
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Resolving the complexity in human milk oligosaccharides using pure shift NMR methods and CASPER†
Marshall J.
Smith
 a,
Emma L.
Gates
a,
Göran
Widmalm
b,
Ralph W.
Adams
a,
Gareth A.
Morris
a and
Mathias
Nilsson
*a
a,
Emma L.
Gates
a,
Göran
Widmalm
b,
Ralph W.
Adams
a,
Gareth A.
Morris
a and
Mathias
Nilsson
*a
aDepartment of Chemistry, University of Manchester, Manchester, M13 9PL, UK. E-mail: mathias.nilsson@manchester.ac.uk
bDepartment of Organic Chemistry, Arrhenius Laboratory, Stockholm University, Stockholm, Sweden
First published on 14th April 2023
Abstract
Human milk oligosaccharides belong to an important class of bioactive molecules with diverse effects on the development of infants. NMR is capable of providing vital structural information about oligosaccharides which can aid in determining structure–function relationships. However, this information is often concealed by signal overlap in 1H spectra, due to the narrow chemical shift range and signal multiplicity. Signal overlap in oligosaccharide spectra can be greatly reduced, and resolution improved, by utilising pure shift methods. Here the benefits of combining pure shift methods with the CASPER computational approach to resonance assignment in oligosaccharides are demonstrated.
Introduction
Human Milk Oligosaccharides (HMOs) are the third most abundant component of human breast milk and have diverse effects on the development of infants, including stimulation of the intestinal barrier function and anti-pathogenic effects.1HMOs are structurally diverse, with each monosaccharide residue potentially linking to multiple residues in a variety of ways. Detailed information on the structures of HMOs is key to elucidating their structure–function relationships.2 NMR has proven to be vital in providing this knowledge, allowing insight into the stereochemistry, linkage type, and conformational preferences of HMOs.3 However, the limited chemical shift range and prominent signal multiplicity exhibited in 1H spectra of oligosaccharides often cause signals to overlap in the 1H domain, impeding access to such information.
Pure shift methods4,5 aim to alleviate spectral overlap by suppressing the effects of homonuclear scalar couplings (JHH) within a spectrum. Each chemical shift is then, ideally, represented by a well-resolved singlet. This is usually achieved by using a J-refocusing element to refocus the effects of JHH. A variety of methods have been proposed for achieving broadband homonuclear decoupling, including Zangger–Sterk (ZS),6 Pure Shift Yielded by CHIRP Excitation (PSYCHE),7 and BIlinear Rotation Decoupling (BIRD).8 Each of these methods splits the 1H spin population into two subpopulations, active spins (those that contribute to the signal to be measured) and passive spins (those that do not directly contribute to the measured signal but are responsible for the multiplet structure of the measured signals).
Homonuclear decoupled data can be collected in real-time (RT)9 or in parameter-time (interferogram) mode.6,10 In real-time acquisition the FID is periodically interrupted by ZS or BIRD J-refocusing elements. Parameter-time methods use a pseudo-2D acquisition scheme in which a J-refocusing element is applied at the midpoint of an evolution time t1. A short chunk of FID is acquired for each value of t1 and these chunks are then combined to generate a time-domain interferogram in which only chemical shift evolution is observed.6 RT acquisition is faster than the interferogram approach, but this comes at the cost of a decrease in resolution caused by relaxation and pulse imperfections in the repeated J-refocusing elements. This significant reduction in resolution often imposes a concomitant sensitivity penalty to set against the time saving.
Even with the increased resolution offered by the pure shift methods advocated in this work, the analysis of HMO spectra can still be time-consuming. Computational approaches, such as Computer Assisted SPectrum Evaluation of Regular polysaccharides (CASPER),11,12 GlyNest,13 and GlycoNMRSearch,14 have been proposed for identifying these saccharides and providing approximate spectral assignments, significantly speeding up analysis. NMR data are well suited to such approaches as the chemical shift is sensitive to the chemical surroundings of an atom, and a particular chemical shift can often be correlated with a single atom within a given structure.
CASPER uses an increment rule-based approach to predict the chemical shifts of an oligosaccharide.11,13 The chemical shifts of glycosyl residues in an oligo- or polysaccharide differ from those in monosaccharides in a predictable manner. Hence, based on connected residues and linkage types, glycosylation shifts can be applied to the chemical shifts of a free monosaccharide to predict a single chemical shift for each atom in an oligosaccharide.13 However, CASPER does not take into account the multiplet structure typically present for 1H NMR signals. To avoid the ambiguity caused by peak multiplicity, the user must either apply a severe time-domain weighting function so that the multiplicity is suppressed (at a high cost in resolution), or manually pick a single chemical shift for each multiplet/chemical environment. This is often time-consuming, limiting the potential of CASPER as an efficient analysis tool. As pure shift methods aim to collapse multiplets into well-resolved singlets, pure shift data are ideal for use in CASPER. Here, we demonstrate the advantages of combining pure shift methods with CASPER, allowing for efficient analysis of three carbohydrates using automatic peak-picking routines.
Results and discussion
The di-, tri-, and hexasaccharides lactose, 3-fucosyllactose (3-FL) and lacto-N-difucohexaose I (LNDFH I), are three prominent oligosaccharides found in human breast milk,15 but even these relatively simple saccharides show significant overlap in the conventional 1H NMR spectrum, as shown in Fig. 1a for LNDFH I. Utilising 1D pure shift methods such as PSYCHE and ZS, the resolution of a spectrum can be increased (by up to an order of magnitude) by suppressing the effects of homonuclear scalar coupling, as shown in Fig. 1b. However, even with this significant increase in resolution, some spectral overlap remains. 2D techniques such as COSY,16 HSQC17 and TOCSY18 can alleviate some of this spectral overlap, but in heavily overlapped regions, e.g. 3.5–4.0 ppm in Fig. 1, information may still be concealed.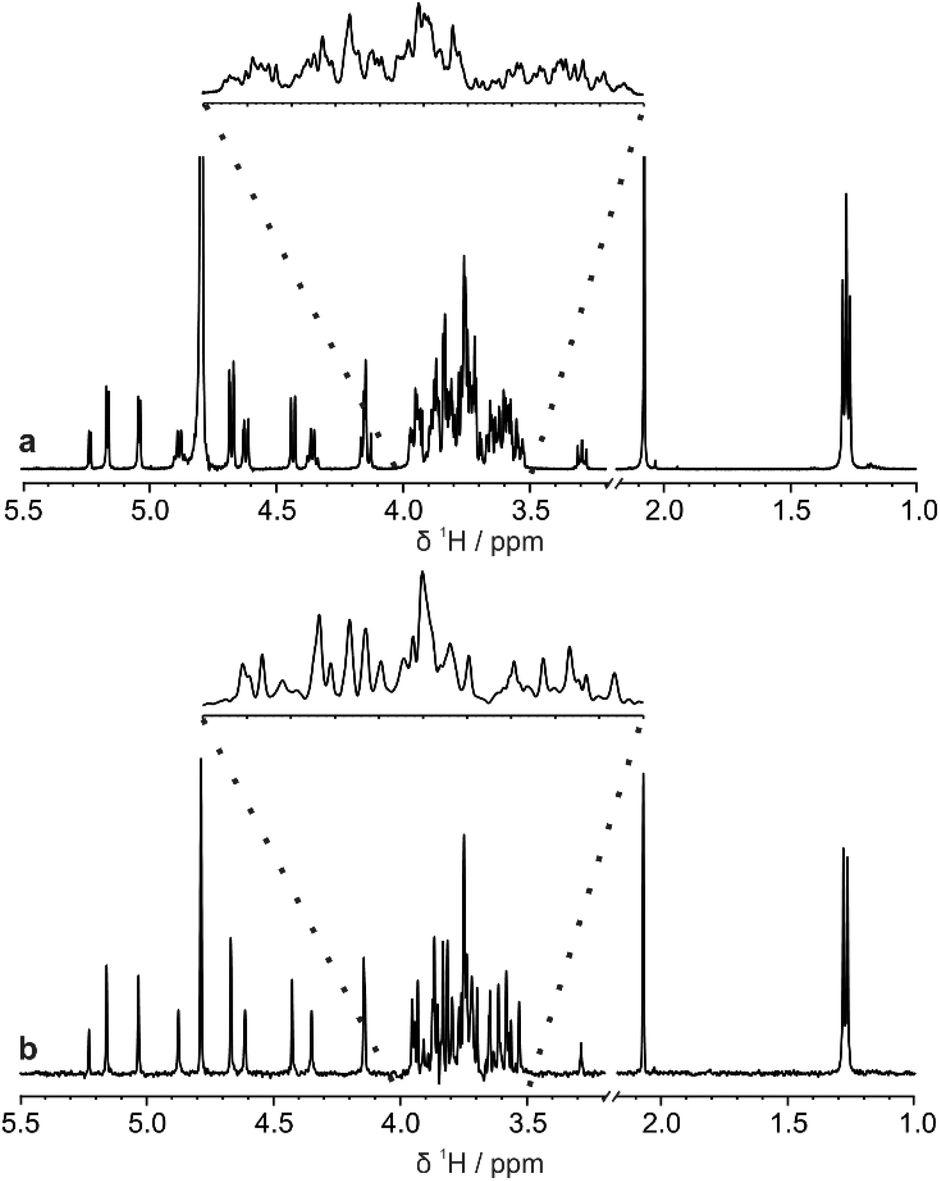 | ||
| Fig. 1 Comparison (a) conventional 1H and (b) pure shift PSYCHE spectra of a sample of 25 mM LNDFH I in D2O acquired on a spectrometer operating at 400 MHz 1H frequency. | ||
The use of additional dimensions (e.g. in 3D experiments) can reduce this overlap by spreading the signals across a higher number of dimensions, but this incurs a very significant time penalty.19
A more efficient approach to reduce spectral overlap is to implement pure shift methods in 2D techniques, to greatly increase the spectral resolution by suppressing the effects of JHH. For instance, real-time BIRD homonuclear decoupling has been integrated into the HSQC experiment, suppressing the effect of JHH on the spectrum at negligible cost in experiment time.20 The active spins selected are those with one-bond couplings to the isotopically dilute 13C atoms. As these spins are just those which are already selected for observation in the conventional HSQC experiment, a major gain in resolution can be obtained with no sensitivity penalty.21 In fact there is typically a gain in sensitivity, as suppressing the effects of JHH means that all the available signal for a given proton is concentrated into a single peak.
Fig. 2 shows a comparison between the conventional multiplicity-edited HSQC (edHSQC) and the multiplicity-edited RT pure shift HSQC (edPSHSQC) spectra for 3-FL. The increased resolution in the edPSHSQC spectrum is evident in the inset of Fig. 2, distinguishing between two signals that are completely overlapped in the conventional edHSQC spectrum. These two signals correspond to H5 of α-L-Fuc in the α- and β-anomeric forms of 3-FL. As most of the signals in the edPSHSQC spectrum of Fig. 2 are collapsed into well-resolved singlets, this is ideal for generating data for CASPER, as each nucleus is represented by a single chemical shift.
