DOI:
10.1039/D3MD00454F
(Research Article)
RSC Med. Chem., 2023,
14, 2611-2624
Discovery of pyrazolopyrimidines that selectively inhibit CSF-1R kinase by iterative design, synthesis and screening against glioblastoma cells†
Received
30th August 2023
, Accepted 9th October 2023
First published on 11th October 2023
Abstract
Glioblastoma multiforme (GBM) is the most aggressive type of brain cancer in adults, with an average life expectancy under treatment of approx. 15 months. GBM is characterised by a complex set of genetic alterations that results in significant disruption of receptor tyrosine kinase (RTK) signaling. We report here an exploration of the pyrazolo[3,4-d]pyrimidine scaffold in search for antiproliferative compounds directed to GBM treatment. Small compound libraries were synthesised and screened against GBM cells to build up structure–antiproliferative activity–relationships (SAARs) and inform further rounds of design, synthesis and screening. 76 novel compounds were generated through this iterative process that found low micromolar potencies against selected GBM lines, including patient-derived stem cells. Phenomics analysis demonstrated preferential activity against glioma cells of the mesenchymal subtype, whereas kinome screening identified colony stimulating factor-1 receptor (CSF-1R) as the lead's target, a RTK implicated in the tumourigenesis and progression of different cancers and the immunoregulation of the GBM microenvironment.
Introduction
Since the 1990's, drug design has been dominated by target-centric strategies focused to discover high-affinity molecules for nominated therapeutic targets.1,2 However, for complex heterogeneous cancers, a single target approach is typically insufficient to discover effective treatments.2–4 In this context, phenotypic screening of compound libraries across cell models that are representative of the disease heterogeneity observed in patients can instead be used to speed up the search for anticancer agents that target key drivers of different disease subtypes.4,5 This is particularly relevant in GBM, a deadly form of brain cancer that is currently treated by surgery, radiotherapy and chemotherapy (temozolomide) to extend average survival from 12 to 15 months,6 which illustrates the urgent need for more effective therapies.7
By exploring the chemical diversity surrounding the pyrazolo[3,4-d]pyrimidine scaffold—a versatile adenine analogue that can be constructed through a multitude of synthetic strategies—8,9 many labs including ours have demonstrated the potential to identify a wide range of bioactive agents, including inhibitors of protein, lipid and atypical kinases.10 The BTK inhibitor ibrutinib (top 3 best-selling oncology drug of 2022), first approved for the treatment of B-cell lymphomas in 2013 and later for various other disorders,11 is the best exemplar of the potential of the pyrazolo[3,4-d]pyrimidine scaffold for the development of anticancer kinase inhibitors.12 In this direction, our lab has primed phenotypic assays in selected cancer models as the main discriminating endpoint to inform the structural evolution of pyrazolopyrimidine derivatives into potent antiproliferative leads, followed by rapid target deconvolution via kinome screening. This biology-driven discovery approach has served to identify different classes of pyrazolopyrimidine-based kinase inhibitors: from RTK inhibitors (FLT3 and AXL)13,14 to selective inhibitors of mTOR15 or SRC family kinases;16 one of which is now in clinical development.17 By selecting cancer cell models of interest without prior nomination of a kinase target, this strategy can potentially uncover both established and novel kinases that drive the survival and proliferation of specific cancer indications and subtypes, which can accelerate the preclinical and clinical development of candidate drugs.
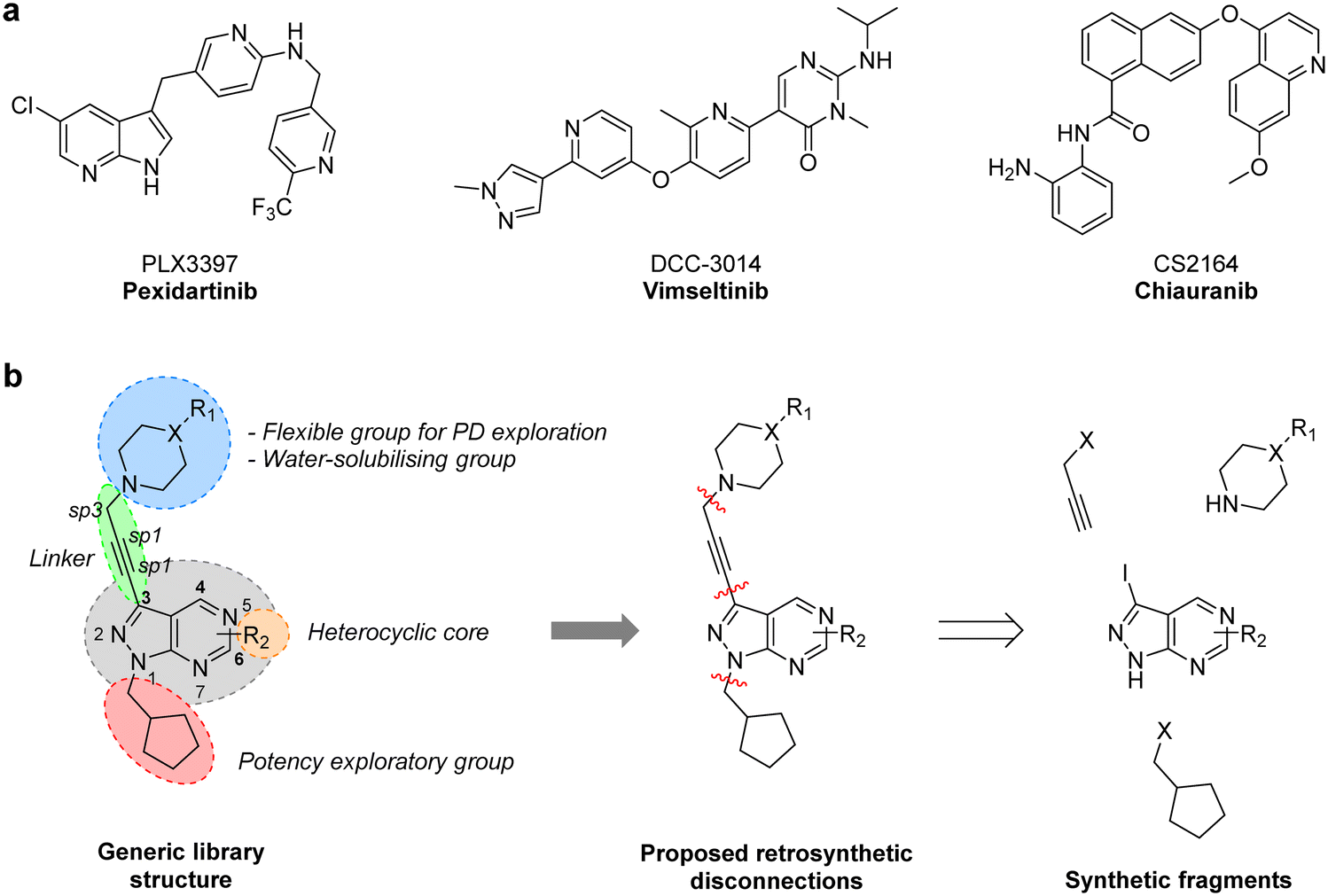
CSF-1R (also known as FMS kinase) is a cell surface receptor that plays an important role in initiating inflammatory, cancer and bone disorders.18,19 CSF-1R is a member of the PDGFR class of kinases and was first identified in sarcoma. Upon binding to its ligand (CSF-1 or IL-34), it dimerises and autophosphorylates to initiate various downstream signalling cascades, including MAPK, PI3K/AKT, SRC signalling pathways.20 Overexpression of this RTK and its ligands is linked with poor prognosis in many cancer types.19 Notably, in GBM, CSF-1R and/or its ligands have been found overexpressed both in cancer cells and tumour-associated macrophages (TAM), having a key regulatory role in the immune microenvironment via microglia and TAMs.19,21 Pexidartinib (PLX3397), a 7-azaindole-based small molecule inhibitor of CSF-1R (as well as KIT and FLT3) was approved in 2019 for the treatment of tenosynovial giant cell tumours and underwent a phase II clinical trial for treating recurrent GBM.22 Although the study did not find a significant improvement of average survival, two patients responded well to the treatment, suggesting that certain subtypes of GBM may benefit from CSF-1R inhibitor therapies.20 A broad number of CSF-1R inhibitors featuring various scaffolds have been reported in recent years (Fig. 1a), including DCC-3014 and CS2164 (both currently in phase III).20 To the best of our knowledge, however, the pyrazolo[3,4-d]pyrimidine scaffold has not yet yielded CSF-1R inhibitors.
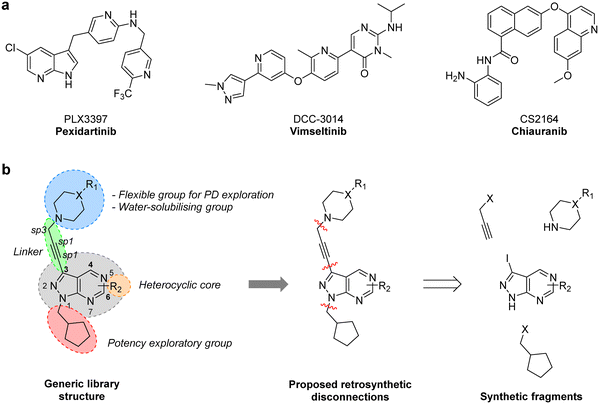 |
| | Fig. 1 a) Structure of CSF-1R inhibitors approved or in late-stage clinical development: pexidartinib, vimseltinib and chiauranib. b) Structural design and retrosynthetic analysis of novel pyrazolo[3,4-d]pyrimidines featuring a diversity of tertiary cyclic amines at the north of the core to explore novel binding affinity profiles and improve physicochemical properties. | |
Results and discussion
Design, synthesis and screening of libraries A1–13 and B1–13
One synthetic advantage of the pyrazolo[3,4-d]pyrimidine scaffold is that facilitates the functionalisation of the positions N1 and C3 of the fused pyrazole ring in an orthogonal manner (Fig. 1b). Previous explorations of the bioactive chemical space provided by this heterocyclic system (see works by Shokat,23 Wang,24–26 Zhang,27 Lindquist,28 and our lab13–16) have focused on introducing a wide range of aromatic moieties at the C3 position of the pyrazolopyrimidine scaffold, usually by palladium-catalyzed Suzuki–Miyaura cross-couplings. This strategy aimed at accessing hydrophobic regions typically conserved across various kinase families. In our search to explore additional binding interactions, we opted to incorporate a set of cyclic tertiary amines at the C3 position of the scaffold through an acetylene linker via palladium-catalyzed Sonogashira cross-coupling. The goal of this strategy was not only to probe the impact of flexible sp3-C-rich rings with different functionalities at the top of the scaffold, but also to introduce weakly-basic lipophilic groups that could increase water solubility without compromising permeability,29 an essential requirement for the stringent physicochemical properties demanded for CNS penetrance.30
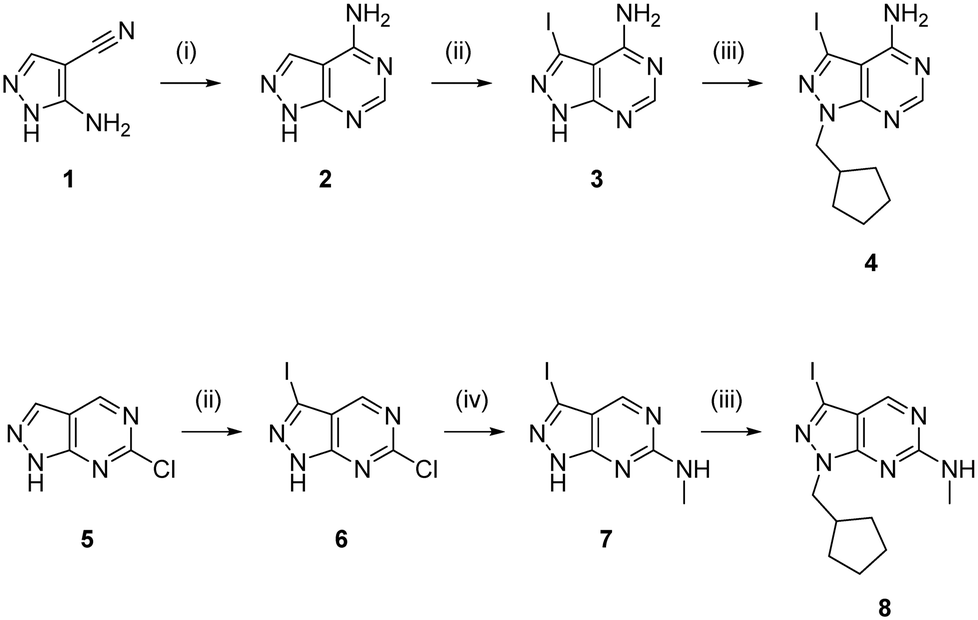
To expand the pharmacodynamic (PD) scope of the scaffold, two pyrazolopyrimidine cores were investigated: 4-amino and 6-methylamino-1H-pyrazolo[3,4-d]pyrimidine, A and B, respectively. Analogues of core A have shown tendency to inhibit nonreceptor tyrosine kinases, lipid and atypical kinases,15,16,23 whereas compounds derived from core B have displayed preferential targeting of RTKs,13,14,24–26 typically through a different binding mode than A. The A core was synthesised from 5-amino-1H-pyrazole-4-carbonitrile, 1, starting with a condensation reaction with formamide to yield the pyrazolopyrimidine intermediate 2, which was selectively iodinated at the C3-position using N-iodosuccinimide to give 3 (Scheme 1). Alkylation of the N1-position of 3 with iodo(cyclopentylmethyl) yielded the desired intermediate 4. The B core was synthesised by iodination of 6-chloro-1H-pyrazolo[3,4-d]pyrimidine, 5, followed by a regioselective nucleophilic aromatic substitution (SNAr) reaction with methylamine at the 6-position to give intermediate 7. Subsequent alkylation of the N1-position of 7 using the same conditions as above yielded the desired intermediate 8. The initial use of the cyclopentylmethyl group at N1 responded to prior studies13,15 that have shown the effect of this moiety in promoting cell activity. Both synthetic schemes make use of microwave-irradiation to raise reaction yields and reduce reaction times. The cores were synthesised at gram scale in good yields and reactions were consistently reproducible, allowing the generation of further starting material as and when required during the project.
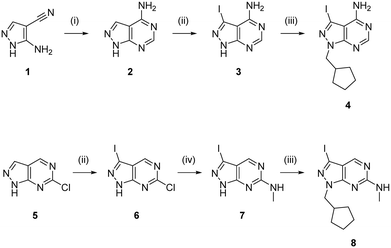 |
| | Scheme 1 Synthesis of intermediates 4 and 8. Reagents and conditions: (i) formamide, 180 °C, μw, 75 min, 84%; (ii) NIS, DMF, 120 °C, μw, 65 min, 58% for 3 and 76% for 6; (iii) (iodomethyl)cyclopentane, NaH, DMF, 150 °C, μw, 85 min, 68% for 4 and 69% for 8; (iv) methylamine, THF, 150 °C, μw, 60 min, 77%. | |
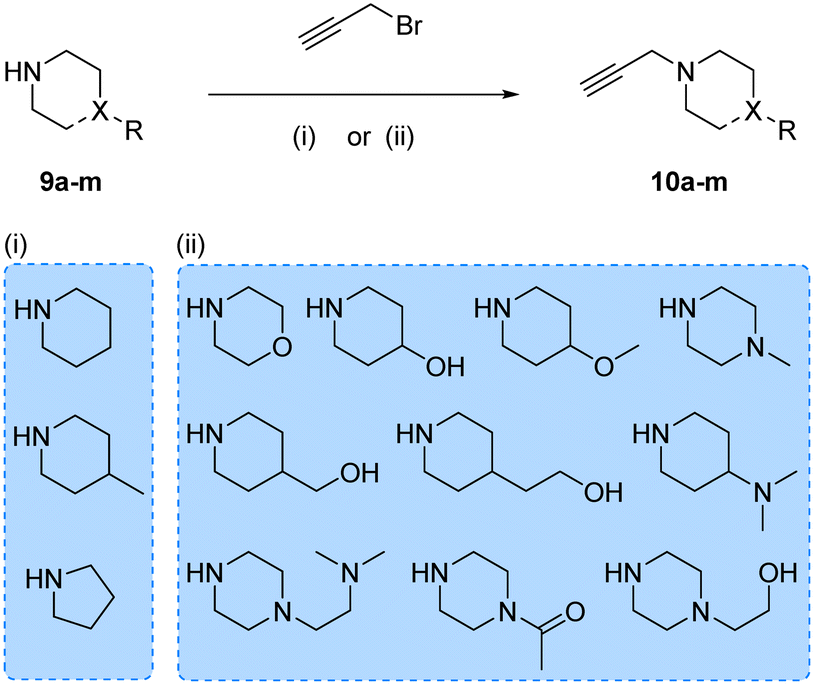
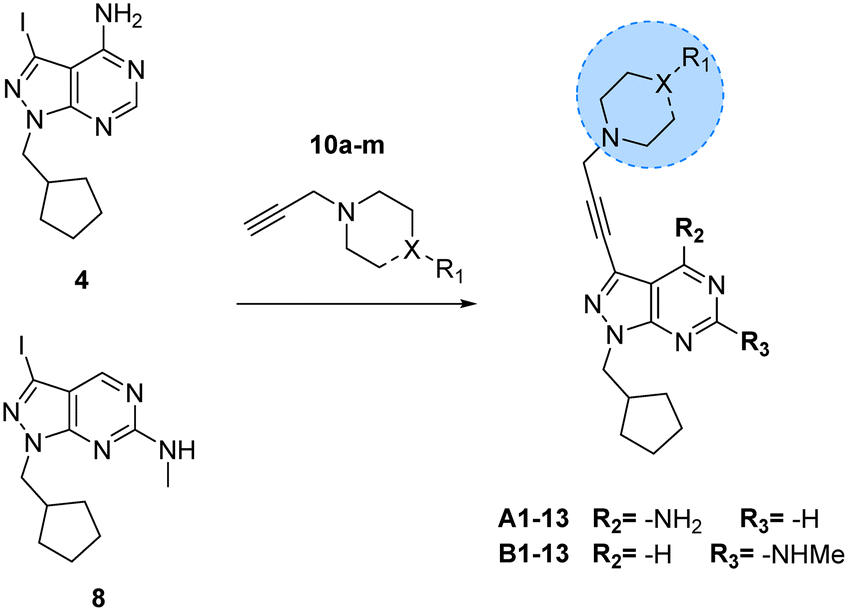
Prior works from Grøtli31 and Zhang27 have studied the incorporation of aromatic rings through a rigid acetylene linker using the Sonogashira reaction to extend their reach into the hydrophobic pocket of the N-lobe of the kinase domain, yielding potent RET inhibitors. On the contrary, in the present work we sought to explore alternative non-hydrophobic interactions using a more flexible linkage. Therefore, we introduced a free-to-rotate methylene group to connect the planar core to a selection of cyclic tertiary amines (see Fig. 1b). To this end, we prepared a library of N-propargylated piperidines, piperazines and morpholine by reacting the corresponding cyclic tertiary amine with propargyl bromide (Scheme 2). Subsequently, using either intermediate 4 or 8, and N-propargyl cyclic amines 10a–m, two 13-member libraries were combinatorially generated by Sonogashira cross-coupling in low to moderate yields (Scheme 3).
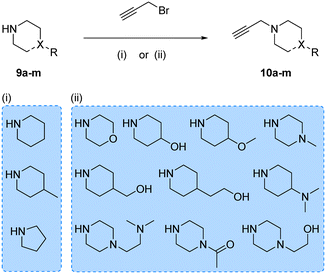 |
| | Scheme 2 Synthesis of N-propargyl cyclic amines 10a–m. Reagents and conditions: (i) K2CO3, DEE, 0–25 °C, 12 h, 14–87%; (ii) K2CO3, THF, reflux, 4–16 h, 2–45%. | |
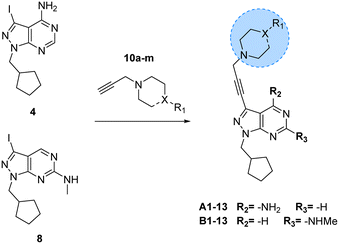 |
| | Scheme 3 Synthesis of A1–13 and B1–13. Reagents and conditions: Pd(PPh3)2Cl2, CuI, TEA, THF, 70 °C, μw, 2 h, 14–86%. | |
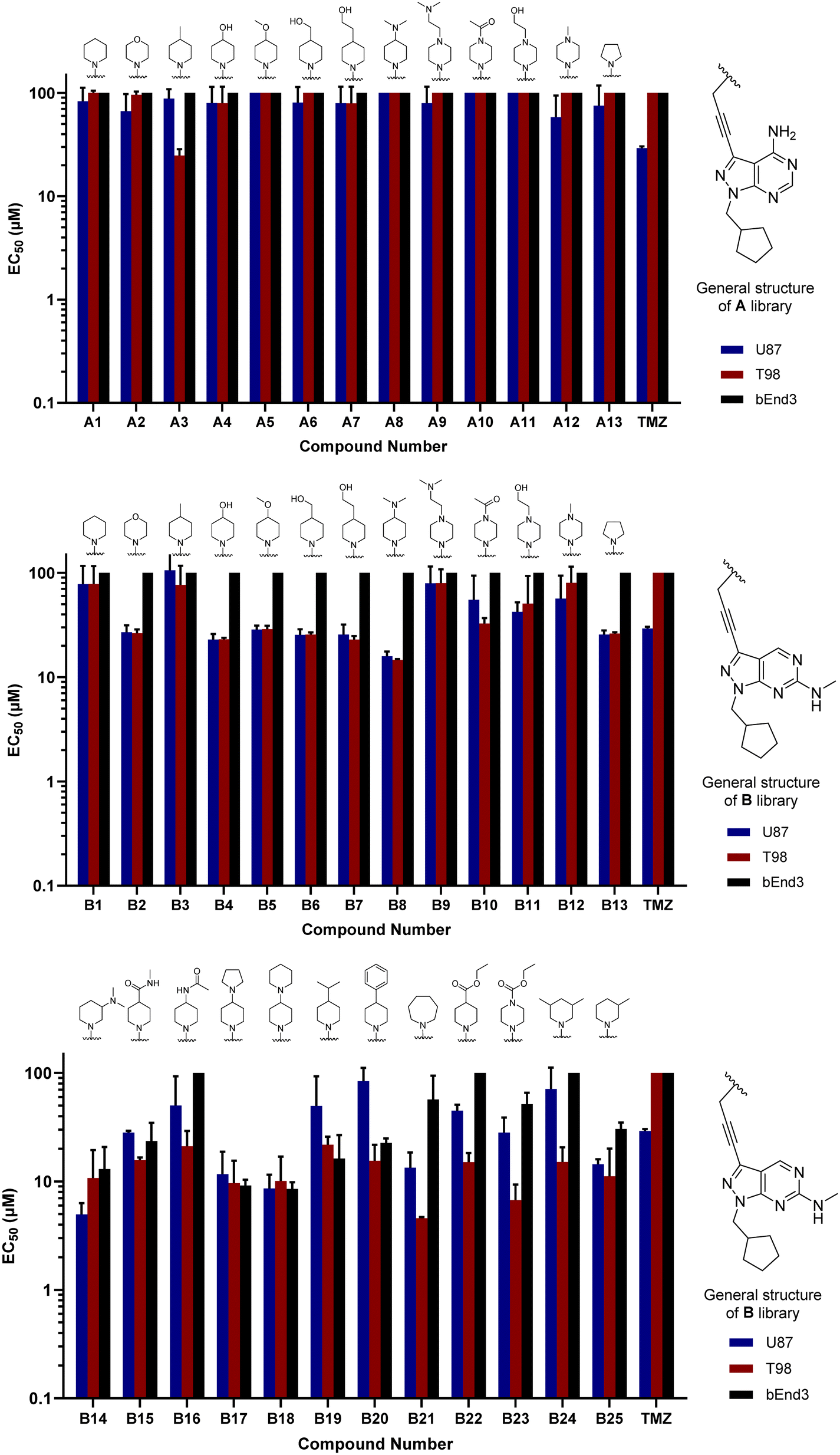
Compounds A1–13 and B1–13 were then tested against two human glioma cell models: epithelial-type U87 cells, characterised by KRAS mutation and lack of PTEN,32 and the T98 cell line, which exhibits the typical expression profile of the mesenchymal-like GBM phenotype, including the expression of growth factor genes.33 In addition, we used the immortalised murine brain endothelial line bEnd.3 as a Pgp-expressing non-glioma cell control.34 Inhibition of cell growth was used as the primary output of the screening. Half-maximal effective concentration (EC50) values were calculated for all library members and temozolomide (TMZ, positive control) using a 3-point dose–response study (3, 10 and 30 μM). Cell viability was determined at day 5 using the PrestoBlue reagent, and fluorescence measured using a microplate reader. Curves were fitted using GraphPad Prism software and normalised relative to the untreated cells to estimate EC50 values. As shown in Fig. 2, the derivatives featuring the A core were the least potent compounds of the assay, with only A3 (4-methylpiperidinyl analogue) exhibiting an EC50 value below 30 μM. On the contrary, most compounds of the B library displayed activity against glioma cells, highlighting the potency of B8 (4-(dimethylamino)piperidinyl analogue) with EC50 values of 15.9 and 14.8 μM against U87 and T98, respectively. Of note, TMZ only showed some level of antiproliferative activity against U87 cells, in agreement with prior studies.35
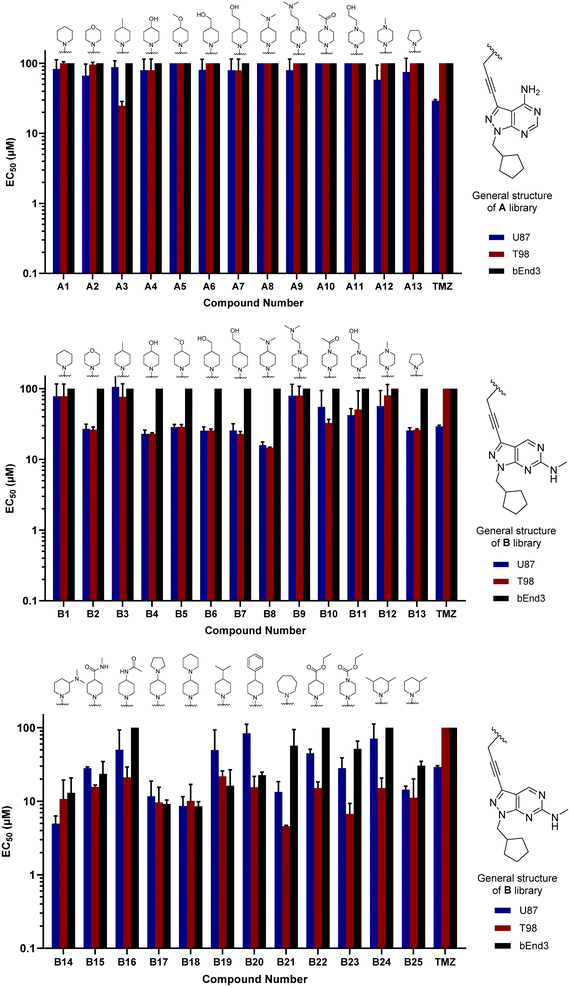 |
| | Fig. 2 Cell screening of initial libraries A and B. EC50 values calculated for A1–13 (top), B1–13 (middle) and B14–25 (bottom), and positive control TMZ against U87, T98 and bEnd.3 cells. Data obtained from a cell viability assay where PrestoBlue® reagent was added after drug treatment for 5 d at 30, 10 and 3 μM doses. Error bars: ±SD from n = 3. | |
Hit-to-lead optimisation of core B
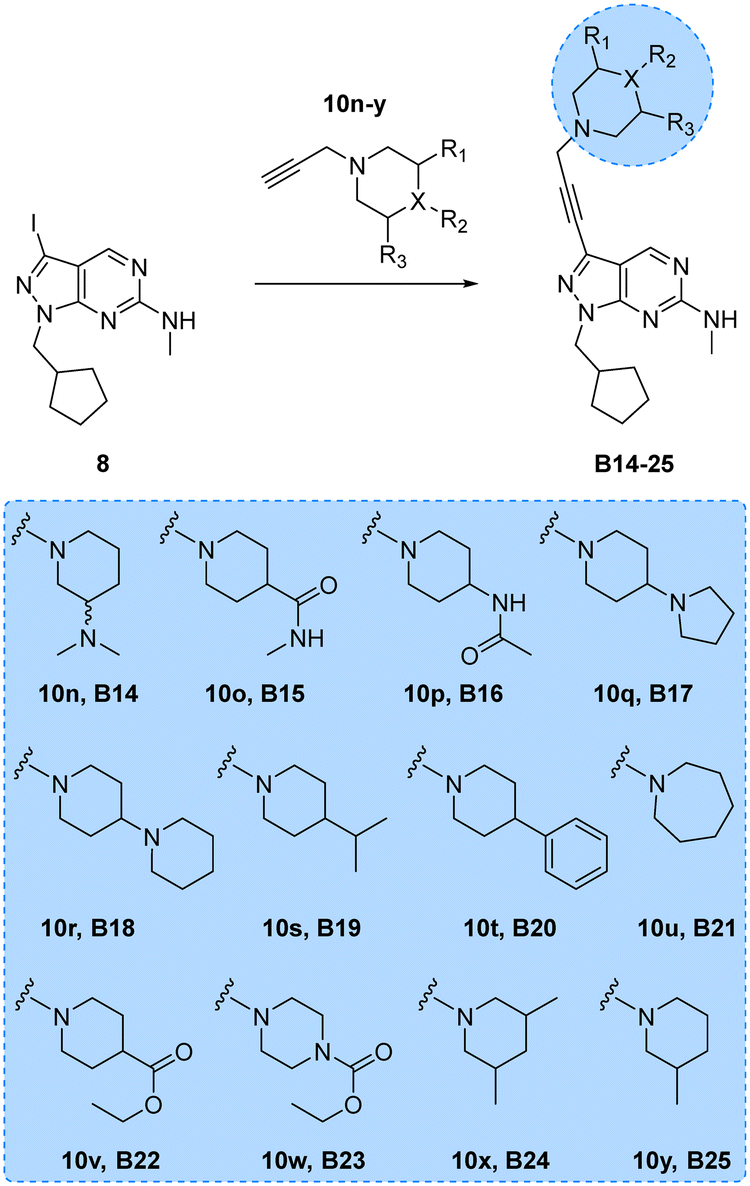
Encouraged by the superior activity of the compounds with the B core, a new set of N-propargyl cyclic amines, 10n–w, were prepared as previously described and cross-coupled with 8 to generate compounds B14–25 (see Scheme 4). As SAARs indicated that the most activity-enhancing moieties were those with different degrees of polarity at the 4-position of the piperidine ring, a range of polar and non-polar groups were included in the cyclic amine ring to evaluate their effect in glioma cell proliferation. As before, the anticancer activity of the second library of B analogues was assessed in 3-point (3–30 μM) cell viability assays against the three cell lines; U87, T98 and bEnd.3. Results from this screening are shown in Fig. 2 (bottom panel). Notably, several compounds exhibited activities below the 10 μM threshold. Derivative B14, where the racemic 3-(dimethylamino)piperidinyl was coupled to the core, was the most potent hit of the library, displaying an EC50 value against U87 and T98 of 5.0 and 10.8 μM, respectively. Another potent hit identified was B21, which features an expanded ring size (azepane) at the top of its structure and displayed an EC50 value against T98 of 4.6 μM. Of note, this hit and others such as B16, B22 and B23 showed a selective antiproliferative profile against the T98 cell line.
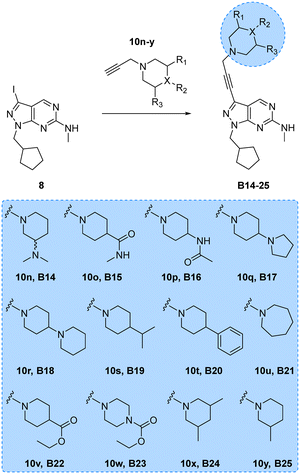 |
| | Scheme 4 Synthesis of B14–25. Reagents and conditions: Pd(PPh3)2Cl2, CuI, TEA, THF, 70 °C, μw, 2 h, 9–51%. | |
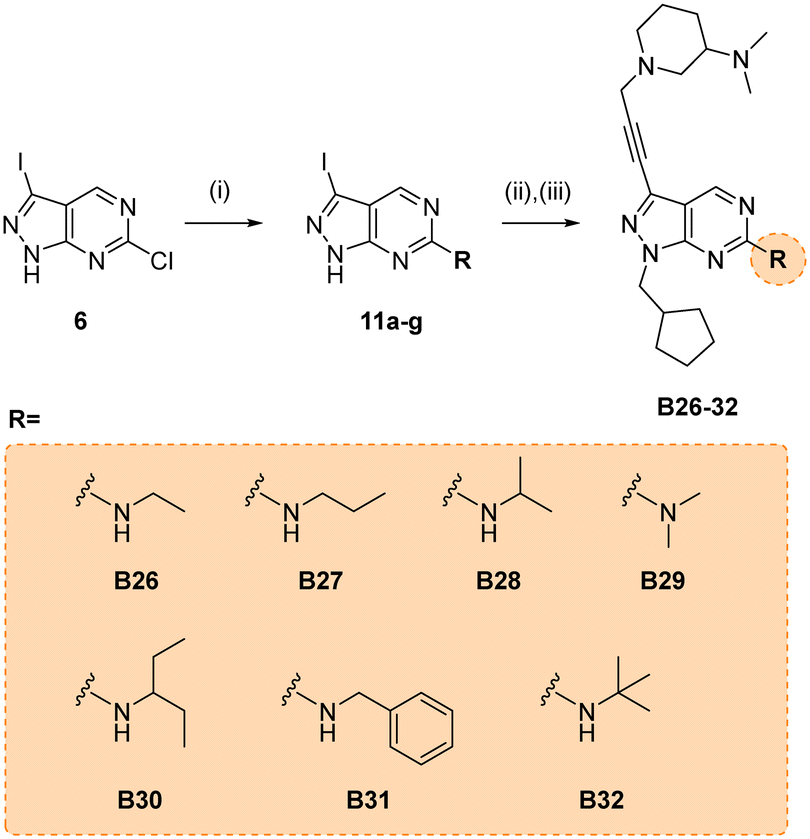
Due to its potency, the 3-(dimethylamino)piperidinyl group of compound B14 was selected as the common motif to build a new library of compounds to study the optimal alkyl substituent/s of the amine group at the position 6 of the core (see synthesis in Scheme 5).
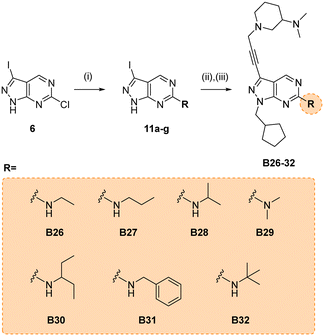 |
| | Scheme 5 Synthesis of B26–32. Reagents and conditions: (i) alkylamine, THF, 150 °C, μw, 60 min, 46–73%; (ii) (iodomethyl)cyclopentane, NaH, DMF, 150 °C, μw, 85 min, 37–68%; (ii) 10n, Pd(PPh3)2Cl2, CuI, TEA, THF, 70 °C, μw, 2 h, 26–59%. | |
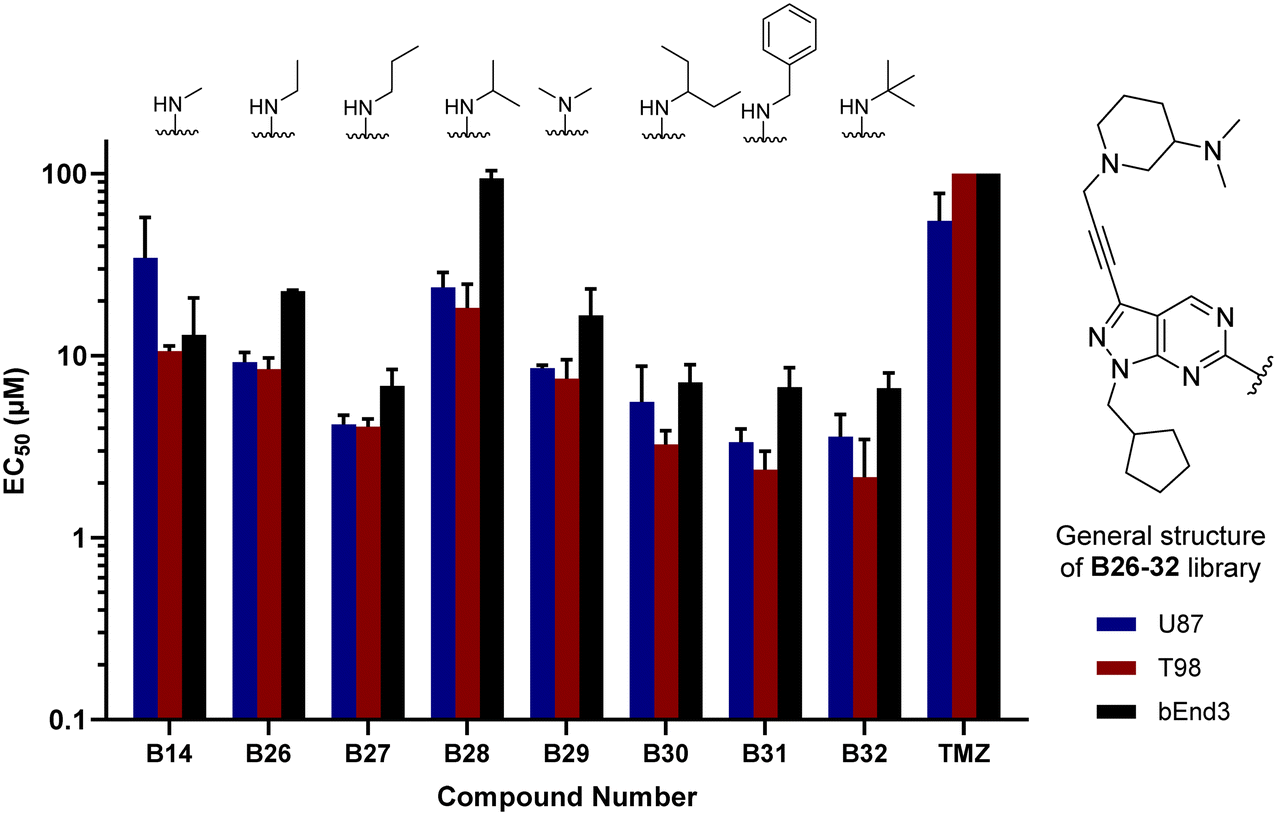
The glioma cell screening of compounds B26–32 was performed as previously described against U87, T98 and bEnd.3 cells, using B14 and TMZ as reference controls. Results clearly showed that the increment of the size of the alkyl groups had a positive impact on the antiproliferative activity of the compounds. B27 (propyl), B30 (3-pentyl), B31 (benzyl) and B32 (tert-butyl) displayed the most potent activities, with EC50 values below 4 μM against the mesenchymal-like T98 cells (Fig. 3).
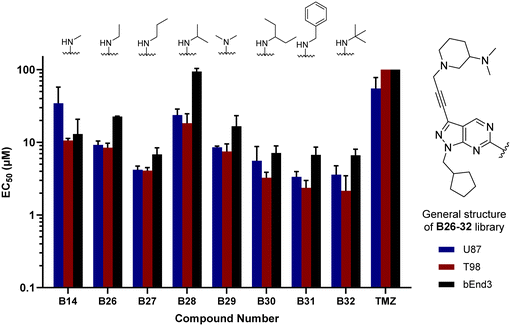 |
| | Fig. 3 Cell screening of B26–32. EC50 values calculated for B26–32 and reference controls B14 and TMZ against U87, T98 and bEnd.3 cells. Data obtained from a cell viability assay where PrestoBlue® reagent was added after drug treatment for 5 d at 30, 10 and 3 μM doses. Error bars: ±SD from n = 3. | |
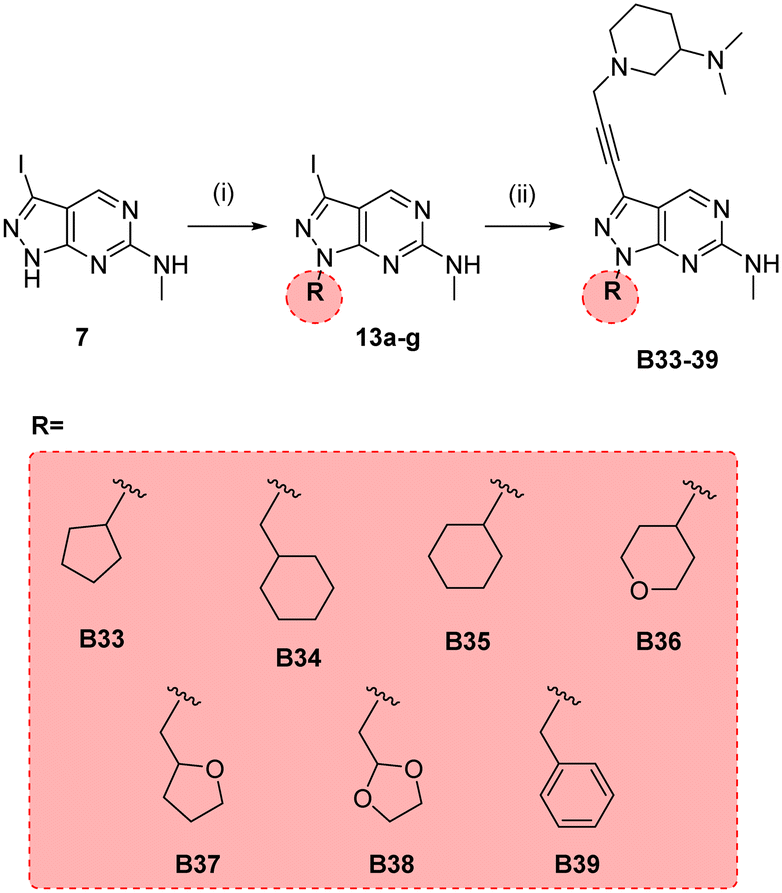
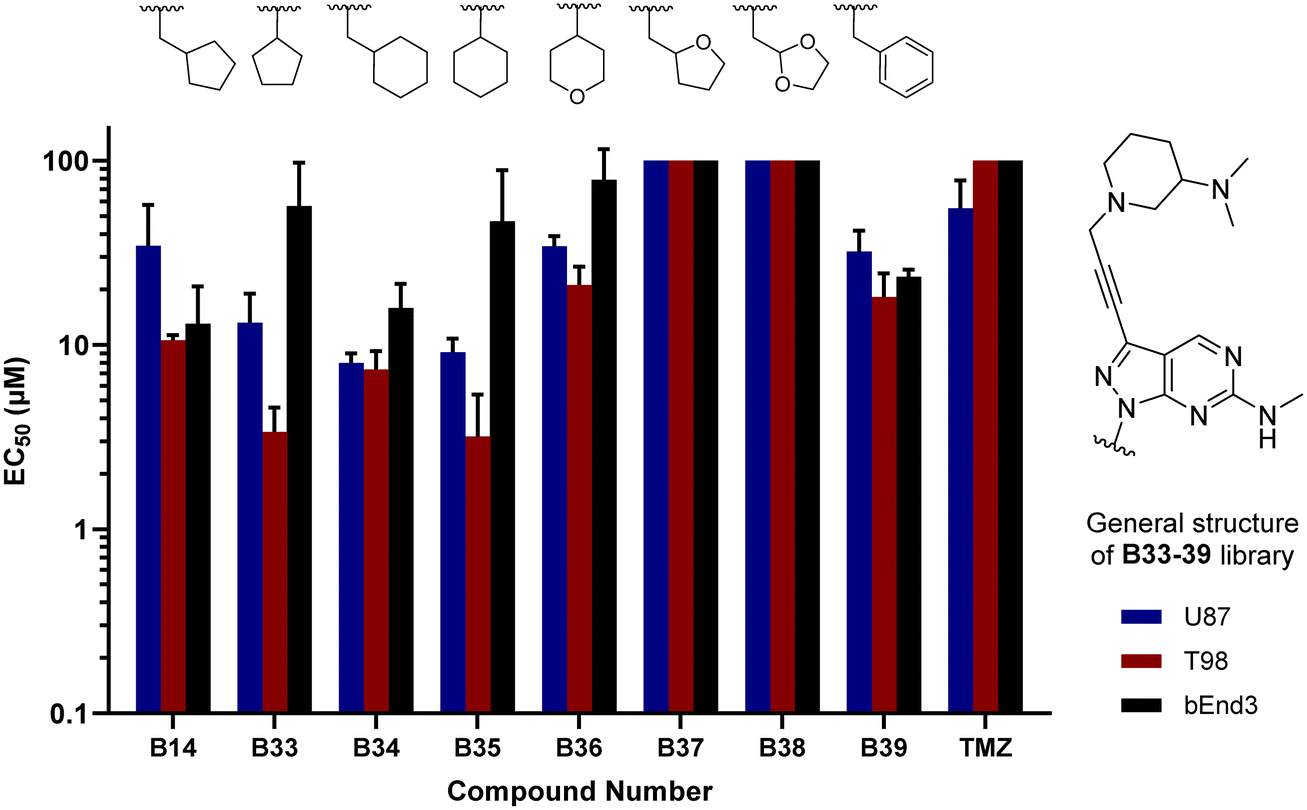
In parallel, we synthesised an additional library of compounds to study different substituents at the N1 position of the B core, by exchanging the methylcyclopentyl group with a range of cycloalkyl-containing moieties (see synthesis in Scheme 6). As shown in Fig. 4, the screening against glioma cell lines evidenced that the incorporation of oxygen atoms in the aliphatic rings significantly reduced analogues' activity. Three compounds, B33 (cyclopentyl), B34 (methylcyclohexyl) and B35 (cyclohexyl) displayed the highest activities against the T98 cell line.
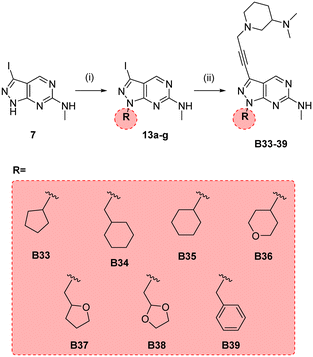 |
| | Scheme 6 Synthesis of B33–39. Reagents and conditions: (i) alkyl bromide, NaH, DMF, 150 °C, μw, 85 min, 24–82%; (ii) 10n, Pd(PPh3)2Cl2, CuI, TEA, THF, 70 °C, μw, 2 h, 26–44%. | |
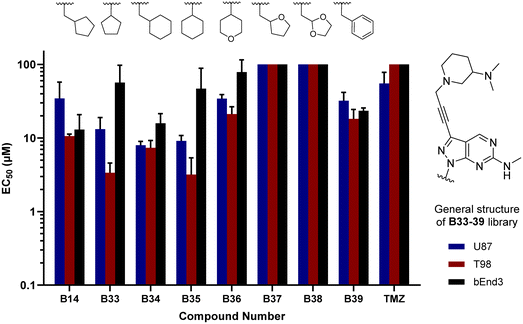 |
| | Fig. 4 Cell screening of B33–39. EC50 values calculated for B33–39 and reference controls B14 and TMZ against U87, T98 and bEnd.3 cells. Data obtained from a cell viability assay where PrestoBlue® reagent was added after drug treatment for 5 d at 30, 10 and 3 μM doses. Error bars: ±SD from n = 3. | |
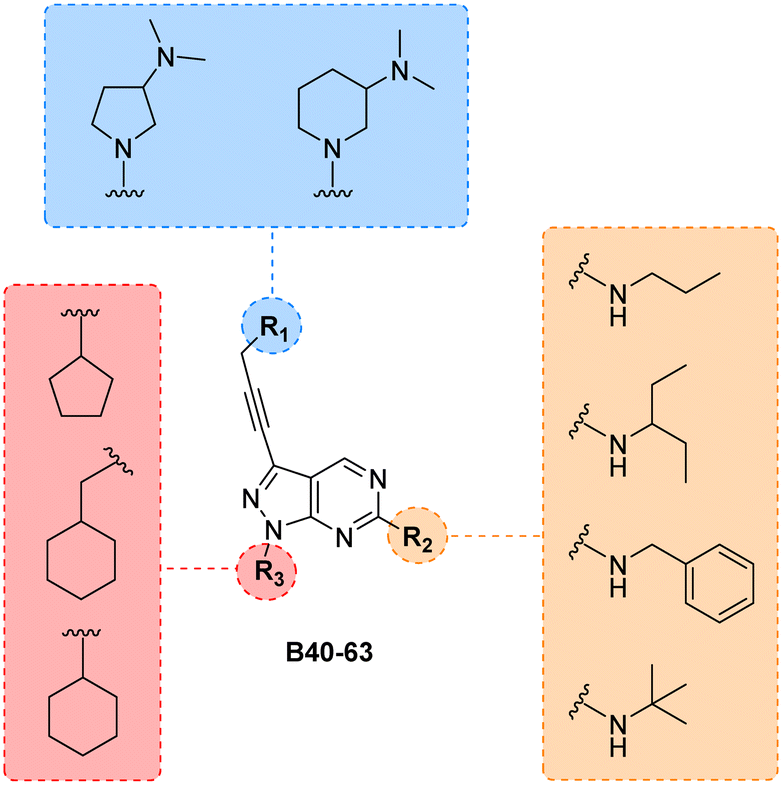
Finally, making use of the SAARs generated by the campaign to optimise the alkylamino group and the cycloalkyl moiety at the C6 and N1 positions of core B, respectively, we prepared a library of derivatives featuring two structurally-similar cyclic amines at C3 position of the scaffold: 3-(dimethylamino)piperidine and 2-(dimethylamino)pyrrolidine (see the combinatorial library in Fig. 5). 24 new compounds (B40 to B63), 12 featuring each cyclic amine, were synthesised from common intermediate 6 as previously described (see Schemes 4–6) in moderate yields (18–52%).
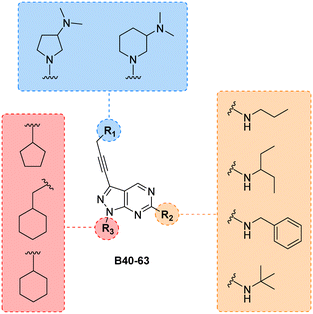 |
| | Fig. 5 Final combinatorial library of the B core. All permutations yield 24 new structures. Compounds were synthesised following the synthetic routes previously outlined. | |
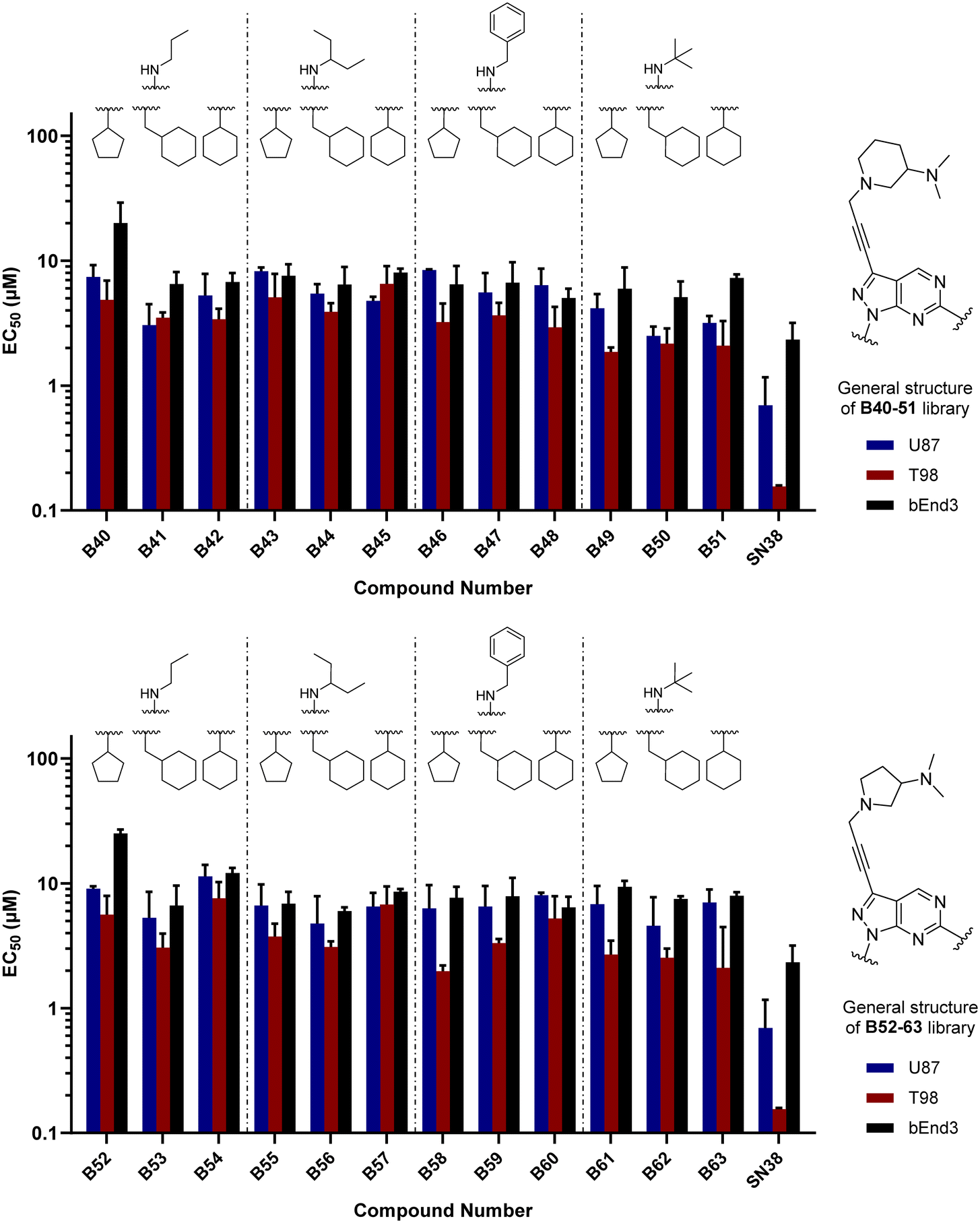
As per previous libraries, the 24-member combination library was first evaluated by a three-point dose–response study against U87, T98 and bEnd.3 cells (Fig. 6). Cells were treated with the compounds in triplicate and incubated for 5 d, using cells treated with DMSO only as negative control and, instead of TMZ, the potent cytotoxic drug SN-38 (active metabolite of irinotecan)36 as positive control. From the compounds featuring a 3-(dimethylamino)piperidine moiety (B40–51) at the north of the scaffold, the analogues with a tert-butylamino group at position C6 (B49, B50, B51) displayed the highest antiproliferative activities, being B49 the most potent analogue of the series. On the other hand, from the 2-(dimethylamino)pyrrolidinyl derivatives, compound B58 (which contains a benzylamino group at C6 and a cyclopentyl group at N1) exhibited an EC50 value around 2 μM against the mesenchymal-like T98 glioma cells, being the most potent antiproliferative agent found in this study. To ensure calculation of accurate EC50 values, the three most potent compounds of the combinatorial library (B49, B58 and B63) were further evaluated in a full dose–response study against T98 and U87. EC50 values were determined using a semi-log 7-point dose response experiment (from 0.03 to 30 μM). B58 was confirmed as the most potent derivative against T98 cells, with an EC50 value of 2.41 μM (see Fig. S1, ESI†). Interestingly, B63 displayed equivalent potency against U87 and T98 cells (EC50 = 2.71 and 2.54 μM, respectively).
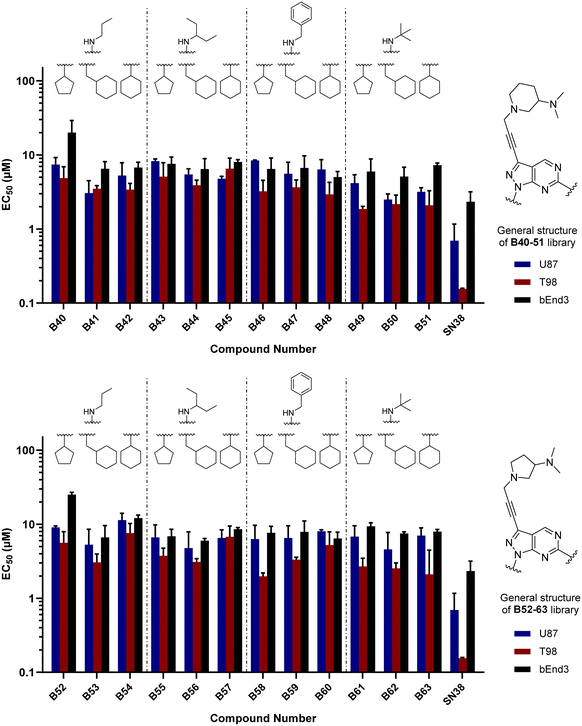 |
| | Fig. 6 Cell screening of B52–63. EC50 values calculated for B33–39 and positive control SN38 against U87, T98 and bEnd.3 cells. Data obtained from a cell viability assay where PrestoBlue® reagent was added after drug treatment for 5 d at 30, 10 and 3 μM doses. Error bars: ±SD from n = 3. | |
Phenomics evaluation in patient-derived glioma cells
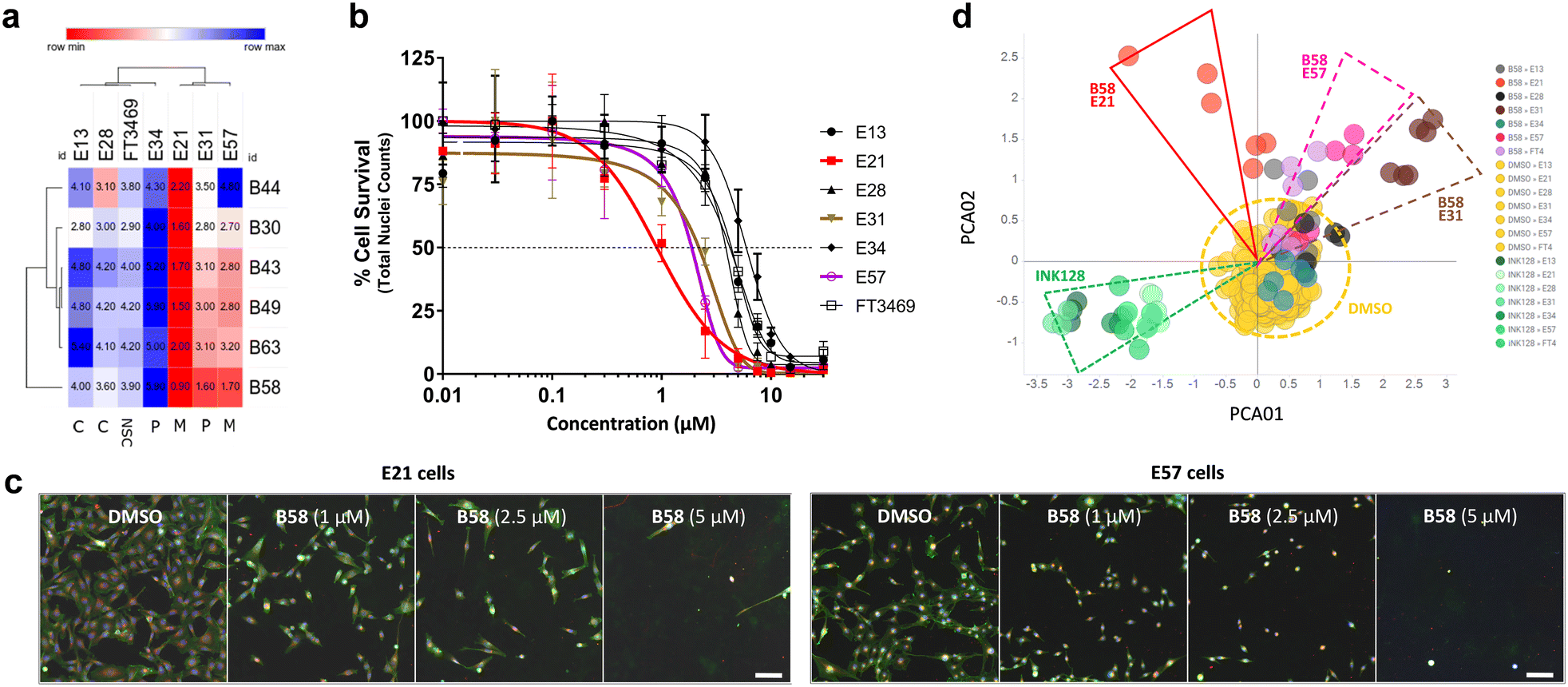
While the use of commercial cell lines has been invaluable for the purpose of our drug discovery strategy, further analysis across a panel of well-characterised patient-derived cell models selected to represent the heterogeneity of GBM tumors can facilitate the evaluation of clinical utility and development of personalised medicine strategies. Studies based on bulk gene expression profiles of patient tumours suggest that at least three GBM cell subtypes exist: proneural (P), classical (C) and mesenchymal (M).37 In addition, GBM is known to hijack mechanisms of neural development to produce glioma stem cells (GSCs) that contribute to therapy resistance and relapse. Recent advances have established defined culture conditions that maintain and expand GBM stem cells in vitro.38 Therefore, we applied these protocols and an established multiparametric high-content Cell Painting assay39 to phenotypically profile the biological activity of the most potent derivatives, including B49, B58 and B63, across a panel of patient-derived GSCs representing the major proneural, classical and mesenchymal disease subtypes. Reference controls included SN-38 and kinase inhibitors dinaciclib (CDKs) and INK128 (mTOR), among others. High-content Cell Painting analysis provided rich phenotypic profiles, including automatic quantification of nuclear counts, which served as a surrogate measure of % cell survival and compound EC50 across the GBM stem cell panel. In agreement with the superior potency observed by B58 against the mesenchymal-type T98 cell line, the two mesenchymal GSCs E21 and E57 displayed high sensitivity to the lead compound (see Fig. 7a–c), highlighting the antiproliferative activity against E21 cells with an EC50 value of 0.9 μM (see dose response curves for all compounds in Fig. S2, ESI†). Principle component analysis (PCA) on high-content phenotypic profiles show strong B58-induced effects on E21, E57 and E31 glioma stem cells, which are phenotypically distinct from those induced by the pyrazolopyrimidine-based mTOR inhibitor INK128 (Fig. 7d). Further PCA analysis by compound and representative high-content images from the Cell Painting assay are shown in the ESI† (Fig. S3–S10).
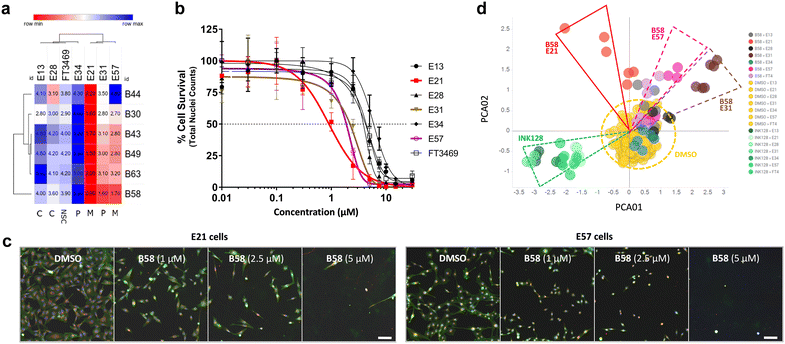 |
| | Fig. 7 Dose response and phenotypic profile of B58 on patient-derived GSCs. a) EC50 data table and hierarchical clustering of compounds B44, B30, B43, B49, B63 and B58 on a glioma stem cell panel. Data was derived from nuclei counts (Hoechst stain) and normalised to DMSO to give % cell survival. Values in μM. b) Plot of B58 dose response across glioma cells. Includes non-transformed neural stem cells FT3469 (NSC). c) Representative images of the phenotypic effect of B58 on E21 (left) and E57 (right) cells at different concentrations versus DMSO control. Scale bar represents 100 μm. d) PCA plot of B58 effects on glioma stem cells from Cell Painting images. Comparison of B58 phenotypic response indicated on E21 (red), E57 (purple) and E31 cells (brown) at 1 and 2.5 μM versus DMSO (yellow) and INK128 (green). | |
Kinome profiling and cytokine microarray analysis
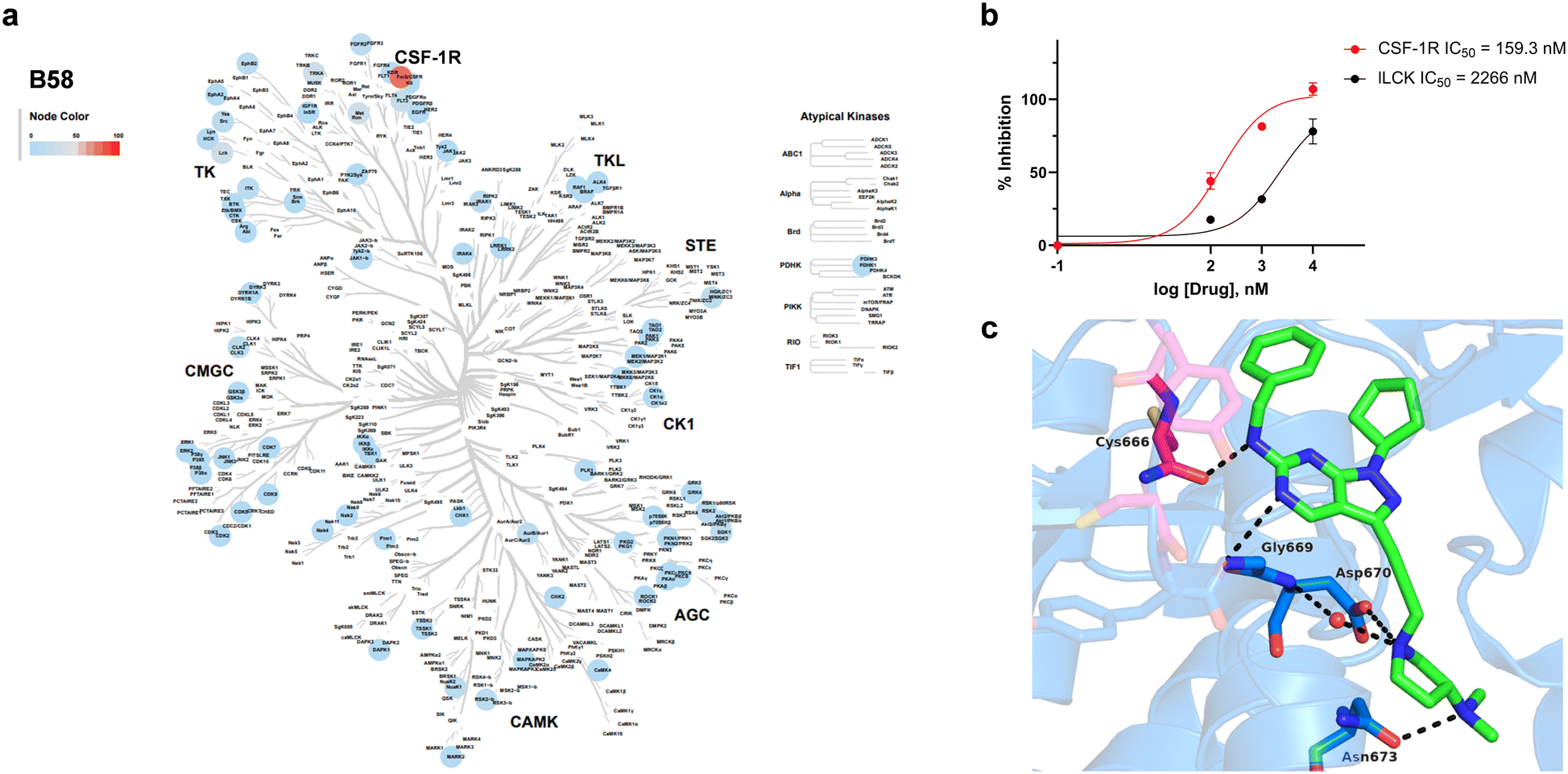
Encouraged by the information generated by the phenomics study, B58 was next screened across a broad panel of 74 kinases to identify its potential target/s and determine its general selectivity profile. The lead compound was screened at 3-point dose–response (0.1, 1 and 10 μM) using the Z′-LYTE assay (ThermoFischer Scientific). Z′-LYTE is a ratiometric assay that employs synthetic peptide substrates conjugated with a FRET pair at each end and is based on the differential sensitivity of phosphorylated and non-phosphorylated peptides to proteolytic cleavage. Remarkably, the screening identified CSF-1R as the kinase most highly inhibited of the panel, with an IC50 value of 0.159 μM (Fig. 8a and b). The IC50 value for the second most inhibited kinase, LCK, was significantly higher (2.27 μM), whereas most of the kinases tested showed nil to low levels of inhibition, even at 10 μM (see Table S1, ESI†).
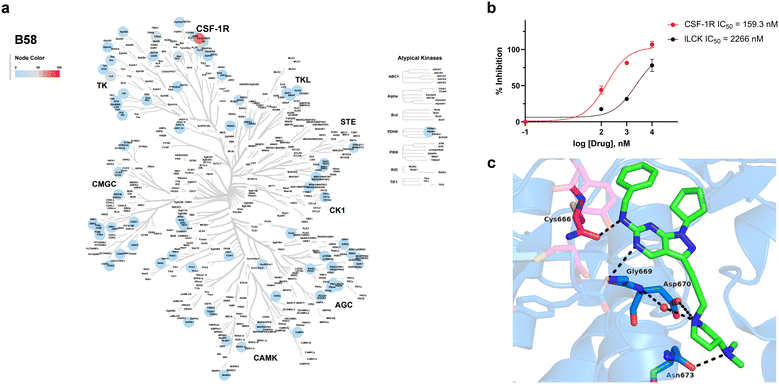 |
| | Fig. 8 Kinase inhibition profile B58 and computational modelling of CSF1R binding. a) Kinome phylogenetic tree showing the enzymatic inhibition mediated B58 across a broad panel of 74 protein kinases. The colour of the spots relates to the % inhibition of the corresponding kinase tested at a dose of 1 μM. Kinome tree plotted using Coral.40 b) Dose response curves and IC50 values of the inhibition of CSF-1R and LCK by B58. c) In silico docking of B58 in the ATP pocket of CSF-1R (PDB: 6T2W) visualised with PyMOL. Key interactions between B58 and protein residues are shown as dotted black lines. | |
Given the selective activity showed by B58 for the immunoregulatory RTK CSF-1R and its distinct antiproliferative profile against patient-derived glioma cells, we further characterised the GSC panel at the immune modulatory level. We performed a cytokine microarray analysis on cell supernatants to study the basal production of immune modifying cytokines and growth factors, including CSF-1 ligand (Fig. S11, ESI†). The results showed that E21 exhibits high production of CSF-1 ligand and, uniquely, IL-6 overexpression relative to the other cell lines. The inhibition of CSF-1R has been shown to reduce IL-6 in microglia,41 suggesting a potential biomarker for CSF-1R inhibitor response in GBM. These results not only provide evidence of the role of autocrine CSF-1R stimulation in E21 glioma cells, but also suggests that the immune deregulation mediated by CSF-1R and its ligands in GBM may be linked to or more prevalent in the mesenchymal-like GBM subtype.
In silico docking study and DMPK profiling
Capitalising on the availability of the crystal structure of CSF-1R in complex with a dihydropurinone inhibitor (PDB entry 6T2W),42B58 structure was docked in its target to gain insights into its mode of binding. B58 was first modelled using Avogadro,43 and the optimised structure docked into the kinase domain of the protein using GOLD and visualised with PyMOL. Optimal poses strongly indicated that B58 binds the ATP binding pocket of CSF-1R. As shown in Fig. 8c, the amino at C6 position is predicted to form an H-bond with the backbone of Cys666 in the kinase hinge and the N5 establishes a second H-bond with Gly669. Asp670 is predicted to interact with the N atom of the pyrrolidine by forming a water-mediated H-bond with the backbone and/or by an ionic interaction with its carboxylate group, and the dimethylamino group forms an H-bond with Asn673. In addition, it is predicted that the lipophilic benzyl and cyclopentyl groups of B58 are buried in a large hydrophobic pocket formed by Leu588, Val596, Ala614, Val747, Leu785 and Phe797 (see Fig. S12, ESI†). The docking study agrees with the affinity of B58 for CSF-1R and, importantly, suggests that there is room for improvement.
Finally, we performed a computational assessment of the physicochemical and DMPK properties of B58.44,45 As shown in Table 1, the lead compound is predicted to display high solubility under physiological conditions, good Caco-2 cell permeability and high intestinal absorption. SwissADME predicts that the compound will be able to cross the blood brain barrier, a critical requirement for the development of drugs to treat GBM. In addition, B58 is also expected to have a relatively low unbound fraction in plasma (approx. 10%) and a moderate intrinsic clearance. This preliminary assessment, along with its potency against mesenchymal GSCs, supports the selection of B58 as a promising candidate for lead optimisation.
Table 1
B58 physicochemical and PK predictions
|
DruMap.
SwissADME.
|
| Molecular weight |
443.59 g mol−1 |
| Num. rotable bonds |
6 |
| Num. H-bond acceptors |
6 |
| Num. H-bond donors |
1 |
| TPSA |
62.11 Å2 |
clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P P |
3.58 |
| Predicted solubility (pH = 7.4)a |
>10 μg mL−1 |
| Papp (A-to-B, Caco-2)a |
>1 × 10−6 cm s−1 |
| BBB penetrantb |
Yes |
| Intestinal absorption (human, % absorbed)b |
93.5% |
| fu, plasma (human)a |
0.097 |
| CLint (human)a |
45.87 μL min−1 mg |
Conclusions
Performing an exploratory investigation through the bioactive structural space around the pyrazolopyrimidine scaffold, we have identified novel analogues that potently inhibit glioma cell proliferation. B58 (a.k.a. eDB333), the most potent antiproliferative compound of the study, was identified as a selective inhibitor of the RTK CSF-1R. Remarkably, cell assays and phenomics screening across different subtypes of established and patient-derived glioma cells showed that the lead compound displays preferential antiproliferative activity against glioma cells of the mesenchymal subtype, one of the most aggressive and invasive of all GBM subclasses. The most sensitive cell line, E21, displays high expression of the CSF-1 ligand, and also IL-6, which is characteristic of a pro-inflammatory TAM phenotype. To the best of our knowledge, this is the first study to connect CSF-1/CSF-1R axis inhibition and the mesenchymal-like GBM subtype, and eDB333 is the first pyrazolopyrimidine-based CSF-1R inhibitor to be identified. Given the prominent role of CSF-1R in cancer immune evasion in GBM and other diseases, this work provides strong foundation for future medchem campaigns and cancer biology research on route to the development of novel personalised targeted therapies against GBM, the most life-threatening form of brain cancer.
Experimental section
Synthetic procedures
General methods.
Chemicals and reagents were purchased from Sigma-Aldrich and Alfa-Aesar. Flash column chromatography was performed with silica gel (220–440 mesh). NMR spectra were recorded on a Bruker 500 MHz spectrometer at 300 K and referenced relative to the solvent residual peaks with chemical shifts (δ) reported in ppm. Multiplicity is reported: s, singlet; d, doublet. Coupling constants (J) are shown in hertz. HRMS was obtained using a Bruker 3.0 T Apex II Spectrometer under electron spray ionization conditions. Compound 4 used in the biological experiments was >95% pure by HPLC measured using Waters 600E (100 μL) gradient pump using a 717 plus autosampler and a Waters 996 Photodiode Array Detector (210–400 nm) equipped with a Phenomenex Luna C18(2), 5 μm, 250 × 4.6 mm column at a flow rate of 1 mL min−1 with an injection volume of 10 μL. Method: eluent A, water with TFA (0.1%); eluent B, acetonitrile with TFA (0.1%): A/B = 95:5 to 5:95 in 30 min, 5:95 isocratic 5 min, 5:95 to 95:5 in 5 min, 95:5 isocratic 10 min.
Synthesis of lead compound B58 (eDB333)
6-Chloro-3-iodo-1H-pyrazolo[3,4-d]pyrimidine (6).
6-Chloro-1H-pyrazolo[3,4-d]pyrimidine (5) (2.02 g, 13.1 mmol) was suspended in DMF (15 mL) in a 20 mL microwave vial equipped with stirrer bar. The suspension was stirred, and N-iodosuccinimide (3.84 g, 17.1 mmol, 1.3 eq.) was added portion-wise. The vial was sealed and heated to 120 °C while stirring in a microwave reactor for 65 min. The resultant deep red reaction mixture was partitioned between EtOAc (50 mL) and water (50 mL). The organic layer was collected, and the aqueous layer was washed with EtOAc (3 × 50 mL). The organic layers were combined and washed with water (2 × 20 mL) and brine (50 mL). The organic fraction was then dried over MgSO4 and concentrated in vacuo to give the crude product, a deep red solid. The crude product was purified by flash column chromatography on silica using a 0–10% MeOH/DCM eluent gradient. The appropriate fractions by TLC were combined and concentrated under reduced pressure to yield the product (2.81 g, 10.0 mmol, 76%) as a yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 14.63 (s, 1H), 9.01 (s, 1H). 13C NMR (126 MHz, DMSO-d6) δ 157.8 (C), 155.6 (CH), 155.2 (C), 117.6 (C), 94.1 (C). LRMS (ESI +ve) [M + H] 281.00.
N-Benzyl-3-iodo-1H-pyrazolo[3,4-d]pyrimidin-6-amine (11f).
6-Chloro-3-iodo-1H-pyrazolo[3,4-d]pyrimidine (1.0 mmol) was added to a 20 mL microwave vial equipped with stirrer bar. The solid was suspended in THF (1.5 mL) and dissolved through sonication. The mixture was stirred, and amine solution (2 M in THF, 3.0 eq.) was added. The vial was sealed with septum cap and heated to 150 °C while stirring in a microwave reactor for 60 min under microwave irradiation. The reaction vial was removed from the microwave and allowed to cool to ambient temperature, resulting the formation of a precipitate, which was collected by vacuum filtration. The solid was washed with water (3 × 4 mL) and dried to yield the product (273.3 mg, 0.778 mmol, 62%) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 13.34 (br. s., 1H), 8.50 (s, 1H), 8.14 (br. s., 1H), 7.27–7.33 (m, 4H), 7.18–7.23 (m, 1H), 4.54 (br. s., 2H). 13C NMR (126 MHz, DMSO-d6) δ 161.7 (C), 156.7 (CH), 153.7 (2 × C), 128.2 (2 × C), 126.9 (2 × C), 126.5 (C), 93.0 (C), 44.1 (CH2). LRMS (ESI +ve) [M + H] 351.90.
N-Benzyl-1-cyclopentyl-3-iodo-1H-pyrazolo[3,4-d]pyrimidin-6-amine (12f).
N-Benzyl-3-iodo-1H-pyrazolo[3,4-d]pyrimidin-6-amine (1.0 mmol) was added to a 20 mL microwave vial equipped with a stirrer bar. The solid was suspended in DMF (10 mL) and stirred to a homogeneous solution. Sodium hydride (1.5 eq., 60% dispersion in mineral oil) was added, and the reaction was stirred for 30 min for gas evolution to subside. Bromo cycloalkane (1.5 eq.) was added, and the vial was sealed with a septum cap and placed into a microwave reactor. The reaction was heated to and stirred at 150 °C for 90 min under microwave irradiation, and the reaction was cooled whilst stirring overnight. The reaction mixture was partitioned between water (50 mL) and EtOAc (50 mL). The organic layer was collected, and the aqueous layer was washed with further EtOAc (4 × 50 mL). The organic fractions were collected, combined, washed with water (100 mL) and brine (100 mL). The resultant organic layer was dried over MgSO4 and concentrated in vacuo to give the crude product. The crude was purified by flash column chromatography on silica using a 0–8% MeOH/DCM eluent system. The appropriate fractions by TLC were combined and concentrated to give the product (180.0 mg, 0.429 mmol, 50%) as a cream-coloured solid. 1H NMR (500 MHz, DMSO-d6) δ 8.45 (br. s., 1H), 8.23 (br. s., 1H), 7.35 (br. s., 2H), 7.29 (t, J = 7.53 Hz, 2H), 7.17–7.24 (m, 1H), 4.99 (quin, J = 7.41 Hz, 1H), 4.54 (br. s., 2H), 2.00 (br. s., 2H), 1.88–1.96 (m, 2H), 1.82 (br. s., 2H), 1.57–1.70 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 153.9 (2 × C), 148.4 (CH), 128.1 (3 × CH), 127.4 (2 × CH), 126.6 (CH), 113.0 (C), 101.9 (C), 44.3 (CH2), 31.4 (CH), 30.8 (2 × CH2), 24.1 (2 × CH2). LRMS (ESI +ve) [M + H] 419.90.
N,N-Dimethyl-1-(prop-2-yn-1-yl)-pyrrolidin-3-amine (10z).
3-Dimethylamino-pyrrolidine (6 mmol) was added to a 50 mL round-bottomed flask equipped with stirrer bar. The amine was suspended in THF (50 mL). Potassium carbonate (12 mmol, 2 eq.) was added, and the reaction mixture was stirred at ambient temperature for 5 min. Propargyl bromide (9 mmol, 80% in toluene, 1.5 eq.) was added dropwise to the stirring reaction mixture. The reaction was fitted with a condenser and stirred at reflux (68 °C) for 8 h. The reaction was removed from the heat and allowed to cool while stirring overnight. The solvent was removed in vacuo and the crude was partitioned between water (40 mL) and DCM (50 mL). The organic layer was collected, and the aqueous layer was washed with DCM (2 × 25 mL). The organic fractions were combined and dried over MgSO4, before being concentrated under reduced pressure to give the crude product. The crude product was purified by flash column chromatography, using an eluent system of 0–20% MeOH in DCM. The appropriate fractions were combined and concentrated to give the product (82.2 mg, 0.54 mmol, 5%) as a dark brown oil. 1H NMR (500 MHz, DMSO-d6) δ 3.19 (t, J = 2.36 Hz, 1H), 2.89 (d, J = 10.01 Hz, 1H), 2.59–2.65 (m, 1H), 2.53 (d, J = 5.28 Hz, 6H), 2.20–2.32 (m, 1H), 2.15 (t, J = 9.58 Hz, 1H), 1.83–1.90 (m, 1H), 1.69–1.77 (m, 1H), 1.40–1.50 (m, 1H), 1.30–1.39 (m, 1H), 1.24 (s, 1H). 13C NMR (126 MHz, DMSO-d6) δ 79.0 (CH), 76.1 (C), 61.1 (CH2), 51.2 (CH2), 46.2 (CH2), 40.7 (CH2), 24.7 (CH), 22.8 (2 × CH3).
N-Benzyl-1-cyclopentyl-3-(3-(3-(dimethylamino) pyrrolidine-1-yl)prop-1-yn-1-yl)-1H-pyrazolo[3,4-d]pyrimidin-6-amine (B58, eDB333).
N-Benzyl-1-cyclopentyl-3-iodo-1H-pyrazolo[3,4-d]pyrimidin-6-amine (0.170 mmol) was added to a 5 mL microwave vial equipped with a stirrer bar. To the vial were added, N,N-dimethyl-1-(prop-2-yn-1-yl)-pyrrolidin-3-amine (1.2 eq.), bis(triphenylphosphine) palladium(II) dichloride (6 mol%) and copper(I) iodide (15 mol%). The reagents were suspended in THF (4 mL) and stirred to a homogeneous solution. Triethylamine (2.0 eq.) was added, and the vial was sealed with septum cap and placed into a microwave reactor. The reaction was heated to and stirred at 70 °C for 2 h under microwave irradiation, and the reaction was cooled while stirring at room temperature overnight. The reaction was partitioned between water (40 mL) and EtOAc (50 mL). The organic layer was collected, and the aqueous layer was extracted with further EtOAc (4 × 50 mL). The organic extracts were combined and washed with water (100 mL) and brine (100 mL). The resultant phase was dried over MgSO4 and concentrated in vacuo to give the crude product. The crude product was purified by preparative-TLC on silica using a 10% MeOH/DCM eluent system. The appropriate dual active band by UV and KMnO4 visualisation was scraped from the plate and the silica was washed with 20% MeOH/DCM solution. The filtrate was collected and concentrated in vacuo to give the product (28.3 mg, 0.065 mmol, 38%) as a light brown solid. 1H NMR (500 MHz, DMSO-d6) δ 8.72 (s, 1H), 8.20 (br. S., 1H), 7.36 (br. S., 2H), 7.30 (t, J = 7.57 Hz, 2H), 7.19–7.23 (m, 1H), 5.02 (quin, J = 7.33 Hz, 1H), 4.54 (br. S., 2H), 3.66 (s, 2H), 2.78–2.84 (m, 1H), 2.67–2.78 (m, 2H), 2.60–2.66 (m, 1H), 2.44–2.48 (m, 1H), 2.11 (s, 6H), 1.97–2.07 (m, 2H), 1.88–1.96 (m, 3H), 1.77–1.87 (m, 2H), 1.58–1.69 (m, 3H). 13C NMR (126 MHz, DMSO-d6) δ 160.8 (CH), 152.9 (2 × C), 139.8 (C), 128.1 (4 × CH), 127.5 (2 × C), 126.6 (CH), 89.5 (C), 75.7 (C), 65.1 (2 × CH2), 57.1 (CH), 56.0 (CH2), 51.2 (2 × CH2), 44.3 (CH), 43.3 (CH2), 42.9 (CH2), 31.4 (CH2), 28.6 (CH2), 24.2 (2 × CH3). LRMS (ESI +ve) [M + H] 444.20. HRMS (ESI +ve) [M + H] 444.2870 ([C26H33N7]+, calc. exact mass: 443.2800).
Biological studies
General methods.
U87, T98 and bEnd.3 cells (ATCC) were cultured in DMEM (Gibco), supplemented with 10% (v/v) FBS (Gibco) and 2 mM L-glutamine (Gibco) in a standard incubator (95% humidity, 5% CO2), and sub-cultured twice per week through trypsinisation. Glioma stem cells were detached using Accutase solution (Sigma) and subcultured once per week.
Dose–response cell viability assay.
Cells (1 × 103 U87 cells per well, 5 × 102 T98 cells per well and 1 × 104 bEnd3 cells per well) were plated in 96-well plates in 100 μL of DMEM medium containing 10% FBS and 2 mM L-glutamine and incubated for 48 h in an incubator at 37 °C and 5% CO2. After 48 h, the media was aspirated from each well and replaced with 95 μL of fresh medium. Compounds, including DMSO, were prepared at 20× in DMEM medium in a separate 96-well intermediate plate. 5 μL from the intermediate plate was then added to each well containing cells. Untreated cells were incubated with DMSO (0.1% v/v). After 5 d, PrestoBlue cell viability reagent (10 μL) was added to each well, and the plates incubated for 60–90 min. Fluorescence emission was detected using an Envision fluorescence plate reader (excitation 540 nm, emission 590 nm). All conditions were normalised to the untreated cells (100%), and curves were fitted using a four parameter logistic fit with minimum value constrained to zero using GraphPad Prism software, to calculate EC50 values.
Patient derived GSC culture.
Cells were established by and obtained from the Glioma Cellular Genetics Resource (http://gcgr.org.uk) funded by Cancer Research UK. Ethics approval for use of patient-derived cells and early access confirmation to reagents from the Glioma Cellular Genetics Resource assured that all ethical issues according to UK legislation were fulfilled. Cells were maintained in DMEM/HAMS-F12 media (Sigma, D8437) supplemented with glucose (8 mM, Sigma G8644), beta-mercaptoethanol (100 μM, Ginco, 31350-010), MEM non-essential amino acids (Gibco, 11140-035), BSA (0.015%, Gibco 15260-037), recombinant hFGF basic (12 ng mL−1, Peprotech 100-18b), recombinant mEGF (12 ng mL−1, Peprotech, 315-09) and laminin (4–6 μg mL−1, R&D Stems 3446-0050-01), B27 (1:100, Gibco 17504-044) and N2 supplements (1:200, Gibco 17502-048). Cells were plated onto 384-well microplates (Greiner Bio-One, 781091), after coating with laminin (10 μg mL−1), at 500–1500 cells per well. After overnight incubation, cells were dosed using a D300 digital dispenser (Tecan) with varied concentrations as indicated, in triplicate. All wells were backfilled to final concentration of 0.1% DMSO (v/v). The plates were incubated for 72 h at 37 °C and 5% CO2 prior to live MitoTracker staining and fixation.
Cell painting assay and analysis
Cells were stained with a protocol modified from Bray et al.46 MitoTracker DeepRed (5 μL per well, 3 μM, final 300 nM) was added to each well using a multidrop dispenser (Thermo). The plates were incubated for 30 min at 37 °C and 5% CO2 and then fixed by the addition of 15% formaldehyde (20 μL per well, final conc. 4% HCHO). The remaining staining and imaging steps were performed as previously described.47 Images were analysed via a custom CellProfiler pipeline (http://cellprofiler.org) and dimensionality reduction was performed on StratomineR high content analysis platform (http://corelifeanalytics.com). Outputs were analysed/visualised using Spotfire Analyst (Tibco), GraphPad Prism (http://graphpad.com) and Morpheus (http://software.broadinstitute.org/morpheus/).
Cytokine microarray
Forward-phase protein array basal cytokine analysis. Conditioned medium was collected after 72 h incubation. Microarrays were generated using an in-house Aushon BioSystems 2470 array printing platform. The arrays were blocked for 1 h with SuperG Blocking Buffer (Grace Bio Labs) at rt. Conditioned media from glioma stem cell samples were centrifuged at 1000 × g for 5 min at 4 °C. Supernatants were collected and added to microarrays followed by incubation for 12 h at 4 °C. The arrays were washed three times for 5 min in TBS-Tween (0.1% v/v) and blocked for 10 min with SuperG Blocking Buffer at rt on an orbital shaker, then washed again washed three times for 5 min in TBS-Tween (0.1% v/v). Detection antibody mixtures (1:500 antibody dilutions in 5% bovine serum albumin/phosphate buffered saline Tween-20 0.1% (BSA/PBST), 1% SuperG Blocking Buffer) were made in microplates. Microarrays were clamped and 50 μL of each antibody was added to corresponding microarray wells. Microarrays were incubated for 1 h on a flat surface. Clamps were removed and microarrays were washed three times for 5 min in TBS-Tween (0.1% v/v). Microarrays were then blocked for 10 min with SuperG Blocking Buffer at room temperature and again washed three times for 5 min in PBST. 3 mL of IRDye 800CW streptavidin (LI-COR Biosciences) was diluted 1:5000 in PBST supplemented with 5% BSA, 1% SuperG Blocking Buffer. Microarrays were covered and incubated with IRDye on a rocker at room temperature for 45 min, then washed for 5 min, three times in PBST followed by three 5 min PBS washes and finally washed with distilled water. Microarrays were dried then scanned on the InnoScan 710 high-resolution microarray scanner (Innopsys Life Sciences). The data was normalised for protein concentration and background fluorescence via Microsoft Excel templates.
Kinome profiling
Kinase inhibition screening was performed by ThermoFischer Scientific using the ratiometric assay Z′-LYTE. 74 wild type kinases were tested at three doses (0.1, 1 and 10 μM), in duplicate. Final DMSO concentration in each well was 1%. ATP concentration was the Km app (previously determined using a Z′-LYTE assay). Maximum emission ratio (= 100% inhibition) is established in the absence of ATP (no kinase activity). 0% inhibition is established with a synthetically phosphorylated peptide of the same sequence as the substrate.
In silico docking studies
The crystal structure of CSF-1R in complex with a dihydropurinone inhibitor was downloaded from the RCSB PDB website (PDB entry 6T2W). DMSO and water molecules were removed. Avogadro was used to generate idealised 3D starting conformations of B58 using the MMFF94 force field. The docking was performed with GOLD 5.8.1. The binding site was set at the same location of the dihydropurinone inhibitor and the size of the binding site enlarged 10 Å around it. “Chemscore_kinase” was used as docking protocol and Piecewise Linear Potential (ChemPLP) as the fitness function. 50 poses of B58 were generated and the high-scored representative and consistent docking result was shown in Fig. 8c and the ESI.†
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
We are grateful to Medical Research Scotland (PHD-1046-2016) for funding and a joint Cancer Research UK (A28596) and The Brain Tumour Charity award (GN-000676) to N. O. C. T. V. thanks EU Horizon 2020 research and innovation programme under the Marie Skłodowska Curie grant agreement No. 749299. We also acknowledge Professor Steven Pollard and Dr Gillian Morrison and the CRUK Glioma Cellular Genetic resource for the provision of patient derived GSCs. We also thank Kenneth G. Macleod for his assistance with performing cytokine microarray. We also thank MSD for funding the kinome screening.
Notes and references
- D. C. Swinney, Clin. Pharmacol. Ther., 2013, 93, 299–301 CrossRef CAS PubMed.
- C. H. Emmerich, L. M. Gamboa, M. C. J. Hofmann, M. Bonin-Andresen, O. Arbach, P. Schendel, B. Gerlach, K. Hempel, A. Bespalov, U. Dirnagl and M. J. Parnham, Nat. Rev. Drug Discovery, 2021, 20, 64–81 CrossRef CAS PubMed.
- S. J. Warchal, A. Unciti-Broceta and N. O. Carragher, Future Med. Chem., 2016, 8, 1331–1347 CrossRef CAS PubMed.
- W. Zheng, N. Thorne and J. C. McKew, Drug Discovery Today, 2013, 18, 1067–1073 CrossRef CAS PubMed.
- F. Vincent, A. Nueda, J. Lee, M. Monica Schenone, M. Prunotto and M. Mercola, Nat. Rev. Drug Discovery, 2022, 21, 899–914 CrossRef CAS PubMed.
- B. Oronsky, T. R. Reid, A. Oronsky, N. Sandhu and S. J. Knox, Front. Oncol., 2021, 10, 574012 CrossRef PubMed.
- W. Wu, J. L. Klockow, M. Zhang, F. Lafortune, E. Chang, L. Jin, Y. Wu and H. E. Daldrup-Link, Pharmacol. Res., 2021, 171, 105780 CrossRef CAS PubMed.
- A. E. Kassab, Arch. Pharm., 2023, 356, e2200424 CrossRef PubMed.
- M. A. Abdelgawad, N. A. A. Elkanzi, A. Musa, N. H. Alotaibi, W. A. A. Arafa, S. M. Gomha and R. B. Bakr, Arabian J. Chem., 2022, 15, 103781 CrossRef CAS.
- D. J. Baillache and A. Unciti-Broceta, RSC Med. Chem., 2020, 11, 1112–1135 RSC.
- P. N. Munot, S. Mirgh, N. Mehra and B. P. Bagal, Cancer Research, Statistics, and Treatment, 2020, 3, 767–784 CrossRef.
- C. C. Ayala-Aguilera, T. Valero, A. Lorente-Macías, D. J. Baillache, S. Croke and A. Unciti-Broceta, J. Med. Chem., 2022, 65, 1047–1131 CrossRef CAS PubMed.
- S. H. Myers, C. Temps, D. R. Houston, V. G. Brunton and A. Unciti-Broceta, J. Med. Chem., 2018, 61, 2104–2110 CrossRef CAS PubMed.
- O. Cruz-López, C. Temps, B. Longo, S. H. Myers, F. Franco-Montalban and A. Unciti-Broceta, ACS Omega, 2020, 4, 21620–21626 CrossRef PubMed.
- C. Fraser, N. O. Carragher and A. Unciti-broceta, Med. Chem. Commun., 2016, 7, 471–477 RSC.
- C. Fraser, J. C. Dawson, R. Dowling, D. R. Houston, J. T. Weiss, A. F. Munro, M. Muir, L. Harrington, S. P. Webster, M. C. Frame, V. G. Brunton, E. E. Patton, N. O. Carragher and A. Unciti-Broceta, J. Med. Chem., 2016, 59, 4697–4710 CrossRef CAS PubMed.
- C. Temps, D. Lietha, E. R. Webb, X.-F. Li, J. C. Dawson, M. Muir, K. G. Macleod, T. Valero, A. F. Munro, R. Contreras-Montoya, J. R. Luque-Ortega, C. Fraser, H. Beetham, C. Schoenherr, M. Lopalco, M. J. Arends, M. C. Frame, B.-Z. Qian, V. G. Brunton, N. O. Carragher and A. Unciti-Broceta, Cancer Res., 2021, 81, 5438–5450 CrossRef CAS PubMed.
- M. I. El-Gamal, S. K. Al-Ameen, D. M. Al-Koumi, M. G. Hamad, N. A. Jalal and C. H. Oh, J. Med. Chem., 2018, 61, 5450–5466 CrossRef CAS PubMed.
- S. Chockalingam and S. S. Ghosh, Tumor Biol., 2014, 35, 10635–10644 CrossRef CAS PubMed.
- J. Wen, S. Wang, R. Guo and D. Liu, Eur. J. Med. Chem., 2023, 245, 114884 CrossRef CAS PubMed.
- K. N. Green, J. D. Crapser and L. A. Hohsfield, Trends Immunol., 2020, 41, 771–784 CrossRef CAS PubMed.
- N. Butowski, H. Colman, J. F. De Groot, A. M. Omuro, L. Nayak, P. Y. Wen, T. F. Cloughesy, A. Marimuthu, S. Haidar, A. Perry, J. Huse, J. Phillips, B. L. West, K. B. Nolop, H. H. Hsu, K. L. Ligon, A. M. Molinaro and M. Prados, Neuro. Oncol., 2016, 18, 557–564 CrossRef PubMed.
- B. Apsel, J. Blair, B. Gonzalez, T. M. Nazif, M. E. Feldman, B. Aizenstein, R. Hoffman, R. L. Williams, K. M. Shokat and Z. A. Knight, Nat. Chem. Biol., 2008, 4, 691–699 CrossRef CAS PubMed.
- J. Liu, C. Yang, C. Simpson, D. Deryckere, A. Van Deusen, M. J. Miley, D. Kireev, J. Norris-Drouin, S. Sather, D. Hunter, V. K. Korboukh, H. S. Patel, W. P. Janzen, M. Machius, G. L. Johnson, H. S. Earp, D. K. Graham, S. V. Frye and X. Wang, ACS Med. Chem. Lett., 2012, 3, 129–134 CrossRef CAS PubMed.
- J. Liu, W. Zhang, M. A. Stashko, D. Deryckere, C. T. Cummings, D. Hunter, C. Yang, C. N. Jayakody, N. Cheng, C. Simpson, J. Norris-Drouin, S. Sather, D. Kireev, W. P. Janzen, H. S. Earp, D. K. Graham, S. V. Frye and X. Wang, Eur. J. Med. Chem., 2013, 65, 83–93 CrossRef CAS PubMed.
- W. Zhang, D. DeRyckere, D. Hunter, J. Liu, M. A. Stashko, K. A. Minson, C. T. Cummings, M. Lee, T. G. Glaros, D. L. Newton, S. Sather, D. Zhang, D. Kireev, W. P. Janzen, H. S. Earp, D. K. Graham, S. V. Frye and X. Wang, J. Med. Chem., 2014, 57, 7031–7041 CrossRef CAS PubMed.
- C. Wang, H. Liu, Z. Song, Y. Ji, L. Xing, X. Peng, X. Wang, J. Ai, M. Geng and A. Zhang, Bioorg. Med. Chem. Lett., 2017, 27, 2544–2548 CrossRef CAS PubMed.
- S. A. Ramachandran, P. S. Jadhavar, M. P. Singh, A. Sharma, G. N. Bagle, K. P. Quinn, P. Y. Wong, A. A. Protter, R. Rai, S. M. Pham and J. N. Lindquist, Bioorg. Med. Chem. Lett., 2017, 27, 750–754 CrossRef CAS PubMed.
- S. L. Degorce, M. S. Bodnarchuk, I. A. Cumming and J. S. Scott, J. Med. Chem., 2018, 61, 8934–8943 CrossRef CAS PubMed.
- T. P. Heffron, J. Med. Chem., 2016, 59, 10030–10066 CrossRef CAS PubMed.
- P. Dinér, J. P. Alao, J. Söderlund, P. Sunnerhagen and M. Grøtli, J. Med. Chem., 2012, 55, 4872–4876 CrossRef PubMed.
- M. Allen, M. Bjerke, H. Edlund, S. Nelander and B. Westermark, Sci. Transl. Med., 2016, 8, 354re3 Search PubMed.
- L. N. Kiseleva, A. V. Kartashev, N. L. Vartanyan, A. A. Pinevich and M. P. Samoilovich, Cell Tissue Biol., 2016, 10, 341–348 CrossRef.
- R. Park, S. Y. Kook, J. C. Park and I. Mook-Jung, Cell Death Dis., 2014, 5, e1299 CrossRef CAS PubMed.
- T. Valero, D. J. Baillache, C. Fraser, S. H. Myers and A. Unciti-Broceta, Bioorg. Med. Chem., 2020, 28, 115215 CrossRef CAS PubMed.
- C. Adam, A. M. Pérez-López, L. Hamilton, B. Rubio-Ruiz, T. L. Bray, D. Sieger, P. M. Brennan and A. Unciti-Broceta, Chem. – Eur. J., 2018, 24, 16783–16790 CrossRef CAS PubMed.
- R. G. Verhaak, K. A. Hoadley, E. Purdom, V. Wang, Y. Qi, M. D. Wilkerson, C. R. Miller, L. Ding, T. Golub, J. P. Mesirov, G. Alexe, M. Lawrence, M. O'Kelly, P. Tamayo, B. A. Weir, S. Gabriel, W. Winckler, S. Gupta, L. Jakkula, H. S. Feiler, J. G. Hodgson, C. D. James, J. N. Sarkaria, C. Brennan, A. Kahn, P. T. Spellman, R. K. Wilson, T. P. Speed, J. W. Gray, M. Meyerson, G. Getz, C. M. Perou and D. N. Hayes, Cancer Cell, 2010, 17, 98–110 CrossRef CAS PubMed.
- S. M. Pollard, K. Yoshikawa, I. D. Clarke, D. Danovi, S. Stricker, R. Russell, J. Bayani, R. Head, M. Lee, M. Bernstein, J. A. Squire, A. Smith and P. Dirks, Cell Stem Cell, 2009, 4, 568–580 CrossRef CAS PubMed.
- S. M. Gustafsdottir, V. Ljosa, K. L. Sokolnicki, J. A. Wilson, D. Walpita, M. M. Kemp, K. P. Seiler, H. A. Carrel, T. R. Golub, S. L. Schreiber, P. A. Clemons, A. E. Carpenter and A. F. Shamji, PLoS One, 2013, 8, e80999 CrossRef PubMed.
- K. S. Metz, E. M. Deoudes, M. E. Berginski, I. Jimenez-Ruiz, B. A. Aksoy, J. Hammerbacher, S. M. Gomez and D. H. Phanstiel, Cell Syst., 2018, 7, 347–350.e1 CrossRef CAS PubMed.
- N. Hagan, J. L. Kane, D. Grover, L. Woodworth, C. Madore, J. Saleh, J. Sancho, J. Liu, Y. Li, J. Proto, M. Zelic, A. Mahan, M. Kothe, A. A. Scholte, M. Fitzgerald, B. Gisevius, A. Haghikia, O. Butovsky and D. Ofengeim, Cell Death Dis., 2020, 11, 904 CrossRef CAS PubMed.
- F. W. Goldberg, M. R. V. Finlay, A. K. T. Ting, D. Beattie, G. M. Lamont, C. Fallan, G. L. Wrigley, M. Schimpl, M. R. Howard, B. Williamson, M. Vazquez-Chantada, D. G. Barratt, B. R. Davies, E. B. Cadogan, A. Ramos-Montoya and E. Dean, J. Med. Chem., 2020, 63, 3461–3471 CrossRef CAS PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, Aust. J. Chem., 2012, 4, 17 CAS.
- Y. Natsume-kitatani, R. Ohashi, H. Komura and K. Mizuguchi, J. Med. Chem., 2023, 66, 9697–9709 CrossRef PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- M. A. Bray, S. Singh and H. Han,
et al.
, Nat. Protoc., 2016, 11, 1757–1774 CrossRef CAS PubMed.
- R. E. Hughes, R. J. R. Elliott and A. F. Munro,
et al.
, SLAS Discovery, 2020, 25, 770–782 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Methods, characterisation of intermediates and final compounds, NMR spectra of compounds B40–63, Table S1 and Fig. S1–S11. See DOI: https://doi.org/10.1039/d3md00454f |
| ‡ Current affiliation: Department of Medicinal and Organic Chemistry and Unit of Excellence in Chemistry Applied to Biomedicine and Environment, Faculty of Pharmacy, Campus Cartuja s/n, University of Granada, 18071 Granada, Spain; GENYO, Pfizer/University of Granada/Andalusian Regional Government, PTS Granada, Avda. Ilustración 114, 18016 Granada, Spain; and Instituto de Investigación Biosanitaria ibs.GRANADA, Granada, Spain. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Teresa
Valero‡
ab,
Teresa
Valero‡