
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStretchable, recyclable thermosets via photopolymerization and 3D printing of hemiacetal ester-based resins†
You-Chi Mason
Wu
 a,
Gloria
Chyr
b,
Hyunchang
Park
a,
Anna
Makar-Limanov
c,
Yuran
Shi
ac,
Joseph M.
DeSimone
ad and
Zhenan
Bao
*a
a,
Gloria
Chyr
b,
Hyunchang
Park
a,
Anna
Makar-Limanov
c,
Yuran
Shi
ac,
Joseph M.
DeSimone
ad and
Zhenan
Bao
*a
aDepartment of Chemical Engineering, Stanford University, Stanford, CA 94305, USA. E-mail: zbao@stanford.edu
bDepartment of Materials Science and Engineering, Stanford University, Stanford, CA 94305, USA
cDepartment of Chemistry, Stanford University, Stanford, CA 94305, USA
dDepartment of Radiology, Stanford University, Stanford, CA 94305, USA
First published on 25th October 2023
Abstract
Achieving a circular plastics economy is one of our greatest environmental challenges, yet conventional mechanical recycling remains inadequate for thermoplastics and incompatible with thermosets. The next generation of plastic materials will be designed with the capacity for degradation and recycling at end-of-use. To address this opportunity in the burgeoning technologies of 3D printing and photolithography, we report a modular system for the production of degradable and recyclable thermosets via photopolymerization. The polyurethane backbone imparts robust, elastic, and tunable mechanical properties, while the use of hemiacetal ester linkages allows for facile degradation under mild acid. The synthetic design based on hemiacetal esters enables simple purification to regenerate a functional polyurethane diol.
Introduction
The increasing production and disposal of plastics poses a serious threat to the environment and human wellbeing.1–3 Although conventional mechanical recycling of thermoplastics, such as polyethylene, has been promoted as a solution to the plastics crisis, this strategy is limited by a number of drawbacks (Fig. 1a). The mechanical recycling process causes scission of polymer chains, which quickly deteriorates the material properties of these plastics.4 Furthermore, the collection and sorting of plastic waste is logistically challenging, and mismanaged waste inevitably leaks into the environment,2 where the chemical persistence of these plastics remains the problem. On the other hand, mechanical recycling is impossible when polymer chains are covalently crosslinked, yet this represents a large class of important materials known as thermosets, which includes silicones and rubbers (Fig. 1b). Given the deficiencies of our current materials economy, it is imperative to make the shift to a circular model, where materials are designed and built to exhibit triggered degradation at their end-of-use.5 Besides reducing waste, circular life cycles significantly decrease both cost and emissions from production.6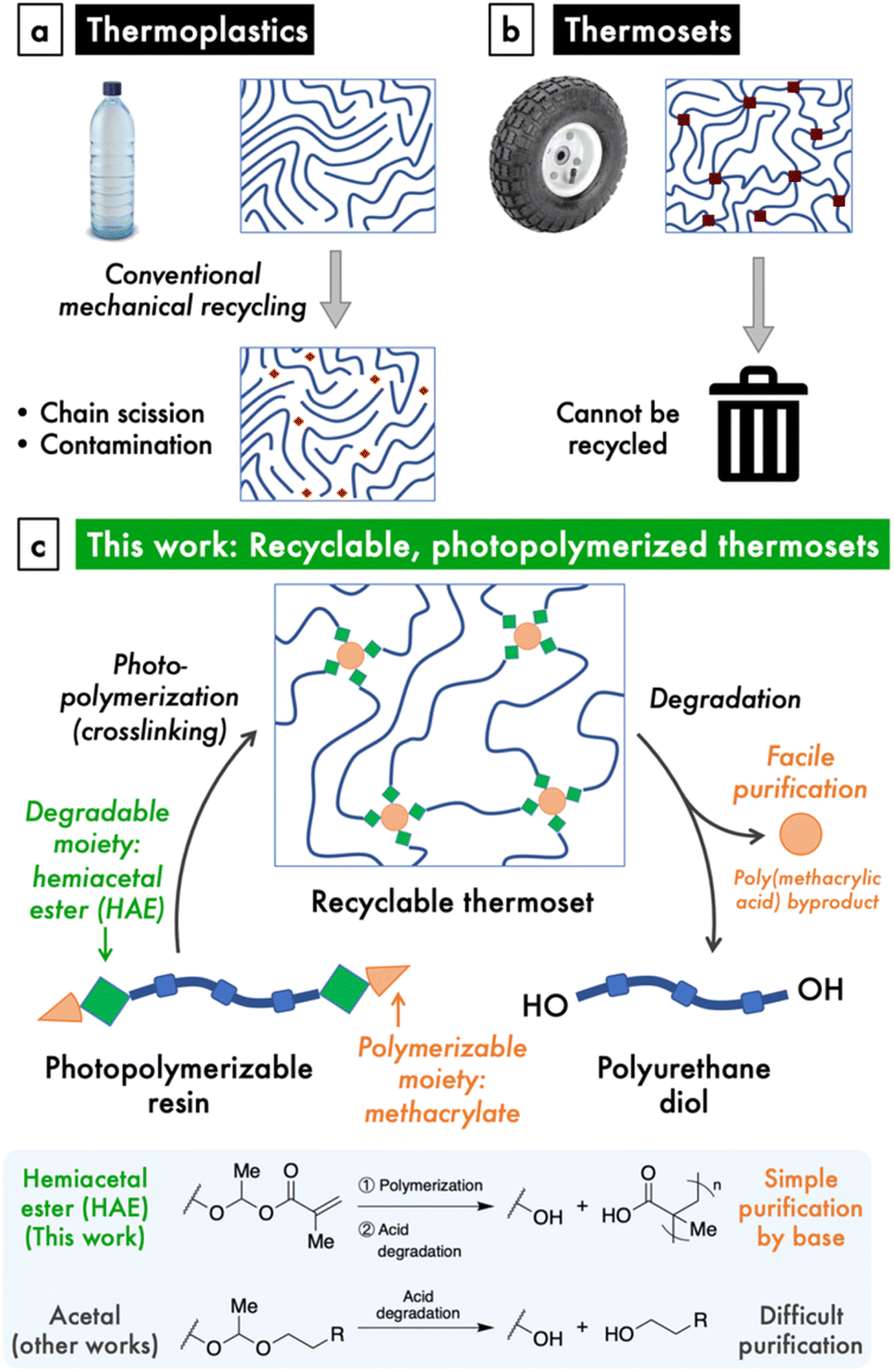 | ||
| Fig. 1 (a) Conventional mechanical recycling of thermoplastics remains a limited solution to plastic waste. (b) Thermosets are covalently crosslinked and thus impossible to reprocess or recycle. (c) In this work, we report a modular design for a degradable and recyclable photopolymerized thermoset based on chemically labile hemiacetal ester (HAE) linkages and polyurethane backbones. The HAE linkage enables facile purification after mild acid degradation. | ||
For traditionally non-recyclable thermosets, the advancement toward recyclability requires the introduction of stimuli-responsive, degradable bonds within these polymers. This paradigm shift is urgently needed for a particular class of thermosets that is garnering increasing attention: photopolymerizable resins that are used in 3D printing and photolithography. 3D printing, or additive manufacturing (AM), enjoys notable advantages in the manufacture process, including speed, customizability, ease of producing complex objects, and high spatial and temporal resolution, as well as reduced waste and greenhouse gas emissions through increased efficiency.7,8 As such, AM is challenging the dominance of traditional injection molding-related manufacturing methods, which comprise a $330 billion industry.7 This inflection point presents an opportunity to direct nascent AM technologies, particularly those based on photopolymerization of thermosets, onto a more sustainable path.
A handful of reports have described photoresins for 3D printing that employ dynamic bonding to impart either degradability or reprocessability to the photocrosslinked materials.9,10 These studies utilize covalent adaptable networks based on established dynamic covalent chemistries, such as transesterification or Diels–Alder reactions, to produce materials with the capacity for repair and reprocessing.11–22 Some of these materials that have cleavable crosslinks are able to be degraded or recycled as well, but often under impractical conditions such as excess chemical reagents.16,23–25 In a related strategy based on reversible hindered-urea bonds, the crosslinked thermoset can be directly reprocessed into a polymerizable resin by reaction with the blocking/crosslinking agent, but the byproduct of photocrosslinking is not removed.19,20 On the other hand, the incorporation of cleavable comonomers into photopolymerized networks allows for the breakdown of the backbone into oligomeric fragments.26,27 Overall, the development of photopolymerized thermoset materials that enjoy facile degradation at end-of-use remains an area that needs further study.
Results and discussion
In order to design a recyclable, photocrosslinkable thermoset, we sought to combine a chemically labile linkage and a polymerizable group within a polymer material. Hemiacetal esters (HAEs) are an intriguing functional group that undergo both acid-catalyzed hydrolysis, similarly to the more common acetals, as well as thermal dissociation via a retro-ene reaction. A few studies incorporated HAEs into either the main chain or side chains of polymers and observed thermal dissociation at high temperatures, typically with an onset of around 160–200 °C,28,29 as well as acidic hydrolysis.30,31 Thermosets crosslinked through HAE moieties have also been shown to degrade under heating32–34 or with a photoacid generator, the latter in the context of photolithography.35 Nonetheless, the use of HAEs to realize a photopolymerizable material with useful mechanical properties and that is recyclable under convenient acidic conditions has not been reported. Yet, we saw the chemical structure of the HAE moiety as a unique opportunity to combine the property of acid lability with the methacrylate groups that are ubiquitous in photopolymerizable materials. Another significant advantage of a HAE over an acetal group is that both the synthesis and degradation of HAEs involve excess/byproduct carboxylic acids whose acidic nature enables simple purification (Fig. 1c).To demonstrate this HAE-based recyclable platform (Fig. 1c), we targeted polyurethane-based materials for their superior and versatile mechanical properties,36 which would enable mechanically robust and stretchable thermosets. To do so, we designed a modular synthetic scheme to prepare polyurethanes terminated with hemiacetal methacrylate (HAMA) groups. These resins can be photocrosslinked to produce thermosets with high stretchabilities and tunable moduli, and are amendable to 3D-printing into complex shapes. Lastly, the HAE-based thermosets are easily de-crosslinked by dilute, catalytic amounts of acid, after which the small amount of poly(methacrylic acid) (PMAA) byproduct is easily removed by either ion-exchange resin or aqueous base wash to regenerate a functional polyurethane material.
Fig. 2 outlines the synthetic design of this recyclable platform. By reacting an oligomeric diol with an excess of a diisocyanate, an isocyanate-terminated polyurethane is produced that is then end-capped with ethylene glycol vinyl ether to furnish vinyloxy chain ends, which are further converted to HAMA groups by acid-catalyzed addition of methacrylic acid. This three-step, modular synthetic scheme affords many handles for tuning the composition of the resin, including the type and molecular weight of the oligomeric diol, the type of diisocyanate, and the ratio of diol to diisocyanate.
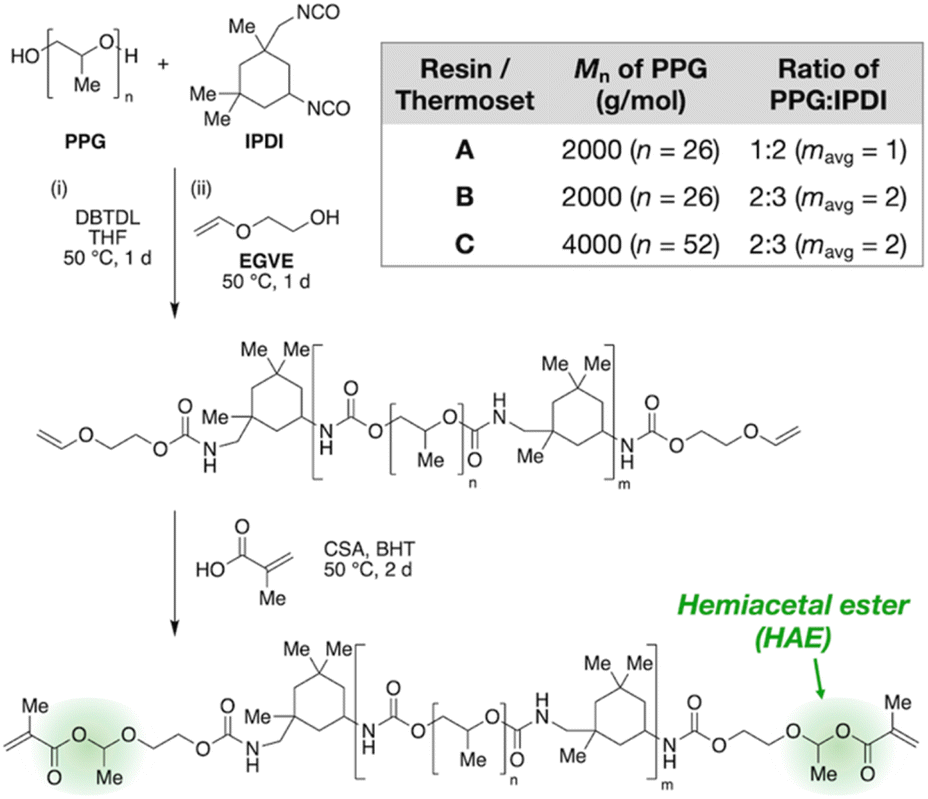 | ||
| Fig. 2 Tunable synthetic design of HAE-based photopolymerizable polyurethanes. DBTDL, dibutyltin dilaurate; CSA, rac-camphorsulfonic acid; BHT, butylated hydroxytoluene. Inset table: composition of Resins/Thermosets A, B, and C studied in this work. | ||
In this work, we focus on polypropylene glycol (PPG) as the diol and isophorone diisocyanate (IPDI) as the diisocyanate to obtain liquid resins suitable for 3D printing. We varied molecular weight and diol/diisocyanate ratio to produce three variations, which we name Resins A, B, and C (Fig. 2 table). After the second reaction step (end-capping by ethylene glycol vinyl ether), we used 1H NMR to verify vinyloxy termination of the polyurethane (Fig. S1–S3 in the ESI†) and FT-IR to confirm full consumption of isocyanates (Fig. S8–S10†). Size-exclusion chromatography (SEC) corroborates increasing average chain lengths from A to B to C, as expected from their synthetic compositions (see Table S2† for tabulated values). Next, NMR characterization of the final products is consistent with the HAMA-terminated polyurethanes (Fig. S4–S6†). Finally, the resins were crosslinked by UV irradiation (365 nm) after addition of 1 wt% of a radical photoinitiator, diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide, with FT-IR confirming full conversion of the methacrylate groups (Fig. S11–S13†).
Thermal characterization of these HAE-based Thermosets A, B, and C by differential scanning calorimetry (DSC) reveals glass transition temperatures around −60 °C for all three (Fig. S14†), corresponding to the PPG backbone. Thermal dissociation of the HAE group via a retro-ene reaction is evinced by an endothermic peak with an onset of around 170 °C, consistent with literature reports.32 Thermal gravimetric analysis (TGA) demonstrates high thermal stability, with all three thermosets maintaining at least 99% weight at 200 °C (Fig. S15†). The initial weight loss increases in rate from C to B to A, correlating with an increasing density of HAEs, presumably as further decomposition reactions occur after thermal dissociation of the HAE group.34
HAE-based Thermosets A, B, and C exhibit robust mechanical properties, with good flexibility and stretchability (Fig. 3a). Tensile tests reveal the stress–strain behavior of these materials, with representative curves in Fig. 3b, showing purely elastic deformation without yielding or plastic deformation, consistent with a crosslinked material. We find that the decrease in crosslink density from A to B to C correlates with a decrease in Young's modulus (Fig. 3b labels) but an increase in strain at break, with C exhibiting up to a remarkable 250% strain at break. Thermoset B has the highest ultimate tensile strength of the three, at 2.1 MPa. As a control experiment, we synthesized a non-polyurethane, HAMA-terminated PPG resin (PPG-HAMA) with a similar average molecular weight as B (see ESI† for synthetic details). Without hydrogen-bonding urethane groups like in B, crosslinked PPG-HAMA suffers from inferior mechanical properties such as strain at break, ultimate tensile strength, and toughness (Fig. 3b). Thus, the polyurethane-based synthetic design is crucial to creating mechanically useful photocrosslinked thermosets.
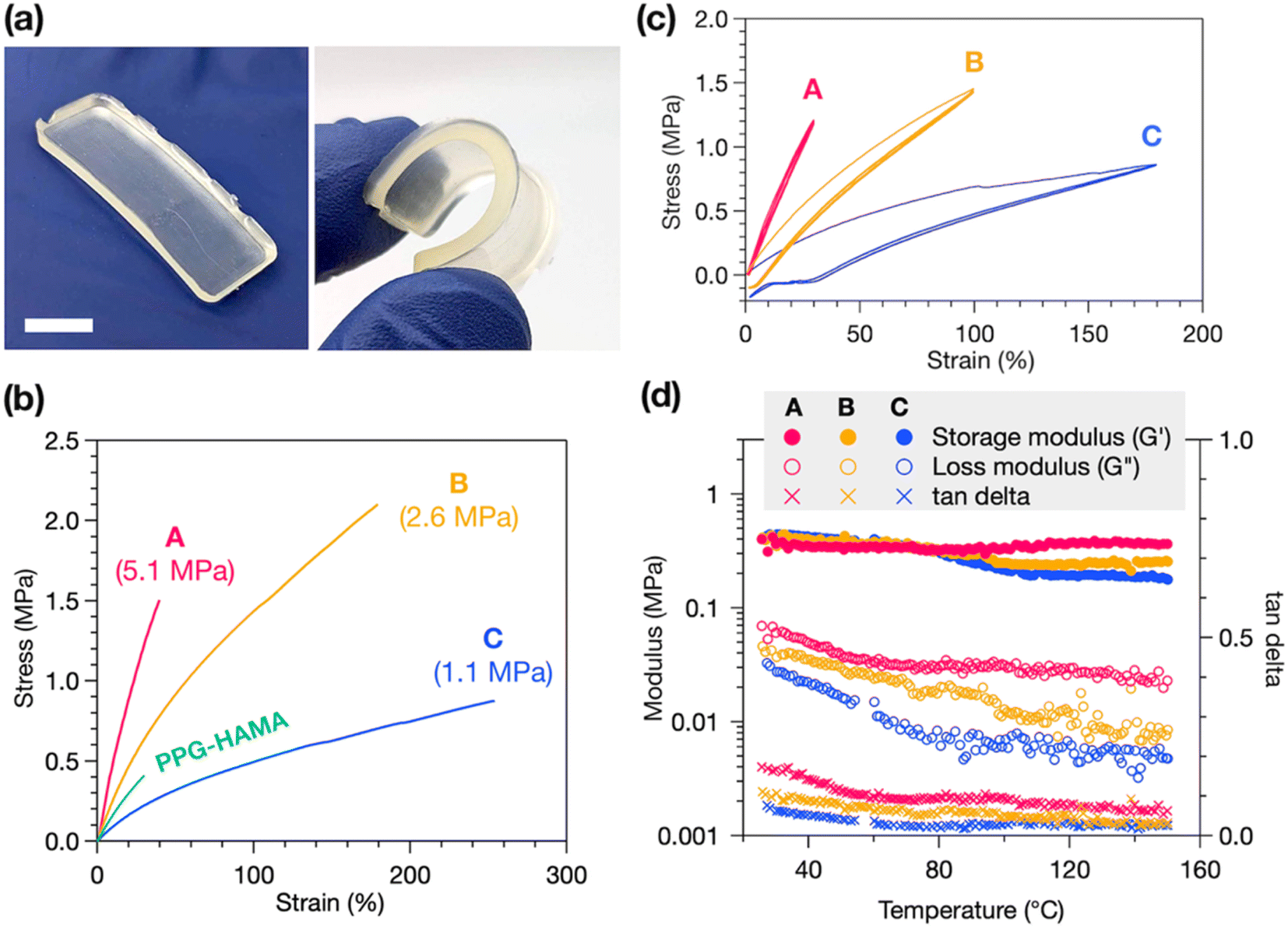 | ||
| Fig. 3 (a) Representative images of Thermoset B. Scale bar = 1 cm. (b) Representative stress–strain curves of tensile tests of Thermosets A, B, and C, as well as a non-polyurethane control, PPG-HAMA, with similar molecular weight as B. Young's moduli shown in parentheses in the labels (averaged over 3 trials with the following standard deviations: 0.2, 0.13, and 0.14 MPa, respectively). (c) Stress–strain cycling tests of Thermosets A, B, and C, showing robust elasticity and minimal plastic deformation. (d) Rheometric measurements of Thermosets A, B, and C from 25 to 150 °C, showing that mechanical stability is maintained up to high temperatures. | ||
In addition to tensile tests, stress–strain cycling tests were performed to demonstrate the elasticity of these materials (Fig. 3c). All three thermosets experience very stable stress–strain behavior over three cycles, indicating good elasticity and minimal plastic deformation, flow, or breakage of crosslinks. The occurrence of hysteresis after only the first elongation, and not in subsequent cycles, suggests this is due to slippage of the sample in the instrument and not deformation. Lastly, the temperature stability of the mechanical properties of the thermosets was probed by rheometry. We measured the storage and loss moduli of each thermoset at 1 Hz oscillatory strain while heating from 25 to 150 °C. As shown in Fig. 3d, the storage moduli of all three thermosets remain generally stable up to 150 °C, indicating high thermal stability, while the slight decrease in storage and loss moduli with increasing temperature suggests dissociation of hydrogen bonds at higher temperatures.37 From C to B to A we find progressively smaller decreases in storage moduli upon heating, with A experiencing a slight increase at high temperature. We attribute this to a greater contribution of entropic elasticity, which causes an increase in storage modulus at higher temperatures, at higher crosslink densities.
We next explored the viability of Resins A, B, and C for 3D printing in a Continuous Liquid Interface Production (CLIP) system.38 In the CLIP system, resins must be able to flow to maintain the continuous liquid interface between a printed part and the window.39 The resins demonstrated Newtonian flow, with the viscosities measured to be in the range of 10–19 Pa s (see Fig. S18†), indicating suitability for printing. We used an in-house-built printer with 30 μm pixel size (see ESI† for detailed methods).40 Using the CLIP system, we were able to achieve the printing of various complex geometries, demonstrating the versatility and precision of this fabrication method (Fig. 4). Rheometric measurements show similar storage moduli of the 3D-printed thermosets compared to that of the bulk UV-cured samples (Fig. S16†).
 | ||
| Fig. 4 Thermosets A, B, and C 3D-printed by the CLIP system. Scale bar = 1 cm. | ||
Lastly, to establish the recyclability of these materials, we investigated their chemical degradation behavior and material recovery. As shown in Fig. 5a, acid-catalyzed degradation of the HAE linkages was performed by soaking the thermoset in dilute acidic methanol, which results in a polyurethane diol and a small amount of PMAA byproduct (the latter comprising a mass fraction of 2.5–8.0% of the thermoset based on stoichiometric calculations). Notably, this degradation strategy does not require harsh conditions or excess chemical reagents. Degradation was demonstrated on the 3D-printed thermosets, resulting in full dissolution within hours in 0.05 M HCl in methanol (Fig. 5b for B and S19† for A and C). We quantified the degradation process by monitoring the residual dry weights of the solids over time, alongside control experiments in pure methanol, 0.05 M HCl in water, and pure water. As shown in Fig. 5c, 2 × 4 × 4 mm pieces of the thermosets completely degraded and dissolved in acidic methanol within 1–3 hours, with degradation rate increasing from A to B to C. SEC analysis shows minimal change in molecular weights pre-crosslinking vs. post-degradation (Fig. S20†), indicating negligible degradation of the polyurethane backbones. On the other hand, in the control experiments, the thermosets remained stable for up to 2 weeks, with ≤2% weight loss under acidic or neutral aqueous conditions (Fig. 5c inset), suggesting good resistance to moisture. In pure methanol, ≥90% weight was maintained after 2 weeks. As most of this weight loss occurs within the first day and then slows down, we attribute this primarily to swelling in methanol and subsequent diffusion of small amounts of residual non-crosslinked chains out of the material, in addition to slow hydrolysis of the HAE group (see Table S1† for swelling ratios of the thermosets).
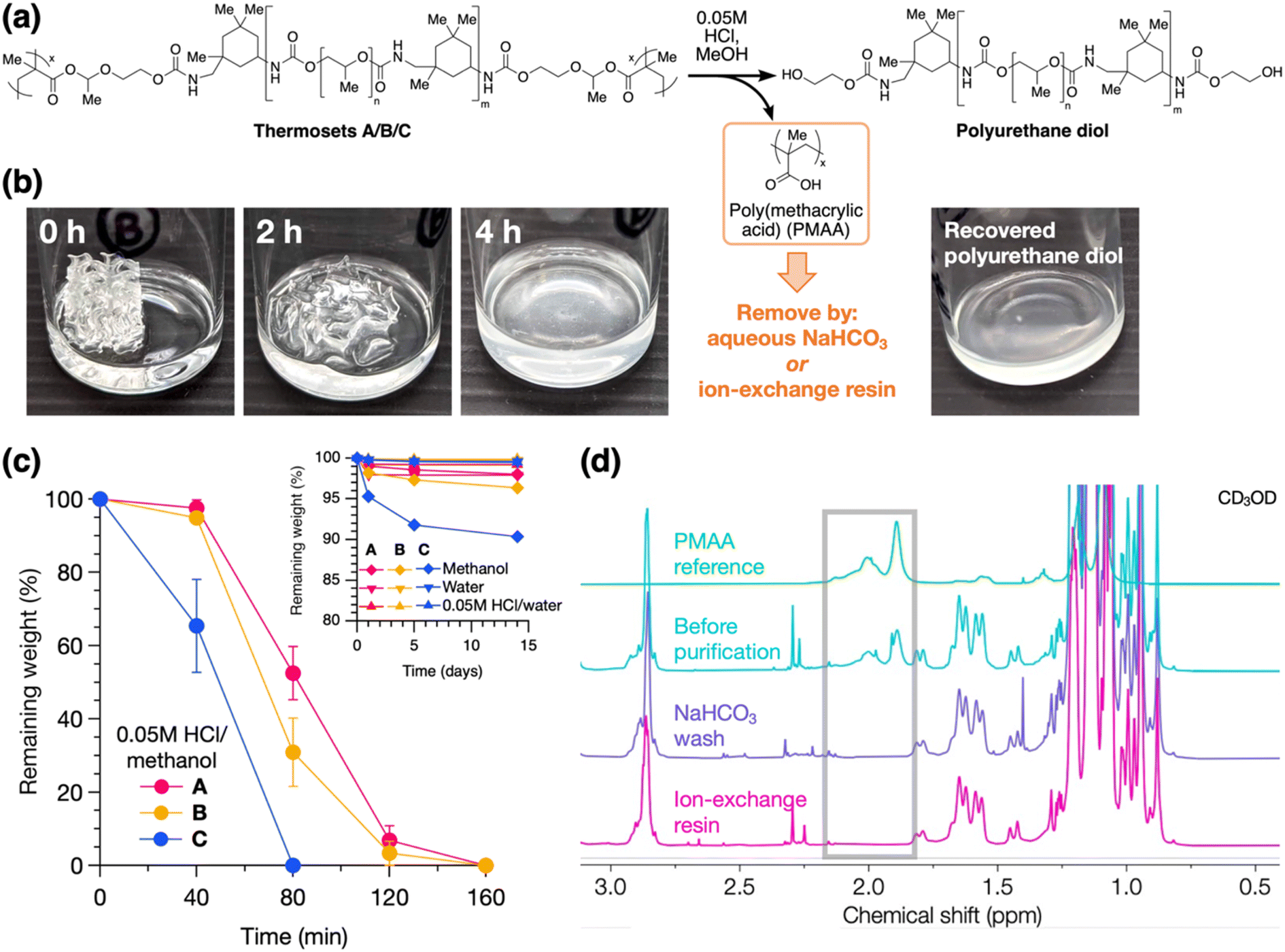 | ||
| Fig. 5 (a) Schematic of the acid-catalyzed degradation of Thermosets A, B, and C, whereby cleavage of the HAE linkage produces a polyurethane diol, which can be recycled into other functional materials, and a poly(methacrylic acid) (PMAA) byproduct, which is easily removed by either aqueous base wash or ion-exchange resin. (b) Timelapse photos of the degradation of Thermoset B in 0.05 M HCl in methanol, along with the recovered polyurethane diol. (c) Residual dry weights of the thermosets during the degradation process in 0.05 M HCl in methanol over time. Averaged over 3 trials; dimensions of samples are 2 × 4 × 4 mm. Inset graph: control experiments in pure methanol, pure water, and 0.05 M HCl in water. All lines are to guide the eye. (d) 1H NMR spectra showing complete removal of the PMAA byproduct from the recycled polyurethane diol by either NaHCO3 wash or ion-exchange resin, as evidenced by the disappearance of the CH2 peaks of PMAA (1.8–2.2 ppm) after purification. B is used as the example. | ||
With the use of HAE as the degradable linkage, the byproduct PMAA is easily removed by taking advantage of its acidic nature (Fig. 5a), whereas this is not possible with an analogous acetal group. This purification process recovers a polyurethane diol that can be recycled into other functional materials (Fig. 5b). We compared two routes for the removal of PMAA: (1) by an ion-exchange resin, Amberlyst A21, containing tertiary amine groups, or (2) by aqueous base wash using NaHCO3 (see ESI† for detailed procedures). Both methods effect complete removal of PMAA from the degraded material, as shown by NMR analysis (Fig. 5d). Yields of the recycled polyurethanes are higher with the ion-exchange resin method (94–97%) compared to the base wash method (85% for B; see Table S4† for tabulated data), and the ion-exchange resin can be regenerated using NaOH for a circular process.41 Full NMR spectra of the recycled polyurethanes from A, B, and C are shown in Fig. S21–S23.†
Conclusions
In conclusion, we have developed a modular, recyclable platform for the design and construction of photocrosslinked thermosets based on an acid-labile HAE linkage. The use of a polyurethane backbone produces materials with high flexibility and stretchability, and we investigate the influence of their tunable composition on thermomechanical properties. Acid-catalyzed degradation occurs readily in dilute acid, and the PMAA byproduct is easily removed either by ion-exchange resin or aqueous base wash, recovering a polyurethane diol that can be reincorporated into functional materials. By merging traditionally nonrecyclable thermoset materials with degradable bonds, we have demonstrated a material design with applicability in the light-based 3D printing of flexible, stretchable, and complex objects. Further studies will explore the incorporation of a wider range of polymer substrates, such as polysiloxanes and fluorinated polyethers, as well as the development of linker chemistries that can realize fully closed-loop materials.Data availability
Data available in the ESI† (ESI): general methods, synthetic procedures, spectral, thermal, mechanical, swelling, SEC, and viscosity characterization, and recycling studies.Author contributions
YMW: project conceptualization, experimental work, manuscript writing and editing; GC: experimental work (3D printing, viscometry), manuscript writing; HP: experimental work (rheometry), manuscript editing; YS: experimental work (part of DMA); AML: experimental work (3D printing); JMD: supervision; ZB: project conceptualization, supervision, manuscript editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Stanford Precourt Pioneering Project on Plastic Innovation. YMW acknowledges postdoctoral fellowship support from the Taiwan Science and Technology Hub at Stanford. Part of this work was performed at the Stanford Nano Shared Facilities (SNSF), supported by the National Science Foundation under award ECCS-1542152.Notes and references
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- J. R. Jambeck, R. Geyer, C. Wilcox, T. R. Siegler, M. Perryman, A. Andrady, R. Narayan and K. L. Law, Science, 2015, 347, 768–771 CrossRef CAS PubMed.
- C. Campanale, C. Massarelli, I. Savino, V. Locaputo and V. F. Uricchio, Int. J. Environ. Res. Public Health, 2020, 17, 1212 CrossRef CAS PubMed.
- Z. O. G. Schyns and M. P. Shaver, Macromol. Rapid Commun., 2021, 42, 2000415 CrossRef CAS PubMed.
- L. T. J. Korley, T. H. Epps, B. A. Helms and A. J. Ryan, Science, 2021, 373, 66–69 CrossRef CAS PubMed.
- N. Vora, P. R. Christensen, J. Demarteau, N. R. Baral, J. D. Keasling, B. A. Helms and C. D. Scown, Sci. Adv., 2021, 7, eabf0187 CrossRef CAS PubMed.
- G. Chyr and J. M. DeSimone, Green Chem., 2023, 25, 453–466 RSC.
- M. Gebler, A. J. M. Schoot Uiterkamp and C. Visser, Energy Policy, 2014, 74, 158–167 CrossRef CAS.
- V. S. D. Voet, ACS Mater. Au, 2023, 3, 18–23 CrossRef CAS.
- E. M. Maines, M. K. Porwal, C. J. Ellison and T. M. Reineke, Green Chem., 2021, 23, 6863–6897 RSC.
- B. Zhang, K. Kowsari, A. Serjouei, M. L. Dunn and Q. Ge, Nat. Commun., 2018, 9, 1831 CrossRef PubMed.
- E. Rossegger, R. Höller, D. Reisinger, M. Fleisch, J. Strasser, V. Wieser, T. Griesser and S. Schlögl, Polymer, 2021, 221, 123631 CrossRef CAS.
- W. Zhao, L. An and S. Wang, Polymers, 2021, 13, 296 CrossRef CAS PubMed.
- C. Hao, T. Liu, S. Zhang, W. Liu, Y. Shan and J. Zhang, Macromolecules, 2020, 53, 3110–3118 CrossRef CAS.
- C. Cui, L. An, Z. Zhang, M. Ji, K. Chen, Y. Yang, Q. Su, F. Wang, Y. Cheng and Y. Zhang, Adv. Funct. Mater., 2022, 32, 2203720 CrossRef CAS.
- J. J. Hernandez, A. L. Dobson, B. J. Carberry, A. S. Kuenstler, P. K. Shah, K. S. Anseth, T. J. White and C. N. Bowman, Macromolecules, 2022, 55, 1376–1385 CrossRef CAS.
- A. Durand-Silva, K. P. Cortés-Guzmán, R. M. Johnson, S. D. Perera, S. D. Diwakara and R. A. Smaldone, ACS Macro Lett., 2021, 10, 486–491 CrossRef CAS PubMed.
- L. L. Robinson, J. L. Self, A. D. Fusi, M. W. Bates, J. Read de Alaniz, C. J. Hawker, C. M. Bates and C. S. Sample, ACS Macro Lett., 2021, 10, 857–863 CrossRef CAS PubMed.
- J. Poelma, M. S. Zhang, X. Gu, J. P. Rolland and J. M. DeSimone, US Pat., 20210246300A1, 2021 Search PubMed.
- J. Poelma, J. P. Rolland, R. H. Curvers and J. M. DeSimone, US Pat., 20210394399A1, 2021 Search PubMed.
- H. Li, B. Zhang, R. Wang, X. Yang, X. He, H. Ye, J. Cheng, C. Yuan, Y. Zhang and Q. Ge, Adv. Funct. Mater., 2022, 32, 2111030 CrossRef CAS.
- X. Li, R. Yu, Y. He, Y. Zhang, X. Yang, X. Zhao and W. Huang, ACS Macro Lett., 2019, 8, 1511–1516 CrossRef CAS PubMed.
- Y. Xu, K. Odelius and M. Hakkarainen, ACS Sustain. Chem. Eng., 2020, 8, 17272–17279 CrossRef CAS.
- K. P. Cortés-Guzmán, A. R. Parikh, M. L. Sparacin, A. K. Remy, L. Adegoke, C. Chitrakar, M. Ecker, W. E. Voit and R. A. Smaldone, ACS Sustain. Chem. Eng., 2022, 10, 13091–13099 CrossRef.
- C. Wang, T. M. Goldman, B. T. Worrell, M. K. McBride, M. D. Alim and C. N. Bowman, Mater. Horiz., 2018, 5, 1042–1046 RSC.
- N. Gil, C. Thomas, R. Mhanna, J. Mauriello, R. Maury, B. Leuschel, J. Malval, J. Clément, D. Gigmes, C. Lefay, O. Soppera and Y. Guillaneuf, Angew. Chem., Int. Ed., 2022, 61, e202117700 CrossRef CAS PubMed.
- S. C. Leguizamon, K. Lyons, N. T. Monk, M. T. Hochrein, B. H. Jones and J. C. Foster, ACS Appl. Mater. Interfaces, 2022, 14, 51301–51306 CrossRef CAS PubMed.
- W. Van Camp, F. E. Du Prez and S. A. F. Bon, Macromolecules, 2004, 37, 6673–6675 CrossRef.
- H. Otsuka and T. Endo, Macromolecules, 1999, 32, 9059–9061 CrossRef CAS.
- A. Kazama and Y. Kohsaka, Polym. Chem., 2019, 10, 2764–2768 RSC.
- A. E. Neitzel, L. Barreda, J. T. Trotta, G. W. Fahnhorst, T. J. Haversang, T. R. Hoye, B. P. Fors and M. A. Hillmyer, Polym. Chem., 2019, 10, 4573–4583 RSC.
- Y. Nakane, M. Ishidoya and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 609–614 CrossRef CAS.
- E. Khosravi, F. Iqbal and O. M. Musa, Polymer, 2011, 52, 243–249 CrossRef CAS.
- X. Zhang, G. Chen, A. Collins, S. Jacobson, P. Morganelli, Y. L. Dar and O. M. Musa, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1073–1084 CrossRef CAS.
- D. Matsukawa, T. Mukai, H. Okamura and M. Shirai, Eur. Polym. J., 2009, 45, 2087–2095 CrossRef CAS.
- J. O. Akindoyo, M. D. H. Beg, S. Ghazali, M. R. Islam, N. Jeyaratnam and A. R. Yuvaraj, RSC Adv., 2016, 6, 114453–114482 RSC.
- E. Wittenberg, A. Meyer, S. Eggers and V. Abetz, Soft Matter, 2018, 14, 2701–2711 RSC.
- J. R. Tumbleston, D. Shirvanyants, N. Ermoshkin, R. Janusziewicz, A. R. Johnson, D. Kelly, K. Chen, R. Pinschmidt, J. P. Rolland, A. Ermoshkin, E. T. Samulski and J. M. DeSimone, Science, 2015, 347, 1349–1352 CrossRef CAS PubMed.
- G. Lipkowitz, T. Samuelsen, K. Hsiao, B. Lee, M. T. Dulay, I. Coates, H. Lin, W. Pan, G. Toth, L. Tate, E. S. G. Shaqfeh and J. M. DeSimone, Sci. Adv., 2022, 8, eabq3917 CrossRef CAS PubMed.
- B. J. Lee, K. Hsiao, G. Lipkowitz, T. Samuelsen, L. Tate and J. M. DeSimone, Addit. Manuf., 2022, 55, 102800 CAS.
- D. Guimarães and V. A. Leão, J. Hazard. Mater., 2014, 280, 209–215 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: general methods, synthetic procedures, spectral characterization, thermal characterization, SEC characterization, and recycling studies (PDF). See DOI: https://doi.org/10.1039/d3sc03623e |
| This journal is © The Royal Society of Chemistry 2023 |