthesis of benzylboronates by reaction of dialkyl ketone N-tosylhydrazones with arylboronic acids coud be also accomplished under continuous flow as shown in the preparation of 5c, 5d, 5e and 5g. In particular, the synthesis of 5k, which was obtained with very low yield under batch conditions could be performed in flow with high yield. Finally, the carboborylation of a dialkylketone N-tosylhydrazone with an alkylboronic acid is also possible under flow conditions as illustrated by the synthesis of 5v.
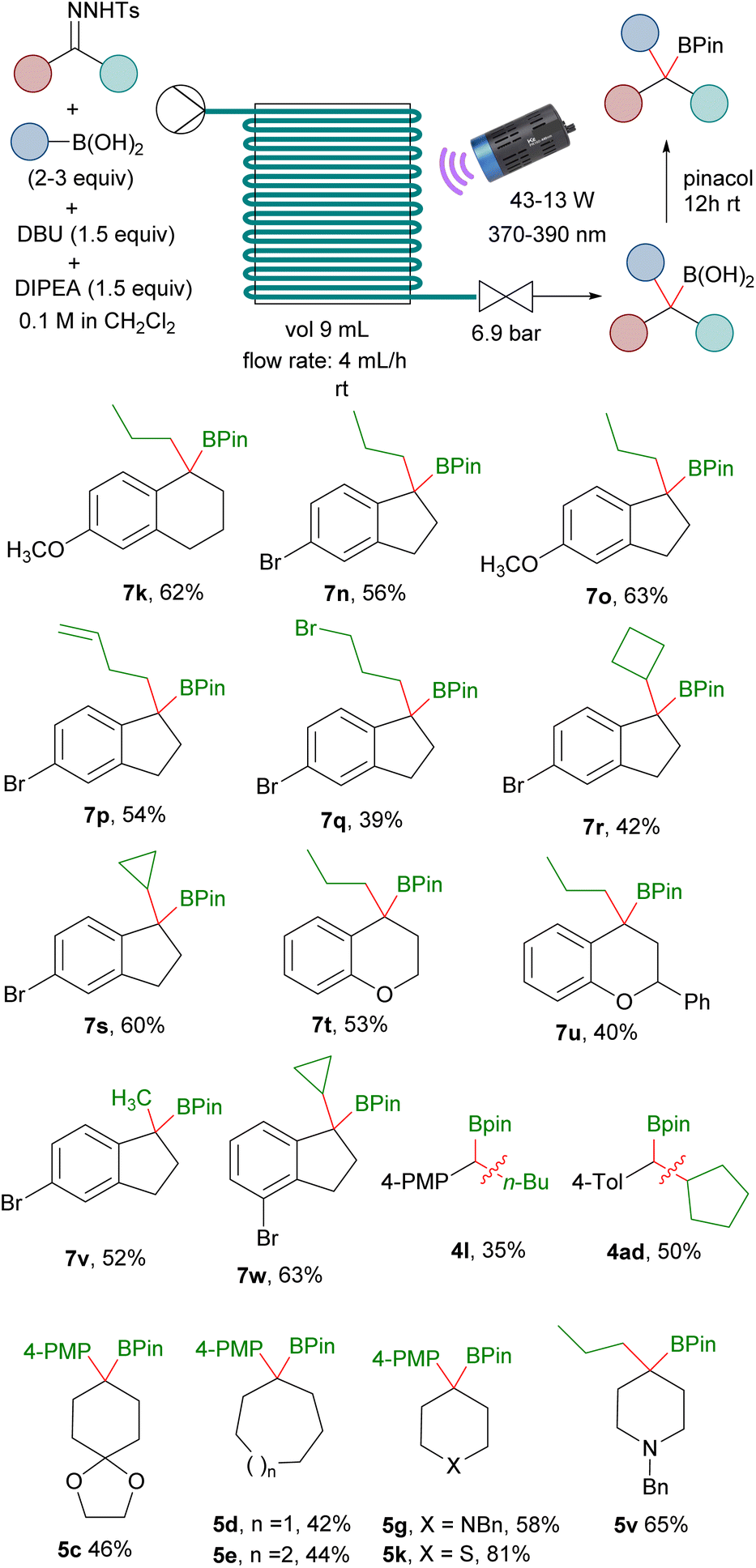 | ||
| Scheme 6 Continuous flow synthesis of benzylboronates. See ESI† for the specific details of each reaction. | ||
It must be emphasized that although unstabilized diazoalkanes have been previously generated under continuous flow conditions from other precursors, such as unprotected NH2 hydrazones and oxadiazolines,26 the particularly easy availability and stability of N-tosylhydrazones makes this methodology a highly promising new transformation.
Alkyl boronic acids and their pinacol esters are highly valuable intermediates in organic synthesis, and a myriad of useful transformations have been described including protodeboronation, oxidation, amination and C–Csp3 bond forming reactions.3–5,27 Interestingly, depending on the workup once the photochemical reaction has concluded, two different products could be obtained, as illustrated by the reaction of N-sulfonylhydrazone 1d with 3-butenylboronic acid: treatment with H2O2 for 1 h or stirring at the open air gives the corresponding alcohol 9, and as already mentioned, treatment with pinacol leads to the pinacolboronate 7p (Scheme 7a). Furthermore, the spirocyclic tetrahydrofuran 10 could be directly obtained in the reaction of 1d with 3-bromopropylboronic acid upon treatment with H2O2 and 1 M NaOH overnight, providing a novel approach towards this interesting class of heterocycles (Scheme 7b).
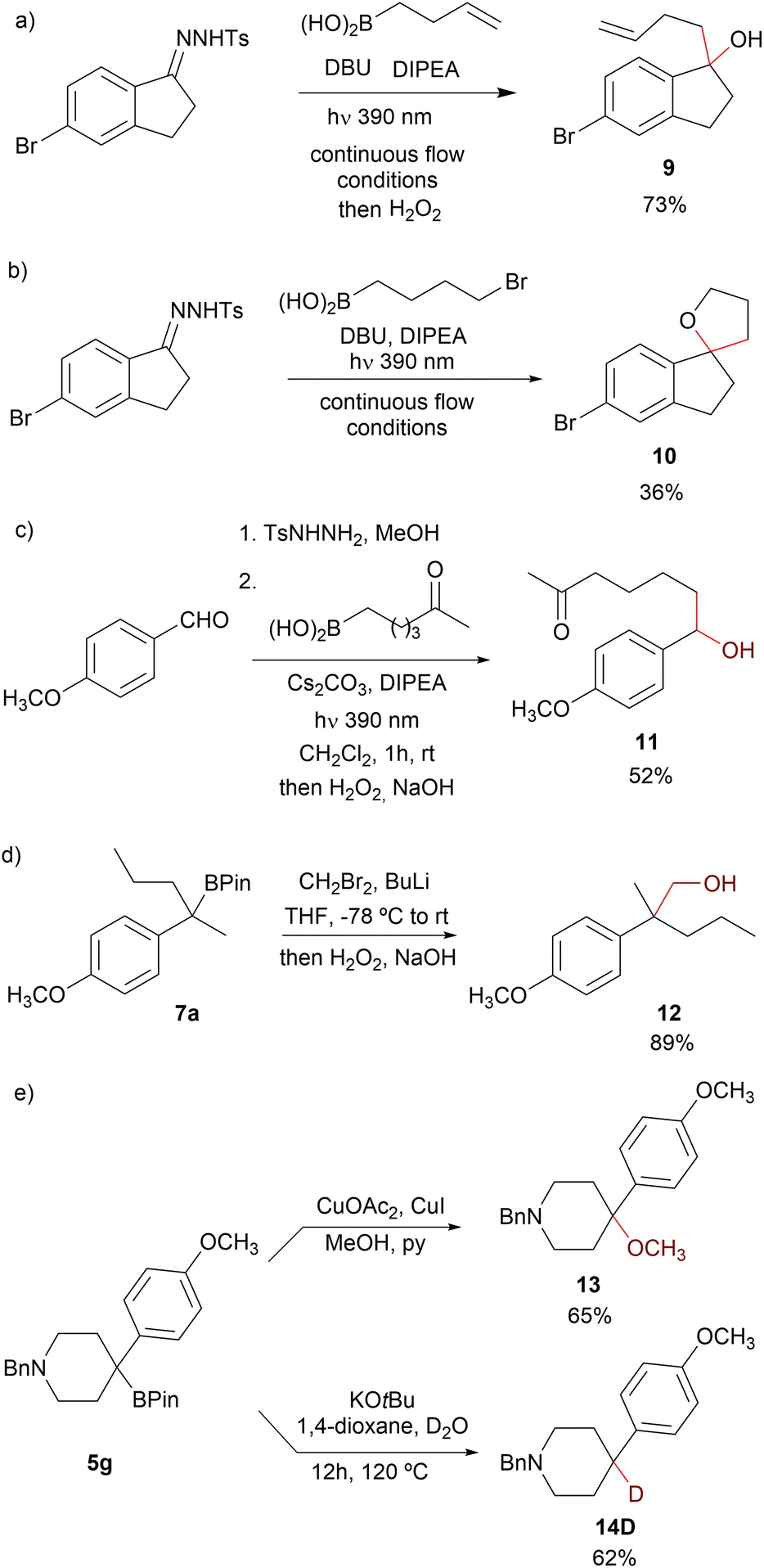 | ||
| Scheme 7 Some synthetic applications of benzyl boronic acids and pinacol boronic esters. | ||
On the other hand, taking advantage of the functional group tolerance of the carboborylation, the direct oxidation of the homologated boronic acid allowed for the synthesis of hydroxyketone 11 without the need of protective groups (Scheme 7c). This synthesis of benzyl boronic ester features great potential towards the geminal disubstitution on carbonyls. For instance, one carbon Matteson homologation28 of the pinacol boronic ester 7a by treatment with in situ generated bromomethyl lithium followed by oxidation led to the primary alcohol 12 (Scheme 7d). Considering the overall transformation, two Csp3–Csp3 bonds have been formed on the carbonylic carbon of the precursor ketone. Additionally, the boronic ester 5g could be methoxylated under Chan–Lam conditions to give tertiary methoxy ether 13.