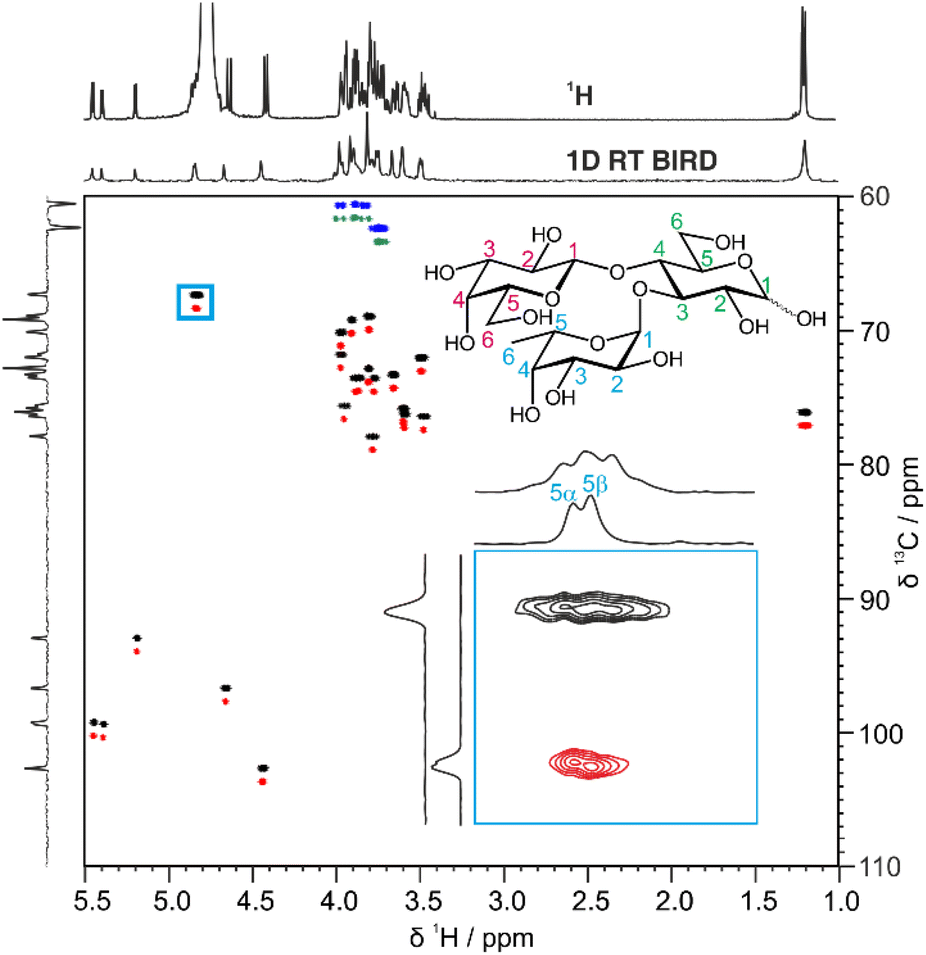 | ||
| Fig. 2 Comparison between the conventional multiplicity-edited HSQC spectrum (blue and black contours) and the pure shift real-time BIRD HSQC spectrum (green and red contours) for a sample of 54 mM 3-FL in D2O. The top 1D spectra are separately acquired 1H and RT pure shift BIRD spectra. The inset is an expansion of the region defined by the blue box. The two 2D spectra were acquired with similar experiment times. The cross-peaks in the pure shift multiplicity edited real-time BIRD HSQC spectrum have been offset vertically by +1 ppm. The peaks at 1.2 ppm have been aliased in F1 (the 13C dimension). | ||
Automatic peak-picking routines can then be used to efficiently generate CASPER-compatible data. However, one limitation of BIRD pure shift methods is that geminal couplings are not suppressed (because BIRD cannot discriminate between protons that are connected to the same 13C atom). Hence, non-equivalent methylene protons in oligosaccharides remain as doublets in the edPSHSQC spectrum. Although some methods have been proposed for removing this residual multiplicity, using the perfectBIRD element22 or an extra J-resolved dimension,23 these methods both require a three-dimensional acquisition scheme, at a significant cost in experiment time. As the geminal couplings between methylene protons in saccharides are well understood,24 and methylene signals are easily identified as they appear with opposite phase to methine and methyl protons in multiplicity-edited spectra, an automatic peak-picking macro was written (see ESI†) to automatically assign a single chemical shift to each proton in a methylene group. The user defines a threshold for the maximum splitting to be recognised as a geminal coupling, and the macro then returns a single correlation for signals that have the same 13C chemical shift, a negative phase in the multiplicity-edited spectrum, and a splitting that is less than the user-defined threshold. Hence, a single 1H and 13C chemical shift pair is generated for each CH fragment in an oligosaccharide. This information is exported in a user-friendly format for input into CASPER, producing an efficient workflow.
CASPER generates ranked possible structure assignments for a given experimental dataset. It is often necessary to acquire further data to distinguish between the top-ranked possibilities. TOCSY experiments (giving information on contiguous scalar coupling networks within a molecule) are particularly useful in oligosaccharides, as the sub-spectra of the constituent residues can often be extracted. However, due to the limited chemical shift range of monosaccharide residues, overlap often remains in the resultant sub-spectra. Again, using pure shift methods in combination with TOCSY experiments increases the resolution of the spectrum. F1-PSYCHE-TOCSY25 suppresses the effect of JHH in the indirect dimension (Fig. S4†). Covariance processing is a useful tool, especially when signals are resolved in the homodecoupled dimension, to produce a spectrum which is pure shift (i.e. fully decoupled) in both the direct and indirect dimensions, as shown in Fig. 3 for 3-FL. In cases where peaks overlap, care should be taken, as spurious cross-peaks can occur in covariance spectra. The inset in Fig. 3 exemplifies the advantages of the increased resolution, as a distinction can be made between the correlations between protons H5 and H6 in the fucosyl residues of the α- and β-anomeric forms of 3-FL.
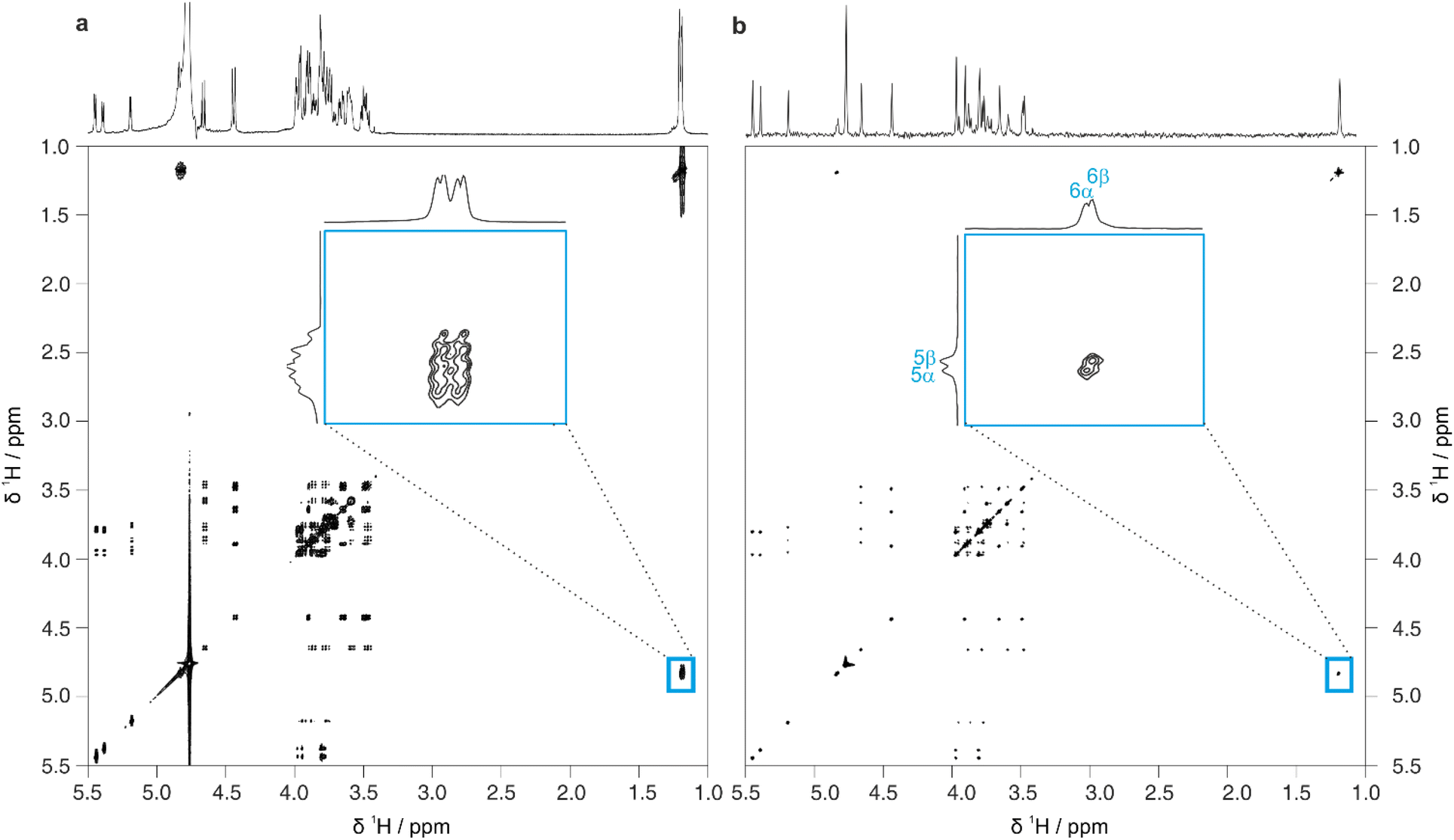 | ||
| Fig. 3 Comparison of 2D spectra obtained using (a) conventional TOCSY and (b) pure shift TOCSY using PSYCHE homonuclear decoupling in F1 and covariance processing in F2, for a sample of 54 mM 3-FL in D2O. The inset is an expansion of the region defined by the blue box. The pure shift TOCSY spectrum without covariance processing and full experimental details are provided in the ESI. | ||
In homonuclear 2D proton NMR experiments, signals that do not correlate with any individual oligosaccharide signal may be present, for example the water solvent peak or, in the case of pure shift experiments, strong coupling artefacts. Conventional peak-picking may report these signals, requiring the user to remove them before inputting the data to CASPER. To streamline oligosaccharide analysis, a further macro has been developed that edits the pure shift TOCSY (PSTOCSY) peak list using the 1H correlations in the edPSHSQC peak list. Signals that are in the PSTOCSY peak list but do not show a 13C correlation in the edPSHSQC spectrum are removed automatically, reducing the number of spurious peaks identified in the F1-PSYCHE-TOCSY spectrum.
Full 2D spectra contain extensive information on the full scalar coupling network in a molecule, but this level of detail may not be needed to distinguish between the top-ranked structure assignment predictions generated by CASPER. Identifying TOCSY correlations from a limited number of signals is often sufficient. This information can be rapidly obtained using 1D pure shift selective TOCSY, where the signals observed are restricted to those within the same spin system as a chosen spin.26 This is particularly useful in cases where a limited number of anomeric signals are present that are well-resolved, and can therefore be selected individually by frequency-selective pulses. Even in cases where multiplets overlap, a GEMSTONE selective excitation element can be used to select individual signals by chemical shift.27
Fig. 4 shows a comparison between 1D selective TOCSY and pure shift selective TOCSY data for lactose. Additional responses (denoted by a blue asterisk) can be observed in the pure shift selective TOCSY spectra. These do not correspond to any individual nuclei in the molecule but are caused by coherence transfer between strongly coupled nuclei during the J-refocusing element. Such responses often occur midway between two strongly coupled spins. To avoid these artefacts being identified in the peak-picking routine, a 1D peak-picking macro was created to ensure once again that only 1H signals that are present in the edPSHSQC spectrum are retained. Signals H5 and H6 of the galactose residue are not observed in these TOCSY spectra because the small JHH value between H4 and H5 limits the effectiveness of the TOCSY transfer.
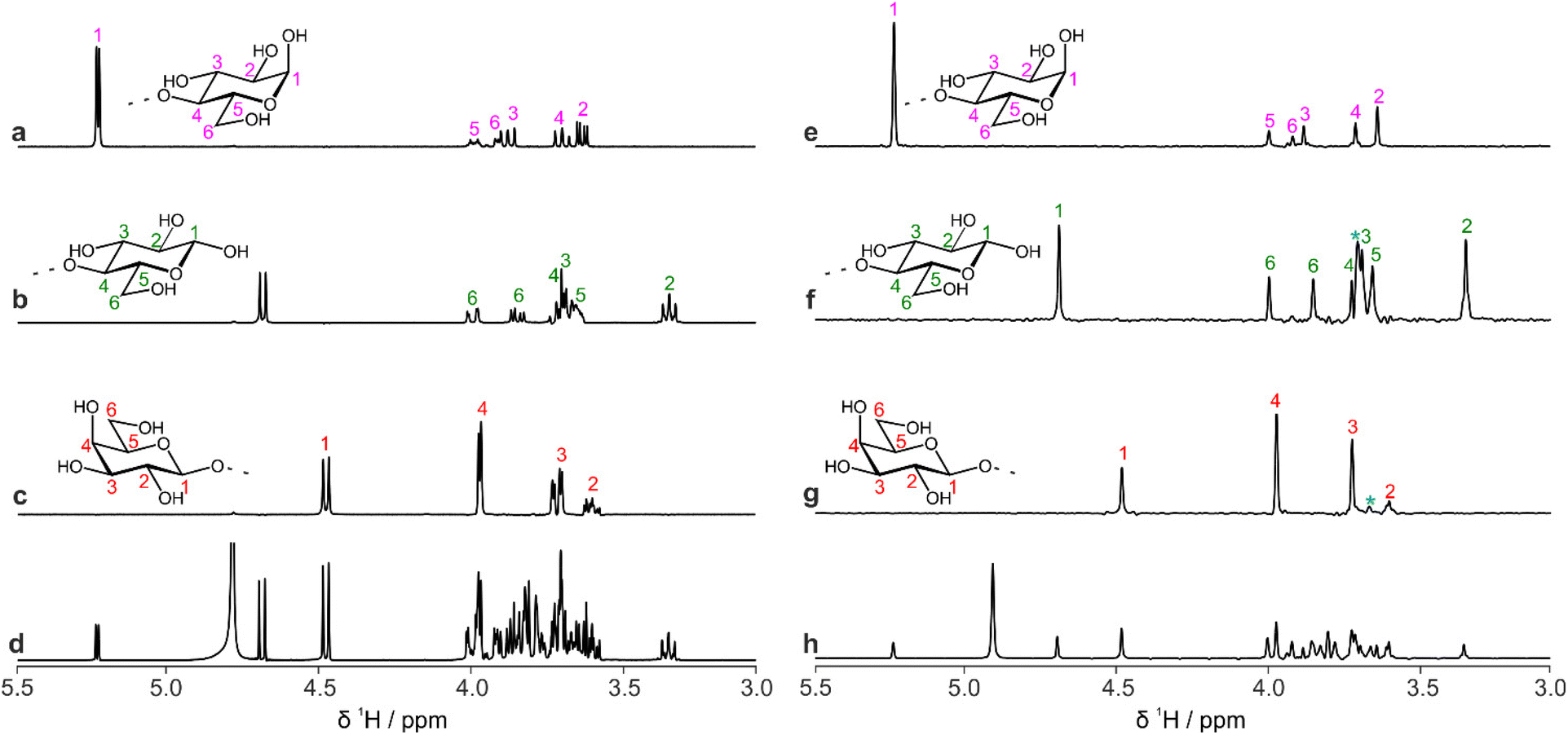 | ||
| Fig. 4 Comparison of 1D proton spectra of lactose obtained by (a–c) selective TOCSY and (e–g) pure shift selective TOCSY, using PSYCHE as the J-refocusing element; (a and e) reducing end α-D-Glcp; (b and f) reducing end β-D-Glcp; (c and g) terminal β-D-Galp residue. A 150 ms DIPSI-2 isotropic mixing duration was used in all cases. The blue asterisks indicate artefacts due to strong coupling, for example between H2 and H3 in the galactose residue. Spectra (d) and (h) are conventional 1H and PSYCHE spectra, respectively. | ||
The data that are generated by pure shift experiments require little, if any, user intervention for use with CASPER.28,29Table 1 shows the structure assignments suggested by CASPER on inputting the data generated by the edPSHSQC and PSTOCSY macros for lactose, 3-FL and LNDFH I. Fig. 5 shows a comparison between the literature assignments for 3-FL30 and the corresponding CASPER assignments, for (a) 1H and (b) 13C. The almost perfect correlation between the two datasets shows that the chemical shifts suggested by CASPER agree closely with those in the literature. The outlier in Fig. 5b corresponds to C4 of the L-Fuc residue. Multiple-bond 1H–13C correlations in 2D NMR spectra were consistent with the proposed CASPER assignment of this signal, suggesting that the literature assignment should be revisited. In cases with small differences in 13C chemical shift, some assignments may be reversed, but the tentative assignments generated form the basis for additional NMR experiments.
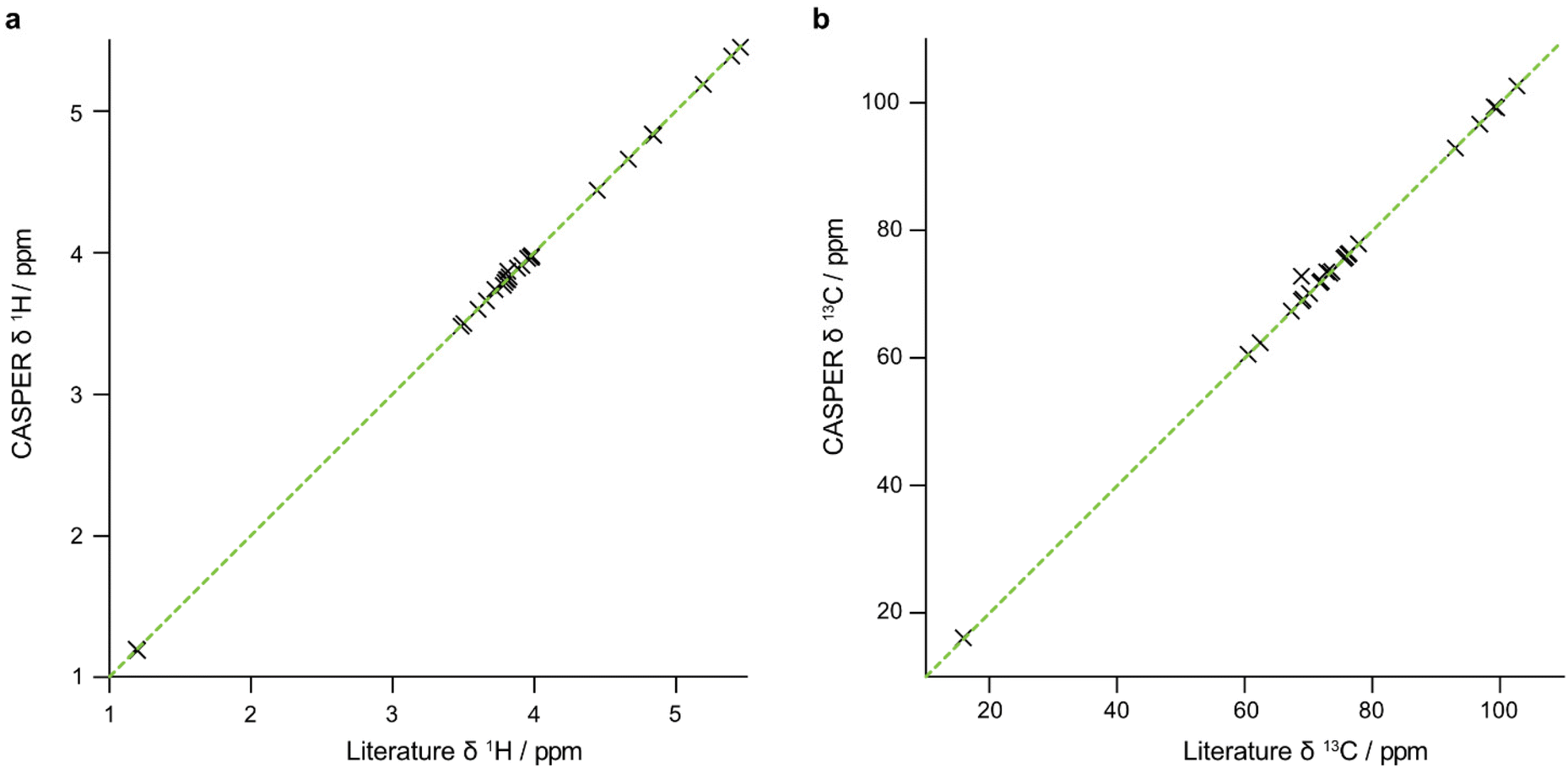 | ||
| Fig. 5 Comparison between the chemical shifts, (a) 1H and (b) 13C of 3-FL reported in the literature and generated by CASPER. An appropriate displacement was applied to the 13C NMR chemical shifts to account for the difference in sample temperature and chemical shift referencing between the two datasets. The dotted green diagonal has unit slope. | ||
| a α-Anomeric form of glucosyl residue. b β-Anomeric form of glucosyl residue. c (1 → 4)-linked. d (1 → 2)-linked. | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| (a) β-D-Gal-(1 → 4)-α/β-D-Glc | |||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 6′ | |||
| α-D-Glc | 1H/13C | 5.23/92.81 | 3.59/72.14 | 3.84/72.41 | 3.65/79.45 | 3.96/71.10 | 3.88/60.98 | 3.88/60.98 | |
| β-D-Glc | 1H/13C | 4.67/96.73 | 3.29/74.81 | 3.65/75.35 | 3.67/79.33 | 3.60/75.79 | 3.96/61.10 | 3.80/61.10 | |
| β-D-Gal | 1H/13C | 4.45/103.91 | 3.55/71.95 | 3.67/73.52 | 3.93/69.55 | 3.73/76.33 | 3.78/62.03 | 3.78/62.03 | |
| (b) α-L-Fuc-(1 → 3) [β-D-Gal-(1 → 4)]-α/β-D-Glc | |||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 6′ | |||
| α-D-Glc | 1H/13C | 5.19/92.93 | 3.77/73.54 | 3.95/75.59 | 3.87/73.48 | 3.97/71.79 | 3.89/60.57 | 3.89/60.57 | |
| β-D-Glc | 1H/13C | 4.66/96.68 | 3.48/76.41 | 3.78/77.88 | 3.88/73.54 | 3.59/76.24 | 3.82/60.66 | 3.98/60.66 | |
| β-D-Gal | 1H/13C | 4.44/102.65 | 3.49/72.02 | 3.66/73.28 | 3.91/69.18 | 3.60/75.80 | 3.74/62.36 | 3.74/62.36 | |
| α-L-Fuca | 1H/13C | 5.39/99.34 | 3.80/68.92 | 3.97/70.12 | 3.81/72.81 | 4.84/67.31 | 1.22/16.09 | ||
| α-L-Fucb | 1H/13C | 5.45/99.23 | 3.80/68.92 | 3.97/70.12 | 3.81/72.81 | 4.83/67.33 | 1.20/16.06 | ||
| (c) α-L-Fuc-(1 → 2)-β-D-Gal-(1 → 3) [α-L-Fuc-(1 → 4)]-β-D-GlcNAc-(1 → 3)-β-D-Gal-(1 → 4)-α/β-D-Glc | |||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 6′ | CH3(NAc) | ||
| α-D-Glc | 1H/13C | 5.23/92.55 | 3.59/71.87 | 3.84/72.09 | 3.65/78.96 | 3.95/70.89 | 3.91/60.22 | 3.91/60.22 | |
| β-D-Glc | 1H/13C | 4.67/96.46 | 3.29/74.54 | 3.65/75.06 | 3.66/78.92 | 3.61/75.55 | 3.81/60.82 | 3.95/60.82 | |
| β-D-Gal | 1H/13C | 4.43 /103.71 | 3.57/70.91 | 3.72/82.28 | 4.15/69.35 | 3.72/75.55 | 3.78/61.69 | 3.78 /61.69 | |
| β-D-GlcNAc | 1H/13C | 4.62/103.97 | 3.86/56.48 | 4.14/75.23 | 3.75/72.71 | 3.53/75.91 | 3.86/60.69 | 3.86/60.69 | 2.07/22.90 |
| α-L-Fucc | 1H/13C | 5.03/98.52 | 3.82/68.53 | 3.93/69.84 | 3.82/72.72 | 4.87/67.74 | 1.28/16.06 | ||
| β-D-Gal | 1H/13C | 4.67/101.35 | 3.62/77.23 | 3.82/74.38 | 3.87/69.47 | 3.58/75.49 | 3.75/62.33 | 3.75/62.33 | |
| α-L-Fucd | 1H/13C | 5.16/100.28 | 3.77/69.00 | 3.70/70.19 | 3.74/72.52 | 4.35/66.98 | 1.27/16.07 | ||
Pure shift methods allow CASPER-compatible data to be generated more efficiently, significantly speeding up the analysis of oligosaccharides. Further information to distinguish between the top-ranked CASPER structure assignments may be obtained by performing additional experiments such as PSYCHE-CPMG-HSQMBC to detect long-range 1H–13C correlations, as previously applied to a heparin analogue trisaccharide.31 Other useful experiments include pure shift NOESY32, ROESY33 and COSY32,34
Experimental
All experiments were performed at 298 K on a Bruker AVII 400 spectrometer using a 5 mm BBO probe with a nominal maximum z-gradient strength of 0.535 T m−1. The HMOs were obtained from BioCarb Chemicals, Lund, Sweden and Kemikaliebolaget KEBO AB, Stockholm, Sweden. The three oligosaccharides 3-fucosyllactose, lacto-N-difucohexaose I, and lactose were dissolved in D2O with concentrations of 54 mM, 25 mM and 450 mM, respectively. 1H and 13C chemical shifts were referenced to TSP at δH 0.0 ppm and δC at a nominal 60 ppm, as spectral aliasing was used in the indirect dimension to maximise resolution. 13C chemical shifts were corrected to account for the differences in referencing and sample temperature between the acquired data and those in the CASPER database. The PSYCHE spectrum (Fig. 1b) was acquired with 32 20 ms chunks of data, 16 transients, a 15 ppm spectral width, 2048 complex points and a recovery delay of 2 s. A saltire pulse with 70 ms duration, a 10 kHz bandwidth and a 20° flip angle was used for J-refocusing.The multiplicity-edited HSQC and real-time BIRD multiplicity-edited HSQC spectra were acquired with 4 transients, 512 increments, 2048 complex points, and spectral widths of 12 ppm and 60 ppm in the direct and indirect dimensions, respectively. A recovery delay of 3.5 s was used, 16 chunks were acquired with a 25.6 ms duration employing a BIRD J-refocusing element with a total duration of 6.8 ms; 1JCH was set to 145 Hz. TOCSY and F1-PSYCHE-TOCSY data were acquired with 4 transients, 2048 increments, 4096 points in the direct dimension, and a spectral width of 7 ppm in both dimensions. DIPSI-2 isotropic mixing was employed, with a duration of 120 ms. The pure shift selective TOCSY data were acquired using a 20 Hz RSNOB pulse to select the signals at (a) 5.23 ppm, (b) 4.67 and (c) 4.45 ppm. The PSYCHE element used a 70 ms, 10 kHz saltire pulse with a 20° flip angle applied simultaneously with a 3% gradient. The DIPSI-2 isotropic mixing duration was 150 ms, and 32 chunks were acquired with durations of 20 ms. Further information is provided in the ESI.†
Conclusions
Pure shift methods greatly improve the resolution in oligosaccharide spectra and facilitate identification of the chemical shifts of correlated spin pairs. Automatic peak-picking routines have been introduced to identify a single chemical shift for each distinct nucleus within a molecule in a multiplicity-edited pure shift HSQC spectrum, and to remove spurious signals from F1-PSYCHE-TOCSY data. These tools facilitate compatibility with the CASPER program, which was used to generate chemical shift assignments for lactose, 3-FL and LNDFH I that are in good agreement with those reported in the literature.19,30,35 All experimental data, pulse sequence code and macros in Bruker format are freely available at https://doi.org/10.48420/22270126Author contributions
All experiments were performed by MJS. All authors contributed to the analysis of results and writing of the manuscript and Electronic ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the Engineering and Physical Sciences Research Council (grant numbers EP/R018790/1 and EP/R513131/1 studentship number 2297284) and the Swedish Research Council (grant number 2017-03703) for supporting this work. For the purpose of open access, the authors have applied a Creative Commons Attribution (CC BY) licence to any Author Accepted Manuscript version arising.References
- L. Cheng, R. Akkerman, C. Kong, M. T. C. Walvoort and P. de Vos, Crit. Rev. Food Sci. Nutr., 2021, 61, 1184–1200 CrossRef CAS PubMed.
- J. T. Smilowitz, C. B. Lebrilla, D. A. Mills, J. B. German and S. L. Freeman, Annu. Rev. Nutr., 2014, 34, 143–169 CrossRef CAS PubMed.
- A. Maliniak and G. Widmalm, in Food Oligosaccharides, 2014, pp. 320–349, DOI:10.1002/9781118817360.ch18.
- L. Castañar, Magn. Reson. Chem., 2017, 55, 47–53 CrossRef PubMed.
- K. Zangger, Prog. Nucl. Magn. Reson. Spectrosc., 2015, 86–87, 1–20 CrossRef CAS PubMed.
- K. Zangger and H. Sterk, J. Magn. Reson., 1997, 124, 486–489 CrossRef CAS.
- M. Foroozandeh, R. W. Adams, N. J. Meharry, D. Jeannerat, M. Nilsson and G. A. Morris, Angew. Chem., Int. Ed., 2014, 53, 6990–6992 CrossRef CAS PubMed.
- J. R. Garbow, D. P. Weitekamp and A. Pines, Chem. Phys. Lett., 1982, 93, 504–509 CrossRef CAS.
- A. Lupulescu, G. L. Olsen and L. Frydman, J. Magn. Reson., 2012, 218, 141–146 CrossRef CAS PubMed.
- J. A. Aguilar, S. Faulkner, M. Nilsson and G. A. Morris, Angew. Chem., Int. Ed., 2010, 49, 3901–3903 CrossRef CAS PubMed.
- M. Lundborg and G. Widmalm, Anal. Chem., 2011, 83, 1514–1517 CrossRef CAS PubMed.
- A. Furevi, A. Ruda, T. Angles d'Ortoli, H. Mobarak, J. Ståhle, C. Hamark, C. Fontana, O. Engström, P. Apostolica and G. Widmalm, Carbohydr. Res., 2022, 513, 108528 CrossRef CAS PubMed.
- A. Loss, R. Stenutz, E. Schwarzer and C. W. Von Der Lieth, Nucleic Acids Res., 2006, 34, W733–W737 CrossRef CAS PubMed.
- P. Klukowski and M. Schubert, Bioinformatics, 2019, 35, 293–300 CrossRef CAS PubMed.
- V. Ayechu-Muruzabal, A. H. van Stigt, M. Mank, L. E. M. Willemsen, B. Stahl, J. Garssen and B. van't Land, Front. Pediatr., 2018, 6, 239 CrossRef PubMed.
- W. P. Aue, E. Bartholdi and R. R. Ernst, J. Chem. Phys., 1976, 64, 2229–2246 CrossRef CAS.
- G. Bodenhausen and D. J. Ruben, Chem. Phys. Lett., 1980, 69, 185–189 CrossRef CAS.
- L. Braunschweiler and R. R. Ernst, J. Magn. Reson., 1983, 53, 521–528 CAS.
- Q. Shi, J. Yan, B. Jiang, X. Chi, J. Wang, X. Liang and X. Ai, Carbohydr. Polym., 2021, 267, 118218 CrossRef CAS PubMed.
- L. Paudel, R. W. Adams, P. Király, J. A. Aguilar, M. Foroozandeh, M. J. Cliff, M. Nilsson, P. Sándor, J. P. Waltho and G. A. Morris, Angew. Chem., Int. Ed., 2013, 52, 11616–11619 CrossRef CAS PubMed.
- P. Kiraly, M. Nilsson and G. A. Morris, Magn. Reson. Chem., 2018, 56, 993–1005 CrossRef CAS PubMed.
- L. Kaltschnee, A. Kolmer, I. Timári, V. Schmidts, R. W. Adams, M. Nilsson, K. E. Kövér, G. A. Morris and C. M. Thiele, Chem. Commun., 2014, 50, 15702–15705 RSC.
- P. Sakhaii, B. Bohorc, U. Schliedermann and W. Bermel, Sci. Rep., 2021, 11, 21566 CrossRef CAS PubMed.
- M. Tafazzoli and M. Ghiasi, Carbohydr. Res., 2007, 342, 2086–2096 CrossRef CAS PubMed.
- M. Foroozandeh, R. W. Adams, M. Nilsson and G. A. Morris, J. Am. Chem. Soc., 2014, 136, 11867–11869 CrossRef CAS PubMed.
- G. Dal Poggetto, L. Castañar, G. A. Morris and M. Nilsson, RSC Adv., 2016, 6, 100063–100066 RSC.
- P. Kiraly, N. Kern, M. P. Plesniak, M. Nilsson, D. J. Procter, G. A. Morris and R. W. Adams, Angew. Chem., Int. Ed., 2021, 60, 666–669 CrossRef CAS PubMed.
- J. Ståhle and G. Widmalm, in NMR in Glycoscience and Glycotechnology, ed. K. Kato and T. Peters, The Royal Society of Chemistry, 2017, pp. 335–352, 10.1039/9781782623946-00335.
- M. Lundborg and G. Widmalm, in Glycoinformatics, ed. T. Lütteke and M. Frank, Springer New York, New York, NY, 2015, pp. 29–40, DOI:10.1007/978-1-4939-2343-4_3.
- Y. Ishizuka, T. Nemoto, M. Fujiwara, K.-I. Fujita and H. Nakanishi, J. Carbohydr. Chem., 1999, 18, 523–533 CrossRef CAS.
- I. Timári, L. Szilágyi and K. E. Kövér, Chem. – Eur. J., 2015, 21, 13939–13942 CrossRef PubMed.
- J. A. Aguilar, A. A. Colbourne, J. Cassani, M. Nilsson and G. A. Morris, Angew. Chem., Int. Ed., 2012, 51, 6460–6463 CrossRef CAS PubMed.
- J. Ilgen, J. Nowag, L. Kaltschnee, V. Schmidts and C. M. Thiele, J. Magn. Reson., 2021, 324, 106900 CrossRef CAS PubMed.
- V. M. R. Kakita and J. Bharatam, Magn. Reson. Chem., 2018, 56, 963–968 CrossRef CAS PubMed.
- S. S. van Leeuwen, B. J. H. Kuipers, L. Dijkhuizen and J. P. Kamerling, Carbohydr. Res., 2014, 400, 54–58 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Containing further experimental data, macros and guidance. See DOI: https://doi.org/10.1039/d3ob00421j |
| This journal is © The Royal Society of Chemistry 2023 |