
Revitalizing zinc-ion batteries with advanced zinc anode design
Shuwei
Chen†
a,
Huibo
Wang†
ab,
Mengyu
Zhu
a,
Fan
You
 a,
Wang
Lin
d,
Dan
Chan
a,
Wanxin
Lin
a,
Peng
Li
a,
Yuxin
Tang
*ac and
Yanyan
Zhang
*a
a,
Wang
Lin
d,
Dan
Chan
a,
Wanxin
Lin
a,
Peng
Li
a,
Yuxin
Tang
*ac and
Yanyan
Zhang
*a
aCollege of Chemical Engineering, Fuzhou University, Fuzhou 350116, P. R. China. E-mail: yxtang@fzu.edu.cn; zyanyan@fzu.edu.cn
bInstitute of Applied Physics and Materials Engineering, University of Macau, Macau 999078, P. R. China
cQingyuan Innovation Laboratory, Quanzhou 362801, P. R. China
dArmy Logistics Academy, Chongqing 401311, P. R. China
First published on 4th October 2022
Abstract
Rechargeable aqueous zinc-ion batteries (AZIBs) have attracted significant attention in large-scale energy storage systems due to their unique merits, such as intrinsic safety, low cost, and relatively high theoretical energy density. However, the dilemma of the uncontrollable Zn dendrites, severe hydrogen evolution reaction (HER), and side reactions that occur on the Zn anodes have hindered their commercialization. Herein, a state-of-the-art review of the rational design of highly reversible Zn anodes for high-performance AZIBs is provided. Firstly, the fundamental understanding of Zn deposition, with regard to the nucleation, electro-crystallization, and growth of the Zn nucleus is systematically clarified. Subsequently, a comprehensive survey of the critical factors influencing Zn plating together with the current main challenges is presented. Accordingly, the rational strategies emphasizing structural design, interface engineering, and electrolyte optimization have been summarized and analyzed in detail. Finally, future perspectives on the remaining challenges are recommended, and this review is expected to shed light on the future development of stable Zn anodes toward high-performance AZIBs.
1. Introduction
Rechargeable secondary batteries, as highly efficient and convenient green energy storage devices, provide a promising solution for renewable energy storage. Among the energy storage devices, lithium-ion batteries (LIBs) are the most mature electrochemical energy storage systems with the advantages of high efficiency, convenience, and stability.1–7 Nonetheless, the predicament of the limited lithium reserves, high preparation cost, safety issues arising from the flammable and explosive organic electrolytes, and environmental concerns are challenging their market dominance. Herein, the exploration of safe and inexpensive aqueous batteries is crucial to the upgradation of the energy storage industry. Aqueous zinc ion batteries (AZIBs) are promising for large-scale energy storage devices because of the abundant Zn metal reserves, environmental friendliness, excellent compatibility with aqueous solutions, low redox potential (−0.76 V vs. standard hydrogen electrode, SHE), and high theoretical capacity (820 mA h g−1 or 5855 mA h cm−3).8–14 Although great progress has been made in the research on cathode materials for AZIBs, such as polyanion compounds, manganese dioxide, vanadium oxide and Prussian blue,15–17 the notorious bottlenecks such as Zn dendrite growth, hydrogen evolution reaction (HER), and side reactions that occur on the surface of Zn anode severely deteriorate the Coulombic efficiency (CE) and performance of AZIBs, limiting their further commercialization.18–23 Regarding the Zn dendrites, which probably pierce the separator, cause short-circuit and fall off from the Zn anodes inducing “dead zinc”.24–26 It is known that Zn nucleation thermodynamics depends on the critical nucleation overpotential (η), and a smaller nucleation size would be created with higher η, benefitting the uniform and fine-grained Zn deposition.27–31 Moreover, the inhomogeneous electric field and Zn2+ ion distribution on the surface of the Zn anode would inevitably induce uneven Zn deposition and finally trigger the formation of Zn dendrites under the “tip effect”. Hence, a consistent electric field and homogeneous Zn2+ ion distribution are required to alleviate and suppress the Zn dendrite formation and growth.22,32–35 Besides, the HER (0 V vs. SHE) taking place during the process of Zn reduction would increase the local concentration of OH− ions, further accelerating the corrosion and passivation of the Zn anode.8,9,18,36 In turn, such continuous corrosion would make an even rougher Zn anode surface, promoting the H2 evolution as well as the uneven Zn plating. Therefore, an in-depth investigation and an understanding of these issues should be implemented to achieve an effective remedy for highly stable Zn anodes.In this review, we aim to provide a concise design strategies for stabilizing Zn anodes. We first interpret the electrochemical deposition processes of Zn in terms of nucleation, ion transport, and electro-crystallization. Then, the critical factors that influence Zn deposition, conjugating with the major challenges, are pointed out. Furthermore, the protection strategies are discussed with an emphasis on structural design, interface engineering, and electrolyte optimization, to provide a reasonable design concept for Zn anodes with high stability (Fig. 1). Finally, ourperspectives on future research opportunities targeting the remaining challenges are elaborated to promote the large-scale implementation of AZIBs.
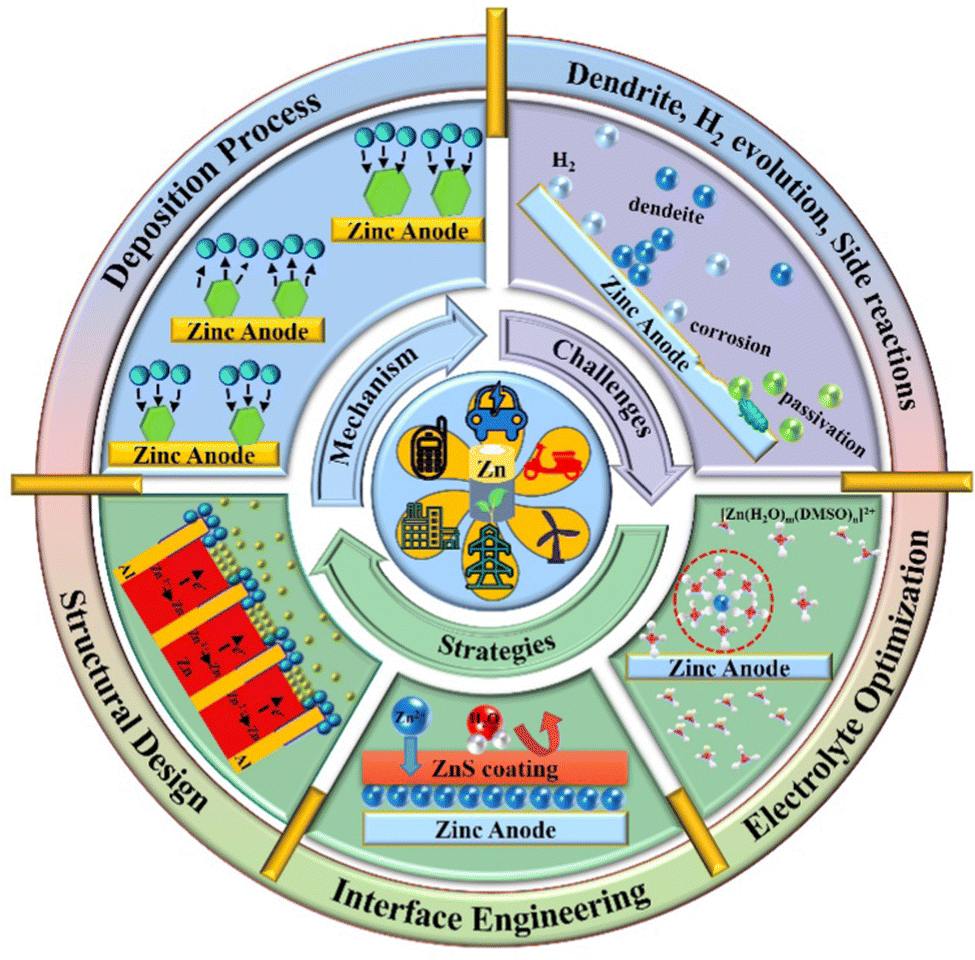 | ||
| Fig. 1 An overview of current challenges and corresponding strategies of AZIBs. | ||
2. Electrochemical deposition of Zn anode
The electrochemical behavior of Zn deposition is essentially composed of the process of Zn2+ ion reduction and the electro-crystallization of the newly-born Zn atoms; while the former process follows the thermodynamic rules like the nucleation overpotential, the latter is mainly governed by the electrochemical reaction kinetics involving the diffusion of Zn2+ ions and newly-born Zn atoms.34 Moreover, it is known that the Zn metal electrodeposition is a complex process with several sequential or simultaneous interfacial reaction procedures, including liquid phase mass transfer (migration of Zn2+ ions to the electrode surface), pre-transformation (dehydration or rearrangement of hydrated Zn2+ ions), charge transfer (Zn2+ ions are reduced to adsorbed Zn atoms), and electro-crystallization (adsorbed Zn atoms form crystals by integrating into the lattice or aggregating with other atoms).20,33,37–39 Since these steps jointly determine the morphology of the deposited Zn layer, a thorough understanding of Zn electrochemical deposition is essential.In a neutral/mild acidic environment, Zn2+ ions normally occur in the hydrated state ([Zn(H2O)6]2+ ions) and will undergo the so-called desolvation process, where the coordinated water molecules rearrange and further decrease, to be recovered as Zn2+ ions during their migration from the electrolyte to the near electrode surface. Subsequently, the transformed Zn2+ ions obtain electrons at the interface (Zn2+ + 2e− → Zn) and then generate unstably adsorbed Zn atoms that can effortlessly migrate over the crystal's surface. Furthermore, the adsorbed Zn atoms migrate to suitable locations (growth points) throughout the surface and undergo a crucial electro-crystallization process including integration into the lattice or aggregation with other newly-born Zn atoms to further grow.8,40 The detailed electro-crystallization processes can be expressed as follows:
(I) Integration into the lattice. The Zn growth process is related to two basic theories including the epitaxial growth theory and the screw dislocation growth theory. The former refers to the direct growth of nucleating particles into the crystal lattice to achieve the outward extension of the original crystal, while the latter refers to nucleating particles growing at defect sites such as dislocation positions, grain boundaries, and base crystal corners.41 In actuality, the crystal is imperfect, with multiple kinds of imperfections on its surface, which is more conducive to the growth of screw dislocations. Specifically, the transition from the initial [Zn(H2O)6]2+ state to the final Zn (kink) state can be achieved through the step-edge ion transfer mechanism and terrace ion transfer mechanism (Fig. 2a), which realize the continuous growth on the original metal lattice.10,39 The former refers to adsorbed Zn atoms diffusing along the step edges and finally reaching the kink positions. The latter is considered as the process where adsorbed Zn atoms diffuse through the paths with the lowest energy from the flat surface of the terrace region to the step edges, and finally reach kink sites to integrate into the crystals.
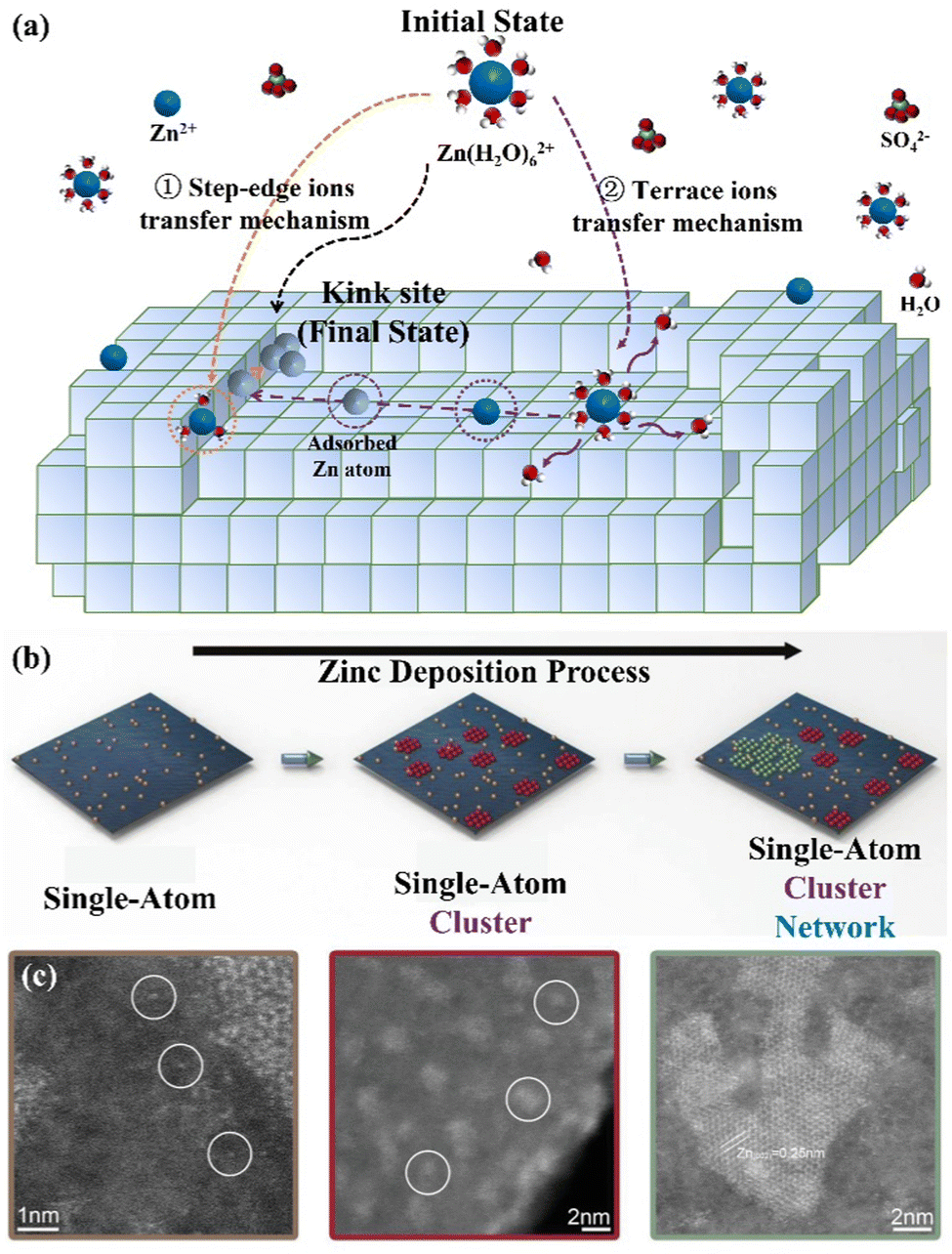 | ||
| Fig. 2 Schematic diagrams of the (a) step-edge ion transfer mechanism and terrace ion transfer mechanism, and (b) Zn nucleation evolution during deposition: single-atom Zn (brown); Zn cluster (red); Zn porous network (green). (c) The HAADF-STEM images of different forms of Zn atoms on the carbon host. Reproduced with permission from ref. 42. Copyright 2021, Wiley-VCH. | ||
(II) Agglomeration and growth. The initially adsorbed Zn atoms tend to form single-atom sites with weak binding energy. To enhance the stability, more Zn atoms would aggregate around these single-atom positions to form an atomic cluster, which can further expand to the mesoporous Zn network (Fig. 2b).42–44 In this regard, the high-resolution high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images have demonstrated the presence of these three forms during Zn deposition, including single atoms, clusters, and porous networks, as shown in Fig. 2c.
In this process, it was found that the mass transfer process of Zn deposition can be explained by the Nernst–Planck (eqn (1)):11,45
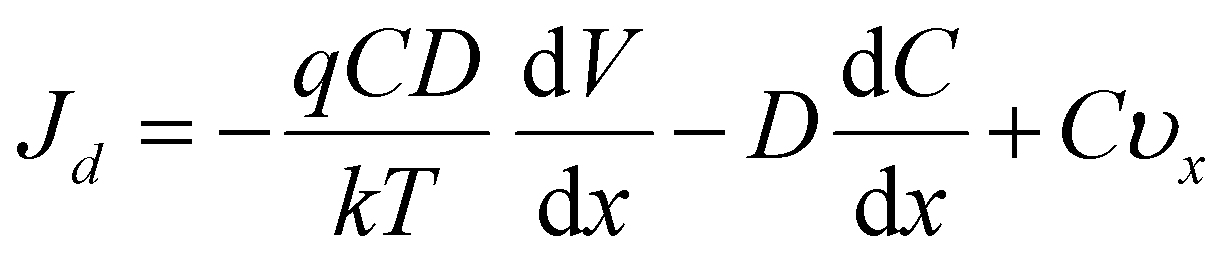 | (1) |
Zn deposition is prone to form a dendritic morphology under the irregular electric field distribution and uneven Zn2+ ions distribution near the anode surface. The finite-difference simulations illustrated how the factors like electric field and ion distribution influence the deposition process (Fig. 3a and b).11,12,48,49 To be more precise, Zn2+ ions are adsorbed on the Zn anode's surface before aggregating at the original nucleation sites with 2D-directional diffusion. “Tips” will form since the electric fields at the nucleation sites are substantially stronger than the other sites of the substrate. The sharp tips further exacerbate the irregular electric field and the Zn2+ ion concentration polarization on the electrode surface.20 Finally, vertical Zn deposition and Zn dendrite formation are promoted under the tip effect. (Fig. 3c).26 The concave edge has a larger electric field as well, which in turn induces the growth of Zn dendrites (Fig. 3d).31
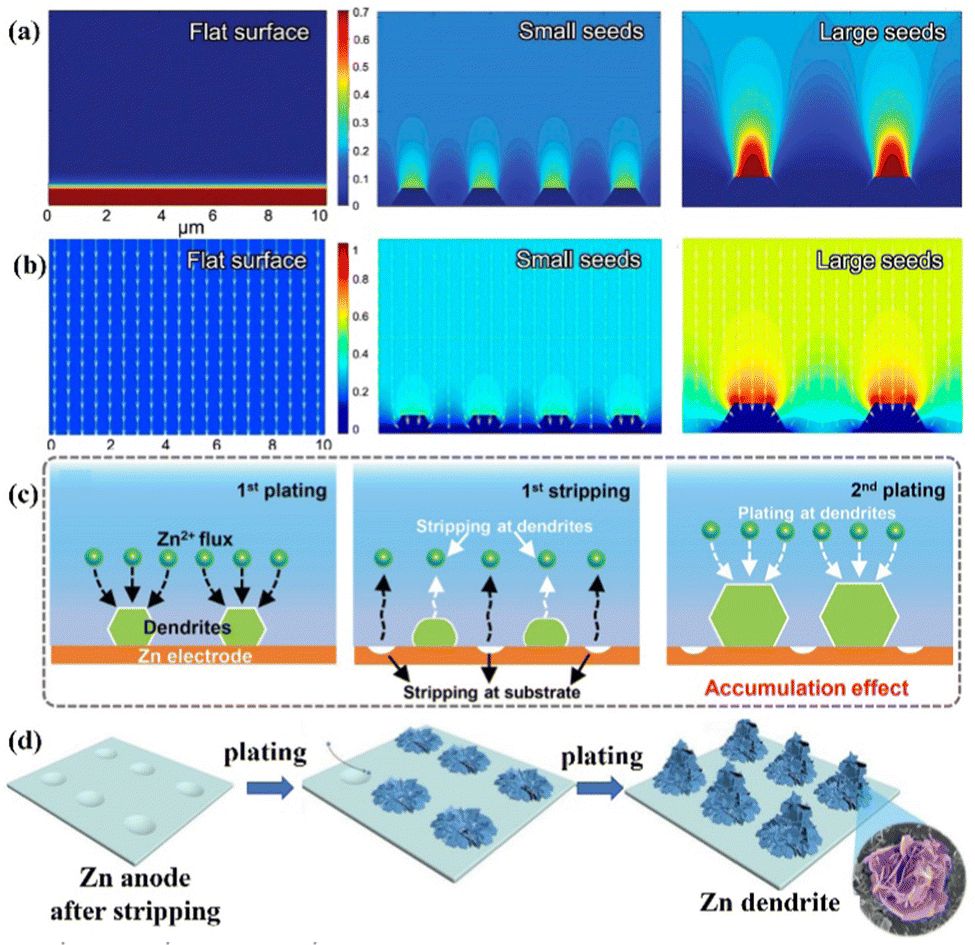 | ||
| Fig. 3 Schematic diagrams of the finite-difference simulations for (a) Zn2+ ions distribution and (b)the electric field under various conditions. Reprinted with permission from ref. 49. Copyright 2020, American Chemical Society. (c) Repeated plating/stripping of Zn at the tip positions. Reproduced with permission from ref. 26. Copyright 2020, Wiley-VCH. (d) A schematic diagram of Zn deposition at the concave edge. Reproduced with permission from ref. 31. Copyright 2021, The Royal Society of Chemistry. | ||
Accordingly, electric field and ion distribution significantly affect Zn nucleation and growth. Zn2+ ions tend to accumulate in the nucleation sites under a greater electric field, specifically the surface tip and the concave edge of the Zn anode, and then deposit along the vertical direction to form Zn dendrites under the so-called tip effect. Due to the large Young's modulus of Zn (EZn ≈ 108 Gpa), the separator will be quickly penetrated by Zn dendrites, which causes an internal short circuit.9,50
The plating/stripping curve is an effective tool for studying the process of Zn plating/stripping, which can determine the kinetic and thermodynamic processes of Zn deposition, including Zn nucleation, nuclei growth, Zn stripping and the soft shorts (SS). Firstly, the time–voltage curve (Fig. 4) can accurately reflect the plating/stripping process. In detail, the potential of the working electrode will drop to the nucleation overpotential (ηn) firstly to provide sufficient energy to drive nucleation (Fig. 4a, point i), and then the Zn deposition will occur on the Zn anode. Subsequently, the overpotential decreases because the energy barrier for nuclei growth is less than that for nucleation. Therefore, a rebound (Fig. 4a, curve ii–iii) representing the continued process of nuclei growth is observed.51,52
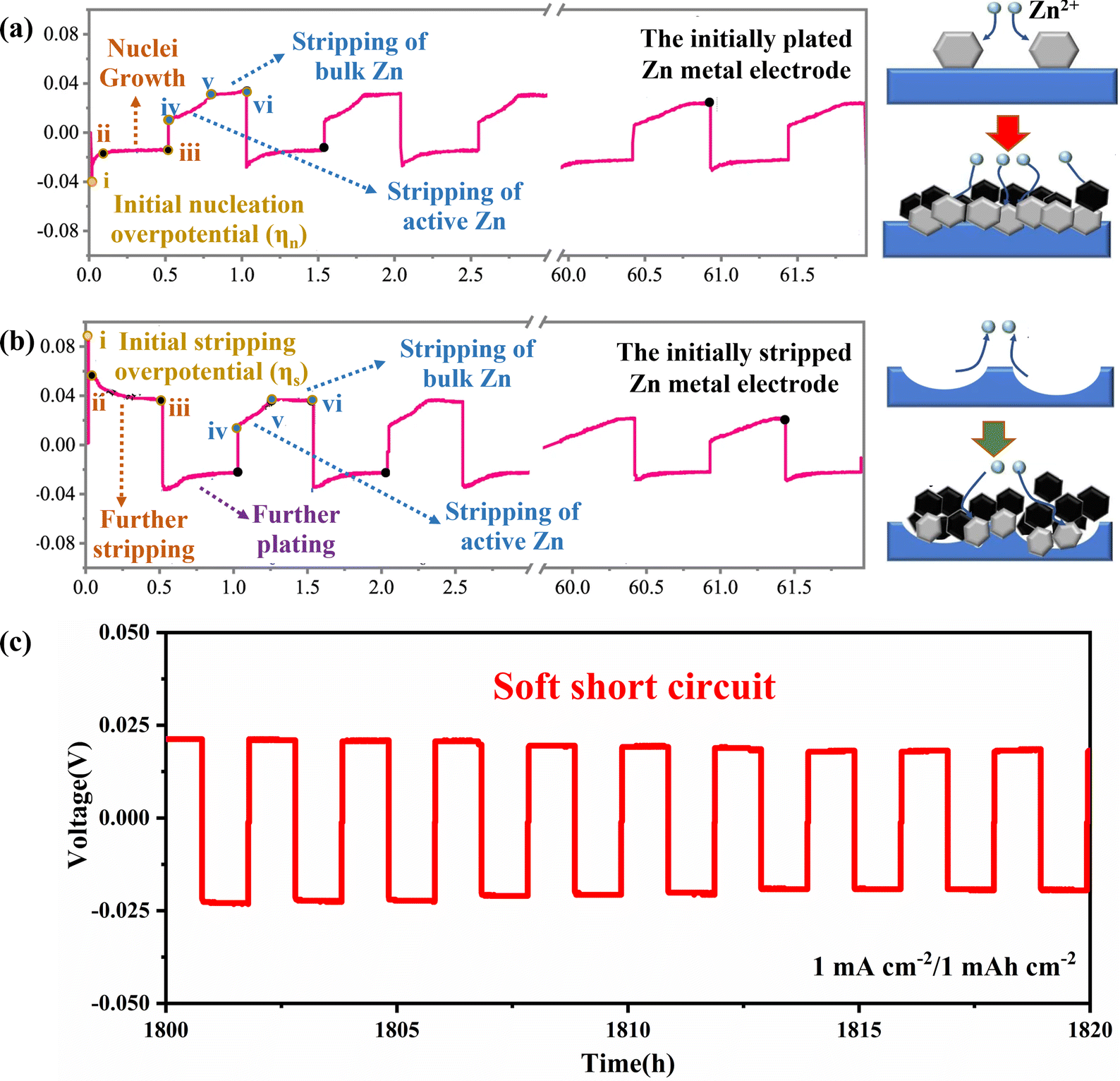 | ||
| Fig. 4 The time–voltage curves and corresponding schematic diagram of Zn plating/stripping behaviour of (a) P-Zn, and (b) S-Zn. Reproduced with permission from ref. 52. Copyright 2022, Springer Nature. (c) “Soft Short” phenomenon in symmetric cells. | ||
The differences and effects of the initial plating and initial stripping process of the Zn anode have been studied in depth as shown in Fig. 4a and b. It was found that the two different initial processes of plating and stripping have different morphological evolution of zinc metal anode. The initial plating process includes the nucleation stage (Fig. 4a, i) and the growth of nuclei (Fig. 4a, ii–iii), while the initial stripping process includes the initial stripping of bulk Zn in Zn foil (Fig. 4b, i) and further stripping of Zn at dissolution sites (Fig. 4b, ii–iii). The morphological evolution of the initial Zn plating electrode (P-Zn) and the initial Zn stripping electrode (S-Zn) during the cycling process can be elucidated by combining voltage profiles and in situ morphological observations (Fig. 4a and b). During the cycling process, S-Zn first appears concave, and then more Zn dendrites gradually form at the concave position. P-Zn produces shallower pits and less bulk Zn loss during the stripping process, which is more favorable for uniform Zn plating/stripping behavior.52,53
It can also be found through the time–voltage curve that the cycle life of symmetric batteries is generally overestimated, and the root cause of this illusion may be the occurrence of SS, as shown in Fig. 4c. SS refers to a localized electrical contact between two electrodes, allowing direct electron transfer and interfacial reactions to coexist. When the overpotential of the symmetrical cell is very low and the voltage curve is very flat, it means that SS may have occurred. In addition, there are two main methods to detect SS: (1) monitoring the impedance difference after low current density and high current density cycling tests, and (2) comparing the activation energy (Ea) of the dynamic interfacial reaction.54
Given the severe destructiveness of dendrites, the comprehensive investigation of the effects of various factors on the Zn electrodeposition process should be strictly executed, providing theoretical guidance for the preparation of Zn anodes with high reversibility.
3. Influential factors on Zn deposition
The dilemma of Zn dendrites, HER, and corrosion has severely obstructed the large-scale application of AZIBs (Fig. 5a).55–58 In detail, the non-uniform distribution of the electric field and Zn2+ ions trigger the inhomogeneous Zn deposition and dendrite growth. Owing to the weak adhesion to the Zn anode, the Zn dendrites are easily detached from the bulk electrode, resulting in “dead zinc”. Moreover, from the thermodynamic perspective, the HER would inevitably occur and raise the local concentration of OH− ions, causing the corrosion and production of inert by-products (e.g., Zn4SO4(OH)6·xH2O (ZSH)), aggravating the interfacial concentration polarization and promoting the growth of Zn dendrites.26 Since the electrochemical deposition of Zn is affected and induces morphological changes in the plated Zn, this section will describe in detail the influential factors, such as the pH value of the electrolyte, current density, temperature, and surface morphology and composition, to comprehensively understand the Zn plating processes.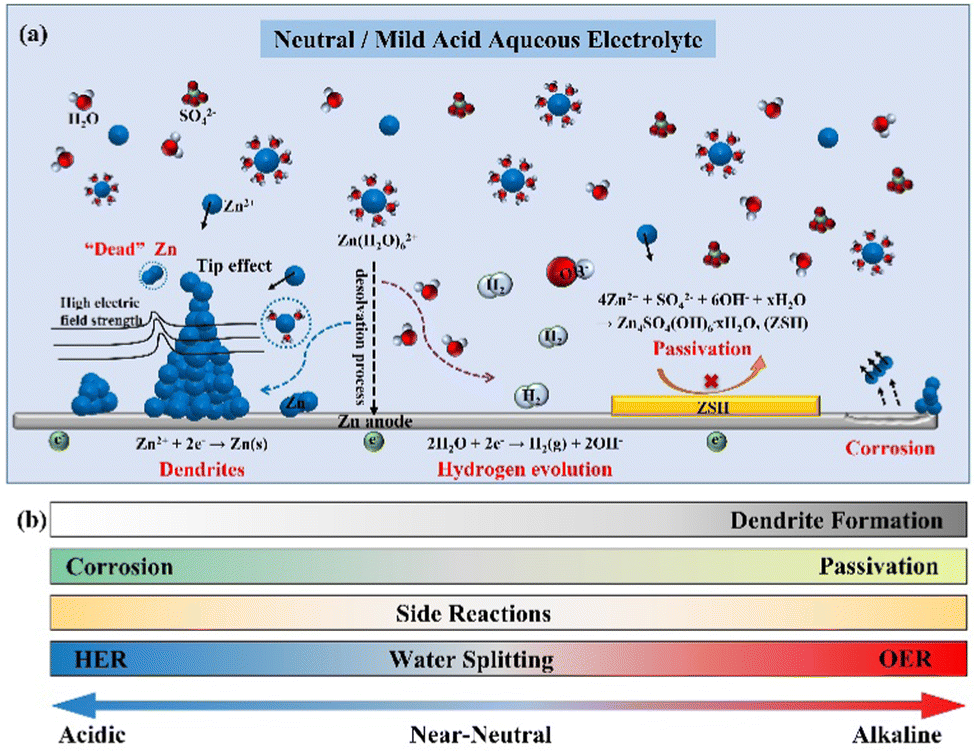 | ||
| Fig. 5 Schematic diagrams of (a) the mechanism of dendrite growth, HER, and side reactions for the Zn anode; (b) the effect of pH value on zinc anode. | ||
3.1 The effect of the pH of the electrolyte on the deposition process
The stability of the Zn deposition chemistry and the thermodynamic stability of the Zn anode are significantly affected by the pH value of the electrolyte.33,59 Much effort has been made regarding these studies,8,33,35,40,60–62 and a thorough explanation should be given about how the pH value of the electrolyte affects the stability of the Zn anode interface.In alkaline electrolytes, Zn2+ ions exist in the form of thermodynamically unstable Zn(OH)42− ions. When the Zn(OH)42− ions reach the limiting concentration, they will be converted into the electrochemically inert byproduct ZnO, resulting in the passivation of the Zn anode.8,59,63
While in the neutral/mild acidic electrolyte, the Zn species will present as [Zn(H2O)6]2+ ions and be reduced to Zn atom by gaining electrons at the electrode surface (Eq. 2, φ1 is the standard reduction potential of Zn2+/Zn). Moreover, the concomitant HER (Eq. 3, φ2 is the standard reduction potential of HER) deeply depends on the pH of the electrolyte according to the equation for the H2 evolution potential (Eq. 4, EHER is the hydrogen evolution overpotential). It is known that the lower the pH value of the electrolyte (the higher the H+ ion concentration), the more probably the HER will occur.8 For example, the potential of HER is −0.41 V vs. SHE (neutral environment, pH =7), which is higher than the standard reduction potential of Zn2+/Zn (−0.76 V vs. SHE), indicating that the HER is thermodynamically more favorable.8,11 Therefore, decreasing the potential of HER (i.e., increasing the absolute value of EHER) through increasing the pH value is an effective way to reduce H2 evolution. However, the HER leads to the consumption of H+ ions in the electrolyte, originating from the decomposition of H2O molecules, and causes an increase in the local OH− ions concentration.64 It is simple to determine that the lower pH value will increase the HER potential based on Eq. (4), which exacerbates the instability of the Zn anode surface. Taking the ZnSO4 electrolyte as an example, as the production of OH− ions originates from the HER, the electrochemically inert by-product ZSH will be formed when Zn2+ ions, SO42− ions, and OH− ions participate in the reaction (5), which leads to the corrosion and passivation of the electrode surface.65,66 Therefore, the pH value also affects the degree of corrosion and passivation on the Zn anode surface, which drastically limits the reversibility of Zn.
| Zn2+ + 2e− → Zn(s), φ1 = −0.76 V vs. SHE | (2) |
| 2H+ + 2e− → H2(g), φ2 = 0 V vs. SHE | (3) |
| EHER = Eθ – 0.0592 × pH (Eθ = SHE) | (4) |
| 4Zn2+ + SO42− + 6OH− + xH2O → Zn4SO4(OH)6·xH2O | (5) |
The pH value also impacts the side reactions, which in turn affects the formation of Zn dendrites. Corrosion will make an irregular electrode surface and cause uneven electric field distribution, while the presence of a passivation layer hinders the transport of Zn2+ ions and intensifies the ion concentration polarization at the interface, thus leading to the generation of Zn dendrites. Dendrites, in turn, expand the area of contact between the electrode and the electrolyte, amplifying the HER even more. The dendrite growth, HER, and side reactions are closely linked and interact with each other.
In conclusion, as the pH value of the solution increases (pH > 7), the solid precipitates (e.g., ZnO and Zn(OH)2) will easily form on the Zn anode, leading to electrode passivation. The thermodynamic stability of Zn decreases as the pH value of the solution decreases (pH < 7), and the hydrogen evolution reactions become easy, causing electrode corrosion and uneven Zn deposition. The formation of a passivation layer will impede the transport of Zn2+ ions between the electrode and the electrolyte (Fig. 5b), thus severely affecting Zn deposition and leading to severe dendrites. It is crucial to investigate the appropriate pH value to enhance the stability of the Zn anode.8,26
Therefore, selecting an electrolyte solution with a suitable pH value is one of the key factors to guide the uniform and reversible Zn deposition.
3.2 The influence of current density
It has been demonstrated through the simulation results and experimental evidence that the current density plays a significant role in the formation and growth of dendrites during the deposition process.67,68 Several simulation models, such as the space charge model, the stress-driven dendrite growth model, the nonuniform nucleation model, and the deposition–dissolution model have been employed to explain the formation and early growth of dendrites. Of these, the space charge model theory has emerged as the most well-acknowledged demonstration.69,70 Brissot and Chazalviel et al.24,71 revealed that the dendrite growth time “Sand's time (τs)” is inversely proportional to the square of the current density, which is described as following:70,72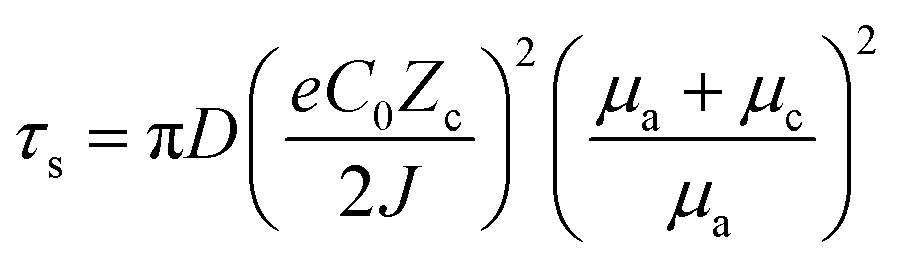 | (6) |
The nucleation-growth process controlled by nucleation rate (νn) and critical radius (rc) is also closely related to J (eqn (7) and (8)), as shown in Fig. 6a.72 The νn and rc for the formation of thermodynamically stable nuclei related to the J are as follows:
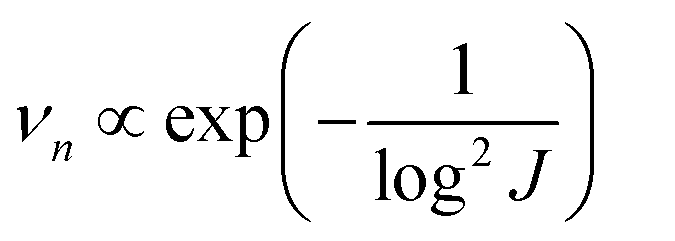 | (7) |
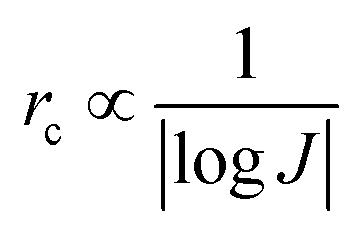 | (8) |
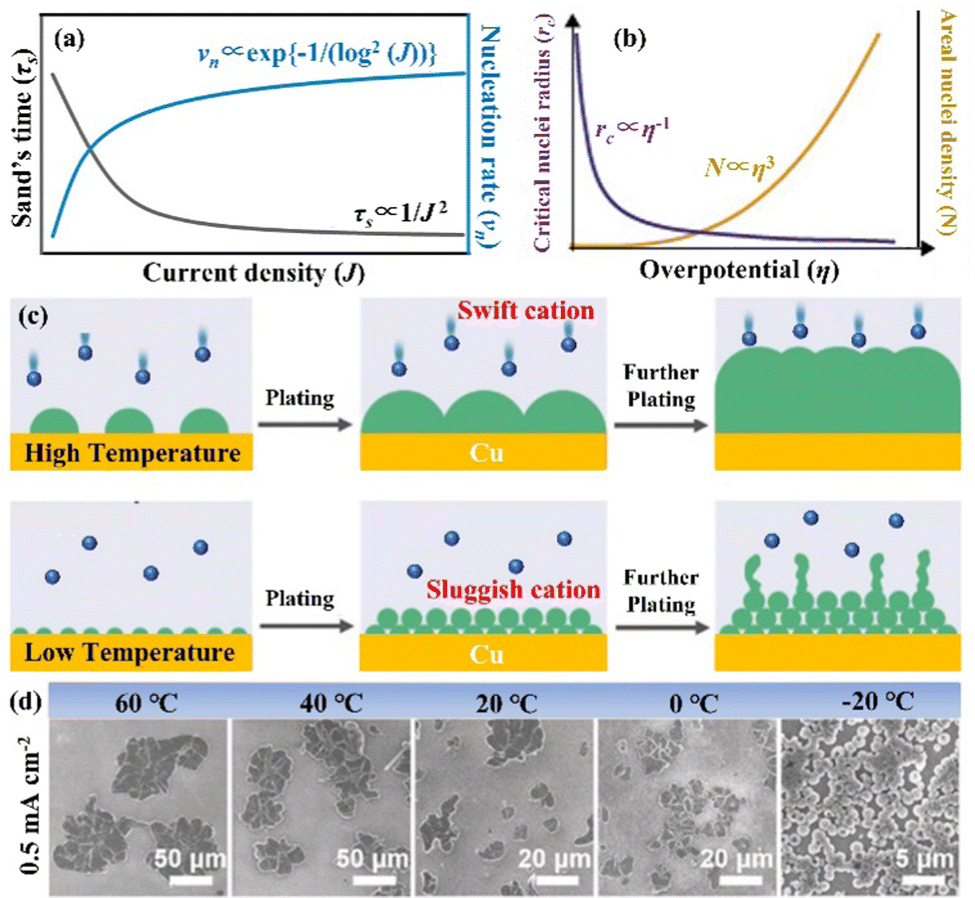 | ||
| Fig. 6 (a) Theoretical correlation between τs/νn and J. Reproduced with permission from ref. 72. Copyright 2021, Wiley-VCH. (b) The relationship between the critical nucleation radius, areal nucleation density, and nucleation overpotential. Reproduced with permission from ref. 28. Copyright 2017, American Chemical Society. (c) Schematic diagram of nuclei generation and growth mechanism under high and low temperature. (d) SEM images of nuclei layers under varied temperature conditions. Reproduced with permission from ref. 78. Copyright 2019, Wiley-VCH. | ||
From eqn (7) and (8), it can be concluded that νn is proportional to J, while rc is inversely proportional to J. Therefore, a larger J increases vn and decreases rc as compared to a smaller J. The increased J facilitates more abundant small-size nuclei, which significantly inhibits dendrite growth and achieves dense Zn deposition. These phenomena could be explained by Gibbs free energy and classical nucleation theory where the nucleation and growth of metals are not a spontaneous process and require additional energy to overcome the barriers (nucleation overpotential).10,12,74 he relationship between the nucleation η and νn and rc are shown in eqn (9) and (10) and Fig. 6b.11,28,30,72
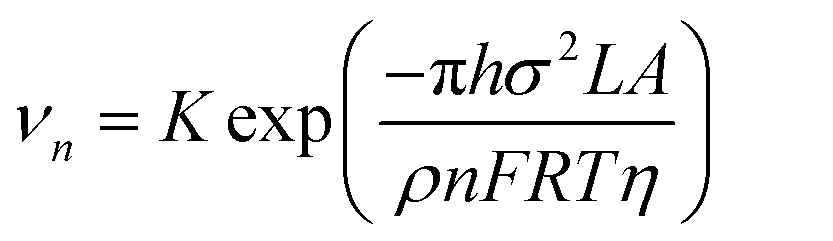 | (9) |
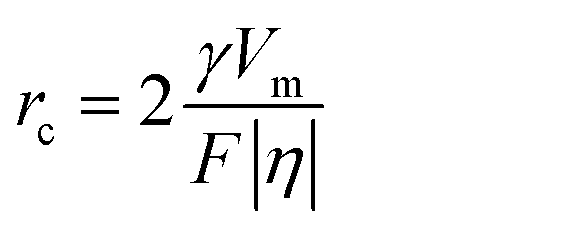 | (10) |
Based on the above theoretical analysis, the current density (J) affects Zn deposition from two aspects (kinetics and thermodynamics). In terms of kinetics, τs is correlated with J. A higher J leads to a shorter τs, which increases the growth of Zn dendrites and determines the stability of the Zn anode. Whereas, in thermodynamics, a higher J provides more additional energy for nucleation, which reduces the nucleation radius and increases the nucleation density, contributing to a uniform and dense Zn deposition. Therefore, a rational experimental design is needed to optimize J to balance the kinetically relevant τs with the thermodynamically relevant nucleation processes (νn and rc).72,75,76
3.3 The influence of temperature
According to previous research, surface free energy between the electrolyte and electrode is strongly dependent on the temperature, where a lower temperature can increase the surface free energy.77 In addition, the Gibbs free energy of nucleation ΔG is related to the surface free energy as follows: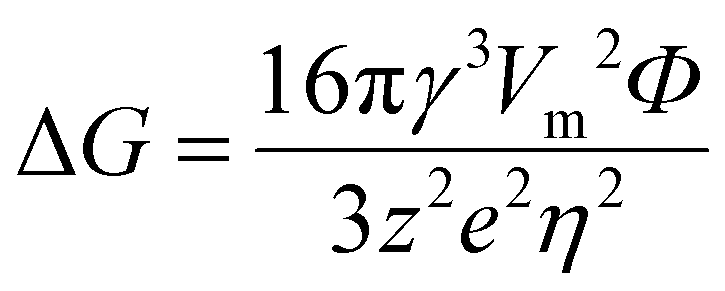 | (11) |
3.4 The influence of surface morphology and composition
The surface morphology and composition of the electrode have a tremendous impact on Zn deposition. Zn foil is used as the anode in AZIBs due to its advantages of easy large-scale preparation, however, surface defects such as surface roughness, microcracks, and scratches generated during the manufacturing process can lead to unfavorable factors such as local tip and local stress variations. The so-induced uneven electric field distribution at the electrode/electrolyte interface in turn causes local dendrite growth and a significant decrease in the cycling stability of the Zn anode.81,82 Fortunately, the polishing process can effectively reduce surface defects and mitigate dendrite growth. Moreover, replacing the planar Zn foil with 3D porous electrodes can also greatly uniformize the charge distribution and interfacial electric field, achieving a dendrite-free anode.83–85 Besides, it was shown that Zn5(CO3)2(OH)6 and ZnO passivation layers were formed on the surface of Zn metal exposed to air, while this passivation layer hindered the migration and deposition of Zn2+ ions on the electrode surface, as well as failed to protect the corrosion of Zn anode in mild acidic electrolytes.86,87 Therefore, it is essential to add a stable SEI layer to the anode surface. On the other hand, Zn atoms have varied binding energies to different interfaces, and the zincophilic interface with greater binding energy is advantageous to Zn nucleation and guides the orderly growth of Zn.87–89In conclusion, dendrite growth, HER, and side reactions like corrosion and passivation are all interrelated, according to the above analysis. Numerous factors control the formation of Zn dendrites, including the pH of the electrolyte, current density, and temperature, among others. Zn dendrites are only weakly adsorbed to the electrode and may slip away from the bulk electrode during cycling, resulting in “dead Zn” and a reduction in Zn utilization. What's worse, Zn dendrimers with a high young's modulus might penetrate the separator easily, resulting in internal circuit failure. To produce dendrite-free Zn anodes and high-performance AZIBs, it is advantageous to reconstruct a uniform electric field and ion distribution on the electrode surface.8,9,40 The challenges of HER, corrosion, and passivation also seriously affect the stability of the Zn anode. Accordingly, increasing the HER overpotential of the Zn anode (in terms of thermodynamic aspects) and reducing the water molecule content/activity on the Zn surface (from kinetic aspects) are the keys to limiting H2 evolution. Since some metals have a higher HER overpotential than that of Zn, alloyed electrode design is an effective strategy to limit H2 evolution and enhance corrosion resistance.33 In addition, the introduction of interfacial coatings or changes in the solventized structure of Zn2+ ions can efficiently avoid direct interaction of a large number of active water molecules with the Zn anode, achieving the inhibition of the HER and side reactions.87
4. Mitigation strategy
For Zn anodes, the challenges of Zn dendrites, the HER, and side reactions in aqueous electrolytes severely limit the practical application of AZIBs. These problems could be efficiently overcome by controlling the uniform distribution of the electric field and ions, lowering the number of water molecules that are active on the Zn surface, and decreasing the corrosion overpotential and/or increasing the HER overpotential, based on an understanding of the intrinsic failure mechanism. Hence, we have summarized various types of protection strategies in this section, which include electrode structural design, interface engineering, and electrolyte optimization, to achieve high-stability Zn anodes for AZIBs.4.1 Structure design
The structure design is an efficient technology for obtaining an extremely stable Zn anode. The 3D porous structure greatly increases the contact area between the electrode and electrolyte, reduces the local current density and promotes homogeneous Zn deposition. Additionally, combining Zn metal with metals like Mg, Al, Cu, In, and Ga to create an alloy solid solution can significantly increase the electrode surface's resistance to corrosion. This method optimizes the behavior of Zn deposition and growth as well, resulting in the creation of a fairly reversible Zn anode.(i) 3D structured Zn anode. The issue of Zn dendrites caused by strong local currents is successfully resolved by designing the 3D porous construction with a large specific surface area.90,91 Rolison's group92 reported a 3D sponge structure Zn anode with a high specific surface area, which greatly increased Zn nucleation sites and reduced the local current density on the Zn anode surface, resulting in a uniform and dense Zn deposition (Fig. 7a). This 3D sponge Zn anode effectively solved the problems of passivation and dendrites, and achieved thousands of stable cycles in alkaline nickel–zinc batteries. With the treatment of oxidation, electrodeposition, and high-temperature calcination, Pu's group84 developed a dual-channel 3D nano-porous Zn anode (denoted as DCP-Zn) for high-speed ion/electron transfer. The DCP-Zn anode's continuous porous conducting skeleton enabled a homogeneous electric field on its surface, and the nano-pores are beneficial for uniform Zn deposition, thereby inhibiting the dendrite growth and alleviating the volume variations (Fig. 7b). Moreover, the dual-channel porous structure also facilitated high rates of electron and ion transport, reducing internal polarization and “dead zinc”. Ultimately, the DCP-Zn symmetric cell could cycle stably for more than 1400 h and 200 h at 0.1 mA h cm−2 and 10 mA h cm−2, respectively, and the DCP-Zn//V2O5 full cell had a high capacity of 443.4 mA h g−1 and stable cycling performance (more than 500 cycles). Xu's group93 reported a 3D porous copper skeleton Zn anode. As a carrier for Zn deposition, the porous copper skeleton possessed strong electrical conductivity and an open architecture that could guarantee uniform Zn deposition. Usually, the Zn foil anode often serves as both the active component and the carrier, which leads to significant volume variations and instability during charging and discharging. The robust and flexible porous copper skeleton can withstand such volume changes, thus improving cycling performance (Fig. 7c). The results showed that the 3D copper skeleton structured Zn anode had stable cycling performance, and the 3D skeleton-Zn//ultrathin MnO2 nanosheet full cell demonstrated good cycling stability with a retained capacity of 173 mA h g−1 after 300 cycles at 0.4 A g−1. Qian's group94 designed 3D porous Ti skeletons and 3D porous Ti–TiO2 skeletons to increase the internal Zn deposition space, limit Zn dendrite formation and mitigate large volume changes.95,96 As shown in Fig. 7d, the Zn deposition on the porous Ti skeleton was more uniform as compared to that of Zn foil. However, the Zn deposition and growth on the Ti skeleton surface would still lead to the formation of localized submicron dendrites due to the weak zincophilicity property of Ti.97 Therefore, further construction of a porous Ti–TiO2 skeleton zinc anode with zincophilicity could achieve a long cycle performance of 2000 h, and the 3D Ti–TiO2–Zn/S-Mxene@MnO2 full cell could retain 95.46% of the capacity after 500 cycles at 5 A g−1.
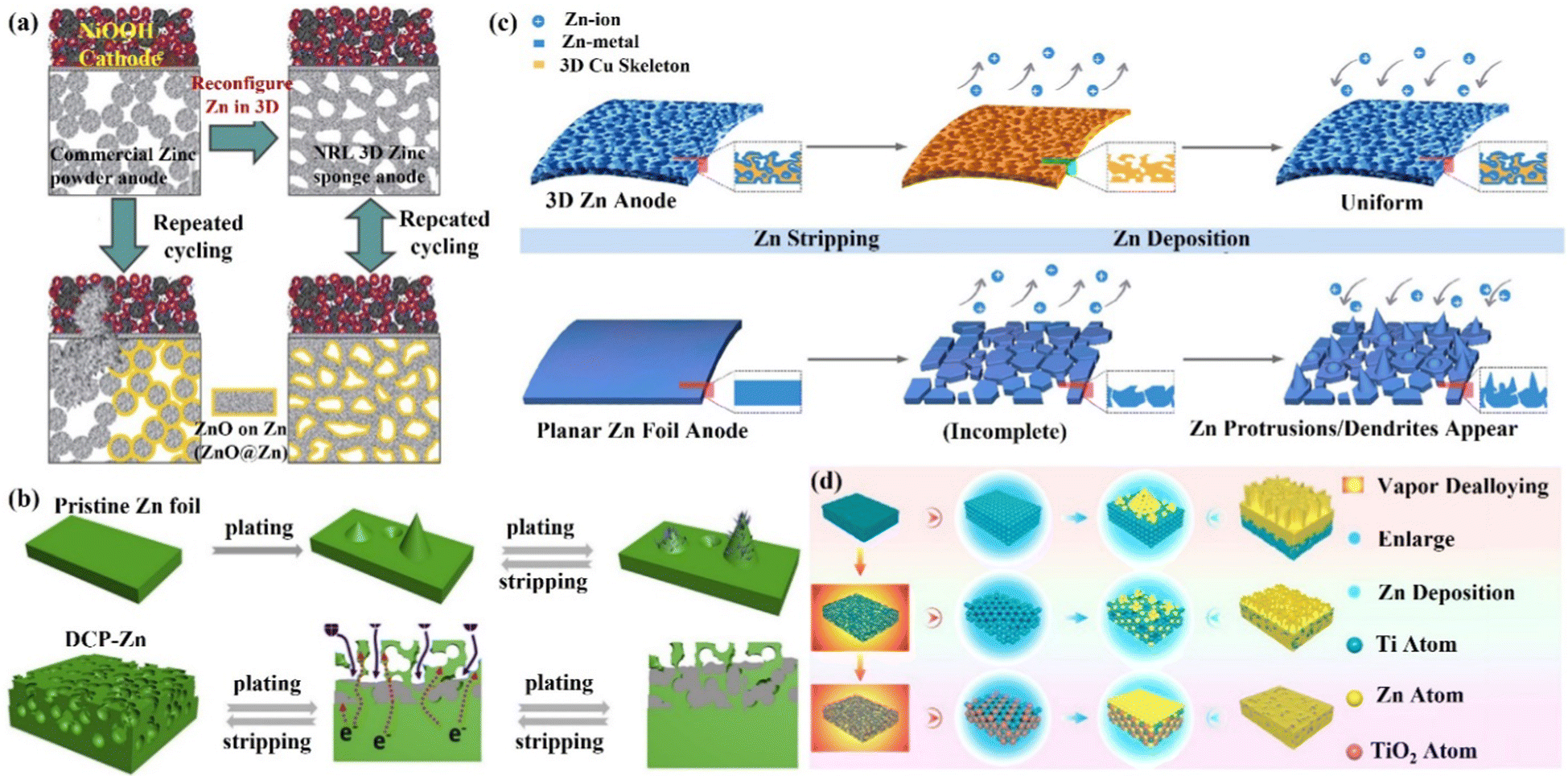 | ||
| Fig. 7 Schematic diagram of (a) controllable Zn deposition on the designed 3D sponge Zn anode in a Zn–Ni cell. Reproduced with permission from ref. 92. Copyright 2017, The American Association for the Advancement of Science. (b) Repeated Zn plating/stripping behaviors of Zn foil and DCP-Zn. Reproduced with permission from ref. 84. Copyright 2020, Elsevier Inc. (c) The corresponding Zn deposition and stripping process when 3D porous copper skeleton and flat Zn foil are used as electrodes, respectively. Reproduced with permission from ref. 93. Copyright 2019, American Chemical Society. (d) Planar Ti, 3D Ti skeleton, and 3D Ti–TiO2 skeleton as carriers and the corresponding Zn deposition process. Reproduced with permission from ref. 94. Copyright 2021, American Chemical Society. | ||
(ii) 3D carbon material skeleton Zn anode. Zincophilic materials can promote nucleation as well as further dense Zn deposition, which can effectively suppress the undesirable effects arising from dendrites. Therefore, the introduction of zincophilic sites can effectively solve the kinetics of Zn deposition. However, issues such as the uneven distribution of zincophilic sites and coverage of zincophilic sites still lead to the failure of this strategy. Chao's group89 proposed a hierarchical confinement strategy (Fig. 8a) that used zincophilic sites to capture Zn to induce uniform nucleation, and then spatially “enclosed” the subsequently deposited Zn in porous carbon cages and the 3D networks, thus effectively solving the kinetic and stability compatibility problems (Fig. 8b). By combining the zincophobic porous carbon with the zincophilic sites, the direct exposure of the zincophilic sites was prevented to the greatest extent, and the stability of the zincophilic sites was improved while reducing the possibility of water splitting. This structure could also reduce the local current density, and uniform Zn flux, as well as alleviate the volume expansion problem during cycling. In this case, the CoCC-Zn symmetrical cell could maintain stable cycling for more than 800 cycles at 20 mA cm−2 (polarization potential of only 65 mV), and the CoCC-Zn//MnO2 full cell achieved a long cycle performance of up to 2000 cycles at 2 A g−1. Li's group85 prepared a Zn/carbon nanotube (Zn/CNT) foam anode with high conductivity by chemical vapor deposition. During the cycling process, the deposition behavior of Zn occurred mainly inside the carbon nanotube foam network (Fig. 8c), which promoted ion transport and avoided the formation of dendrites. The capacity retention of the Zn/CNT foam symmetric cell remained at 100% after 250 and 100 cycles at 40% and 81% depth of discharge (DODZn), respectively. The Zn/CNT foam//MnO2 full cell had a long life of up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 minutes at 3 mA cm−2, and it had outstanding rate performance and stability. With a capacity retention rate of 81.9%, the capacity was 172 mA h g−1 after 2500 cycles at 10 C and 83 mA h g−1 after 5000 cycles at 30 C. This yields an energy density of 169 W h kg−1 and a power density of 3.1 kW kg−1. By integrating a 3D framework (carbon nanotubes, CNT) as a Zn deposition carrier with the substrate of carbon cloth (CC), Lu's group98 was able to create a high-performance flexible AZIB with good cycling stability by introducing a 3D framework structure of carbon nanotubes (CNT) as a Zn deposition/dissolution carrier. Through this modification, the production of Zn dendrites and by-products was successfully reduced (Fig. 8d). A more homogenous electric field and a significantly smaller Zn nucleation overpotential were obtained by the 3D CNT framework, making the interface more favorable for rapid migration and the uniform nucleation of Zn2+ ions.83,99–102 The Zn/CNT symmetric cell could achieve a stable cycle of 200 h with a CE of 97.9% at a discharge depth of 28%, while the Zn/CNT//MnO2 full cell maintained the capacity retention of 88.7% after 1000 cycles, exhibiting exceptional mechanical flexibility.
000 minutes at 3 mA cm−2, and it had outstanding rate performance and stability. With a capacity retention rate of 81.9%, the capacity was 172 mA h g−1 after 2500 cycles at 10 C and 83 mA h g−1 after 5000 cycles at 30 C. This yields an energy density of 169 W h kg−1 and a power density of 3.1 kW kg−1. By integrating a 3D framework (carbon nanotubes, CNT) as a Zn deposition carrier with the substrate of carbon cloth (CC), Lu's group98 was able to create a high-performance flexible AZIB with good cycling stability by introducing a 3D framework structure of carbon nanotubes (CNT) as a Zn deposition/dissolution carrier. Through this modification, the production of Zn dendrites and by-products was successfully reduced (Fig. 8d). A more homogenous electric field and a significantly smaller Zn nucleation overpotential were obtained by the 3D CNT framework, making the interface more favorable for rapid migration and the uniform nucleation of Zn2+ ions.83,99–102 The Zn/CNT symmetric cell could achieve a stable cycle of 200 h with a CE of 97.9% at a discharge depth of 28%, while the Zn/CNT//MnO2 full cell maintained the capacity retention of 88.7% after 1000 cycles, exhibiting exceptional mechanical flexibility.
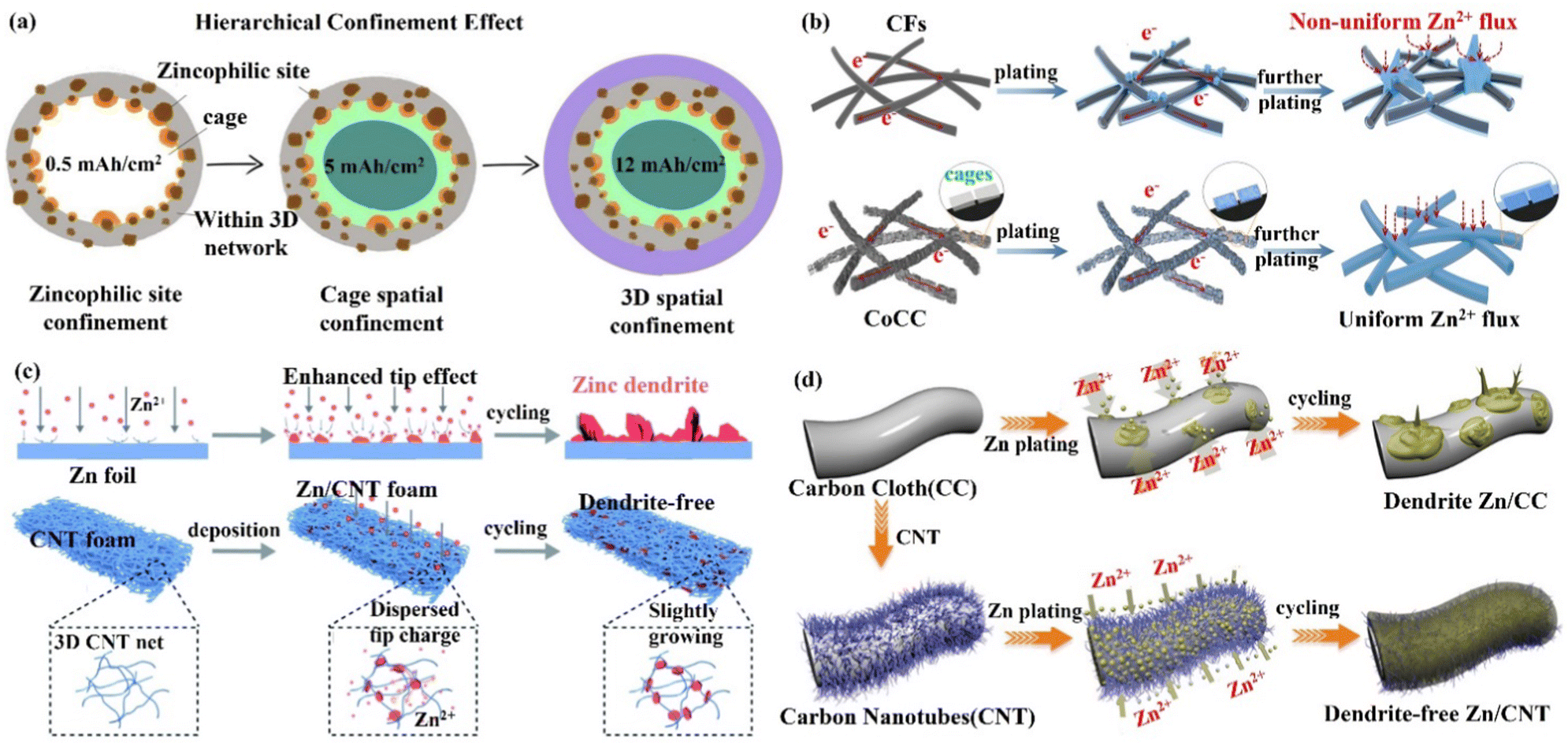 | ||
| Fig. 8 Schematic diagrams of (a) the hierarchical confinement effect upon Zn deposition, (b) the carbon fiber (CF) skeleton network and the Co-embedded carbon cage (CoCC) skeleton network, which are used as carriers, and the accompanying behaviors of Zn deposition. Reproduced with permission from ref. 89. Copyright 2022, American Chemical Society. (c) Zn dendrite growth on the surface of pristine Zn foil and uniform Zn plating/stripping inside the 3D foam-structured Zn/CNT anode. Reproduced with permission from ref. 85. Copyright 2020, The Royal Society of Chemistry. (d) Zn deposited on carbon cloth (CC) electrode to form dendrites, while on carbon nanotube (CNT) electrode to achieve dendrite-free growth. Reproduced with permission from ref. 98. Copyright 2019, Wiley-VCH. | ||
(iii) 3D layered anode and alloyed Zn anode. The layered structure and alloyed structure enhance the wettability of the electrode–electrolyte interface, and the high ion transport, and uniform Zn deposition endow the electrode with excellent performance. Qian's group103,104 constructed a flexible 3D layered Ti3C2Tx MXene@Zn paper as the electrode body, which had high electrical conductivity (15000 S cm−1), good wettability, and great toughness.105,106 The layered structure, as illustrated in Fig. 9a, would provide fast electron/ion migration channels; the high surface area and internal space could accomplish uniform charge distribution and prevent the formation of dendrites. Liu's group107 produced a Zn–Ag alloy electrode by using Zn-soluble metallic silver as a seed material on a carbon substrate to build an alloy solid solution surface layer (ZnxAg1−x) for heterogeneous seed growth and homogeneous Zn deposition (Fig. 9b), and for the first time, used optical microscopy to examine the deposition process of the Zn seed crystals. The majority of Zn was deposited on the silver seeds, which made Zn nucleation and growth more easily regulated in space. Furthermore, Zn2+/ZnxAg1−x had a greater redox electrochemical potential than Zn2+/Zn, which improved the corrosion resistance of the Zn–Ag alloy anode. Ultimately, the introduction of metallic silver seeds greatly improved the stability of the Zn–Ag alloy electrode.
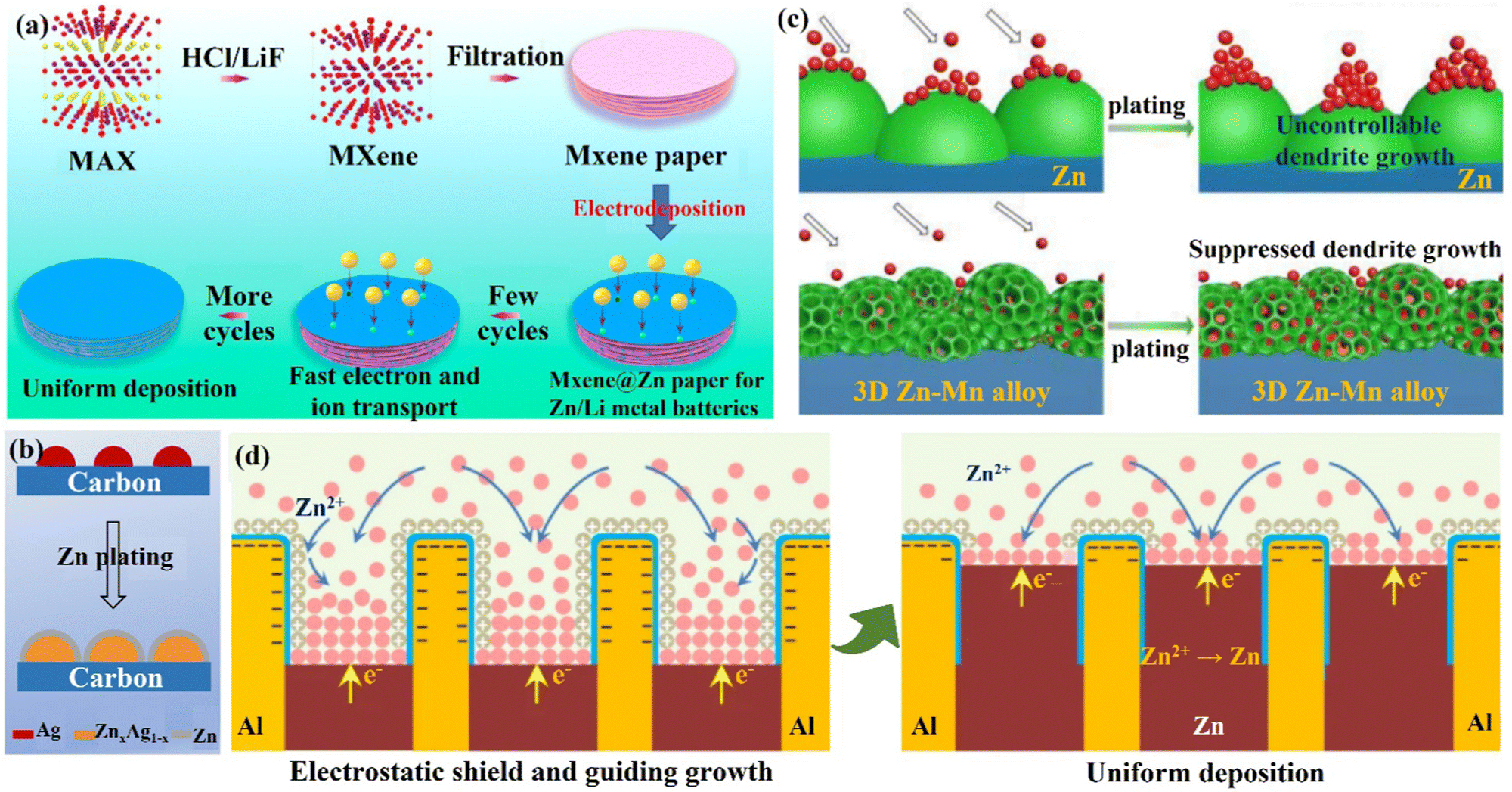 | ||
| Fig. 9 Schematic diagram of (a) the preparation process of the Ti3C2Tx MXene@Zn electrode and its layered structure with fast electron/ion transport channels to promote a uniform Zn deposition process. Reproduced with permission from ref. 103. Copyright 2019, American Chemical Society. (b) Metallic silver as crystalline seed material to construct an alloy solid solution surface layer (ZnxAg1−x) for homogeneous Zn deposition. Reproduced with permission from ref. 107. Copyright 2021, American Chemical Society. (c) Zn deposition at the tip position on the Zn foil anode to form dendrites (top), while it occurs at the grooves and internal nano-pores on the Zn–Mn alloy electrode (bottom). Reproduced with permission from ref. 19. Copyright 2021, Springer Nature. (d) The eutectic Zn–Al alloy anode guiding uniform Zn deposition. Reproduced with permission from ref. 109. Copyright 2020, Springer Nature. | ||
Yang's group19 created a 3D nanostructured Zn–Mn alloy anode that could withstand thousands of cycles even under extreme electrochemical conditions. The Zn–Mn alloy (Zn3Mn) electrode had a cauliflower-like microscopic appearance with a large number of internal pores and a super-hydrophilic surface that allowed for mass transfer and homogeneous deposition (Fig. 9c). Atomic force microscopy (AFM), simulation, and in situ optical microscopy demonstrated that Zn deposition occurred in internal nanopores and grooves between Zn/Mn particles, effectively inhibiting dendrite growth.108 In addition, the 3D Zn–Mn nanoalloy anode was found to have high corrosion resistance, supporting more than 1000 h of stable charge/discharge cycle performance even under seawater electrolyte and high current density (80 mA cm−2). Jiang's group109 designed a Zn–Al alloy (Zn88Al12) anode with an alternating Zn and Al lamellar nanostructure (layer spacing of roughly 450 nm). The Al layer acted as a two-dimensional skeleton capable of generating Al2O3 with insulating properties in situ, forming an Al/Al2O3 core/shell structure.110 When Zn2+ ions come in contact with the Al skeleton, Zn2+ ions are repelled and fall uniformly between the layers of the Al skeleton due to the electrostatic shielding effect of the Al/Al2O3 layer, thus leading to uniform Zn deposition (Fig. 9d). The presence of Al2O3 also prevents the dissolution of the Al layer and the generation of various by-products, which significantly improves the performance of the Zn–Al alloy electrode.
4.2 Interface engineering
Interface engineering is one of the most important ways to solve the poor stability of Zn anode electrodes. The stability on the electrode–electrolyte interface can be significantly improved by constructing functional material coatings, such as inorganic-, organic- and organic/inorganic hybrid coatings on the surface of the Zn anode. Functional coatings address the challenges facing Zn anodes by (1) improving the interfacial wettability to make the Zn2+ ion flux distribution uniform; (2) avoiding direct contact between electrodes and the electrolyte to inhibit the HER and side reactions on the Zn anode; (3) inducing uniform and dense Zn deposition to effectively extend the cycle life of Zn anodes.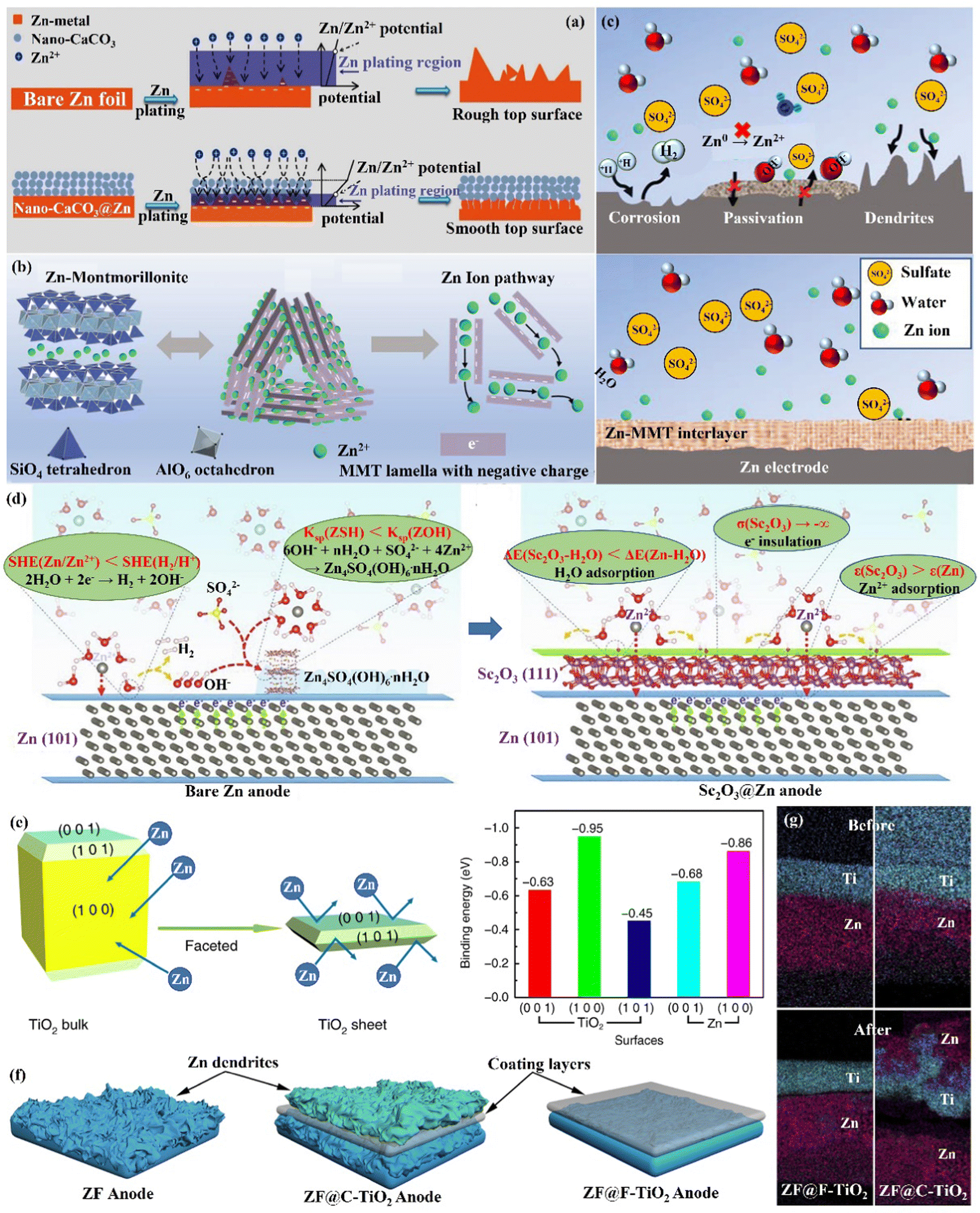 | ||
| Fig. 10 Schematic diagrams of (a) morphological changes in bare Zn and nano-CaCO3@Zn anode surfaces during Zn deposition, and the corresponding mechanisms. Reproduced with permission from ref. 88. Copyright 2018, Wiley-VCH. (b) Crystal structure of Zn-based MMT, the charge distribution in MMT lamella, and the interlamellar pathway for Zn2+ ion migration. (c) The Zn-based MMT layer promotes uniform deposition while suppressing severe side reactions. Reproduced with permission from ref. 111. Copyright 2021, Wiley-VCH. (d) The modification mechanism of the Zn anode by Sc2O3 coating. Reproduced with permission from ref. 65. Copyright 2021, Elsevier Inc. (e) The Calculated Zn atom binding energies with various TiO2 facets. (f) The behavior of Zn deposition in the presence of various TiO2 layers. (g) The EDS mapping images of F-TiO2@Zn and C-TiO2@Zn before and after 10 cycles. Reproduced with permission from ref. 112. Copyright 2020, Springer Nature. | ||
(ii) Zn compound coating. By introducing inorganic elements (S, P, F, O, etc.) to form a zinc compound coating with Zn, the charge distribution at the interface could be significantly changed, and the diffusion of Zn2+ ions could be promoted to alleviate the concentration polarization on the electrode surface. Guo's group86 formed a dense artificial SEI layer (ZnS) in situ with high mechanical strength, high stability, high adhesion, and high ion diffusion rate on the Zn foil surface by a vapor–solid strategy (Fig. 10a). Due to charge migration, S atoms formed Zn–S polar bonds with Zn atoms, which enhanced the adhesion of the ZnS layer to Zn foil. The unbalanced charge distribution also facilitated Zn2+ ion diffusion between the ZnS and Zn phases, resulting in a high ionic conductivity (tZn2+,ZnS@Zn = ∼0.78 vs. tZn2+,Zn = ∼0.33). The ZnS protective layer acted as a physical barrier, isolating the Zn foil anode from direct contact with the electrolyte, and guiding a uniform interlayer plating/stripping process through its insulating property (Fig. 10b). The ZnS@Zn symmetric cell exhibited a life higher than 1100 h at 2 mA h cm−1, and the ZnS@Zn//MnO2 full cell achieved high cycle stability (>2500 cycles) and high coulombic efficiency (>99.8%). Lee's group114 prepared a new type of zinc metal electrode with controllable surface composition and morphology by a periodic anodization technique, which was denoted as a zinc hexagonal pyramid array electrode coated with a functionalized ZnO layer (ZnO@Zn HPA). The current supply and interruption processes were repeated in a constant cycle to dissolve ZnO in NH4Cl/H2O2 solution and deposit it on the electrode, resulting in the formation of the ZnO coating (Fig. 11c). Through experimental optimization, the best performance of the ZnO@Zn HPA electrode was determined when the “splitting” value was 2 (i.e., the ratio of current disconnection time to current on time was 2). HPA increased the specific surface area, which reduced the local current density and facilitated the uniform distribution of Zn2+ ions. the functionalized ZnO passivation layer had a gradient thickness (thicker at the top than at the edges and bottom, with higher interfacial resistance), which could guide the deposition of Zn on the sides and bottom (Fig. 11c and d). After 1000 cycles at 9.0 A g−1 (29.22C), the ZnO@Zn//MnO2 full cell continued to operate excellently, with the CE remaining above 99%. Yang's group115 prepared a Zn–P solid solution alloy coating with corrosion resistance by electrochemical deposition. The ZnP layer was able to maintain a certain electrochemical activity and capacity contribution, which was beneficial for enhancing the energy density. The addition of P atoms significantly increased the binding energy with Zn atoms (Fig. 11e), which promoted the transfer and deposition of Zn2+ ions. The ZnP@Zn symmetric cell was cycled for over 300 h at 20 mA cm−2/30 mA h cm−2, and the ZnP@Zn//MnO2 full cell still had a discharge-specific capacity of 154.4 mA h g−1 after 1000 cycles at 1 A g−1. A 3D interconnected ZnF2 matrix was created by Yang's group116 to serve as a multipurpose protective layer (Fig. 11f), which redistributed the Zn2+ ion flux and was able to significantly reduce the activation energy of [Zn(H2O)6]2+ desolvation, resulting in more stable Zn deposition kinetics. The ZnF2 layer also allowed the insertion of Zn2+ ions and induced uniform Zn deposition behavior under the layer through an interstitial diffusion mechanism, which inhibited the growth of dendrites.117–120 Moreover, the properties of ZnF2 coating such as corrosion resistance, inhibition of HER, and better wetting made the ZnF2@Zn anode more stable. A ZnF2@Zn symmetric cell was obtained with a long cycle duration of 800 h and a high CE of 99.5%. The ZnF2@Zn//V2O5 full cell was stabilized at 279.2 mA h g−1 after 2000 cycles at 10 A g−1, with a capacity retention rate of 96%.
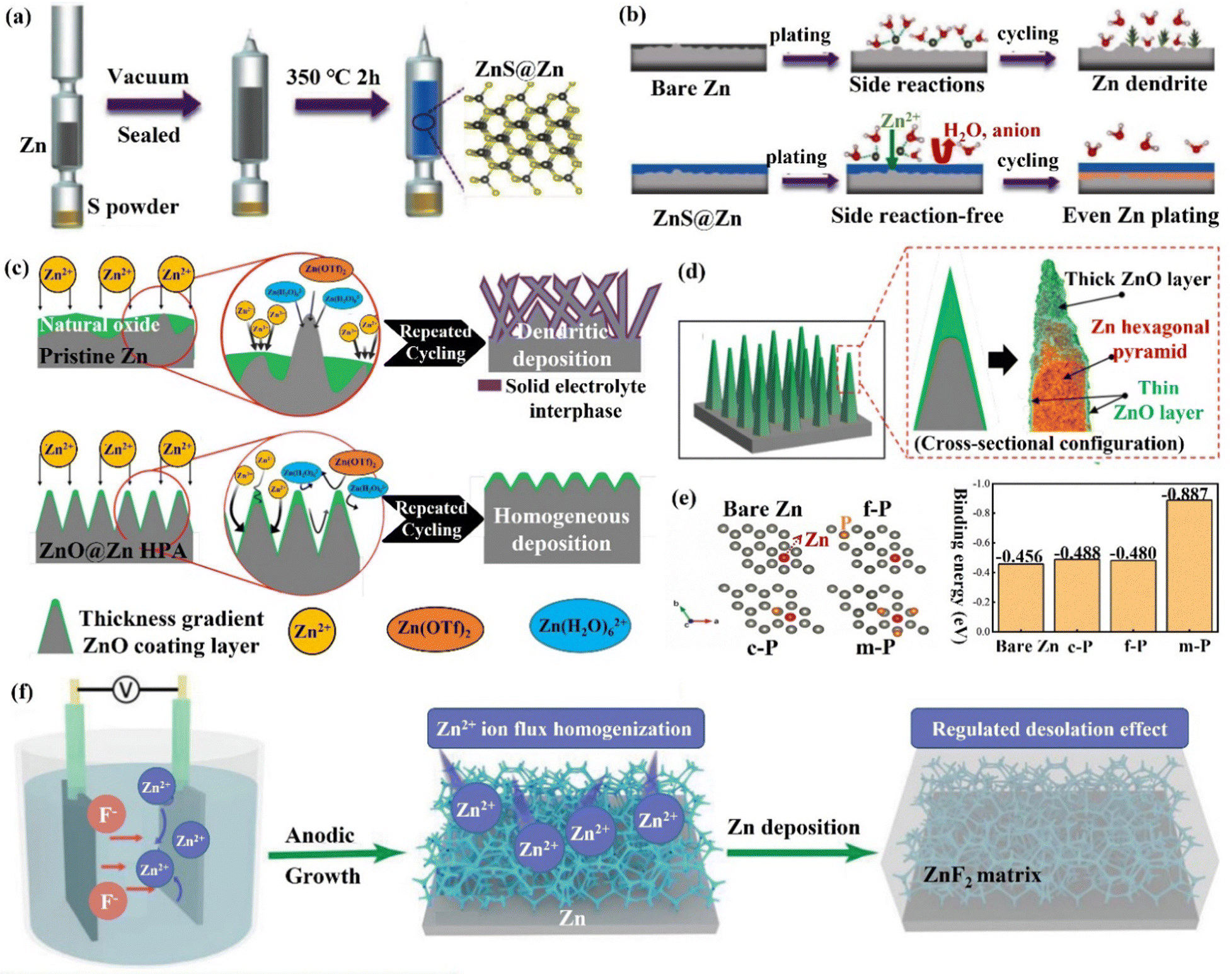 | ||
| Fig. 11 Schematic diagrams of (a) an in situ method for adding the ZnS layer to the Zn metal substrate, (b) Zn deposition behavior with/without ZnS layer. Reproduced with permission from ref. 86. Copyright 2020, Wiley-VCH. (c) Zn deposited on a pristine Zn anode and the ZnO@Zn HPA anode, (d) the cross-sectional configuration of the dendrite-free “functionalized ZnO layer” coated on a Zn HPA. Reproduced with permission from ref. 114. Copyright 2020, Wiley-VCH. (e) Zn was adsorbed on the bare Zn and ZnP layer at different adsorption sites, and the corresponding binding energy was calculated by the DFT. Reproduced with permission from ref. 115. Copyright 2021, Wiley-VCH. (f) The fabrication of the ZnF2@Zn anode and corresponding Zn deposition behavior. Reproduced with permission from ref. 116. Copyright 2021, Wiley-VCH. | ||
(iii) Metal coating. Metallic materials have recently been widely used in Zn anode interface modification strategies due to their high electrical conductivity and low impact on the battery as an anode surface coating. Tu's group121 formed a strong metallic tin (Sn) coating on the surface of Zn by chemical vapor deposition, which effectively inhibited the HER and corrosion.122,123 Moreover, the Sn nanoparticles formed on the Sn coating could also guide the uniform deposition of Zn2+ ions and avoid the formation of dendrites. With this modification, the Sn@Zn symmetric cell could cycle steadily for more than 500 h. Yang's group18,124 coated a layer of indium metal (In) with dual functions of corrosion inhibitor and nucleation agent to achieve the simultaneous inhibition of corrosion and dendritic problems at the Zn anode (Fig. 12a). In had zincophilicity, and the adsorption energy of In to Zn atoms was higher than that of Zn foil, making Zn2+ ions preferentially deposited on the In substrate.125 Moreover, the higher roughness and specific surface area of the In layer led to lower current density and uniform Zn2+ ion flux for uniform Zn deposition. The In@Zn symmetric cell could then achieve a stable cycle of 1500 h with a polarization voltage of 54 mV. Furthermore, Ji's group126 and Zhang's group127 were inspired by the gallium–indium–zinc phase diagram to design electrochemically inert gallium–indium (Ga–In) liquid alloy coatings, which effectively suppressed corrosion, HER and dendrite growth under the synergistic liquid alloying-diffusion strategy. The liquid Ga–In alloy had high zincophilicity and high charge conductivity. When the liquid Ga–In surface was newly generated with Zn atoms, the Zn atoms rapidly diffused into the Ga–In alloy interlayer through the zincation of the binary alloy phase. Once the Zn ratio exceeded the limit of the solid phase line of the alloy, the excess Zn atoms spontaneously grew inward and quickly “sank” into the liquid intermediate layer, and then nucleated uniformly near the Zn foil side (Fig. 12b). The stability of the Zn anode was significantly increased by the liquid Ga–In alloy coating, and the Ga–In@Zn symmetric cell was able to cycle steadily for more than 2100 h at 0.25 mA cm−2 with a polarization voltage of just 24 mV, and the Ga–In@Zn//MnO2 full cell was cycled more than 1000 times at 1 A g−1. The storage mechanism of the aqueous zinc–iodine (Zn//I2) battery with high energy density depends on the dissolution–deposition reaction. However, the monomeric I2 cathode tends to form the highly soluble I3− ions dissolving in the electrolyte, and the soluble ions will penetrate the separator and react on the surface of the Zn anode (I3− + Zn → Zn2+ + I−), leading to problems such as self-discharge and low CE.128–131 Isolating the direct contact between Zn anode and the I3− ions is one of the most promising strategies for enhancing the stability of the Zn//I2 battery. Therefore, the Sn protective layer constructed on the surface of the Zn anode in situ could isolate the direct contact between the Zn anode and the dissolved I3− ions. Consequently, the electrochemical performance of the Zn//I2 battery would be greatly improved.128,131
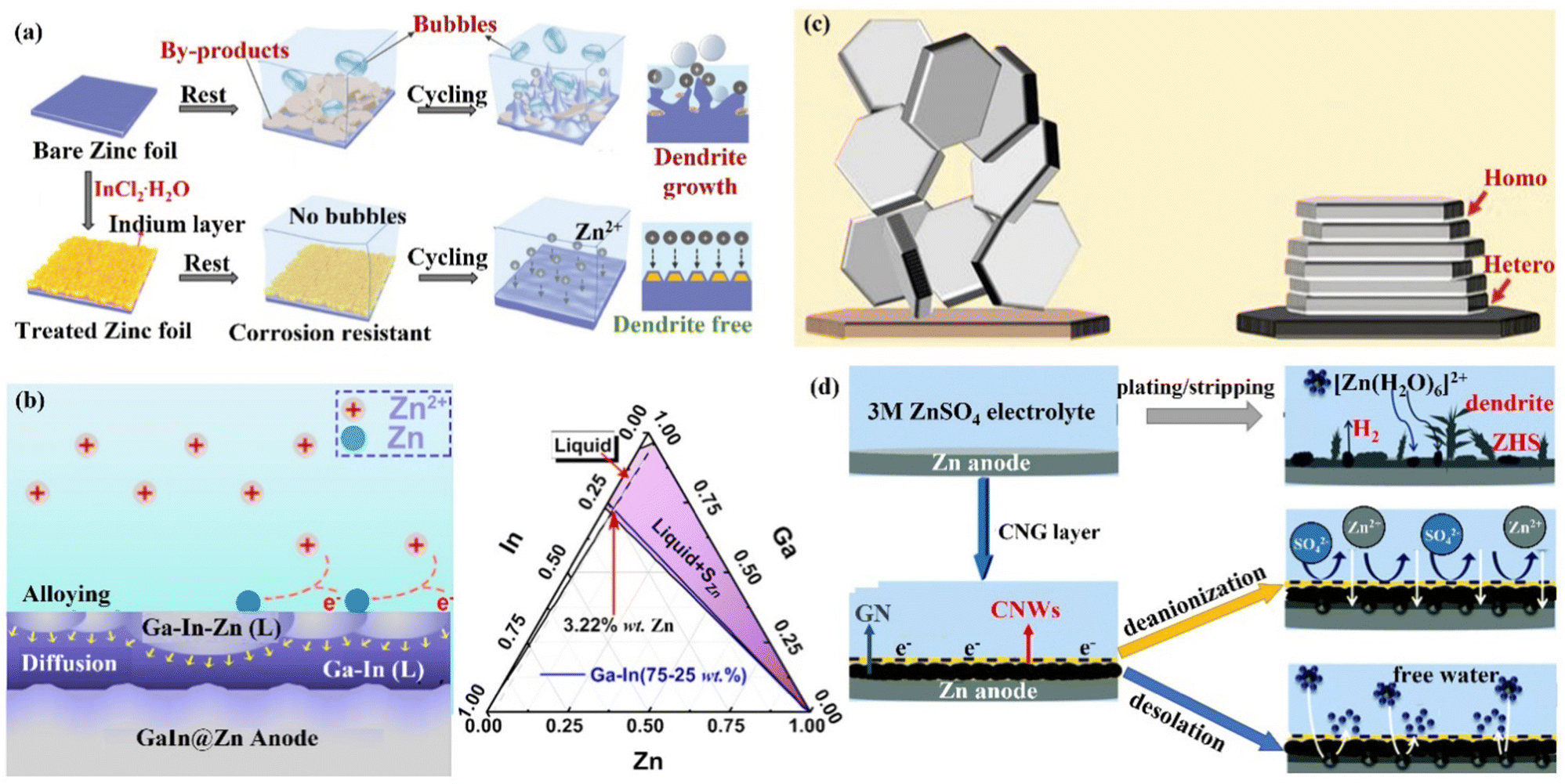 | ||
| Fig. 12 A schematic diagram of (a) Zn deposition on bare Zn and In@Zn anode. Reproduced with permission from ref. 18. Copyright 2020, Wiley-VCH. (b) The Ga–In–Zn phase diagram, and alloying-diffusion mechanism for dendrite-free Zn deposition. Reproduced with permission from ref. 126. Copyright 2021, American Chemical Society. (c) Using graphene (002) as a substrate to achieve the epitaxial deposition of Zn. Reproduced with permission from ref. 133. Copyright 2019, The American Association for the Advancement of Science. (d) Zn deposition behavior in the presence/absence of CNG film. Reproduced with permission from ref. 134. Copyright 2021, The Royal Society of Chemistry. | ||
(iv) Carbon material coating. Carbon materials have the advantages of high electrical conductivity, good chemical stability, and rich pore structure, which can effectively enhance the performance of ZIBs when compounded with the ZIBs cathode active material or Zn anode.132 Archer's group133 developed a dendrite-free Zn anode with an epitaxial deposition mechanism by introducing a graphene (002) substrate (a low lattice mismatch of 7% between graphene and Zn metal). “Epitaxial deposition” could effectively drive Zn deposition with a locked crystal orientation relationship, fundamentally changing the irregular and non-planar electrodeposition of Zn2+ ions at the liquid–solid interface. During the epitaxial deposition process, Zn was nucleated along the graphene (002) crystal plane orientation, forming an epitaxial layer with the same crystal orientation as the graphene substrate (heterogeneous epitaxial process). When the surface of the graphene substrate was completely covered by Zn deposition, a homogeneous epitaxial process would occur, and finally, a platelet stacking structure Zn deposition would be obtained (Fig. 12c). Electrochemical studies revealed that the epitaxial Zn anode's reversibility was greatly enhanced, with a CE of more than 99% even after thousands of cycles. Additionally, Li's group134 assembled graphene (GN) and cellulose nano-whiskers (CNW) through CH–π interactions to construct a bifunctional cellulose nano-whisker-graphene (CNG) membrane. The CNG membrane had strong ductility and puncture resistance and could adapt to the volume change during plating/stripping. Moreover, through the attraction of CNW to water molecules and the repulsion of GN to water molecules, the CNG film could be used as a desolvation layer to promote the desolvation process of [Zn(H2O)6]2+. Further, the CNG film with low electronic conductivity and high ionic conductivity had a negatively charged surface, shielding anions while promoting the diffusion of Zn2+ ions through the CNG layer (Fig. 12d) and achieving (002) crystal-oriented Zn deposition. Under this synergy, the CNG@Zn symmetric cell achieved an ultra-long lifetime of 5500 h at 0.25 mA cm−2/0.5 mA h cm−2, which was more than 27 times that of the bare Zn anode. The CNG@Zn//MnO2 full cell has a discharge capacity of up to 307 mA h g−1 and a capacity retention rate of 87.8% even after 5000 cycles at 5C.
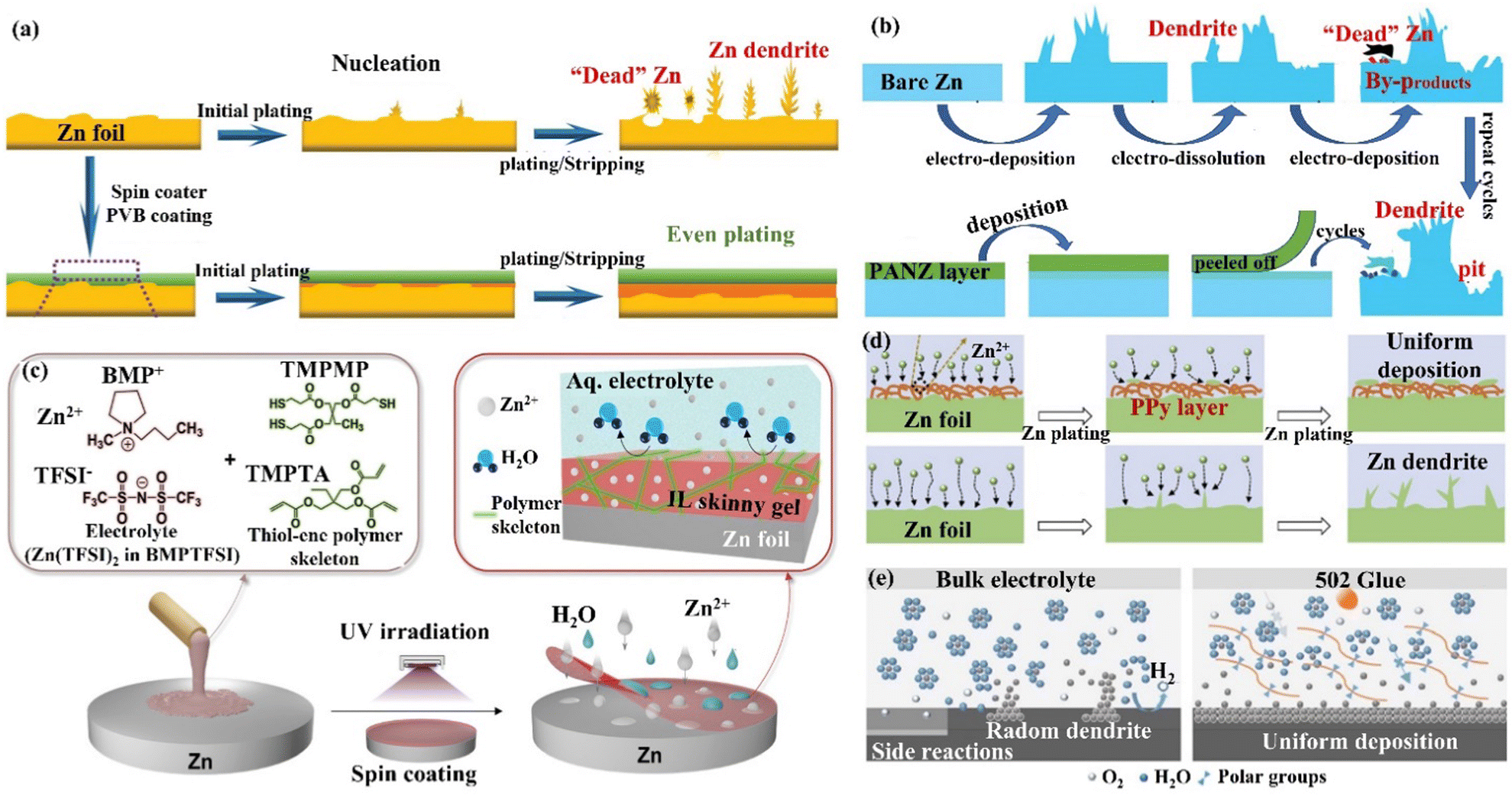 | ||
| Fig. 13 A schematic diagram of (a) the morphological evolution of bare Zn and the PVB@Zn anode during repeated cycles. Reproduced with permission from ref. 135. Copyright 2020, Wiley-VCH. (b) Zn deposition behavior on bare Zn and the PANZ@Zn anode. Reproduced with permission from ref. 136. Copyright 2021, Wiley-VCH. (c) The process of preparation of IL thin gels, and the chemical composition and protection mechanism of IL coating. Reproduced with permission from ref. 137. Copyright 2021, Wiley-VCH. (d) Zn deposition process on the Zn foil and the PPy@Zn anode. Reproduced with permission from ref. 138. Copyright 2020, Elsevier Inc. (e) The behaviors of Zn deposition and side reactions on Zn foil and 502-glue@Zn surfaces, and the modification mechanism of 502 coatings. Reproduced with permission from ref. 139. Copyright 2020, Elsevier Inc. | ||
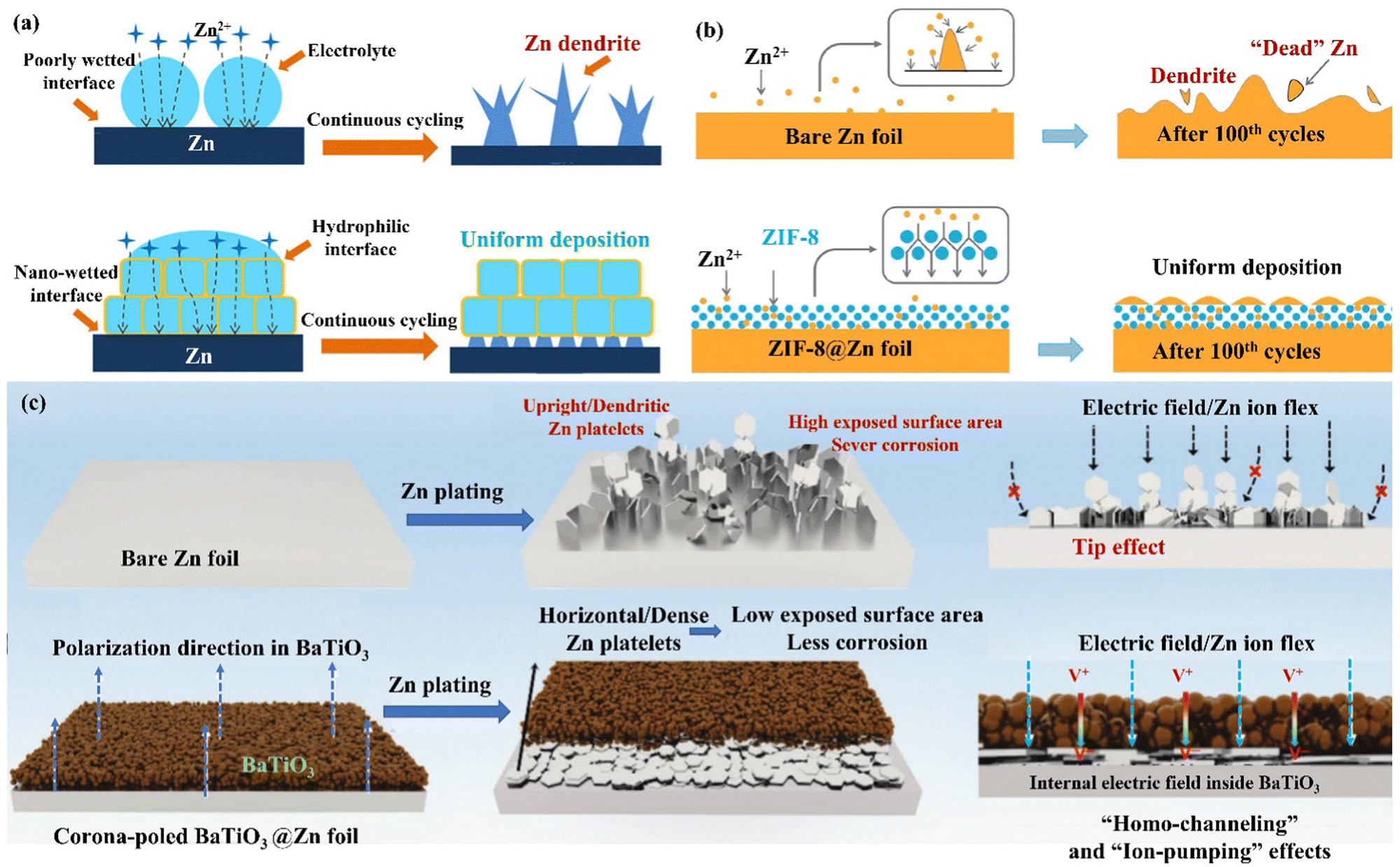 | ||
| Fig. 14 (a) A schematic diagram of the Zn deposition mechanism on bare Zn and MOF−PVDF@Zn. Reproduced with permission from ref. 146. Copyright 2019, American Chemical Society. (b) Morphology evolutions for Zn foil and the ZIF-8@Zn electrode during repeated Zn plating/stripping processes. Reproduced with permission from ref. 147. Copyright 2020, Springer Nature. (c) A schematic diagram of Zn deposition morphology, electric field, and ion flux distribution on bare Zn and poled BTO-coated Zn. Reproduced with permission from ref. 148. Copyright 2021, Wiley-VCH. | ||
4.3 Electrolyte optimization
The stability of the Zn anode can be successfully improved by optimizing the electrolyte, except for the electrode material protection approaches. The presence of electrolytes provides an effective path for the transmission of Zn2+ ions in the battery system, which is one of the important aspects that affect the energy storage of the battery system. Compared with organic electrolytes, the cost of aqueous electrolytes is lower, and safety and ionic conductivity are significantly improved. However, aqueous electrolytes also face some challenges such as narrow electrochemically stable potential window (ESPW) (∼1.23 V), Zn2+ ions forming a solvated structure Zn(H2O)62+ with water molecules, and the decomposition of water molecules leads to side reactions such as HER and corrosion.149 Therefore, when optimizing the electrolyte, the following points must be met: (i) improve the transfer of Zn2+ ions and induce uniform Zn deposition. (ii) Diminish the content of active water in the electrolyte to inhibit side reactions. (iii) Widen the electrochemical stable potential window (ESPW) so that the electrode material can work at high voltage. (iv) Change the structure of the Zn solvate and reduce the impact of the desolvation process. Some of the additives are able to adsorb on the surface of the Zn electrode or react with the Zn electrode to form a compositionally controlled and structurally stable SEI layer in situ. This SEI layer significantly enhances the stability of the electrode–electrolyte interface. This section optimizes the types of Zn salts and electrolyte additives to improve battery performance.ZnSO4 with a regular tetrahedral structure has a strong polar S–O bond, and good stability.150 However, during the charging and discharging process, serious side reactions still occur, producing insulation by-products such as ZHS and Zn4(OH)6SO4·nH2O. The insulation by-products covered the surface of the Zn electrode, leading to an increase in overpotential and hindering the migration of Zn2+ ions, and caused a decline in electrochemical performance and stability. The Zn symmetric cell using ZnSO4 as the electrolyte could cycle steadily for 70 h while maintaining a CE of 99.1% at 0.5 mA cm−2.
In contrast, the presence of larger CF3SO3− and TFSI− anions in Zn(CH3COO)2 and Zn(TFSI)2 reduces the number of coordinated water molecules surrounding Zn2+ ions, decreases the degree of solvation with H2O, and facilitates the migration of Zn2+ ions, which alleviated the side reaction to achieve higher reversibility.150,157 However, the dendrite growth caused by an uneven electric field and ion distribution was not significantly improved, and the Zn symmetric cell with Zn(CF3SO3)2 and Zn(TFSI)2 electrolytes could be cycled stably for 220 h and 175 h, respectively.
Through the continuous development of high-performance zinc salt electrolytes, Sun's group150 found that Zn(ClO4)2 electrolytes could significantly improve the electrochemical performance of AZIBs. During the charging and discharging process, ClO4− anions were reduced to form a thin protective layer containing Cl− ions, which inhibited the side reactions. The Zn symmetric cell containing the Zn(ClO4)2 electrolyte had excellent performance and achieved a high stability performance of more than 3500 h. However, at high temperatures, Zn(ClO4)2 electrolytes may pose safety risks, which limits its large-scale application in battery systems. Given the instability of the electrolyte in the cell, other methods are needed for optimization, such as adjusting the concentration of the electrolyte and introducing electrolyte additives.
(i) The alteration of Zn2+ ions solvation structure. In the aqueous electrolyte, Zn2+ ions will coordinate with water molecules to form a Zn[H2O]62+ solvation structure, which additionally increases the energy required for the solvent removal process. In addition, the free water molecules removed during the desolvation process will be decomposed and reduced in contact with the Zn electrode, leading to side reactions such as corrosion, HER, passivation, and the generation of dendrites, which diminish the utilization rate and cycle life of Zn. Mai's group66 simultaneously controlled the solvation structure of Zn2+ ions and the uniform distribution of ions and electric field at the interface by introducing a trace amount of glucose molecules (low-cost and low concentration) into the ZnSO4 electrolyte to construct a dendrite-free ZIBs (Fig. 15a). By replacing the water molecules in the solvation layer, glucose molecules effectively limited the occurrence of HER, Zn metal corrosion, and the generation of by-products. The organic molecular additives are promising for designing an advanced electrolyte. The polar groups can establish strong interactions with Zn2+ ions, water molecules and Zn surfaces, reconstruct the solvated structure of Zn2+ ions, build a hydrogen bond network, and preferentially adsorb on the Zn anode to form a zincophilic layer.158 The glucose molecules adsorbed on the electrode surface also helped to shield the electric field at the tip, which prevented the dendrite growth. With this trace amount of glucose additives, the Zn symmetric cell could cycle steadily for more than 2000 h and 270 h at 1 mA cm−2/1 mA h cm−2 and 5 mA cm−2/5 mA h cm−2, respectively. The Zn//MnO2 full cell could be stable for more than 1000 cycles at 10 C, and the capacity can reach 265.6 mA h g−1 at 1C. Pan's group32 regulated the coordination environment of Zn2+ ions by introducing alcohol additives, thereby inhibiting the growth of Zn dendrites (Fig. 15b). The research results showed that the polyol had a good coordination ability with Zn2+ ions, which was beneficial for controlling the growth of Zn dendrites.159 For example, after adding ethylene glycol (EG) additives, part of the water molecules in the solvated [Zn(H2O)6]2+ structure were replaced by EG molecules to form the [Zn(H2O)m(EG)n]2+ structure. This coordination structure reduced the desolvation energy of Zn2+ ions, and its larger size had a steric hindrance effect on the diffusion of Zn2+ ions. In the alcohol/water mixed electrolyte, the crystal grains of the deposition product were refined and the dendrite growth process was significantly inhibited. The Zn symmetric cell with 3 M ZnSO4/H2O/68 vol% EG electrolyte achieved a long cycle life (2668 h stable cycle at 0.5 mA cm−2). Qiao's group160 proposed a bifunctional electrolyte additive (ethylenediaminetetraacetic acid tetrasodium salt, Na4EDTA) to improve the reversibility of the Zn anode. the EDTA anion preferentially adsorbed and covered the Zn anode surface, reducing the active sites of the HER (the hydrogen production of ZnSO4 electrolyte containing Na4EDTA additive was only 1/4 of the original electrolyte). The EDTA-adsorbed layer formed Zn2+ ion diffusion channels at the interface and the polar functional groups facilitated the desolvation process of Zn(H2O)62+, which allowed uniform Zn deposition (Fig. 15c). With this bifunctional electrolyte additive, the Zn symmetric cell achieved a CE of 99.5% after 2500 stable cycles at 5 mA cm−2/2 mA h cm−2, and the Zn//VO2 full cell showed almost no capacity decay within 2000 cycles at 4 A g−1. Wu's group36 changed the solvation structure of Zn2+ ions and reconstructed the hydrogen bond (S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯HO) by adding dimethyl sulfoxide (DMSO) additive to the ZnSO4 electrolyte, significantly improving the battery performance and low-temperature applications. Compared with H2O, the binding energy between DMSO and Zn2+ ions was greater than the Zn2+ ion's coordination to form a solvated structure ([Zn(H2O)m(DMSO)n]2+), which increased the nucleation overpotential and corrosion potential of Zn2+ ions, improved the deposition kinetics of Zn2+ ions and inhibited the corrosion of Zn metal.155 In addition, DMSO induced the deposition of Zn2+ ions on the (002) crystal plane, forming a hexagonal structure without dendrites (Fig. 15d). With the DMSO additive, the Zn symmetric cell was stable when cycled for more than 2100 h and 1200 h at 20 °C and −20 °C, respectively. The Zn//MnO2 full cell could work stably for more than 3000 cycles and 300 cycles at 20 °C and −20 °C, respectively, and could maintain a reversible capacity of 140 mA h g−1 even at 10C (20 °C). With additives such as carbonyl-containing organic solvents (N-methyl-2-pyrrolidone, NMP),161 polyhydroxy organics (maltose),162 and phosphorus-functional group-containing molecules (TCEP-HCl),163 and so on, the solvated structural rearrangement of Zn and the stabilization of the hydrogen bond network were facilitated. This synergistic effect inhibited the formation of dendrites and side reactions caused by water splitting, which improved the Zn anode stability. These additives also provide new insights into the design of aqueous electrolytes.
O⋯HO) by adding dimethyl sulfoxide (DMSO) additive to the ZnSO4 electrolyte, significantly improving the battery performance and low-temperature applications. Compared with H2O, the binding energy between DMSO and Zn2+ ions was greater than the Zn2+ ion's coordination to form a solvated structure ([Zn(H2O)m(DMSO)n]2+), which increased the nucleation overpotential and corrosion potential of Zn2+ ions, improved the deposition kinetics of Zn2+ ions and inhibited the corrosion of Zn metal.155 In addition, DMSO induced the deposition of Zn2+ ions on the (002) crystal plane, forming a hexagonal structure without dendrites (Fig. 15d). With the DMSO additive, the Zn symmetric cell was stable when cycled for more than 2100 h and 1200 h at 20 °C and −20 °C, respectively. The Zn//MnO2 full cell could work stably for more than 3000 cycles and 300 cycles at 20 °C and −20 °C, respectively, and could maintain a reversible capacity of 140 mA h g−1 even at 10C (20 °C). With additives such as carbonyl-containing organic solvents (N-methyl-2-pyrrolidone, NMP),161 polyhydroxy organics (maltose),162 and phosphorus-functional group-containing molecules (TCEP-HCl),163 and so on, the solvated structural rearrangement of Zn and the stabilization of the hydrogen bond network were facilitated. This synergistic effect inhibited the formation of dendrites and side reactions caused by water splitting, which improved the Zn anode stability. These additives also provide new insights into the design of aqueous electrolytes.
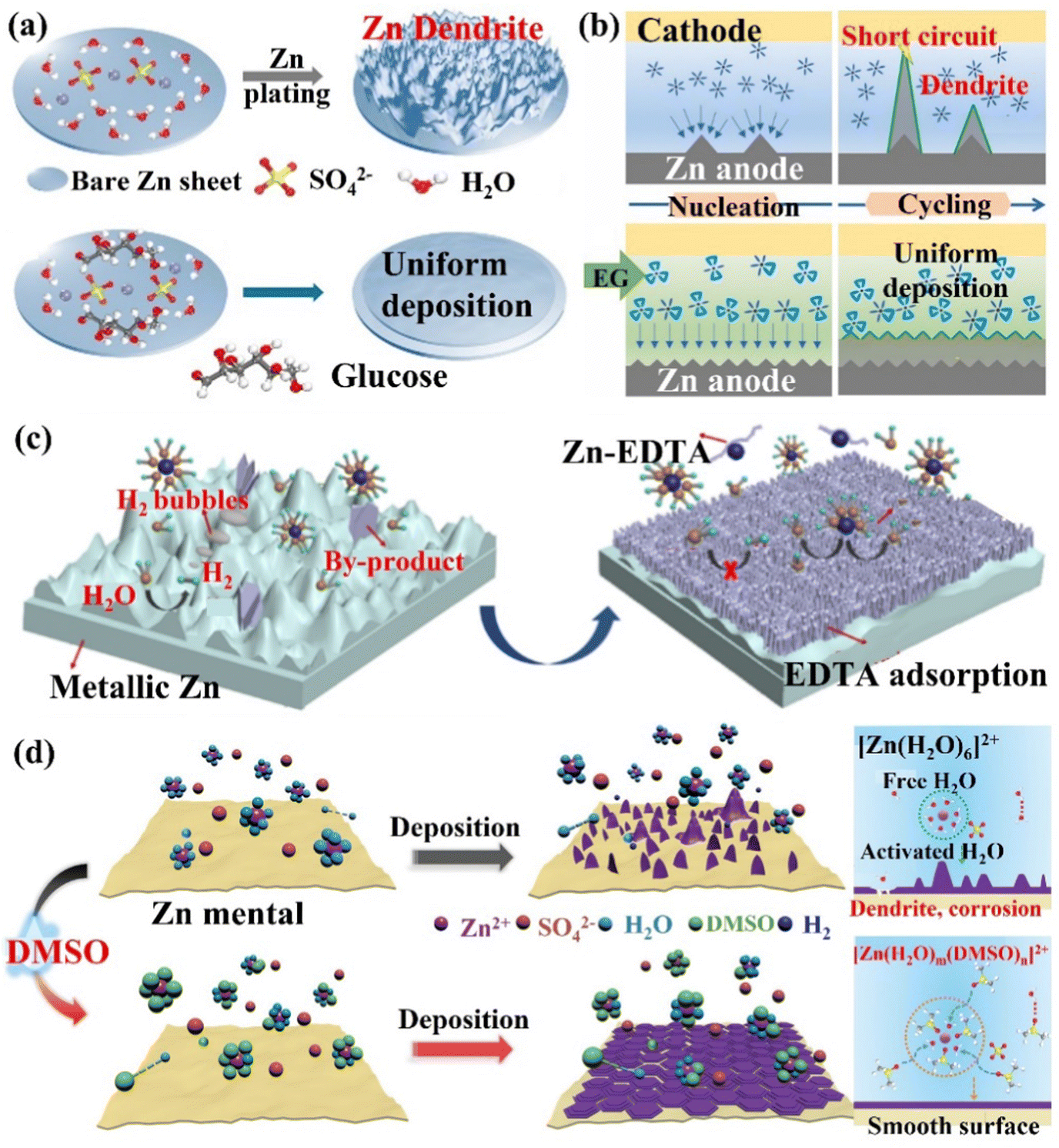 | ||
| Fig. 15 The schematic diagram of (a) Zn deposition behaviors in ZnSO4 and ZnSO4–glucose electrolytes. Reproduced with permission from ref. 66. Copyright 2021, Wiley-VCH. (b) A schematic diagram of dendrite inhibition of ZnSO4/H2O/EG hybrid electrolytes. Reproduced with permission from ref. 32. Copyright 2020, Elsevier Inc. (c) A schematic of Zn/electrolyte interphase behaviors during Zn plating in ZnSO4 electrolyte and EDTA-functionalized ZnSO4 electrolyte. Reproduced with permission from ref. 160. Copyright 2021, Wiley-VCH. (d) A diagram of the Zn2+ ions solvation structure, the corresponding nucleation and deposition behaviors in 2 M ZnSO4 and DMSO-containing hybrid electrolytes. Reproduced with permission from ref. 36. Copyright 2021, Wiley-VCH. | ||
(ii) The in situ formation of the SEI layer. The establishment of SEI can effectively block the contact between the electrolyte and the Zn anode surface to suppress side reactions. However, because of significant volume fluctuations during electrochemical cycling, these brittle SEI layers may be harmed and separated from the Zn surface. Moreover, the low ionic conductivity, Zn2+ ion migration number of artificial SEI layers and poor interfacial contact with Zn also lead to their limited enhancement for the stability of the Zn anode, and the formation of the in situ SEI layer can effectively solve these problems.164–166 Cui's group156,157 reported an acetamide (Ace) – Zn(TFSI)2 eutectic electrolyte. TFSI− was able to coordinate with Zn2+ ions to form zinc complexes ([ZnTFSIm(Ace)n](2−m)+), significantly reducing the decomposition energy of the TFSI− anion (Fig. 16a). This caused the reductive decomposition process of TFSI− to occur preferentially to the Zn deposition process (0.37 V vs. −0.76 V), favoring the in situ formation of mechanically rigid SEI layers (ZnF2) (Fig. 16b). Through this improved zinc salt electrolyte, the Zn symmetric cell could maintain an average CE of 99.7% and achieve a stable cycle of more than 1000 h. The negatively charged MXene nanosheets could form a stable protective film, which might create a durable protective layer that would balance out the distribution of ions and reduce the path of Zn2+ ions during their diffusion.167 Moreover, ample functional groups and the high electrical conductivity of the Mxene layer allowed for uniform Zn nucleation, the inhibition of dendritic growth, and the reduction of byproducts (Fig. 16c). Through the MXene nanosheet electrolyte additive, the Zn symmetric cell could be cycled stably for 1180 h, and the CE was as high as 99.7%. The Zn//V2O5 full cell exhibited high cycle stability with more than 300 cycles and maintained a reversible capacity of 192.9 mA h g−1 at 1 A g−1. Wang's group154 added 0.05 m SnCl2 additive to the ZnCl2 electrolyte to form a eutectic electrolyte, which effectively overcame the two challenges of disordered dendrite growth and poor low-temperature performance. The eutectic electrolyte could be reduced and decomposed to form a zincophilic Sn/zincophobic Zn5(OH)8Cl2·H2O double-layer interface phase in situ (Fig. 16d), and realize a stable performance at low temperature. The zincophilic Sn layer could reduce the overpotential of Zn deposition and promote uniform Zn deposition, while the zincophobic Zn5(OH)8Cl2·H2O top layer inhibited the growth of Zn dendrites. In addition, because the solvated Zn2+ ions and Cl− ions disrupted the hydrogen bond network, the eutectic electrolyte had a high ionic conductivity of 0.8 mS cm−1 even at a temperature of −70 °C. Thanks to the design of this eutectic electrolyte, the Zn symmetric cell could be stably cycled for more than 500 h at 3 mA cm−2 with an overpotential of only 8 mV. Moreover, even at −50 °C, the Zn//VOPO4 full cell maintained a capacity of over 95% and a CE of 99.9% after 200 cycles. Guo's group165 added 0.025 M Zn(H2PO4)2 to 1 M Zn(CF3SO3)2 to form a dense and uniform Zn phosphate SEI layer (2H2PO4− + 4OH− + 3Zn2+ → Zn3(PO4)2·4H2O↓, ∼140 nm). This Zn phosphate SEI layer had good interface stability, high ion mobility, and ion conductivity, ensuring a uniform and rapid diffusion process for Zn2+ ions, thereby achieving dendrite-free Zn deposition (Fig. 16e). This electrolyte design idea was also applicable to the ZnSO4 system electrolyte. Through this in situ formation of SEI layer Zn(H2PO4)2, the stable cycle performance of the Zn//V2O5 battery was significantly improved, and the capacity retention efficiency could still be as high as 94.4% after 500 cycles. Additives such as alkylammonium salts (Me3EtNOTF),118 ZnSe,168 and ZnF2169,170 could also help to form a composite interface in situ with high conductivity and waterproof performance, suppressing the HER, increasing the ECSW of the electrolyte, and achieving a highly reversible stable cycling performance.
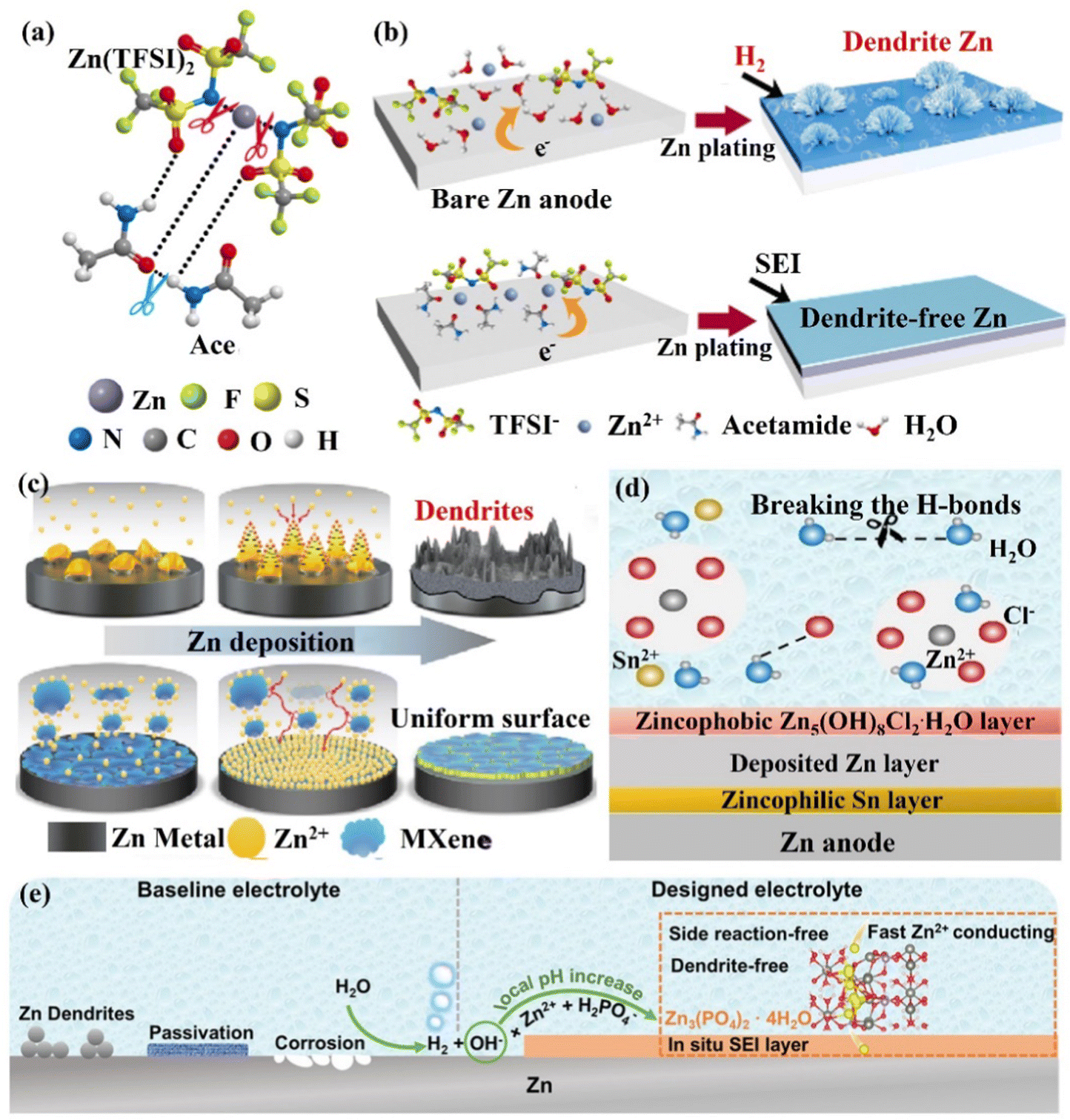 | ||
| Fig. 16 Schematic diagrams of (a) Zn2+ ions, TFSI− ions, and Ace interacting to generate eutectic solutions, (b) Zn deposition in 1 M Zn(TFSI)2 and the regulatory mechanism for achieving uniform Zn deposition with the SEI layer. Reproduced with permission from ref. 156. Copyright 2019, Springer Nature. (c) The regulation of Zn deposition by MXene additive. Reproduced with permission from ref. 167. Copyright 2021, Springer Nature. (d) The formation of SEI layers in the presence of SnCl2 additive and its regulatory mechanism for Zn deposition. Reproduced with permission from ref. 154. Copyright 2021, Wiley-VCH. (e) The mechanism of zinc phosphate SEI layer generation and the control of Zn anode surface. Reproduced with permission from ref. 165. Copyright 2021, Wiley-VCH. | ||
(i) All-solid-state polymer electrolyte. High mechanical strength is an inherent advantage of SPEs, which effectively solves the short-circuit problem caused by Zn dendrites in aqueous electrolytes. Currently, SPEs based on polyethylene oxide (PEO), polyacrylonitrile (PAN), and polyvinyl chloride (PVC) have been widely studied due to their high mechanical strength, chemical stability, and flexibility. However, the low ionic conductivity (<0.1 mS cm−1) and poor interfacial contact of SPEs at room temperature limit their further development.171,174,175 Research has shown that the crystallinity of SPEs could be significantly reduced and ionic conductivity greatly enhanced by (1) adding inorganic nanofillers (Al2O3, SiO2, TiO2, ZnO) to lower the phase transition temperature,176–179 and (2) blending with low molecular weight polymers likes polyvinylidene fluoride (PVDF)23 and branched aramid nanofibers.180 Zhi's group23,179 prepared a polyvinylidene fluoride-hexafluoropropylene/polyethylene oxide (PVDF-HFP/PEO) all-solid polymer electrolyte with high ionic conductivity (16.9 mS cm−1), excellent flexibility and mechanical properties, which significantly inhibited both the HER and dendrite growth. Solid-state ZIBs based on PVDF-HFP/PEO exhibit excellent cycling stability and rate capability, enabling the operation in a wide temperature range from −20 °C to 70 °C. However, lower ionic conductivity and unstable interface still restrict the application of SPEs.
(ii) Hydrogel electrolytes (HEs). HEs exhibit excellent performance since they possess the properties of both liquid and solid materials. At present, several hydrogels have been successfully applied in functional AZIBs, such as polyvinyl alcohol (PVA),181 polyacrylamide (PAM),182–186 polyacrylic acid (PAA),187 sodium polyacrylate (PANa),158,188 zwitterionic sulfobetaine189 and so on. These HEs exhibit high ionic conductivity from 10−3 to 10−1 S cm−1, which is higher than that of SPEs. Compared with the aqueous electrolytes, HEs have many advantages, such as (1) strong hygroscopicity and water retention, which can effectively alleviate the loss of electrolyte; (2) uniformly distributed internal 3D network structure that facilitates the rapid transport of Zn2+ ions and dense Zn deposition; (3) reduced water content that significantly inhibits side reactions and dissolution of the active material.190
However, HEs are extremely easy to freeze at low temperatures, leading to the disappearance of the inherent advantage (flexibility) and the degradation of battery performance. Zhi's group184 designed a glycol/polyacrylamide-based oil–water double-crosslinked hydrogel that exhibited a low freezing point, high ionic conductivity, high salt resistance, and strong mechanical stability (can pull stretch ∼900%). The ether groups introduced into the hydrogel matrix can form strong hydrogen bonds with free water molecules, effectively reducing the freezing point of the hydrogel and maintaining high ionic conductivity. Through this modification, the assembled flexible Zn//MnO2 battery maintained a discharge capacity of 146 mA h g−1 at −20 °C, and the capacity retention rate was 72.54% of the initial capacity after 600 cycles. Nevertheless, HEs still face many challenges that need to be addressed. First, the inhomogeneous quasi-solid–electrode interface inevitably leads to large resistance, which requires improved preparation processes. Secondly, the mechanical properties of HEs are much inferior to SPEs, so a rational double cross-linking design is required to achieve HEs with both flexibility and mechanical stability. Furthermore, it is still necessary to improve the stability of HEs over a wide temperature range.
(iii) Other non-liquid electrolytes. In addition to SPEs and HEs, other materials have been explored for non-liquid electrolytes. For example, quasi-solid electrolytes (PVC, PVDF, PVDF–HFP, polyethylene glycol diglycidyl ether (PEGDGE), tetraethylene glycol diacrylate (TEGDA)) consisting of liquid electrolyte (solvent) and polymer matrix and new inorganic high concentration colloidal electrolytes (HCCE). These have been extensively investigated to achieve excellent properties by tuning the compositions.158,171,173,191
5. Conclusions and outlook
The AZIBs are promising for application in the field of energy storage due to their inherent advantages such as abundant Zn metal reserves, environmental friendliness, low redox potential (−0.76 V vs. SHE), and high theoretical capacity (820 mA h g−1 or 5855 mA h cm−3). However, the poor reversibility of the Zn anode seriously hinders their practical large-scale applications. In this review, the intrinsic failure mechanisms of the Zn anode, influential factors on Zn deposition, and the state-of-the-art protection strategies for highly reversible Zn anode design are systematically summarized. Although significant progress has been achieved in the improvement of high-performance AZIBs, there are still some challenges for their future practical applications, and even some key issues that need attention have often been overlooked in the previous research. More attention should be paid to the zinc utilization, cost and battery energy density to meet the growing demand for the commercialization of zinc ion batteries. The future perspectives and development direction for the high-performance AZIBs are as follows:5.1 Modulating the relationship between the depth of discharge (DOD)/CE and the reversibility of the Zn anode
Currently, the utilization rate of a Zn anode and the Coulombic efficiency (CE) of the electrochemical performance of batteries is still low for practical applications. The DOD represents the percentage of the Zn anode participating in the electrochemical reaction as compared to the total capacity of the entire Zn electrode. Battery life is affected by the DOD of the anode, which is an important parameter for measuring the utilization of electrode materials. This parameter is related to battery capacity decay and energy density. CE is also another important indicator for evaluating the cycle life and utilization of Zn anodes. At present, there is no unified standard for quantifying the CE and DOD of the Zn anode, and many studies have obtained high CE under the low DOD of the Zn anode, which lacks practical guiding significance for the development of AZIBs with high energy density. Therefore, it is significant to develop the Zn anodes with high CE under the high DOD for the development and application of AZIBs.5.2 Selecting suitable types of zinc anode for ZIBs
Most zinc−ion batteries generally choose Zn flake as the anode and a low-loading mass of cathode for the related research. The test conditions are relatively mild (high N/P ratio, low areal capacity, and so on), thus the actual energy density of the total battery system is very low, and it is far away from meeting the needs of practical applications. Although great progress has been made for the Zn flake anode in the lab, it has some shortcomings that are difficult for the Zn anode towards the commercialization of ZIBs as follows: (1) low utilization of Zn, which is not conducive to the construction of practical high-energy density electronic devices; (2) serious dendrite growth, which can easily pierce the separator and cause an internal short circuit; (3) repeated plating/stripping processes lead to the overall pulverization of the electrode, which results in the failure of the battery. Compared with Zn flake, Zn powder has more industrial application prospects as a Zn anode material. First, the loading quality of the Zn powder can be adjustable, which is conducive to the preparation of high-utilization Zn anodes and reduces the capacity ratio of negative/positive (N/P) electrodes to meet actual needs. Secondly, Zn powder has a more diversified processing technology, which also provides favorable conditions for constructing a dendrite-free Zn anode. However, the Zn powder anode increases the contact area between the electrode and the electrolyte, which inevitably leads to problems such as HER and side reactions. It is expected to facilitate the commercial application of Zn powder anodes by improving Zn powder binders, current collectors, and hybrid structures.5.3 Application under extreme conditions
The ability to maintain high performance under extreme operating conditions (lean electrolyte, high current density, and wide temperature range, etc.) is very important for the commercialization of AZIBs. The electrolyte added to the ZIBs button cells assembled in the experimental stage is usually excessive, which not only seriously reduces the energy density of the battery but also can not directly reflect the problem of electrolyte consumption due to the occurrence of side reactions. In the case of ensuring the normal operation of the battery, the smaller the amount of electrolyte added, the higher the energy density of the AZIBs that can be achieved. Under the conditions of high current density, side reactions and other undesirable failures inside the battery will be more obvious, resulting in unstable battery performance and even short-circuit failure. In addition, aqueous electrolytes will freeze at low temperatures, resulting in decreased conductivity and reduced battery performance. These are the problems that need to be solved urgently in the process of commercial application of AZIBs. Seeking low-cost electrolytes or additives to maintain stable performance under extreme working conditions is also the main theme for the development of AZIBs.5.4 Developing the flexible and stretchable wearable electronic devices
Based on the inherent safety performance of AZIBs, it is promising for flexible and stretchable electronics applications. Gel polymer electrolytes provide a great boost for the development of flexible aqueous electronic devices as a result of their great wettability, high flexibility and excellent mechanical strength. However, in order to ensure the performance and energy density of the battery, the thickness and weight of the gel electrolyte must also be fully considered. In addition, the self-healing substrates developed so far typically suffer from short lifespans due to the dehydration in air. More attention should be directed towards improving the resistance of water loss or developing new suitable packaging materials. Moreover, the effects of components such as flexible and stretchable separators, current collectors, and binders on the performance of AZIBs are still not studied enough, and are also crucial for the improvement of the performance of AZIBs.5.5 Designing the battery automatic detection device
Real-time detection of battery electrochemical performance during charge and discharge can accurately reflect the stability of battery material. Therefore, it is necessary to design an in situ automatic battery detection device to precisely forecast the “battery failure time” which guarantees that the battery pack operates within a safe range. This rational device can implement battery parameter monitoring, battery state estimation, online fault diagnosis, and charging control for the battery, which can also respond in a timely manner when a failure occurs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21875040, 21902027 & 21905051) and Engineering Research Center of Clinical Functional Materials and Diagnosis & Treatment Devices of Zhejiang Province (Grant No. WIUCASK20002).References
- S. Ma, M. Jiang, P. Tao, C. Song, J. Wu, J. Wang, T. Deng and W. Shang, Prog. Nat. Sci., 2018, 28, 653–666 CrossRef CAS.
- Y. Chen, T. Wang, H. Tian, D. Su, Q. Zhang and G. Wang, Adv. Mater., 2021, 33, 2003666 CrossRef CAS PubMed.
- M. Chen, Y. Zhang, G. Xing, S.-L. Chou and Y. Tang, Energy Environ. Sci., 2021, 14, 3323–3351 RSC.
- X. Wu, J. Dong, M. Qiu, Y. Li, Y. Zhang, H. Zhang and J. Zhang, Nanoscale Horiz., 2020, 5, 359–365 RSC.
- H. Li and A. Y. Elezzabi, Nanoscale Horiz., 2020, 5, 691–695 RSC.
- E. Cha, M. Patel, S. Bhoyate, V. Prasad and W. Choi, Nanoscale Horiz., 2020, 5, 808–831 RSC.
- G. Liu, H.-H. Wu, Q. Meng, T. Zhang, D. Sun, X. Jin, D. Guo, N. Wu, X. Liu and J.-K. Kim, Nanoscale Horiz., 2020, 5, 150–162 RSC.
- C. Li, L. Wang, J. Zhang, D. Zhang, J. Du, Y. Yao and G. Hong, Energy Storage Mater., 2022, 44, 104–135 CrossRef.
- W. Lu, C. Zhang, H. Zhang and X. Li, ACS Energy Lett., 2021, 6, 2765–2785 CrossRef CAS.
- T. Wang, C. Li, X. Xie, B. Lu, Z. He, S. Liang and J. Zhou, ACS Nano, 2020, 14, 16321–16347 CrossRef CAS PubMed.
- F. Wan, X. Zhou, Y. Lu, Z. Niu and J. Chen, ACS Energy Lett., 2020, 5, 3569–3590 CrossRef CAS.
- Q. Zhang, J. Luan, Y. Tang, X. Ji and H. Wang, Angew. Chem., Int. Ed., 2020, 59, 13180–13191 CrossRef CAS PubMed.
- Y. Tian, Y. An, C. Wei, B. Xi, S. Xiong, J. Feng and Y. Qian, Adv. Energy Mater., 2021, 11, 2002529 CrossRef CAS.
- D. Selvakumaran, A. Pan, S. Liang and G. Cao, J. Mater. Chem. A, 2019, 7, 18209–18236 RSC.
- X. Li, Z. Chen, Y. Yang, S. Liang, B. Lu and J. Zhou, Inorg. Chem. Front., 2022, 9, 3986–3998 RSC.
- Z. Liu, L. Qin, B. Lu, X. Wu, S. Liang and J. Zhou, ChemSusChem, 2022, 15, e202200348 CAS.
- Z. Liu, Y. Yang, S. Liang, B. Lu and J. Zhou, Small Struct., 2021, 2, 2100119 CrossRef CAS.
- D. Han, S. Wu, S. Zhang, Y. Deng, C. Cui, L. Zhang, Y. Long, H. Li, Y. Tao, Z. Weng, Q. H. Yang and F. Kang, Small, 2020, 16, 2001736 CrossRef CAS PubMed.
- H. Tian, Z. Li, G. Feng, Z. Yang, D. Fox, M. Wang, H. Zhou, L. Zhai, A. Kushima, Y. Du, Z. Feng, X. Shan and Y. Yang, Nat. Commun., 2021, 12, 237 CrossRef CAS PubMed.
- Y. Zuo, K. Wang, P. Pei, M. Wei, X. Liu, Y. Xiao and P. Zhang, Mater. Today Energy, 2021, 20, 100692 CrossRef CAS.
- B. Li, X. Zhang, T. Wang, Z. He, B. Lu, S. Liang and J. Zhou, Nanomicro Lett., 2021, 14, 6 Search PubMed.
- A. Naveed, A. Ali, T. Rasheed, X. Wang, P. Ye, X. Li, Y. Zhou, S. Mingru and Y. Liu, J. Power Sources, 2022, 525, 231122 CrossRef CAS.
- L. Ma, S. Chen, N. Li, Z. Liu, Z. Tang, J. A. Zapien, S. Chen, J. Fan and C. Zhi, Adv. Mater., 2020, 32, 1908121 CrossRef CAS PubMed.
- M. Rosso, C. Brissot, A. Teyssot, M. Dollé, L. Sannier, J.-M. Tarascon, R. Bouchet and S. Lascaud, Electrochim. Acta, 2006, 51, 5334–5340 CrossRef CAS.
- Y. Wang, Y. Wang, C. Chen, X. Chen, Q. Zhao, L. Yang, L. Yao, R. Qin, H. Wu, Z. Jiang and F. Pan, Chem. Commun., 2021, 57, 5326–5329 RSC.
- Q. Yang, Q. Li, Z. Liu, D. Wang, Y. Guo, X. Li, Y. Tang, H. Li, B. Dong and C. Zhi, Adv. Mater., 2020, 32, 2001854 CrossRef CAS PubMed.
- J. Su, X. Yin, H. Zhao, H. Yang, D. Yang, L. He, M. Wang, S. Jin, K. Zhao, Y. Wang and Y. Wei, Nano Lett., 2022, 22, 1549–1556 CrossRef CAS PubMed.
- A. Pei, G. Zheng, F. Shi, Y. Li and Y. Cui, Nano Lett., 2017, 17, 1132–1139 CrossRef CAS PubMed.
- P. Biswal, S. Stalin, A. Kludze, S. Choudhury and L. A. Archer, Nano Lett., 2019, 19, 8191–8200 CrossRef CAS PubMed.
- S. Wang, Z. Wang, Y. Yin, T. Li, N. Chang, F. Fan, H. Zhang and X. Li, Energy Environ. Sci., 2021, 14, 4077–4084 RSC.
- Y. Li, P. Wu, W. Zhong, C. Xie, Y. Xie, Q. Zhang, D. Sun, Y. Tang and H.-Y. Wang, Energy Environ. Sci., 2021, 14, 5563–5571 RSC.
- R. Qin, Y. Wang, M. Zhang, Y. Wang, S. Ding, A. Song, H. Yi, L. Yang, Y. Song, Y. Cui, J. Liu, Z. Wang, S. Li, Q. Zhao and F. Pan, Nano Energy, 2021, 80, 105478 CrossRef CAS.
- C. Han, W. Li, H. K. Liu, S. Dou and J. Wang, Nano Energy, 2020, 74, 104880 CrossRef CAS.
- K. Wang, P. Pei, Z. Ma, H. Xu, P. Li and X. Wang, J. Power Sources, 2014, 271, 65–75 CrossRef CAS.
- P. Chen, Y. Wu, Y. Zhang, T.-H. Wu, Y. Ma, C. Pelkowski, H. Yang, Y. Zhang, X. Hu and N. Liu, J. Mater. Chem. A, 2018, 6, 21933–21940 RSC.
- D. Feng, F. Cao, L. Hou, T. Li, Y. Jiao and P. Wu, Small, 2021, 17, 2103195 CrossRef CAS PubMed.
- J. W. Diggle, A. R. Despic and J. O. M. Bockris, J. Electrochem. Soc., 1969, 116, 1503 CrossRef CAS.
- M. Fayette, H. J. Chang, I. A. Rodri Guez-Perez, X. Li and D. Reed, ACS Appl. Mater. Interfaces, 2020, 12, 42763–42772 CrossRef CAS PubMed.
- H. E. M. Hussein, R. J. Maurer, H. Amari, J. J. P. Peters, L. Meng, R. Beanland, M. E. Newton and J. V. Macpherson, ACS Nano, 2018, 12, 7388–7396 CrossRef CAS PubMed.
- J. Hao, X. Li, X. Zeng, D. Li, J. Mao and Z. Guo, Energy Environ. Sci., 2020, 13, 3917–3949 RSC.
- F. R. McLarnon and E. J. Cairns, J. Electrochem. Soc., 1991, 138, 645–656 CrossRef CAS.
- F. Xie, H. Li, X. Wang, X. Zhi, D. Chao, K. Davey and S.-Z. Qiao, Adv. Energy Mater., 2021, 11, 2003419 CrossRef CAS.
- Q. Zhu, W. A. Saidi and J. C. Yang, J. Phys. Chem. C, 2017, 121, 11251–11260 CrossRef CAS.
- H. Wang, D. Ning, L. Wang, H. Li, Q. Li, M. Ge, J. Zou, S. Chen, H. Shao, Y. Lai, Y. Zhang, G. Xing, W. K. Pang and Y. Tang, Small, 2022, 18, 2107491 CrossRef CAS PubMed.
- H. Jia, Z. Wang, B. Tawiah, Y. Wang, C.-Y. Chan, B. Fei and F. Pan, Nano Energy, 2020, 70, 104523 CrossRef CAS.
- Q. Li, H. Pan, W. Li, Y. Wang, J. Wang, J. Zheng, X. Yu, H. Li and L. Chen, ACS Energy Lett., 2018, 3, 2259–2266 CrossRef CAS.
- C. Li, X. Xie, S. Liang and J. Zhou, Energy Environ. Mater., 2020, 3, 146–159 CrossRef CAS.
- Y. Yin, S. Wang, Q. Zhang, Y. Song, N. Chang, Y. Pan, H. Zhang and X. Li, Adv. Mater., 2020, 32, 1906803 CrossRef CAS PubMed.
- Q. Yang, G. Liang, Y. Guo, Z. Liu, B. Yan, D. Wang, Z. Huang, X. Li, J. Fan and C. Zhi, Adv. Mater., 2019, 31, 1903778 CrossRef CAS PubMed.
- Z. Zhao, J. Zhao, Z. Hu, J. Li, J. Li, Y. Zhang, C. Wang and G. Cui, Energy Environ. Sci., 2019, 12, 1938–1949 RSC.
- L. Qian, W. Yao, R. Yao, Y. Sui, H. Zhu, F. Wang, J. Zhao, C. Zhi and C. Yang, Adv. Funct. Mater., 2021, 31, 2105736 CrossRef CAS.
- Q. Li, A. Chen, D. Wang, Y. Zhao, X. Wang, X. Jin, B. Xiong and C. Zhi, Nat. Commun., 2022, 13, 3699 CrossRef CAS PubMed.
- P. Zengxia, Nano Res. Energy, 2022, 1, e9120023 CrossRef.
- Q. Li, A. Chen, D. Wang, Z. Pei and C. Zhi, Joule, 2022, 6, 273–279 CrossRef.
- W. Du, E. H. Ang, Y. Yang, Y. Zhang, M. Ye and C. C. Li, Energy Environ. Sci., 2020, 13, 3330–3360 RSC.
- C. Cao, F. Liang, W. Zhang, H. Liu, H. Liu, H. Zhang, J. Mao, Y. Zhang, Y. Feng, X. Yao, M. Ge and Y. Tang, Small, 2021, 17, 2102233 CrossRef CAS PubMed.
- J. Zhao, Z. Cong, J. Hu, H. Lu, L. Wang, H. Wang, O. I. Malyi, X. Pu, Y. Zhang, H. Shao, Y. Tang and Z. L. Wang, Nano Energy, 2022, 93, 106893 CrossRef CAS.
- M. Zhu, H. Wang, W. Lin, D. Chan, H. Li, K. Wang, Y. Tang, T. Hao, S. Chen, O. I. Malyi, Y. Tang and Y. Zhang, Small Struct., 2022, 3, 2200016 CrossRef CAS.
- Y. Liu, X. Lu, F. Lai, T. Liu, P. R. Shearing, I. P. Parkin, G. He and D. J. L. Brett, Joule, 2021, 5, 2845–2903 CrossRef CAS.
- A. Konarov, N. Voronina, J. H. Jo, Z. Bakenov, Y.-K. Sun and S.-T. Myung, ACS Energy Lett., 2018, 3, 2620–2640 CrossRef CAS.
- Z. Zhou, Y. Zhang, P. Chen, Y. Wu, H. Yang, H. Ding, Y. Zhang, Z. Wang, X. Du and N. Liu, Chem. Eng. Sci., 2019, 194, 142–147 CrossRef CAS.
- B. E. Hawkins, D. E. Turney, R. J. Messinger, A. M. Kiss, G. G. Yadav, S. Banerjee and T. N. Lambert, Adv. Energy Mater., 2022, 12, 2103294 CrossRef CAS.
- L. Hu, P. Xiao, L. Xue, H. Li and T. Zhai, Energy Chem., 2021, 3, 100052 CrossRef CAS.
- Q. Yang, L. Li, T. Hussain, D. Wang, L. Hui, Y. Guo, G. Liang, X. Li, Z. Chen, Z. Huang, Y. Li, Y. Xue, Z. Zuo, J. Qiu, Y. Li and C. Zhi, Angew. Chem., Int. Ed., 2022, 61, e202112304 CAS.
- M. Zhou, S. Guo, G. Fang, H. Sun, X. Cao, J. Zhou, A. Pan and S. Liang, J. Energy Chem., 2021, 55, 549–556 CrossRef CAS.
- P. Sun, L. Ma, W. Zhou, M. Qiu, Z. Wang, D. Chao and W. Mai, Angew. Chem., Int. Ed., 2021, 60, 18247–18255 CrossRef CAS PubMed.
- X.-B. Cheng, R. Zhang, C.-Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS PubMed.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 RSC.
- Y. Okajima, Y. Shibuta and T. Suzuki, Comput. Mater. Sci., 2010, 50, 118–124 CrossRef CAS.
- X. Zhang, A. Wang, X. Liu and J. Luo, Acc. Chem. Res., 2019, 52, 3223–3232 CrossRef CAS PubMed.
- C. Brissot, M. Rosso, J. N. Chazalviel, P. Baudry and S. Lascaud, Electrochim. Acta, 1998, 43, 1569–1574 CrossRef CAS.
- Z. Hou, Y. Gao, R. Zhou and B. Zhang, Adv. Funct. Mater., 2022, 32, 2107584 CrossRef CAS.
- M. K. Aslam, Y. Niu, T. Hussain, H. Tabassum, W. Tang, M. Xu and R. Ahuja, Nano Energy, 2021, 86, 106142 CrossRef CAS.
- X. Gao, Y.-N. Zhou, D. Han, J. Zhou, D. Zhou, W. Tang and J. B. Goodenough, Joule, 2020, 4, 1864–1879 CrossRef CAS.
- H. Glatz, E. Tervoort and D. Kundu, ACS Appl. Mater. Interfaces, 2020, 12, 3522–3530 CrossRef CAS PubMed.
- J. Zhi, S. Li, M. Han and P. Chen, Sci. Adv., 2022, 6, eabb1342 CrossRef PubMed.
- Y. Han, Y. Jie, F. Huang, Y. Chen, Z. Lei, G. Zhang, X. Ren, L. Qin, R. Cao and S. Jiao, Adv. Funct. Mater., 2019, 29, 1904629 CrossRef CAS.
- K. Yan, J. Wang, S. Zhao, D. Zhou, B. Sun, Y. Cui and G. Wang, Angew. Chem., Int. Ed., 2019, 58, 11364–11368 CrossRef CAS PubMed.
- A. C. Thenuwara, P. P. Shetty and M. T. McDowell, Nano Lett., 2019, 19, 8664–8672 CrossRef CAS PubMed.
- H. Li, H. Wang, Z. Xu, K. Wang, M. Ge, L. Gan, Y. Zhang, Y. Tang and S. Chen, Small, 2021, 17, 2103679 CrossRef CAS PubMed.
- S. Zhang, Z. Zhang, Y. Si, B. Li, F. Deng, L. Yang, X. Liu, W. Dai and S. Luo, ACS Nano, 2021, 15, 15238–15248 CrossRef CAS PubMed.
- M. Ge, C. Cao, F. Liang, R. Liu, Y. Zhang, W. Zhang, T. Zhu, B. Yi, Y. Tang and Y. Lai, Nanoscale Horiz., 2020, 5, 65–73 RSC.
- Q. Cao, H. Gao, Y. Gao, J. Yang, C. Li, J. Pu, J. Du, J. Yang, D. Cai, Z. Pan, C. Guan and W. Huang, Adv. Funct. Mater., 2021, 31, 2103922 CrossRef CAS.
- W. Guo, Z. Cong, Z. Guo, C. Chang, X. Liang, Y. Liu, W. Hu and X. Pu, Energy Storage Mater., 2020, 30, 104–112 CrossRef.
- Y. Zhou, X. Wang, X. Shen, Y. Shi, C. Zhu, S. Zeng, H. Xu, P. Cao, Y. Wang, J. Di and Q. Li, J. Mater. Chem. A, 2020, 8, 11719–11727 RSC.
- J. Hao, B. Li, X. Li, X. Zeng, S. Zhang, F. Yang, S. Liu, D. Li, C. Wu and Z. Guo, Adv. Mater., 2020, 32, 2003021 CrossRef CAS PubMed.
- J. Hao, X. Li, S. Zhang, F. Yang, X. Zeng, S. Zhang, G. Bo, C. Wang and Z. Guo, Adv. Funct. Mater., 2020, 30, 2001263 CrossRef CAS.
- L. Kang, M. Cui, F. Jiang, Y. Gao, H. Luo, J. Liu, W. Liang and C. Zhi, Adv. Energy Mater., 2018, 8, 1801090 CrossRef.
- H. Li, C. Guo, T. Zhang, P. Xue, R. Zhao, W. Zhou, W. Li, A. Elzatahry, D. Zhao and D. Chao, Nano Lett., 2022, 22, 4223–4231 CrossRef CAS PubMed.
- P. Liu, X. Ling, C. Zhong, Y. Deng, X. Han and W. Hu, Front. Chem., 2019, 7, 656 CrossRef CAS PubMed.
- J. Zhao, H. Lu, Y. Zhang, S. Yu, O. I. Malyi, X. Zhao, L. Wang, H. Wang, J. Peng, X. Li, Y. Zhang, S. Chen, H. Pan, G. Xing, C. Lu, Y. Tang and X. Chen, Sci. Adv., 2021, 7, eabd6978 CrossRef CAS PubMed.
- J. F. Parker, C. N. Chervin, I. R. Pala, M. Machler and D. R. Rolison, Science, 2017, 356, 415–418 CrossRef CAS PubMed.
- Z. Kang, C. Wu, L. Dong, W. Liu, J. Mou, J. Zhang, Z. Chang, B. Jiang, G. Wang, F. Kang and C. Xu, ACS Sustainable Chem. Eng., 2019, 7, 3364–3371 CrossRef CAS.
- Y. An, Y. Tian, S. Xiong, J. Feng and Y. Qian, ACS Nano, 2021, 15, 11828–11842 CrossRef CAS PubMed.
- M. Ge, Y. Tang, O. I. Malyi, Y. Zhang, Z. Zhu, Z. Lv, X. Ge, H. Xia, J. Huang, Y. Lai and X. Chen, Small, 2020, 16, 2002094 CrossRef CAS PubMed.
- Y. Tang, Y. Zhang, O. I. Malyi, N. Bucher, H. Xia, S. Xi, Z. Zhu, Z. Lv, W. Li, J. Wei, M. Srinivasan, A. Borgna, M. Antonietti, Y. Du and X. Chen, Adv. Mater., 2018, 30, 1802200 CrossRef PubMed.
- Y. An, Y. Tian, L. Ci, S. Xiong, J. Feng and Y. Qian, ACS Nano, 2018, 12, 12932–12940 CrossRef CAS PubMed.
- Y. Zeng, X. Zhang, R. Qin, X. Liu, P. Fang, D. Zheng, Y. Tong and X. Lu, Adv. Mater., 2019, 31, 1903675 CrossRef PubMed.
- H. Gan, J. Wu, R. Li, B. Huang and H. Liu, Energy Storage Mater., 2022, 47, 602–610 CrossRef.
- Y. Hao, J. Zhou, G. Wei, A. liu, Y. Zhang, Y. Mei, B. Lu, M. Luo and M. Xie, ACS Appl. Energy Mater., 2021, 4, 6364–6373 CrossRef CAS.
- J. Zhou, M. Xie, F. Wu, Y. Mei, Y. Hao, R. Huang, G. Wei, A. Liu, L. Li and R. Chen, Adv. Mater., 2021, 33, 2101649 CrossRef CAS PubMed.
- P. Xue, C. Guo, L. Li, H. Li, D. Luo, L. Tan and Z. Chen, Adv. Mater., 2022, 34, 2110047 CrossRef CAS PubMed.
- Y. Tian, Y. An, C. Wei, B. Xi, S. Xiong, J. Feng and Y. Qian, ACS Nano, 2019, 13, 11676–11685 CrossRef CAS PubMed.
- Y. Tian, Y. An, C. Liu, S. Xiong, J. Feng and Y. Qian, Energy Storage Mater., 2021, 41, 343–353 CrossRef.
- Y. Zhang, Z. Cao, S. Liu, Z. Du, Y. Cui, J. Gu, Y. Shi, B. Li and S. Yang, Adv. Energy Mater., 2022, 12, 2103979 CrossRef CAS.
- D. Sha, C. Lu, W. He, J. Ding, H. Zhang, Z. Bao, X. Cao, J. Fan, Y. Dou, L. Pan and Z. Sun, ACS Nano, 2022, 16, 2711–2720 CrossRef CAS PubMed.
- Y. Zhang, J. D. Howe, S. Ben-Yoseph, Y. Wu and N. Liu, ACS Energy Lett., 2021, 6, 404–412 CrossRef CAS.
- H. Chen, C. Dai, F. Xiao, Q. Yang, S. Cai, M. Xu, H. J. Fan and S.-J. Bao, Adv. Mater., 2022, 34, 2109092 CrossRef CAS PubMed.
- S. B. Wang, Q. Ran, R. Q. Yao, H. Shi, Z. Wen, M. Zhao, X. Y. Lang and Q. Jiang, Nat. Commun., 2020, 11, 1634 CrossRef PubMed.
- J. Zheng, Z. Huang, Y. Zeng, W. Liu, B. Wei, Z. Qi, Z. Wang, C. Xia and H. Liang, Nano Lett., 2022, 22, 1017–1023 CrossRef CAS PubMed.
- H. Yan, S. Li, Y. Nan, S. Yang and B. Li, Adv. Energy Mater., 2021, 11, 2100186 CrossRef CAS.
- Q. Zhang, J. Luan, X. Huang, Q. Wang, D. Sun, Y. Tang, X. Ji and H. Wang, Nat. Commun., 2020, 11, 3961 CrossRef CAS PubMed.
- G. Liu, H. G. Yang, X. Wang, L. Cheng, H. Lu, L. Wang, G. Q. Lu and H.-M. Cheng, J. Phys. Chem. C, 2009, 113, 21784–21788 CrossRef CAS.
- J. Y. Kim, G. Liu, G. Y. Shim, H. Kim and J. K. Lee, Adv. Funct. Mater., 2020, 30, 2004210 CrossRef CAS.
- P. Cao, X. Zhou, A. Wei, Q. Meng, H. Ye, W. Liu, J. Tang and J. Yang, Adv. Funct. Mater., 2021, 31, 2100398 CrossRef CAS.
- Y. Yang, C. Liu, Z. Lv, H. Yang, Y. Zhang, M. Ye, L. Chen, J. Zhao and C. C. Li, Adv. Mater., 2021, 33, 2007388 CrossRef CAS PubMed.
- J. Han, H. Euchner, M. Kuenzel, S. M. Hosseini, A. Groß, A. Varzi and S. Passerini, ACS Energy Lett., 2021, 6, 3063–3071 CrossRef CAS.
- L. Cao, D. Li, T. Pollard, T. Deng, B. Zhang, C. Yang, L. Chen, J. Vatamanu, E. Hu, M. J. Hourwitz, L. Ma, M. Ding, Q. Li, S. Hou, K. Gaskell, J. T. Fourkas, X.-Q. Yang, K. Xu, O. Borodin and C. Wang, Nat. Nanotechnol., 2021, 16, 902–910 CrossRef CAS PubMed.
- D. Wang, D. Lv, H. Peng, N. Wang, H. Liu, J. Yang and Y. Qian, Nano Lett., 2022, 22, 1750–1758 CrossRef CAS PubMed.
- G. Liang, J. Zhu, B. Yan, Q. Li, A. Chen, Z. Chen, X. Wang, B. Xiong, J. Fan, J. Xu and C. Zhi, Energy Environ. Sci., 2022, 15, 1086–1096 RSC.
- W. Guo, Y. Zhang, X. Tong, X. Wang, L. Zhang, X. Xia and J. Tu, Mater. Today Energy, 2021, 20, 100675 CrossRef CAS.
- S. Zhai, X. Shi, K. Jiang, X. Tan, W. Zhang, J. Zhang, H. Zhang and Z. Li, Chem. Eng. J., 2022, 437, 135246 CrossRef CAS.
- H. Yu, Y. Zeng, W. Li Nian, D. Luan, L. Yu and W. Lou Xiong, Sci. Adv., 2022, 8, eabm5766 CrossRef CAS PubMed.
- Y. Shi, Y. Chen, L. Shi, K. Wang, B. Wang, L. Li, Y. Ma, Y. Li, Z. Sun, W. Ali and S. Ding, Small, 2020, 16, 2000730 CrossRef CAS PubMed.
- K. Ouyang, D. Ma, N. Zhao, Y. Wang, M. Yang, H. Mi, L. Sun, C. He and P. Zhang, Adv. Funct. Mater., 2022, 32, 2109749 CrossRef CAS.
- C. Liu, Z. Luo, W. Deng, W. Wei, L. Chen, A. Pan, J. Ma, C. Wang, L. Zhu, L. Xie, X.-Y. Cao, J. Hu, G. Zou, H. Hou and X. Ji, ACS Energy Lett., 2021, 6, 675–683 CrossRef CAS.
- H. Jia, Z. Wang, M. Dirican, S. Qiu, C. Y. Chan, S. Fu, B. Fei and X. Zhang, J. Mater. Chem. A, 2021, 9, 5597–5605 RSC.
- J. Ma, M. Liu, Y. He and J. Zhang, Angew. Chem., Int. Ed., 2021, 60, 12636–12647 CrossRef CAS PubMed.
- D. Lin, D. Rao, S. Chiovoloni, S. Wang, J. Q. Lu and Y. Li, Nano Lett., 2021, 21, 4129–4135 CrossRef CAS PubMed.
- W. Shang, Q. Li, F. Jiang, B. Huang, J. Song, S. Yun, X. Liu, H. Kimura, J. Liu and L. Kang, Nanomicro Lett., 2022, 14, 82 CAS.
- T. Yadong, C. Song, H. Yulong, C. Qianwu, Z. Lili and Z. Jintao, Nano Res. Energy, 2022 DOI:10.26599/NRE.2022.9120025.
- L. Wu and Y. Dong, Energy Storage Mater., 2021, 41, 715–737 CrossRef.
- J. Zheng, Q. Zhao, T. Tang, J. Yin, C. D. Quilty, G. D. Renderos, X. Liu, Y. Deng, L. Wang, D. C. Bock, C. Jaye, D. Zhang, E. S. Takeuchi, K. J. Takeuchi, A. C. Marschilok and L. A. Archer, Science, 2019, 366, 645–648 CrossRef CAS PubMed.
- X. Zhang, J. Li, D. Liu, M. Liu, T. Zhou, K. Qi, L. Shi, Y. Zhu and Y. Qian, Energy Environ. Sci., 2021, 14, 3120–3129 RSC.
- J. Hao, X. Li, S. Zhang, F. Yang, X. Zeng, S. Zhang, G. Bo, C. Wang and Z. Guo, Adv. Funct. Mater., 2020, 30, 2001263 CrossRef CAS.
- P. Chen, X. Yuan, Y. Xia, Y. Zhang, L. Fu, L. Liu, N. Yu, Q. Huang, B. Wang, X. Hu, Y. Wu and T. van Ree, Adv. Sci., 2021, 8, 2100309 CrossRef CAS PubMed.
- D. Lee, H. I. Kim, W. Y. Kim, S. K. Cho, K. Baek, K. Jeong, D. B. Ahn, S. Park, S. J. Kang and S. Y. Lee, Adv. Funct. Mater., 2021, 31, 2103850 CrossRef CAS.
- F. Zhang, C. Wang, J. Pan, F. Tian, S. Zeng, J. Yang and Y. Qian, Mater. Today Energy, 2020, 17, 100443 CrossRef.
- Z. Cao, X. Zhu, D. Xu, P. Dong, M. O. L. Chee, X. Li, K. Zhu, M. Ye and J. Shen, Energy Storage Mater., 2021, 36, 132–138 CrossRef.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- Z. Zhao, R. Wang, C. Peng, W. Chen, T. Wu, B. Hu, W. Weng, Y. Yao, J. Zeng, Z. Chen, P. Liu, Y. Liu, G. Li, J. Guo, H. Lu and Z. Guo, Nat. Commun., 2021, 12, 6606 CrossRef CAS PubMed.
- J. H. Park, M.-J. Kwak, C. Hwang, K.-N. Kang, N. Liu, J.-H. Jang and B. A. Grzybowski, Adv. Mater., 2021, 33, 2101726 CrossRef CAS PubMed.
- J. Zhao, Y. Ying, G. Wang, K. Hu, Y. D. Yuan, H. Ye, Z. Liu, J. Y. Lee and D. Zhao, Energy Storage Mater., 2022, 48, 82–89 CrossRef.
- C. Guo, J. Zhou, Y. Chen, H. Zhuang, Q. Li, J. Li, X. Tian, Y. Zhang, X. Yao, Y. Chen, S.-L. Li and Y.-Q. Lan, Angew. Chem., Int. Ed., 2022, 12, 6606 Search PubMed.
- R. Zhao, Y. Yang, G. Liu, R. Zhu, J. Huang, Z. Chen, Z. Gao, X. Chen and L. Qie, Adv. Funct. Mater., 2021, 31, 2001867 CrossRef CAS.
- M. Liu, L. Yang, H. Liu, A. Amine, Q. Zhao, Y. Song, J. Yang, K. Wang and F. Pan, ACS Appl. Mater. Interfaces, 2019, 11, 32046–32051 CrossRef CAS PubMed.
- X. Pu, B. Jiang, X. Wang, W. Liu, L. Dong, F. Kang and C. Xu, Nanomicro Lett., 2020, 12, 152 CrossRef CAS PubMed.
- P. Zou, R. Zhang, L. Yao, J. Qin, K. Kisslinger, H. Zhuang and H. L. Xin, Adv. Energy Mater., 2021, 11, 2100982 CrossRef CAS.
- X. Jia, C. Liu, Z. G. Neale, J. Yang and G. Cao, Chem. Rev., 2020, 120, 7795–7866 CrossRef CAS PubMed.
- L. Wang, Y. Zhang, H. Hu, H.-Y. Shi, Y. Song, D. Guo, X.-X. Liu and X. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 42000–42005 CrossRef CAS PubMed.
- G. Yang, J. Huang, X. Wan, B. Liu, Y. Zhu, J. Wang, O. Fontaine, S. Luo, P. Hiralal, Y. Guo and H. Zhou, EcoMat, 2022, 4, e12165 CrossRef CAS.
- S. Huang, L. Hou, T. Li, Y. Jiao and P. Wu, Adv. Mater., 2022, 34, 2110140 CrossRef CAS PubMed.
- C. Zhang, J. Holoubek, X. Wu, A. Daniyar, L. Zhu, C. Chen, D. P. Leonard, I. A. Rodríguez-Pérez, J.-X. Jiang, C. Fang and X. Ji, Chem. Commun., 2018, 54, 14097–14099 RSC.
- L. Cao, D. Li, F. A. Soto, V. Ponce, B. Zhang, L. Ma, T. Deng, J. M. Seminario, E. Hu, X.-Q. Yang, P. B. Balbuena and C. Wang, Angew. Chem., Int. Ed., 2021, 60, 18845–18851 CrossRef CAS PubMed.
- L. Cao, D. Li, E. Hu, J. Xu, T. Deng, L. Ma, Y. Wang, X. Q. Yang and C. Wang, J. Am. Chem. Soc., 2020, 142, 21404–21409 CrossRef CAS PubMed.
- Z. Peng, Q. Wei, S. Tan, P. He, W. Luo, Q. An and L. Mai, Chem. Commun., 2018, 54, 4041–4044 RSC.
- H. Qiu, X. Du, J. Zhao, Y. Wang, J. Ju, Z. Chen, Z. Hu, D. Yan, X. Zhou and G. Cui, Nat. Commun., 2019, 10, 5374 CrossRef PubMed.
- H. Dong, J. Li, J. Guo, F. Lai, F. Zhao, Y. Jiao, D. J. L. Brett, T. Liu, G. He and I. P. Parkin, Adv. Mater., 2021, 33, 2007548 CrossRef CAS PubMed.
- J. Hao, L. Yuan, C. Ye, D. Chao, K. Davey, Z. Guo and S.-Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 7366–7375 CrossRef CAS PubMed.
- S.-J. Zhang, J. Hao, D. Luo, P.-F. Zhang, B. Zhang, K. Davey, Z. Lin and S.-Z. Qiao, Adv. Energy Mater., 2021, 11, 2102010 CrossRef CAS.
- T. C. Li, Y. Lim, X. L. Li, S. Luo, C. Lin, D. Fang, S. Xia, Y. Wang and H. Y. Yang, Adv. Energy Mater., 2022, 12, 2103231 CrossRef CAS.
- W. Chen, S. Guo, L. Qin, L. Li, X. Cao, J. Zhou, Z. Luo, G. Fang and S. Liang, Adv. Funct. Mater., 2022, 32, 2112609 CrossRef CAS.
- M. S. Gonzalez, Q. Yan, J. Holoubek, M. Li, Z. Wu, H. Zhou, S. Kim, H. Liu, B. Y. Jung, S. W. Lee, Z. Chen and P. Liu, Adv. Funct. Mater., 2021, 31, 2102198 CrossRef CAS.
- X. Guo, Z. Zhang, J. Li, N. Luo, G.-L. Chai, T. S. Miller, F. Lai, P. Shearing, D. J. L. Brett, D. Han, Z. Weng, G. He and I. P. Parkin, ACS Energy Lett., 2021, 6, 395–403 CrossRef CAS.
- X. Zeng, J. Mao, J. Hao, J. Liu, S. Liu, Z. Wang, Y. Wang, S. Zhang, T. Zheng, J. Liu, P. Rao and Z. Guo, Adv. Mater., 2021, 33, 2007416 CrossRef CAS PubMed.
- Y. Liu, J. Hu, Q. Lu, M. Hantusch, H. Zhang, Z. Qu, H. Tang, H. Dong, O. G. Schmidt, R. Holze and M. Zhu, Energy Storage Mater., 2022, 47, 98–104 CrossRef.
- C. Sun, C. Wu, X. Gu, C. Wang and Q. Wang, Nanomicro Lett., 2021, 13, 89 CAS.
- C. Huang, X. Zhao, Y. Hao, Y. Yang, Y. Qian, G. Chang, Y. Zhang, Q. Tang, A. Hu and X. Chen, Adv. Funct. Mater., 2022, 32, 2112091 CrossRef CAS.
- Y. An, Y. Tian, K. Zhang, Y. Liu, C. Liu, S. Xiong, J. Feng and Y. Qian, Adv. Funct. Mater., 2021, 31, 2101886 CrossRef CAS.
- Y. Chu, S. Zhang, S. Wu, Z. Hu, G. Cui and J. Luo, Energy Environ. Sci., 2021, 14, 3609–3620 RSC.
- Y. Lv, Y. Xiao, L. Ma, C. Zhi and S. Chen, Adv. Mater., 2022, 34, 2106409 CrossRef CAS PubMed.
- C. Liu, X. Xie, B. Lu, J. Zhou and S. Liang, ACS Energy Lett., 2021, 6, 1015–1033 CrossRef CAS.
- J. Yang, B. Yin, Y. Sun, H. Pan, W. Sun, B. Jia, S. Zhang and T. Ma, Nanomicro Lett., 2022, 14, 42 CAS.
- J. Zhu, M. Yao, S. Huang, J. Tian and Z. Niu, Angew. Chem., Int. Ed., 2020, 59, 16480–16484 CrossRef CAS PubMed.
- P. Yang, C. Feng, Y. Liu, T. Cheng, X. Yang, H. Liu, K. Liu and H. J. Fan, Adv. Energy Mater., 2020, 10, 2002898 CrossRef CAS.
- X. Lin, C. Chu, Z. Li, T. Zhang, J. Chen, R. Liu, P. Li, Y. Li, J. Zhao, Z. Huang, X. Feng, Y. Xie and Y. Ma, Nano Energy, 2021, 89, 106351 CrossRef CAS.
- Z. Lei, J. Shen, J. Wang, Q. Qiu, G. Zhang, S.-S. Chi, H. Xu, S. Li, W. Zhang, Y. Zhao, Y. Deng and C. Wang, Chem. Eng. J., 2021, 412, 128733 CrossRef CAS.
- X. C. Chen, Y. Zhang, L. C. Merrill, C. Soulen, M. L. Lehmann, J. L. Schaefer, Z. Du, T. Saito and N. J. Dudney, J. Mater. Chem. A, 2021, 9, 6555–6566 RSC.
- D. Wang, X. Guo, Z. Chen, Y. Zhao, Q. Li and C. Zhi, ACS Appl. Mater. Interfaces, 2022, 14, 27287–27293 CrossRef CAS PubMed.
- M. Wang, A. Emre, S. Tung, A. Gerber, D. Wang, Y. Huang, V. Cecen and N. A. Kotov, ACS Nano, 2019, 13, 1107–1115 CAS.
- Y. Zeng, X. Zhang, Y. Meng, M. Yu, J. Yi, Y. Wu, X. Lu and Y. Tong, Adv. Mater., 2017, 29, 1700274 CrossRef PubMed.
- L. Ma, S. Chen, H. Li, Z. Ruan, Z. Tang, Z. Liu, Z. Wang, Y. Huang, Z. Pei, J. A. Zapien and C. Zhi, Energy Environ. Sci., 2018, 11, 2521–2530 RSC.
- Z. Liu, Q. Yang, D. Wang, G. Liang, Y. Zhu, F. Mo, Z. Huang, X. Li, L. Ma, T. Tang, Z. Lu and C. Zhi, Adv. Energy Mater., 2019, 9, 1902473 CrossRef CAS.
- F. Mo, G. Liang, Q. Meng, Z. Liu, H. Li, J. Fan and C. Zhi, Energy Environ. Sci., 2019, 12, 706–715 RSC.
- H. Li, C. Han, Y. Huang, Y. Huang, M. Zhu, Z. Pei, Q. Xue, Z. Wang, Z. Liu, Z. Tang, Y. Wang, F. Kang, B. Li and C. Zhi, Energy Environ. Sci., 2018, 11, 941–951 RSC.
- Y. Huang, M. Zhong, F. Shi, X. Liu, Z. Tang, Y. Wang, Y. Huang, H. Hou, X. Xie and C. Zhi, Angew. Chem., Int. Ed., 2017, 56, 9141–9145 CrossRef CAS PubMed.
- Z. Pei, Z. Yuan, C. Wang, S. Zhao, J. Fei, L. Wei, J. Chen, C. Wang, R. Qi, Z. Liu and Y. Chen, Angew. Chem., Int. Ed., 2020, 59, 4793–4799 CrossRef CAS PubMed.
- Y. Tang, X. Li, H. Lv, D. Xie, W. Wang, C. Zhi and H. Li, Adv. Energy Mater., 2020, 10, 2000892 CrossRef CAS.
- F. Mo, Z. Chen, G. Liang, D. Wang, Y. Zhao, H. Li, B. Dong and C. Zhi, Adv. Energy Mater., 2020, 10, 2000035 CrossRef CAS.
- Z. Wang, H. Li, Z. Tang, Z. Liu, Z. Ruan, L. Ma, Q. Yang, D. Wang and C. Zhi, Adv. Funct. Mater., 2018, 28, 1804560 CrossRef.
- J. Gao, X. Xie, S. Liang, B. Lu and J. Zhou, Nanomicro Lett., 2021, 13, 69 Search PubMed.
Footnote |
| † Shuwei Chen and Huibo Wang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |