
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitric oxide/paclitaxel micelles enhance anti-liver cancer effects and paclitaxel sensitivity by inducing ferroptosis, endoplasmic reticulum stress and pyroptosis†
Huilan Li‡
a,
Xiaoyu Deng‡b,
Ziwei Zhangb,
Zunhua Yang b,
Hesong Huanga,
Xide Ye*b,
Linyun Zhongb,
Guoliang Xub,
Ronghua Liub and
Yuanying Fang*a
b,
Hesong Huanga,
Xide Ye*b,
Linyun Zhongb,
Guoliang Xub,
Ronghua Liub and
Yuanying Fang*a
aNational Engineering Research Center for Manufacturing Technology of TCM Solid Preparation, Jiangxi University of Chinese Medicine, Nanchang 330006, China. E-mail: fangyuanying@163.com
bCollege of Pharmacy, Jiangxi University of Chinese Medicine, Nanchang 330004, China. E-mail: 20070994@jxutcm.edu.cn
First published on 30th October 2023
Abstract
The objective of this study was to investigate the anticancer activities of biodegradable polymeric micelles composed of monomethoxy poly(ethylene glycol), polylactic acid, and nitric oxide (mPEG-PLA-NO) loaded with paclitaxel (PTX) as a nanomedicine delivery system. We aimed to compare the anticancer effects of these NO/PTX micelles with PTX alone and elucidate their mechanism of action. We evaluated the impact of NO/PTX and PTX on cell viability using Cell Counting Kit-8 (CCK8) assays conducted on the Bel-7402 liver cancer cell line. Additionally, we employed H22 xenografted mice to assess the in vivo tumor growth inhibitory activity of NO/PTX. To examine the cytotoxicity of NO/PTX, the intracellular levels of reactive oxygen species (ROS), and the expression of ferroptosis-related proteins, we conducted experiments in the presence of the ferroptosis inhibitor ferrostatin-1 (Fer-1) or the ROS inhibitor N-acetyl cysteine (NAC). Furthermore, we investigated the expression of endoplasmic reticulum stress (ERS) and apoptosis-associated proteins. Our results demonstrated that NO/PTX exhibited enhanced anticancer effects compared to PTX alone in both Bel-7402 cells and H22 xenografted mice. The addition of Fer-1 or NAC reduced the anticancer activity of NO/PTX, indicating the involvement of ferroptosis and ROS in its mechanism of action. Furthermore, NO/PTX modulated the expression of proteins related to ERS and apoptosis, indicating the activation of these cellular pathways. The anticancer effects of NO/PTX in liver cancer cells were mediated through the induction of ferroptosis, pyroptosis, ERS, and apoptosis-associated networks. Ferroptosis and pyroptosis were activated by treatment of NO/PTX at low concentration, whereas ERS was induced to trigger apoptosis at high concentration. The superior anti-tumor effect of NO/PTX may be attributed to the downregulation of a multidrug resistance transporter and the sensitization of cells to PTX chemotherapy. In summary, our study highlights the potential of mPEG-PLA-NO micelles loaded with PTX as a nanomedicine delivery system for liver cancer treatment. The observed enhancement in anticancer activity, combined with the modulation of key cellular pathways, provides valuable insights into the therapeutic potential of NO/PTX in overcoming resistance and improving treatment outcomes in liver cancer patients.
1. Introduction
Hepatocellular carcinoma (HCC) ranks as the sixth most common malignancy and carries the third-highest mortality rate among all tumors, which presents a serious threat to human health.1 Currently, transcatheter arterial chemoembolization or systemic chemotherapy are the primary treatments for HCC patients who are not amenable to surgical intervention.2,3 Due to occurrence of acquired drug resistance, novel strategies and more effective agents need to be developed to improve therapy for liver cancer. Sensitizing chemotherapy represents a promising approach for the treatment of HCC.4–6Paclitaxel (PTX) is a clinical first-line anti-tumor chemotherapeutic drug, but it often leads to drug resistance through various mechanisms. Overexpression of ATP binding cassette transporter P-glycoprotein (P-gp) alters the efflux capacity of PTX.7,8 In addition, the mutation of β-tubulin, the primary target of PTX, may affect the assembly, movement and stability of microtubules to cause PTX resistance.9–12 Furthermore, several important apoptosis regulators (such as p53, BCL-2 and others) can interact with tubulin and impact the function of microtubules, leading to PTX resistance.13,14 Nitric oxide (NO) plays an important role in anti-tumor activity and has been found to be a pivotal factor in the chemosensitization of tumor cells to various chemotherapeutic drugs.15–18 NO can reverse drug resistance by inhibiting the ATPase activity of P-gp in human multi-drug resistant cancer cells.19 NO-mediated inhibition of NF-κB activity downstream from its anti-apoptotic targets can increase the sensitivity to chemotherapeutic drugs. Gene transfer of iNOS was reported to enhance the anti-tumor activity of cisplatin in vitro and in vivo.20 Frederiksen et al. demonstrated that NO increased anti-prostate cancer efficacy by sensitizing cells to doxorubicin and inducing apoptosis.21 In addition, NO promoted tumor vessel normalization by diminishing vessel diameter and vascular permeability and increasing the extent of perivascular cell coverage.22,23
Recently, additional cell death modes except apoptosis have been discovered which are involved in the actions of various anticancer agents.24 Ferroptosis is an iron-dependent non-apoptotic cell death mode accompanied by lipid reactive oxygen species (ROS).25 Lipid peroxidation, down-regulation of glutathione peroxidase 4 (GPX4) and glutathione consumption are three key features of ferroptosis.26 A series of studies have confirmed the key role of ferroptosis in killing tumor cells and suppressing tumor growth.27–29 NO causes nitrosative stress by forming the highly reactive nitrogen species peroxynitrite (ONOO˙), which can lead to cell death via ferroptosis.30
The endoplasmic reticulum (ER) is an essential site for regulation of cellular homeostasis and is a vital cellular compartment for protein synthesis and maturation.31,32 It has other functions as well including calcium storage and maintenance of Ca2+ homeostasis, steroid synthesis and lipid and glycogen synthesis. Glucose-regulated protein 78 (GRP78) serves as a marker for ER-mediated stress (ERS) and initiation of the unfolded protein response.33 Increasing evidence suggests that ERS represents a potential therapeutic target in tumors.
Pyroptosis is a newly discovered mode of inflammatory programmed cell death relying on caspase-1.34,35 Caspase-1, an interleukin (IL)-1β invertase, can exhibit pro-inflammatory activity by transforming Pro-IL-1β and Pro-IL-18 to their active forms IL-1β and IL-18. Inflammasomes break down pro-caspase-1 to cleaved-caspase-1, resulting in the cell membrane pore formation, cell swelling and rupture. As a result, intracellular inflammatory factors such as IL-1β and IL-18 are released and induce cell death.36,37
To enhance the water solubility, bioavailability, and reduce toxicity of taxanes, various pharmaceutical formulation techniques such as liposomes, polymeric micelles, and other carriers have been developed as nanomedicine delivery systems.38–40 Building upon the synergistic anti-tumor effect of NO and PTX and with the goal of improving the pharmacokinetic profile of PTX, we previously designed and synthesized an NO-donating polymer, consisting of monomethoxy poly(ethylene glycol) and polylactic acid (mPEG-PLA-NO). This polymer was used to generate biodegradable polymeric micelles loaded with PTX (NO/PTX) in a nanoparticle delivery system (Fig. 1). The structure and molecular weight of polymer mPEG-PLA-NO were determined by 1H-NMR and gel permeation chromatography. Characterization of the NO/PTX nanoparticle was performed using dynamic light scattering and transmission electron microscopy.41
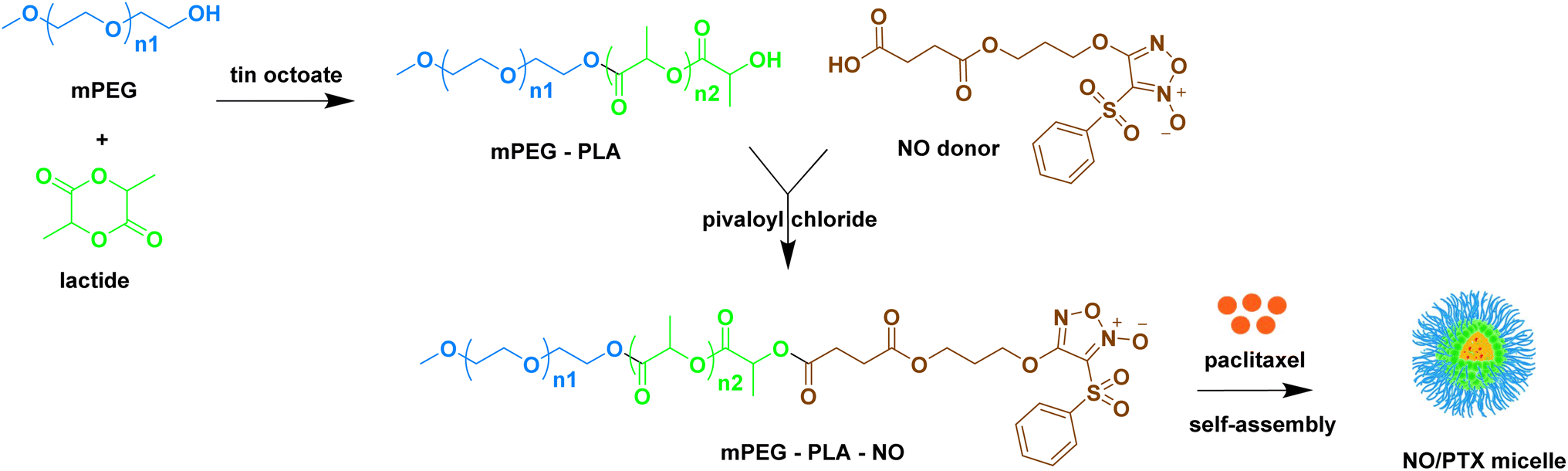 | ||
| Fig. 1 Preparation of paclitaxel-loaded micelles containing NO-releasing moiety (NO/PTX). | ||
During the anticancer evaluation of NO/PTX, we found its specifically high potential on inhibition of liver cancer. Thus, our focus is on comparing the anti-liver cancer effects of these NO/PTX micelles with PTX alone and elucidate their mechanism of action. In the present study, we evaluated the anti-hepatic cancer effects of NO/PTX in vitro and in vivo, the modes of action of NO/PTX, and PTX sensitivity in the liver tumor cell line Bel-7402 using western blot analysis.
2. Materials and methods
2.1. Materials
The HCC cell line Bel-7402 and H22 cancer cells were obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). KM mice (4–6 weeks old, 18–22 g) were provided by Hunan SJA Laboratory Animals (Hunan, China). The animal experiment was performed in accordance with China SAC/TC 281 Laboratory Animal-Guideline for Ethical Review of Animal Welfare (GB/T 35892-2018) and was approved by the Experimental Animal Ethics Committee of Jiangxi University of Chinese Medicine (Nanchang, China). All mice were kept in the specific pathogen free (SPF) animal laboratory under rearing conditions of room temperature 22 ± 1 °C and 12 h/12 h light/night cycle. NO/PTX micelles were prepared and characterized by the pharmaceutics group of Jiangxi University of Chinese Medicine. PTX injections were purchased from Sichuan Sunnyhope Pharmaceutical Co., Ltd (Chengdu, China).2.2. Cytotoxicity of NO/PTX alone and with ferroptosis inhibitor or ROS inhibitor
The HCC cell line Bel-7402 was used for the in vitro cytotoxicity study. Firstly, the antiproliferative effect of PTX (positive control) and NO/PTX were assessed using 10-fold dilutions ranging from 0.0001 up to 10 μg mL−1 with Cell Counting Kit-8 (CCK8) cytotoxicity detection kits (Shanghai Bestbio Biotechnology Co., Ltd, Shanghai, China). Furthermore, the cytotoxicity of NO/PTX in combination with the ferroptosis inhibitor, ferrostatin-1 (Fer-1, 5 μM), or ROS inhibitor N-acetyl cysteine (NAC, 5 mM) were also tested against Bel-7402 cells. Each experiment was independently performed a minimum of three times. SPSS 13.0 software (IBM, Armonk, NY, USA) was used to determine the 50% inhibitory concentration (IC50).Bel-7402 cells were treated with PTX, NO/PTX, NO/PTX + Fer-1 or NO/PTX + NAC, with PTX concentrations of 0.1, 1 or 10 μg mL−1, incubated with Hoechst 33258 dye (Beyotime Biotech, Shanghai, China) and photographed by a fluorescence microscope, which showed the nuclei of apoptotic cells in bright blue.
2.3. In vivo tumor growth inhibitory assay
Male KM mice were injected subcutaneously with H22 cell suspension in saline to establish a tumor bearing mouse model. PTX and NO/PTX were injected intraperitoneally at doses of 30 and 45 mg per kg body weight, respectively, on days 0, 3 and 6, with saline as control. The selection of doses was based on our previous assay of acute toxicity.41 On day 9, the mice were euthanatized and the tumors, livers, spleens and thymus glands were harvested and weighed.2.4. NO/PTX-induced ferroptosis, pyroptosis, ERS and apoptosis
To investigate whether the anticancer activity of the NO/PTX is induced by ferroptosis, the intracellular ROS of Bel-7402 cells were determined after treatment with PTX, NO/PTX or NO/PTX + Fer-1 (0.1 μg mL−1 for PTX). A 2′,7′-dichlorofluorescin diacetate (DCFH-DA) fluorescence probe (#D6883; Sigma, St. Louis, MO, USA) was used and the stained cells were imaged using a Leica fluorescent microscope (Model: DMI 300B, magnification ×200) and analyzed by densitometer software ImageJ. After treatment with PTX, NO/PTX or NO/PTX + Fer-1 (0.1 or 1 μg mL−1), the levels of antioxidant parameters in Bel-7402 cells including total superoxide dismutase (SOD), glutathione peroxidase (GSH-Px) and malondialdehyde (MDA), were determined with commercial reagent kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). The expression of ferroptosis-related proteins, including GPX4, p-ERK1/2, and BCL-2, was assessed using western blotting with an infrared laser imaging system (Biored, Model: Universal hood) and quantified using ImageJ.To investigate the correlation between the anticancer efficacy of NO/PTX and pyroptosis, ERS, and apoptosis, we analyzed the expression of key proteins in Bel-7402 cells after treatment with varying concentrations of NO/PTX. These proteins included caspase-1 and IL-1β (pyroptosis-related), GRP78 and phosphorylated inositol-requiring enzyme 1a (p-IRE1a) (ERS-related), P-gp and β3-tubulin (drug resistance-related), as well as BCL-2, nuclear NF-κB, caspase-3, and cleaved caspase-3 (apoptosis-related).
To explore the mechanism responsible for the cytotoxic effects of PTX on Bel-7402 cells, the apoptosis rates of cells treated with PTX or NO/PTX were measured using an annexin V-FITC apoptosis detection kit (BD Biosciences, San Jose, CA, USA) and analyzed by flow cytometry (FACScan, BD Biosciences).
2.5. Statistical analysis
Results were expressed as the mean ± SD of triplicate experiments with similar patterns. T-test was used to evaluate the statistical differences between samples. Differences were considered to be significant when P < 0.05.3. Results and discussion
3.1. Cytotoxicity of NO/PTX
To evaluate the antiproliferative potential of NO/PTX, we conducted CCK8 assays on the human liver cancer cell line Bel-7402 with PTX as positive control. Six increasing concentrations of NO/PTX and PTX were tested, and the results are depicted in Fig. 2A. It was observed that both NO/PTX and PTX exhibited a dose-dependent inhibitory effect on cell proliferation. Remarkably, NO/PTX demonstrated a significantly higher potency compared to PTX alone, displaying approximately 2-fold stronger activity. The calculated IC50 values for NO/PTX and PTX were 3.7 μg mL−1 and 7.8 μg mL−1, respectively. To further validate these findings, Hoechst 33258 staining was performed to detect dead cells. The cell density and grade of blue color reflected (Fig. 2A) by staining were consistent with the CCK8 assays. These combined results provide strong evidence supporting the enhanced anti-proliferative activity of NO/PTX compared to PTX alone in Bel-7402 cells.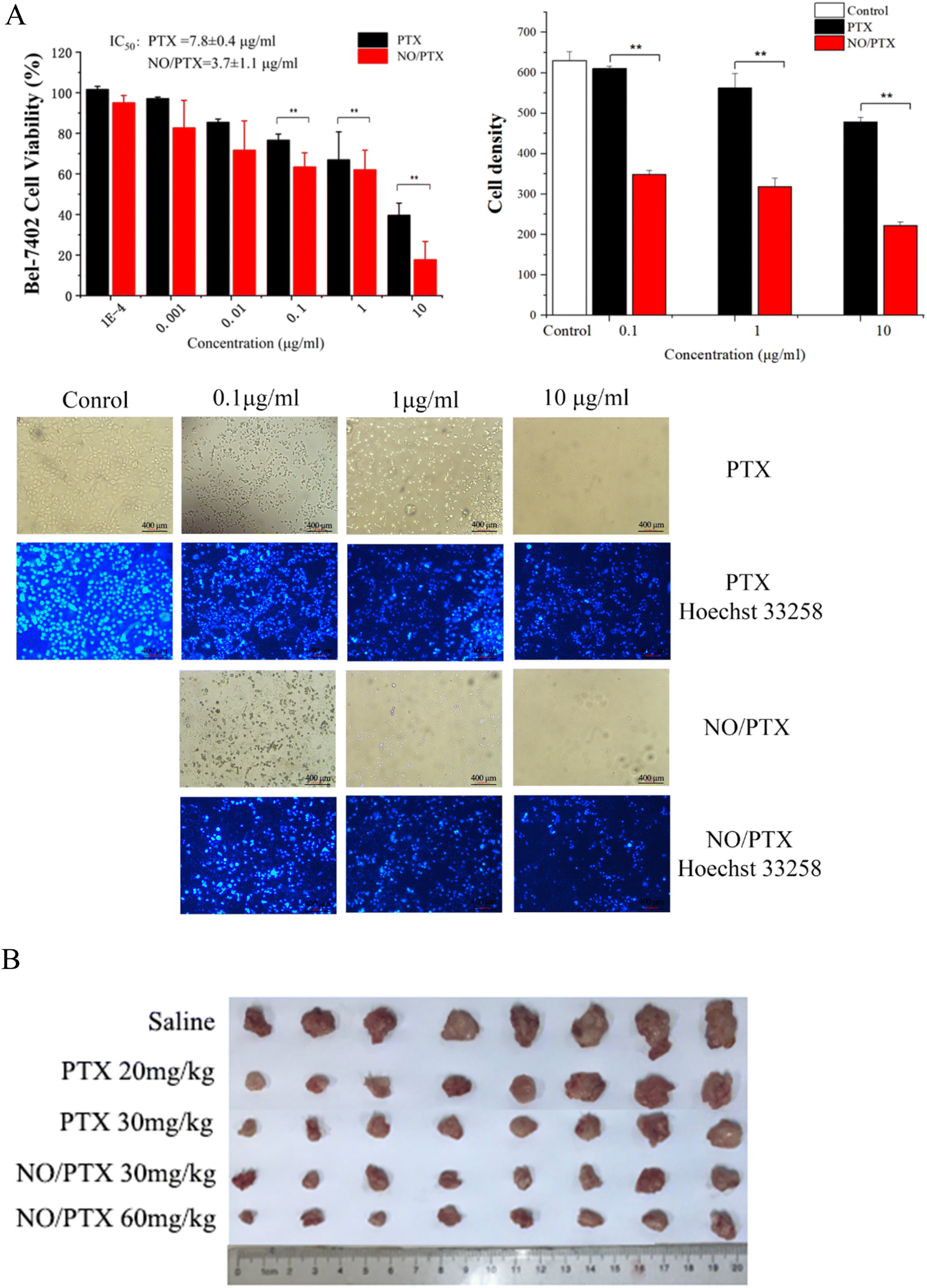 | ||
| Fig. 2 (A) Cell viability, density and Hoechst 33258 staining of cytotoxic assay by NO/PTX and PTX against Bel-7402 cells (**P < 0.01). (B) In vivo tumor growth inhibitory effect of NO/PTX and PTX in the H22 HCC model of KM mice. | ||
3.2. In vivo tumor growth inhibitory activity
In order to evaluate the antitumor efficacy of NO/PTX in comparison to PTX alone, we conducted experiments using the H22 hepatocellular carcinoma (HCC) model in KM mice. The tumor volume was measured after tumor implantation to assess the therapeutic effects. Additionally, the thymus and spleen indices were utilized to investigate the immune-modulating effects of the treatments. The results, presented in Table 1 and Fig. 2B, demonstrated that both PTX and NO/PTX exhibited dose-dependent antitumor effects. At a dosage of 30 mg kg−1, NO/PTX exerted a stronger inhibitory effect on tumor growth compared to PTX alone, with a tumor inhibition rate of 74.0% versus 68.8%, respectively. These findings indicated that NO/PTX possessed enhanced tumor growth inhibitory properties. Furthermore, the administration of NO/PTX demonstrated the ability to modulate the immune response in tumor-bearing mice, as evidenced by higher thymus and spleen indices compared to PTX treatment. Importantly, throughout the duration of the experiment, no mortality was observed among the animals treated with either NO/PTX or PTX. These results collectively highlighted the superior antitumor efficacy of NO/PTX, as well as its immunomodulatory potential, without inducing any significant adverse effects on the experimental animals.| Group | Dosage (mg kg−1) | Body weight (g) | Tumor weight (g) | Tumor inhibition rate (%) | Spleen indices | Thymus indices |
|---|---|---|---|---|---|---|
| a P < 0.01, downregulated (n = 8).b P < 0.01, upregulated (n = 8). | ||||||
| Saline | — | 37.0 ± 3.5 | 1.61 ± 0.51 | — | 0.55 ± 0.16 | 0.36 ± 0.11 |
| PTX | 20 | 34.2 ± 2.8 | 0.63 ± 0.39a | 58.2 ± 24.0 | 0.47 ± 0.15 | 0.12 ± 0.07a |
| PTX | 30 | 32.6 ± 3.6 | 0.50 ± 0.26a | 68.8 ± 16.4 | 0.45 ± 0.25 | 0.12 ± 0.05a |
| NO/PTX | 30 | 32.0 ± 2.1 | 0.42 ± 0.22a | 74.0 ± 13.6 | 0.62 ± 0.18b | 0.14 ± 0.08a |
| NO/PTX | 60 | 30.1 ± 2.4 | 0.39 ± 0.15a | 75.8 ± 9.5 | 0.61 ± 0.16b | 0.13 ± 0.06a |
3.3. NO/PTX-induced ferroptosis
In order to explore the correlation between ferroptosis and the anticancer efficacy of the NO/PTX treatment, we conducted a series of experiments on the Bel-7402 cell line. Firstly, we assessed the cell viability of the tumor cells treated with NO/PTX, both in the presence and absence of the ferroptosis inhibitor Fer-1 and the reactive oxygen species (ROS) scavenger N-acetylcysteine (NAC). Additionally, we performed an analysis of the intracellular ROS levels to evaluate oxidative stress, as well as measured the activities of glutathione peroxidase (GSH-Px), levels of malondialdehyde (MDA), and superoxide dismutase (SOD) to assess the cellular antioxidant defense system. Furthermore, we investigated the expression of glutathione peroxidase 4 (GPX4), a key enzyme involved in the regulation of ferroptosis, using western blotting techniques. Through these comprehensive experiments, we aimed to gain insights into the interplay between ferroptosis induction, ROS modulation, and the anti-tumor effects of NO/PTX treatment in the Bel-7402 cell line.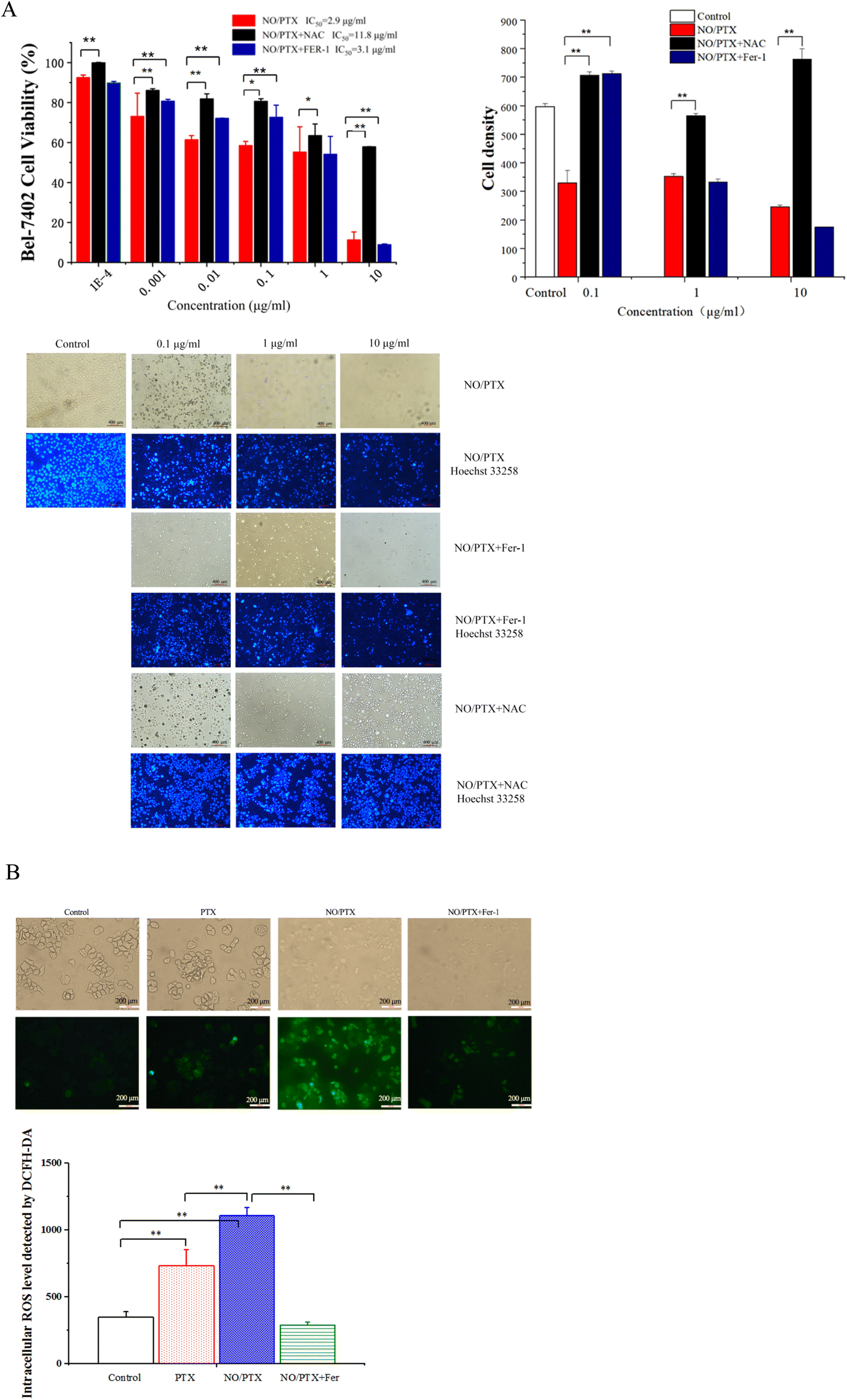 | ||
| Fig. 3 (A) Cell viability, density and Hoechst 33258 staining of NO/PTX-treated cells influenced by Fer-1 or NAC (*P < 0.05, **P < 0.01). (B) The intracellular level of ROS in the cells treated with PTX, NO/PTX or NO/PTX + Fer-1 (**P < 0.01). | ||
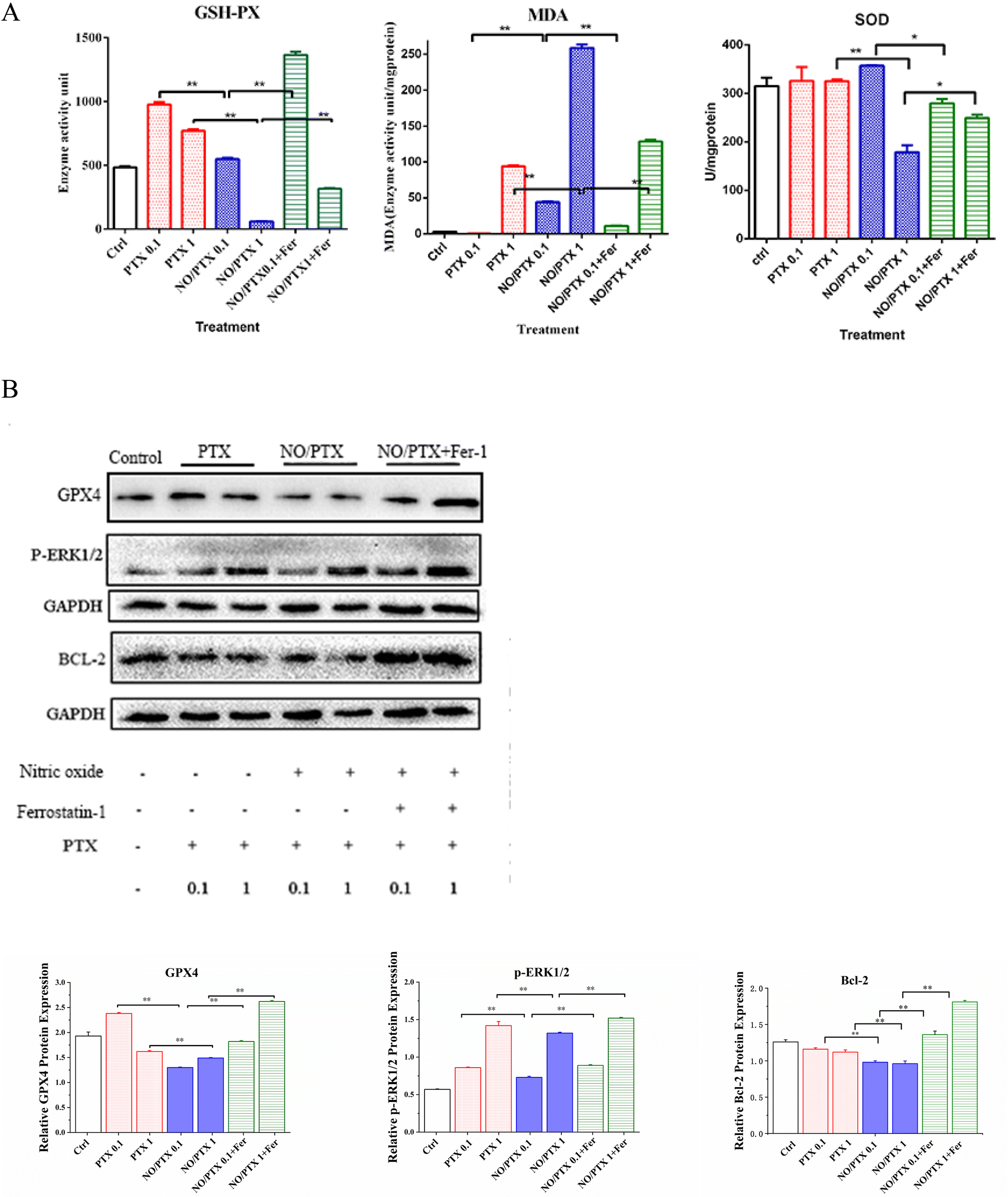 | ||
| Fig. 4 (A) The intracellular GSH-Px, MDA and SOD levels in the cells treated with PTX, NO/PTX or NO/PTX + Fer-1 (*P < 0.05, **P < 0.01). (B) Expression of ferroptosis-related proteins in the cells treated with PTX, NO/PTX or NO/PTX + Fer-1 (**P < 0.01). | ||
The increase of MDA reflects the level of lipid peroxidation and triggers ferroptosis in the cell. The MDA content of the blank group remained at a low level. After administration of NO/PTX, the MDA content increased significantly. However, after adding Fer-1, the intracellular MDA content of the NO/PTX group decreased accordingly.
SOD can effectively remove superoxide anion free radicals in the human body. The results showed that the group treated with 1 μg mL−1 of NO/PTX had a significant decrease in SOD compared with the blank group and the PTX group. After adding Fer-1, the SOD content increased (P < 0.05).
3.4. NO/PTX-induced pyroptosis
Caspase-1 mediates two types of programmed cell death: pyroptosis and apoptosis.44 Compared with the PTX group, NO/PTX increased the expression of caspase-1 and IL-1β protein in Bel-7402 cells (Fig. 5A). In addition, living cells in the control group displayed round and regular nuclei, while only a few cells with condensed and/or fragmented nuclei characteristic of apoptotic cells were present at low concentrations in the NO/PTX group, showing the pyroptotic characteristics of nuclei expansion, while high concentrations caused pyknosis of the nucleus and the appearance of apoptotic bodies (Fig. 5B). Therefore, pyroptosis may be activated by low concentrations of NO/PTX, whereas high concentrations of NO/PTX may activate apoptosis.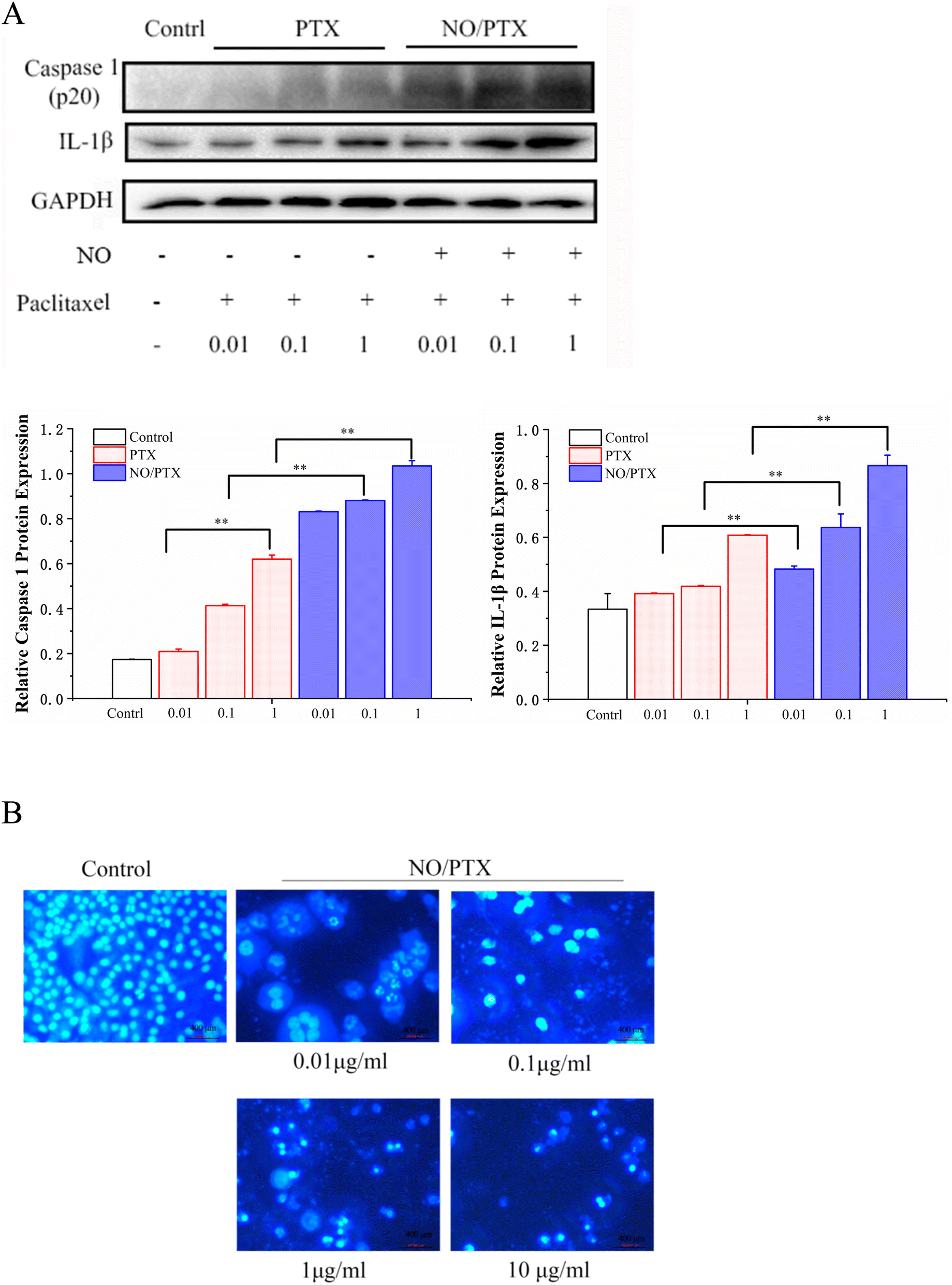 | ||
| Fig. 5 (A) Expression of pyroptosis-related proteins in the cells treated with PTX or NO/PTX (**P < 0.01). (B) Morphological change of cells treated with NO/PTX at various concentrations by Hoechst 33258 staining. | ||
3.5. NO/PTX induced ERS
To validate the endoplasmic reticulum stress (ERS) effect of the NO/PTX treatment in Bel-7402 cells, a series of experiments were conducted. The cells were exposed to different concentrations of PTX or NO/PTX, and subsequent analysis was performed to assess the impact of NO/PTX on the expression of hepatic ERS-related proteins. The results, depicted in Fig. 6, revealed a significant increase of ERS-related proteins, including glucose-regulated protein 78 (GRP78) and phosphorylated inositol-requiring enzyme 1 alpha (p-IRE1a), in the NO/PTX-treated cells comparing to PTX group. These findings strongly suggested that NO/PTX treatment induced ERS, thereby triggering apoptosis in the Bel-7402 cells. The upregulation of GRP78 and p-IRE1a further supported the involvement of the ERS pathway in mediating the apoptotic effects of NO/PTX treatment.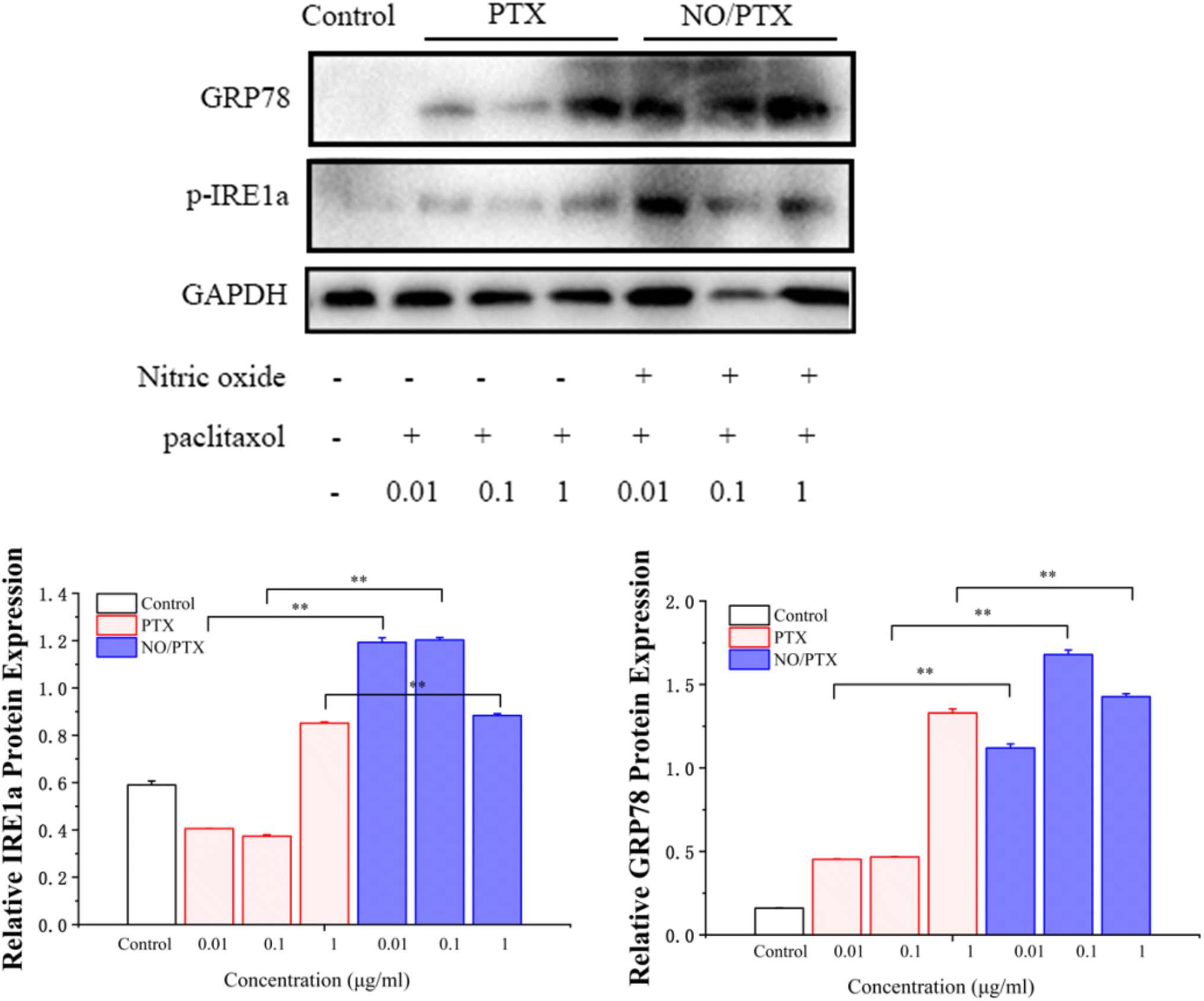 | ||
| Fig. 6 Expression of ER-stress-related proteins in the cells treated with PTX or NO/PTX (*P < 0.05, **P < 0.01). | ||
3.6. Expression of P-gp, β3-tubulin and apoptosis-related proteins in Bel-7402 cells
The mechanism of PTX resistance in tumor cells is mainly related to increased expression of efflux protein and β3-tubulin.45–47 To explore whether NO/PTX increases the sensitivity of PTX against HCC cells, we examined the expression of P-gp, β3-tubulin and apoptosis-related proteins by western blot. The results showed that compared with the PTX group, NO/PTX reduced the expression of P-gp, β3-tubulin, BCL-2 and nuclear NF-κB, while increasing the expression of caspase-3 and cleaved caspase-3 in Bel-7402 cells (Fig. 7A and B). These data indicated that NO/PTX could suppress the efflux efficacy of PTX and sensitize PTX chemotherapy against Bel-7402 cells.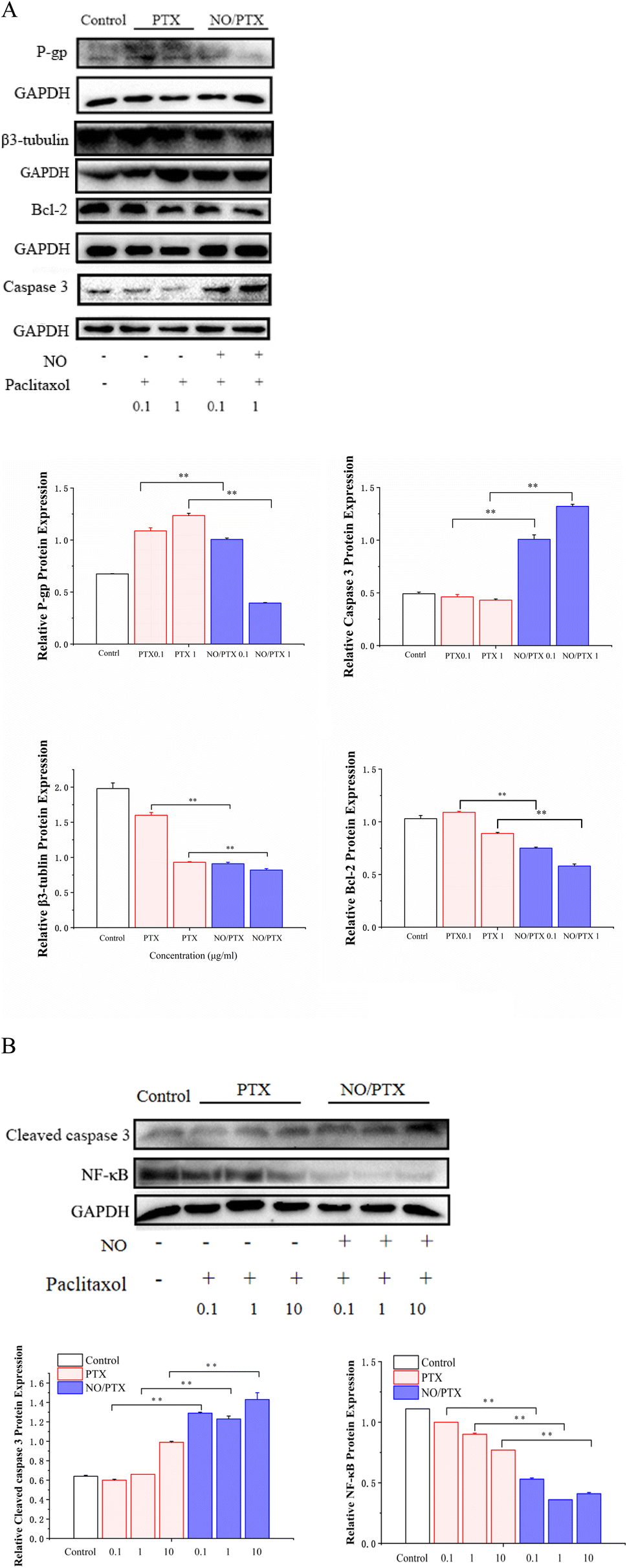 | ||
| Fig. 7 (A) Expression of P-gp, β3-tubulin, BCL-2 and caspase-3 in Bel-7402 cells treated with PTX or NO/PTX (**P < 0.01). (B) Expression of cleaved Caspase-3 and nuclear NF-κB in Bel-7402 cells treated with PTX or NO/PTX (**P < 0.01). | ||
3.7. Apoptosis assay
In order to explore the underlying mechanism responsible for the cytotoxic effects of PTX on Bel-7402 cells, an apoptosis analysis using flow cytometry was conducted. The results revealed that PTX treatment led to a dose-dependent induction of apoptosis in Bel-7402 cells. Moreover, in the presence of nitric oxide (NO), the treatment of NO/PTX exhibited a significantly higher rate of apoptotic cells compared to both the control group and the PTX treatment alone group. These findings strongly indicated that the addition of NO enhanced the cytotoxicity of PTX against Bel-7402 cells. The data, as depicted in Fig. 8, clearly demonstrated the potent apoptotic effects of NO/PTX, further supporting the hypothesis that apoptosis played a crucial role in accounting for the observed cytotoxicity of PTX in Bel-7402 cells.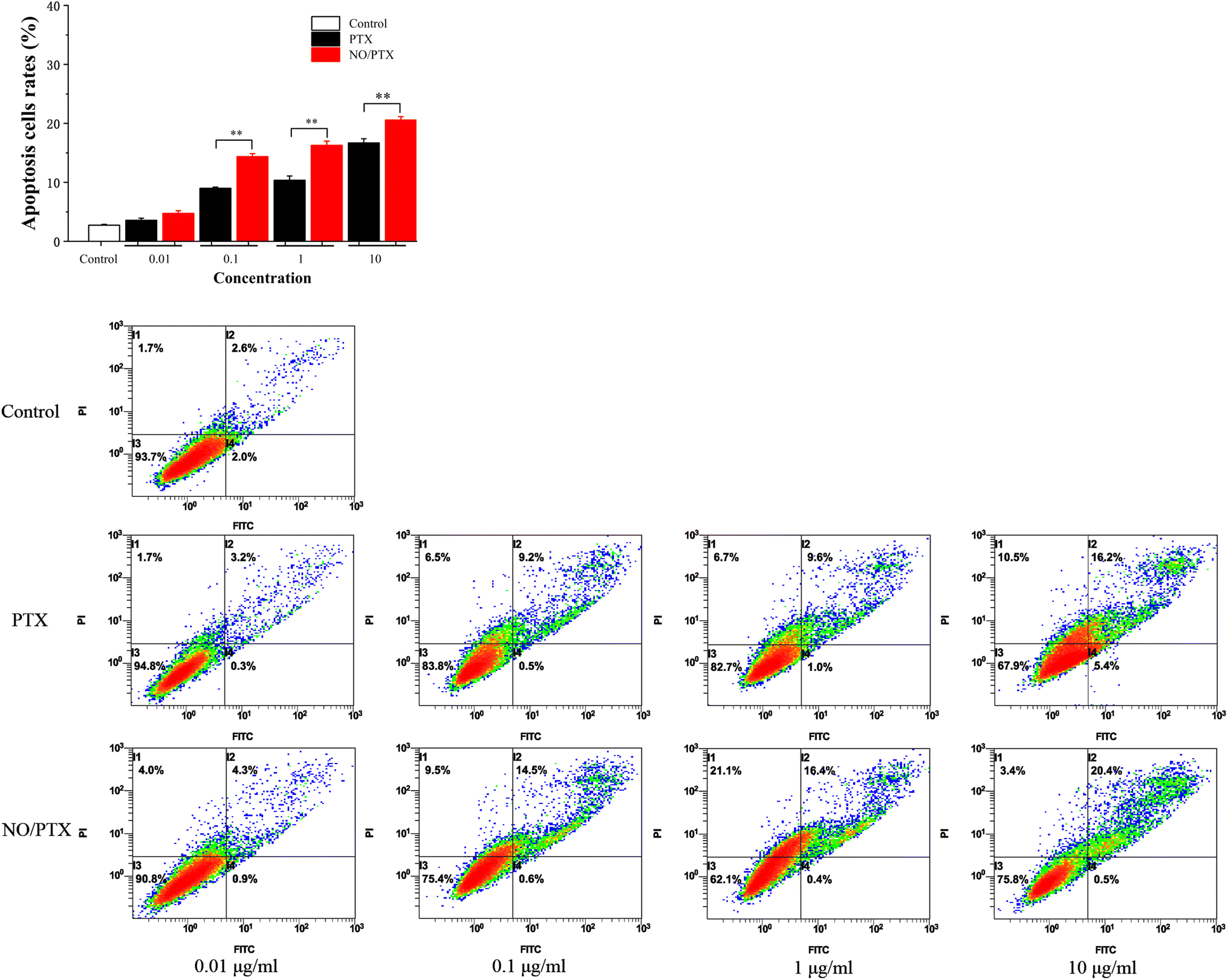 | ||
| Fig. 8 Bel-7402 cells apoptosis induced by PTX and NO/PTX (**P < 0.01). | ||
4. Conclusion
Our findings revealed that NO/PTX displayed 2-fold stronger activity than PTX with IC50 of 3.7 vs. 7.8 μg mL−1, which indicated that NO/PTX exhibited more potent cytotoxicity against Bel-7402 cells than did PTX. Moreover, in an in vivo model, NO/PTX demonstrated superior tumor growth inhibitory effect compared to PTX, with a tumor inhibition rate of 74.0% vs. 68.8%, respectively, when administered at a dosage of 30 mg kg−1.To elucidate the mode of action, we explored several cellular processes. NO/PTX induced ferroptosis by elevating the expression level of ROS and MDA while reducing GSH-Px, SOD and GPX4 levels. In addition, NO/PTX triggered pyroptosis by upregulating caspase-1 and inflammatory cytokine IL-1β. Ferroptosis and pyroptosis were primarily activated at lower concentrations of NO/PTX. Furthermore, NO/PTX induced ERS and apoptosis-associated networks, as evidenced by the upregulation of proteins IRE1a, GRP78, caspase-3, and cleaved caspase-3 in a dose-dependent manner. Conversely, NO/PTX downregulated BCL-2 and NF-κB, promoting apoptotic cell death. Importantly, NO/PTX reduced the expression of P-gp and β3-tubulin in Bel-7402 cells, enhancing the sensitivity of PTX chemotherapy-induced apoptosis. This enhancement was evident as NO/PTX demonstrated greater anti-liver cancer activity than PTX (Fig. 9).
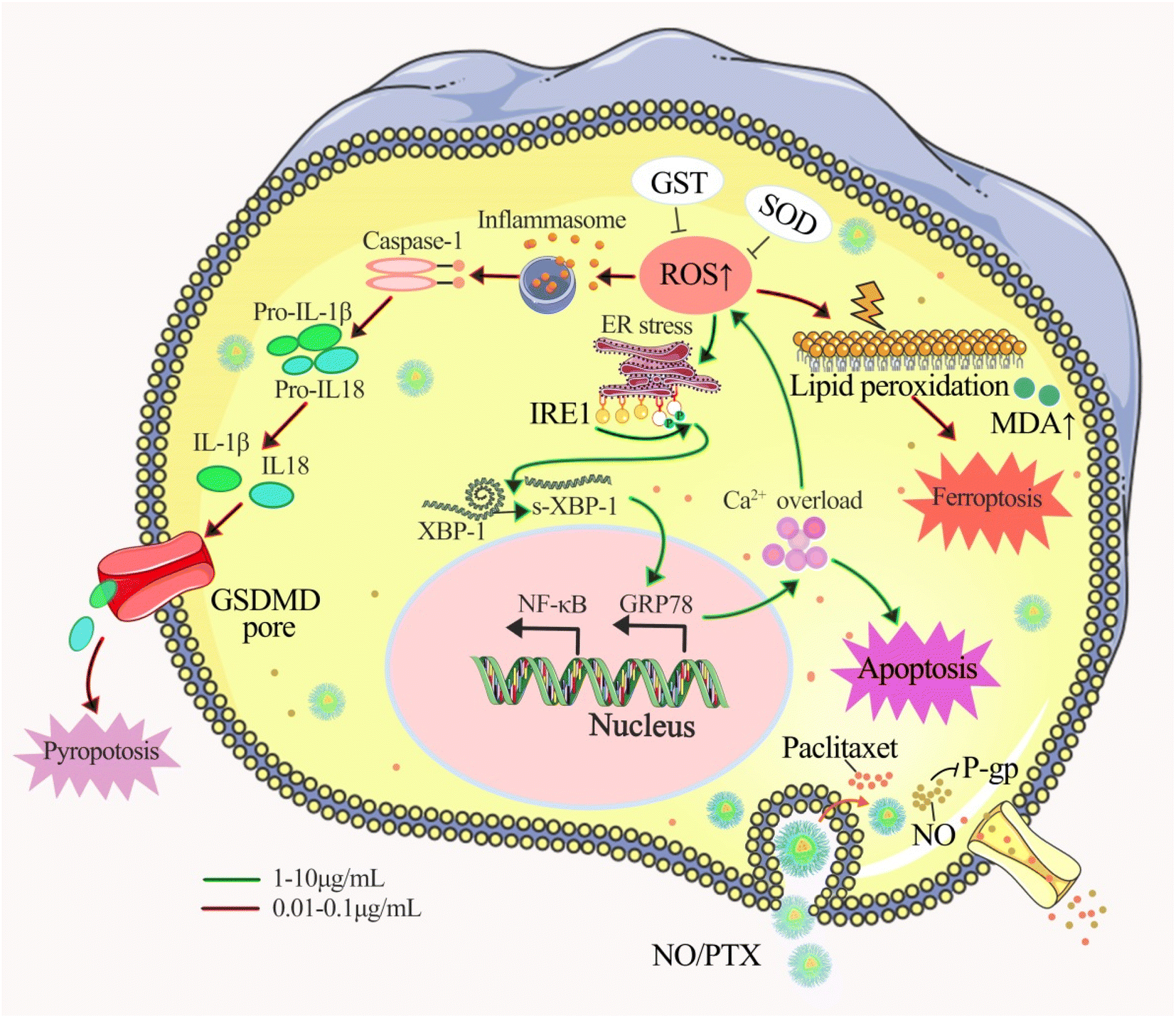 | ||
| Fig. 9 Proposed anticancer mechanism of NO/PTX. | ||
Taken together, our study provides valuable insights into the potential clinical applications of nanoparticle-mediated combination therapy utilizing NO and PTX for liver cancer treatment. The enhanced cytotoxicity, induction of ferroptosis and pyroptosis, activation of ERS and apoptosis-associated networks, and downregulation of drug resistance-related proteins collectively contribute to the superior anti-tumor effects of NO/PTX compared to PTX alone. These findings pave the way for further exploration of this therapeutic approach and its translation into clinical practice, offering new possibilities for improving liver cancer treatment outcomes.
Author contributions
Conceptualization, Xide Ye and Yuanying Fang; data curation, Huilan Li, Ziwei Zhang and Xiaoyu Deng; formal analysis, Huilan Li and Hesong Huang; funding acquisition, Zunhua Yang, Xide Ye and Yuanying Fang; investigation, Huilan Li, Xiaoyu Deng, Hesong Huang and Guoliang Xu; methodology, Huilan Li, Ziwei Zhang and Xiaoyu Deng; resources, Zunhua Yang, Guoliang Xu and Linyun Zhong; supervision, Xide Ye, Ronghua Liu and Yuanying Fang; validation, Huilan Li and Yuanying Fang; writing – original draft, Huilan Li; writing – review & editing, Xiaoyu Deng, Zunhua Yang, Xide Ye and Yuanying Fang.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This study was supported by National Natural Science Foundation of China (82160663), Jiangxi Provincial Department of Science and Technology (20212BAB206015, 20212BAB206016, 20232ACB216016, 20232BCJ22054), Education Department of Jiangxi Province (GJJ201211) and Innovative Group of Jiangxi University of Chinese Medicine (CXTD22003).References
- J. U. Marquardt and S. S. Thorgeirsson, Cancer Cell, 2014, 25, 550 CrossRef CAS PubMed.
- E. Manieri, L. Herrera-Melle, A. Mora, A. Tomás-Loba, L. Leiva-Vega, D. I. Fernández, E. Rodríguez, L. Morán, L. Hernández-Cosido, J. L. Torres, L. M. Seoane, F. J. Cubero, M. Marcos and G. Sabio, J. Exp. Med., 2019, 216, 1108–1119 CrossRef CAS PubMed.
- S. A. Hussain, D. R. Ferry, G. El-Gazzaz, D. F. Mirza, N. D. James, P. McMaster and D. J. Kerr, Ann. Oncol., 2001, 12, 161–172 CrossRef CAS PubMed.
- M. Tong, N. Che, L. Zhou, S. T. Luk, P. W. Kau, S. Chai, E. S. Ngan, C. M. Lo, K. Man, J. Ding, T. K. Lee and S. Ma, J. Hepatol., 2018, 69, 826–839 CrossRef CAS PubMed.
- F. M. Suk, C. L. Liu, M. H. Hsu, Y. T. Chuang, J. P. Wang and Y. J. Liao, Sci. Rep., 2019, 9, 17259 CrossRef PubMed.
- S. Ma, J. Sun, Y. Guo, P. Zhang, Y. Liu, D. Zheng and J. Shi, Theranostics, 2017, 7, 3228–3242 CrossRef CAS PubMed.
- J. P. Gillet and M. M. Gottesman, Methods Mol. Biol., 2010, 596, 47–76 CrossRef CAS PubMed.
- R. W. Robey, K. M. Pluchino, M. D. Hall, A. T. Fojo, S. E. Bates and M. M. Gottesman, Nat. Rev. Cancer, 2018, 18, 452–464 CrossRef CAS PubMed.
- V. K. Kashyap, Q. Wang, S. Setua, P. K. B. Nagesh, N. Chauhan, S. Kumari, P. Chowdhury, D. D. Miller, M. M. Yallapu, W. Li, M. Jaggi, B. B. Hafeez and S. C. Chauhan, J. Exp. Clin. Cancer Res., 2019, 38, 29 CrossRef PubMed.
- K. Verma and K. Ramanathan, J. Cell. Biochem., 2015, 116, 1318–1324 CrossRef CAS PubMed.
- S. Tripathi, G. Srivastava and A. Sharma, Biochem. Biophys. Res. Commun., 2016, 476, 273–279 CrossRef CAS PubMed.
- M. Hari, F. Loganzo, T. Annable, X. Tan, S. Musto, D. B. Morilla, J. H. Nettles, J. P. Snyder and L. M. Greenberger, Mol. Cancer Ther., 2006, 5, 270–278 CrossRef CAS PubMed.
- Y. Tabuchi, J. Matsuoka, M. Gunduz, T. Imada, R. Ono, M. Ito, T. Motoki, T. Yamatsuji, Y. Shirakawa, M. Takaoka, M. Haisa, N. Tanaka, J. Kurebayashi, V. C. Jordan and Y. Naomoto, Int. J. Oncol., 2009, 34, 313–319 CAS.
- R. T. Penson, E. Oliva, S. J. Skates, T. Glyptis, A. F. Fuller, A. Goodman and M. V. Seiden, Gynecol. Oncol., 2004, 93, 98–106 CrossRef CAS PubMed.
- E. Hays and B. Bonavida, Antioxidants, 2019, 89, 407 CrossRef PubMed.
- S. Huerta-Yepez, M. Vega, A. Jazirehi, H. Garban, F. Hongo, G. Cheng and B. Bonavida, Oncogene, 2004, 23, 4993–5003 CrossRef CAS PubMed.
- J. Kim, B. Yung, W. Kim and X. Chen, J. Controlled Release, 2017, 263, 223–230 CrossRef CAS PubMed.
- B. Bonavida and H. Garban, Redox Biol., 2015, 6, 486–494 CrossRef CAS PubMed.
- B. K. Sinha, C. D. Bortner, R. P. Mason and R. E. Cannon, Biochim. Biophys. Acta, Gen. Subj., 2018, 1862, 2806–2814 CrossRef CAS PubMed.
- C. Adams, H. O. McCarthy, J. A. Coulter, J. Worthington, C. Murphy, T. Robson and D. G. Hirst, J. Gene Med., 2009, 11, 160–168 CrossRef CAS PubMed.
- L. J. Frederiksen, D. R. Siemens, J. P. Heaton, L. R. Maxwell, M. A. Adams and C. H. Graham, J. Urol., 2003, 170, 1003–1007 CrossRef CAS PubMed.
- Y. C. Sung, P. R. Jin, L. A. Chu, F. F. Hsu, M. R. Wang, C. C. Chang, S. J. Chiou, J. T. Qiu, D. Y. Gao, C. C. Lin, Y. S. Chen, Y. C. Hsu, J. Wang, F. N. Wang, P. L. Yu, A. S. Chiang, A. Y. Wu, J. J. Ko, C. P. Lai, T. T. Lu and Y. Chen, Nat. Nanotechnol., 2019, 14, 1160–1169 CrossRef CAS PubMed.
- S. Kashiwagi, K. Tsukada, L. Xu, J. Miyazaki, S. V. Kozin, J. A. Tyrrell, W. C. Sessa, L. E. Gerweck, R. K. Jain and D. Fukumura, Nat. Med., 2008, 14, 255–257 CrossRef CAS PubMed.
- E. Koren and Y. Fuchs, Cancer Discovery, 2021, 11, 245–265 CrossRef CAS PubMed.
- J. Zheng and M. Conrad, Cell Metab., 2020, 32, 920–937 CrossRef CAS PubMed.
- D. Tang, X. Chen, R. Kang and G. Kroemer, Cell Res., 2021, 31, 107–125 CrossRef CAS PubMed.
- Y. Tsai, C. Xia and Z. Sun, Front. Pharmacol., 2020, 11, 598555 CrossRef PubMed.
- S. D. Markowitsch, P. Schupp, J. Lauckner, O. Vakhrusheva, K. S. Slade, R. Mager, T. Efferth, A. Haferkamp and E. Juengel, Cancers, 2020, 12, 3150 CrossRef CAS PubMed.
- B. Zhou, J. Y. Zhang, X. S. Liu, H. Z. Chen, Y. L. Ai, K. Cheng, R. Y. Sun, D. Zhou, J. Han and Q. Wu, Cell Res., 2018, 28, 1171–1185 CrossRef CAS PubMed.
- A. F. Vanin, Cell Biochem. Biophys., 2019, 77, 279–292 CrossRef CAS PubMed.
- X. Chen and J. R. Cubillos-Ruiz, Nat. Rev. Cancer, 2021, 21, 71–88 CrossRef CAS PubMed.
- N. E. Karagas and K. Venkatachalam, Cells, 2019, 8, 1232 CrossRef CAS PubMed.
- A. S. Lee, Cancer Res., 2007, 67, 3496–3499 CrossRef CAS PubMed.
- Z. Tang, L. Ji, M. Han, J. Xie, F. Zhong, X. Zhang, Q. Su, Z. Yang, Z. Liu, H. Gao and G. Jiang, Life Sci., 2020, 257, 118065 CrossRef CAS PubMed.
- I. Jorgensen, M. Rayamajhi and E. A. Miao, Nat. Rev. Immunol., 2017, 173, 151–164 CrossRef PubMed.
- W. Dong, Q. Zhu, B. Yang, Q. Qin, Y. Wang, X. Xia, X. Zhu, Z. Liu, E. Song and Y. Song, Chem. Res. Toxicol., 2019, 32, 1051–1057 Search PubMed.
- M. Jiang, L. Qi, L. Li and Y. Li, Cell Death Discovery, 2020, 6, 112 Search PubMed.
- E. Bernabeu, M. Cagel, E. Lagomarsino, M. Moretton and D. A. Chiappetta, Int. J. Pharm., 2017, 526, 474–495 CrossRef CAS PubMed.
- D. L. Priwitaningrum, K. Pednekar, A. V. Gabriël, A. A. Varela-Moreira, S. Le Gac, I. Vellekoop, G. Storm, W. E. Hennink and J. Prakash, Drug Delivery Transl. Res., 2023, 13, 1470–1483 CrossRef CAS PubMed.
- B. Sun, J. F. Lovell and Y. Zhang, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2023, 15, e1854 CAS.
- H. Li, Y. Fang, X. Li, L. Tu, G. Xu, Y. Jin, R. Liu and Z. Yang, New J. Chem., 2021, 45, 13763–13774 RSC.
- B. A. Zachara, J. Trace Elem. Electrolytes Health Dis., 1992, 63, 137–151 Search PubMed.
- C. Wu, W. Zhao, J. Yu, S. Li, L. Lin and X. Chen, Sci. Rep., 2018, 8, 574 CrossRef PubMed.
- Z. Liu, C. Wang, J. Yang, Y. Chen, B. Zhou, D. W. Abbott and T. S. Xiao, Immunity, 2020, 53, 106–114 CrossRef CAS PubMed.
- A. Ganguly, H. Yang and F. Cabral, Oncotarget, 2011, 25, 368–377 CrossRef PubMed.
- M. S. Jin, M. L. Oldham, Q. Zhang and J. Chen, Nature, 2012, 490, 566–569 CrossRef CAS PubMed.
- P. P. Gan, E. Pasquier and M. Kavallaris, Cancer Res., 2007, 67, 9356–9363 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra04861f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |