
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFlavonoids and related privileged scaffolds as potential urease inhibitors: a review
Munirah M. Al-Rooqia,
Ehsan Ullah Mughal
 *b,
Qandeel Alam Rajab,
Essam M. Husseinac,
Nafeesa Naeemb,
Amina Sadiqd,
Basim H. Asghara,
Ziad Moussae and
Saleh A. Ahmed
*ac
*b,
Qandeel Alam Rajab,
Essam M. Husseinac,
Nafeesa Naeemb,
Amina Sadiqd,
Basim H. Asghara,
Ziad Moussae and
Saleh A. Ahmed
*ac
aDepartment of Chemistry, Faculty of Applied Sciences, Umm Al-Qura University, 21955 Makkah, Saudi Arabia. E-mail: saahmed@uqu.edu.sa
bDepartment of Chemistry, University of Gujrat, Gujrat-50700, Pakistan. E-mail: ehsan.ullah@uog.edu.pk
cChemistry Department, Faculty of Science, Assiut University, 71516 Assiut, Egypt
dDepartment of Chemistry, Government College Women University, Sialkot-51300, Pakistan
eDepartment of Chemistry, College of Science, United Arab Emirates University, P.O. Box 15551, Al Ain, Abu Dhabi, United Arab Emirates
First published on 23rd January 2023
Abstract
Infections caused by bacteria are a significant issue on a global scale, and imperative action is required to discover novel or improved therapeutic agents. Flavonoids are a class of plant-derived compounds that have a variety of potentially useful bioactivities. These activities include immediate antimicrobial properties, synergistic effect with antimicrobials, ferocious repression of pathogenicity, anti-urease activity etc. This review summarizes current studies concerning anti-urease actions of flavonoids as well as structural–activity correlation investigations of the flavonoid core structure. It is possible that if researchers investigate the many structural changes that may be made in flavonoid rings, they'll be able to build up novel compounds that have powerful and effective anti-urease properties.
 Ehsan Ullah Mughal | Ehsan Ullah Mughal is a Tenured Associate Professor of Organic Chemistry at the Department of Chemistry in University of Gujrat, Gujrat, Pakistan. He obtained his Doctorate (Dr rer. nat.) from Bielefeld University, Germany under the supervision of Prof. Dr Dietmar Kuck. For postdoctoral studies, he joined the group of Prof. Dr Xinliang Feng, Max-Planck-Institute for Polymer Research, Mainz, Germany. His current research interests include the design and synthesis of bioactive heterocycles, synthetic flavonoids, transition metals-based terpyridine complexes and their fabrication in dye-sensitized solar cells as well as efficient photo-catalysts and nanographenes synthesis for applications in organic electronics. |
 Saleh A. Ahmed | Saleh A. Ahmed received his PhD in photochemistry under the supervision of Prof. Heinz Dürr in Saarland University, Saarbrücken, Germany. He has more than 20 years of experience as a postdoctoral fellow, senior researcher and visiting professor in France, Japan, Germany, Italy and the USA. Since 2010 he is currently a senior professor of organic chemistry at Umm Al-Qura University, Saudi Arabia and Assiut University, Egypt. He had successfully published more than 220 publications in high-ranked journals and more than 10 US patents. His current research interests include the synthesis and photophysical properties of novel organic compounds, the development of synthetic methodologies for the synthesis of novel organic compounds with unique theoretical and biological applications. |
1. Introduction
Urease (urea amidohydrolase; EC 3.5.1.5) is a multi-subunit, nickel containing enzyme that catalyzes the hydrolysis of urea to carbamate and ammonia as a result of catabolism of nitrogen-containing compounds (Scheme 1).1 Carbamate is converted naturally to ammonia and carbonic acid. Carbonic acid, like the two ammonia molecules that are protonated to generate ammonium and hydroxide ions, equilibrates in water. As a consequence of this reaction, the pH of the reaction environment increases.2 The following is a summary of the response: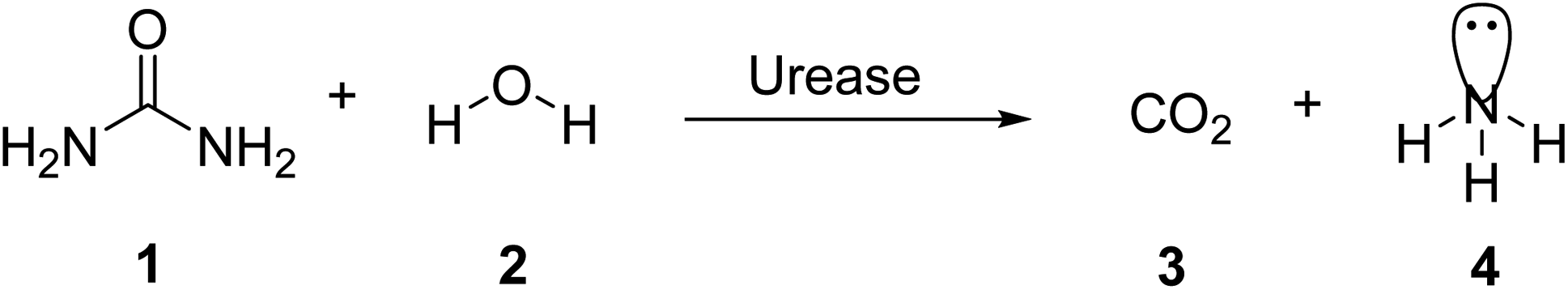 | ||
| Scheme 1 Hydrolysis of urea via urease. | ||
In aqueous solutions, urea is stable. The uncatalyzed reaction is quite sluggish and results in isocyanate and ammonia in an elimination reaction.3
The jack bean urease (JBU) has been studied the most (Canavalia ensiformis). JBU was the first enzyme to be crystallized in 1926. To better understand the biological properties of plant ureases including the mechanism of insecticidal activity, we initiated the structural studies on some of them. Here, we report the crystal structure of JBU, the first plant urease structure, at 2.05 A resolution.4 The scaffold of urease from Klebsiella aerogenes was unravelled in 1995, and subsequently, at that time, numerous other scaffolds, including those from Bacillus pasteurii and Helicobacter pylori, have been described.5 Ureases in plants and fungi are homo-oligomeric proteins with 90 kDa alike subunits, whereas ureases in bacteria are multiverse with binary or tertiary subunit complexes. The sequences of bacterial and plant ureases are quite similar. The active-site architecture of jack bean urease resembles that of binickel-containing bacterial ureases. Although plant and bacterial ureases have similar amino acid sequences, their biological activities differ significantly.6 In divergence to bacterial ureases, which are made up of two or three polypeptides labelled and plant ureases fabricated of a single-chain polypeptide. The subunit contains active sites for all known ureases. The active site amino acid sequences are largely well-preserved in entirely identified ureases, and the catalytic pathway of action is thought to be identical.7
Plants, fungi, bacteria, invertebrates and algae all have ureases.8 Bacteria, yeast, fungi, and plants yield them by catalyzing the degradation of urea to provide these entities with a nitrogen source for development. Urease catalyzes urea integration after uptake into plant cells and participates in the metabolism of N-containing scaffolds in plants (Canavalia ensiformis, Glycine max).9
For bacteria that may invade the human body, urea provides a nitrogen supply. A large amount of the urea generated in the liver is excreted in the intestines, where it may be digested and absorbed by a variety of ureolytic bacteria (Fig. 1).10 Streptococcus salivarius, a common oral bacterium, may also utilize urea as a major nitrogen source for growth. In terms of pathophysiology, ammonia release seems to be the most important factor. Ammonium hydroxide, which is harmful to mammalian cells, may be formed during ureolysis.11 In the context of an oxidative burst caused by immune cells, ammonia may be converted to monochloramine, which has been demonstrated to cause DNA damage. The pH of the reaction environment may rise to 9.21 as a consequence of this. H. pylori is a ureolytic bacterium that causes gastric and duodenal ulcers in the intestine.12 Ammonia production raises pH, allowing bacteria to thrive in such environment. Urinary tract infections are caused by ureolytic bacteria such as13 Proteus vulgaris and Proteus mirabilis.14 Urinary stones are formed when the pH of the urine rises due to the precipitation of usually soluble polyvalent ions in the urine. Ammonia has an undeviating cytostatic impact on epithelial cells as well. Urease inhibition prevents these bacteria from alkalinizing their surroundings.13,15–23 Hydroxamic acids and their derivatives, which are well-known inorganic urease inhibitors, have been demonstrated to be revocable, slow-binding inhibitors of mutually microbial and plant urease. Unfortunately, acetohydroxamic acid has a long list of negative side effects. Plant extracts or natural substances are a superior option since they have fewer side effects and are well tolerated.24
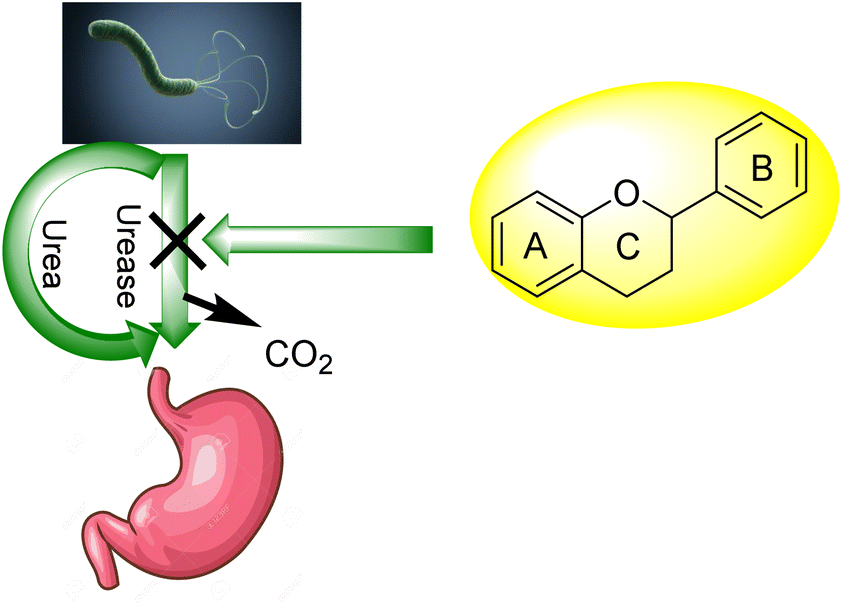 | ||
| Fig. 1 Flavonoid activity against H. Pylori. | ||
In this perspective, using medicinal plants to cure and prevent a variety of diseases is growing around the entire globe, and natural products are regaining space and relevance in the therapeutic sector as springs of new potentially active chemicals.25 Several plants have been described as potent in inhibiting urease in scientific trials.7,26–28 The presence of flavonoids is primarily responsible for many plants' therapeutic benefits.29
Flavonoids30 5 are a varied group of secondary metabolites with over 9000 structures discovered to date.31 They are the most numerous and significant polyphenolic chemicals found in plants.32 Entire vascular plants, in addition to certain mosses, contain these chemicals.33 Plant pigments, usually generated from benzo-pyrone, are referred to as flavonoids (rings A and C in Fig. 2).34
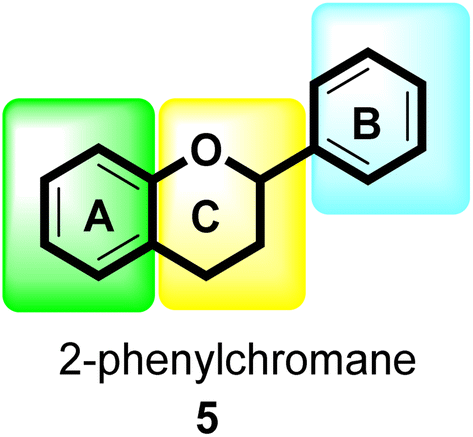 | ||
| Fig. 2 General flavonoid (2-phenylchromane) scaffold. | ||
Malonyl-CoA and p-coumaroyl-CoA are the basic metabolites that deliver the 15-carbon scaffolds (C6–C3–C6) to flavonoids.35 The abridgment of 3 molecules of malonyl-CoA with p-coumaroyl-CoA to produce a chalcone 6 intermediate is their key biosynthetic reaction.36 Chalcones are also key precursors for various plant flavonoids. Most flavanones 7 are 6-membered heterocycles synthesized by conjugate addition of a phenol on an enone.37 Chalcone synthase catalyzes the initial phase in the flavonoid mechanism (CHS; see Scheme 2). The aurones, a flavonoid sub-category present in some plant classes, can then be converted from chalcones.
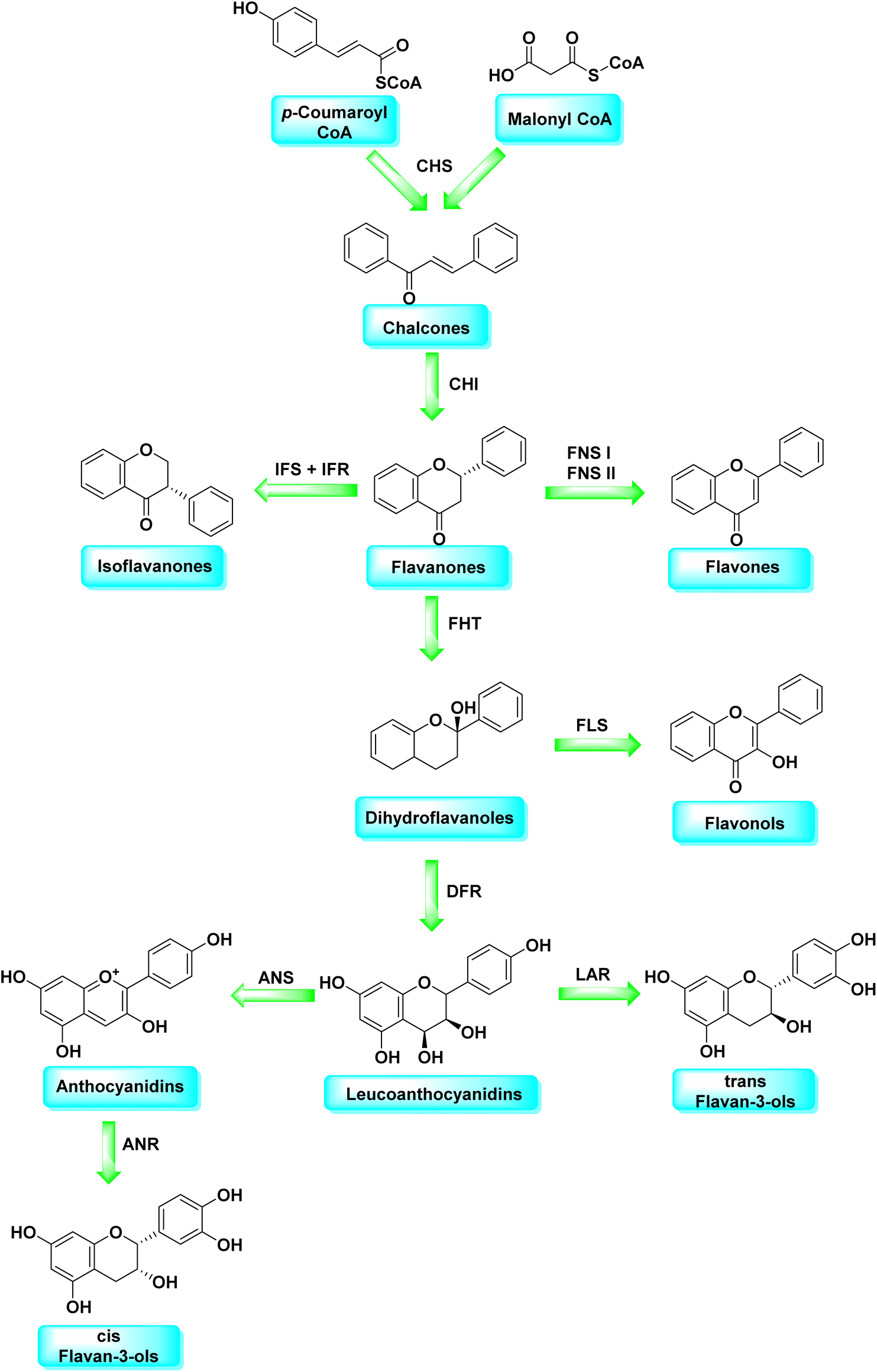 | ||
| Scheme 2 Flavonoids biosynthetic pathway. | ||
Following CHS, stereospecific cycloisomerization catalyzed by chalcone isomerase (CHI) generates the 2-S-flavanones 7, which is a phase mutual in flavonoid biosynthesis paths. Since the isomerization of such scaffolds produces the other class of flavonoids, flavanones characterize the utmost imperative diverging point in flavonoid metabolism. However, the chemical synthesis of hydroxyacetophenone is primarily accomplished through cyclization and condensation.38 The flavonoids are allocated into 14 sets based on the structure of the scaffold and the substitution pattern on rings A, B, and C.39 Flavones 9, flavonols 11, flavanones 7, isoflavones 8, flavanols 12, trans flavan-3-ols 10, cis flavan-3-ols 14, anthocyanins 15 and anthocyanidins 13 are the most well-known of these groups (Fig. 3).40–42 Flavonoids are phytochemicals, which are chemicals derived from plant material and have the potential to improve human health. Many traditional medicines' therapeutic effects may be linked to the presence of these polyphenols in many cases.43 Antiviral,44 antiallergic,45 antiplatelet,46 anticancerogenic, antiestrogenic, anti-inflammatory, antiangiogenic, antiproliferative, and antioxidant activities, for example, have been reported for these substances,47 and their incorporation typically yields little or no toxicity.48 Flavonoids have been shown to have antispasmodic, anti-secretory, antidiarrheal, and antiulcer properties in the gastrointestinal tract.49
 | ||
| Fig. 3 Chemical structures of different subclasses of flavonoids. | ||
2. Urease inhibition via flavonoids
The complex pathophysiology of peptic ulcers has been emphasized by recent advancements in our knowledge, gastric acid secretion is quite the culprit of this condition, so antacids, H2-receptor inhibitors such as famotidine, ranitidine, anticholinergics like telezipine, pirenzepin, or proton pump inhibitors such as lansoprazole, omeprazole, and others are used to control this secretion.50 However, gastric ulcer treatment presently has a significant limitation in that most of the medications on the market have poor efficiency against gastrointestinal disorders and are frequently linked with adverse effects.51,52 In scientific studies, some plants have been proven to be treat gastroduodenal ailments.53,54 Additional organic and inorganic scaffold-like alkaloids, coumarins, terpenoids, phenolic acids, tannins, and antioxidant micronutrients such as Mn, Zn, and Cu may also play a part in the medicinal characteristics of many plants.55Ninety-five flavonoids were found and documented in this literature review, with gastroprotective properties ranging from moderate to active and even robust. Here, 42 flavonoids were shown not to be active; however, dormancy may fluctuate extensively based on the kind of experiment, animal, method of administration, and dose. Flavonols such as robinin, kaempferol, and dactailin, for example, had no gastroprotective impact in investigational models of reserpine56 and restraint stress-induced ulcers in mice,57 but kaempferol showed gastroprotective activity at dosages of 50 and 100 mg kg−1, but no activity at 250 mg kg−1.58 In a model of induced ulcers, nobeletin, a flavone, was demonstrated to protect the stomach mucosa of rats from damage produced by EtOH and HCl/EtOH at dosages of 8 and 25 mg kg−1, respectively, but was mildly effective at a dose of 50 mg kg−1.59 Although many flavonoids' pharmacological and metabolic effects are owing to their antioxidant characteristics,60 high levels of inactivity may be due to flavonoids' propensity to behave as pro-oxidants. Flavonoids such as myricetin, quercetin, and kaempferol cause a concentration-dependent decline in nuclear glutathione (GSH) and glutathione S-transferase (GST) activities in secluded rat liver nuclei which may damage DNA oxidatively.61 This could be accountable for their carcinogenicity and mutagenicity; this effect could be described via pro-oxidant effects. However, the structural properties that may influence these compounds' pro-oxidant activity are unclear.
2.1 Chalcones as urease inhibitors
In 2005, Ansari and co-workers presented a protocol where they synthesized a new series of chalcones and studied their activity as a urease inhibitor (Fig. 4).62 Flex docking was investigated in 2008 as a follow-up to their prior work into the inhibition of urease by various chalcones to get a better understanding of the mechanism behind their inhibitory impact. In silico analysis of these compounds revealed that the activity is dependent on the ligand's interaction with the nickel metallocentre, two amino acid residues, Asp224 and Cys322, and the orientation of rings A and B in the enzyme's catalytic core. The most active chemical forms a strong bond with Ni701 and Ni702 via a network of interactions. Several hydrogen bonds and hydrophobic interactions with the nearby amino acid residues make up this structure. It has been shown that the difference in the activity of certain diastereomers is configuration-dependent for their reduced analogs. This could be primarily because of the differences in how the two stereoisomers' rings B 16 are oriented and how much they interact with Asp224 and Cys322 in the enzyme's catalytic core.63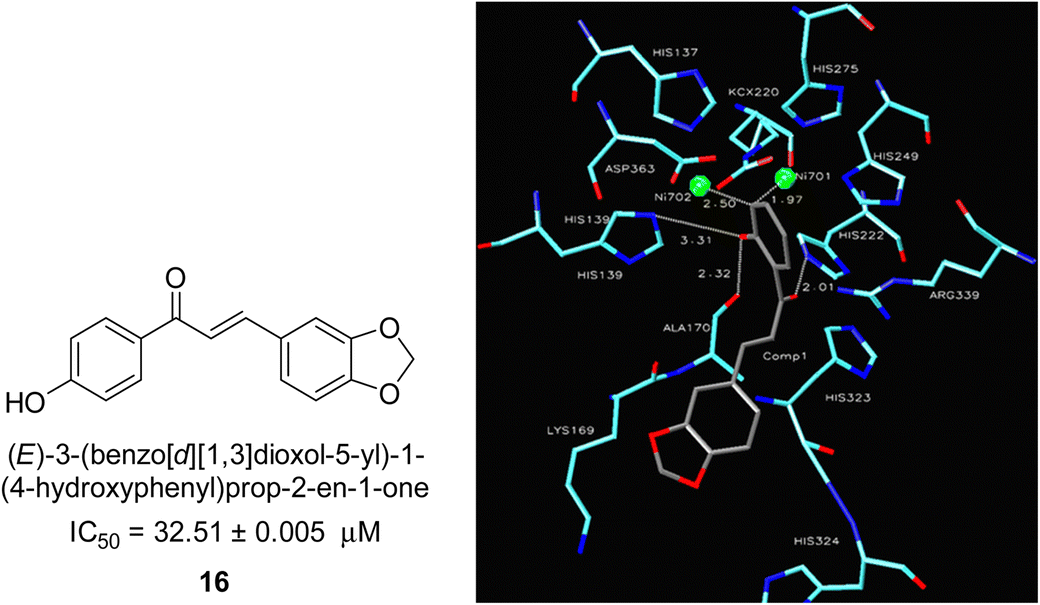 | ||
| Fig. 4 Chemical structure and docking image of chalcone 16 and its urease inhibition. The molecule in gray is the ligand with the active site residues. Metals are represented in green.63 | ||
A novel technique for the production of β-aryl β-mercapto ketone derivatives as possible urease inhibitors was developed in 2013 by Mahdi and his coworkers. The objective was to develop a novel scaffold of substances having anti-urease action. A novel and simple procedure for the synthesis of derivatives of β-aryl β-mercapto ketone based on Michael reaction was created by improving the thiophenol addition to chalcones in an ionic liquid as a solvent. The products were produced in yields ranging from average to excellent, and they were characterized by spectroscopic techniques and elemental analyses. They were also quite pure.64 For the purpose of finding novel inhibitors of jack bean urease, the activities of the scaffolds that were assessed. All the 22 synthetic compounds 17–38 have shown an inhibitory action in the micromolar range, with the most effective molecule having an IC50 value of 6 μM. For comparison, the hydroxyurea used as a reference inhibitor has an IC50 value of 100 μM.
An altered version of the Berthelot reaction was employed to evaluate the level of urease activity by monitoring the amount of ammonia formed. In a buffer consisting of 100 μM sodium phosphate and containing 50 μM urea, 100 μL (2 μg mL−1) of JBC, and 100 μL of the test chemicals at a range of concentrations, the reaction mixture was as follows: (pH 7.6). Following a preliminary incubation period of 30 minutes at 37 °C, the reaction was terminated by pouring 500 milliliters of a solution containing 0.5% PhOH and 0.0025% sodium nitroprusside. Next, 500 μL of a solution containing 0.25% NaOH and 0.21% NaClO was added and incubated for thirty minutes at a temperature of 37 °C so that the color could develop. This method is based on the reaction between the released ammonia (NH3) and the hypochlorite (OCl−), which results in the formation of a monochloramine. The absorbance at 625 nm was utilized to obtain the value. Every result was determined by running the experiment three times. They were successful in developing a method that was efficient, economical, and practical for the synthesis of a series of β-aryl-β-mercapto ketone derivatives. The method involved using [omim]Cl, which is not only inexpensive but also readily available. It was carried out under mild conditions and did not require any acidic or basic catalysts. Compounds that were synthesized were investigated for their potential to deactivate the urease of jack bean. All compounds exhibited inhibitory effects when tested with urease. In this series, compound 19 demonstrated the best inhibition compared to the other compounds. Understanding this possible inhibitor's binding pattern, which is confined to the active site, could help us get a better handle on the inhibitory effect it exerts. It is possible to use the scaffold of β-aryl-β-mercapto ketone urease inhibitors as basis for further optimization to increase potency and selectivity by making modifications in the basic skeleton (Table 1).65
| Compound no. | Chemical structures & IUPAC names | IC50 (μM) |
|---|---|---|
| 17 | 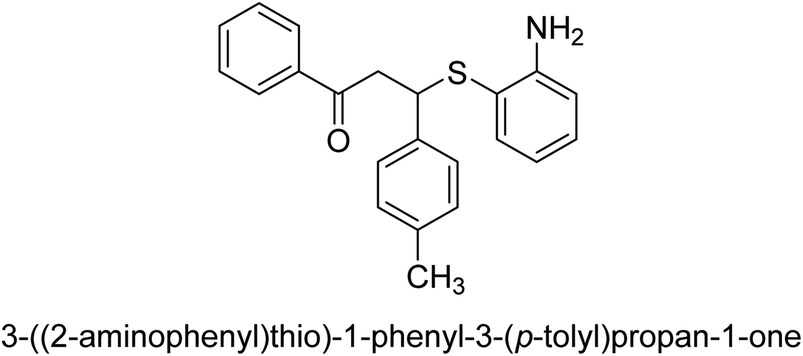 |
28 |
| 18 | 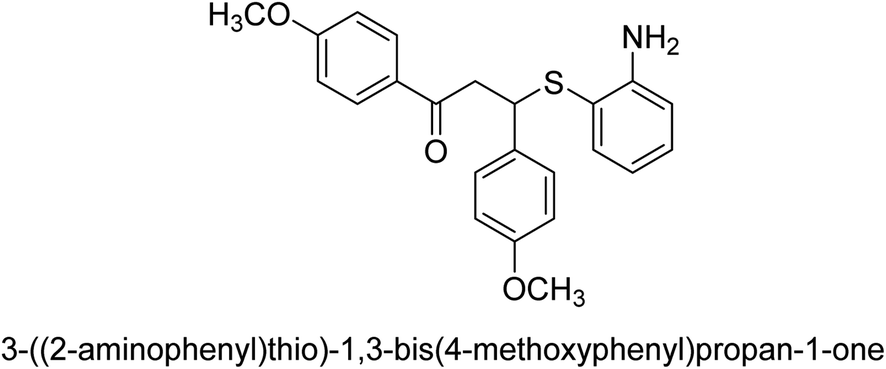 |
12 |
| 19 | 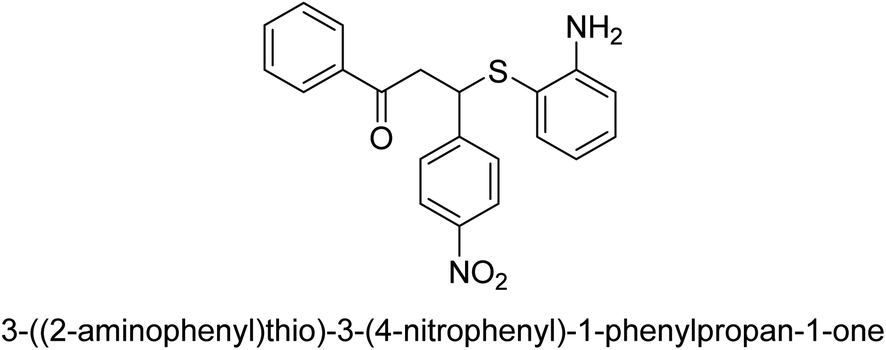 |
6 |
| 20 | 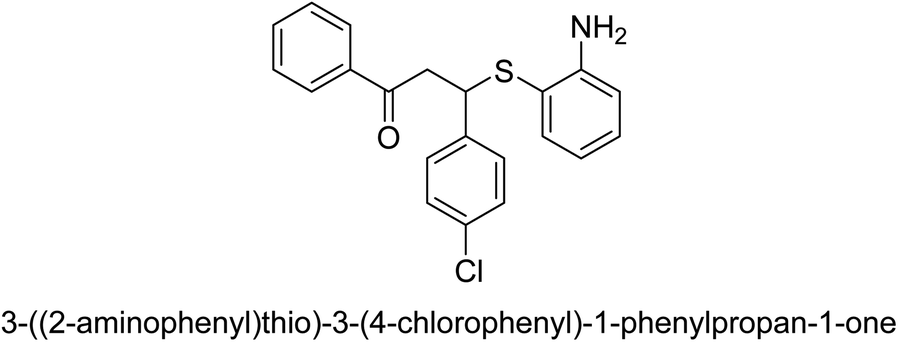 |
102 |
| 21 | 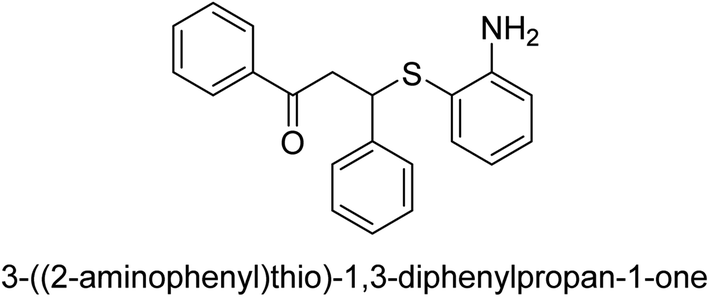 |
56 |
| 22 | 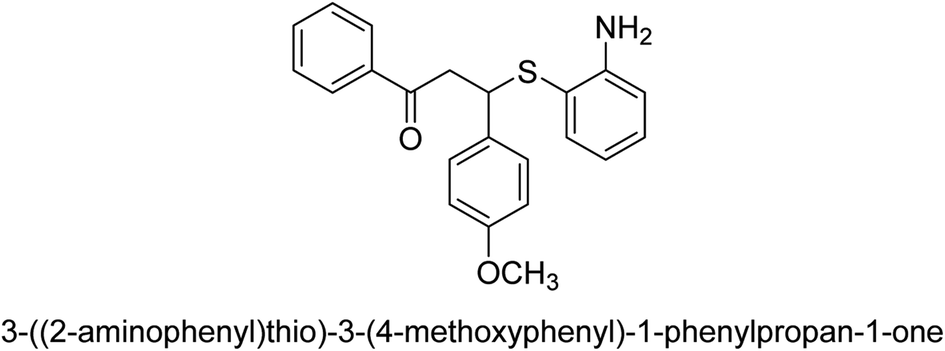 |
63 |
| 23 | 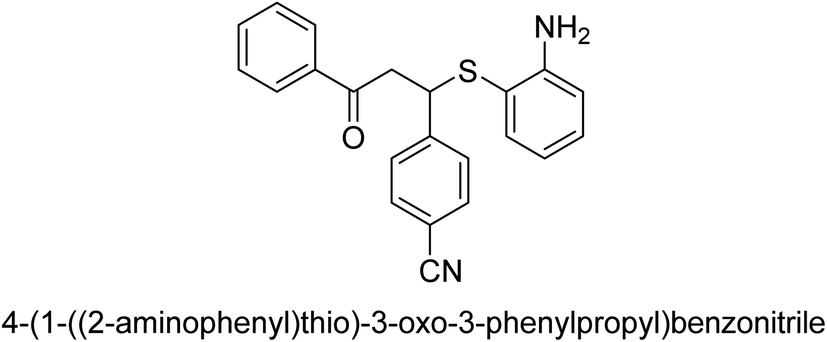 |
18 |
| 24 | 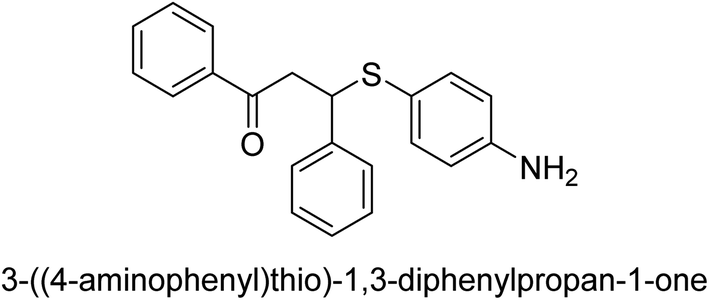 |
127 |
| 25 | 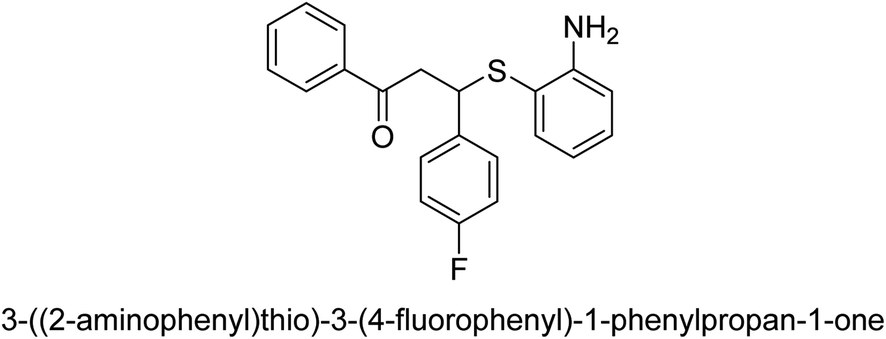 |
16.6 |
| 26 | 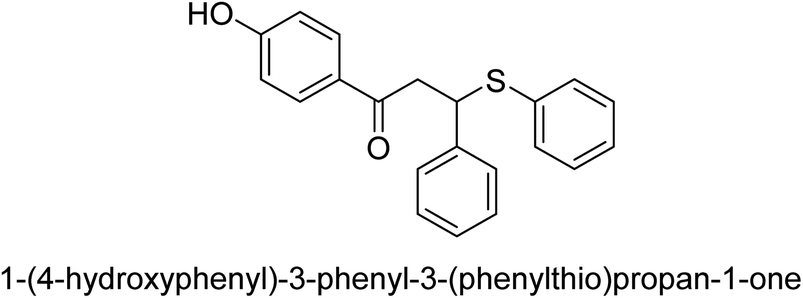 |
35 |
| 27 | 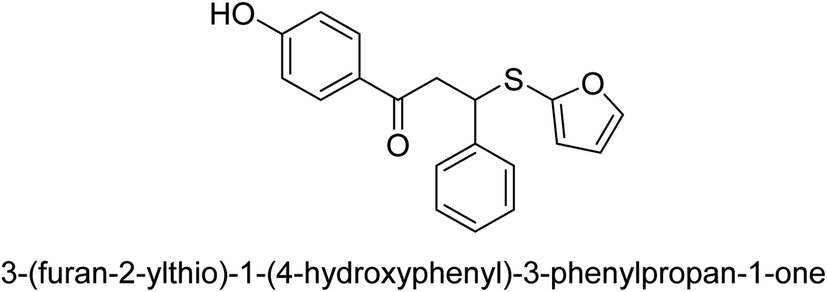 |
25 |
| 28 | 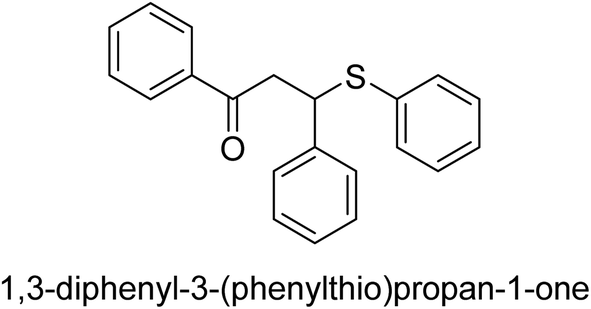 |
85 |
| 29 | 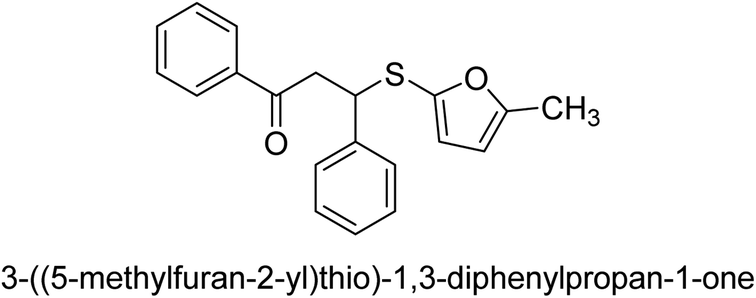 |
83 |
| 30 | 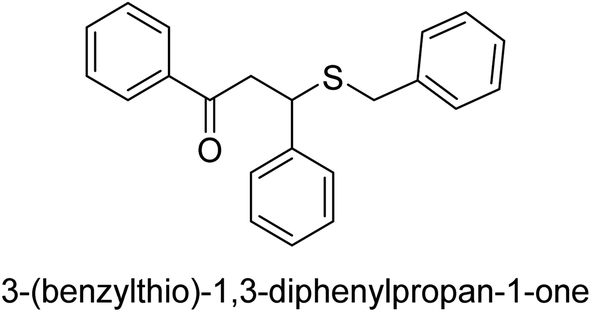 |
151 |
| 31 | 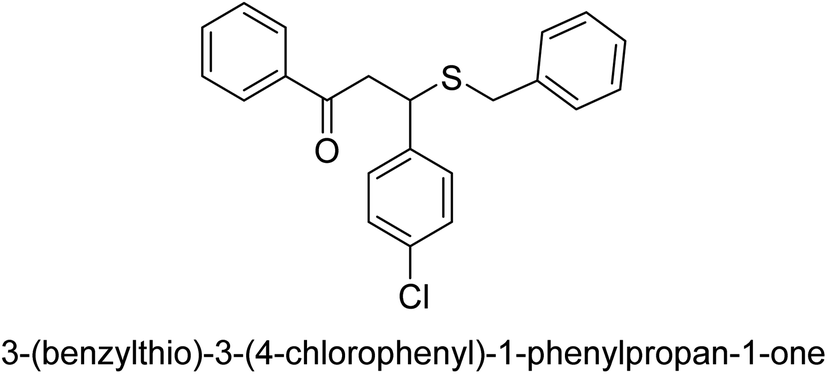 |
97 |
| 32 | 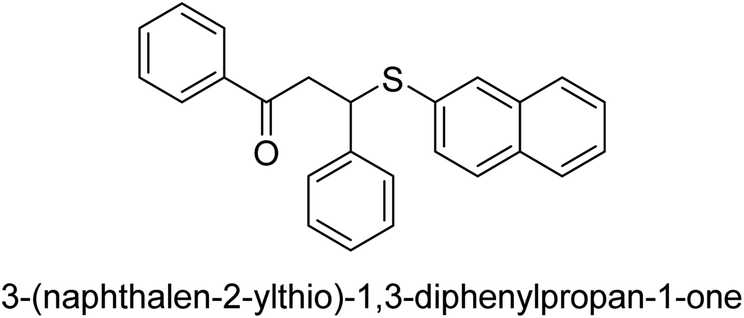 |
16.3 |
| 33 | 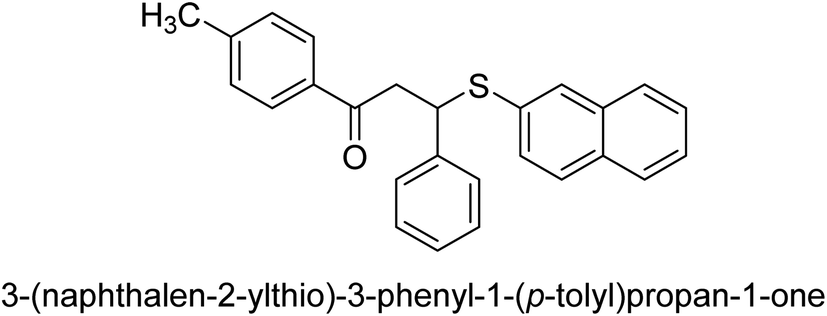 |
14 |
| 34 | 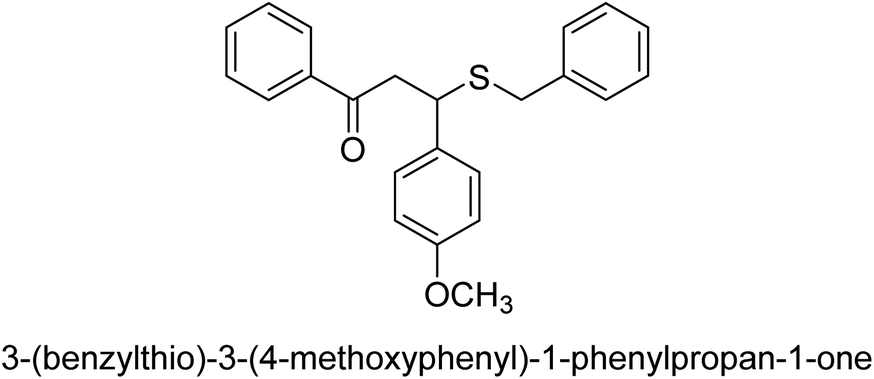 |
167 |
| 35 | 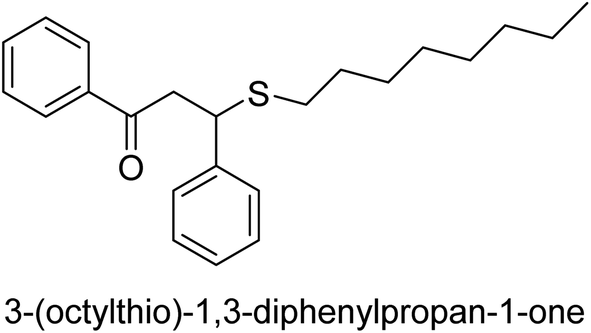 |
125 |
| 36 | 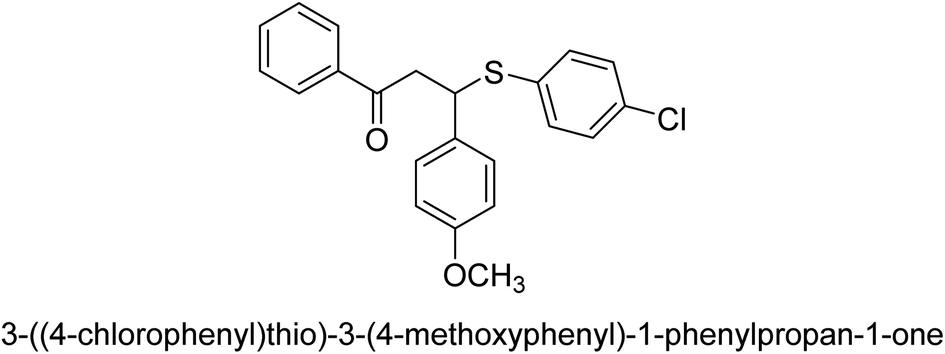 |
71 |
| 37 | 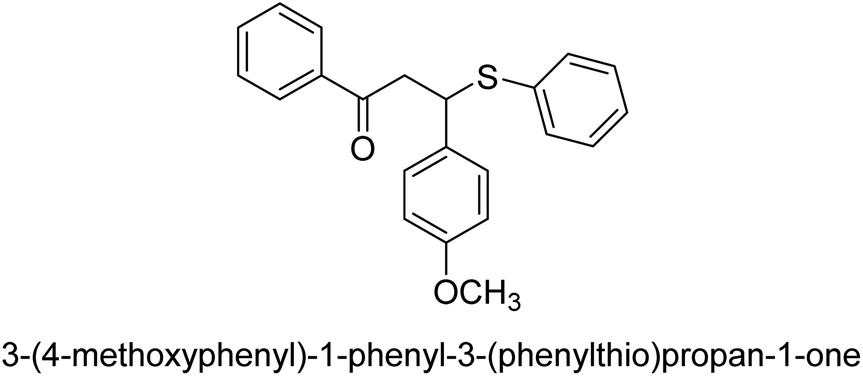 |
135 |
| 38 | 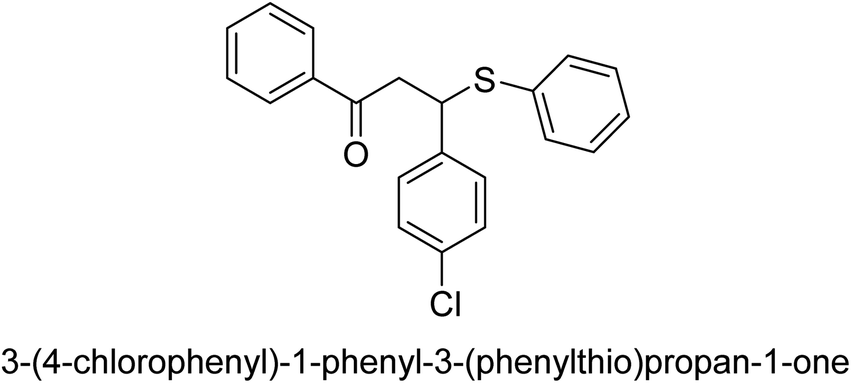 |
181 |
In 2016, Song and co-workers described the enantioselective Michael reaction and biological activity of MeNO2 to chalcone-comprising pyridine.66 In the year 2020, Acevedo and his colleagues conducted in vitro and molecular docking experiments on chalcones prepared by silica-supported heterogeneous catalysts as powerful urease blockers. In this study, the authors developed an easy methodology that produced a good yield of the chalcones. The newly synthesized chemicals were put through a biological test to see how effective they were as urease inhibitors. It was discovered that the majority of the compounds have strong urease-inhibiting action. The chalcone 3-(3-hydroxyphenyl)-1-phenylpropenone was determined as best with a % inhibition of 86.17 ± 0.89 and an IC50 value of 11.51 ± 0.03 μM. This result indicates that it inhibits 86.17 ± 0.89 μM of the target enzyme.
It is essential to take into account the fact that electron donating groups may significantly boost urease inhibition. However, the nature of the substitution, as well as its structural location, are important considerations. The observed sequence of their activities, in terms of para-substituted ligands, is as follows: 4′-OH–, 4-OCH3 → 4′-OH–, 2-furyl → 4′-CH3–, 4-(CH3)2NPh → 4′-CH3–, Ph → 4′-CH3–, 2-furyl → 4′-OH–, Ph–. In a similar vein, in terms of the m-substituted ligands, the 3′-OH–, Ph– has shown a higher level of urease inhibition in comparison to the 3′-OH–, 2-furyl. In contrast, it was discovered that electron-withdrawing groups such as OH- and CH3-groups, particularly when they were in the meta position, reduced the urease inhibition. Despite this, it is essential to determine the structure–activity relationship of chalcones 39–47 as shown in Table 2 that were studied. This could be because of a series of factors, such as the polarizability, sizing, structure, and electronegativity of a ligand, which all play an essential part in the inhibition of an enzyme.
| Compound no. | Chemical structures & IUPAC names | IC50 (μM) |
|---|---|---|
| 39 | 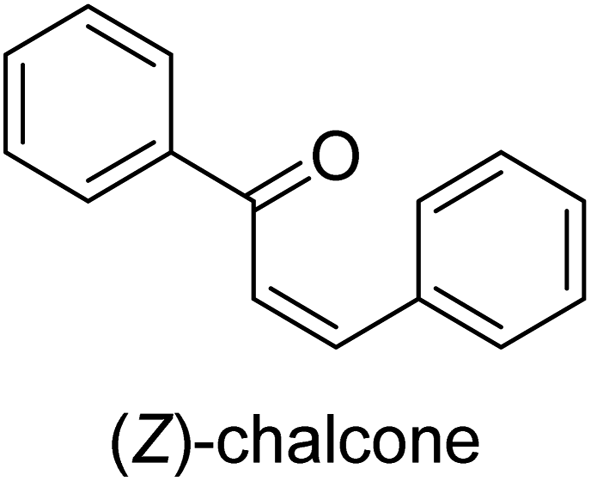 |
28.41 ± 0.09 |
| 40 |  |
27.11 ± 0.03 |
| 41 | 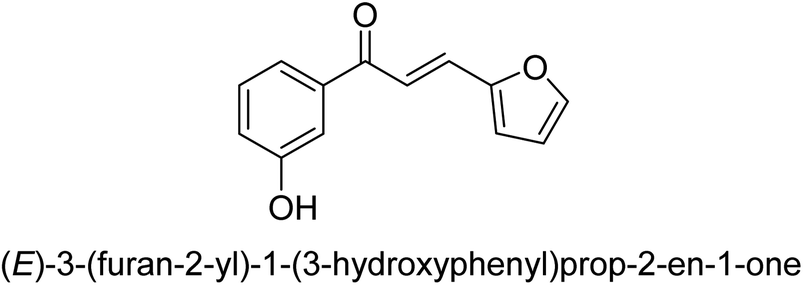 |
58.81 ± 0.11 |
| 42 | 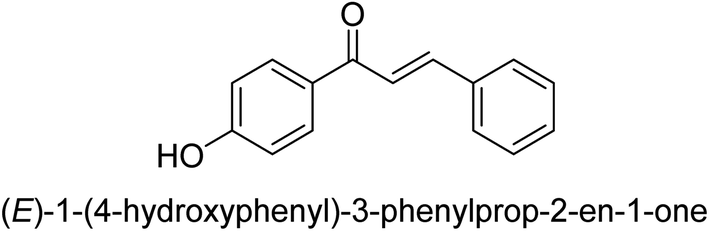 |
74.35 ± 0.19 |
| 43 | 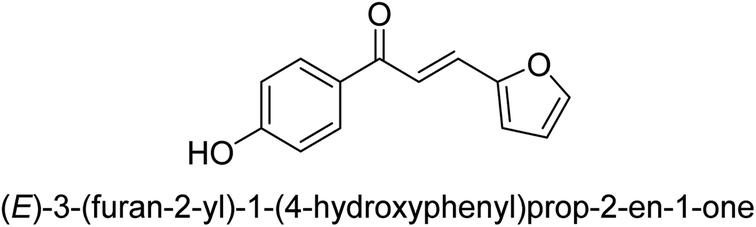 |
14.62 ± 0.01 |
| 44 | 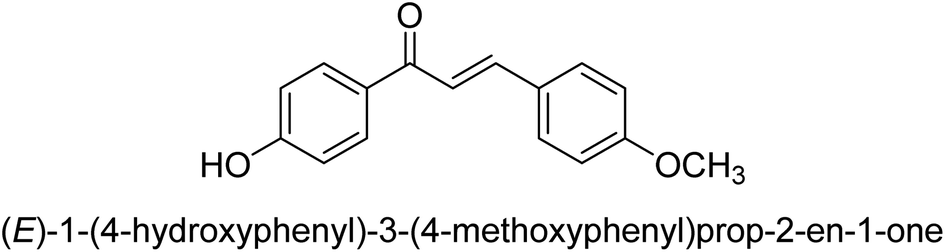 |
11.51 ± 0.03 |
| 45 | 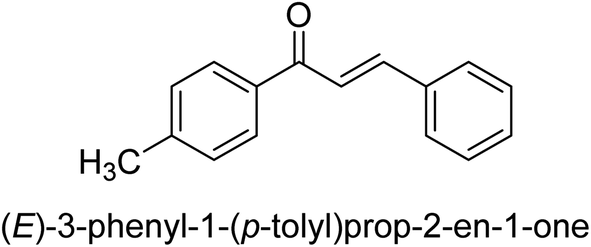 |
25.78 ± 0.11 |
| 46 | 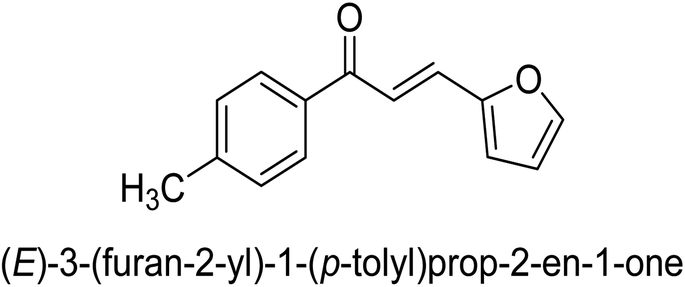 |
35.44 ± 0.11 |
| 47 | 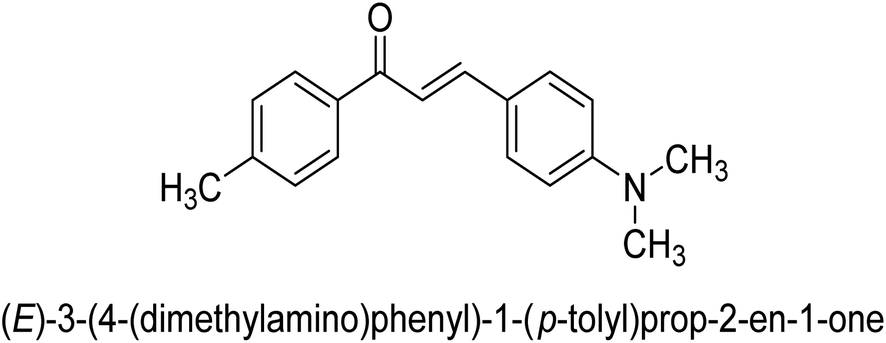 |
23.52 ± 0.01 |
Ligand 44 showed best % inhibition of 86.17 ± 0.89 μM and an IC50 of 11.51 ± 0.03 μM. According to the findings of the molecular docking investigation, the identical ligands 43, 44, and 47 produced better docking results than thiourea (control) and demonstrated significant urease affinity. Compared to the control ligand thiourea, which had a score of 1600 and an ACE value of 103.71 kJ mol−1, ligands 43, 44, and 47 proved as highly capable inhibitors of urease. They also exhibited better docking scores of 5718, 5940, and 5596, and an ACE of 246.66, 244.79, and 243.06 kJ mol−1. This is in line with the observation that urease inhibition values of 43, 44 and 47 are even better than those of thiourea. According to the findings from both, the in silico and in vitro research, such compounds are deemed efficient urease inhibitors and effective anti-urease drugs.67
In 2021, Khalid and co-workers devised a novel strategy for the synthesis of 3-(3-bromo-phenyl)-1-(2-trifluoromethylphenyl)-propenone and 3-(3-bromo-5-chloro-phenyl)-1-(2-trifluoromethylphenyl)-propenone and reported their DNA binding, urease deactivation, molecular docking and DFT investigations. Through a base-catalyzed condensation process, two novel chalcones, 48 and 49, were created from fluorinated acetophenone and aldehydes with various substituents. Routine spectroscopic methods provided the structural validation of the produced compounds. Using ultraviolet-visible spectroscopy, the interaction of 48 and 49 with salmon sperm DNA (SS-DNA) was investigated. Experimental evaluations of the antioxidant and urease inhibition potentials were also conducted, and these results were confirmed by molecular docking investigations. At the B3LYP/6-311 G(d,p) level of the DFT, calculations of the frontier molecular orbitals (FMOs), natural population analysis (NPA), natural bond orbitals (NBOs), and nonlinear optical (NLO) analysis of 48 and 49 were made. According to the findings, chemicals 48 and 49 significantly interact with SS-DNA in an intercalation way. Furthermore, DFT research demonstrated that interactions with hyper conjugates stabilize 49 more strongly than they stabilize 48 (Fig. 5).
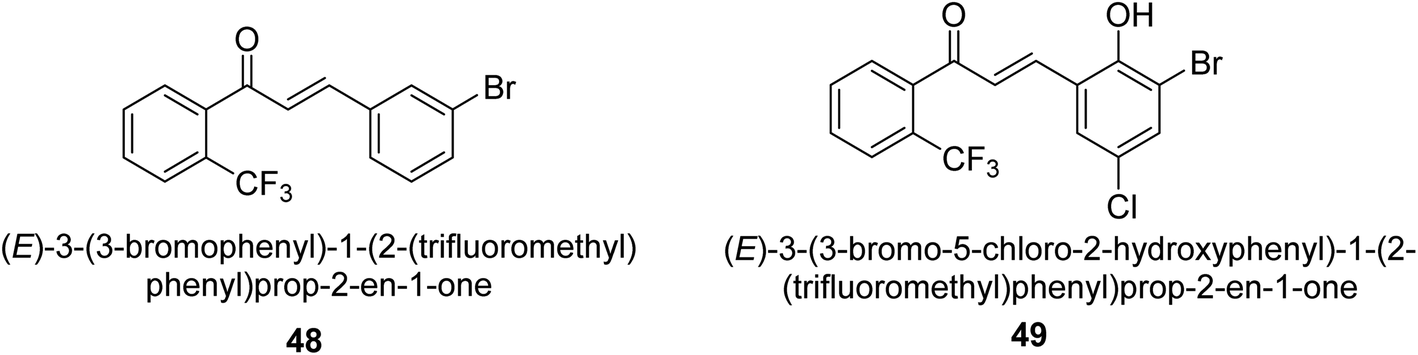 | ||
| Fig. 5 Structures of chalcone-based urease inhibitors. | ||
By using the indophenol technique, the urease enzyme inhibition of produced compounds was evaluated. Using this technique, the quantity of ammonia created throughout the operation may be quantified. A 96-well plate was filled with phosphate buffer (0.01 M), ethylenediaminetetraacetic acid (1 μM), and lithium chloride (0.01 M) as a blank, inhibitor, and control, respectively. Compounds 1 and 2 (5 mL) were each combined with 10 mL of the jack-bean urease enzyme, with concentration ranges of 5–500 μM. Incubation was subsequently carried out at 30 °C for 15 min with thiourea as a standard. The following liquids were added to wells: PhOH (50 mL, 1 percent w/v), sodium hypochlorite (70 mL, 0.1 percent w/v), sodium nitroprusside (50 mL, 0.005 percent w/v), and sodium hydroxide (70 mL, 0.5 percent w/v). The absorbance at 630 nm was recorded at intervals of 50 minutes utilizing a microtiter plate reader.68
2.2 Flavanol as urease inhibitors
Catechin is a flavan-3-ol, a kind of secondary metabolite found in plants that has antioxidant properties. It belongs to the flavonoid's subgroup of polyphenols. In 1999, Mabe and his colleagues conducted research on the anti-Helicobacter pylori properties of tea catechins in vitro and in vivo settings. The catechin known as epigallocatechin gallate had the highest level of action against Helicobacter pylori out of the six tea catechins that were investigated (the minimum inhibitory concentration, or MIC, for fifty percent of the strains investigated, was eight mg mL−1).69In 2003, green tea was used to isolate catechins 50, 51, 52, and 53. Fig. 6 depicts the structure of epigallocatechin gallate. These compounds exhibited strong anti-H. pylori urease action in vivo, achieving IC50 values of 2.2, 9.8, 8.7, and 19.6 μM. According to the results of several experiments using molecular docking, each of these substances acts as a competitive inhibitor.70
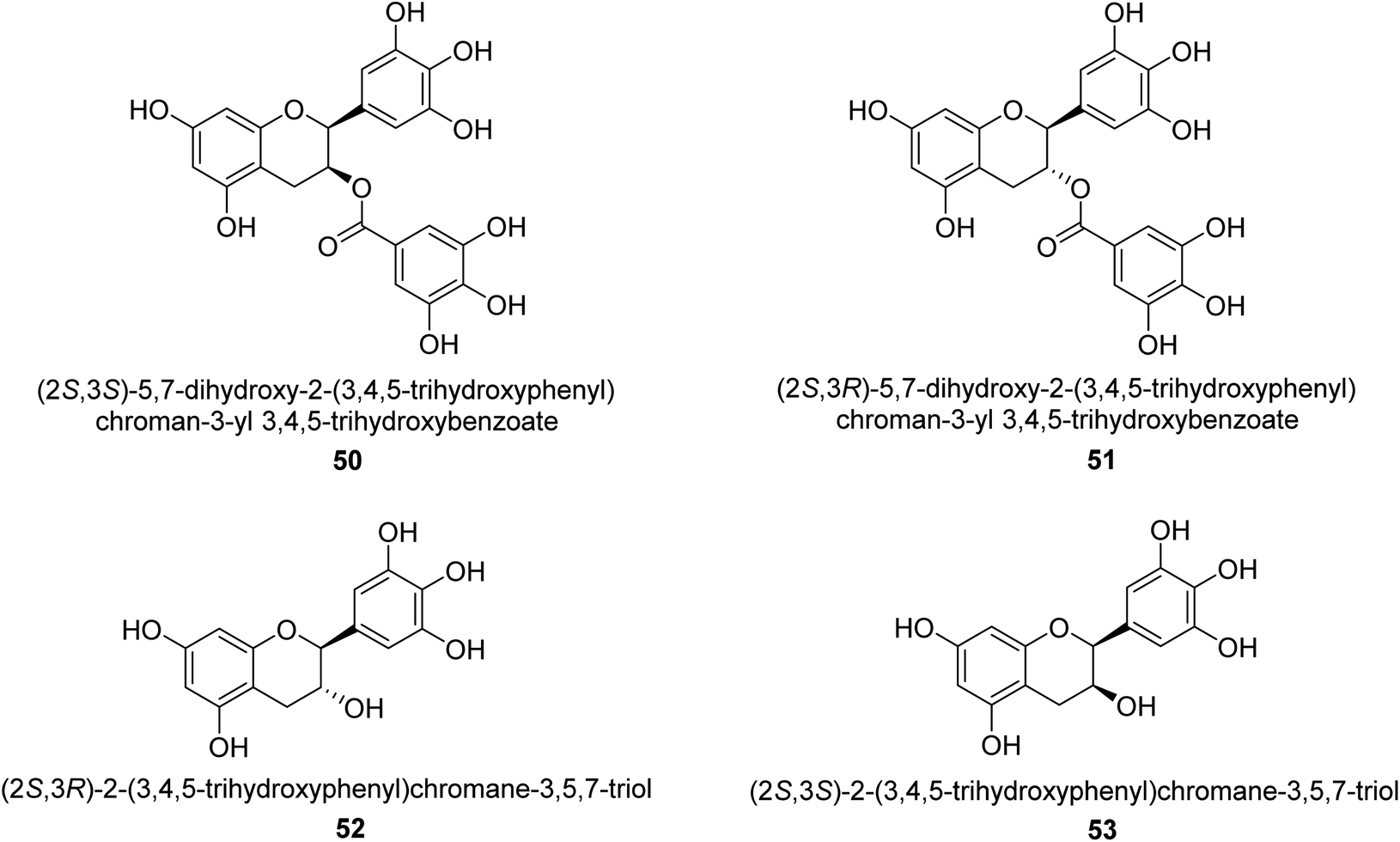 | ||
| Fig. 6 Structures of catechins-based urease inhibitors. | ||
Ordouzadeh and his colleagues began their research on the in vitro suppression of Helicobacter pylori urease by non and partially fermented Camellia sinensis in the year 2003. Through the use of GC, a comparison was made between the extracts of nonfermented and semifermented C. sinensis L. from the Iranian city of Lahijan. As a consequence of catechins undergoing a process of semi-fermentation, the levels of practically all of the volatile components, including alcoholic and aromatic compounds, that were hydrolyzed by the enzyme increased. The catechin content of C. sinensis L. is very high, including that of epicatechin, epigallocatechin, epicatechin gallate, and epigallocatechin gallate. Catechins are destroyed during the semi-fermentation process, hence the nonfermented form of C. sinensis has a higher concentration of them than the fermented form dose. Polyphenolic catechins, in particular epigallocatechin gallate and epicatechin gallate, have been shown by research carried out over the last 20 years to have the ability to suppress the development of a broad variety of bacterial species, both Gram-positive and Gram-negative, to a modest degree.71
In 2013, Loes and his colleagues focused on inhibiting the urease enzyme of the urinary tract bacteria Staphylococcus saprophyticus. The Gram-positive bacterium causes infections in the urinary system and produces urease as a virulence factor. They used soluble extracts of the aforementioned strain in order to investigate the degree to which this enzyme is susceptible to chemical inhibition. Both acetohydroxamic acid and DL-phenylalanine hydroxamic acid were able to block urease activity, but their Ki values were significantly different. Acetohydroxamic acid's Ki value was 8.2 μg mL−1, whereas DL-phenylalanine hydroxamic acid's Ki value was 21 μg mL−1. However, the competitive inhibition brought on by the phosphorodiamidate fluorofamide (Ki = 0.12 μg mL−1 = 0.553 μmol−1 = 0.000553 mmol−1) was not brought on by the imidazole omeprazole. (+)-Catechin hydrate (Ki = 357 μg mL−1 = 1.23 mmol−1) and (−)-epigallocatechin gallate (Ki = 210 μg mL−1 = 0.460 mmol L−1) are two of the flavonoids that may be detected in green tea extract. Both of these flavonoids provided a mixed inhibition. The urease activity of whole cells of strains ATCC 15305, ATCC 35552, and ATCC 49907 cultured in either a rich medium or an artificial urine medium was suppressed by fluorofamide, (−)-epigallocatechin gallate, DL-phenylalanine hydroxamic acid, (+)-catechin hydrate, and acetohydroxamic acid. The rise in pH is typically caused by the development of S. saprophyticus in cultures grown on an artificial urine medium was slowed down by the addition of acetohydroxamic acid or fluorofamide. Based on these findings, it seems that urease inhibitors could act as alternative cure for urinary tract infections brought on by S. saprophyticus.72
In 2014, Pastene and colleagues conducted research on the catechin-based procyanidins found in the aqueous extract of Peumus boldus Mol. They found that these procyanidins inhibited Helicobacter pylori urease and adenocarcinoma stomach cell adhesion. The effectiveness of dried leaves aqueous extract of Peumus boldus Mol. (Monimiaceae) against Helicobacter pylori was studied in this particular work. This extract showed significant effectiveness in inhibiting the urease produced by H. pylori. As a result, a bioassay-guided fractionation technique was used in order to get clarity on the types of chemicals that are responsible for producing such an impact. The most effective inhibitor of H. pylori urease was found in the aqueous extract fraction designated as F5 (mDP = 7.8), which had an IC50 value of 15.9 g gallic acid equivalents (GAE) per milliliter. Our findings, taken together, lead us to believe that boldo extract exhibits powerful anti-urease capability and anti-adherent impact against H. pylori. Both of these characteristics are directly related to the presence of catechin-derived proanthocyanidins.73
2.3 Flavones as urease inhibitors
It was discovered that the flavonol quercetin 54 derived from the plant Psidium guajava had anti-JBU action with an IC50 value of 80 mM.74 Quercetin is a powerful antioxidant flavonol. Myricetin 55 and luteolin 56, both of which were isolated from Lonicera japonica Thunb., have been shown in another study to possess anti-H. pylori urease activity. Both of these substances exhibited significant inhibitory effects, with their IC50 values coming in at 77.2 and 35.5 mM, in that order.75 5,7-Dihydroxyflavone 57, was extracted from the medicinal plant Daphne retusa. This chemical showed considerable inhibition of JBU with an IC50 value of 60.4 mM.76 The EtOAc fraction of Datisca cannabina Linn was used to isolate the powerful anti-JBU inhibitor known as datisdirin 58, which has an IC50 value of 83.8 mM.77 Both dimethoxyflavone 59 and trimethoxy flavone 60 were shown to be powerful anti-UIs after being discovered by Muhammad et al.78 Their IC50 values were 118.4 and 144.7 μM, respectively. Their findings demonstrated that the presence of hydroxyl groups in these compounds is responsible for their inhibitory effects. Hydroxyl groups can chelate very readily with nickel, which is present in the active site of urease. The anti-JBU activity of 3′-methylquercetin 61, which was isolated from V. cinerascens, was determined to be efficacious (IC50 = 75.6 μM) (Fig. 7).79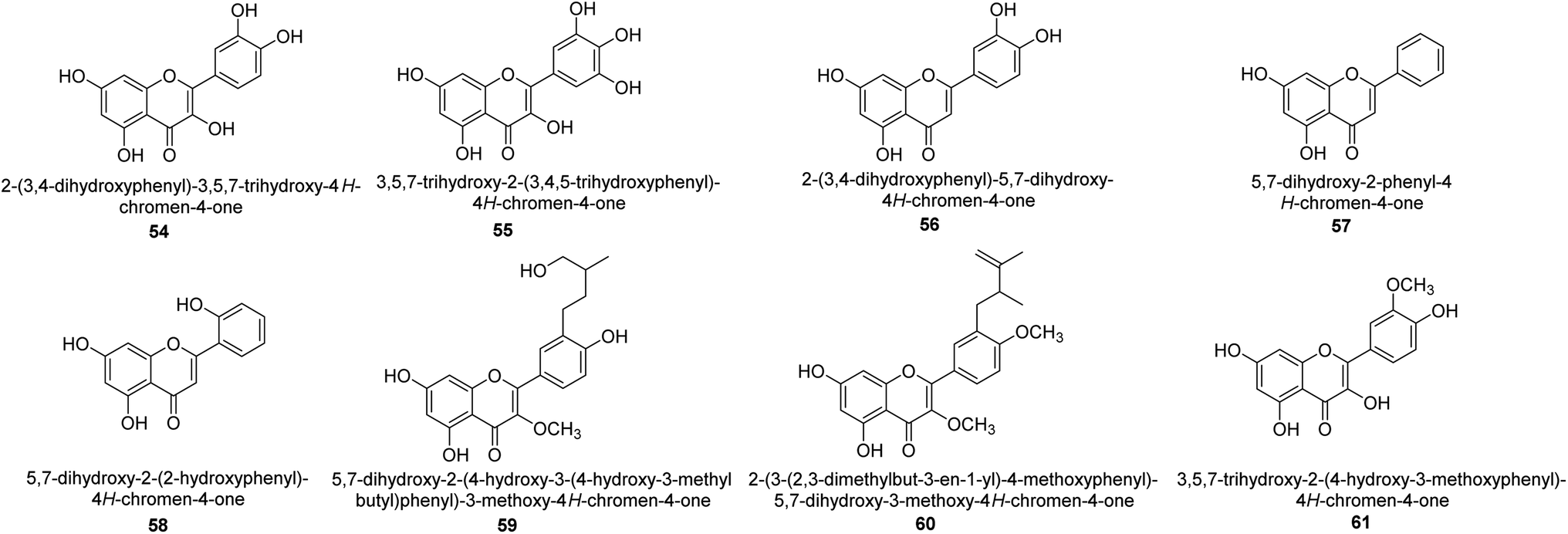 | ||
| Fig. 7 Structures of flavone-based urease inhibitors. | ||
Baicalin (BA) 62, also known as flavone glucuronide, is extracted, and refined from the Scutellaria baicalensis plant's dried roots. Using a method that targeted –SH functions in the active region of urease and having an IC50 value of 270 mM, researchers discovered that BA acts as a non-competitive inhibitor of JBU.80 Scutellarin (SL) 63, also known as a flavone glucuronide, is extracted from the Erigeron breviscapus plant that is native to China. In a manner that was both concentration- and time-dependent, SL was shown to possess inhibitory action in relation to JBU (IC50 value of 1350 μM). Accordingly, SL is a potent inhibitor because it binds to sulfhydryl functions in the enzyme's active region in a slow-binding, reversible, and concentration-dependent way.81 Recently, Yu et al.82 compared standard AHA (IC50 = 140 μM) and stated that BA and SL (isolated from S. baicalensis Georgi) successfully repress H. pylori urease in time-independent and dose-dependent ways. However, the kinetic study showed that the two species are non-competitive inhibitors with Ki values = 140. The anti-human urease inhibitory effect of rubranonoside 64, which was isolated from P. rubra, was shown by an IC50 value of 212.3 μM (Fig. 8).
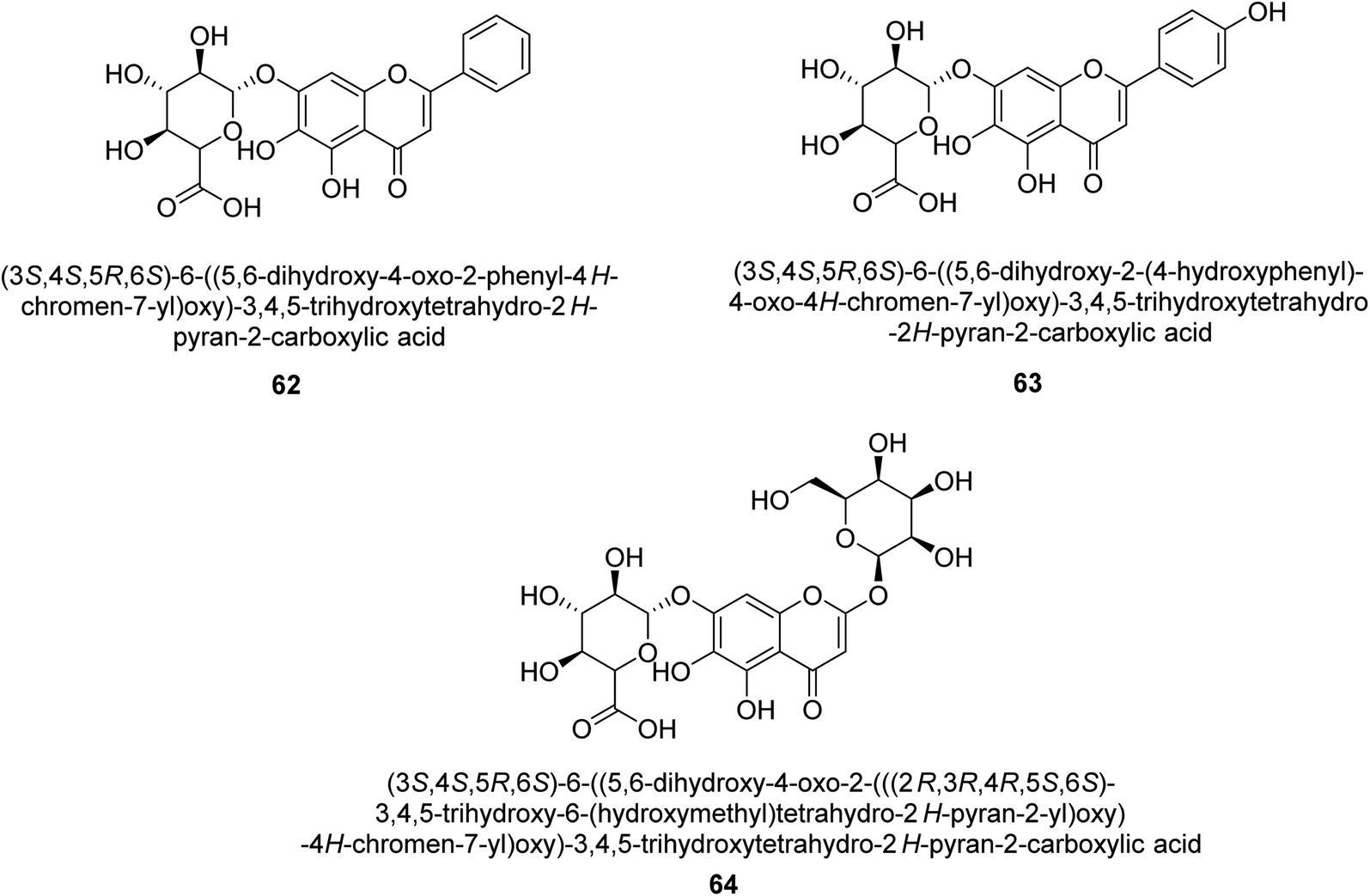 | ||
| Fig. 8 Structures of flavone glucuronide-based urease inhibitors. | ||
2.4 Flavanones as urease inhibitors
Ponciretin 65 (Fig. 9) is the primary metabolite of poncirin. It was isolated from an aqueous extract of the fruits of Poncirus trifoliate and is metabolized by bacteria that live in the human intestinal tract. The IC50 value for ponciretin's anti-H. pylori urease activity was 15.8 μM, indicating that it was an effective agent. In addition to that, it has been shown that this substance directly inhibits the bacterium H. pylori. This particular species acts as a non-competitive blocker of urease. Kim et al. studied the substituted ponciretin 66 as an inhibitor of H. pylori. They isolated this species from fruit and analyzed the activity of flavanone against the urease enzyme and it was proven a potent growth inhibitor of the urease enzyme in human intestinal bacteria.83 Kim et al. also investigated the inhibition of Helicobacter pylori development by flavonoids and phenolic acids, which may be generated from flavonoids by human intestinal microbiota (HP). Inhibitors of HP included hesperetin, ponciretin, naringenin, and diosmetin. The most effective of them all, with a MIC of 10–20 μg mL−1, was ponciretin 67. However, the urease activity of HR was hardly at all inhibited by these HP-active substances.84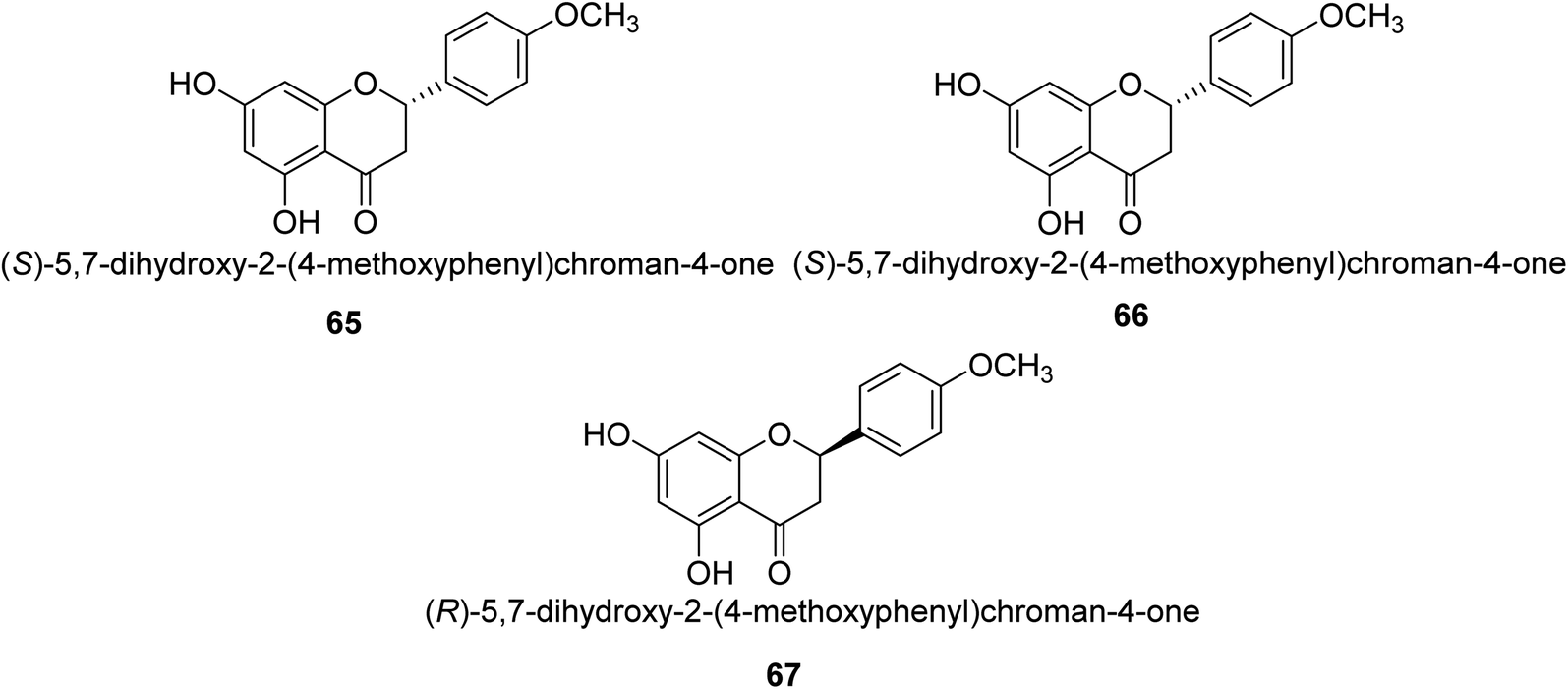 | ||
| Fig. 9 Structures of substituted flavanone-based urease inhibitors. | ||
Commonly referred to as Indian gooseberry, Emblica officinalis Gaertn. is highly valued in traditional systems of medicine. In recent research, Patel and colleagues identified a large number of phytochemicals from E. officinalis and briefly discussed and recapitulated their molecular processes, ethnomedical applications, and pharmacological potentials. They used the phenol-hypochlorite method to test E. officinalis for its impact on jack-bean urease and discovered that extracts from the leaves or even fruit itself are anti-urease with an IC50 range of 0.74–4.54 mg mL−1 and displayed anti-urease action against a number of bacteria, including Staphylococcus aureus 109, Staphylococcus aureus 3160, Pse60, Pseudominas aeruginosa, Proteus vulgaris and Klebsiella pneumonia.85
Natesan and colleagues investigated how naringin 68 (Fig. 10) affected hyperammonemic rats brought on by ammonium chloride. Ammonia damages the central nervous system (CNS) in animals, which results in liver damage and urea cycle disorders. An increase in ammonia build-up affects the central nervous system in a variety of ways, involving both excitatory and inhibitory neurotransmission. The purpose of Natesan's probe was to find out if naringin exhibits protective properties against hyperammonemia in rats stimulated by ammonium chloride. Specifically, naringin 80 mg kg−1 b.w. showed the most successful antihyperammonemic action of the three dosages (40 mg kg−1, 80 mg kg−1, and 160 mg kg−1 of body weight). Therefore, Natesan's work established that naringin has a protective effect in hyperammonemic rats generated by NH4Cl in a seemingly dose-dependent way.86
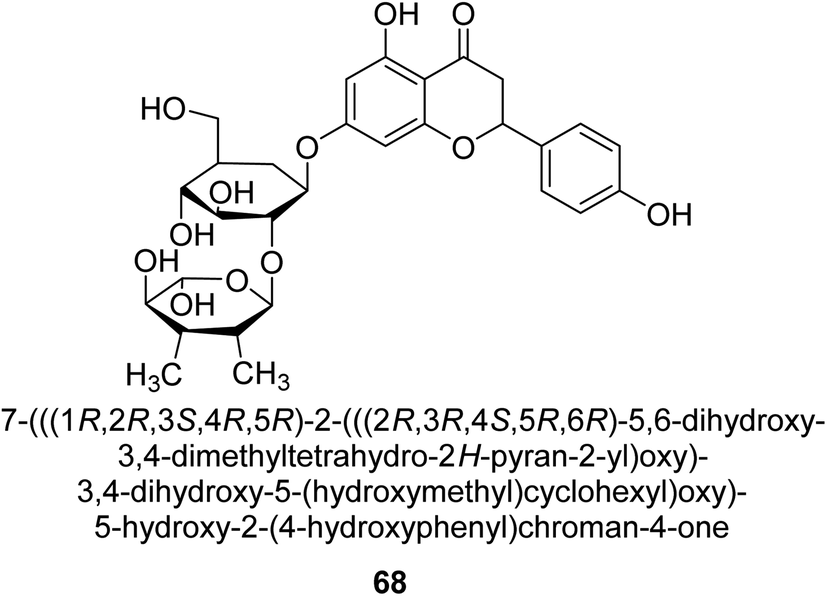 | ||
| Fig. 10 Structure of flavanone-based urease inhibitor. | ||
Naringin, a bioflavonoid, was also investigated by Sing et al. for its potential to protect rats' kidneys from ischemia-reperfusion (I/R) damage. The possibility that harmful oxygen radicals are implicated in the pathophysiology of (I/R) damage to the kidney is growing. The work aimed to find out how naringin (Ng), a bioflavonoid, affected rat kidney failure brought on by I/R. Using histological and biochemical measures, the protecting impact of naringin against the destruction caused by reactive oxygen species (ROS) during renal I/R was examined in Sprague-Dawley rats. In one set of trials, animals had unilateral nephrectomies and underwent a 45 minutes blockage of the left renal pedicle; in a different group, both renal pedicles were blocked for 45 minutes before being repercussed for 24 hours. 60 minutes before the ischemia, naringin (400 mg kg−1, p.o.) was given. Rats were slaughtered after the reperfusion period. In renal tissue, the amounts of thiobarbituric acid reactive species (TBARS), reduced glutathione (GSH), catalase (CAT), and superoxide dismutase (SOD) enzymes were measured. The results suggest that ROS is a contributing factor in I/R-stimulated renal damage and naringin has renoprotective impact likely due to its antioxidant and radical scavenging properties.87
2.5 Isoflavonoids as urease inhibitors
The Pueraria thunbergiana plant is a well know source of isoflavonoids like genistein 69 (Fig. 11) which is possible to extract from the rhizomes and flowers. The IC50 value of this species' inhibition of H. pylori urease was 1.6 mM,88 and it was shown to be effective. In another investigation, Xiao and co-workers89 examined the ability of 20 polyphenols based on isoflavones to inactivate the urease of H. pylori. These polyphenols are naturally found in various plants used for medicinal purposes and consumed by humans. According to the findings, isoflavonoids 70 was best inhibitor of H. pylori urease in a time-dependent fashion, with an IC50 of 140 μM. The results of the kinetic study showed that OH functions are very significant to the anti-urease action of this molecule. The anti-H. pylori urease potential of 19 flavonoids produced from plants was investigated and analyzed by Xiao et al.90 Isoflavone 71 was top effective UI, with an IC50 that was 0.85 μM greater than that of AHA, which had an IC50 of 18.2 μM. Further, molecular docking research was performed on this molecule, and the results revealed that it acts as a competitive inhibitor of H. pylori urease, with a Ki value of 0.64 μM. Researchers determined that the presence of the hydroxyl group was responsible for its anti-urease action.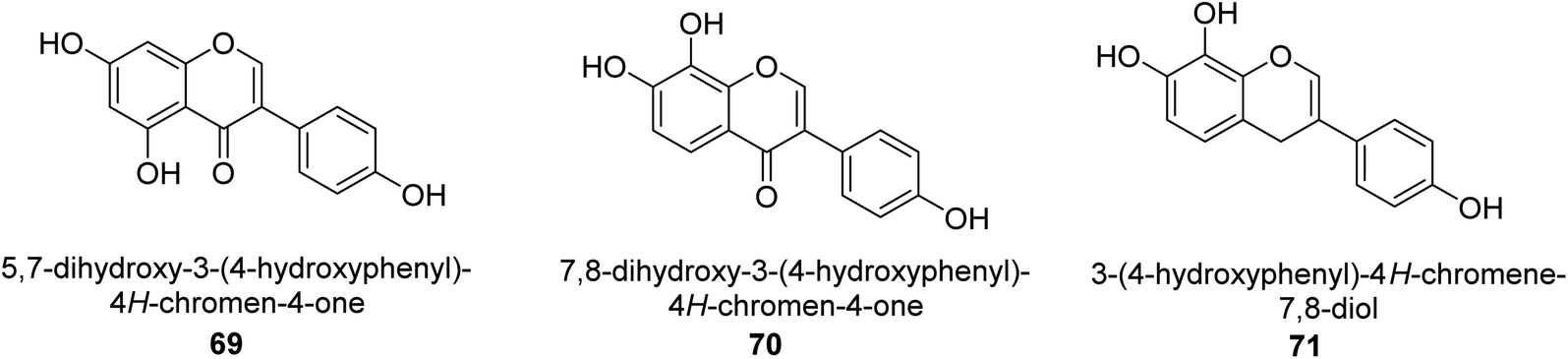 | ||
| Fig. 11 Structures of isoflavonoid-based urease inhibitors. | ||
As urease inhibitors for Helicobacter pylori, polyphenols based on isoflavones 72–75 were investigated by Zhu et al. Twenty polyphenols were synthesized, and their impact on Helicobacter pylori urease was assessed. Among them, 7,8,4′-trihydroxyisoflavone 74 (IC50 = 0.14 μM) showed strong inhibitory effects and inhibited the urease of H. pylori in a time-dependent manner. According to the structure–activity assessment of these polyphenols, the two ortho hydroxyl groups were crucial for the polyphenol's inhibitory effect as shown in Table 3. The inhibitory action of the isoflavone significantly diminished when the C-ring was disrupted. The carboxyl group was harmful to deoxybenzoin.89
| Compound no. | Chemical structures & IUPAC names | % Inhibition |
|---|---|---|
| 72 | 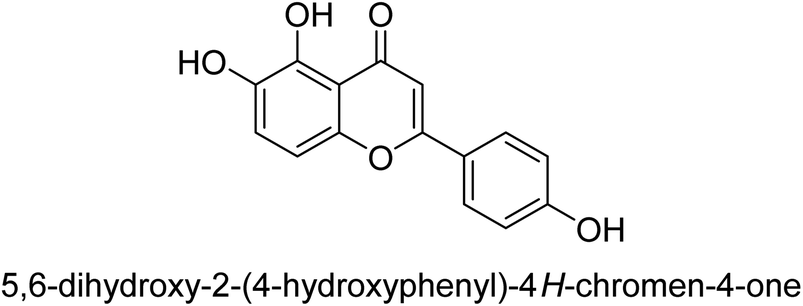 |
81.7 |
| 73 | 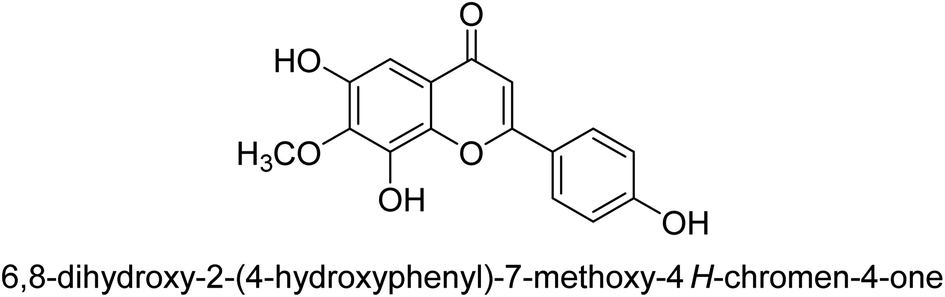 |
36 |
| 74 | 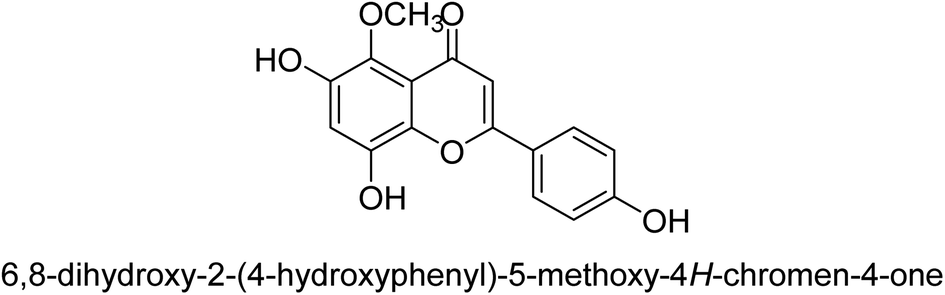 |
23 |
| 75 | 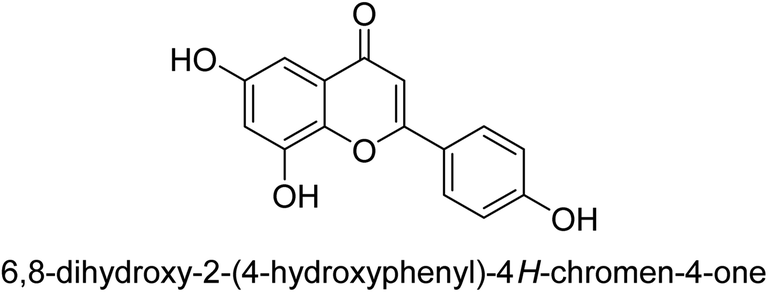 |
— |
Kai and his co-workers investigated the potential inhibitory activities of the urease enzyme as shown in Table 4. Okara, a by-product of the soybean industry, can be used to process bioactive substances sustainably and create functional foods. The enzyme-inhibitory properties of Okara's isoflavones were investigated. Genistin 76 (2.15 mg/100 g), glycitin 77 (1.98 mg/100 g), daidzin 78 (5.85 mg/100 g), genistein 79 (3.65 mg/100 g), glycitein 80 (1.17 mg/100 g) and daidzein 81 (6.20 mg/100 g) were found in Okara by high-performance liquid chromatography analysis. With IC50 values for urease of 41 ± 1 to 65 ± 2 μg mL−1 for Okara and 10 ± 1 to 21 ± 1 μg mL−1 for isoflavones, with the controls SC(NH2)2 (7.1 ± 0.1 g mL−1) and 7.3 ± 0.9 to 16 ± 1 for isoflavones, with the controls allopurinol (5.4 ± 0.1) g mL−1, both Okara extracts according to the findings, Okara offers isoflavones and nutraceuticals with possible enzyme repression properties (Table 4). Single isoflavones from Okara may be investigated as potential useful components that function as organic cures for xanthine oxidase and urease-related illnesses.91
| Compound no. | Chemical structures & IUPAC names | IC50 (μg mL−1) |
|---|---|---|
| 76 | 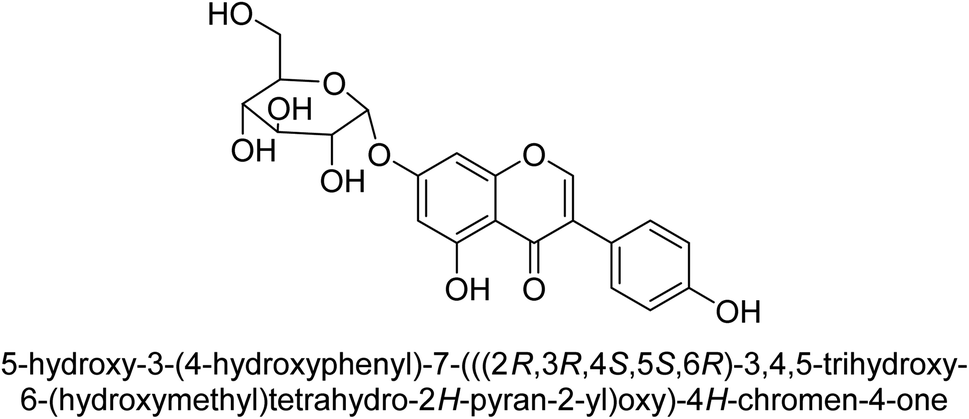 |
14 ± 1 |
| 77 | 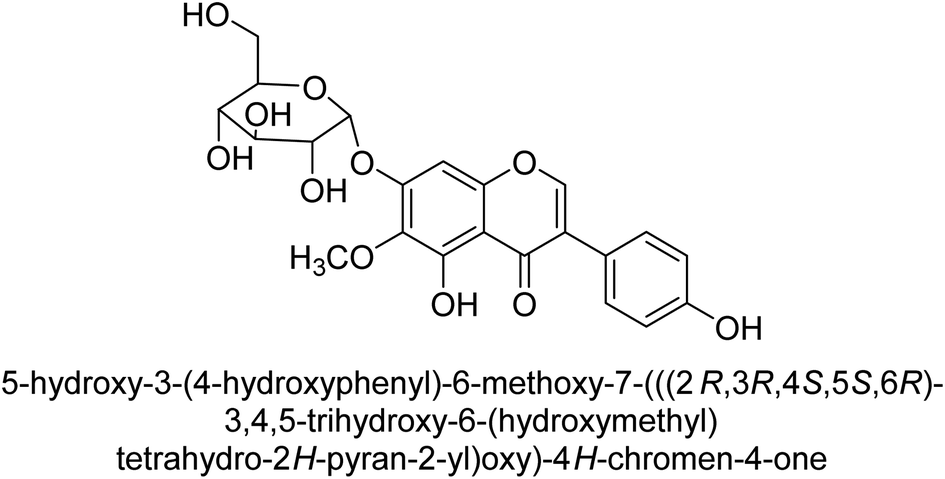 |
18 ± 1 |
| 78 | 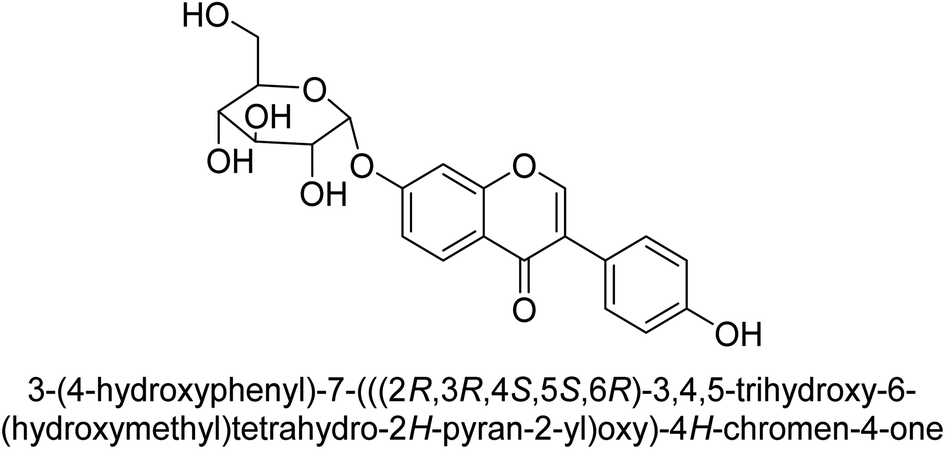 |
17 ± 1 |
| 79 | 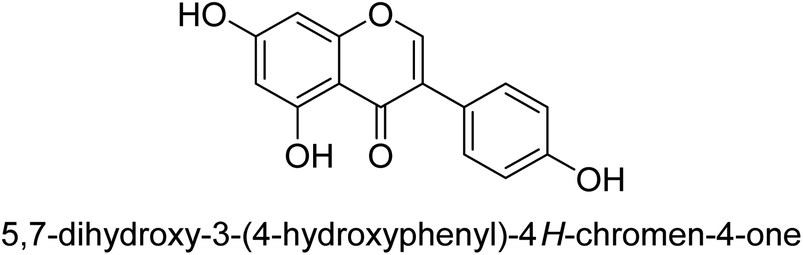 |
10 ± 1 |
| 80 | 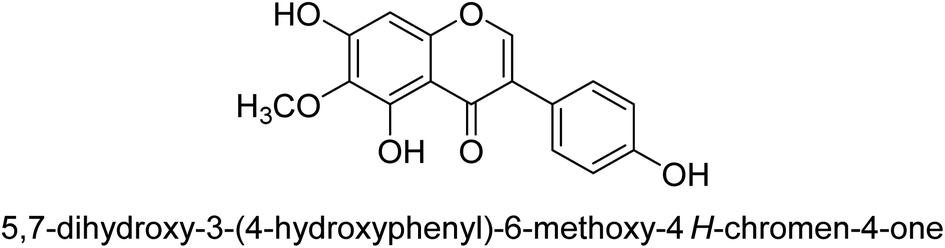 |
21 ± 1 |
| 81 | 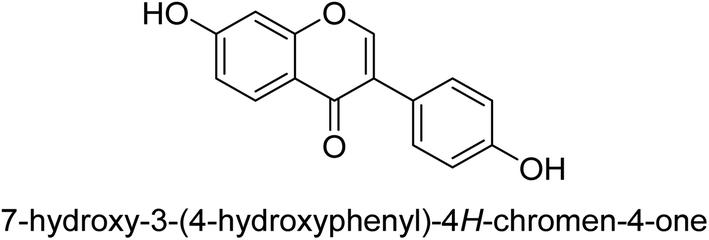 |
11 ± 1 |
Keskin and colleagues investigated the phenolic makeup and inhibitory capabilities of the therapeutically significant enzyme urease from H. lupulus. The hop plant, Humulus lupulus L., is mostly utilized in the brewing business. Hops are known to have certain secondary metabolites with important biological functions. This is the first research that we are aware of that reports hop extracts' ability to inhibit urease. The extracts' ability to inhibit clinically significant urease enzymes was investigated. TPC was discovered in methanol extracts of hop cone and leaf at 7.12 ± 0.09 and 6.86 ± 0.05 mg GAE per g, respectively. Hop cone and leaf methanol extracts were shown to have potent urease inhibitory effects (IC50 of 0.58 ± 0.02 and 0.87 ± 0.02 mg mL−1, in that order). Methanol extract of H. lupulus may be an option to cure peptic ulcer.92
2.6 Flavonoid glycosides as urease inhibitors
The efforts to investigate the inhibitory potential of flavonoids were kicked into high gear as a direct result of these studies. As a result, 11 naturally occurring compounds and 19 synthetic ones were assessing for their capability to impede H. pylori urease. These ranged from moderately competitive (in the μM range) to weak blockers, with quercetin 84, 82 and 83 being the most active (Fig. 12).93 The mode of binding of flavonoid 82 was elucidated using the AutoDock programme to dock it into the crystal structure of H. pylori urease. This revealed the interactions of the inhibitor. In more specific terms, the compound is arranged so that its benzopyrone is near the urea binding cavity.94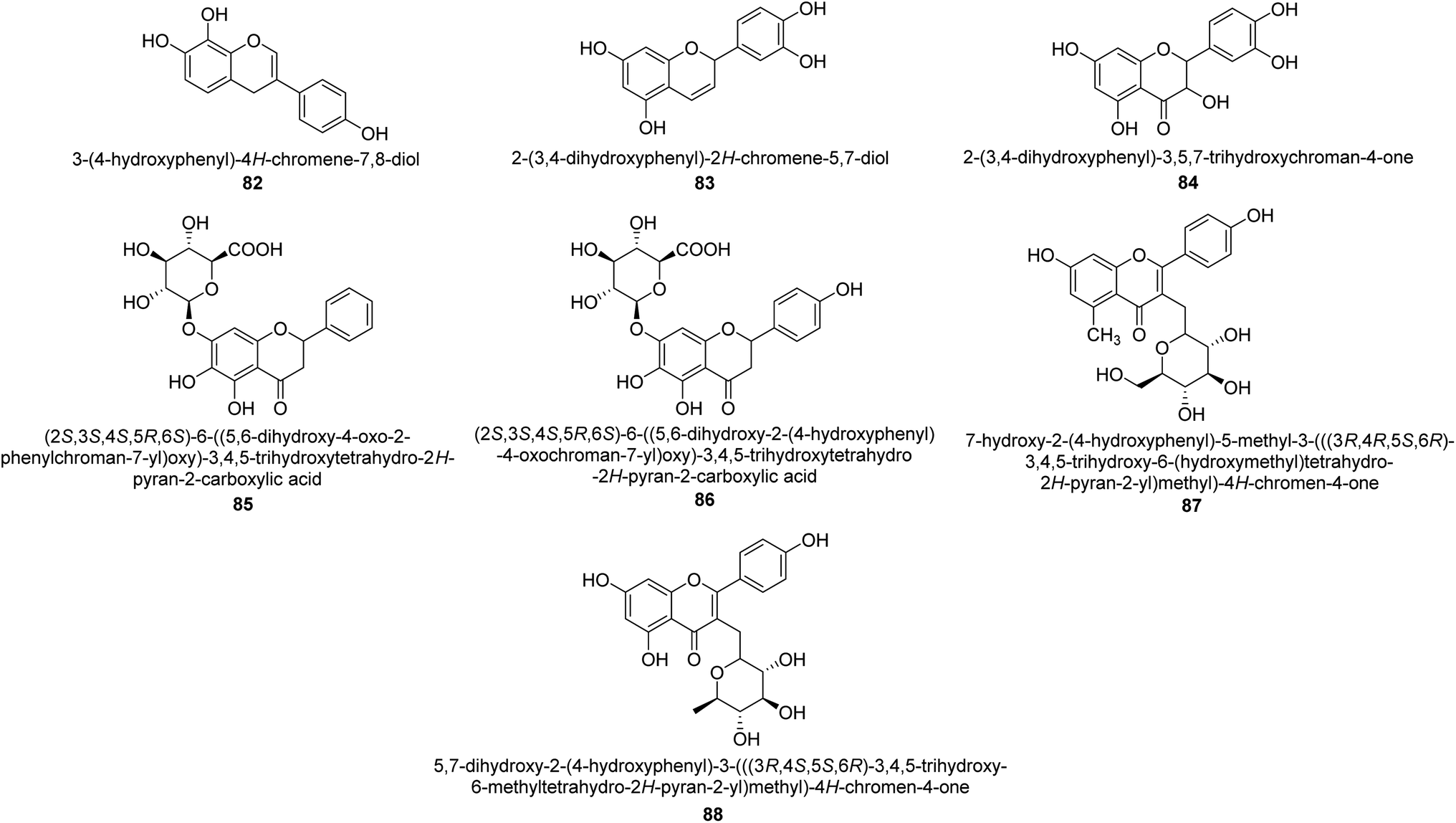 | ||
| Fig. 12 Flavonoid glycosides-based urease inhibitors. | ||
Radix scutellariae, sometimes referred to as Huang-Qin in Chinese, is derived from the Scutellaria baicalensis root that has been dried and powdered. Flavone glycosides like baicalin and scutellarin make up the majority of their bioactive components (Fig. 12, compounds 85 and 86).95 It was discovered that baicalin acts as a competitive, slow-binding, and concentration-dependent blocker of ureases produced by both jack bean and H. pylori.96 Sequestered from the fruits of Syzygium alternifolium, glucopyranoside 87 and rhamnopyranoside 88 (Fig. 12) proved to be more powerful inhibitors of the H. pylori enzyme.28
2.7 Anthocyanins as urease inhibitors
A more contemporary method for treating specific diseases and conditions, including as diabetes, Alzheimer's disease, skin hyperpigmentation, and some microbiological infections, is key-enzyme inhibition. Flavonoid-rich extracts from peels of Ficus carica fruits were evaluated concurrently against several enzymes in the search for possible natural multi-target inhibitors, and several techniques were utilized to gauge their antioxidant efficacy. According to phytochemical research, considerable amounts of flavonoids, mostly flavonols, were extracted from the peels (up to 50.4 mg g−1) (up to 81 percent of total flavonoids). A high synergy was seen for enzyme inhibition and metal chelation due to the unique extract composition, which included 2 fractions (flavonoids and citric acid). Extracts from F. carica that are high in flavonoids might be employed as multipurpose bioactive components to prevent stomach ulcers. Following Giusti and Wrolstad's instructions, the pH-differential technique was used to determine the anthocyanin content (At).97 The findings suggested that the extracts' anthocyanin content may be largely responsible for the inhibitory actions against urease.98Here, novel urease inhibitors of agricultural importance based on red grape pomace polyphenols and deep eutectic solvents (DES) were tested (jack bean urease, JBU). In order to extract and transport polyphenols, DES-based on choline chloride (CHO) and betaine (BET) was coupled with (CH2OH)2 (EG), HOC(CH2CO2H)2 (CA), and CO(NH2)2 (U), serving as an active component of the formulations developed. The urea- and citric acid-based DES combinations performed highest in terms of polyphenol extraction, 1.2–1.4 folds better than the hydroalcoholic mixture. Grape pomace has a complicated profile that comprises flavonol glycosides, phenolic acids and alcohols, catechins, stilbenes, hydroxycinnamic acids, proanthocyanidines, and anthocyanins.99,100 Grape pomace is distinguished by its elevated phenolic content The CHO–EG–PF formulation's notable inclusion of catechins, condensed-catechins (proanthocyanidins), anthocyanins, and gallic acid may account for its potent anti-urease efficacy.101 The most common anthocyanins are pelargonidin 89,102 petunidin 90,103 delphinidin 91,104 cyanidin 92,105 malvidin 93 (ref. 106) and peonidin 94 (ref. 107) as shown in Fig. 13.
 | ||
| Fig. 13 Structures of assorted anthocyanins-based urease inhibitors. | ||
Metwaly and colleagues created a highly sensitive viscose (Vis) fabric for visual colorimetric identification of fluid urea. A natural anthocyanin (Ac) spectroscopic probe from red cabbage was co-encapsulated with the urease enzyme as a catalyst in the calcium alginate biopolymer matrix, then consequently immobilized in place into viscose fibers as a host matrix. Employing the anthocyanin extract and urease as the inside components and the crosslinked calcium alginate as the shell, calcium alginate nanocapsules were produced on viscose surfaces. The paddry-cure approach was used in situ to perform the co-encapsulation procedure under ambient circumstances. This anthocyanin probe was ideal for encapsulating into calcium alginate nanoparticles biosensor assay due to its high sensitivity and tiny molecular size (Vis–Ac). When the overall amount of urea was increased, the anthocyanin receptor embedded in calcium alginate showed ratiometric alterations in the absorbance spectra to the extent of 127 nm hypsochromic blue-shift from 567 nm to 440 nm. As the quantity of urea rose, the colorimetric variations of Vis–Ac were seen between purple and greenish-yellow. The biochromic sensor barcode used demonstrated a rather rapid response time (6–9 min) and a detection limit of 300–1000 ppm. To gain insight into the comfortability of the treated viscose, its bend length, air permeability, and fastness qualities were investigated. Proton transfer from the anthocyanin dye to ammonium ions produced as a result of the hydrolysis-catalyzed interaction of urea with urease allows for the detection of urea. An interaction between urea, urease and the anthocyanin chromophore 95–98 is thought to be the mechanism for the anthocyanin chromophore's reactivity, which is the basis for this ecologically beneficial biochromic sensor. Following the release of NH3 from the urease-catalyzed hydrolysis process of urea shown in Scheme 3, the color changed from purple to greenish-yellow.108
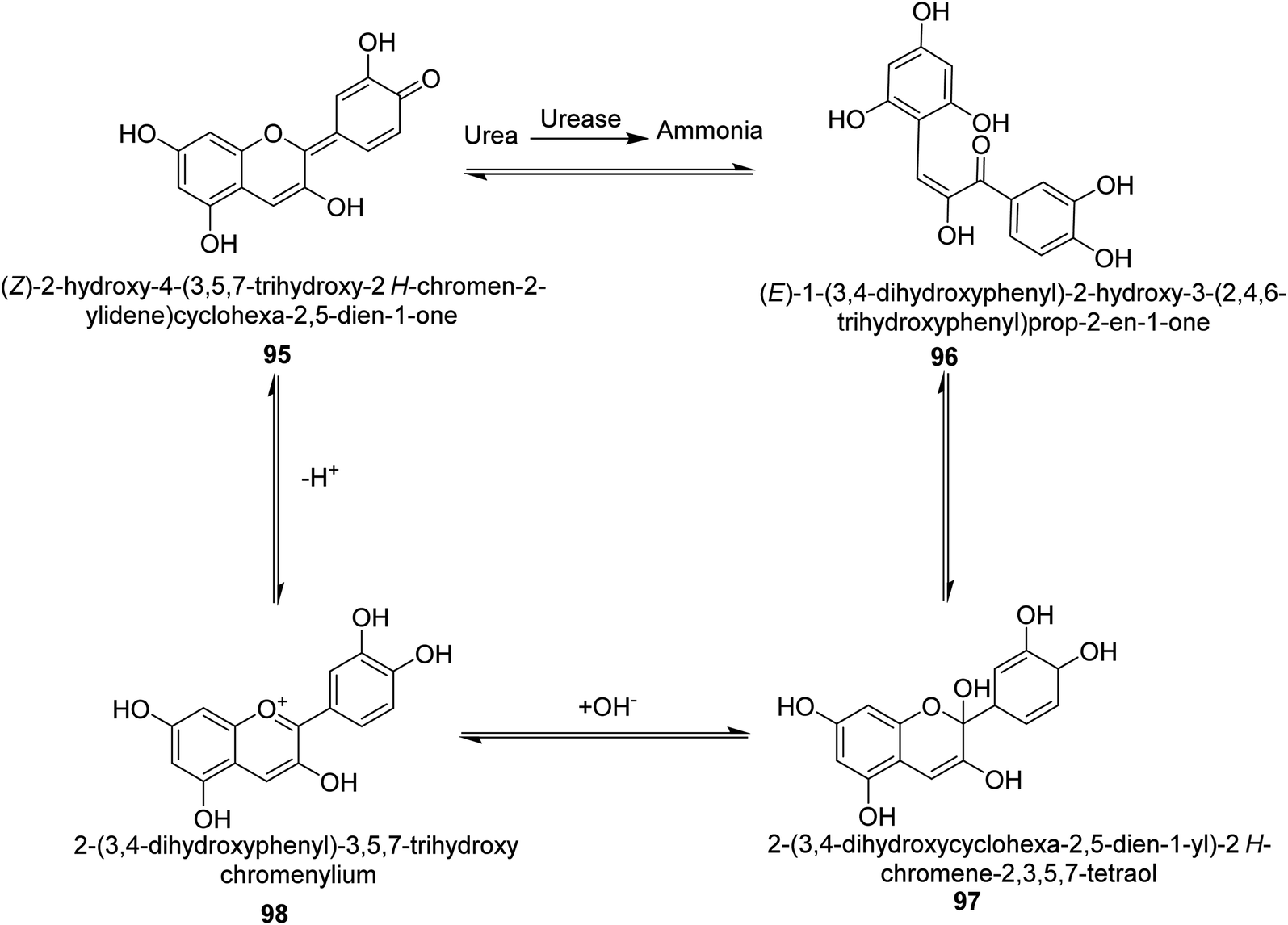 | ||
| Scheme 3 A mechanism for the colorimetric detection of urea using anthocyanin extract from red-cabbage. | ||
Sahin's study's objective was to assess potential novel natural sources of urease blockers. To ascertain the inhibitory effects of both enzymes, chestnut, oak, and polyfloral honey extracts were utilized. Using tests for total phenolic content (TPC) and ferric reducing antioxidant power (FRAP) of these honeys were also investigated. Chestnut and oak honey are discovered to be potent sources for inhibiting both enzymes as they contain elevated phenolic content. Particularly, chestnut, and oak honey had 0.012–0.021 g mL−1 IC50 values for urease inhibition, respectively. This honey may help avoid pathological conditions caused by reactive oxygen species and stomach ulcers resulting from Helicobacter pylori.109
2.8 Miscellaneous flavonoids as urease inhibitors
From the EtOAc extract of the roots of Alhagi maurorum, Laghari and co-workers isolated a the flavanenol 99 (Fig. 14). When tested against H. pylori urease, this chemical had an inhibitory activity that was 24 percent more potent than that of the conventional inhibitor protocatechuic acid (at a dosage of 20.8 mM).110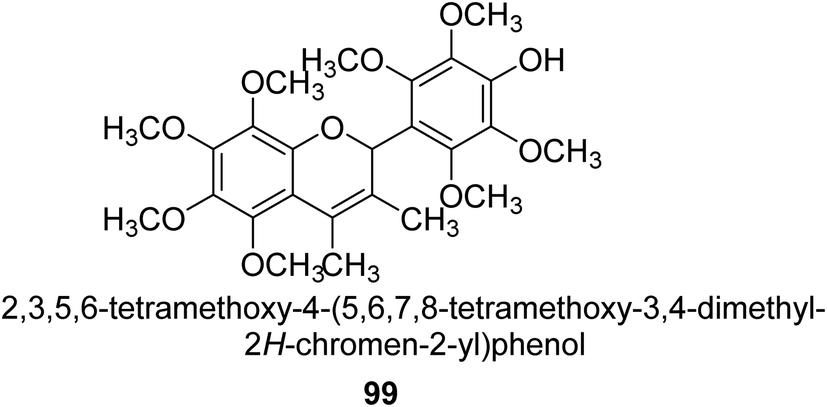 | ||
| Fig. 14 Structure of flavanenol-based urease inhibitor. | ||
Saleem et al. examined the chemical, biological, and in silico features of MeOH and CH2Cl2 fractions of the roots of Alhagi maurorum in terms of antioxidant, enzyme inhibition, and phytochemical content. The ability of an enzyme to block urease enzymes was researched. The overall phenolic (105.91 mg GAE per g extract) and flavonoid (2.27 mg RE per g extract) contents of the MeOH extract were greater, which is connected to its greater potential and enzyme inhibition. Emmotin A, luteolin 5,3′-dimethyl ether, and preferrugone were three substances that were further explored for their in silico molecular docking properties against the examined enzymes. The chosen substances exhibited a greater level of binding contact with the enzyme. The findings of the current investigation have shown that A. mauroram is a leading source of naturally occurring enzyme inhibitor chemicals.111
Alhagi is a genus of plants in the Fabaceae family that is extensively dispersed in several Asian, Australian, and European nations. Alhagi, also known as camel thorn, contains several kinds that are well-known for usage as feed and traditional medicines. Different portions of the Alhagi species have been shown to contain a variety of pharmacoactive secondary metabolites, including phospholipids, alkaloids (alhacidin and alhacin), steroids, flavonoids, pseudalhagin A, and polysaccharides. Another species well known for its strong urease inhibitory action is Alhagi maurorum. It has a great nutritional value because of its abundance in various minerals, oils, and lipids. The urease inhibiting octamethoxyflavanenol 99 was recently discovered in an ethyl acetate fraction of A. maurorum roots.112
It is common knowledge that the structural variety and complexity present in natural products encourages research into the possibility of using these substances as potential lead compounds for treating a variety of diseases. It is common practice to cure gastritis and urinary tract infections with extracts from a variety of plants, like green tea and cranberries, amongst others. It is believed that the activity of (+)-catechin and (−)-epigallocatechin gallate as urease blockers are the cause of this effect.113 Also, other plant flavonoids, such as Daphne retusa (daphnretusic acid), Pistacia atlantica (transilitin and dihydro luteolin), and cotton (gossypol, gossypolone, and apogossypol), proved as μM-range blockers of urease from jack bean.114
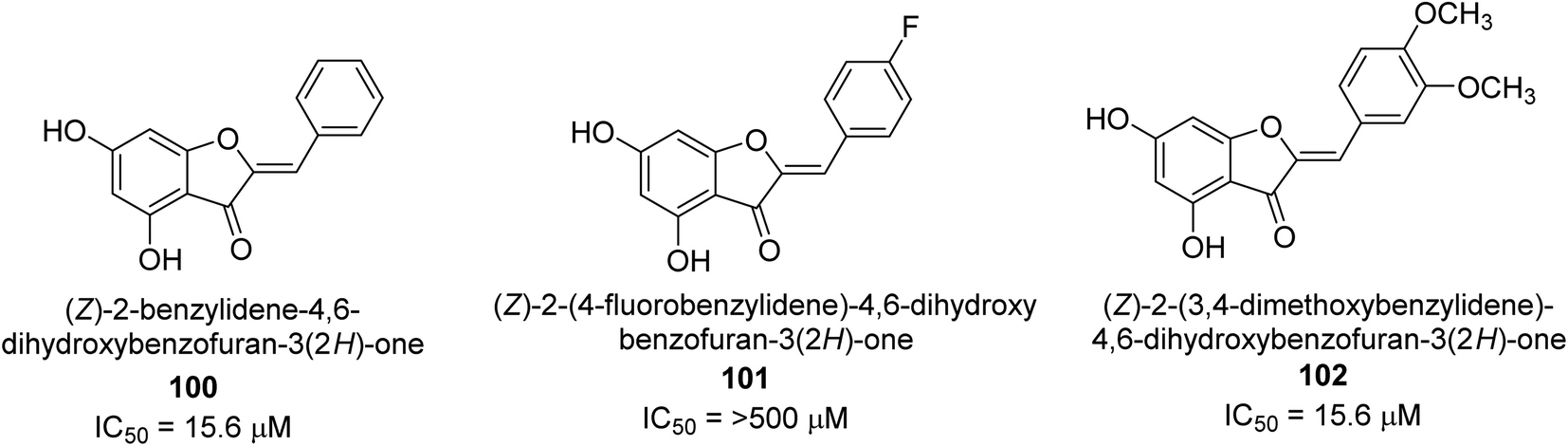
A collection of aurones with diverse substitutes was created and tested against H. pylori. When tested against a wide variety of bacterial (Gram-negative) strains, including resistant ones, compounds 100, 101, and 102 showed little cytotoxicity (Fig. 15). The antibacterial action of at least some of the aurones might be connected to changes in the bacterial membrane, according to a permeabilization study. Overall, this work supports using the aurone scaffold to create novel, powerful, and targeted antibacterial compounds.115
 | ||
| Fig. 15 Structures of aurone-based urease inhibitors. | ||
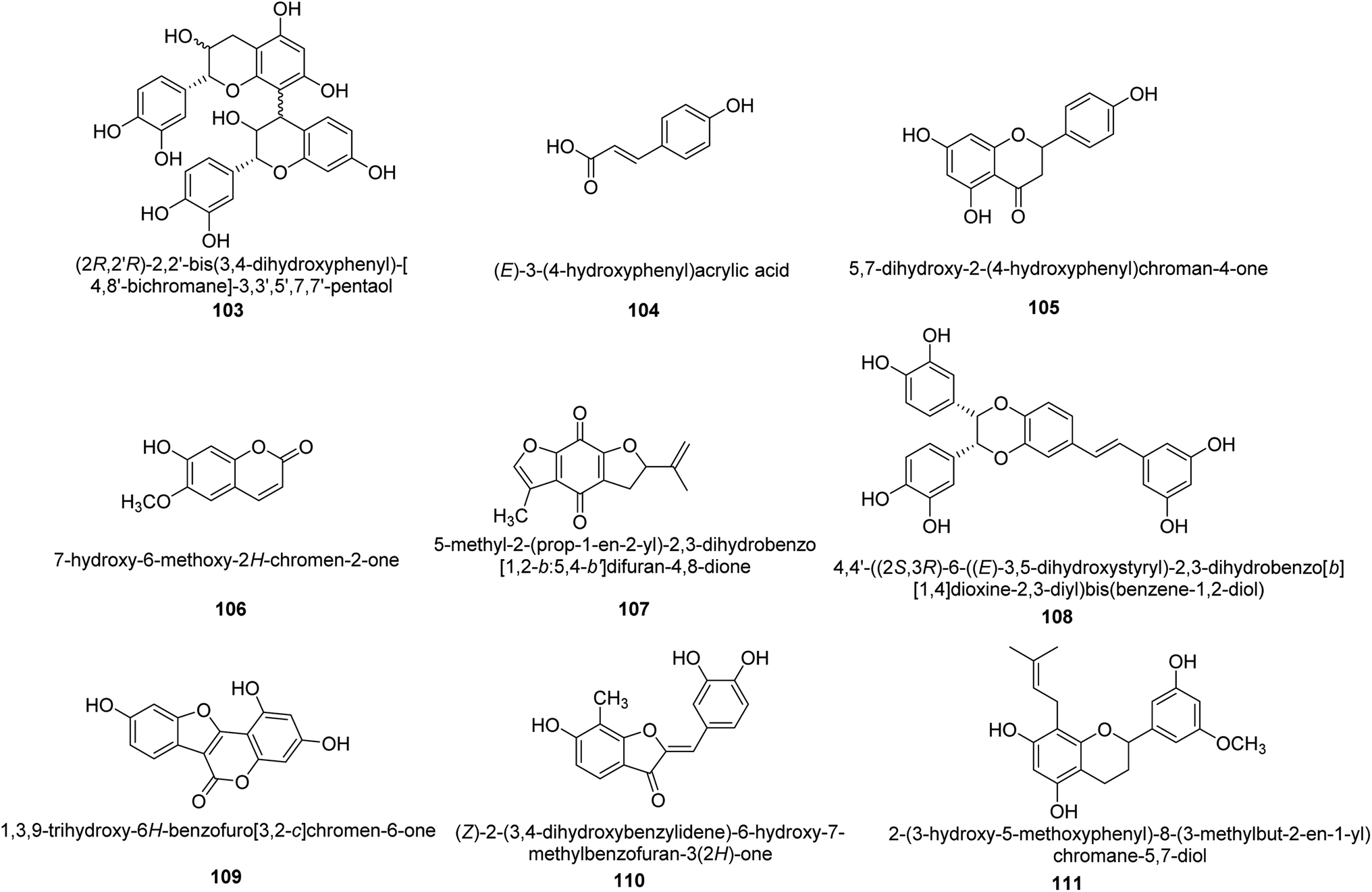
Worldwide, Cyperus has been widely utilized as a multipurpose medicinal herb. One goals of a recent work was to identify the various compounds in the methanol extract of Cyperus conglomeratus Rottb. and to evaluate its in vivo gastroprotective efficacy in a rat model of EtOH-promoted gastric ulcer. TNF- and galactin-3 serum levels were used as biochemical indicators. Complete metabolites analysis of the extract using UPLC-qTOF-MS/MS was used to profile active substances. The discovered metabolites were categorized as organic acids, phenolic acids and derivatives of cinnamic acid, flavonoids, stilbenes, aurones, quinones, terpenes, and steroids. Fig. 16 provides the chemical structures of a few chosen metabolites 103–111 found in the C. conglomeratus extract. The most prevalent class was phenolic acids.116
 | ||
| Fig. 16 Structures of various urease inhibitors. | ||
2.9 Structure–activity relationship
Investigations on enzyme inhibition continue to be an important subject in the field of drug development since this research has resulted in the discovery of new bioactive compounds that are helpful in curing various illnesses. Plant metabolites have played an important part in pharmaceutical research for many decades. This research has the potential to yield many different therapeutic leads, each of which has a unique chemical structure and set of biological activities. There has been a recent interest spike in the research and development of antiulcer drugs, sparking a quest for physiologically promising new flavonoids. Although many different commercial medications are currently being used to cure stomach and urinary tract infections, various obstacles need to be overcome to develop novel antiulcer therapeutic candidates. This is since most of the treatments that are currently available are limited by the presence of unwanted effects, poisonousness, and low stability. The present review highlighted and summarised relevant research from the previous two decades concerning the top promising species originating from plants as well as synthesized in the laboratory which can be utilized as efficient drugs to cure illnesses triggered by urease-manufacturing bacteria; these are in vitro and in vivo investigations centred on numerous different assay techniques. The results that were described in this review demonstrated a startling influence of structural changes, and the SAR study showed that the inhibitory action profile of plant-derived species depended on the location and type of functional groups. Both findings are a product of this research. Fig. 17 is a summary of the most potent plant-derived flavonoids, together with the kind of inhibition they exert and the most active moieties they contain. This information was collected through SAR investigations and the type of inhibition exerted by the flavonoids.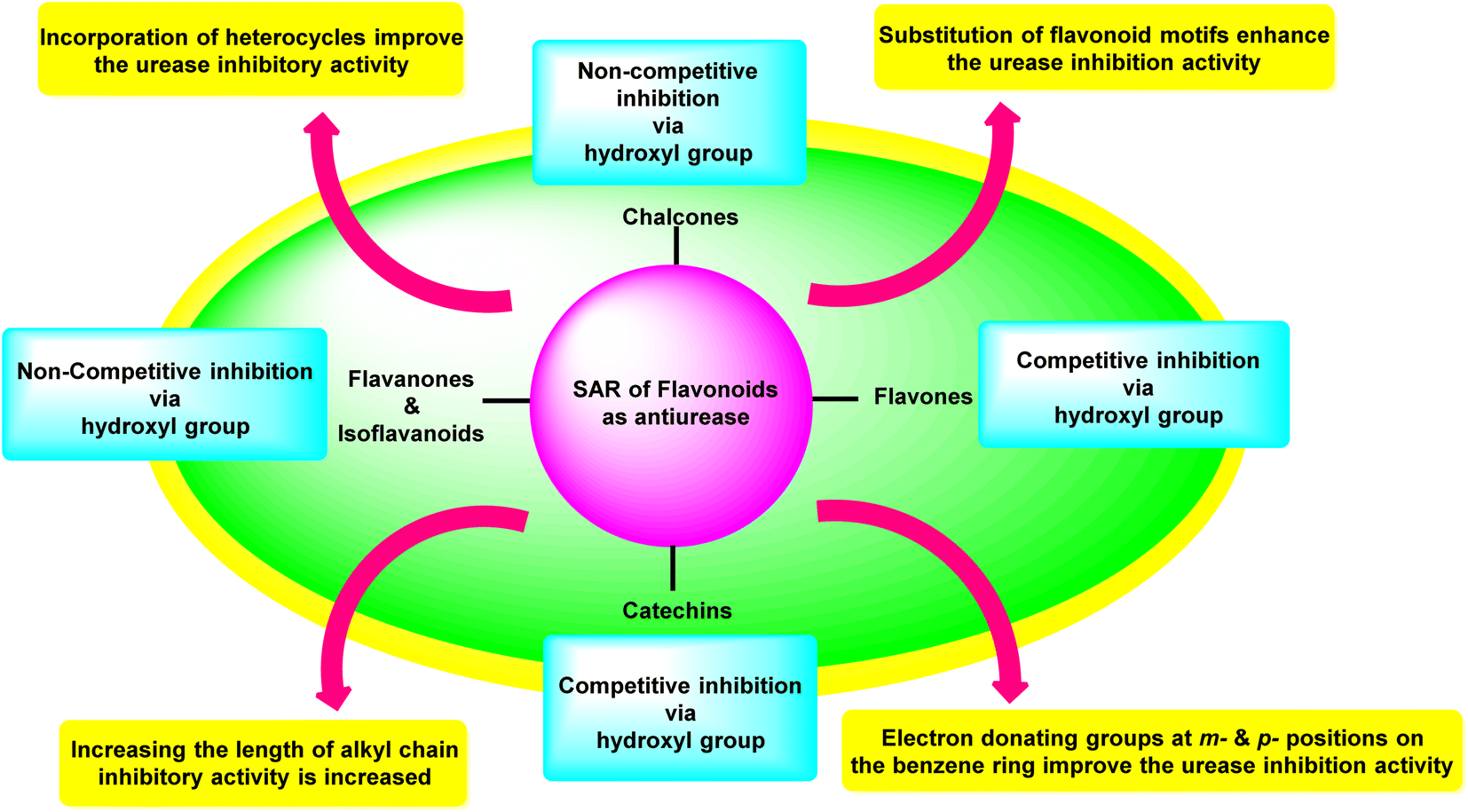 | ||
| Fig. 17 Structure–activity relationship of flavonoids as anti-urease agents. | ||
3. Conclusions and future perspectives
Flavonoids are an interesting class of secondary metabolites with the potential to benefit human health. These compounds protect the mucosa of the gastrointestinal system against ulcers and necrotic agents in a range of experimental ulcer types. A multitude of pathways might be responsible for this protective effect. Chalcones have cytoprotective characteristics, boosting mucosal blood flow, encouraging muco substance synthesis in the gastrointestinal mucosa, and raising PG levels, especially those possessing multiple isoprenyloxyl groups. The anti-urease properties of flavonoids, which prevent urea formation in the small intestine, represents the most crucial mechanism of action for their anti-ulcer efficacy. Furthermore, the two initial polyphenolic compounds show anti-H. pylori action and might be used as a supplement or replacement for existing treatment. As a result, flavonoids may offer the optimal therapeutic solution to cure gastrointestinal illnesses, notably peptic ulcers, that is more effective and less harmful. Furthermore, flavonoids have been found to have inhibitory activity against urease enzyme and different classes of flavonoids have different level of inhibitory activity, such as flavones have moderate, flavonols and flavanones have strong, and anthocyanins have weak activity.In the future, flavonoids-based urease inhibitors are likely to be explored further as a potential treatment for kidney stones, as well as for reducing the environmental impact of ammonium-based fertilizers. Researchers may also investigate the use of flavonoids-based urease inhibitors in combination with other therapies, such as antibiotics, to improve the effectiveness of treatment for conditions such as urinary tract infections. Additionally, new methods of extraction and purification of flavonoids from plant sources may be developed to improve their bioavailability and efficacy as urease inhibitors. In agriculture field, research may focus on exploring the potential of flavonoids-based urease inhibitors to improve the efficiency of fertilizers and reduce the environmental impact of ammonium-based fertilizers. This could lead to the development of new fertilizers and farming methods that are more sustainable and environmentally friendly. Overall, flavonoids-based urease inhibitors are an area of active research with potential applications in medicine and agriculture, and further research is likely to uncover new uses and improve the effectiveness of these compounds. The collected information is expected to provide rational guidance and effective strategy to develop novel, potent, and safe urease inhibitors for better practical applications in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the Deanship of Scientific Research at Umm Al-Qura University, for supporting this work by grant code: 22UQU4220005DSR01. Dr Ziad Moussa is grateful to the United Arab Emirates University (UAEU) and to the Research Office for supporting the research developed in his laboratory and reported herein (SUREPLUS grant code G00003918). The financial support by the Higher Education Commission of Pakistan (HEC) under project no. NRPU-15800 is also gratefully acknowledged.References
- H. Mobley, M. D. Island and R. P. Hausinger, Microbiol. Rev., 1995, 59, 451–480 CrossRef CAS PubMed.
- R. A. Burne and Y.-Y. M. Chen, Microbes Infect., 2000, 2, 533–542 CrossRef CAS PubMed.
- B. Krajewska, J. Mol. Catal., 2009, 59, 9–21 CrossRef CAS.
- A. Balasubramanian and K. Ponnuraj, J. Mol. Biol., 2010, 400, 274–283 CrossRef CAS PubMed.
- H. Carlsson and E. Nordlander, Bioinorg. Chem. Appl., 2010, 20(10), 549–560 Search PubMed.
- S. Benini, W. R. Rypniewski, K. S. Wilson, S. Miletti, S. Ciurli and S. Mangani, Structure, 1999, 7, 205–216 CrossRef CAS PubMed.
- H. Mobley and R. Hausinger, Microbiol. Rev., 1989, 53, 85–108 CrossRef CAS PubMed.
- Y. Qin and J. M. Cabral, Biocatal. Biotransform., 2002, 20, 1–14 CrossRef CAS.
- P. Kafarski and M. Talma, J. Adv. Res., 2018, 13, 101–112 CrossRef CAS PubMed.
- S. Odake, T. Morikawa, M. Tsuchiya, L. Imamura and K. Kobashi, Biol. Pharm. Bull., 1994, 17, 1329–1332 CrossRef CAS PubMed.
- K. Stingl and H. De Reuse, Int. J. Med. Microbiol., 2005, 295, 307–315 CrossRef CAS PubMed.
- R. Pounder and D. Ng, Aliment. Pharmacol. Ther., 1995, 9, 33–39 Search PubMed.
- T. Abu-Izneid, A. Rauf, M. Saleem, N. Mansour, M. I. Abdelhady, M. M. Ibrahim and S. Patel, Nat. Prod. Res., 2020, 34, 553–557 CrossRef CAS PubMed.
- G. Sachs and D. R. Scott, F1000Medicine Reports, 2012, 4(7), 1–5 Search PubMed.
- M. Aljabria, J. Doughtyc and R. J. Scottc, Journal of Umm Al-Qura University for Applied Science, 2020, 6, 1–5 Search PubMed.
- U. Shaheen, N. A. Shoeib, A. Temraz and M. I. Abdelhady, Pharmacogn. Mag., 2017, 13, S484 CrossRef PubMed.
- N. Malafronte, A. Vassallo, F. Dal Piaz, A. Bader, A. Braca and N. De Tommasi, Phytochem. Lett., 2012, 5, 621–625 CrossRef CAS.
- K. Bashir, B. Ahmad, A. Rauf, S. Bawazeer, K. Ur Rahman, T. Rehman, M. Saleem, R. S. Ahmed, H. Linfang and R. Ikram, Biomed. Res., 2017, 28(22), 10026–10032 CAS.
- A. Naseer, F. A. Osra, A. N. Awan, A. Imran, A. Hameed, S. A. Ali Shah, J. Iqbal and Z. A. Zakaria, Pharmaceuticals, 2022, 15, 1288 CrossRef CAS PubMed.
- A. Rauf, M. Raza, M. Humayun Khan, H. A. Hemeg, Y. S. Al-Awthan, O. Bahattab, S. Bawazeer, S. Naz, F. Basoglu and M. Saleem, Ann. Med., 2022, 54, 495–506 CrossRef CAS PubMed.
- A. Bader, T. Tuccinardi, C. Granchi, A. Martinelli, M. Macchia, F. Minutolo, N. De Tommasi and A. Braca, Phytochemistry, 2015, 116, 262–268 CrossRef CAS PubMed.
- V. Amirkhanov, A. Rauf, T. B. Hadda, V. Ovchynnikov, V. Trush, M. Saleem, M. Raza, T. Rehman, H. Zgou and U. Shaheen, Mini-Rev. Med. Chem., 2019, 19, 1015–1027 CrossRef CAS PubMed.
- H. Elabidi, Journal of Umm Al-Qura University for Applied Science, 2020, 6, 25–38 Search PubMed.
- N. Broutet, A. Marais, H. Lamouliatte, A. de Mascarel, R. Samoyeau, R. Salamon and F. Mégraud, J. Clin. Microbiol., 2001, 39, 1319–1322 CrossRef CAS PubMed.
- A. Tonkic, M. Tonkic, P. Lehours and F. Mégraud, Helicobacter, 2012, 17, 1–8 CrossRef PubMed.
- B. Khameneh, M. Iranshahy, V. Soheili and B. S. Fazly Bazzaz, Antimicrob. Resist. Infect. Contr., 2019, 8, 1–28 CrossRef PubMed.
- S. T. Hassan and M. Žemlička, Arch. Pharm., 2016, 349, 507–522 CrossRef CAS PubMed.
- M. Amin, F. Anwar, F. Naz, T. Mehmood and N. Saari, Molecules, 2013, 18, 2135–2149 CrossRef CAS PubMed.
- A. F. Cutler, S. Havstad, C. K. Ma, M. J. Blaser, G. I. Perez-Perez and T. T. Schubert, Gastroenterology, 1995, 109, 136–141 CrossRef CAS PubMed.
- R. J. Obaid, E. U. Mughal, N. Naeem, A. Sadiq, R. I. Alsantali, R. S. Jassas, Z. Moussa and S. A. Ahmed, RSC Adv., 2021, 11, 22159–22198 RSC.
- S. B. A. Ghani, L. Weaver, Z. H. Zidan, H. M. Ali, C. W. Keevil and R. C. Brown, Bioorg. Med. Chem. Lett., 2008, 18, 518–522 CrossRef PubMed.
- C. A. Williams and R. J. Grayer, Nat. Prod. Rep., 2004, 21, 539–573 RSC.
- J. B. Harborne and C. A. Williams, Phytochemistry, 2000, 55, 481–504 CrossRef CAS PubMed.
- A. Hässig, W. Linag, H. Schwabl and K. Stampfli, Med. Hypotheses, 1999, 52, 479–481 CrossRef PubMed.
- K. S. de Lira Mota, G. E. N. Dias, M. E. F. Pinto, Â. Luiz-Ferreira, A. R. Monteiro Souza-Brito, C. A. Hiruma-Lima, J. M. Barbosa-Filho and L. M. Batista, Molecules, 2009, 14, 979–1012 CrossRef PubMed.
- S. Martens and A. Mithöfer, Phytochemistry, 2005, 66, 2399–2407 CrossRef CAS PubMed.
- P. M. Dewick, Medicinal natural products: a biosynthetic approach, John Wiley & Sons, 2002 Search PubMed.
- P. Khurana, D. Vishnudasan and A. K. Chhibbar, Physiol. Mol. Biol. Plants, 2008, 14, 277–298 CrossRef CAS PubMed.
- B. H. Havsteen, Pharmacol. Ther., 2002, 96, 67–202 CrossRef CAS PubMed.
- G. R. Beecher, J. Nutr., 2003, 133, 3248S–3254S CrossRef CAS PubMed.
- J. A. Ross and C. M. Kasum, Annu. Rev. Nutr., 2002, 22, 19–34 CrossRef CAS PubMed.
- C. Yang, Annu. Rev. Nutr., 2022, 21, 381–406 CrossRef PubMed.
- C. Manach, C. Morand, C. Demigné, O. Texier, F. Régérat and C. Rémésy, FEBS Lett., 1997, 409, 12–16 CrossRef CAS PubMed.
- J. W. Critchfield, S. T. Butera and T. M. Folks, AIDS Res. Hum. Retroviruses, 1996, 12, 39–46 CrossRef CAS PubMed.
- H. Cheong, S.-Y. Ryu, M.-H. Oak, S.-H. Cheon, G.-S. Yoo and K.-M. Kim, Arch. Pharmacal Res., 1998, 21, 478–480 CrossRef CAS PubMed.
- A. Carotenuto, E. Fattorusso, V. Lanzotti, S. Magno, V. De Feo and C. Cicala, Phytochemistry, 1997, 44, 949–957 CrossRef CAS PubMed.
- J. T. Lima, J. R. Almeida, J. M. Barbosa-Filho, T. S. Assis, M. S. Silva, E. V. da-Cunha, R. Braz-Filhod and B. A. Silva, Zeitschrift für Naturforschung B, 2005, 60, 1093–1100 CrossRef CAS.
- G. Di Carlo, G. Autore, A. Izzo, P. Maiolino, N. Mascolo, P. Viola, M. Diurno and F. Capasso, J. Pharm. Pharmacol., 1993, 45, 1054–1059 CrossRef CAS PubMed.
- C. La Casa, I. Villegas, C. A. De La Lastra, V. Motilva and M. M. Calero, J. Ethnopharmacol., 2000, 71, 45–53 CrossRef CAS PubMed.
- C. V. Rao, S. Ojha, K. Radhakrishnan, R. Govindarajan, S. Rastogi, S. Mehrotra and P. Pushpangadan, J. Ethnopharmacol., 2004, 91, 243–249 CrossRef PubMed.
- D. Bandyopadhyay, K. Biswas, M. Bhattacharyya, R. J. Reiter and R. K. Banerjee, NISCAIR-CSIR, 2002, 20, 678–689 Search PubMed.
- D. E. Mendonça and S. B. Onofre, Rev. Bras. Farmacogn., 2009, 19, 577–581 CrossRef.
- J. Kanner and T. Lapidot, Free Radical Biol. Med., 2001, 31, 1388–1395 CrossRef CAS PubMed.
- I. Gürbüz, Ç. Akyüz, E. Yeşilada and B. Şener, J. Ethnopharmacol., 2000, 71, 77–82 CrossRef PubMed.
- E. Czinner, K. Hagymasi, A. Blazovics, A. Kery, É. Szőke and E. Lemberkovics, J. Ethnopharmacol., 2001, 77, 31–35 CrossRef CAS PubMed.
- O. Barnaulov, O. Manicheva, V. Shelyuto, M. Konopleva and V. Glyzin, Pharm. Chem. J., 1984, 18, 544–549 CrossRef.
- O. Barnaulov, O. Manicheva and N. Komissarenko, Pharm. Chem., 1983, 17, 946–951 CAS.
- A. Izzo, G. D. Carlo, N. Mascolo, F. Capasso and G. Autore, Phytother. Res., 1994, 8, 179–181 CrossRef CAS.
- H. Hirano, H. Takase, K. Yamamoto, K. Abe and J. Saito, Jpn. Kokai Tokkyo Koho, 1994, 72, 870 Search PubMed.
- R. A. Larson, Phytochemistry, 1988, 27, 969–978 CrossRef CAS.
- S. C. Sahu and G. C. Gray, Cancer Lett., 1996, 104, 193–196 CrossRef CAS PubMed.
- F. L. Ansari, S. Umbreen, L. Hussain, T. Makhmoor, S. A. Nawaz, M. A. Lodhi, S. N. Khan, F. Shaheen and M. I. Choudhary, Chem. Biodiversity, 2005, 2, 487–496 CrossRef CAS PubMed.
- F. L. Ansari, A. Wadood, A. Ullah, F. Iftikhar and Z. Ul-Haq, J. Enzyme Inhib. Med. Chem., 2009, 24, 151–156 CrossRef CAS PubMed.
- N. Al Somal, K. Coley, P. Molan and B. Hancock, J. R. Soc. Med., 1994, 87, 9 CrossRef CAS PubMed.
- M. M. Ahari-Mostafavi, A. Sharifi, M. Mirzaei and M. Amanlou, J. Iran. Chem. Soc., 2014, 11, 1113–1119 CrossRef CAS.
- G. Zhang, C. Zhu, D. Liu, J. Pan, J. Zhang, D. Hu and B. Song, Tetrahedron, 2017, 73, 129–136 CrossRef CAS.
- A. Sultan, S. Shajahan, T. Ahamad, S. M. Alshehri, N. Sajjad, M. H. U. Rehman, L. Torun, M. Khalid and R. Acevedo, Monatsh. Chem., 2020, 151, 123–133 CrossRef CAS.
- F. Rasool, M. Khalid, M. Yar, K. Ayub, M. Tariq, A. Hussain, M. Lateef, M. Kashif and S. Iqbal, J. Mol. Liq., 2021, 336, 116302 CrossRef CAS.
- K. Mabe, M. Yamada, I. Oguni and T. Takahashi, Antimicrob. Agents Chemother., 1999, 43, 1788–1791 CrossRef CAS PubMed.
- S. Matsubara, H. Shibata, F. Ishikawa, T. Yokokura, M. Takahashi, T. Sugimura and K. Wakabayashi, Biochem. Biophys. Res. Commun., 2003, 310, 715–719 CrossRef CAS PubMed.
- A. S. Hassani, N. Ordouzadeh, A. Ghaemi, N. Amirmozafari, K. Hamdi and R. Nazari, Indian J. Med. Microbiol., 2009, 27, 30–34 CrossRef PubMed.
- A. Loes, L. Ruyle, M. Arvizu, K. Gresko, A. Wilson and C. Deutch, Lett. Appl. Microbiol., 2014, 58, 31–41 CrossRef CAS PubMed.
- E. Pastene, V. Parada, M. Avello, A. Ruiz and A. García, Phytother. Res., 2014, 28, 1637–1645 CrossRef CAS PubMed.
- S. Shabana, A. Kawai, K. Kai, K. Akiyama and H. Hayashi, Biosci., Biotechnol., Biochem., 2010, 74, 878–880 CrossRef CAS PubMed.
- Z.-P. Xiao, X.-D. Wang, Z.-Y. Peng, S. Huang, P. Yang, Q.-S. Li, L.-H. Zhou, X.-J. Hu, L.-J. Wu and Y. Zhou, J. Agric. Food Chem., 2012, 60, 10572–10577 CrossRef CAS PubMed.
- F. Mansoor, I. Anis, A. Khan, B. P. Marasini, M. I. Choudhary and M. R. Shah, J. Asian Nat. Prod. Res., 2014, 16, 210–215 CrossRef CAS PubMed.
- W. Yang, Q. Feng, Z. Peng and G. Wang, Eur. J. Med. Chem., 2022, 114273 CrossRef CAS PubMed.
- A. Muhammad, I. Anis, A. Khan, B. P. Marasini, M. I. Choudhary and M. R. Shah, Arch. Pharmacal Res., 2012, 35, 431–436 CrossRef CAS PubMed.
- I. Ahmad, M. Ashraf, B. Chaudhary, K. H. Janbaz and M. Uzair, J. Chem. Soc. Pak., 2011, 33, 114–117 CAS.
- L. Tan, J. Su, D. Wu, X. Yu, Z. Su, J. He, X. Wu, S. Kong, X. Lai and J. Lin, Sci. World J., 2013, 20(13), 440–452 Search PubMed.
- D.-W. Wu, X.-D. Yu, J.-H. Xie, Z.-Q. Su, J.-Y. Su, L.-R. Tan, X.-Q. Huang, J.-N. Chen and Z.-R. Su, Fitoterapia, 2013, 91, 60–67 CrossRef CAS PubMed.
- X.-D. Yu, R.-B. Zheng, J.-H. Xie, J.-Y. Su, X.-Q. Huang, Y.-H. Wang, Y.-F. Zheng, Z.-Z. Mo, X.-L. Wu and D.-W. Wu, J. Ethnopharmacol., 2015, 162, 69–78 CrossRef CAS PubMed.
- D.-H. Kim, E.-A. Bae and M. J. Han, Biol. Pharm. Bull., 1999, 22, 422–424 CrossRef CAS PubMed.
- E.-A. Bae, M. J. Han and D.-H. Kim, Planta Med., 1999, 65, 442–443 CrossRef CAS PubMed.
- B. C. Variya, A. K. Bakrania and S. S. Patel, Pharmacol. Res., 2016, 111, 180–200 CrossRef CAS PubMed.
- R. Arumugam, V. Natesan and R. Mani, J. Acute Med., 2016, 6, 55–60 CrossRef.
- D. Singh and K. Chopra, Pharmacol. Res., 2004, 50, 187–193 CrossRef CAS PubMed.
- E.-A. Bae, M. J. Han and D.-H. Kim, Planta Med., 2001, 67, 161–163 CrossRef CAS PubMed.
- Z.-P. Xiao, D.-H. Shi, H.-Q. Li, L.-N. Zhang, C. Xu and H.-L. Zhu, Bioorg. Med. Chem., 2007, 15, 3703–3710 CrossRef CAS PubMed.
- Z.-P. Xiao, Z.-Y. Peng, J.-J. Dong, J. He, H. Ouyang, Y.-T. Feng, C.-L. Lu, W.-Q. Lin, J.-X. Wang and Y.-P. Xiang, Eur. J. Med. Chem., 2013, 63, 685–695 CrossRef CAS PubMed.
- S. H. Nile, A. Nile, J.-W. Oh and G. Kai, Food Biosci., 2020, 38, 100778 CrossRef CAS.
- Ş. Keskin, Y. Şirin, H. Çakir and M. Keskin, S. Afr. J. Bot., 2019, 120, 170–174 CrossRef.
- S. Mahernia, K. Bagherzadeh, F. Mojab and M. Amanlou, Iran. J. Pharm. Res., 2015, 14, 943 CAS.
- L. Mazzei, M. Cianci, F. Musiani, G. Lente, M. Palombo and S. Ciurli, J. Inorg. Biochem., 2017, 166, 182–189 CrossRef CAS PubMed.
- B. W. Lee, I.-H. Park, D. Yim and S. S. Choi, Nat. Prod. Sci., 2017, 23, 46–52 CrossRef CAS.
- T. M. C. Babu, S. S. Rajesh, B. V. Bhaskar, S. Devi, A. Rammohan, T. Sivaraman and W. Rajendra, RSC Adv., 2017, 7, 18277–18292 RSC.
- L. E. Rodriguez-Saona and R. E. Wrolstad, Current Protocols in Food Analytical Chemistry, 2001, F1.1.1–F1.1.11 Search PubMed.
- L. Meziant, M. Bachir-bey, C. Bensouici, F. Saci, M. Boutiche and H. Louaileche, Eur. J. Integr. Med., 2021, 43, 101272 CrossRef.
- D. Kammerer, A. Claus, R. Carle and A. Schieber, J. Agric. Food Chem., 2004, 52, 4360–4367 CrossRef CAS PubMed.
- M. Cotoras, H. Vivanco, R. Melo, M. Aguirre, E. Silva and L. Mendoza, Molecules, 2014, 19, 21154–21167 CrossRef PubMed.
- C. Samorì, L. Mazzei, S. Ciurli, G. Cravotto, G. Grillo, E. Guidi, A. Pasteris, S. Tabasso and P. Galletti, ACS Sustainable Chem. Eng., 2019, 7, 15558–15567 CrossRef.
- Y. Noda, T. Kaneyuki, A. Mori and L. Packer, J. Agric. Food Chem., 2002, 50, 166–171 CrossRef CAS PubMed.
- M. Nagaoka, T. Maeda, S. Moriwaki, A. Nomura, Y. Kato, S. Niida, M. C. Kruger and K. Suzuki, Int. J. Mol. Sci., 2019, 20, 2795 CrossRef CAS PubMed.
- L. J. Porter, L. N. Hrstich and B. G. Chan, Phytochemistry, 1985, 25, 223–230 CrossRef.
- F. C. Stintzing, A. S. Stintzing, R. Carle, B. Frei and R. E. Wrolstad, J. Agric. Food Chem., 2002, 50, 6172–6181 CrossRef CAS PubMed.
- R. Brouillard and B. Delaporte, J. Am. Chem. Soc., 1977, 99, 8461–8468 CrossRef CAS.
- P.-N. Chen, S.-C. Chu, H.-L. Chiou, C.-L. Chiang, S.-F. Yang and Y.-S. Hsieh, Nutr. Cancer, 2005, 53, 232–243 CrossRef CAS PubMed.
- S. D. Al-Qahtani, H. K. Alzahrani, O. A. Azher, Z. O. Owidah, M. Abualnaja, T. M. Habeebullah and N. M. El-Metwaly, J. Environ. Chem. Eng., 2021, 9, 105493 CrossRef CAS.
- H. Sahin, J. Enzyme Inhib. Med. Chem., 2016, 31, 490–494 CAS.
- A. H. Laghari, S. Memon, A. Nelofar, K. M. Khan, A. Yasmin, M. N. Syed and A. Aman, J. Mol. Struct., 2010, 965, 65–67 CrossRef CAS.
- H. Saleem, M. Sarfraz, K. M. Khan, M. A. Anwar, G. Zengin, I. Ahmad, S.-U. Khan, M. F. Mahomoodally and N. Ahemad, Drug Dev. Ind. Pharm., 2020, 46, 861–868 CrossRef CAS PubMed.
- G. Muhammad, M. A. Hussain, F. Anwar, M. Ashraf and A. H. Gilani, Phytother. Res., 2015, 29, 1–13 CrossRef PubMed.
- G. Uddin, I. Ismail, A. Rauf, M. Raza, H. Khan, N. Uddin, M. Khan, U. Farooq, A. Khan and A. Ullah, Nat. Prod. Res., 2016, 30, 1411–1416 CrossRef CAS PubMed.
- Y. Chen, J. Liao, M. Chen, Q. Huang and Q. Lu, J. Chem. Pharm. Res., 2015, 7, 10–15 CAS.
- H. Olleik, S. Yahiaoui, B. Roulier, E. Courvoisier-Dezord, J. Perrier, B. Pérès, A. Hijazi, E. Baydoun, J. Raymond and A. Boumendjel, Eur. J. Med. Chem., 2019, 165, 133–141 CrossRef CAS PubMed.
- A. I. Elshamy, A. R. H. Farrag, I. M. Ayoub, K. A. Mahdy, R. F. Taher, A. E.-N. G. E. Gendy, T. A. Mohamed, S. S. Al-Rejaie, Y. A. Ei-Amier and A. M. Abd-EIGawad, Molecules, 2020, 25, 4234 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |