
Covalent adaptive networks with repairable, reprocessable, reconfigurable, recyclable, and re-adhesive (5R) performance via dynamic isocyanate chemistry
Jialiang
Lai†
a,
Xijin
Xing†
b,
Huanzhi
Feng
b,
Zhanhua
Wang
 *a and
Hesheng
Xia
*a
*a and
Hesheng
Xia
*a
aState Key Laboratory of Polymer Materials Engineering, Polymer Research Institute, Sichuan University, Chengdu 610065, China. E-mail: zhwangpoly@scu.edu.cn; xiahs@scu.edu.cn
bCNOOC Research Institute Co., Ltd, Beijing 100028, China
First published on 7th September 2023
Abstract
Polymer materials with covalent adaptive networks (CANs) structures have attracted considerable attention in recent years due to their excellent repairable, reprocessable, reconfigurable, recyclable, and re-adhesive (5R) performance. Many types of CANs based on reversible dissociation or association reactions have been developed. Of these, CANs via dynamic isocyanate chemistry have made significant progress on the creation of smart polyurethane (PU) and polyurea (PUR) materials. Herein, we provide a comprehensive review on the recent development of CANs via dynamic isocyanate chemistry. First, we provide a brief introduction of dynamic isocyanate chemistry. Second, several categories of dynamic isocyanate chemistry (and the mechanism behind them) are discussed in detail. Third, we focus on the characterization of CANs via dynamic isocyanate chemistry by physical and chemical approaches. Fourth, we focus on novel types of “smart” polymer materials containing a CANs structure with 5R properties via dynamic isocyanate chemistry. The influence of different categories of dynamic isocyanate chemistry on the stress relaxation and 5R performance are summarized in detail in this part. The advantages and disadvantages of different types of dynamic isocyanate chemistry for 5R applications are also discussed. Finally, conclusions and the outlook on the development and challenges of CANs via dynamic isocyanate chemistry are provided.
 Jialiang Lai | Jialiang Lai received bachelor and master degrees in polymer materials and engineering at Xi'an Technological University under the supervision of Professor Hongwei Zhou in 2018 and 2021, respectively. He has been pursuing his doctorate in Sichuan University under the supervision of Professor Zhanhua Wang since 2021. His research interests lie in self-healing elastomers and flexible electronic devices. |
 Xijin Xing | Xing Xijin graduated from Southwest Petroleum University in 2008 with a master's degree. In the same year, he joined CNOOC Research Institute. He is now a senior engineer in oilfield chemistry. His research focuses on polymer materials for drilling fluid, shape-memory lost-circulation materials, and self-degrading functional materials. |
 Huanzhi Feng | Huanzhi Feng is a senior oilfield chemistry engineer at CNOOC Research Institute. His research interest mainly focuses on deep-water drilling and well integrity. |
 Zhanhua Wang | Zhanhua Wang received his doctorate in polymer chemistry and physics from Jilin University under the supervision of Professor Bai Yang in 2011. He worked as a postdoctoral fellow with Professor Marek Urban at the University of Southern Mississippi and Clemson University. He moved to Wageningen University as a postdoctoral researcher and worked with Professor Han Zuilhof until 2016. He is a professor at the Polymer Research Institute of Sichuan University. His scientific interests focus on bio-inspired anti-fouling coatings, self-healing, and 3D printing of polymer materials. |
 Hesheng Xia | Hesheng Xia is a Professor and Deputy Director of State Key Laboratory of Polymer Materials Engineering (China) within Sichuan University. He received his bachelor and master degrees from Northeastern University in 1994 and 1997, respectively, and doctorate in materials science in Sichuan University under the supervision of Professor Qi Wang in 2001. In 2003–2005, he worked as a Research Associate in the Department of Materials, Loughborough University, UK. His main research interests are graphene/carbon nanotubes polymer composites, self-healing polymer materials, dynamic polymer materials for 3D printing, and ultrasound mechanochemistry. |
1. Introduction
Covalent adaptable networks (CANs; also known as vitrimers) are covalently crosslinked polymers that can be reshaped via crosslinking and/or strand exchange at high temperatures.1–4 These materials possess excellent repairable, reprocessable, reconfigurable, recyclable and re-adhesive (5R) properties.5–11 CANs can undergo dissociative or associative reversible reactions upon external stimuli (Fig. 1).12,13 Dynamic chemical bonds or moieties such as Diels–Alder (DA) bonds,14,15 urethane groups,16,17 urea bonds,18,19 alkyl anilinium or triazolium salt,20,21 alkoxyamines,22,23 and reversible homolytic reaction of nonpolar bonds24,25 have been introduced into polymer materials to prepare dissociative CANs. Disulfide bonds,26,27 trithioester bonds,28 transesterification,29,30 imine bonds (Schiff bases),31,32 vinyl urethane/urea,33–35 silyl ether bond,36,37 and metathesis of alkene and boronate bonds38–40 are the most rapidly developing dynamic covalent bonds for making associative CANs. The possibility of using dynamic isocyanate chemistry to make functional polymer materials with a CANs structure has been studied. Isocyanate chemistry is a very important reaction for the polymer industry due to the wide application of polyurethane (PU) and polyurea (PUR) materials.41 One way to develop PU/PUR materials with a CANs structure is by introducing dynamic covalent bonds into PU networks. The drawback of this strategy is that the introduction of additional dynamic groups warrants a complicated synthesis and may alter the original property of PU/PUR materials. Making urethane/urea bonds dynamic can overcome this change because extra events are not involved. Therefore, great efforts have been devoted to develop CANs via dynamic isocyanate chemistry. This is a rational approach to develop PU/PUR with 5R performance to meet with the high criteria of sustainable development of polymer materials. | ||
| Fig. 1 Dynamic process of dissociative and associative CANs with 5R performance formed by different categories of dynamic isocyanate chemistry where X denotes O, S, NH, or NR groups (schematic). Reproduced from ref. 166 with permission from the American Association for the Advancement of Science, copyright 2018. Reproduced from ref. 87 with permission from the American Chemical Society, copyright 2020. Reproduced from ref. 67 with permission from the Royal Society of Chemistry, copyright 2020. | ||
Dynamic isocyanate chemistry can involve dissociative or associative exchange during thermal processing (Fig. 1). In theory, the crosslinking density remains constant for associative CANs via dynamic isocyanate chemistry during the dynamic process if side reactions are absent. There is virtually no difference in the rate constant of the forward reaction and reversible reaction because the reactant and product are usually the same type of compound (Fig. 1). The dynamic process is a kinetic control step dependent only upon temperature. An obvious exchange reaction induces network rearrangement above the topological temperature, leading to a 5R performance.42–45 As for dissociative CANs via dynamic isocyanate chemistry, the rate constant of the forward reaction should be larger than the reversible reaction; and then stable polymer materials can be prepared (Fig. 1).13 The crosslinking density decreases obviously with increasing temperature, leading to the destruction of polymer networks and decrease in the system viscosity, and resulting in 5R performance.46
One review summarized recent developments on dynamic covalent polymers enabled by reversible isocyanate chemistry. Different preparation approaches of dynamic covalent polymers based on reversible isocyanate chemistry were described for applications in self-healing materials, recycling, shape-memory polymers, and three-dimensional (3D) printing.47 Different from that review article, herein we focus on recent developments on CANs via dynamic isocyanate chemistry and the derived “smart” polymer materials with 5R performance.
First, we provide a brief introduction of dynamic isocyanate chemistry. Second, three main categories of dynamic isocyanate chemistry and the dynamic mechanisms behind them are discussed. Third, we focus on characterization of CANs via dynamic isocyanate chemistry by physical and chemical approaches. Fourth, we focus on novel kinds of smart polymer materials with 5R properties obtained via dynamic isocyanate chemistry. The relationship between the stress-relaxation character and 5R performance is discussed in detail in this part. The influence of different parameters on the stress-relaxation characteristics of CANs via dynamic isocyanate chemistry is discussed in this part. In addition, the advantages and disadvantages of different types of dynamic isocyanate chemistry for 5R applications are discussed. Finally, conclusions and the outlook for the development of CANs via dynamic isocyanate chemistry are provided.
2. Dynamic mechanism of isocyanate chemistry by study of models
The reactivity of the isocyanate group with a linear structure composed of two double bonds (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO) is based on the polarization induced by the high electronegativity of nitrogen and oxygen atoms. The electron-density distribution and geometry of the isocyanate group are poorly studied, but the carbon atom possesses the minimum electron density. Due to the resonance effect, the addition reaction between the isocyanate group and active nucleophilic reagent occurs readily through the nucleophilic center attacking the electrophilic carbon of isocyanate. The most important reactions involving isocyanate are addition reactions with alcohol, thiol, or amine groups (Fig. 1). Whether these addition reactions can dissociate into isocyanate and the nucleophilic reagents is dependent upon the magnitude of charge separation between the carbonyl carbon and nucleophilic-center atom. This parameter is determined by the: isocyanate structures and nucleophile agent; relative rate of forward and reversible reactions of the nucleophile agent with the isocyanate; polarity and hydrogen-bonding potential of the reaction medium; type and concentration of catalysts; environmental temperature.48 Electron-donating substituents in the nucleophilic agent will strengthen the labile bond. Electron-withdrawing substituents in the nucleophilic agent will weaken the bond, resulting in a low dissociation temperature. In general, if steric factors are neglected, any electron-withdrawing group attached on the isocyanate group will increase the positive charge on the carbon atom and enhance the reactivity.49 The degree of dynamism of this reaction is dependent upon the electronegativity and spatial hindrance of the neighboring group of X and isocyanate. Spatial hindrance weakens the bond energy of the formed product, making the addition reaction dynamic. The influence of electronegativity on the dynamic property is more complicated and is discussed in detail below. In the presence of an XH group, the dynamic exchange reaction between XH and urethane/urea/thiourethane groups also occurs (Fig. 1). Usually, extra energy must be inputted for this reaction. The content and position of the XH group in the polymer network have a big influence on this reaction. If the X1 group is more active than the X2 group, then the forward reaction also occurs, but making this reaction reversible is difficult.
C and CO) is based on the polarization induced by the high electronegativity of nitrogen and oxygen atoms. The electron-density distribution and geometry of the isocyanate group are poorly studied, but the carbon atom possesses the minimum electron density. Due to the resonance effect, the addition reaction between the isocyanate group and active nucleophilic reagent occurs readily through the nucleophilic center attacking the electrophilic carbon of isocyanate. The most important reactions involving isocyanate are addition reactions with alcohol, thiol, or amine groups (Fig. 1). Whether these addition reactions can dissociate into isocyanate and the nucleophilic reagents is dependent upon the magnitude of charge separation between the carbonyl carbon and nucleophilic-center atom. This parameter is determined by the: isocyanate structures and nucleophile agent; relative rate of forward and reversible reactions of the nucleophile agent with the isocyanate; polarity and hydrogen-bonding potential of the reaction medium; type and concentration of catalysts; environmental temperature.48 Electron-donating substituents in the nucleophilic agent will strengthen the labile bond. Electron-withdrawing substituents in the nucleophilic agent will weaken the bond, resulting in a low dissociation temperature. In general, if steric factors are neglected, any electron-withdrawing group attached on the isocyanate group will increase the positive charge on the carbon atom and enhance the reactivity.49 The degree of dynamism of this reaction is dependent upon the electronegativity and spatial hindrance of the neighboring group of X and isocyanate. Spatial hindrance weakens the bond energy of the formed product, making the addition reaction dynamic. The influence of electronegativity on the dynamic property is more complicated and is discussed in detail below. In the presence of an XH group, the dynamic exchange reaction between XH and urethane/urea/thiourethane groups also occurs (Fig. 1). Usually, extra energy must be inputted for this reaction. The content and position of the XH group in the polymer network have a big influence on this reaction. If the X1 group is more active than the X2 group, then the forward reaction also occurs, but making this reaction reversible is difficult.
2.1 Reversible chemistry between hydroxyl and isocyanate groups
Usually, the dissociation reaction of carbamate groups to isocyanates and hydroxyl groups occurs at high temperatures (>200 °C) and is accompanied by inevitable side reactions.50 The dissociation temperature of carbamate groups can be reduced markedly if a secondary alcohol with an electron-withdrawing group is used as the monomer.51 For example, the bond formed between 1,3-dichloro-2-propanol and isocyanates becomes very labile because the highly electro-negative character of the chlorine atom drains the electron density present in the oxygen atom of the blocking agent.51 Similarly, the carbamate bonds formed by the reaction between isocyanates and oximes groups are stable at room temperature, but can undergo a reversible decomposition reaction after being heated due to the strong electron-withdrawing effect of the imine bond.48,52 Oxime-carbamate groups undergo a five-membered cyclic intramolecular transition during the dissociation process (Fig. 2A).16,53,54 The degree of dissociation can be tuned readily by varying the structure of oximes and isocyanates. Introduction of electron-withdrawing groups at the alpha position of oxime groups or increasing the steric effect of isocyanate groups can reduce the decomposition temperature.48 The rate and extent of this reversible reaction is also dependent on solvents, catalysts, and temperature.55 | ||
| Fig. 2 Possible mechanism for the reversible reaction of oxime-carbamate (A), and the forward (B) and reversible (C) reaction between phenol and isocyanate with tertiary amine as a catalyst (schematic). Reproduced from ref. 56 with permission from the Royal Society of Chemistry, copyright 2016. | ||
Although phenols react more slowly with isocyanates than alcohols, the formed phenol-carbamate moieties dissociate at lower temperatures than aliphatic urethanes, which is consistent with the slower rate of the reverse reaction. Amine bases are effective catalysts for forward and reverse reactions (Fig. 2B and C).56 The dissociation temperature and rate depend on the electronic effects and acidity values of the nucleophilic agents. The phenolate anion is bonded loosely with the carbonyl group with a phenol compound possessing high acidity. The nucleophilic agent dissociates at lower temperatures with electron-withdrawing substituents such as chlorine, trifluoromethyl and ester groups, and at higher temperatures with electron-releasing substituents such as methyl and methoxy groups.56–58 Similar to phenol, urethane formed by pyridinol and isocyanate is also a type of dynamic covalent bond.59 The electron-withdrawing substituent on the ortho position reduces the electron density and nucleophilicity of the hydroxyl group. Thus, the urethane bond will be more labile and the dissociation temperature will be lower. In addition, high steric hindrance blocks the hydroxyl group to attack the NCO group, so the urethane bond becomes more labile.60
The dissociation transcarbamoylation reaction also occurs. The exchange rate is highly dependent on the catalyst category and content. The DBTDL catalyst used for the formation of carbamate bonds also promotes its dynamic exchange at high temperatures.61,62 Whether the carbamate exchange reaction undergoes an associative or dissociative process depends on the catalyst, temperature, and position of the free hydroxyl.34,63–65 Dissociative exchange dominates the carbamate bonds exchange reaction in the presence of stannous octanoate irrespective of whether a free hydroxyl group is left (Fig. 3).66 If a free hydroxyl group is involved in the transcarbamoylation reaction, the dissociative process is much slower due to the competitive coordination of the free hydroxyl group (Fig. 3A). In the absence of a free hydroxyl group, the urethane group can bind more freely to the catalyst (stannous octanoate) to induce a rapid dissociation reaction (Fig. 3B). p-Toluenesulfonic acid also catalyzes the transcarbamoylation reaction via associative exchange in the presence of a free hydroxyl group.67 If DBTDL is used as a catalyst, associative and dissociative exchange reactions occur for carbamate bonds.67 If the hydroxyl group is located at the β-position of the carbamate moiety, an obvious exchange reaction occurs at 140 °C.64 The transcarbamoylation reaction via an associative process can be activated by mechanical stress due to the twisting of nitrogen lone pairs out of conjugation with the π orbitals of the carbonyl group. Hence, whether carbamate bonds underdo associative or dissociative exchange reactions is a complicated process, and can be influenced strongly by the: nature of the isocyanate group; catalyst species and content; free hydroxyl content and position; temperature. This challenge is hindered further due to the challenging capture of isocyanate species during the dynamic process because the regenerated isocyanate species are transient at most temperatures.67
 | ||
| Fig. 3 Transcarbamoylation reaction with Sn(Oct)2 as a catalyst in the presence (A) or absence (B) of free hydroxyl groups (schematic). Reproduced from ref. 66 with permission from the American Chemical Society, copyright 2019. | ||
2.2 Reversible chemistry between amine and isocyanate groups
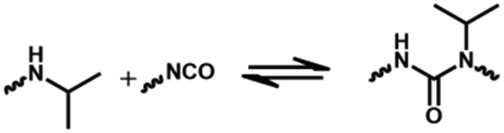
The reaction between an amine compound and isocyanate is much faster than that for an alcohol compound, and a catalyst is not needed for this reaction. The addition product of a primary amine and isocyanate is highly stable due to the resonance effect, and making it dynamic is difficult. Wang et al. found that the dissociation rate of this reaction could be accelerated in the presence of a Lewis acid.19 Density functional theory (DFT) calculations have demonstrated that the presence of a zinc ion accelerates the dissociation of urea by two orders of magnitude via the formation of O-bound Zn complexes, indicating the dynamic nature of the urea bond formed by the primary amine and isocyanate with a zinc ion as a catalyst (Fig. 4C). The reaction starts with a hydrogen transfer from one nitrogen to the other nitrogen atoms, followed by the cleavage of the C–N bond, finally leading to the formation of isocyanate and amine groups. They also found that normal urea bonds incorporated into polymer networks could be made dynamic in the absence of a catalyst due to the entropy changes of the polymer system,68 which was due to the entropic gain on debonding of the network.69 When an aromatic monomer is used for preparing normal urea, an associative dynamic process with an amine–urea exchange reaction occurs. The exchange reaction is based on an addition–fragmentation process in which a nucleophilic attack of the free amine to the carbonyl group takes place. DFT calculations have revealed that the opening reaction of urea needs more energy than the amine–urea exchange reaction.70 Meanwhile, this amine–urea exchange process is highly dependent upon regioisomerism. If an ortho-phenylenediamine is used as the monomer, this exchange reaction occurs accompanied by side reactions.71 The exchange chemistry of different diaryl urea groups occurs at 120 °C. This reaction rate can be accelerated by introducing electron-donating groups into the phenyl group.72 | ||
| Fig. 4 Possible dynamic mechanism of urea bonds (schematic). A: Urea formed by a secondary amine with a bulky substitution group. Reproduced from ref. 18 with permission from the Springer Nature, copyright 2014. B: Urea formed by pyrazole. Reproduced from ref. 74 with permission from the Springer Nature, copyright 2019. C: Urea formed by a primary aliphatic amine with zinc acetate as a catalyst. Reproduced from ref. 19 with permission from the Royal Society of Chemistry, copyright 2019. | ||
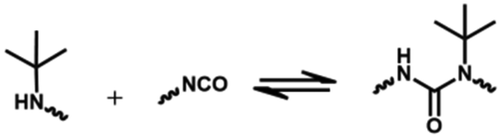
The dissociation temperature of the urea bond formed by a secondary amine is much lower than that of the primary amine due to the steric effect of the substituent,48,73 and decreases with an increase in the substituent volume.18 The reversible dissociation and reformation process of urea bonds bearing a hindered group on the nitrogen atom is shown in Fig. 4A. Dissociation of the urea bond formed by pyrazole and isocyanate is promoted by undergoing an intermediate five-membered-ring state (Fig. 4B). Theoretical calculations indicate that the pathway with nucleophilic addition is a rate-limiting step that accords with first-order kinetics. The resonance energy in pyrazole–urea bonds is lower than that of normal urea bonds formed by a primary amine, but is higher than that of hindered urea bonds. Therefore, the stability and reversibility of a pyrazole–urea moiety originates from the aromatic character of pyrazole moderately weakening the resonance stabilization and the presence of an adjacent nitrogen atom facilitating intramolecular 1,4-hydrogen transfer.74 The dissociation rate of the pyrazole–urea moiety increases if the α-carbon is substituted by groups with high steric hindrance. The kd increases from 0.00654 s−1 to 0.0457 s−1 if the neighboring group of α-carbon is changed from hydrogen to the tert-butyl group.75
2.3 Reversible chemistry between thiol and isocyanate groups
Thiourethane formed by the addition reaction between thiols and isocyanates is considered to be a type of click chemistry due to the fast reaction rate and highly efficient conversion.76 Ainara and coworkers proposed the dynamic character of the thiourethane group in a patent, but they did not explain the detailed mechanism.77 In a later study, they reported that crosslinked polythiourethane materials formed by aromatic thiol and isocyanate groups could be reprocessed, indicating the dynamic nature of thiourethane chemistry.78 Torkelson et al. used small-molecule model systems to confirm that thiourethanes could undergo exchange reactions with free thiol groups at room temperature and a dissociation reaction to regenerate thiols and isocyanates after being heated.79 Serra and coworkers proposed that the thiourethane exchange is a associative process in the presence of a catalyst, whereas associative and dissociative reactions can occur at different temperatures without a catalyst.80 Bowman and coworkers reported that the thiourethane exchange proceeds via an associative pathway involving the attack of a thiolate anion on the carbonyl carbon and regeneration of a thiocarbamate and thiolate anion in the presence of a base catalyst (Fig. 5A, route B).81 In the presence of a strong nucleophile, the thiourethane exchange reaction proceeds through a dissociative pathway which involves the attack of a nucleophile on the carbonyl group and regeneration of the thiolate anion and isocyanate (Fig. 5A, route A).81 If a nucleophilic and basic dual catalyst (DBN: 1,5-diazabicyclo[4.3.0]non-5-ene) are used, association and dissociation exchanges occur (Fig. 5B).81 | ||
| Fig. 5 Possible thiourethane exchange mechanisms (schematic). A: Exchange mechanisms occurring via nucleophilic (route A) and basic (route B) catalysis. B: Thiourethane exchange mechanism for a combined nucleophilic and basic catalyst, DBN. Reproduced from ref. 80 with permission from Elsevier, copyright 2020. | ||
2.4 Other types of dynamic chemistry with involvement of isocyanate groups
Urethane compounds with a β-OH group can decompose to amine and five-membered ring carbonates at 140 °C, which can reform into carbamate bonds at 80 °C with a tertiary amine as a catalyst.64 The reaction between the six-membered-ring carbonate and amine group can also be utilized to synthesize polyhydroxylurethane. This achieves network rearrangement through transcarbamoylation between the carbamate group and pendant hydroxyl group, but the reversible reaction for the carbamate bond dissociating into an amine and six-membered-ring carbonate is difficult.63,823. Characterization of CANs via dynamic isocyanate chemistry
In part 2, we have summarized different categories of dynamic isocyanate chemistry and clarified the dynamic mechanism behind them by a model study of small compounds. If dynamic isocyanate chemistry is incorporated into a polymer matrix to develop CANs materials, then the dynamic process is quite complicated and deserves exploration. A well-used method for characterizing the CANs structure in a polymer network is stress relaxation conducted by dynamic mechanical analysis (DMA) or rheology. Different form traditional thermosets, CANs can relax to zero during thermal processing due to the dynamic exchange reaction (Fig. 6A). A longer time is needed for a lower relaxation temperature. CANs via associative dynamic isocyanate chemistry can maintain the original crosslinking density of the network throughout the exchange process. Therefore, associative CANs undergo a gradual Arrhenius-like reduction in viscosity at high temperatures while maintaining mechanical and network integrity. Conversely, dissociative isocyanate chemistry reduces the crosslinking density with an increase of temperature due to the decomposition of dynamic moieties. However, within a limited temperature range, only a small degree of crosslinking dissociation occurs. The stress-relaxation behavior is limited by the weak bond exchange in this situation, and so also follows the Arrhenius relationship.83 Hence, the stress relaxation of associative and dissociative CANs via dynamic isocyanate chemistry would obey the Arrhenius relationship,83 which can be used for confirming if the network structure is reversible for chemically crosslinked polymer materials. | ||
| Fig. 6 A: Stress-relaxation curves commonly used to monitor the network exchange of CANs. B: Arrhenius plot of ln(τ*) against the inverse of temperature. | ||
The relaxation active energy (Ea) can be calculated from the Arrhenius plot of ln(τ*) against the inverse of temperature (Fig. 6B). Theoretically, bond-exchange reactions should be the primary contribution to Ea.84 The Ea can be affected by several aspects of the network, including polymer identity, topology, and crosslinking density. As such, CANs with the same dynamic chemistry may demonstrate different values of Ea in different systems.83 The temperature range used for the stress-relaxation experiment varies. In principle, the start temperature should be higher than the glass transition temperature (Tg) because stress relaxation can occur due to segment movement. If side reactions are absent, this temperature range should be very wide for associative CANs because the crosslinking density is well maintained during the exchange process. With respect to dissociative CANs, this temperature range should be between Tg and the gel-to-sol transition temperature (Tgel). This is because a sufficient number of crosslinking points will be broken above Tgel, which leads to thermoplastic-like segment flow and the Arrhenius law is no longer obeyed.79 Therefore, to ascertain of CANs undergo dissociative or associative isocyanate chemistry, a wide temperature range for stress-relaxation tests should be used. In principle, a linear function between relaxation time and inverse of the temperature should be obtained in a wide temperature range for CANs via associative isocyanate chemistry due to the maintained crosslinking density. Conversely, this range is relatively narrow for dissociative CANs. A further increase in the temperature will result in a gel-to-sol transition, and finally leads to the recovery of monomers or the production of homo-polymers with low molecular weight.
Investigation of evolution of the storage modulus with temperature by DMA or a rheology experiment will show a rubber plateau for associative CANs in a wide range of temperatures due to the maintained crosslinking density. The plateau modulus (G0) varies very weakly with temperature. In comparison, an obvious rubber plateau or a relatively narrow rubber state is not observed for CANs via dissociative isocyanate chemistry. In most cases, the modulus deceases with an increase in temperature due to the equilibrium shifting towards the dissociation of the product. This is because the strong increase in temperature leads to disruption of the network, which results in a decrease in crosslinking density and, finally, Tgel is obtained from the DMA curve. This obvious G0 variation with a temperature increase and generation of Tgel can be used to determine the dynamic mechanism. For example, Rowan and coworker developed a series of CANs by hindering the urea bond. The rubber state became narrower with an increase in hindrance. When the urea bond was connected with the 2,2,6,6-tetramethyl-4-piperidinol group, almost no rubber state was observed, which confirmed the dissociation mechanism.85
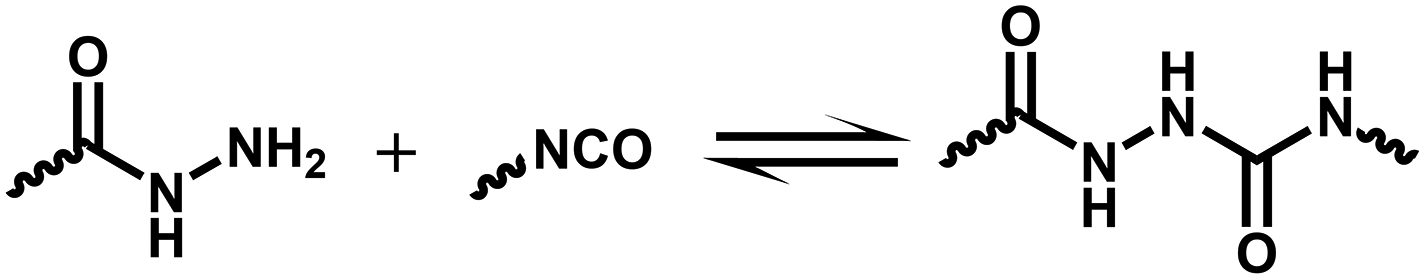
Similar to stress relaxation, creep experiments can also determine if the dynamic crosslinked network structure is associative or dissociative. Creep experiments lead directly to obtaining the shear viscosity, which can be used to calculate the viscous flow energy. As expected from networks maintaining a constant crosslinking density, comparable values for viscous-flow activation energy (Ea) and kinetic Ea can be obtained for associative CANs via dynamic isocyanate chemistry. In theory, in the plateau temperature range, a linear relationship can be obtained between shear viscosity and inverse of the temperature. For dissociative systems, this situation varies differently due to the change in crosslinking density. Montarnal et al. proposed that dissociative CANs display four distinct viscoelastic regimes.86 The crosslinking density decreases dramatically between Tg and Tgel, and the shear viscosity exhibits a stronger temperature dependence. Although a linear function between the shear viscosity and inverse of the temperature has also been reported, the temperature range was very narrow. A comparable Ea from stress-relaxation and creep experiments has been reported for dissociative CANs, but the temperature range varied differently. Therefore, conducting a creep experiment in a wide temperature range is essential to judge if a CAN undergoes an associative or dissociative process. For example, a CAN containing reversible acylsemicarbazide moieties displays almost the same stress-relaxation Ea (100.0 kJ mol−1) and viscous-flow Ea (99.6 kJ mol−1), but it dissociates with the temperature increase, which has been verified by the in situ IR analysis.87
Another common approach for characterizing the dynamic mechanism is through spectroscopy. The most popular form spectroscopy is in situ FTIR. The obvious signal change at 2250–2270 cm−1 attributed to the isocyanate group in IR spectra can show whether CANs undergo a dissociative process. Obtaining the threshold temperature for the dissociation of urethane, urea, or thiourethane groups is difficult because a small part of the NCO group in the IR spectra cannot be captured readily.
4. CANs with 5R performance via dynamic isocyanate chemistry
Many kinds of dynamic isocyanate chemistry have been employed to develop CANs with excellent 5R performance (Fig. 1): reversible addition of hydroxyl and isocyanate groups; exchange reaction between hydroxyl and urethane groups; carbamate exchange;133–135 reversible dissociation–reformation of urea groups;18 exchange reaction between amine and urea groups; exchange reaction between thiol and thiourethane groups; thioureathane exchange reactions.130,136–138 As discussed in parts 2 and 3, the dynamic mechanism can be verified by model study of a small compound or by rheology study of CANs materials.In this part, we discuss how these different categories of dynamic isocyanate chemistry affect the 5R performance through varying the polymer identity, catalyst, free hydroxyl group, and relaxation temperature. Also, because stress relaxation is used to study the network arrangement of CANs, we also discuss the correlation between stress relaxation and the 5R performance.
4.1 Repairable performance
Tables 1 and 2 show various repairable polymer materials based on different categories of dynamic isocyanate chemistry. The first repairable polymer materials via dynamic isocyanate chemistry were reported by Cheng and coworkers in 2004 based on hindered urea bonds.18 A secondary amine monomer with tert-butyl as a substituent was used to synthesize poly(urethane–urea), which displayed an excellent self-healing property at 37 °C (Fig. 7A and B). The tert-butyl group weakened the urea bond due to steric hindrance, which made the formed urea dynamic at a slightly elevated temperature, leading to self-healing. Similarly, Hager and coworker synthesized poly(methacrylate)-based thermosets containing sterically hindered urea leakages. The resultant materials could heal scratches at temperatures above 70 °C multiple times, and their mechanical properties can be partially regenerated.139 Cheong and coworkers reported a type of polyacrylate-based crosslinked network bearing reversible bulky urea bonds and displaying fully scratch healing in air for 24 h or in water for 1 h.140 These examples clearly show that polymer materials with repairable performance at mild conditions (or even at room temperature) can be prepared by hindering urea bonds. | ||
| Fig. 7 A and B: Schematic illustration and optical images showing of the self-healing mechanism for polyurea containing tert-butyl as a hindered group. B: Preparation and reprocessing of crosslinked polyurea containing tert-butyl as a hindered group (schematic). Reproduced from ref. 18 with permission from Springer Nature, copyright 2014. C: Schematic illustration of the dynamic mechanism of the polyurea formed by a primary amine and isocyanate. Optical images showing the damged (D1–D2) and repaired (D3–D4) polyurea formed by a primary amine and isocyanate. Reproduced from ref. 68 with permission from the Royal Society of Chemistry, copyright 2020. | ||
| Dynamic reaction | Temperature/°C | E a/kJ mol−1 | Conditions | 5R performance | Ref. |
|---|---|---|---|---|---|

|
110–150 | 114 | 130 °C 40 min | Reconfiguration | 61 |

|
140–160 | 143–188 | 160 °C 8 MPa 12 min | Reprocessable | 88 |

|
160–200 | 140 | 160 °C, 6 MPa 0.5 h | Reprocessable | 89 |

|
120–160 | 110 | 150 °C 60 min | Reconfiguration | 90 |

|
110–150 | 139–165 | — | — | 66 |

|
170–190 | 109–196 | 160 °C 30 MPa 4 h | Reprocessable | 91 |
| 160 °C 30 MPa 1 h | Repairable (with pressure) | ||||

|
110–140 | 143 | 160 °C 2.9 MPa 12 min | Reprocessable | 92 |

|
110–140 | 146 | 160 °C 3.5 MPa 12 min | Reprocessable | 92 |

|
110–140 | 146 | 160 °C 3.5 MPa 12 min | Reprocessable | 92 |

|
140–170 | 184 | 160 °C 40 min | Reconfiguration | 93 |

|
170–190 | 99–134 | 160 °C 4 MPa 4 min annealed 225–580 min | Reprocessable | 82 |

|
140–170 | 121 | — | — | 94 |

|
140–160 | 91–127 | 160 °C 4 MPa | Reprocessable | 95 |

|
150–180 | 86–105 | 200 °C 10 MPa 3 h | Reprocessable and recyclable | 96 |

|
140–170 | 85–131 | 140 °C 10 MPa 2 h | Reprocessable | 97 |

|
150–165 | 82–135 | 150 °C MPa 3 h | Reprocessable and reconfiguration | 98 |
| 150 °C 4 h | |||||

|
110–170 | 92 | 130 °C 10 MPa 10 min | Reprocessable | 99 |

|
150–165 | 77 | 150 °C 10 MPa 6 h | Reprocessable | 100 |
| 150 °C 5 h | Reconfiguration | ||||

|
60–120 | 55 | 100 °C, 130 °C 5–10 MPa 2 h, | Repairable, reprocessable and recyclable | 101 |

|
160 | — | 160 °C 0.2 MPa | Reprocessable | 102 |
| 160 °C 3 h | Reconfiguration | ||||

|
120–160 | 135 | 140 °C 11 MPa 2 h | Reprocessable | 103 |

|
110–115 | 118 | 130 °C 1.5–2 Mt 30 min | Reprocessable | 104 |

|
80–120 | 124 | 110 °C 10 MPa 30 min, 110 °C 30 min | Reprocessable and repairable (scratch healing) | 17 |

|
80–130 | 98–110 | 120 °C 10 MPa 30 min, 120 °C 10 min | Reprocessable and repairable | 16 |

|
120–160 | 145 | 130 °C 5 MPa 1 h, 80 °C 15 h | Reprocessable and repairable | 105 |

|
80–90 | 105 | 120 °C 5 MPa 10 min | Reprocessable | 106 |

|
140–150 | 183 | 140 °C 5 MPa 10 min | Reprocessable | 106 |

|
130–170 | 110 | 100 °C 5 MPa 30 min, 80 °C 10 s | Reprocessable and reconfiguration | 107 |

|
170–190 | 60–117 | 140 °C 4 MPa 40 min, 808 nm NIR laser | Reconfiguration and re-adhesive | 108 |

|
40–80 | 93 | 100 °C 5 MPa 1 min, 80 °C 120 min | Reprocessable and repairable | 109 |

|
180–240 | 66–107 | 130 °C 5 MPa 1 h, 140 °C 0.5 h | Reprocessable and repairable | 110 |

|
70–120 | 100–120 | 130 °C 15 MPa 20 min | Reprocessable | 111 |

|
60–100 | 60–128 | 100 °C 10 MPa 10 min, 100 °C, 2 h | Reprocessable and repairable | 112 |

|
70–120 | 101 | 90 °C−140 °C 5 min 5 MPa | Reprocessable | 113 |
| Annealed 40 °C for 12 h | |||||

|
70–100 | 52 | 90 °C 24 h | Repairable | 19 |

|
100–140 | 29 | NIR 5 min | Reprocessable and repairable | 68 |

|
125–145 | 120 | 130 °C 1 h 20 MPa | Reprocessable and repairable (with solvent) | 114 |
|
140–160 | 100 | 140 °C 15 MPa 1 h | Reprocessable | 87 |
| 120 °C 1 h DMF | Repairable (with solvent) | ||||

|
90–130 | 109 | 130 °C 10 MPa 30 min | Reprocessable | 74 |

|
90–130 | 81 | 120 °C 10 MPa 30 min | Reprocessable and repairable | 75 |

|
110–140 | 80 | 120 °C 10 MPa 30 min | Reprocessable and repairable | 75 |

|
120–160 | 57 | 120 °C 10 MPa 30 min | Reprocessable and repairable | 75 |

|
130–155 | 158 | 150 °C 5 MPa 2 h | Reprocessable and repairable | 115 |

|
90–110 | 182 | 150 °C 5 MPa 2 h | Reprocessable and repairable (partial scratch healing) | 115 |

|
140–165 | 66 | 150 °C 5 MPa 2 h | Reprocessable and repairable (partial scratch healing) | 115 |

|
90–110 | 85 | 150 °C 5 MPa 2 h | Reprocessable and repairable (partial scratch healing) | 115 |
|
120–160 | 91–98 | 130 °C 0.5 MPa 5 min | Reprocessable and repairable | 116 |
| 130 °C 0.5 MPa 10 min | |||||

|
120–150 | 101 | 150 °C 10 min | Reconfiguration | 117 |

|
130–160 | 74 | 80 °C 16 MPa 1 h | Reprocessable | 118 |

|
35–65 | 2.5–46 | RT 200 kPa, 35 °C 24 h | Reprocessable and repairable | 119 |

|
70–100 | 119 | 80 °C 60 min | Repairable, reprocessable | 120 |
| 60 °C 10 MPa 10 min | Re-adhesive | ||||
| 80 °C 60 min | |||||

|
90–130 | 92 | 110 °C 12 h–48 h, 110 °C 15 MPa 1 h | Repairable and reprocessable | 121 |

|
70–120 | 52–69 | 80 °C 3 MPa 60 min, 40 °C 12 h | Reprocessable, repairable and reconfiguration | 122 |
|
80–170 | 109–113 | 120 °C 4 tons 25 min | Reprocessable | 123 |

|
110–180 | 59 | — | — | 85 |

|
110–180 | 130 | — | — | 85 |

|
60–100 | 132 | — | — | 85 |

|
130–170 | 105–128 | 170 °C 20 MPa 60 min, 40 °C 12 h | Reprocessable, re-adhesive | 124 |

|
130–170 | 104 | 180 °C 25 min | Reprocessable | 125 |

|
130–170 | 115 | 180 °C 25–40 min | Reprocessable | 125 |

|
130–170 | 123 | 180 °C 40 min | Reprocessable | 125 |

|
110–150 | 85 | 120 °C 5 MPa 30 min, 130 °C 30 min | Reprocessable and reconfiguration | 126 |

|
110–180 | 34 | 150 °C 300 kPa 2 h, 130 °C 30 min | Reprocessable, repairable | 127 |

|
100–130 | 71–229 | 100 °C 12 MPa 120 min | Reprocessable | 72 |

|
110–130 | 121 | 120 °C 10 MPa 30 min | Reprocessable, repairable | 128 |
| 120 °C 60 min | Reconfiguration | ||||
| 120 °C 30 min | |||||

|
140–170 | 96–125 | 150 °C 10 MPa 40 min | Reprocessable | 129 |

|
130–200 | 138 | 150 °C 100 bar 1 h | Reprocessable | 70 |

|
140–200 | 122–147 | 160 °C 100 bar 1 h | Reprocessable | 71 |

|
120–180 | 119 | 140 °C 100 bar 1 h | Reprocessable and repairable (scratch healing) | 78 |

|
150–180 | 75 | 80 °C 3 MPa 30 min | Reprocessable | 130 |
| 100 °C 3 MPa 30 min | Re-adhesive | ||||
| 100 °C 3 MPa 30 min | Repairable (with pressure) | ||||

|
160–185 | 72–102 | 180 °C 40 min | Reconfiguration | 131 |

|
160–185 | 186 | 165 °C 8 MPa 2.5 h | Reprocessable | 80 |

|
160–185 | 91–107 | 165 °C 8 MPa 2.5 h | Reprocessable | 80 |

|
170–190 | 137–192 | — | — | 132 |
| Dynamic motif | Mechanical properties | Healing conditions | Healing efficiency/% | Ref. | ||
|---|---|---|---|---|---|---|
| Tensile stress/MPa | Break strain/% | Modulus/MPa | ||||
| HDI: hexamethylene diisocyanate, IPDI: isophorone diisocyanate, RT: room temperature, DMF: N,N-dimethylformamide, NIR: near infrared, DOU: dimethylglyoxime–urethane. | ||||||
| Hindered urea bonds | 0.9 | 300 | 1 | 37 °C, 12 h | 87 | 18 |
| Hindered urea bonds | HDI 8 | 145 | — | 60 °C 4 d | 89 | 140 |
| IPDI 9 | 105 | — | 60 °C 4 d | 87 | ||
| Normal urea bonds | HDI 0.36 | 330 | — | 90 °C 24 h | 99 | 19 |
| IPDI 7 | 650 | — | 110 °C 16 h | 90 | ||
| Hindered urea bonds | 3.8 | 570 | — | 60 °C 0.5 h | 92 | 145 |
| Hindered urea bonds | 17 | — | 600 | 140 °C 6 d | 40 | 146 |
| Hindered urea bonds | 14 | 140 | 21.7 | 75 °C 7 d | 82 | 147 |
| Hindered urea bonds | 8 | 2200 | 27 | RT | 62 | 148 |
| Acylsemicarbazide bonds | 22.8 | 285 | 77.8 | 75 °C 7 d | 57 | 65 |
| Hindered urea bonds | 2.31 | 529 | 1.52 | 40 °C 12 h | 97 | 122 |
| Normal urea bonds | 5 | 820 | — | NIR 5 min | 90 | 68 |
| Hindered urea bonds | 24 | 650 | — | 100 °C 0.5 h | 95 | 149 |
| Hindered urea bonds | 5.9 | 997 | — | 70 °C 5 h | 99 | 150 |
| Hindered urea bonds | >90 | 3–4 | 1006 | 70 °C 1 h | >95 | 151 |
| Hindered urea bonds | >7 | >2600 | — | 90 °C 10 h | 96.5 | 152 |
| Hindered urea bonds | 5.3 | 500 | — | 80 °C 6 d | 100 | 153 |
| Hindered urea bonds | — | — | — | 75 °C 24 h | 100 | 154 |
| Hindered urea bonds | 5.4 | 200 | — | 120 °C 0.5 h | 94 | 155 |
| Hindered urea bonds | 7.8 | 600 | — | 50 °C 24 h | 95 | 156 |
| Hindered urea bonds | 17.3 | — | 340 | 100 °C 72 h | 50 | 139 |
| Hindered urea bonds | 10 | 150 | — | 60 °C 96 h | 75–95 | 140 |
| Hindered urea bonds | 39.5 | 3.2 | 1900 | 100 °C 0.5 h 30 kPa | 95 | 157 |
| Phenol-carbamate bonds | 14 | 550 | — | 140 °C 0.5 h | 96.5 | 158 |
| Phenol-carbamate bonds | 10.4 | — | 250 | 150 °C 48 h | 85 | 58 |
| Pyrazole urea bonds | 3.5 | 185 | 3.1 | 120 °C 1 h | 99 | 75 |
| Phenol-carbamate bonds | 2.8 | 240 | — | 80 °C 2 h | 98 | 109 |
| Phenol-carbamate bonds | 52 | 12 | — | 110 °C 0.5 h | >95 | 159 |
| Phenol-carbamate bonds | 11 | 600 | — | 150 °C 8 h | 68 | 160 |
| Phenol-carbamate bonds | 5.2 | 510 | — | 120 °C 8 h | 86 | 143 |
| Acylsemicarbazide bonds | 69 | 181 | 1740 | 140 °C DMF 1 h | 99.7 | 87 |
| Acylsemicarbazide bonds | 100 | 5 | 2840 | 120 °C 1 h | 94.4 | 114 |
| Thiourethane bonds | 62.7 | <0.1 | 2000 | 120 °C 3 MPa 1 h | 99 | 130 |
| Phenol-carbamate bonds | 46.4 | 615 | — | 100 °C 2 h | 93 | 161 |
| Oxime-carbamate bonds | 5.38 | 620 | — | 90 °C 1.5 h | 94 | 162 |
| Oxime-carbamate bonds | 13.5 | 812 | 10.9 | 100 °C 2 h | 99 | 17 |
| Oxime-carbamate bonds | 7.43 | 677 | 4.79 | RT 75 min | 94 | 141 |
| Hydroxyl-urethane exchange | 25.6 | 513 | 13.2 | 110 °C 0.5 h | 92 | 163 |
Most of healable polymer materials can repair only micro/nano-scratches; achieving repair of macro-scratches has rarely been achieved. Wang and coworkers developed a type of dynamic crosslinked PUR–polydimethylsiloxane elastomer in which polydopamine nanoparticles were introduced to induce rapid photothermal transition. The obtained composite elastomers exhibited a rapid solid–liquid transition under NIR exposure because of the fast dissociation rate of the high-density dynamic urea bonds present in the polymer network, and so demonstrated excellent macro-damage repair. A macro-size mechanical scratch (width: 1 mm; depth: 0.25 mm) can be repaired under NIR exposure for 5 min (Fig. 7C and D).68 High energy or catalysis is required to induce decomposition of the primary aliphatic urea bond to achieve repair, but recovery of the mechanical property can be obtained in minutes due to the rapid reformation of the urea linkage from the cleaved amine and isocyanate compound. The temperature generated by photothermal transition during repair far surpasses the temperature range (90–120 °C) used to determine the stress-relaxation Ea, which results in very fast segment relaxation, and leads to flowing of segments to achieve macro-damage repair.
The first typical self-healing PUs via dynamic isocyanate chemistry were developed by Xu and coworkers. They developed a class of poly(oxime-urethanes) prepared from the addition of oxime compounds and diisocyanate at ambient temperature.16 The oxime-carbamate structures could be made after being heated through oxime-enabled transcarbamoylation via a thermally dissociative mechanism, enabling efficient repairable performance. Xia and coworkers extended this dynamic reaction to prepare a self-healing PU elastomer.17 The poly(oxime-urethane) with a crosslink density of 0.2 mmol cm−3 possessed a tensile strength of 13.5 MPa, a breaking strain of 812%, toughness up to 40 MJ m−3, an excellent elastic recovery of 90%, and an average optical transmittance of 86% in the visible range. This material could be healed completely at 110 °C in 0.5 h. The healing temperature used for determining the Ea was 80–120 °C, which demonstrated that the reversible dissociation of the oxime-urethane bond was mainly responsible for the repair process. In situ structural characterizations revealed that the repairable properties originated from the reversibility of oxime-carbamate bonds and hydrogen bonds. You and coworkers reduced the repair temperature to room temperature by introducing a copper catalyst. The latter helped physical crosslinking through metal coordination and weakening the bond strength of oxime-urethane moieties. The as-developed copper-based PU elastomer displayed a tensile strength and toughness of 14.8 MPa and 87.0 MJ m−3, respectively, and could self-heal at room temperature due to the accelerated dissociation rate of the oxime-carbamate bond from the coordination of copper.141 Silica nanoparticles can be incorporated into a poly(oxime-urethane) elastomer to develop damage-tolerant self-healing composites.142 The examples stated above demonstrate that the dissociation reaction of oxime-urethane moieties can be finely adjusted by changing the segment flexibility or introducing metal coordination. Also, the derived reparable polymers can be thermosets or elastomers with a repair temperature spanning from room temperature to >100 °C. Huang and coworkers reported a thermal self-healing PU thermoset based on a dynamic phenolic urethane reaction.143 The phenolic urethane partially decomposed into isocyanates and phenolic hydroxyls above 120 °C and could reform into the phenolic urethane group after cooling down. These properties contributed to the thermal self-repair of the thermosetting PU network. The repaired PU thermoset recovered ∼70% of its tensile strength and 86% of its elongation at break. Different from the urea bond or oxime-urethane bond, the rate of the addition reaction of the phenol group with an isocyanate compound was relatively slow, which resulted in a longer self-healing process and sometimes in sufficient recovery of the mechanical property.
The temperature of self-healing spans from ambient temperature to 150 °C depending on the different categories of dynamic chemistry. Most of the reported self-healing systems can achieve >90% healing with sufficient energy input. Efforts should focus on how to obtain efficient self-healing with minimal energy input (including at a low temperature for a short time). Combination of metal coordination with dynamic isocyanate chemistry has been proposed to weaken the bond strength, leading to a lower self-healing temperature. Among all self-healing materials reported so far, the highest mechanical strength and Young's modulus can reach 100 MPa and 2.8 GPa, respectively. Usually, extra pressure is required to assist the healing process in these systems. NIR or UV irradiation and solvent assistance also facilitates the repair process. Although dynamic isocyanate chemistry is responsible for the healing process, other chemical events that occur during the damage–repair cycle should also be considered. For example, the hydrogen bonding formed among urethane/urea/thiourethane groups contribute significantly to the self-healing process. In addition, polymer self-healing is a complicated process; many chemical and physical events occur apart from the recombination of dynamic covalent bonds. In crosslinked PU networks, damage-induced formation of primary amines can attack urethane bonds if zinc acetate is the catalyst, which would lead to self-healing.65 Damage to part of PU networks containing methyl-α-D-glucopyranoside can lead to reactions with atmospheric CO2 and H2O, thus reforming covalent linkages to repair the cleaved network segments and resulting in self-healing.144 For materials with a designed architecture, energy input and the healing rate should be balanced to achieve self-healing while maintaining the geometry. For example, Sun et al. realized the repair of a 3D-printed sole without damaging the porous structure (Fig. 11C and D).
Tables 1 and 2 suggest that only dissociative chemistry via carbamate or urea moieties (including phenol-carbamate bonds, oxime-carbamate bonds, hindered urea bonds, and metal-catalyzed normal aliphatic urea bonds) has been employed to prepare repairable materials. This is because the dissociation of the dynamic moiety accelerates movement of the segment, thereby facilitating repairable processes. Most of these systems exhibit a stress-relaxation Ea <120 kJ mol−1 or <100 kJ mol−1 (Table 1). The repairable efficiency and rate depends heavily on the equilibrium constant of isocyanate chemistry. An increase in temperature will shift the equilibrium to the decomposition of carbamate or urea moieties, which enables disruption of the network and rapid movement of the segment, thereby filling the damage to realize repair. Rapid reformation of cleaved carbamate or urea moieties should also be considered to fix the shape of the damaged area and recover the mechanical performance, otherwise fast flowing of the segment may result in the loss of materials. If dynamic chemistry via a primary aliphatic urea bond is utilized to develop repairable materials, then fast and highly efficient repair can be achieved. On the contrary, the phenol-carbamate bond dissociates at a lower temperature than the primary aliphatic urea bond, but the longer repair time is due to the its slower rate of combination between phenol and isocyanate. Therefore, the forward and reverse reaction rates of dynamic isocyanate chemistry should be considered when designing “ideal” repairable materials. Compared with dissociative dynamic isocyanate chemistry, the associative dynamic chemistry between hydroxyl and carbamate groups or exchange reaction between carbamate groups to develop repairable materials have rarely been reported. Usually, the stress-relaxation Ea of these two reactions is >100 kJ mol−1 (Table 1), which produces relatively slow movement of the segment, thereby blocking the repair process. This phenomenon explains why repairable performance has been realized via the dissociative primary aliphatic urea bond, whereas reparable materials have not been obtained via the primary aromatic urea bond because it undergoes an associative process.
4.2 Reprocessable performance
Traditional thermoset polymer materials cannot be reprocessed readily due to their permanent crosslinked network structure. This challenge can be overcome by incorporation of dynamic covalent bonds into polymer networks. For example, thermoset PUR prepared through the reaction between 2-(tert-butylamino) ethanol and a trifunctional homopolymer of hexamethylene diisocyanate can be reprocessed by hot pressing (Fig. 8A) due to the dynamic exchange of hindered urea bonds. An obvious decrease in mechanical performance was not observed ever after five reprocessing cycles.157 Xu and colleagues developed a type of dynamic covalent PUR prepared by the addition reaction of pyrazoles and diisocyanates,74 which could be reprocessed upon heating. Fu et al. found that acylsemicarbazide (ASC) moieties can dissociate reversibly into isocyanate and hydrazide at high temperatures, thereby exhibiting dynamic reversibility. A transparent PASC possessing a Young's modulus of 1.74 GPa, yield strength of 63.6 MPa, tensile strength of 69 MPa, strain at break of 181%, and toughness of 98 MJ m−3 could be reprocessed four times.87 This excellent mechanical performance is due to the disordered multiple hydrogen bonds formed by ASC moieties. PASC can maintain its superior optical transparency, modulus, and yield strength during reprocessing (Fig. 8C and D), which can be attributed to the side reaction of regenerated isocyanates at high temperature. Rekondo et al. reported a reprocessable crosslinked poly(urea–urethane) CAN containing aromatic urea groups with free amines “dangling” in the network. Model studies of small-molecular compounds by nuclear magnetic resonance (NMR) spectroscopy and DFT calculations confirmed that the exchange reaction between the aromatic amine and urea was responsible for these reprocessable properties.70 The contribution of the hydrogen bond to the mechanical performance and remolding process is important for reprocessing PUR. Strong hydrogen bonding formed, for example by ASC groups, can improve the mechanical performance, but extra energy is needed to achieve reprocessing. Metal coordination with the urea bond can induce further physical crosslinking and lead to improvement of mechanical performance. Metal coordination can weaken the bonding strength of urea bonds, leading to an improved dynamic property. Hence, incorporation of metal coordination can have a dual role: collaborative improvement of reprocessable and mechanical performances. | ||
| Fig. 8 Optical images showing the reprocessing process of a polyurea containing tert-butyl as a hindered group (A) and acylsemicarbazide group. Reproduced from ref. 157 with permission from John Wiley and Sons, copyright 2016. (B) Optical (C) and mechanical (D) properties of a polyacylsemicarbazide after being reprocessed four times. Reproduced from ref. 87 with permission from the American Chemical Society, copyright 2020. | ||
Fortman et al. reported a class of polyhydroxyurethane (PHU) CANs derived from the attack of amines towards six-membered cyclic carbonates.63 PHU networks can be reprocessed at 160 °C at a pressure of 4 MPa for 8 h in the absence of an external catalyst, and recover 75% of their as-synthesized values after reprocessing. A stress relaxation occurs through an associative process which undergoes nucleophilic addition of free hydroxyl groups to carbamate groups. DFT calculations revealed that the transcarbamoylation could be attributed to the twisting of nitrogen lone pairs out of conjugation with the π orbitals of the carbonyl group. In a subsequent study, they systematically investigated the reprocessability of crosslinked PHUs synthesized from five- or six-membered cyclic carbonates.82 They concluded that the higher thermodynamic stability of five-membered cyclic carbonates led to reversion and subsequent decomposition of PHUs at high temperatures, but this decomposition was not observed in networks derived from six-membered cyclic carbonates. Torkelson and coworkers reported another type of reprocessable PHU CAN obtained from five-membered cyclic carbonates, which displayed full recovery of properties due to concurrent associative and dissociative dynamic chemistry via transcarbamoylation and reversible cyclic carbonate aminolysis.64 The cyclic carbonate contents in the network increased after each reprocessing confirmed the reverse reaction of aminolysis under reprocessing conditions, which indicated that the de-crosslinking caused by reverse cyclic carbonate aminolysis could be fully reversed. Reprocessing took 2 h at 140 °C to complete. The examples given above demonstrate that the reprocessing of PHUs via the exchange reaction of hydroxyl and urethane groups require a high temperature and long time to achieve recovery of mechanical performance. This harsh processing condition may cause side reactions and limits the application of PHUs in daily life.
Thermal pressing is the widely used method for the characterization of CANs reprocessing. Injection and extrusion are popular processing techniques for making polymer parts, and deserve to be studied for CANs reprocessing. For example, Sheppard et al. realized the reprocessing of PU foam into bulk materials by introducing a catalyst (DBTDL) through twin-screw extrusion via a carbamate exchange reaction.88 The relaxation time of the reprocessed PU films was 28 s at 160 °C, which demonstrated very fast exchange reactions among carbamate groups to be responsible for this reprocessing. Whether the amount of metal catalyst needed accords with real applications warrants investigation.
Table 1 suggests that dissociative and associative dynamic isocyanate chemistry can be employed to develop reprocessable materials. External pressure can boost the melt fusion and bonding together via dynamic isocyanate chemistry. One must balance the reprocessing temperature and degree of side reactions. Increasing the temperature can accelerate the rate of bond exchange whether the dynamic isocyanate chemistry undergoes a dissociative or associative process, thereby facilitating reprocessing. The bond exchange reaction for associative isocyanate chemistry is a kinetic process. The reprocessing temperature should be higher than the topology freezing transition temperature for associative isocyanate chemistry. For slower-exchange isocyanate chemistry, a catalyst is needed to achieve excellent reprocessing. For example, DBTDL and Fe(acac)3 have been used as catalysts to realize the reprocessing of normal PU through the bond exchange reaction between carbamate groups. Dissociative isocyanate chemistry can induce gel-to-sol transition through breaking the network into monomers or oligomers, strongly reducing the melt viscosity. Therefore, reprocessing can be realized without shear force. For example, shear-free reprocessing can be obtained for PUR formed by primary aliphatic amine and aliphatic isocyanate compounds.
Stress relaxation has a major influence on reprocessing. For example, thermoset PU elastomers with 1 wt% DBTDL as a catalyst displaying an Ea of 173 kJ mol−1 can be hot-pressed at 160 °C and 30 MPa for 4 h to achieve a good reprocessing performance.91 Similarly, another type of PU exhibiting an Ea of 140 kJ mol−1 (ref. 89) and 146 kJ mol−1 (ref. 92) needs to be reprocessed at 160 °C and 6 MPa for 0.5 h and at 160 °C and 5–10 MPa for 12 min, respectively. Increasing the content of soft segments72 or the chain length between the crosslinking points,115 or the dynamic moiety content110 can accelerate the relaxation process via the fast bond exchange reaction, which reduces Ea and facilitates reprocessing.
4.3 Reconfiguration performance
The elasticity and plasticity of a material are often inversely related. An elastic material displays low plasticity, and vice versa.164 Macroscopic deformation corresponding to changes in chain conformation can be generated if chemically crosslinked polymers are subjected to an external force, which results in entropy reduction. They would recover back after the force is released if the polymer is an elastomer or the materials possess shape-memory behavior. This entropy-driven shape recovery is commonly referred to as “elasticity”. If CANs are activated under certain conditions, deformation under an external force leads to topological rearrangement of the network with no entropy gain during macroscopic deformation. In essence, the network is permanently deformed from the standpoint of thermodynamics. This is defined as “plasticity” as opposed to elasticity.Xie and coworkers found that the stress relaxation induced by the DBTDL-catalyzed carbamate exchange reaction enabled the intrinsic plasticity of classical thermoset shape-memory PU.61 The Ea determined from 110–150 °C increased from 113.6 to 130.5 kJ mol−1 by varying the amount of aliphatic isocyanate to aromatic isocyanate, indicating slower stress relaxation for aromatic PUs. Incorporating two types of dynamic moieties which display an obvious exchange reaction at different temperatures can tune the stress-relaxation behavior. Without DBTDL, the exchange chemistry for carbonates is very slow, which results in slow stress relaxation and a high Ea (184 kJ mol−1).93 Efficient reconfiguration also needs high energy input to be realized (170 °C). For example, a poly(urethane–urea) copolymer was synthesized by reacting HDI with a mixture of a diol, a hindered amine, and a triol crosslinker, with DBTDL as the catalyst.165 Urethane bond exchange and hindered urea bond exchange occurred at different temperature ranges. Varying their bond ratio could lead to great flexibility in tuning the topological rearrangement kinetics of the network to allow reprocessing and plasticity to be achieved in a wide temperature range. Increase in the ratio between hindered urea and urethane bonds resulted in faster stress relaxation. The dynamic networks exhibited excellent self-healing, reprocessability, and solid-state plasticity with an optimized ratio between hindered urea and urethane bonds.
Jin et al. designed a dynamic polymer network that responded to light and heat.166 Thermally induced transesterification and transcarbamoylation triggered the topological rearrangement of the network. The resulting solid-state plasticity allowed permanent shape reconfiguration by manual folding (e.g., from 2D sheet to 3D airplane) followed by stress relaxation at 140 °C. As shown in Fig. 9, flowers, airplanes, and elephants were heated (80 °C) and cooled to achieve a reversible shape change of a specific part. For photo-reversibility, at the same temperature (80 °C) and with the pre-stretching force imposed, subsequent exposure to light of 312 nm resulted in cinnamate dimerization, which partially fixed the alignment even after the force had been removed. Because of this partial alignment (or network anisotropy), the sample exhibited actuation behavior via a reversible shape-memory mechanism.
 | ||
| Fig. 9 Reconfigured polymers based on dynamic covalent bonding. (A) Thermally induced transesterification, transcarbamoylation, hindered urea bond exchange, and photo-reversible dimerization of nitro-cinnamate. (B) Thermally triggered reversible deformation behavior of 3D flowers. (C) Photograph and thermally triggered reversible shape change of crane and elephant. Reproduced from ref. 166 with permission from the American Association for the Advancement of Science, copyright 2018. (D) Reversible construction of complex shapes with thermal drive. Reproduced from ref. 61 with permission from John Wiley and Sons, copyright 2016. | ||
In principle, dissociative and associative isocyanate chemistry can be employed to construct reconfigurable polymer materials. Obvious bond exchange should occur during the reconfiguration process. For CANs via dissociative isocyanate chemistry, determination of the threshold temperature for reconfiguration is difficult. Normally, the temperature range (Table 1) used for determining the stress-relaxation energy could be a good choice to reshape materials. Within this range, a higher temperature requires a shorter time to fully relax the stress and finish the reshaping process. Cheng and coworkers developed a reconfigurable poly(urea–urethane) thermoset based on hindered urea bonds.117 An Ea of 101.4 kJ mol−1 was obtained at 120–150 °C (Table 1). Full reconfiguration was achieved at 150 °C for 5 min to fully relax the stress. The plasticity temperature should be lower than the Tgel for dissociative CANs, otherwise flowing hampers shape fixing. For CANs via dissociative isocyanate chemistry, reconfiguration occurs above the Tv. Introducing a catalyst into CANs enables tuning of the kinetics of the bond exchange reaction, which can govern the plasticity process. A plasticity time longer than the full relaxation time is required to realize complete reconfiguration. Otherwise partial reshaping or the shape-memory effect will occur. Hence, all the systems listed in Table 1 can be reconfigured at the temperature range used for calculating the Ea. The difference lies in the reshaping time and deformation degree, which can be adjusted by changing the crosslinking density, the ratio between soft and rigid segments, and dynamic moiety content.
4.4 Recyclable performance
To some extent, there is an overlap on the definitions of reprocessable and recyclable. Many chemically crosslinked polymer materials can be reprocessed due to the incorporation of a dynamic linkage in the network, which is also named as “recyclable” polymer materials. Herein, recyclable systems mean that polymer materials can be partially or fully degraded into their original monomers, and the regenerated monomers can be isolated and reused to prepare new materials. Different from other 4R performances, dynamic isocyanate chemistry for achieving recycling occurs in solution. The stress-relaxation behavior listed in Table 1 shows no direct relationship with the recycling process. Regardless of dissociative or associative isocyanate chemistry, the premise of achieving recycling is to degrade the network with an external attacking reagent, including alcohol and amine compounds or water molecules. The attacking of alcohol or amine compounds to carbamate or urea moieties shifts the equilibrium to the dissociative state, resulting in degradation of the network. In this way, the monomer can be recycled from PU and PUR waste. Unfortunately, it is difficult to isolate recycled monomers from the solution because the original monomer and attacking reagent have similar properties. Therefore, the difference in solubility of the generated amine and recycled monomers must be used to isolate the generated monomer from solution. For example, Liao and coworkers developed a type of PU–PUA network with hydroxyl terminated perfluoropolyether (PFPE) as the soft segment (Fig. 10A). This network could be degraded after being heated in diethylene glycol due to the exchange reaction between hydroxyl and carbamate groups (Fig. 10B).167 The PFPE monomer could be separated and purified from other components because of the poor solubility in diethylene glycol and large mass density (Fig. 10C). Using hydroxyl or amine compounds with a low boiling point as the degradation reagent could be another choice. The generated monomer can be recycled through distillation to eliminate the remaining degradation reagent. For example, PHUs synthesized by a ring-opening reaction of a six-membered cyclic carbonate with an amine compound as an attacking reagent can be degraded via alcoholysis in NaOH solution. The bi(1,3-diol) compound can be regenerated by evaporating the remaining degradation reagent to realize chemical recycling.96 | ||
| Fig. 10 A: Preparation of PU–PUA materials with PFPE as a soft segment (schematic). B and C: Schematic illustration and optical photograph showing the degradation of PU–PUA materials in diethylene glycol. Reproduced from ref. 167 with permission from the Royal Society of Chemistry, copyright 2019. | ||
Due to the water sensitivity of isocyanate groups, another alternative route to achieve the degradation of PU/PUR is to hydrolyze these materials in hot water. If the urethane and urea bonds can undergo a dissociation reaction, then the generated isocyanate groups can react with water molecules and generate amine compounds to realize recycling. For example, PUR containing a hindered urea bond formed by t-butyl and isocyanate groups can be fully degraded in PBS within 100 h, whereas no degradation is observed by replacing t-butyl with a n-butyl group.168 Although no heating is needed to degrade hindered urea-based polymers, their long-term stability at ambient temperature under high humidity is a challenge. Heating the dynamic polymer based on other types of dissociative isocyanate chemistry via phenol-carbamate bonds, oxime-carbamate bonds, and secondary aliphatic urea bonds could be alternative approach to develop recycling polymers. Balancing the service stability with a fast degradation rate should be the key feature.
4.5 Re-adhesive performance
The use of isocyanate chemistry in adhesives began in 1980.169 Isocyanate-based adhesives are used to connect metals, wood, and polymers, particularly for wood bonding.170,171 This approach benefits from the high reactivity between isocyanate groups and active hydrogen-containing molecules.172 Due to the fast development of CANs, this dynamic isocyanate chemistry offers the possibility to prepare elastomers with reversible adhesion properties. CANs can undergo reversible dissociation or exchange under a high temperature, which results in a dramatic decrease in the modulus and viscosity of the material, thereby promoting flow and facilitating material wetting onto a substrate. After subsequent cooling, reformations of the dynamic bonds and polymer network can create rigid adhesive bonds between substrates. To some extent, the reprocessable and re-adhesive performance has some similarity because both require extra pressure to enable the polymer network to bond together via dynamic isocyanate chemistry. Yang et al. developed a tough dynamic polymer network enabled by self-catalytic, cooperative, multiple dynamic moieties containing metal–ligand coordination, multiple hydrogen bonds, and reversible urea bonds. The stress could relax to zero within 10 min at 150 °C. An Ea of 53.9 kJ mol−1 determined in the range 140–160 °C was obtained from the Arrhenius equation, which indicated a very fast exchange reaction at this temperature range. Strong bonding strength should be achieved in this range, and the shear strength reached 3.2 MPa after hot-pressing at 150 °C for 10 min. This value did not decrease dramatically over three re-bonding cycles, demonstrating excellent re-adhesive performance.173 In contrast, a polythiourethane adhesive displayed obvious stress relaxation at 150–180 °C with an Ea of 75 kJ mol−1 due to exchange reactions among dynamic thiocarbamate bonds. A shear strength of ∼5 MPa was obtained after hot-pressing at 80 °C and 3 MPa for 30 min. Although the stress relaxation was very slow at this temperature due to the unobvious thiocarbamate exchange, strong adhesion could be achieved by increasing the pressure and extending the hot-pressing time. Du et al. developed a type of dynamically crosslinked PU hot-melt adhesive constructed with reversible oxime-carbamate bonds. The highest lap shear strength of the adhesive reached up to 6.55 MPa after 30 min of curing. Due to the excellent dynamic property of oxime-carbamate bonds, the lap shear strength remained at 5.56 MPa after seven complete break–repair cycles, which exceeded 80% of the initial lap shear strength.174 Building some reactions between the substrate and CANs via dynamic isocyanate chemistry can improve the bonding strength. A possible approach can be modifying the surface with hydroxyl or amine compounds which can attack the carbamate or urea moieties in CANs during the bonding process. If side reactions are absent, then heating CANs materials above the Tgel enables sufficient contact with the substrate to induce excellent adhesion.Another aspect closely related to re-adhesive performance is 3D printing of CANs. 3D printing of polymer materials is a layer-by-layer process. The interlayer strength is highly dependent upon the adhesive properties between neighboring layers, which determines the mechanical property in the printing direction. Incorporating dynamic covalent bonds into a polymer network could be a solution. Thermal treatment of the printed parts can improve the interlayer bonding strength due to the re-adhesive performance arising from the dynamic covalent bonding linkage. Sun et al. verified this idea by developing PU-based CANs containing dynamic halogenated bisphenol carbamate bonds, which could be processed by selective laser sintering for 3D printing. A dramatic increase in the Z-axis tensile strength of printed parts was achieved through reversible adhesion between neighboring layers via the reversible dissociation and reformation of bisphenol carbamate bonds.175 The relatively slow addition reaction between phenol and isocyanate groups as well as the corresponding dissociation reaction resulted in complete bonding between neighboring layers. Post-treatment by heating the printed parts further strengthened the adhesion between adjacent layers, imparting the printed parts with excellent mechanical performance at different printing orientations. In their further study, PDMS CANs with hindered pyrazole-urea dynamic bonds were developed for selective laser sintering for 3D printing (Fig. 11A).75 The strength of the printed material in the three directions (X, Y, and Z) was essentially identical (Fig. 11B), indicating that the printed parts showed an isotropic mechanical performance in different printing directions. The PDMS CANs exhibited obvious stress relaxation at 90–130 °C with an Ea of 19.4 kcal mol−1, and the relaxation modulus decayed to nearly zero at 120 °C within 3 min. An optimized mild temperature of 80 °C was selected for the thermal treatment of printed parts, which increased the interlayer bonding strength due to reversible dissociation–reformation of the hindered pyrazole urea dynamic bonds, but also maintained the size of the printed parts.
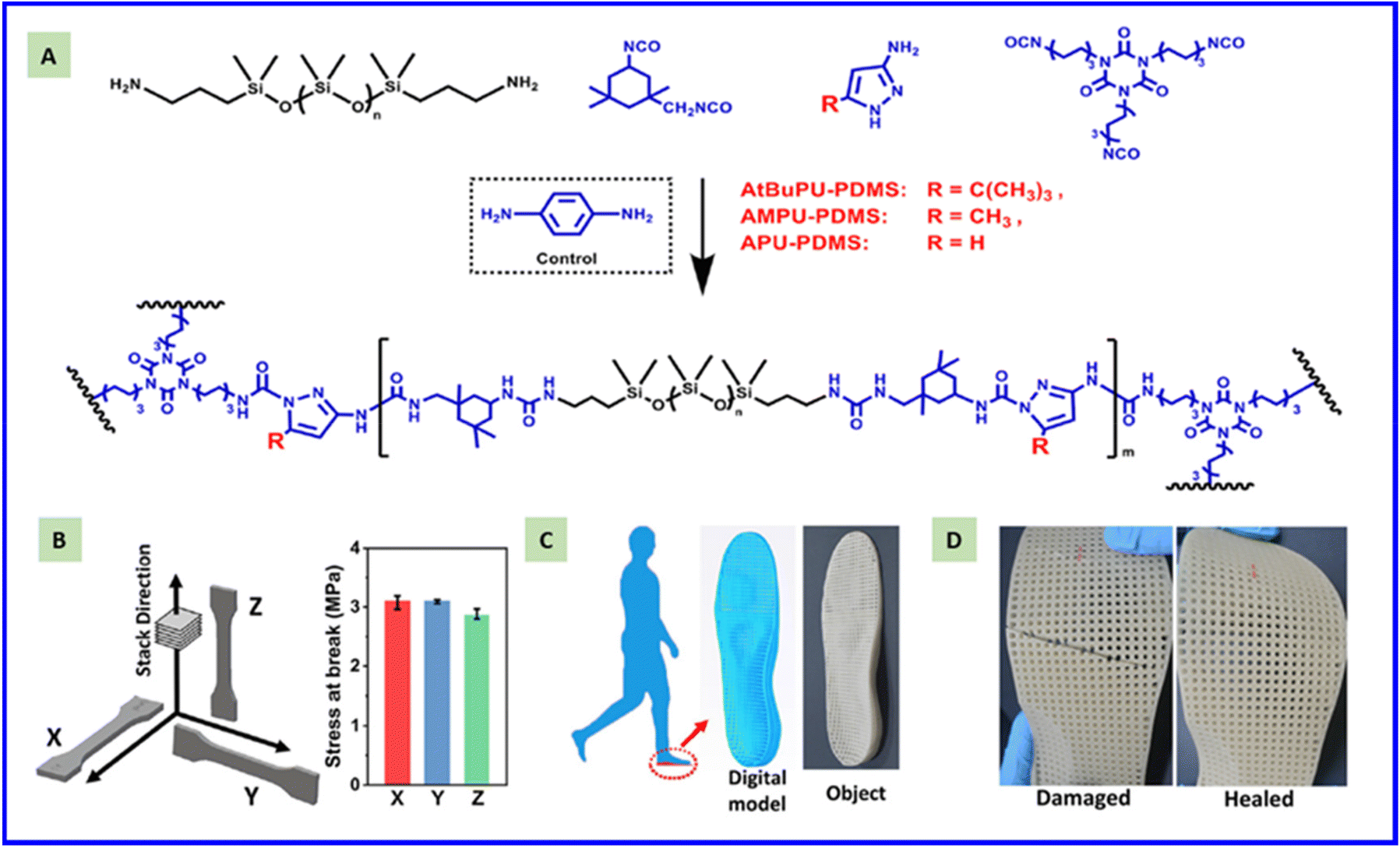 | ||
| Fig. 11 Preparatory structure of a PDMS covalent adaptive network (A). Tensile strength of printed splines in three directions (X, Y, and Z) was compared (B). Shoe soles for printing preparation (C), photograph of a self-healing 3D printed shoe sole (D). Reproduced from ref. 75 with permission from Elsevier, copyright 2021. | ||
4.6 Relationship between Ea and 5R performance
We have discussed the influence of stress-relaxation Ea on the 5R performance. Absolute conclusions about the relationship between the Ea and 5R performance cannot be obtained, but an empirical rule that can be used for guiding how to design a new generation of CANs materials with excellent 5R performance can be created. We have summarized the range of Ea values for CANs with repairable, reprocessing, and reconfiguration performances in Fig. 12. Very few studies have reported CANs with re-adhesive and recyclable properties, so we have not included them in Fig. 12. For CANs undergoing an associative reaction, such as carbamate or hydroxyl-urethane exchange, only reprocessing/reconfiguration performance has been reported, whereas urethane or urea dissociation chemistry can be employed to develop CANs with repairable or reprocessable/reconfiguration performance. Associative CANs maintaining crosslinking density during bond exchange cannot cause sufficient flow at the scale of the damage. On the contrary, dissociative CANs shift the dynamic equilibrium towards bond breaking, which can induce enough segment movement to achieve a repairable performance. If a CAN exhibits an Ea >140 kJ mol−1, repairable performance is hard to be realized only by heating. External pressure or solvent assistance is required for the repairable process. In addition, high temperature (>150 °C) is usually required to achieve a reprocessing performance for these CNAs with a large Ea. In addition, strong segment movement induced by the bond exchange reaction via dynamic isocyanate chemistry is essential to fill the free volume for achieving repairable and reprocessable performances, but macroscopic flow or long-range chain movement is not needed to realize reconfiguration. A deformation force favoring bond exchange occurring at a localized chain movement is sufficient for shape fixing. That is, reconfiguration can occur at a mild dynamic condition even for CANS via dynamic isocyanate chemistry with a higher stress relaxation energy. For example, the carbamate exchange reaction with DBTDL as a catalyst enables excellent reconfiguration of traditional PU elastomers at 130 °C, whereas reprocessing (even under shear) is difficult at this temperature.61 Even if stress relaxation reaches 184 kJ mol−1, excellent reconfiguration can be achieved by relaxing the stress at 160 °C for 40 min.93 | ||
| Fig. 12 Relationship between the activation energy and 5R performance via different categories of dynamic isocyanate chemistry. | ||
5. Conclusions and outlook
The evolution and development of dynamic isocyanate chemistry and the mechanism behind it has been discussed in detail. Four main issues need to be considered regarding the dynamic chemical reaction with isocyanate involvement.First, thermodynamics and kinetics are the most important parameters to determine the reversibility of isocyanate chemistry. They can be adjusted based on electronegativity and steric hindrance. If a chemical group possesses electronegativity and steric hindrance, the dynamic mechanism become very complex. Second, whether dynamic isocyanate chemistry undergoes dissociative or associative process is quite complicated. In most cases, a dissociative process predominates the dynamic process. If free hydroxyl or amine groups dangle in the network, these two processes may occur together, but one of them will predominate. Models studied by NMR spectroscopy, FTIR spectroscopy, and high-performance liquid chromatography can provide evidence of whether the dynamic process is dissociative or associative. DFT can be used to calculate which process has lower energy, further confirming the dynamic reaction. Third, different catalysts, such as acids/bases, have a big influence on the dynamic process of isocyanate chemistry because protonation has a very important role in the mechanism. Studies on acid/base-catalyzed dynamic isocyanate chemistry are warranted. Fourth, although a clear dynamic mechanism form isocyanate chemistry can be verified by model study of small compounds, if this dynamic isocyanate chemistry is incorporated into a polymer matrix to develop CANs materials, the dynamic process remains quite complicated.
Thanks to dynamic isocyanate chemistry, different types of smart polymer materials with 5R performance have been developed. With regard to developing stimuli-responsive polymer materials based on dynamic isocyanate chemistry, six main aspects need to be focused on in future studies.
First, urethane or urea bonds are a hydrogen-bonding motif that can contribute to the mechanical performance of polymers. In some cases, the hydrogen bond will accelerate the dissociation process, which means that the hydrogen bond and dynamic covalent bond work collaboratively. Optimizing the concentration and position of the urea or urethane bonds in the polymer network are crucial for tuning the dynamic properties of the material. Second, the dissociation reaction of some urea or urethane bond occurs at >110 °C, which leads to side reactions of the formed isocyanate group. This phenomenon will influence the thermal stability of the polymer material or result in its degradation. At high humidity, the newly generated isocyanate group will react with water, leading to the degradation of PU or PUR materials. Strategies for protecting the degradation of CANs via dynamic isocyanate chemistry should be considered during the design process. Third, some isocyanate-based chemistry can be made dynamic at room temperature. This is an advantage for preparing smart polymer materials with variable responsive properties at room temperature, such as automatic self-healing and solid reconfiguration. However, the drawback is instability at room temperature, which should be avoided. Fourth, reports about PU or PUR degradation based on dynamic isocyanate chemistry are lacking. The isocyanate group formed during urethane or urea dissociation can be captured by an extra hydrogen-based nucleophilic agent, leading to the degradation of the polymer material. Fifth, in some cases, free hydroxyl/amine groups suppress the reversion of urethane links and minimize the side reactions associated with liberated isocyanate groups under reprocessing conditions to maintain network integrity in the presence of few side reactions.176 Sixth, long-term service stability needs to be improved. For example, CANs via an oxime-urethane moiety can be degraded under UV irradiation.177
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (52173113, 51973137) and CNOOC Research Institute in Beijing (CCL2020RCPS0384PSN).References
- Y. H. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- T. Maeda, H. Otsuka and A. Takahara, Prog. Polym. Sci., 2009, 34, 581–604 CrossRef CAS.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS PubMed.
- C. N. Bowman and C. J. Kloxin, Angew. Chem., Int. Ed., 2012, 51, 4272–4274 CrossRef CAS PubMed.
- Z. H. Wang, X. L. Lu, S. J. Sun, C. J. Yu and H. S. Xia, J. Mater. Chem. B, 2019, 7, 4876–4926 RSC.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS PubMed.
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC.
- M. Podgorski, B. D. Fairbanks, B. E. Kirkpatrick, M. McBride, A. Martinez, A. Dobson, N. J. Bongiardina and C. N. Bowman, Adv. Mater., 2020, 32, 1906876 CrossRef CAS PubMed.
- Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446–7467 RSC.
- S. Y. Wang and M. W. Urban, Nat. Rev. Mater., 2020, 5, 562–583 CrossRef CAS.
- Y. Yang, X. C. Ding and M. W. Urban, Prog. Polym. Sci., 2015, 49–50, 34–59 CrossRef CAS.
- N. Zheng, Y. Xu, Q. Zhao and T. Xie, Chem. Rev., 2021, 121, 1716–1745 CrossRef CAS PubMed.
- G. M. Scheutz, J. J. Lessard, M. B. Sims and B. S. Sumerlin, J. Am. Chem. Soc., 2019, 141, 16181–16196 CrossRef CAS PubMed.
- K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS PubMed.
- R. Gheneim, C. Perez-Berumen and A. Gandini, Macromolecules, 2002, 35, 7246–7253 CrossRef CAS.
- W. X. Liu, C. Zhang, H. Zhang, N. Zhao, Z. X. Yu and J. Xu, J. Am. Chem. Soc., 2017, 139, 8678–8684 CrossRef CAS PubMed.
- D. H. Fu, W. L. Pu, Z. H. Wang, X. L. Lu, S. J. Sun, C. J. Yu and H. S. Xia, J. Mater. Chem. A, 2018, 6, 18154–18164 RSC.
- H. Z. Ying, Y. F. Zhang and J. J. Cheng, Nat. Commun., 2014, 5, 3218 CrossRef PubMed.
- Z. H. Wang, S. Gangarapu, J. Escorihuela, G. X. Fei, H. Zuilhof and H. S. Xia, J. Mater. Chem. A, 2019, 7, 15933–15943 RSC.
- M. M. Obadia, A. Jourdain, P. Cassagnau, D. Montarnal and E. Drockenmuller, Adv. Funct. Mater., 2017, 27, 1703258 CrossRef.
- P. Chakma, Z. A. Digby, M. P. Shulman, L. R. Kuhn, C. N. Morley, J. L. Sparks and D. Konkolewicz, ACS Macro Lett., 2019, 8, 95–100 CrossRef CAS PubMed.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2008, 1, 165 CrossRef.
- H. Otsuka, Polym. J., 2013, 45, 879–891 CrossRef CAS.
- A. Takahashi, R. Goseki and H. Otsuka, Angew. Chem., Int. Ed., 2017, 56, 2016–2021 CrossRef CAS PubMed.
- Z. P. Zhang, M. Z. Rong and M. Q. Zhang, Adv. Funct. Mater., 2018, 28, 1706050 CrossRef.
- T. F. Scott, A. D. Schneider, W. D. Cook and C. N. Bowman, Science, 2005, 308, 1615–1617 CrossRef CAS PubMed.
- B. T. Michal, C. A. Jaye, E. J. Spencer and S. J. Rowan, ACS Macro Lett., 2013, 2, 694–699 CrossRef CAS PubMed.
- Y. Amamoto, J. Kamada, H. Otsuka, A. Takahara and K. Matyjaszewski, Angew. Chem., 2011, 123, 1698–1701 CrossRef.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- J. P. Brutman, P. A. Delgado and M. A. Hillmyer, ACS Macro Lett., 2014, 3, 607–610 CrossRef CAS PubMed.
- G. Deng, C. Tang, F. Li, H. Jiang and Y. Chen, Macromolecules, 2010, 43, 1191–1194 CrossRef CAS.
- W. G. Skene and J.-M. P. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 8270–8275 CrossRef CAS PubMed.
- W. Denissen, G. Rivero, R. Nicolay, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef CAS.
- W. Denissen, M. Droesbeke, R. Nicolay, L. Leibler, J. M. Winne and F. E. Du Prez, Nat. Commun., 2017, 8, 14857 CrossRef CAS PubMed.
- W. Denissen, I. De Baere, W. Van Paepegem, L. Leibler, J. Winne and F. E. Du Prez, Macromolecules, 2018, 51, 2054–2064 CrossRef CAS.
- Y. Nishimura, J. Chung, H. Muradyan and Z. B. Guan, J. Am. Chem. Soc., 2017, 139, 14881–14884 CrossRef CAS PubMed.
- T. Debsharma, V. Amfilochiou, A. A. Wroblewska, I. De Baere, W. Van Paepegem and F. E. Du Prez, J. Am. Chem. Soc., 2022, 144, 12280–12289 CrossRef CAS PubMed.
- S. Y. Lee, H. Lee, I. In and S. Y. Park, Eur. Polym. J., 2014, 57, 1–10 CrossRef.
- I. A. I. Mkhalid, D. N. Coventry, D. Albesa-Jove, A. S. Batsanov, J. A. K. Howard, R. N. Perutz and T. B. Marder, Angew. Chem., Int. Ed., 2006, 45, 489–491 CrossRef CAS PubMed.
- M. Rottger, T. Domenech, R. van der Weegen, A. B. R. Nicolay and L. Leibler, Science, 2017, 356, 62–65 CrossRef PubMed.
- B. Nohra, L. Candy, J. F. Blanco, C. Guerin, Y. Raoul and Z. Mouloungui, Macromolecules, 2013, 46, 3771–3792 CrossRef CAS.
- J. Zheng, Z. M. Png, S. H. Ng, G. X. Tham, E. Y. Ye, S. S. Goh, X. J. Loh and Z. B. Li, Mater. Today, 2021, 51, 586–625 CrossRef CAS.
- M. Guerre, C. Taplan, J. M. Winne and F. E. Du Prez, Chem. Sci., 2020, 11, 4855–4870 RSC.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- N. J. Van Zee and R. Nicolay, Prog. Polym. Sci., 2020, 104, 101233 CrossRef CAS.
- P. Chakma and D. Konkolewicz, Angew. Chem., Int. Ed., 2019, 58, 9682–9695 CrossRef CAS PubMed.
- W. X. Liu, S. J. Yang, L. Huang, J. Xu and N. Zhao, Chem. Commun., 2022, 58, 12399–12417 RSC.
- D. A. Wicks and Z. W. Wicks, Prog. Org. Coat., 1999, 36, 148–172 CrossRef CAS.
- R. G. Arnold, J. A. Nelson and J. J. Verbanc, Chem. Rev., 1957, 57, 47–76 CrossRef CAS.
- E. Delebecq, J. P. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2013, 113, 80–118 CrossRef CAS PubMed.
- I. A. Mohammed and G. Sankar, High Perform. Polym., 2011, 23, 535–541 CrossRef CAS.
- L. Z. Zhang and Z. W. You, Chin. J. Polym. Sci., 2021, 39, 1281–1291 CrossRef CAS.
- A. W. Levine and J. Fech, J. Org. Chem., 1972, 37, 1500–1503 CrossRef CAS.
- A. W. Levine and J. Fech, J. Org. Chem., 1972, 37, 2455–2460 CrossRef CAS.
- T. F. Shen, M. G. Lu and L. Y. Liang, Macromol. Res., 2012, 20, 827–834 CrossRef CAS.
- S. Kalaimani, B. M. Ali and A. S. Nasar, RSC Adv., 2016, 6, 106990–107000 RSC.
- A. S. Nasar and S. Kalaimani, RSC Adv., 2016, 6, 76802–76812 RSC.
- N. Kuhl, M. Abend, R. Geitner, J. Vitz, S. Zechel, M. Schmitt, J. Popp, U. S. Schubert and M. D. Hager, Eur. Polym. J., 2018, 104, 45–50 CrossRef CAS.
- S. Guo, J. W. He, W. X. Luo and F. Liu, Materials, 2016, 9, 110 CrossRef PubMed.
- S. Guo, Y. K. Zou, J. W. He and F. Liu, Int. J. Polym. Anal. Charact., 2016, 21, 708–717 CrossRef CAS.
- N. Zheng, Z. Z. Fang, W. K. Zou, Q. Zhao and T. Xie, Angew. Chem., Int. Ed., 2016, 55, 11421–11425 CrossRef CAS PubMed.
- W. F. Liu, C. Fang, S. Y. Wang, J. H. Huang and X. Q. Qiu, Macromolecules, 2019, 52, 6474–6484 CrossRef CAS.
- D. J. Fortman, J. P. Brutman, C. J. Cramer, M. A. Hillmyer and W. R. Dichtel, J. Am. Chem. Soc., 2015, 137, 14019–14022 CrossRef CAS PubMed.
- X. Chen, L. Q. Li, K. L. Jin and J. M. Torkelson, Polym. Chem., 2017, 8, 6349–6355 RSC.
- Y. Yang and M. W. Urban, Polym. Chem., 2017, 8, 303–309 RSC.
- J. P. Brutman, D. J. Fortman, G. X. De Hoe, W. R. Dichtel and M. A. Hillmyer, J. Phys. Chem. B, 2019, 123, 1432–1441 CrossRef CAS PubMed.
- F. Elizalde, R. H. Aguirresarobe, A. Gonzalez and H. Sardon, Polym. Chem., 2020, 11, 5386–5396 RSC.
- Z. H. Wang, M. Y. Yang, X. R. Wang, G. X. Fei, Z. Zheng and H. S. Xia, J. Mater. Chem. A, 2020, 8, 25047–25052 RSC.
- K. K. Oehlenschlaeger, J. O. Mueller, J. Brandt, S. Hilf, A. Lederer, M. Wilhelm, R. Graf, M. L. Coote, F. G. Schmidt and C. Barner-Kowollik, Adv. Mater., 2014, 26, 3561–3566 CrossRef CAS PubMed.
- A. Erice, A. R. de Luzuriaga, J. M. Matxain, F. Ruiperez, J. M. Asua, H. J. Grande and A. Rekondo, Polymer, 2018, 145, 127–136 CrossRef CAS.
- A. Erice, I. Azcune, A. R. de Luzuriaga, F. Ruiperez, M. Irigoyen, J. M. Matxain, J. M. Asua, H. J. Grande and A. Rekondo, ACS Appl. Polym. Mater., 2019, 1, 2472–2481 CrossRef CAS.
- D. Zhang, H. X. Chen, Q. L. Dai, C. X. Xiang, Y. J. Li, X. Xiong, Y. Zhou and J. L. Zhang, Macromol. Chem. Phys., 2020, 221, 1900564 CrossRef CAS.
- Q. Zhang, S. J. Wang, B. Rao, X. X. Chen, L. Ma, C. H. Cui, Q. Y. Zhong, Z. Li, Y. L. Cheng and Y. F. Zhang, React. Funct. Polym., 2021, 159, 104807 CrossRef CAS.
- W. X. Liu, Z. S. Yang, Z. Qiao, L. Zhang, N. Zhao, S. Z. Luo and J. Xu, Nat. Commun., 2019, 10, 4753 CrossRef PubMed.
- S. J. Sun, G. X. Fei, X. R. Wang, M. Xie, Q. F. Guo, D. H. Fu, Z. H. Wang, H. Wang, G. X. Luo and H. S. Xia, Chem. Eng. J., 2021, 412, 128675 CrossRef CAS.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- A. E. Ainara, R. D. L. M. Alaitz, R. G. Alaitz, G. F. Aratz, A. T. Itxaso, G. hans-Jürgen, F. S. M. Mercedes and A. Xabier, Wo. Pat, WO2019063787A1, 2019 Search PubMed.
- A. Erice, A. R. de Luzuriaga, I. Azcune, M. Fernandez, I. Calafel, H. J. Grande and A. Rekondo, Polymer, 2020, 196, 122461 CrossRef CAS.
- L. Q. Li, X. Chen and J. M. Torkelson, Macromolecules, 2019, 52, 8207–8216 CrossRef CAS.
- F. Gamardella, S. De la Flor, X. Ramis and A. Serra, React. Funct. Polym., 2020, 151, 104574 CrossRef CAS.
- Z. B. Wen, X. Han, B. D. Fairbanks, K. K. Yang and C. N. Bowman, Polymer, 2020, 202, 122715 CrossRef CAS.
- D. J. Fortman, J. P. Brutman, M. A. Hillmyer and W. R. Dichtel, J. Appl. Polym. Sci., 2017, 134, 44984 CrossRef.
- B. R. Elling and W. R. Dichtel, ACS Cent. Sci., 2020, 6, 1488–1496 CrossRef CAS PubMed.
- J. B. Wu, S. J. Li, H. Liu, H. J. Qian and Z. Y. Lu, Phys. Chem. Chem. Phys., 2019, 21, 13258–13267 RSC.
- L. H. Zhang and S. J. Rowan, Macromolecules, 2017, 50, 5051–5060 CrossRef CAS.
- A. Jourdain, R. Asbai, O. Anaya, M. M. Chehimi, E. Drockenmuller and D. Montarnal, Macromolecules, 2020, 53, 1884–1900 CrossRef CAS.
- D. H. Fu, W. L. Pu, J. Escorihuela, X. R. Wang, Z. H. Wang, S. Y. Chen, S. J. Sun, S. Wang, H. Zuilhof and H. S. Xia, Macromolecules, 2020, 53, 7914–7924 CrossRef CAS.
- D. T. Sheppard, K. L. Jin, L. S. Hamachi, W. Dean, D. J. Fortman, C. J. Ellison and W. R. Dichtel, ACS Cent. Sci., 2020, 6, 921–927 CrossRef CAS PubMed.
- B. Quienne, F. Cuminet, J. Pinaud, M. Semsarilar, D. Cot, V. Ladmiral and S. Caillol, ACS Sustainable Chem. Eng., 2022, 10, 7041–7049 CrossRef CAS.
- Z. B. Wen, M. K. McBride, X. P. Zhang, X. Han, A. M. Martinez, R. F. Shao, C. H. Zhu, R. Visvanathan, N. A. Clark, Y. Z. Wang, K. K. Yang and C. N. Bowman, Macromolecules, 2018, 51, 5812–5819 CrossRef CAS.
- H. Kim, I. Cha, Y. Yoon, B. J. Cha, J. W. Yang, Y. D. Kim and C. S. Song, ACS Sustainable Chem. Eng., 2021, 9, 6952–6961 CrossRef CAS.
- D. J. Fortman, D. T. Sheppard and W. R. Dichtel, Macromolecules, 2019, 52, 6330–6335 CrossRef CAS.
- N. Zheng, J. J. Hou, Y. Xu, Z. Z. Fang, W. K. Zou, Q. Zhao and T. Xie, ACS Macro Lett., 2017, 6, 326–330 CrossRef CAS PubMed.
- S. M. Hu, X. Chen, M. A. Bin Rusayyis, N. S. Purwanto and J. M. Torkelson, Polymer, 2022, 252, 124971 CrossRef CAS.
- P. C. Miao, X. F. Leng, J. Liu, G. J. Song, M. M. He and Y. Li, Macromolecules, 2022, 55, 4956–4966 CrossRef CAS.
- P. C. Miao, Z. Y. Jiao, J. Liu, M. M. He, G. J. Song, Z. Y. Wei, X. F. Leng and Y. Li, ACS Appl. Mater. Interfaces, 2023, 15, 2246–2255 CrossRef CAS PubMed.
- W. M. Liu, G. H. Hang, H. G. Mei, L. Li and S. X. Zheng, Polymers, 2022, 14, 1331 CrossRef CAS PubMed.
- L. Li, B. J. Zhao, H. M. Wang, Y. Gao, J. W. Hu and S. X. Zheng, Compos. Sci. Technol., 2021, 215, 109009 CrossRef CAS.
- Z. B. Wen, L. Bonnaud, P. Dubois and J. M. Raquez, J. Appl. Polym. Sci., 2022, 139, e52120 CrossRef.
- L. Li, W. M. Ge, B. J. Zhao, M. Adeel, H. G. Mei and S. X. Zheng, Polymer, 2021, 213, 123314 CrossRef CAS.
- H. T. Wu, B. Q. Jin, H. Wang, W. Q. Wu, Z. X. Cao, J. R. Wu and G. S. Huang, Front. Chem., 2020, 8, 585569 CrossRef CAS PubMed.
- B. J. Zhao, K. Wei, L. Wang and S. X. Zheng, Macromolecules, 2020, 53, 434–444 CrossRef CAS.
- X. Chen, L. Q. Li, T. Wei, D. C. Venerus and J. M. Torkelson, ACS Appl. Mater. Interfaces, 2019, 11, 2398–2407 CrossRef CAS PubMed.
- A. Hernandez, H. A. Houck, F. Elizalde, M. Guerre, H. Sardon and F. E. Du Prez, Eur. Polym. J., 2022, 168, 111100 CrossRef CAS.
- Y. C. Cai, C. M. Li, Y. M. Yang, H. N. Li, Y. H. Wang and Q. Y. Zhang, Ind. Eng. Chem. Res., 2021, 60, 13585–13593 CrossRef CAS.
- L. Z. Zhang, Z. H. Liu, L. J. Sun, L. J. Xiao, Q. B. Guan and Z. W. You, Macromolecules, 2021, 54, 4081–4088 CrossRef CAS.
- Z. J. Guo, W. Y. Wang, Y. M. Yang, K. Majeed, B. L. Zhang, F. T. Zhou and Q. Y. Zhang, Polym. Chem., 2021, 12, 2009–2015 RSC.
- X. X. Chen, R. Y. Wang, C. H. Cui, L. An, Q. Zhang, Y. L. Cheng and Y. F. Zhang, Chem. Eng. J., 2022, 428, 131212 CrossRef CAS.
- J. X. Shi, T. Z. Zheng, B. H. Guo and J. Xu, Polymer, 2019, 181, 121788 CrossRef CAS.
- X. Q. Liu, Y. Li, X. L. Xing, G. J. Zhang and X. L. Jing, Polymer, 2021, 229, 124022 CrossRef CAS.
- J. X. Shi, T. Z. Zheng, Y. Zhang, B. H. Guo and J. Xu, Polym. Chem., 2021, 12, 2421–2432 RSC.
- J. Hu, R. H. Yang, L. Zhang, Y. Chen, X. X. Sheng and X. Y. Zhang, Polymer, 2021, 222, 123674 CrossRef CAS.
- J. X. Shi, T. Z. Zheng, Y. Zhang, B. H. Guo and J. Xu, ACS Sustainable Chem. Eng., 2020, 8, 1207–1218 CrossRef CAS.
- S. Wang, D. H. Fu, X. R. Wang, W. L. Pu, A. Martone, X. L. Lu, M. Lavorgna, Z. H. Wang, E. Amendola and H. S. Xia, J. Mater. Chem. A, 2021, 9, 4055–4065 RSC.
- Y. Li, Y. P. Wang, S. Wang, Z. J. Ye, C. Bian, X. L. Xing, T. Hong and X. L. Jing, Macromolecules, 2022, 55, 9091–9102 CrossRef CAS.
- S. W. Xie, D. Wang, S. Zhang, J. H. Xu and J. J. Fu, J. Mater. Chem. A, 2022, 10, 9457–9467 RSC.
- Y. C. Jia, H. Z. Ying, Y. F. Zhang, H. He and J. J. Cheng, Macromol. Chem. Phys., 2019, 220, 1900148 CrossRef.
- M. A. Bin Rusayyis and J. M. Torkelson, ACS Macro Lett., 2022, 11, 568–574 CrossRef CAS PubMed.
- B. J. Zhao, L. Li, J. W. Hu, H. M. Wang, H. G. Mei and S. X. Zheng, Polymer, 2022, 242, 124591 CrossRef CAS.
- J. S. Zhang, C. Q. Zhang, F. Song, Q. Q. Shang, Y. Hu, P. Y. Jia, C. G. Liu, L. H. Hu, G. Q. Zhu, J. Huang and Y. H. Zhou, Chem. Eng. J., 2022, 429, 131848 CrossRef CAS.
- Z. Q. Zhou, Y. N. Zeng, C. L. Yu, H. B. Chen and F. A. Zhang, Smart Mater. Struct., 2020, 29, 115041 CrossRef CAS.
- S. J. Wang, Y. F. Yang, H. Z. Ying, X. L. Jing, B. Wang, Y. F. Zhang and J. J. Cheng, ACS Appl. Mater. Interfaces, 2020, 12, 35403–35414 CrossRef CAS PubMed.
- L. F. Chen, L. H. Zhang, P. J. Griffin and S. J. Rowan, Macromol. Chem. Phys., 2020, 221, 1900440 CrossRef CAS.
- D. M. Xie, Y. X. Zhang, Y. D. Li, Y. X. Weng and J. B. Zeng, Chem. Eng. J., 2022, 446, 137071 CrossRef CAS.
- H. Ding, B. J. Zhao, H. G. Mei, L. Li and S. X. Zheng, Chin. J. Polym. Sci., 2020, 38, 915–920 CrossRef CAS.
- L. Jiang, Y. Lei, Y. Xiao, H. L. Xu, A. Q. Yuan, Z. K. Wei, Y. Chen and J. X. Lei, Chem. Eng. J., 2021, 410, 128354 CrossRef CAS.
- B. F. Chen, X. H. Liu, J. M. Liu, Z. Q. Feng, X. L. Zheng, X. K. Wu, C. L. Yang and L. Y. Liang, React. Funct. Polym., 2022, 172, 105184 CrossRef CAS.
- L. Jiang, Z. M. Liu, Y. Lei, Y. Yuan, B. Wu and J. X. Lei, ACS Appl. Polym. Mater., 2019, 1, 3261–3268 CrossRef CAS.
- Y. Lei, Y. Wang, A. Q. Yuan, S. W. Zhao, Y. Chen, Y. Xiao, L. Jiang and J. X. Lei, J. Mater. Chem. A, 2022, 10, 6713–6723 RSC.
- C. H. Cui, X. X. Chen, L. Ma, Q. Y. Zhong, Z. Li, A. Mariappan, Q. Zhang, Y. L. Cheng, G. He, X. M. Chen, Z. Dong, L. An and Y. F. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 47975–47983 CrossRef CAS PubMed.
- F. Gamardella, F. Guerrero, S. De la Flor, X. Ramis and A. Serra, Eur. Polym. J., 2020, 122, 109361 CrossRef CAS.
- F. Guerrero, S. de la Flor, X. Ramis, J. I. Santos and A. Serra, Eur. Polym. J., 2022, 174, 111337 CrossRef CAS.
- Q. Ma, S. L. Liao, Y. C. Ma, Y. J. Chu and Y. P. Wang, Adv. Mater., 2021, 33, 2102096 CrossRef CAS PubMed.
- P. Y. Yan, W. Zhao, X. W. Fu, Z. M. Liu, W. B. Kong, C. L. Zhou and J. X. Lei, RSC Adv., 2017, 7, 26858–26866 RSC.
- P. Y. Yan, W. Zhao, Y. J. Wang, Y. Y. Jiang, C. L. Zhou and J. X. Lei, Macromol. Chem. Phys., 2017, 218, 1700265 CrossRef.
- S. Wu, M. Luo, D. J. Darensbourg, D. D. Zeng, Y. H. Yao, X. B. Zuo, X. Hu and D. W. Tan, ACS Sustainable Chem. Eng., 2020, 8, 5693–5703 CrossRef CAS.
- J. J. Zhou, H. M. Yue, M. M. Huang, C. B. Hao, S. Q. He, H. Liu, W. T. Liu, C. S. Zhu, X. Dong and D. J. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 43426–43437 CrossRef CAS PubMed.
- C. J. Fan, Z. B. Wen, Z. Y. Xu, Y. Xiao, D. Wu, K. K. Yang and Y. Z. Wang, Macromolecules, 2020, 53, 4284–4293 CrossRef CAS.
- S. Zechel, R. Geitner, M. Abend, M. Siegmann, M. Enke, N. Kuhl, M. Klein, J. Vitz, S. Grafe, B. Dietzek, M. Schmitt, J. Popp, U. S. Schubert and M. D. Hager, NPG Asia Mater., 2017, 9, e420 CrossRef CAS.
- J. I. Park, A. Choe, M. P. Kim, H. Ko, T. H. Lee, S. M. Noh, J. C. Kim and I. W. Cheong, Polym. Chem., 2018, 9, 11–19 RSC.
- L. Z. Zhang, Z. H. Liu, X. L. Wu, Q. B. Guan, S. Chen, L. J. Sun, Y. F. Guo, S. L. Wang, J. C. Song, E. M. Jeffries, C. L. He, F. L. Qing, X. G. Bao and Z. W. You, Adv. Mater., 2019, 31, 1901402 CrossRef PubMed.
- H. Wang, J. H. Xu, D. M. Xing, X. S. Du, H. B. Wang, Z. L. Du and X. Cheng, J. Appl. Polym. Sci., 2022, 139, e52472 CrossRef CAS.
- S. Cao, S. H. Li, M. Li, L. N. Xu, H. Y. Ding, J. L. Xia, M. Zhang and K. Huang, Polym. J., 2017, 49, 775–781 CrossRef CAS.
- Y. Yang and M. W. Urban, Angew. Chem., Int. Ed., 2014, 53, 12142–12147 CrossRef CAS PubMed.
- S. Liu, S. Chen, W. Shi, Z. Peng, K. Luo, S. Xing, J. Li, Z. Liu and L. Liu, Adv. Funct. Mater., 2021, 31, 2102225 CrossRef CAS.
- M. Abend, S. Zechel, U. S. Schubert and M. D. Hager, Molecules, 2019, 24, 3597 CrossRef PubMed.
- H. M. Lee, S. Perumal, G. Y. Kim, J. C. Kim, Y.-R. Kim, M. P. Kim, H. Ko, Y. Rho and I. W. Cheong, Polym. Chem., 2020, 11, 3701–3708 RSC.
- B. Zhao, H. Ding, S. Xu and S. Zheng, ACS Appl. Polym. Mater., 2019, 1, 3174–3184 CrossRef CAS.
- J. Wang, S. Hu, B. Yang, G. Jin, X. Zhou, X. Lin, R. Wang, Y. Lu and L. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 1994–2005 CrossRef CAS PubMed.
- Z. Zhou, S. Chen, W. Li, J. He, C. Yu and F. Zhang, Macromol. Mater. Eng., 2020, 306, 2000527 CrossRef.
- F. Z. Wang, H. Q. Wang, W. T. Gao and C. H. Li, Mater. Chem. Front., 2022, 6, 473–481 RSC.
- X. X. Chen, Q. Y. Zhong, C. H. Cui, L. Ma, S. Liu, Q. Zhang, Y. S. Wu, L. An, Y. L. Cheng, S. B. Ye, X. M. Chen, Z. Dong, Q. Chen and Y. F. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 30847–30855 CrossRef CAS PubMed.
- S. Ahmed, M. J. Bae, S. Jeong, J. H. Lee, J. C. Kim, Y. I. Park and I. W. Cheong, ACS Appl. Polym. Mater., 2022, 4, 8136–8146 CrossRef CAS.
- G. Y. Kim, S. Sung, M. P. Kim, S. C. Kim, S. H. Lee, Y. I. Park, S. M. Noh, I. W. Cheong and J. C. Kim, Appl. Surf. Sci., 2020, 505, 144546 CrossRef CAS.
- L. Zhang, H. Q. Wang, Z. H. Dai, Z. X. Zhao, F. Y. Fu and X. D. Liu, React. Funct. Polym., 2020, 146, 104444 CrossRef CAS.
- H. Y. Ma, Y. T. Liu, C. C. Zhua, Z. Yuan, C. Z. Z. Yan, J. W. Sun, X. Wang, C. H. Gao and Y. M. Wu, React. Funct. Polym., 2019, 137, 79–87 CrossRef CAS.
- Y. F. Zhang, H. Z. Ying, K. R. Hart, Y. X. Wu, A. J. Hsu, A. M. Coppola, T. A. Kim, K. Yang, N. R. Sottos, S. R. White and J. J. Cheng, Adv. Mater., 2016, 28, 7646–7651 CrossRef CAS PubMed.
- D. W. Lee, H. N. Kim and D. S. Lee, Molecules, 2019, 24, 2201 CrossRef PubMed.
- L. Bai and J. P. Zheng, Macromol. Chem. Phys., 2020, 221, 1900493 CrossRef CAS.
- S. Cao, S. H. Li, M. Li, L. N. Xu, H. Y. Ding, J. L. Xia, M. Zhang and K. Huang, J. Appl. Polym. Sci., 2019, 136, 47039 CrossRef.
- J. Hu, R. Yang, L. Zhang, Y. Chen, X. Sheng and X. Zhang, Polymer, 2021, 222, 123674 CrossRef CAS.
- B. Xu, F. Han, X. Pei, S. Zhang and J. Zhao, Ind. Eng. Chem. Res., 2021, 60, 11095–11105 CrossRef CAS.
- S. H. Lee and D. S. Lee, Macromol. Res., 2019, 27, 460–469 CrossRef CAS.
- Z. Z. Ming, Y. Pang and J. Y. Liu, Adv. Mater., 2020, 32, 1906870 CrossRef CAS PubMed.
- Z. Z. Fang, N. Zheng, Q. Zhao and T. Xie, ACS Appl. Mater. Interfaces, 2017, 9, 22077–22082 CrossRef CAS PubMed.
- B. Jin, H. Song, R. Jiang, J. Song, Q. Zhao and T. Xie, Sci. Adv., 2018, 4, eaao3865 CrossRef PubMed.
- S. L. Liao, Y. L. He, Y. J. Chu, H. G. Liao and Y. P. Wang, J. Mater. Chem. A, 2019, 7, 16249–16256 RSC.
- H. Ying and J. Cheng, J. Am. Chem. Soc., 2014, 49, 16974–16977 CrossRef PubMed.
- I. Skeist and J. Miron, J. Macromol. Sci., Part A: Pure Appl.Chem., 2006, 15, 1151–1163 CrossRef.
- M. S. Sánchez-Adsuar, M. M. Pastor-Blas and J. M. Martín-Martínez, J. Adhes., 1998, 67, 327–345 CrossRef.
- C. W. Phetphaisit, R. Bumee, J. Namahoot, J. Ruamcharoen and P. Ruamcharoen, Int. J. Adhes. Adhes., 2013, 41, 127–131 CrossRef.
- C. S. Schollenberger, in Handbook of Adhesives, ed. I. Skeist, Springer US, Boston, MA, 1990, pp. 359–380, DOI:10.1007/978-1-4613-0671-9_20.
- M. Y. Yang, X. L. Lu, Z. H. Wang, G. X. Fei and H. S. Xia, J. Mater. Chem. A, 2021, 9, 16759–16768 RSC.
- L. Du, Z. Z. Liu, Z. X. Ye, X. L. Hao, R. X. Ou, T. Liu and Q. W. Wang, Eur. Polym. J., 2023, 182, 111732 CrossRef CAS.
- S. J. Sun, X. P. Gan, Z. H. Wang, D. H. Fu, W. L. Pu and H. S. Xia, Addit. Manuf., 2020, 33, 101176 CAS.
- X. Chen, S. M. Hu, L. Q. Li and J. M. Torkelson, ACS Appl. Polym. Mater., 2020, 2, 2093–2101 CrossRef CAS.
- Y. Lei, X. W. Fu, L. Jiang, Z. M. Liu and J. X. Lei, Macromol. Rapid Commun., 2022, 43, 2100781 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2023 |