
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advancing mesoporous carbon synthesis for supercapacitors: a systematic investigation of cross-linking agent effects on pore structure and functionality†
Yaoguang
Song
 a,
Xiaolei
Zhang
*b,
Peter A. A.
Klusener
c and
Peter
Nockemann
*a
a,
Xiaolei
Zhang
*b,
Peter A. A.
Klusener
c and
Peter
Nockemann
*a
aThe QUILL Research Centre, School of Chemistry and Chemical Engineering, Queen's University Belfast, BT9 5AG, Belfast, UK. E-mail: p.nockemann@qub.ac.uk
bDepartment of Chemical and Process Engineering, University of Strathclyde, G1 1XJ, Glasgow, UK. E-mail: xiaolei.zhang@strath.ac.uk
cShell Global Solutions International B.V., Energy Transition Campus Amsterdam, Grasweg 31, 1031 HW Amsterdam, The Netherlands
First published on 20th November 2023
Abstract
Soft-templating synthesis provides an effective route to prepare ordered mesoporous carbons (MCs) that can be used for supercapacitors. During this process, the cross-linking of carbon precursors is critical to obtain tailored pore structural MCs, thus careful selection of appropriate cross-linking agents is required. Despite the shift from the prevailing cross-linker formaldehyde to its more environmentally friendly alternatives, detailed understanding on the influence of different cross-linking agents on templating synthesis is still lacking. Therefore, it remains challenging to draw a conclusion regarding which cross-linker can effectively enable an ideal cross-linking and a robust templating synthesis of ordered MCs. This work presents a systematic study, by comparing three typical cross-linkers (formaldehyde, glyoxal, and glyoxylic acid), on the pore architecture, surface functionality, and electrochemical performance of resulting MCs. Both the type of cross-linker and its ratio with precursor monomer were found to be crucial for the pore architecture and electrochemical performance of resulting MCs. Glyoxal showed to be a promising cross-linker for easily generating ordered mesopores between 3.3–6.1 nm when the molar ratio between cross-linker and carbon precursor ranged from 1 to 2, whereas glyoxylic acid and formaldehyde induced interrupted or disordered mesopores. When the resulting MCs were used as supercapacitor electrodes, those cross-linked with glyoxal also led to overall higher capacitance in both 6 M KOH aqueous and ionic liquid [N2220][NTf2]/acetonitrile electrolytes thanks to the dominance of ordered mesopore channels, especially MC prepared at glyoxal/precursor molar ratio of 1.5. These findings on the effect of cross-linking on templating synthesis can be used to guide the customisation of MCs for supercapacitors and other applications by smartly choosing a suitable cross-linking agent and its ratio with the precursor.
Introduction
Mesoporous carbons (MCs), due to their rich porosity and tuneable ordered mesopores, are widely utilised in energy storage devices such as supercapacitors that offer high power density, fast charge and discharge rates, and long cycle life, making them ideal for a wide range of applications including electric vehicles, wearable devices, and portable electronics.1–3 To maximise the double-layer capacitance, the pore structures of MC electrodes must be tailored to the electrolyte ions as both larger and smaller pore diameters can lead to reduced double-layer capacitance.4,5 Templating synthesis provides an effective approach to customise MCs with desired pore structures, including hard-templating by a reverse replication of the topological structures of inorganic templates and soft-templating by a self-assembly of organic amphiphiles. In a typical soft-templating synthesis, templates are expected to form well-ordered columnar micelles during the self-assembly whereby the carbon precursors reside in the hydrophilic phase of the templates via H-bonding interactions, leading the hydrophobic phase to form pore structures. Notably, soft-templating synthesis has been proven to be more advantageous than hard-templating because (1) it avoids the use of corrosive NaOH or HF to remove template as required in the hard-templating route, (2) it is more tractable to tailor mesopore size, and (3) it is more capable of handling a wider range of carbon precursors, such as recalcitrant lignin from biomass.6–12To date, soft-templating synthesis of MCs relies heavily on aromatic carbon precursors, particularly phenolic polymers/resins that are derived from the cross-linking of phenolic monomers (e.g., phenol, resorcinol, and phloroglucinol). However, one of the key challenges during a soft-templating synthesis lies in realising an ideal polymerisation or cross-linking of the carbon precursor as both deficient and excessive cross-linking can hamper the formation of ordered mesoporous structures.13 Generally, small and low molecular-weight carbon precursors are more favourable to attain ordered mesopores.6,14 Therefore, the selection of appropriate cross-linkers is critical to achieve a successful templating synthesis.
Formaldehyde has been a prevailing cross-linker since the first successful practice of MC preparation.14–20 But, its high toxicity and carcinogenicity have aroused much safety concerns thus propelling the search for more eco-friendly substitutes. Foremost, not all templating synthesis using formaldehyde are able to produce ordered mesopore structures. There is still a large uncertainty during templating synthesis if the chosen cross-linker can enable a robust templating synthesis. To address this challenge, various more sustainable cross-linkers have been reported, especially metallic ions (e.g., Ni2+, Mg2+, Fe3+, Co2+, Zn2+, La3+, Ce3+) through coordination reactions.7,8,21,22 In the meanwhile, effort has also been put into exploring new synthesis procedures.23 Liang et al.24 reported the synthesis of bimodal porous carbon monoliths by a dual phase separation process. Solvent-free solid-state synthesis of ordered MCs have also been widely used, where the self-assembly of carbon precursors and templating agents were achieved via mechanochemistry by using ball milling process.21,25,26 Nevertheless, aldehyde based cross-linking via the wet assembly route using volatile organic solvents still dominate the soft-templating of MCs, with glyoxylic acid and glyoxal drawing particular attention.9,12,27–32
Glyoxylic acid is a nature-based cross-linker and was first reported by Ghimbeu et al.27 for catalyst-free soft-templating synthesis of MCs, where glyoxylic acid played a bi-functional role: cross-linker and catalyst due to the existence of both aldehyde and carboxylic acid groups. Resulting MCs exhibited a high porosity up to 800 m2 g−1 and 1.0 cm3 g−1 with tuneable pores between 0.6–7.0 nm. The cross-linking efficiency can be improved through photopolymerisation, whereby the cross-linking can be completed within 1 h at room temperature whilst with traditional thermopolymerisation at around 80 °C it takes at least 12 h.28,29 Later, the use of glyoxylic acid was extended to the preparation of N-doped MCs30 and carbon spheres.31 Despite its growing popularity, glyoxylic acid as cross-linker tends to generate interrupted or disordered mesopores and a minor change of preparation conditions (such as radiation dose and time) may cause significant deterioration in the pore morphologies.27–30
Glyoxal has been reported for producing formaldehyde-free wood adhesives.33,34 It was initially employed as a cross-linker to prepare MCs using the phloroglucinol monomer as carbon precursor where well-defined mesopores of 7.5 nm were obtained.32 Later, the use of glyoxal cross-linker to prepare MCs was extended to other carbon precursors such as lignin.9,12,34 Herou et al.9 prepared ordered hexagonal MC by replacing phloroglucinol with 50 wt% of organosolv lignin; resulting MC showed a mesopore volume and size of 0.5 cm3 g−1 and 3.9 nm with a volumetric capacitance of 90 F cm−3 in 6 M KOH electrolyte. Wang et al.12 achieved 75 wt% replacement whilst generating ordered MC with a surface area, pore volume and size of 607 m2 g−1, 0.47 cm3 g−1, and 5.0 nm, respectively. Ordered MCs have been achieved by using glyoxal as cross-linker, yet the same is true for using other cross-linkers. It remains unclear if glyoxal can enable a robust templating synthesis, especially at different synthesis conditions such as varying cross-linker/precursor mixing ratios.
While there is a clear trend on shifting from the prevailing formaldehyde cross-linker to more environmentally friendly alternatives like glyoxylic acid and glyoxal, however, the experimental conditions to synthesise MCs using different cross-linkers keep changing constantly. So far, there is a lack of comprehensive comparison on how different cross-linkers influence the templating synthesis under the same condition. It is thus challenging to draw a conclusion which cross-linker can potentially enable a robust templating synthesis through an ideal cross-linking reaction thus generate ordered mesopores more effectively. Furthermore, it also remains unclear how different cross-linking affects the capacitive performance of resulting MCs as supercapacitor electrodes. To bridge this research gap, this study compares the three aldehyde-based cross-linkers in templating synthesis, aiming to investigate the following research questions: (1) How do the more sustainable cross-linkers, glyoxylic acid and glyoxal, compare to formaldehyde in terms of their influence on pore architecture, surface functionality, and electrochemical performance of resulting MCs? (2) What are the optimal ratios of cross-linker to carbon precursor for achieving desired pore structures and electrochemical performance in MCs synthesised using glyoxylic acid and glyoxal? By addressing these questions, this work gives comprehensive insight on the role of different cross-linkers in soft-templating synthesis of MCs, thereby reaching a conclusion on which cross-linker can most effectively enable ideal cross-linking and generate ordered mesopores effectively. The findings can be used to guide the customisation of MCs for energy storage as well as other application purposes by smart selection of both the type of cross-linker and its ratio with the precursor.
Experimental
Preparation of mesoporous carbons (MCs)
The preparation of MCs followed an evaporation-induced self-assembly (EISA) technique. Phloroglucinol-derived aromatic polymer was used as carbon precursor as phloroglucinol preferentially resides in the hydrophilic phase of the cylindrical micelles formed by soft template, favouring the formation of mesopore structures.35 The well-established block copolymer Pluronic F127 was used as the soft template in this work. The mass ratio between phloroglucinol and Pluronic F127 was kept around 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, which is an empirical ratio that is able to generate ordered MCs.9,27,36,37
:2, which is an empirical ratio that is able to generate ordered MCs.9,27,36,37
For a typical procedure, 0.82 g (6.50 mmol) of phloroglucinol (1,3,5-trihydroxybenzene, Sigma-Aldrich) and subsequently 1.71 g (0.137 mmol) of Pluronic F127 (Sigma-Aldrich) were dissolved in 40 mL of absolute ethanol at room temperature. After stirring for 30 min, 6.50 mmol of cross-linker (i.e., 0.94 g of glyoxal (40 wt% aq.), 0.53 g of formaldehyde (37 wt% aq.), or 1.50 g of glyoxylic acid monohydrate) was added and vigorous stirring was continued at room-temperature for 2 h. The resulting homogeneous solution was poured into a smooth glass Petri dish (12 cm in diameter) and placed at 30 °C for 24 h to allow ethanol to evaporate slowly. A homogeneous viscous mixture was yielded, then subsequently cured at 85 °C for 24 h under a convection oven of air. After the cross-linking reaction, the resulting transparent polymer film was roughly chopped into chunks and calcinated at 800 °C for 1 h after a heating-up rate of 1 °C min−1 under N2 flow (100 mL min−1). During the preparation, the only variables are the type of cross-linker used and the molar ratio between cross-linker and phloroglucinol (C/P ratio). Resulting MCs were labelled as Gx, Fx, and GAx, where G, F, and GA denote the cross-linkers namely glyoxal, formaldehyde, and glyoxylic acid, respectively, with the letter x indicating the C/P ratio (1 ≤ C/P ≤ 2) in mol mol−1.
Preparation of ionic liquid (IL) electrolyte
IL triethylammonium bis(tetrafluoromethylsulfonyl)amide, [N2220][NTf2], was synthesised using an exothermic acid–base reaction and metathesis reaction.38 Triethylamine (0.5 mol, Sigma-Aldrich) was introduced into a 500 mL three-neck flask, equipped with a reflux condenser and immersed in an ice bath, then HCl (37 wt%, aq., 0.5 mol) was added dropwise under vigorous stir. After further stirring under room temperature for another 2 h, the mixture was diluted with deionised water and added with lithium bis(tetrafluoromethylsulfonyl)amide (Li[NTf2], 0.51 mol). Slight excess of Li[NTf2] was used to guarantee a complete replacement of chloride. After stirring for 2 h, the solution was mixed with chloroform (100 mL) then transferred into a separating funnel, where two phases were obtained with the aqueous phase dumped. The chloroform phase was washed with water several times until the AgNO3 test showed no precipitation in the aqueous phase. Chloroform was removed using a rotavapor, and the product was dried under a vacuum oven until no impurity detected by nuclear magnetic resonance (NMR) spectroscopy. Resulting [N2220][NTf2] was stored in glovebox for use.MCs characterisation
N2 physisorption analysis was carried out at 77 K under a TriStar II 3020 analyser with pore size distribution calculated using the Barrett–Joyner–Halenda (BJH) method from the adsorption branches of N2 isotherms. Pore morphologies were studied by scanning electron microscopy (SEM) with FEI Quanta 250 FEG scanning electron microscope, and transmission electron microscopy (TEM) with TALOS F200X G2 microscope. High-angle annular dark-field (HAADF) imaging technique was used to obtain scanning transmission electron microscopy (STEM) images with the periodicity of mesopore channels measured by HAADF intensity line profile. Infrared (IR) spectroscopy and X-ray photoelectron spectroscopy (XPS) were used to measure surface functionalities, employing a PerkinElmer Spectrum 100 FT-IR Spectrometer with a universal ATR sampling accessory, and a Thermo Scientific™ ESCALAB™ QXi X-ray Photoelectron Spectrometer, respectively. Each XPS spectrum was obtained by averaging at least 4 random independent measurements. The graphitisation degree was measured by Raman spectroscopy on a WlTec Alpha 300 Raman microscope equipped with a 532 nm laser (10 mW laser power). All spectra were averaged from at least 3 random independent accumulations (50 s accumulation time for each).Electrochemical performance analysis
The resulting MCs were evaluated as supercapacitor electrodes in a symmetric two-electrode Swagelok cell device for their electrochemical performance. 6 M KOH and [N2220][NTf2]/acetonitrile (weight fraction of acetonitrile, ACN: 0.5) solutions were chosen as representative electrolytes. Each electrode consisted of 90% of MCs, 5% of PTFE binder, and 5% of multi-walled carbon nanotubes (MWCNT) as conductive material and was formed into a homogeneous, thin, free-standing pellet by pressing for 5 minutes at 2 tons (10 mg in weight, 13 mm in diameter, ∼120 μm in thickness). The electrodes were separated by a GF/F glass microfiber filter (Whattman®, 15 mm in diameter, 420 μm in thickness) soaked with electrolyte. With KOH electrolyte, the cell was assembled and tested in an ambient environment, undergoing 500 cycles of running at 5.0 A g−1 prior to analysis to maximise the accessible pores for electrolyte. With [N2220][NTf2]/ACN electrolyte, the cell was assembled and tested in a glovebox (water content: <0.3 ppm, oxygen content: <0.1 ppm) without initial cycling. The charge/discharge efficiency was expressed by the ratio of discharge time (td) to charge time (tc): η = td/tc. Gravimetric specific capacitance (Cs, F g−1) was calculated from galvanostatic charge–discharge curve: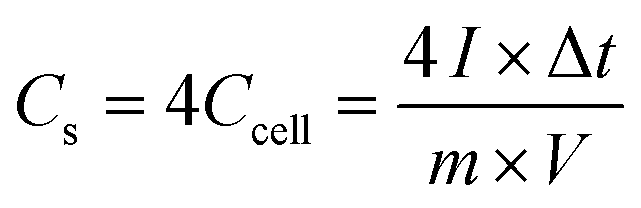
Energy density (E, W h kg−1) and power density (P, W kg−1) were obtained by
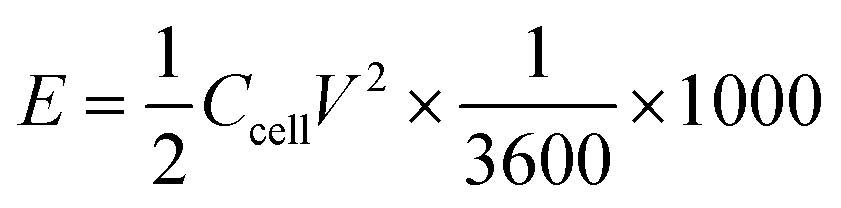
where Δt (s), I (A), m (g), and V (V) represent the discharge time, discharge current, total mass of active materials on both electrodes, and the potential window.
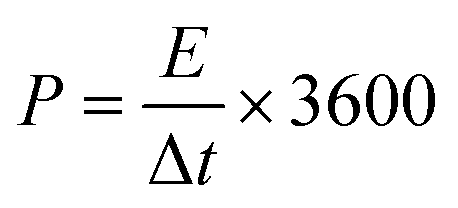
Results and discussion
Pore architecture
The pore architectures of carbon samples, prepared by employing various cross-linkers at different cross-linker/precursor ratios (C/P ratios) were analysed by N2 physisorption analysis and electronic microscopies; the results are summarised in Table 1. Fig. 1 shows the N2 physisorption isotherms and BJH pore size distributions of all the resulting carbons. The shapes of the N2 physisorption isotherms, except that for sample GA1.25 (prepared by using glyoxylic acid as cross-linker at a C/P ratio of 1.25), exhibit typical Type IV isotherms, evidenced by the pronounced hysteresis loops that suggest the presence of dominant mesopores.40 By contrast, sample GA1.25 exhibited a Type I isotherm, indicative of a microporous materials. Moreover, the N2 adsorption volume of the resulting carbons varied significantly depending on the cross-linkers and C/P ratios used. This observation highlights the crucial role of both the cross-linkers and the C/P ratio in the pore architectures of the resulting MCs.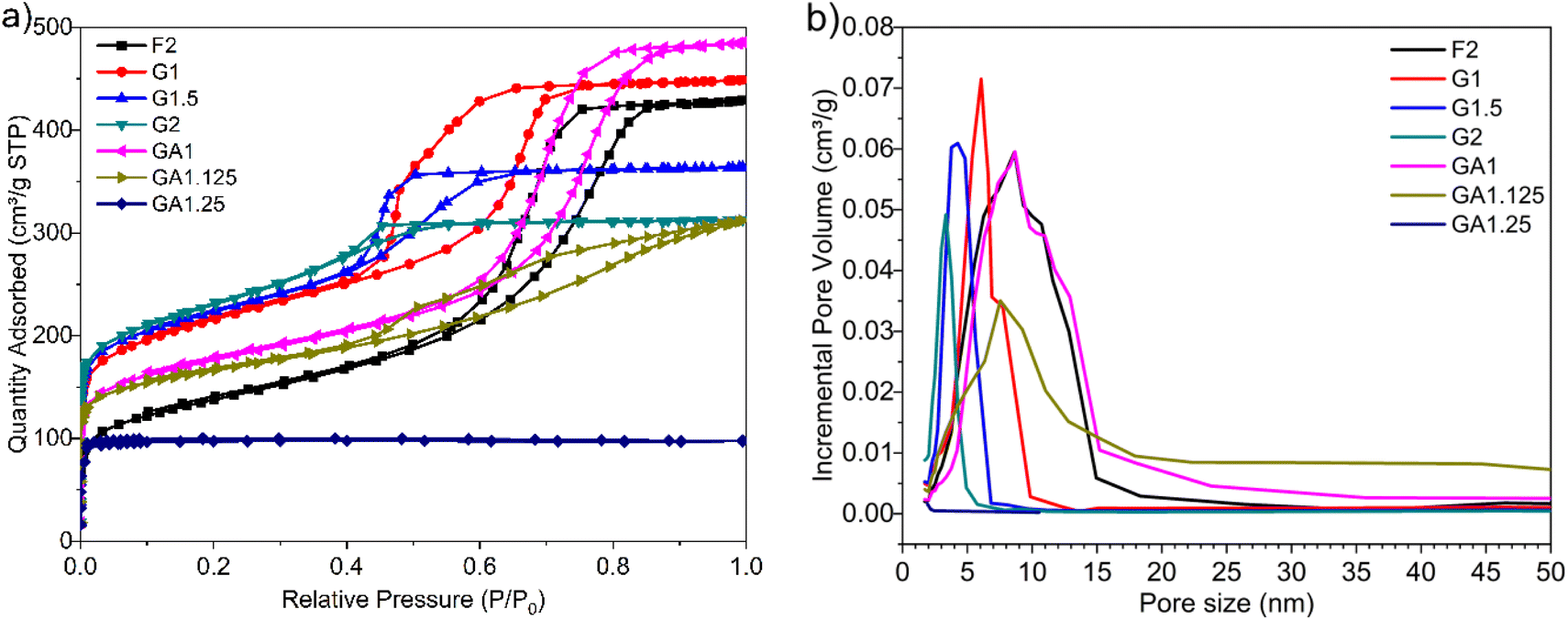 | ||
| Fig. 1 (a) N2 physisorption isotherms and (b) BJH pore size distributions of all resulting MCs. | ||
| Samples | Pore textural properties | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| BET area39 (m2 g−1) | S meso (m2 g−1) | S micro (m2 g−1) | V total (cm3 g−1) | V meso (cm3 g−1) | V micro (cm3 g−1) | V meso/Vtot | Dominant pore sizea (nm) | Pore uniformity | |
| a The dominant pore size is represented by the peak pore size from BJH pore size distribution using the adsorption branches of the N2 isotherms. | |||||||||
| F2 | 478 | 367 | 129 | 0.66 | 0.60 | 0.06 | 91% | 8.6 | Disordered |
| G1 | 753 | 436 | 316 | 0.69 | 0.54 | 0.14 | 78% | 6.1 | Ordered hexagonal |
| G1.5 | 794 | 424 | 367 | 0.56 | 0.38 | 0.16 | 68% | 4.3 | Ordered hexagonal |
| G2 | 821 | 400 | 357 | 0.48 | 0.28 | 0.15 | 58% | 3.3 | Ordered hexagonal |
| GA1 | 622 | 349 | 271 | 0.75 | 0.62 | 0.12 | 83% | 8.7 | Less ordered |
| GA1.125 | 597 | 246 | 332 | 0.48 | 0.32 | 0.14 | 67% | 7.5 | Less ordered |
| GA1.25 | 393 | 6 | 372 | 0.15 | 0.01 | 0.14 | 7% | — | Microporous |
When employing glyoxal as cross-linker, a decrease in the N2 adsorption volume was observed as the C/P ratio increased, demonstrating reduced porosity. As shown in Table 1, the total pore volume of glyoxal-derived MCs decreases from 0.69 to 0.48 cm3 g−1 as the C/P ratio doubled from 1 to 2, although total BET area increased from 753 to 821 m2 g−1. Notably, the mesopore rate decreased from 78% to 58%, accompanied by a reduction in mesopore surface area (from 436 to 400 m2 g−1) and mesopore volume (from 0.54 to 0.28 cm3 g−1). Furthermore, the increasing C/P ratio also induced a shrinkage in the pore size (Fig. 1b), which dropped in the order of G1 > G1.5 > G2, with diameters of 6.1, 4.3, and 3.3 nm, respectively. The narrow pore size distributions (Fig. 1b) imply the formation of uniform and well-ordered mesopores. As shown in Fig. 2 and Fig. S1–S3,† glyoxal-derived MCs showed highly ordered mesopore channels in honeycomb-like hexagonal (p6m) pattern (Fig. 2a–c, Fig. S1a–c†). Sample G1 showed a periodicity of 10.5 nm, with a pore diameter and pore wall of 6.1 and 4.4 nm, respectively (Fig. S3a and b†). When the C/P ratio increased to 1.5, the periodicity decreased to 8.7 nm with a slightly reduced pore diameter of 4.3 nm (Fig. S3c and d†). Sample G2 showed a continued trend of a smaller periodicity of 8.2 nm and narrower pores of 3.3 nm (Fig. S3e and f†). The pore diameters measured by STEM line profile coincide exactly with the calculated pore sizes from N2 physisorption analysis. The observed shrinkage in mesopores is associated with the increasing C/P ratio, which is the only variable. As precursor monomer – phloroglucinol – tends to reside in the hydrophilic outer shell of the micelles formed by templates, leaving the hydrophobic phase to generate pores after carbonisation,13,35 adding more cross-linkers can enhance the polycondensation of precursors; this possibly increased the tractive force uniformly around the hydrophobic phase and eventually caused an overall shrinkage in both the polymer and hydrophobic phase therein (i.e. pores after carbonisation).
 | ||
| Fig. 2 TEM images of MCs: (a–f) G1, G1.5, G2, F2, GA1, and GA1.125, respectively. | ||
When using formaldehyde as cross-linker with C/P ratios below 2, thermopolymerisation at 85 °C for 24 h did not yield homogenous polymer film, but rather a highly viscous matrix (adhesive) at C/P ratio of 1 and 1.5. Formaldehyde possesses only one aldehyde group per molecule, therefore a C/P ratio of 1 is the theoretical threshold to enable the polymerisation, forming linear polymer fragments; a molar ratio between 1 < C/P < 2 enables the bridging of linear fragments, yielding 3D polymer networks. However, even with a C/P ratio of 1.5, the polymers did not fully cross-link within 24 hours. Typically, the polymerisation of phenolic monomer with formaldehyde takes place in the presence of a catalyst.14–16,18,20 Without using a catalyst, a C/P ratio of 1.5 only led to a deficient cross-linking in 24 h, albeit yielding a visually more viscous mixture than a C/P ratio of 1 mol mol−1. As the C/P ratio reached 2, a 3D solid polymer film formed and resulting carbon sample F2 showed a mesopore rate of 91%, with a BET area and pore volume of 478 m2 g−1 and 0.66 cm3 g−1, respectively. However, sample F2 exhibited no uniformity in pore structures but randomly disordered mesopores peaked at 8.6 nm (Fig. 2d, Fig. S1d†). Although the ratio of aldehyde groups used for preparing sample F2 equalled to that for G1, the resulting pore morphologies were much different, namely highly disordered in F2 as opposed to hexagonal ordered in G1. Such difference can be attributed to the inherent structural difference between glyoxal and formaldehyde molecules. The two aldehydes in glyoxal are connected by a C–C bond, therefore the aldehyde group from formaldehyde may have a higher reactivity than those from glyoxal. Glyoxal was implied as a slower reacting aldehyde by stabilising the reaction intermediates during cross-linking.32
Glyoxylic acid also provides only one aldehyde per molecule, but solid polymer films formed after cross-linking within 24 h at lower C/P ratios. By directly comparing the molecular structure of glyoxylic acid with formaldehyde, such faster cross-linking can be attributed to the existence of carboxylic acid which can serve as acid catalyst for the templating synthesis27,41 and promoted the polymerisation process by enhancing the formation of hemiacetals. Besides, as found in glyoxal-derived MCs, total N2 adsorption volume and pore size also followed similar trend in glyoxylic acid-derived MCs and dropped when the C/P ratio slightly increased (Fig. 1). Sample GA1 exhibited a larger adsorption volume thus larger total pore and mesopore volume of 0.75 and 0.62 cm3 g−1. The porosity significantly deteriorated as the C/P ratio increased slightly from 1 to 1.25, where total pore volume and mesopore rate dropped from 0.75 cm3 and 83% to 0.15 cm3 and 7% only and the pore size dropped from 8.7 nm (GA1) to 7.5 nm (GA1.125), further to micropore region (GA1.25). Such strong deterioration in the porosity is associated with the catalytic ability of glyoxylic acid, which may be too strong for the cross-linking, producing over-cross-linked precursors. Excessive cross-linking can force precursors being detached from templates thereby destroying the ordered mesostructures.13 As shown in Fig. 2e and Fig. S1e,† sample GA1 displayed interrupted worm-like cylindrical mesopore channels. Despites being less ordered, mesopores in GA1 were still periodic with an interval of 12.5 nm (Fig. S3g and h†). The estimated pore width was 5.4 nm, smaller than the value of 8.7 nm from N2 physisorption analysis; the difference was mainly caused by the distortion of pore channels or the discontinuity of pore walls. As the C/P ratio increased, sample GA1.125 nearly lost its uniformity in pore channels, displaying disordered mesopores (Fig. 2f, Fig. S1f†). Finally, mesopores were hardly noticeable anymore except micropores in GA1.25 (Fig. S1g and S2†) due to the strong catalytic ability of glyoxylic acid.
Comparing the three cross-linkers at the same C/P ratio of 1, only glyoxal yielded highly ordered mesopores, whilst formaldehyde failed to cross-link precursors sufficiently to produce solid precursor matrix and glyoxylic acid induced slightly over-cross-linked precursors. Although glyoxal provides two aldehydes per molecule, the existence of carboxylic acid in glyoxylic acid escalated the cross-linking reaction and slightly destabilised the ordered mesophase formed after self-assembly, leading to a less ordered mesopores after carbonisation. At higher C/P ratios, glyoxal-derived MCs still maintained highly ordered mesopore channels with rich porosity, as opposed to the random micropores in glyoxylic acid-derived sample and disordered mesopores in formaldehyde-derived MC. From this aspect, glyoxal is proved to be a promising cross-linker to substitute carcinogenic formaldehyde for green synthesis due to its easily generating tuneable ordered mesopores.
Surface functionality
Given the distinct molecular structures of the three aldehyde-based cross-linkers, they can potentially result in different functional groups in the resulting polymer precursor such as hydroxyl, aldehyde, and carboxylic acid groups (Fig. 3). After carbonisation, these organic groups are likely to introduce residual surface functionalities on MCs and cause pseudo-capacitive behaviour when MCs are used as supercapacitor electrode materials. Therefore, the surface environment of all resulting MCs was studied by performing XPS and FTIR analysis, thereby revealing the influence of different cross-linkers on surface functionalities of MCs.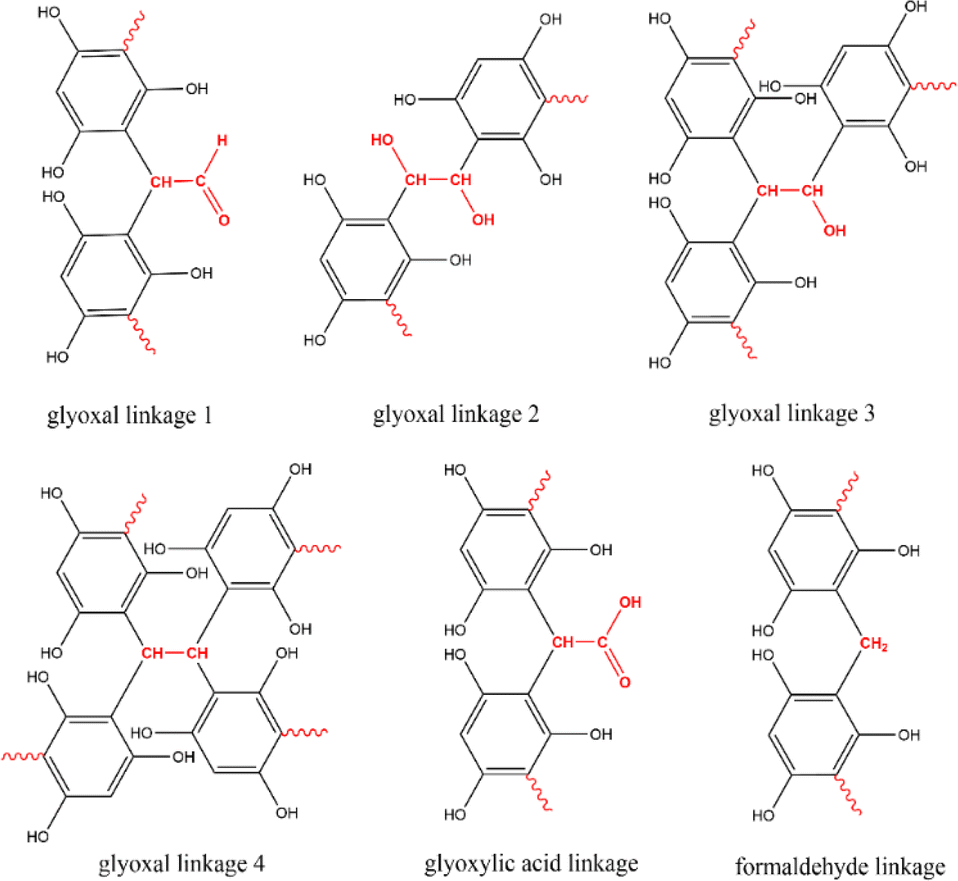 | ||
| Fig. 3 Envisaged linkages formed by different cross-linkers during the cross-linking with phloroglucinol monomer. | ||
The XPS study (Table 2, Fig. S4 and S5†) indicated that all MCs contain a dominant C element of over 93.5% and an O element between 4.2–6.5%. Deconvolution of C 1s and O 1s spectra in XPS spectra (Fig. S4 and S5†), as well as IR spectra (Fig. S6†), indicated that major oxygen-containing functionalities are –C–O, –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and –COO, respectively. With glyoxal as the cross-linker, the peak for –C–O bond at 1020 cm−1 increased (Fig. S6a†) and the content for –C–O bond increased from 4.5% (in G1) to 5.4% (in G2) as the C/P ratio doubled, whilst that for –CO bond at 1560 cm−1 showed an overall reducing trend. This mainly originated from the reaction of the –CO bonds from aldehyde groups, yielding –C–O bonds during cross-linking. With equal molar ratio of aldehyde groups to phloroglucinol, sample F2 showed a higher –C–O content (5.2%) compared with the values for G1 (4.5%). As the two aldehyde groups in glyoxal are bonded together, aldehyde from formaldehyde has higher reactivity during cross-linking,32 thus likely generating more –C–O bonds after the reaction of the –CO bonds. When glyoxylic acid was employed as cross-linker, the peak for –C–O bond in sample GA1 became slightly higher than that for G1 (Fig. S6b†), in line with the higher –C–O content for sample GA1 (4.8%). Although glyoxal provides two aldehydes per molecule, the existence of inherent –COOH in glyoxylic acid not only catalysed more aldehydes to participate in the cross-linking but also resulted in a slightly higher –COO content (6.2%) than G1 (5.6%). Overall, the difference in oxygen-containing groups resulting from different cross-linkers at varying C/P ratios does not appear to be as evident as in pore architectures.
O, and –COO, respectively. With glyoxal as the cross-linker, the peak for –C–O bond at 1020 cm−1 increased (Fig. S6a†) and the content for –C–O bond increased from 4.5% (in G1) to 5.4% (in G2) as the C/P ratio doubled, whilst that for –CO bond at 1560 cm−1 showed an overall reducing trend. This mainly originated from the reaction of the –CO bonds from aldehyde groups, yielding –C–O bonds during cross-linking. With equal molar ratio of aldehyde groups to phloroglucinol, sample F2 showed a higher –C–O content (5.2%) compared with the values for G1 (4.5%). As the two aldehyde groups in glyoxal are bonded together, aldehyde from formaldehyde has higher reactivity during cross-linking,32 thus likely generating more –C–O bonds after the reaction of the –CO bonds. When glyoxylic acid was employed as cross-linker, the peak for –C–O bond in sample GA1 became slightly higher than that for G1 (Fig. S6b†), in line with the higher –C–O content for sample GA1 (4.8%). Although glyoxal provides two aldehydes per molecule, the existence of inherent –COOH in glyoxylic acid not only catalysed more aldehydes to participate in the cross-linking but also resulted in a slightly higher –COO content (6.2%) than G1 (5.6%). Overall, the difference in oxygen-containing groups resulting from different cross-linkers at varying C/P ratios does not appear to be as evident as in pore architectures.
| Samples | O content (at%) | C content (at%) | Different C types (at%) | ||||
|---|---|---|---|---|---|---|---|
| CC |
C–C | C–O | CO |
O–CO |
|||
| F2 | 4.7 | 95.3 | 80.9 | 7.8 | 5.2 | 3.9 | 2.2 |
| G1 | 5.2 | 94.8 | 84.9 | 4.6 | 4.5 | 3.5 | 2.5 |
| G1.5 | 4.8 | 95.2 | 81.5 | 6.6 | 5.0 | 4.1 | 2.8 |
| G2 | 5.6 | 94.4 | 79.8 | 7.9 | 5.4 | 4.2 | 2.7 |
| GA1 | 4.2 | 95.8 | 80.5 | 7.8 | 4.8 | 4.1 | 2.8 |
| GA1.125 | 4.8 | 95.2 | 82.4 | 6.3 | 4.5 | 3.9 | 2.9 |
| GA1.25 | 6.5 | 93.5 | 79.4 | 12.7 | 3.4 | 2.5 | 2.0 |
Electrochemical performance
The electrochemical performance of MCs prepared by employing different cross-linkers at varying C/P ratio was investigated as supercapacitor electrode materials in 6 M KOH and [N2220][NTf2]/acetonitrile (weight fraction of acetonitrile, ACN: 0.5) electrolytes. Aqueous KOH is an extensively studied electrolyte due to its high ionic conductivity, whilst protic IL electrolytes provide larger operating windows and energy density than aqueous electrolytes and better conductivity than aprotic ILs.42–44 Mixing with organic mediators further enhances the charge transfer and ionic conductivity by significantly decreasing the viscosity (e.g. adding 50% of ACN into [N2220][NTf2] delivers a maximum conductivity).38,45 With different types of electrolytes, the role of cross-linking in templating synthesis of MCs for energy storage can be manifested in detail.Cyclic voltammetry (CV) tests were first carried out in 6 M KOH aqueous electrolyte at different scan rates: 5, 10, 20, 50, 100, 200, and 500 mV s−1 (Fig. S7†). As shown in Fig. 4a, at a low scan rate of 5 mV s−1, all resulting MCs except GA1.25 displayed nearly symmetric rectangular curves, demonstrating highly reversible charge–discharge response, which are typical behaviour of electric double-layer capacitors (EDLCs).9,46,47 By contrast, the CV curve for the GA1.25 electrode was non-symmetric triangular rather than rectangular, demonstrating a poor reversibility and poor charge–discharge response. Besides, no obvious redox peaks were observed in the CV curves of all samples; the presence of oxygen-containing surface functionalities apparently did not introduce obvious pseudocapacitive behaviour thus the difference in the content of surface functionalities in each sample could be less significant. Moreover, glyoxal-derived MC electrodes demonstrated higher charging/discharging currents than those prepared with glyoxylic acid and formaldehyde, especially sample G1.5 which exhibited the highest charging/discharging currents thus was expected to deliver the largest capacitance at lower scan rates. Since there was no obvious pseudocapactive behaviour observed, different electrochemical performance can be reasonably attributed to the difference in pore architectures resulting from using different cross-linkers at varying C/P ratios. Amongst the three cross-linkers, only glyoxal resulted in highly ordered mesopore channels, which is mostly likely associated with the better electrochemical performance. Similarly, the poor reversibility and charge–discharge response for sample GA1.25 was likely attributed to its small porosity of 0.15 cm3 g−1. This limited porosity can inhibit the ionic transport within pores and ion adsorption/desorption on pore surface, resulting in lower performance.
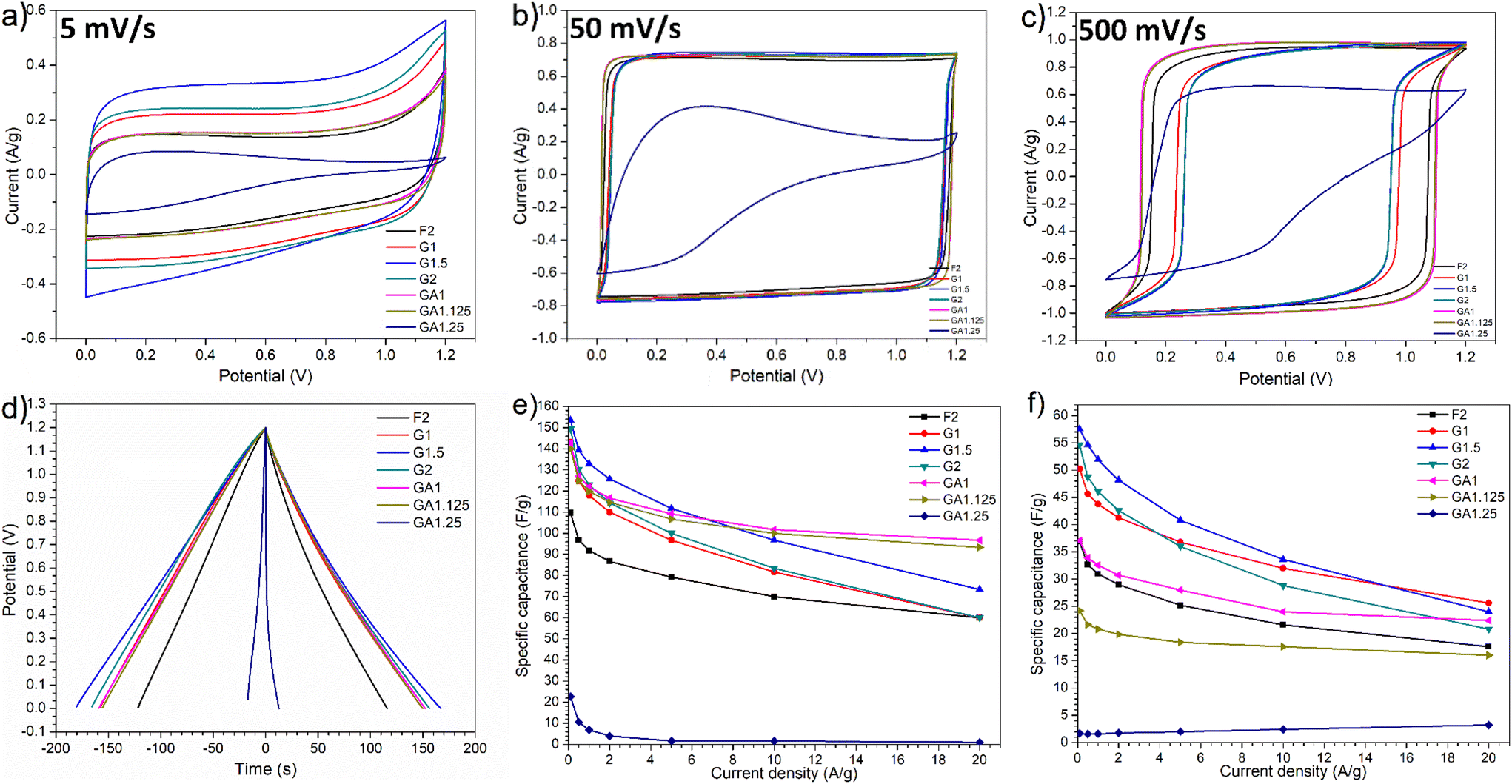 | ||
| Fig. 4 Electrochemical performance of MC electrodes: (a–c) CV test at scan rates of 5, 50, and 500 mV s−1, (d) galvanostatic charge/discharge at 0.5 A g−1, (e) gravimetric capacitance in 6 M KOH electrolyte; (f) gravimetric capacitance in [N2220][NTf2]/ACN electrolyte. | ||
At a higher scan rate of 50 mV s−1, CV curves for MCs, except GA1.25 as electrode, were more rectangular shaped and demonstrated nearly ideal efficient EDLC behaviour with excellent charge–discharge kinetics (Fig. 4b). As the scan rate increased to 500 mV s−1 (Fig. 4c), rectangular shaped CV curves generally remained except for GA1.25, which still showed triangular shape due to low porosity. Noticeably, there was a delayed response in the charge–discharge process of all MC electrodes. Under this circumstance, ionic transport or shuttle in mesopores may not be rapid enough to match up with the fast scan rates, leading to a delayed formation of electric double-layers. Such phenomenon was more evident in glyoxal-derived MC electrodes, where the length of ordered mesopore channels can usually reach a magnitude of a few micrometers with the same orientation (Fig. 5). Compared with shorter mesopores, longer channels were reported to cause a slower mass transfer, higher electrochemical resistance, poor cycling performance, and lower rate capability.48 Therefore, during fast charging/discharging, it takes longer time for ionic transport due to longer penetration depth for the adsorption/desorption of solvated K+ and OH− ions in deeper pores. Moreover, cavitation is likely occurring in glyoxal-derived MCs during fast charging/discharging due to the absence of pore connectivity effect; the constriction of highly ordered mesopore channels with a length of micrometres may lead to pore blocking or cavitation-induced desorption behaviour.49 When glyoxylic acid and formaldehyde were used as the cross-linker, obtained mesopores were either disordered or interrupted thus were well-interconnected with neighbouring pores. This provided an improved in-pore diffusivity and the formation of electric double-layers were faster. Therefore, better electrochemical performance with higher capacitance at faster scan rates was expected for MCs prepared by glyoxylic acid or formaldehyde than those prepared by glyoxal. Existing studies have confirmed this phenomenon by improving the pore network connectivity through additional activation processes, especially CO2 activation, which can effectively unblock and expand the existing porosity, widen narrow mesopores and thus the desorption rate can be enhanced during fast charging/discharging.26,50
 | ||
| Fig. 5 TEM images of ordered mesopore channels in glyoxal-derived sample G1.5 (double blue lines indicate the orientation of 5 neighbouring mesopore channels, which reached a magnitude of micrometres in depth). | ||
Consistent with the CV test, all MC electrodes demonstrated symmetric triangular galvanostatic charge–discharge profiles with fairly linear slope (Fig. 4d), which is a typical feature of EDLCs. At a lower current density of 0.5 A g−1, glyoxal-derived MC electrodes exhibited longer charge–discharge time, particularly for sample G1.5. Fig. 4e summarises the gravimetric specific capacitance of MCs at various current densities. Starting from 0.1 A g−1, glyoxal-derived MC electrodes exhibited higher capacitances with values of 143.1, 153.5, and 149.3 F g−1 for G1, G1.5, and G2, respectively. At higher current densities above 10 A g−1, glyoxylic acid-derived samples GA1 and GA1.125 showed larger capacitance ranging from 93.3 to 101.7 F g−1, because of their faster charge–discharge response to higher scan rate/current density. At a high current density of 20 A g−1, the capacitance for glyoxal-derived MCs dropped drastically with values nearly halved compared with the initial values at 0.1 A g−1, implying a relatively limited rate capability. This could be associated with slower mass transfer hence slower charge–discharge kinetics in longer mesopore channels at higher scan rates or current densities.48 As expected, GA1.25 electrode exhibited lowest capacitance throughout the test. Whereas sample F2 showed a high pore volume with larger mesopore rate, the capacitance was low, which is probably because of its relatively small surface area and disordered large mesopores.
Moussa et al.30 employed glyoxylic acid cross-linker to prepare N-doped MCs with the highest capacitance of 97 F g−1 in H2SO4 electrolyte. In comparison, out work, despite not involving N-doping, showed that the glyoxylic acid-derived carbon GA1 still exhibited a larger capacitance of 146.7 F g−1 in a KOH electrolyte. Herou et al.9 prepared ordered MC from glyoxal–phloroglucinol–polymer and the gravimetric capacitance in KOH electrolyte reached around 75 F g−1; the value was further increased to around 100 F g−1 by replacing phloroglucinol with 50 wt% of lignin. The increase in capacitance is likely due to the decrease in mesopore size from 7.3 to 3.9 nm since replacing phloroglucinol with 50 wt% of lignin also significantly decreased the pore volume and surface area. With the same electrolyte, pore size very close to electrolyte ions could lead to a maximum capacitance.4 Nevertheless, their results are still lower than the value of glyoxal-derived G1.5 (153.5 F g−1) in this work as the pore volume of their sample (0.34 cm3 g−1) is much lower than sample G1.5 (0.56 cm3 g−1). It is worth noting that the record capacitance value of mesoporous carbon electrodes, to our best knowledge, has reached 855 F g−1 in aqueous electrolyte so far,47 which is notably higher than sample G1.5. Since this work primarily emphasise on investigating the role of crosslinking in templated synthesis of mesoporous carbons, further optimisation of their capacitance can be conducted in the future with the goal of surpassing the record value when considering practical applications.
To confirm the role of cross-linking in the pore architecture of MCs and consequent capacitive abilities for energy storage, the electrochemical performance of all MCs prepared with different cross-linkers at varying C/P ratio was also measured in IL triethylammonium bis(tetrafluoromethylsulfonyl)amide/acetonitrile electrolyte, ([N2220][NTf2]/ACN with a weight fraction of ACN: 0.5). Detailed CV curves at various scan rates from 5 to 500 mV of each sample are shown in Fig. S8.† Similarly to the CV test in KOH electrolyte, the nearly rectangular shaped CV profiles suggest excellent reversible EDLC behaviour and no evident pseudo-capacitive behaviour was observed.9,46,47 Despite of a small porosity in GA1.25, CV curves are rectangular shape in IL electrolyte, as opposed to the non-symmetric shape in KOH electrolyte. Glyoxal-derived MCs, thanks to their highly ordered mesopores, allowed for higher charging/discharging currents at various scan rates (Fig. S9†). As expected, G1.5 electrode displayed a highest charging/discharging current whilst GA1.25 electrode displayed the lowest. At a lower scan rate of 5 mV s−1, there was a slight and irreversible oxidative peak over 2.0 V, which may derive from the slow decomposition of acetonitrile due to the effect of polarisation at higher voltages as acetonitrile is quite stable up to 2.5 V with oxidation normally occurring above 2.7 V.38
Brandt et al.51 studied activated carbon-based supercapacitors with a cycling stability over 30000 cycles in [N2220][NTf2] electrolyte, where pseudo-capacitive behaviour was noticed due to the presence of water in electrolyte and the surface groups on activated carbon electrodes. When mixing acetonitrile in [N2220][NTf2], no obvious reversible pseudo-capacitive behaviour was detected in this work although MCs contained oxygen content of 4.2–6.5%. As shown in Fig. 4f, glyoxal-derived MCs still exhibited larger capacitance (than that of glyoxylic acid and formaldehyde), with a maximum value of 57.5 F g−1 for G1.5 at 0.1 A g−1, which dropped by nearly 50% at 20 A g−1. Due to the small porosity, sample GA1.25 barely showed capacitance as achieved in KOH electrolyte.
In neat [N2220][NTf2] electrolyte, Timperman et al.38 reported gravimetric capacitances of 125 F g−1 and 144 F g−1 with activated carbon electrodes when the potential windows were 2 V and 3 V, respectively. Mixing acetonitrile (weight fraction 0.5) in [N2220][NTf2] slightly increased the capacitance to 128 F g−1 with a potential window of 2 V. The larger capacitance values of their work were mainly attributed to the high surface area of electrode materials (1500 m2 g−1) hence a higher potential for charge accumulation. Similarly, Sathyamoorthi et al.45 employed activated carbon electrodes (surface area of 965 m2 g−1) in [N2220][NTf2] electrolyte, and the capacitance and energy density improved from 42 F g−1 and 18.4 W h kg−1 in neat [N2220][NTf2] to 72 F g−1 and 31.2 W h kg−1 when mixed with 0.3 M hydroquinone.
Overall, glyoxal proved its potential as a cross-linker for generating MCs with overall higher capacitance in both aqueous KOH and [N2220][NTf2]/ACN electrolytes, compared to those prepared using formaldehyde and glyoxylic acid. Therefore, the choice of cross-linker is crucial for the soft-templating synthesis of MCs for supercapacitor applications, particularly with respect to the effectively forming of highly ordered mesopore channels.
Whilst using the same cross-linker, the electrochemical performance of ordered MCs prepared at varying C/P ratios was also significantly different. It is envisaged that the C/P ratio used for the cross-linking also plays a crucial role in determining the electrochemical performance of resulting MCs. For glyoxylic acid-derived MCs, a slight increase in C/P ratio from 1 to 1.25 showed a rapid diminishing effect in the porosity, pore morphology, and pore size. Consequently, there was a strong deterioration in the capacitance of resulting carbons, especially with [N2220][NTf2]/ACN electrolyte (Fig. 4e and f). For glyoxal-derived MCs, as the C/P increased from 1 to 2, a steady decrease in the total pore volume, mesopore rate, mesopore size was also noticeable in the order of G1 > G1.5 > G2. Amongst ions within two different electrolytes (namely K+, OH−, [N2220]+, and [NTf2]−), IL anion [NTf2]− has the largest dynamic diameter in the longest dimension of 0.79 nm for the neat ion and 2.24 nm when solvated in acetronitrile.52 Ordered mesopores in sample G1.5 (4.3 nm) may match relatively better with electrolyte ions than those in G1 (6.1 nm), as pores very close to the solvated electrolyte ion size are expected to deliver a maximum double-layer capacitance.4 A similar trend was also reported by Herou et al.,9 where the capacitance increased considerably when the mesopore size decreased, albeit with a substantial decrease in the porosity. Following this trend, sample G2 possessed even smaller mesopores of 3.3 nm to be closer to match both electrolytes than G1.5 but did not exhibit overall higher capacitance. This does not contradict with previous trends; there would be a detrimental effect on the capacitance when the pore diameter continues decreasing to below twice the solvated ion size due to (1) a disrupted compact ion layers on adjacent pore walls and (2) a diminished surface area available for double-layer formation.5 Besides, the pore volume of G2 (0.48 cm3 g−1) was also smaller than G1.5 (0.56 cm3 g−1). Sample G1.5 (4.3 nm) possessed mesopores very close to twice the largest solvated [NTf2]− ion, thus the formation of compact ion layers on both adjacent pore walls may have contributed to its overall larger capacitance. Upon weighing up pore sizes to match with electrolyte ions and pore volume for ion accumulation, sample G1.5 as electrode demonstrated balanced properties thus delivered a relatively higher capacitance in both electrolytes than G1 and G2. As the pore size of MCs is closely associated with the C/P ratio used for the templating synthesis, the effect of C/P ratio need to be considered carefully when optimising ordered MCs for supercapacitors.
Although compact layers of ions or double-layer were able to form in sample G1.5 with both aqueous KOH and [N2220][NTf2]/ACN electrolytes, the electrochemical performance of G1.5 in both electrolytes exhibited notable differences. As shown in Fig. 6a and b, KOH electrolyte showed a narrower potential window but a higher charging/discharging current, longer discharge time (167 s at 0.5 A g−1), and higher efficiency (92.8%) than [N2220][NTf2]/ACN electrolyte (137 s, 84.0%). This discrepancy can be attributed to the smaller ion size and higher mobility of K+ and OH− inside mesopores than [N2220]+ and [NTf2]− ions. KOH electrolyte showed an internal resistance of 0.96 Ω and bulk electrolyte resistance of 0.20 Ω with G1.5 electrode, which were much smaller than the values of 2.10 and 0.62 Ω for [N2220][NTf2]/ACN (Fig. 6c). Moreover, the in-pores diffusivity and dominant capacitive behaviour with KOH electrolyte were slightly better, evidenced by the shorter 45° slope regime and nearly vertical line at intermediate and low frequencies. Given that the solvated ion sizes of K+ and OH− are much smaller than [N2220]+ and [NTf2]−, more charges can accumulate on the pore surface of carbon electrodes in a KOH electrolyte. As a result, the formation of electric double-layer in a KOH electrolyte was faster and higher capacitance was achieved albeit with a drop from 153.5 F g−1 at 0.1 A g−1 to 73.3 F g−1 at 20 A g−1, compared with the values from 57.5 to 24.0 F g−1 in [N2220][NTf2]/ACN electrolyte (Fig. 6d). In contrast, the KOH electrolyte yielded a much smaller energy density with a maximum of 7.7 W h kg−1, compared with 12.5 W h kg−1 in [N2220][NTf2]/ACN electrolyte (Fig. 6e); this is largely attributed to the narrower potential window of KOH electrolyte.
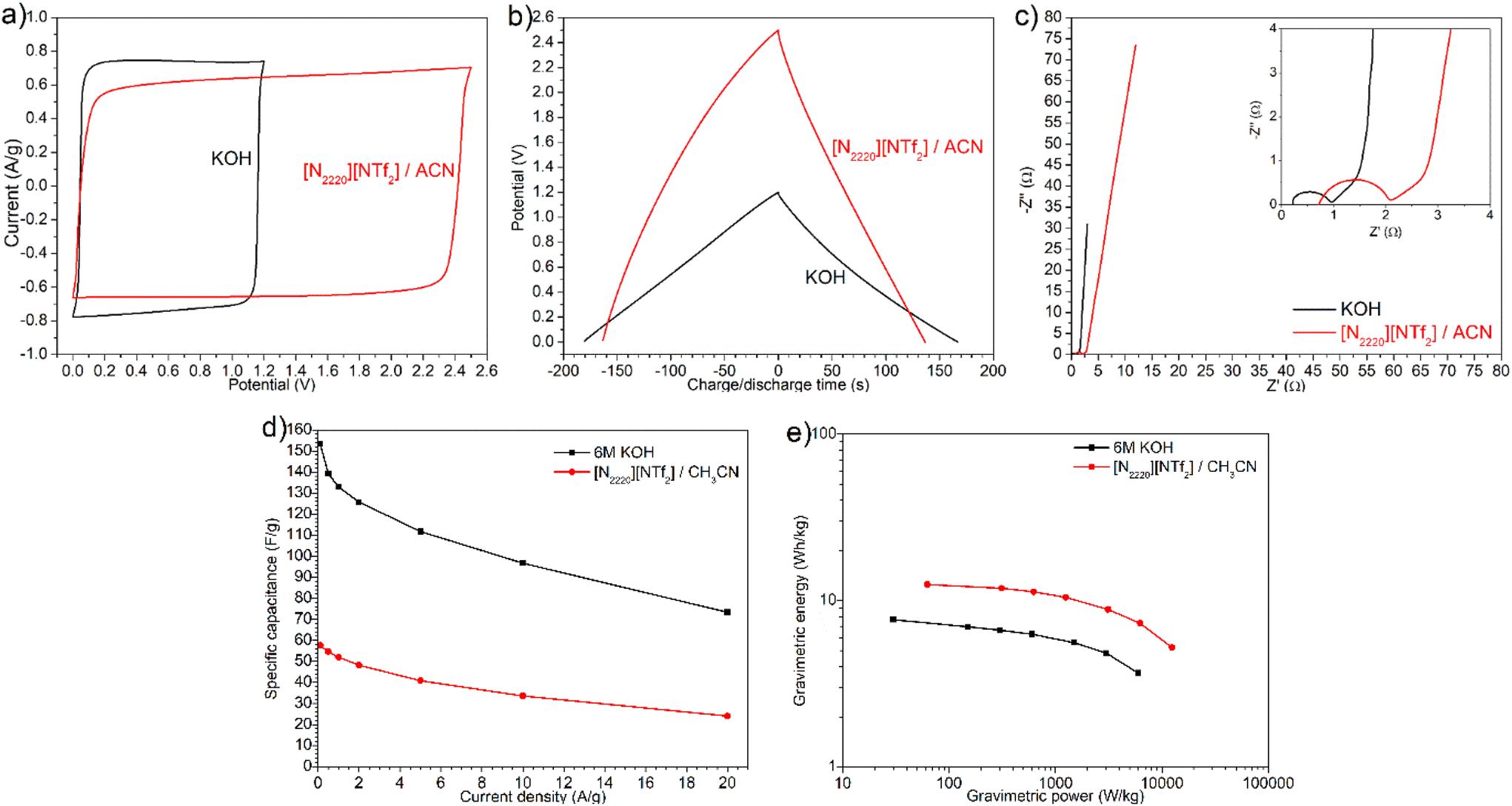 | ||
| Fig. 6 G1.5 electrode in two different electrolytes: (a) CV test at 50 mV s−1, (b) galvanostatic charge/discharge at 0.5 A g−1, (c) Nyquist plot, (d) gravimetric specific capacitance, and (e) energy density, respectively. | ||
Improving the graphitisation degree of electrode materials and incorporating heteroatoms can significantly increase capacitance.47,53,54 As demonstrated in Fig. S10a,† only D band by defected graphite lattice and G band by graphitic structures at around 1350 and 1590 cm−1 were detected for all MC electrodes.46,55 The ratio of the two bands (ID/IG ≈ 1) and the absence of 2D band (at around 2700 cm−1) indicate the lack of long-range ordered graphene-like structures and the amorphous nature of all MCs although local graphite-like crystallites existed (Fig. S10b†). Given the comparable graphitisation degree and surface functionalities, the differing electrochemical performance of MC electrodes mainly stemmed from the diverse pore architectures created by different cross-linking methods.
To further maximise the capacitance and energy density of ordered MCs, future effort can be focused on using glyoxal as cross-linker to customise MCs but with additional measurements including (1) enhancing the graphitisation degree, (2) doping with heteroatoms such as nitrogen, sulphur, and phosphor, (3) improving the interconnectivity of mesopores, and (4) modification of surface functionalities.5,47,53,54 Additional activation processes, especially CO2 activation, can effectively improve the connectivity of the pore networks whilst preserving the 2D hexagonal ordered mesoporous structure, thereby facilitating the desorption and significantly enhancing high-rate capability and stability of MCs.26,50 Even without modifying the pore architectures, incorporating nitrogen doping or increasing the graphitisation degree may lead to a more pronounced improvement in the capacitance; nitrogen-doped ordered mesoporous carbon with graphene-like layers was reported to show extraordinary capacitance of 855 F g−1 with aqueous H2SO4 electrolyte in a three-electrode configuration, much higher than the values for ordered mesoporous graphene-layered carbon without nitrogen doping (325 F g−1) and ordinary ordered MC (135 F g−1).47
Conclusions
The role of cross-linking in templating synthesis of MCs was studied by comparing various cross-linkers, ranging from carcinogenic formaldehyde to more environmentally friendly glyoxylic acid and glyoxal. The type of cross-linker and the ratio between cross-linker and precursor (C/P ratio) play crucial roles in the porosity, pore morphology, and electrochemical performance of resulting MCs, whilst their roles in surface functionalities are less significant.Glyoxal was found as a promising cross-linker to enable a robust templating synthesis at varying cross-linker/precursor mixing ratios thus generated highly ordered mesopores effectively with tuneable size between 3.3 and 6.1 nm. As the ratio of cross-linker/precursor increased, the BET area of glyoxal-derived MCs rose from 753 to 821 m2 g−1 whilst pore volume decreased from 0.69 to 0.48 cm3 g−1. Glyoxylic acid and formaldehyde resulted in interrupted or disordered mesopores. Larger C/P ratios can lead to higher –C–O content. With the same C/P ratio, glyoxylic acid induces higher –C–O content than glyoxal. Given an equal number of aldehydes, formaldehyde yields higher –C–O content than glyoxal due to its higher reactivity. As supercapacitor electrodes, glyoxal-derived MCs exhibited overall higher capacitance than those prepared from glyoxylic acid and formaldehyde, thanks to the highly ordered mesopore channels. With the same cross-linker, different C/P ratio will also influence the electrochemical performance by tuning the pore architectures. By balancing the mesopore size and pore volume, a C/P ratio of 1.5 mol mol−1 with glyoxal cross-linker induced a maximum capacitance of 153.5 and 57.5 F g−1 in 6 M KOH and [N2220][NTf2]/ACN electrolyte, respectively.
This work provided a detailed understanding of the role of cross-linking in determining the pore architecture, surface functionality, and electrochemical performance of resulting MCs. This knowledge effectively customises MCs for optimising the energy storage capabilities or other applications such as catalysis and drug delivery.
Disclaimer
The views and opinions expressed in this work do not necessarily reflect those of the European Commission or the Special EU Programmes Body.Author contributions
Yaoguang Song: Conceptualization, investigation, data curation, formal analysis, methodology, writing – original draft; Xiaolei Zhang: Conceptualization, supervision, funding acquisition, writing – review & editing; Peter Klusener: supervising, writing – review & editing; Peter Nockemann: Conceptualization, supervision, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by The Bryden Centre, which is supported by the European Union's INTERREG VA Programme, managed by the Special EU Programmes Body. The authors acknowledge the facilities and the technical assistance of Dr Alina Schilling and the Ewald Microscopy Facility at Queen's University Belfast. The authors would like to thank Dr Chiranjeevi Maddi and Prof. Davide Mariotti from Ulster University to provide access and useful training on the XPS instrument which is funded by EPSRC (EP/R008841/1), and Dr Shaoliang Guan from Cardiff University for useful discussion on XPS analysis. The authors also thank Mr Hugh O'Connor for helping prepare the Swagelok cell, Mr Marian Borucki for setting up the potentiostat, Dr Chunchun Li for Raman measurement, Dr Josh Bailey for training on the glovebox, and the QUILL Research Centre for providing laboratory instruments.References
- M. R. Benzigar, S. N. Talapaneni, S. Joseph, K. Ramadass, G. Singh, J. Scaranto, U. Ravon, K. Al-Bahily and A. Vinu, Chem. Soc. Rev., 2018, 47, 2680–2721 RSC.
- A. Eftekhari and Z. Fan, Mater. Chem. Front., 2017, 1, 1001–1027 RSC.
- W. Li, J. Liu and D. Zhao, Nat. Rev. Mater., 2016, 1, 16023 CrossRef CAS.
- C. Largeot, C. Portet, J. Chmiola, P. L. Taberna, Y. Gogotsi and P. Simon, J. Am. Chem. Soc., 2008, 130, 2730–2731 CrossRef CAS PubMed.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Science, 2006, 313, 1760–1763 CrossRef CAS PubMed.
- H. Qin, R. Jian, J. Bai, J. Tang, Y. Zhou, B. Zhu, D. Zhao, Z. Ni, L. Wang, W. Liu, Q. Zhou and X. Li, ACS Omega, 2018, 3, 1350–1356 CrossRef CAS PubMed.
- X. Wang, X. Liu, R. L. Smith, Y. Liang and X. Qi, Green Chem., 2021, 23, 8632–8642 RSC.
- Y. Liang, X. Liu and X. Qi, Int. J. Biol. Macromol., 2022, 213, 610–620 CrossRef CAS PubMed.
- S. Herou, M. C. Ribadeneyra, R. Madhu, V. Araullo-Peters, A. Jensen, P. Schlee and M. Titirici, Green Chem., 2019, 21, 550–559 RSC.
- X. Wang, M. Qiu, R. L. Smith, J. Yang, F. Shen and X. Qi, ACS Sustainable Chem. Eng., 2020, 8, 18157–18166 CrossRef CAS.
- S. Wang, G. Sima, Y. Cui, L. Chang and L. Gan, Mater. Lett., 2020, 264, 127318 CrossRef CAS.
- X. Wang, M. Qiu, Y. Tang, J. Yang, F. Shen, X. Qi and Y. Yu, Int. J. Biol. Macromol., 2021, 187, 232–239 CrossRef CAS PubMed.
- D. Saha, R. Zacharia and A. K. Naskar, in Polymer Precursor-Derived Carbon, 2014, vol. 1173, pp. 61–83 Search PubMed.
- Y. Meng, D. Gu, F. Zhang, Y. Shi, L. Cheng, D. Feng, Z. Wu, Z. Chen, Y. Wan, A. Stein and D. Zhao, Chem. Mater., 2006, 18, 4447–4464 CrossRef CAS.
- B. Nagy, E. Geissler and K. László, Microporous Mesoporous Mater., 2020, 294, 1–10 CrossRef.
- S. A. Al-Muhtaseb and J. A. Ritter, Adv. Mater., 2003, 15, 101–114 CrossRef CAS.
- P. Zhang, L. Wang, S. Yang, J. A. Schott, X. Liu, S. M. Mahurin, C. Huang, Y. Zhang, P. F. Fulvio, M. F. Chisholm and S. Dai, Nat. Commun., 2017, 8, 1–10 CrossRef PubMed.
- Y. Meng, D. Gu, F. Zhang, Y. Shi, H. Yang, Z. Li, C. Yu, B. Tu and D. Zhao, Angew. Chem., Int. Ed., 2005, 44, 7053–7059 CrossRef CAS PubMed.
- C. Liang, K. Hong, G. A. Guiochon, J. W. Mays and S. Dai, Angew. Chem., Int. Ed., 2004, 43, 5785–5789 CrossRef CAS PubMed.
- C. Liang, Z. Li and S. Dai, Angew. Chem., Int. Ed., 2008, 47, 3696–3717 CrossRef CAS PubMed.
- P. Zhang, L. Wang, S. Yang, J. A. Schott, X. Liu, S. M. Mahurin, C. Huang, Y. Zhang, P. F. Fulvio, M. F. Chisholm and S. Dai, Nat. Commun., 2017, 8, 1–10 CrossRef PubMed.
- X. Liu, Y. Wang, R. L. Smith, L. Liu and X. Qi, Sci. Total Environ., 2022, 836, 155640 CrossRef CAS PubMed.
- B. Szczȩśniak, J. Choma and M. Jaroniec, Chem. Commun., 2020, 56, 7836–7848 RSC.
- C. Liang and S. Dai, Chem. Mater., 2009, 21, 2115–2124 CrossRef CAS.
- J. Zhao, W. Shan, P. Zhang and S. Dai, Chem. Eng. J., 2020, 381, 122579 CrossRef CAS.
- J. Castro-Gutiérrez, N. Díez, M. Sevilla, M. T. Izquierdo, J. Ghanbaja, A. Celzard and V. Fierro, ACS Sustainable Chem. Eng., 2019, 7, 17627–17635 CrossRef.
- C. M. Ghimbeu, L. Vidal, L. Delmotte, J. M. Le Meins and C. Vix-Guterl, Green Chem., 2014, 16, 3079–3088 RSC.
- M. Sopronyi, F. Sima, C. Vaulot, L. Delmotte, A. Bahouka and C. M. Ghimbeu, Sci. Rep., 2016, 6, 1–13 CrossRef PubMed.
- E. Axente, M. Sopronyi, C. Matei Ghimbeu, C. Nita, A. Airoudj, G. Schrodj and F. Sima, Carbon, 2017, 122, 484–495 CrossRef CAS.
- G. Moussa, S. Hajjar-Garreau, P.-L. Taberna, P. Simon and C. Matei Ghimbeu, J. Carbon Res., 2018, 4, 1–17 Search PubMed.
- A. Maetz, L. Delmotte, G. Moussa, J. Dentzer, S. Knopf and C. M. Ghimbeu, Green Chem., 2017, 19, 2266–2274 RSC.
- R. T. Mayes, C. Tsouris, J. O. Kiggans, S. M. Mahurin, D. W. Depaoli and S. Dai, J. Mater. Chem., 2010, 20, 8674–8678 RSC.
- H. Younesi-Kordkheili, J. Adhes., 2019, 95, 1075–1087 CrossRef CAS.
- M. Siahkamari, S. Emmanuel, D. B. Hodge and M. Nejad, ACS Sustainable Chem. Eng., 2022, 10, 3430–3441 CrossRef CAS.
- Y. Song, F. Norris, D. Hinchcliffe, Y. Xu, X. Zhang and P. Nockemann, Nanoscale, 2022, 14, 14212–14222 RSC.
- D. Saha, K. E. Warren and A. K. Naskar, Carbon, 2014, 71, 47–57 CrossRef CAS.
- C. Tsouris, R. Mayes, J. Kiggans, K. Sharma, S. Yiacoumi, D. Depaoli and S. Dai, Environ. Sci. Technol., 2011, 45, 10243–10249 CrossRef CAS PubMed.
- L. Timperman, P. Skowron, A. Boisset, H. Galiano, D. Lemordant, E. Frackowiak, F. Béguin and M. Anouti, Phys. Chem. Chem. Phys., 2012, 14, 8199–8207 RSC.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- L. Liu, Q. F. Deng, T. Y. Ma, X. Z. Lin, X. X. Hou, Y. P. Liu and Z. Y. Yuan, J. Mater. Chem., 2011, 21, 16001–16009 RSC.
- V. Vijayakumar, M. Ghosh, R. Soni, M. Winter, S. Kurungot, M. Winter and J. R. Nair, Microsupercapacitors, 2021, pp. 47–116 Search PubMed.
- S. Shahzad, A. Shah, E. Kowsari, F. J. Iftikhar, A. Nawab, B. Piro, M. S. Akhter, U. A. Rana and Y. Zou, Global Challenges, 2019, 3, 1–17 CrossRef PubMed.
- L. Yu and G. Z. Chen, Front. Chem., 2019, 7, 1–15 CrossRef PubMed.
- S. Sathyamoorthi, V. Suryanarayanan and D. Velayutham, J. Power Sources, 2015, 274, 1135–1139 CrossRef CAS.
- Y. Song, J. Liu, K. Sun and W. Xu, RSC Adv., 2017, 7, 48324–48332 RSC.
- T. Lin, I. W. Chen, F. Liu, C. Yang, H. Bi, F. Xu and F. Huang, Science, 2015, 350, 1508–1513 CrossRef CAS PubMed.
- B.-J. Lee, H.-Y. Park, D.-S. Yang, T.-H. Kang, S. Hwang and J.-S. Yu, J. Electrochem. Soc., 2019, 166, A5244–A5251 CrossRef CAS.
- J. Castro-Gutiérrez, E. De Oliveira Jardim, R. L. S. Canevesi, J. Silvestre-Albero, M. Kriesten, M. Thommes, A. Celzard and V. Fierro, Carbon, 2021, 174, 413–422 CrossRef.
- J. Castro-Gutiérrez, R. L. S. Canevesi, M. Emo, M. T. Izquierdo, A. Celzard and V. Fierro, Renewable Sustainable Energy Rev., 2022, 167, 112716 CrossRef.
- A. Brandt, J. Pires, M. Anouti and A. Balducci, Electrochim. Acta, 2013, 108, 226–231 CrossRef CAS.
- R. Lin, P. Huang, J. Ségalini, C. Largeot, P. L. Taberna, J. Chmiola, Y. Gogotsi and P. Simon, Electrochim. Acta, 2009, 54, 7025–7032 CrossRef CAS.
- D. Weingarth, M. Zeiger, N. Jäckel, M. Aslan, G. Feng and V. Presser, Adv. Energy Mater., 2014, 4, 1–13 Search PubMed.
- L. L. Zhang, X. Zhao, H. Ji, M. D. Stoller, L. Lai, S. Murali, S. McDonnell, B. Cleveger, R. M. Wallace and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 9618–9625 RSC.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 1–4 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary results. See DOI: https://doi.org/10.1039/d3nr03244b |
| This journal is © The Royal Society of Chemistry 2023 |