Finally, protodeboronation of the boronic ester can be cleanly achieved upon treatment with KOtBu and water, and interestingly, the same reaction in the presence of D2O leads to the selective deuteration of the tertiary benzylic position, as exemplified by the synthesis of the deuterated 4-arylpiperidine 14D from 5g (Scheme 7e).
Mechanistic considerations
König et al. have established that the irradiation of the cesium N-tosylhydrazonate I with 385 nm visible light gives rise to the cleavage of the N–S bond to forge the diazoalkane III and cesium sulfinate17 We have also observed a bathochromic shift in the UV absorption for the solution of N-tosylhydrazone 1a upon treatment with Cs2CO3. The organic base DBU plays a similar role, since a solution of the N-tosylhydrazone of N-benzylpiperidone 1d and DBU (1, 5 equiv.) also showed a significant bathochromic shift when compared with the UV spectrum of the solution of the N-tosylhydrazone in the absence of the base (see ESI†). Thus, photoexcitation of the DBU hydrazonate II might also enable the N–S bond cleavage and the generation of the diazoalkane III.29 Then, reaction with the boronic acid through the boronate species IV followed by 1,2-migration of the organic group provides the homologated boronic acid V, which does not undergo protodeboronation under the mild reaction condition and can be trapped as pinacol boronate or oxidized to render the alcohol (Scheme 8a).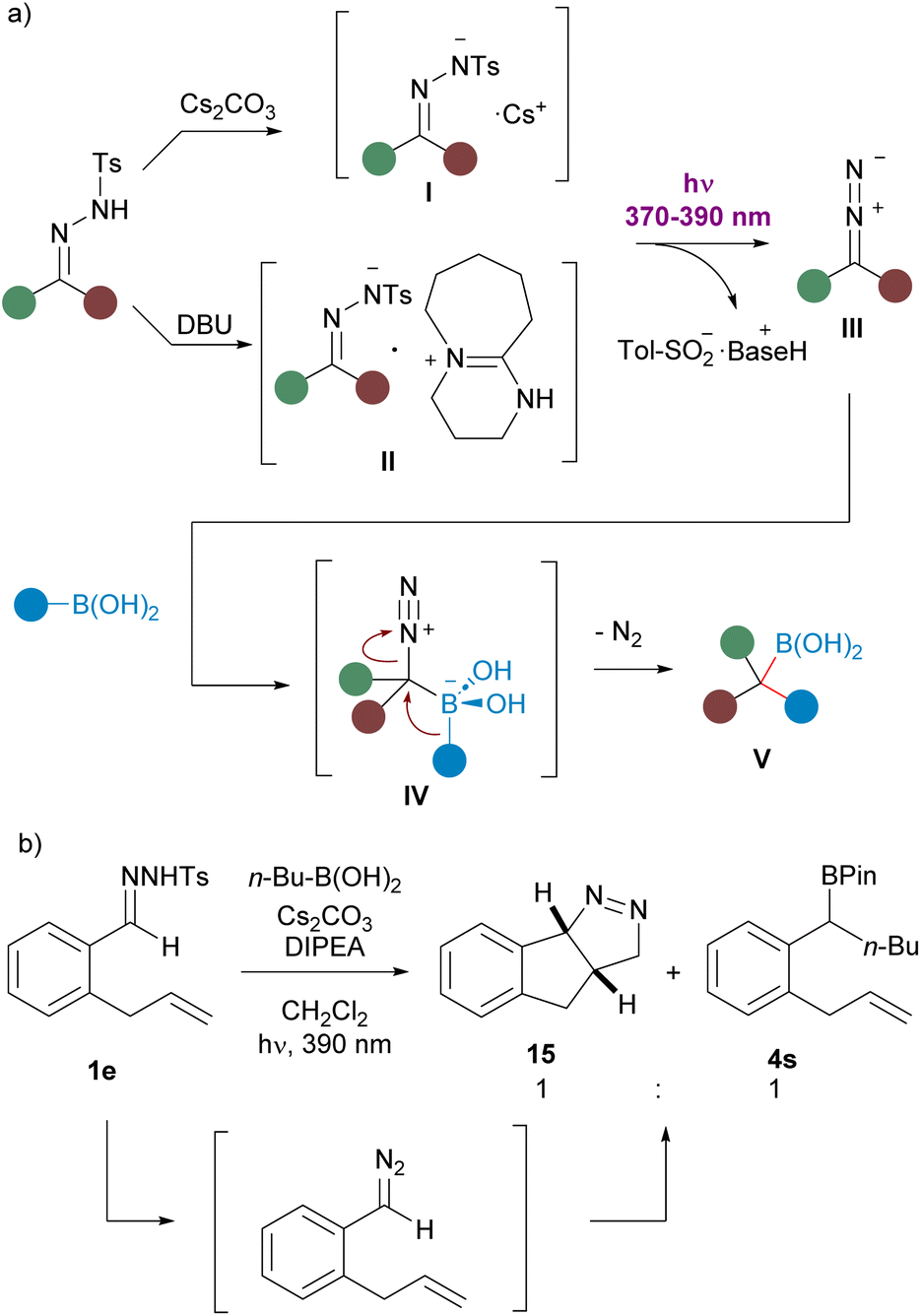 | ||
| Scheme 8 (a) Mechanistic proposal for the photochemically triggered carboborylation. of N-tosylhydrazones. (b) Evidence for the diazoalkane intermediate formation. | ||
Further evidence for the formation of the diazoalkane was provided by the reaction of N-tosylhydrazone 1e with n-butylboronic acid, which afforded a 1:1 mixture of the boronate 4s and the fused pyrazoline 15. Formation of 15 can be explained by the intramolecular 1,3-dipolar cycloaddition between the diazoalkane functionality and the double bond of the allyl moiety (Scheme 8b).
Conclusions
Herein we report a new methodology for the synthesis of secondary and tertiary benzyl boronates by light promoted homologation of boronic acids with N-tosylhydrazones. The new photochemical transformation fills a significant gap, as benzylboronates were not previously accessible under the classical thermal conditions for the reactions between boronic acids and N-sulfonylhydrazones. The transformation features wide scope regarding both coupling partners. Moreover, the reactions have been adapted to continuous flow enabling for better scalability. For these reasons, we believe that this new light promoted transformation may be of high interest as it gives access to a wide structural variety of secondary and tertiary benzylboronates through a straightforward, mild, and experimentally simple procedure. Further applications of this methodology are forthcoming.Data availability
All the necessary data have been included in the ESI.†Author contributions
CV and MP designed the project and supervised the work. AVM, LL and MP carried out the experimental work. All the authors discussed on the manuscript. MP and CV wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support of this work by Ministerio de Ciencia e Innovación of Spain (Agencia Estatal de Investigación: PID2019-107580GB-I00/AEI/10.13039/501100011033). A FICYT (Principality of Asturias) “Margarita Salas Joven” postdoctoral grant to M. P. (AYUD/2021/58397) is gratefully acknowledged.References
- Boronic Acids: Preparation and Applications in Organic Synthesis Medicine and Materials, ed. D. H. Hall, Wiley, Weinheim, 2011 Search PubMed.
- For some recent reviews see: (a) J. W. B. Fyfe and A. J. B. Watson, Chem, 2017, 3, 31 CrossRef CAS; (b) D. M. Volochnyuk, A. O. Gorlova and O. O. Grygorenko, Chem. – Eur. J., 2021, 27, 15277 CrossRef CAS PubMed.
- C–C bond forming reactions: (a) R. P. Sonawane, V. Jheengut, C. Rabalakos, R. Larouche-Gauthier, H. K. Scott and V. K. Aggarwal, Angew. Chem., Int. Ed., 2011, 50, 3760 CrossRef CAS PubMed; (b) A. Bonet, M. Odachowski, D. Leonori, S. Essafi and V. K. Aggarwal, Nat. Chem., 2014, 6, 584 CrossRef CAS PubMed; (c) M. Odachowski, A. Bonet, S. Essafi, P. Conti-Ramsden, J. N. Harvey, D. Leonori and V. K. Aggarwal, J. Am. Chem. Soc., 2016, 138, 9521 CrossRef CAS PubMed; (d) C. M. Wilson, V. Ganesh, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2017, 56, 16318 CrossRef CAS PubMed; (e) M. Takeda, K. Nagao and H. Ohmiya, Angew. Chem., Int. Ed., 2020, 59, 22460 CrossRef CAS PubMed; (f) N. Xu, H. Liang and J. P. Morken, J. Am. Chem. Soc., 2022, 144, 11546 CrossRef CAS PubMed.
- C–N bond forming reactions: (a) J. D. Grayson, F. M. Dennis, C. C. Robertson and B. M. Partridge, J. Org. Chem., 2021, 86, 9883 CrossRef CAS PubMed; (b) P. Xu, M. Zhang, B. Ingoglia, C. Allais, A. M. R. Dechert-Schmitt, R. A. Singer and J. P. Morken, Org. Lett., 2021, 23, 3379 CrossRef CAS PubMed; (c) X. Liu, Q. Zhu, D. Chen, L. Wang, L. Jin and C. Liu, Angew. Chem., Int. Ed., 2020, 59, 2745 CrossRef CAS PubMed.
- C–F bond forming reactions: Z. Li, Z. Wang, L. Zhu, X. Tan and C. Li, J. Am. Chem. Soc., 2014, 136, 16439 CrossRef CAS PubMed.
- J. Barluenga, M. Tomás-Gamasa, F. Aznar and C. Valdés, Nat. Chem., 2009, 1, 494 CrossRef CAS PubMed.
- For some recent reviews see: (a) M. Paraja, M. Plaza and C. Valdés, Synlett, 2017, 28, 2373 CrossRef CAS; (b) C. Valdés, in Homologation Reactions: Reagents, Applications, and Mechanisms, ed. V. Pace, Wiley VCH, Weinheim, 2023, vol 2, pp. 467–511 Search PubMed; (c) Z. Bao and J. Wang, Synlett, 2023, 34, 2071–2078 CrossRef CAS.
- (a) M. C. Pérez-Aguilar and C. Valdés, Angew. Chem., Int. Ed., 2012, 51, 5953 CrossRef PubMed; (b) D. M. Allwood, D. C. Blakemore, A. D. Brown and S. V Ley, J. Org. Chem., 2014, 79, 328 CrossRef CAS PubMed; (c) M. Plaza, M. C. Pérez-Aguilar and C. Valdés, Chem. – Eur. J., 2016, 22, 6253 CrossRef CAS PubMed; (d) R. R. Merchant and J. A. Lopez, Org. Lett., 2020, 22, 2271 CrossRef CAS PubMed; (e) S. Liu, M. Fang, D. Yin, Y. Wang, L. Liu, X. Li and G. Che, Synth. Commun., 2019, 49, 942 CrossRef CAS; (f) G. Wu, Y. Deng, H. Luo, J. Zhou, T. Li, Y. Zhang and J. Wang, Chem. Commun., 2016, 52, 5266 RSC; (g) G. Panda and M. Srinivas Lavanya Kumar, Eur. J. Org. Chem., 2019, 2019, 753 CrossRef CAS; (h) A. Wegener and K. A. Miller, J. Org. Chem., 2017, 82, 11655 CrossRef CAS PubMed; (i) X. Shen, N. Gu, P. Liu, X. Ma, J. Xie, Y. Liu and B. Dai, Chin. J. Chem., 2016, 34, 1033 CrossRef CAS; (j) Y. Ma, B. R. P. Reddy and X. Bi, Org. Lett., 2019, 21, 9860 CrossRef CAS PubMed.
- For the geminal alkylboration of aldehydes: W. Xu, P. Zheng, J. Zhou, Z. Hu and T. XU, Angew. Chem., Int. Ed., 2022, 61, e202214213 CrossRef CAS PubMed.
- (a) M. Plaza and C. Valdés, J. Am. Chem. Soc., 2016, 138, 12061 CrossRef CAS PubMed; (b) M. Plaza, S. Parisotto and C. Valdés, Chem. – Eur. J., 2018, 24, 14836 CrossRef CAS PubMed; (c) M. Plaza, M. Paraja, L. Florentino and C. Valdés, Org. Lett., 2019, 21, 632 CrossRef CAS PubMed.
- (a) L. Florentino, L. López, R. Barroso, M. Cabal and C. Valdés, Angew. Chem., Int. Ed., 2021, 60, 1273 CrossRef CAS PubMed; (b) L. López, M. Cabal and C. Valdés, Angew. Chem., Int. Ed., 2022, 61, e202113370 CrossRef PubMed.
- (a) Y. Yang, J. Tsien, A. Ben David, J. M. E. Hughes, R. R. Merchant and T. Qin, J. Am. Chem. Soc., 2021, 143, 471 CrossRef CAS PubMed; (b) Y. Yang, J. Tsien, J. M. E. Hughes, B. K. Peters, R. R. Merchant and T. Qin, Nat. Chem., 2021, 13, 950 CrossRef CAS PubMed.
- (a) C. Peng, W. Zhang, G. Yan and J. Wang, Org. Lett., 2009, 11, 1667 CrossRef CAS PubMed; (b) C. Wu, G. Wu, Y. Zhang and J. Wang, Org. Chem. Front., 2016, 3, 817 RSC; (c) C. Bomio, M. A. Kabeshov, A. R. Lit, S. H. Lau, J. Ehlert, C. Battilocchio and S. V. Ley, Chem. Sci., 2017, 8, 6071 RSC; (d) S. J. T. Jonker, R. Jayarajan, T. Kireilis, M. Deliaval, L. Eriksson, K. Kaímán and J. S. Szabó, J. Am. Chem. Soc., 2020, 142, 21254 CrossRef CAS PubMed; (e) Q. Wang, L. Eriksson and K. J. Szabo, Angew. Chem., Int. Ed., 2023, 62, e202301481 CrossRef CAS PubMed.
- (a) O. A. Argintaru, D. Ryu, I. Aron and G. A. Molander, Angew. Chem., Int. Ed., 2013, 52, 13656 CrossRef CAS PubMed; (b) G. Wu, Y. Deng, C. Wu, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2014, 53, 10510 CrossRef CAS PubMed; (c) C. Battilocchio, F. Feist, A. Hafner, M. Simon, D. N. Tran, D. M. Allwood, D. C. Blakemore and S. V. Ley, Nat. Chem., 2016, 8, 360 CrossRef CAS PubMed; (d) D. N. Tran, C. Battilocchio, S. B. Lou, J. M. Hawkins and S. V. Ley, Chem. Sci., 2015, 6, 1120 RSC; (e) J. Poh, S. Lau, I. G. Dykes, D. N. Tran, C. Battilocchio and S. V. Ley, Chem. Sci., 2016, 7, 6803 RSC.
- A. Greb, J.-S. Poh, S. Greed, C. Battilocchio, P. Pasau, D. C. Blakemore and S. V. Ley, Angew. Chem., Int. Ed., 2017, 56, 16602 CrossRef CAS PubMed.
- (a) X. Ou, R. Labes, C. Battilocchio and S. V. Ley, Org. Biomol. Chem., 2018, 16, 6652 RSC; (b) J. A. Forni, S. H. Lau, J. S. Poh, C. Battilocchio, S. V. Ley and J. C. Pastre, Synlett, 2018, 29, 825 CrossRef CAS; (c) Y. Chen, D. C. Blakemore, P. Pasau and S. V. Ley, Org. Lett., 2018, 20, 6569–6572 CrossRef CAS PubMed.
- (a) H. Wang, S. Wang, V. George, G. Llorente and B. König, Angew. Chem., Int. Ed., 2022, 61, e202211578 CrossRef CAS PubMed; (b) V. George and B. König, Chem. Commun., 2023, 59, 11835 RSC.
- (a) S. Jana, F. Li, C. Empel, D. Verspeek, P. Aseeva and R. M. Koenigs, Chem. – Eur. J., 2020, 26, 2586 CrossRef CAS PubMed; (b) Y. Xu, G. Lv, K. Yan, H. He, J. Li, Y. Luo, R. Lai, L. Hai and Y. Wu, Chem. – Asian J., 2020, 15, 1945 CrossRef CAS PubMed; (c) K. Yan, H. He, J. Li, Y. Luo, R. Lai, L. Guo and Y. Wu, Chin. Chem. Lett., 2021, 32, 3984 CrossRef CAS.
- The generation of diazoalkanes under mild conditions can be also achieved with N-nosyl and N-triftosylhydrazones: (a) Z. Liu, Q. Li, P. Liao and X. Bi, Chem. – Eur. J., 2017, 23, 4756 CrossRef CAS PubMed; (b) Z. Liu, K. Raveendra Babu, F. Wang, Y. Yang and X. Bi, Org. Chem. Front., 2019, 6, 121 RSC; (c) For a review see: Z. Liu, P. Sivaguru, G. Zanoni and X. Bi, Acc. Chem. Res., 2022, 55, 1763 CrossRef CAS PubMed.
- D. Xia, R. Wu, J. Wang, X. Han, Y. Li, Q. Li, X. Luan, X. Hong, Y. Zhang and W. Zhang, ACS Catal., 2023, 13, 9806 CrossRef CAS.
- For the blue light promoted coupling of arylboronic acids with aryldiazoacetates: A. F. da Silva, M. A. S. Afonso, R. A. Cormanich and I. D. Jurberg, Chem. – Eur. J., 2020, 26, 5648 CrossRef CAS PubMed.
- For photoredox catalytic reactions involving N-sulfonylhydrazones that do not proceed through the formation of a diazoalkane: (a) X. Q. Hu, J. R. Chen, Q. Wei, F. L. Liu, Q. H. Deng, A. M. Beauchemin and W. J. Xiao, Angew. Chem., Int. Ed., 2014, 53, 12163–12167 CrossRef CAS PubMed; (b) X.-Q. Hu, X. Qi, J.-R. Chen, Q.-Q. Zhao, Q. Wei, Y. Lan and W.-J. Xiao, Nat. Commun., 2016, 7, 11188 CrossRef PubMed; (c) E. Brachet, L. Marzo, M. Selkti, B. König and P. Belmont, Chem. Sci., 2016, 7, 5002 RSC; (d) Q. Q. Zhao, X. Q. Hu, M. N. Yang, J. R. Chen and W. J. Xiao, Chem. Commun., 2016, 52, 12749 RSC; (e) Q.-Q. Zhao, J. Chen, D.-M. Yan, J.-R. Chen and W.-J. Xiao, Org. Lett., 2017, 19, 3620 CrossRef CAS PubMed; (f) S. Parisotto, G. Garreffa, C. Canepa, E. Diana, F. Pellegrino, E. Priola, C. Prandi, V. Maurino and A. Deagostino, ChemPhotoChem, 2017, 1, 56 CrossRef CAS; (g) S. Wang, B.-Y. Cheng, M. Sršen and B. König, J. Am. Chem. Soc., 2020, 142, 7524 CrossRef CAS PubMed; (h) P. P. Gao, D. M. Yan, M. H. Bi, M. Jiang, W. J. Xiao and J. R. Chen, Chem. – Eur. J., 2021, 27, 14195 CrossRef CAS PubMed; (i) X. Huang, X. Chen, H. Xie, Z. Tan, H. Jiang and W. Zeng, Org. Lett., 2021, 23, 6784 CrossRef CAS PubMed; (j) Q. S. Gao, Z. Niu, Y. Chen, J. Sun, W. Y. Han, J. Y. Wang, M. Yu and M. D. Zhou, Org. Lett., 2021, 23, 6153 CrossRef CAS PubMed; (k) E. Azzi, G. Ghigo, S. Parisotto, F. Pellegrino, E. Priola, P. Renzi and A. Deagostino, J. Org. Chem., 2021, 86, 3300 CrossRef CAS PubMed; (l) S. Wang and B. König, Angew. Chem., Int. Ed., 2021, 60, 21624 CrossRef CAS PubMed; (m) A. Pulcinella, S. Bonciolini, F. Lukas, A. Sorato and T. Noël, Angew. Chem., Int. Ed., 2023, 62, e202215374 CrossRef CAS PubMed.
- (a) Z. Yang, M. L. Stivanin, I. D. Jurberg and R. M. Koenigs, Chem. Soc. Rev., 2020, 49, 6833 RSC; (b) J. Durka, J. Turkowska and D. Gryko, ACS Sustain. Chem. Eng., 2021, 9, 8895 CrossRef CAS; (c) C. Empel, C. Pei and R. M. Koenigs, Chem. Commun., 2022, 58, 2788 RSC.
- For recent reviews on continuous flow photochemistry: (a) D. Cambié, C. Bottecchia, N. J. V. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276 CrossRef PubMed; (b) L. Buglioni, F. Raymenants, A. Slattery, S. D. A. Zondag and T. Noël, Chem. Rev., 2022, 122, 2752–2906 CrossRef CAS PubMed.
- B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS PubMed.
- For generation and reactions of diazo compounds under continuous flow: (a) K. Donnelly and M. Baumann, J. Org. Chem., 2022, 87, 8279 CrossRef CAS PubMed; (b) K. J. Hock and R. M. Koenigs, Chem. – Eur. J., 2018, 24, 10571 CrossRef CAS PubMed; (c) P. Dingwall, A. Greb, L. N. S. Crespin, R. Labes, B. Musio, J. S. Poh, P. Pasau, D. C. Blakemore and S. V. Ley, Chem. Commun., 2018, 54, 11685 RSC; (d) É. Lévesque, S. T. Laporte and A. B. Charette, Angew. Chem., Int. Ed., 2017, 56, 837–841 CrossRef PubMed; (e) D. N. Tran, C. Battilocchio, S. B. Lou, J. M. Hawkins and S. V. Ley, Chem. Sci., 2015, 6, 1120 RSC; (f) L. Kupracz and A. Kirschning, J. Flow Chem., 2013, 3, 11–16 CrossRef CAS.
- For relevant examples of the synthesis and synthetic applications of secondary and tertiary benzylic boronates see: (a) S. H. Cho and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 8157 CrossRef CAS PubMed; (b) S.-Z. Sun and R. Martin, Angew. Chem., Int. Ed., 2018, 57, 3622 CrossRef CAS PubMed; (c) S. Chakrabarty, H. Palencia, M. D. Morton, R. O. Carr and J. M. Takacs, Chem. Sci., 2019, 10, 4854 RSC; (d) S. Sun, Y. Duan, R. S. Mega, R. J. Somerville and R. Martin, Angew. Chem., Int. Ed., 2020, 59, 4370 CrossRef CAS PubMed; (e) J. Huang, W. Yan, C. Tan, W. Wu and H. Jiang, Chem. Commun., 2018, 54, 1770 RSC; (f) M. W. Campbell, J. S. Compton, C. B. Kelly and G. A. Molander, J. Am. Chem. Soc., 2019, 141, 20069 CrossRef CAS PubMed; (g) G. J. Lovinger and J. P. Morken, J. Am. Chem. Soc., 2017, 139, 17293 CrossRef CAS PubMed; (h) P. Zheng, W. Xu, H. Wang, D. Wang, X. Wu and T. XU, ACS Catal., 2022, 12, 14926 CrossRef CAS; (i) W. Wang, C. Ding and G. Yin, Nat. Catal., 2020, 3, 951 CrossRef CAS; (j) P. Zheng, P. Zhou, D. Wang, W. Xu, H. Wang and T. Xu, Nat. Commun., 2021, 12, 1646 CrossRef CAS PubMed; (k) X. Zhang, C. Gao and J. P. Morken, J. Am. Chem. Soc., 2023, 145, 16344 CrossRef CAS PubMed.
- T. J. Michnick and D. S. Matteson, Synlett, 1991, 631 CrossRef CAS.
- At the time of the submission of our preliminary reprint a paper reporting the photochemical generation of diazo alkanes from N-tosylhydrazones in the presence of DBU was reported: Y. Zhang, Y. Li, S. Ni, J. Li, D. Xia, X. Han, J. Lin, J. Wang, S. Das and W. Zhang, Chem. Sci., 2023, 14, 10411 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, description of the batch and continuous flow photochemical setups, characterization data, and copies of the 1H and 13C NMR spectra. See DOI: https://doi.org/10.1039/d3sc05678c |
| ‡ We have observed that the homologation reaction at rt is quite sensitive to steric hindrance. Reactions with tosylhydrazones bearing larger substituents than 5i at the α position failed to provide the homologated boronate (see ESI for details). |
| This journal is © The Royal Society of Chemistry 2023 |