Theoretical insights into NH3 absorption mechanisms with imidazolium-based protic ionic liquids†
Received
7th November 2022
, Accepted 5th January 2023
First published on 6th January 2023
Abstract
Ionic liquids (ILs) provide a promising way for efficient absorption and separation of ammonia (NH3) due to their extremely low vapor pressures and adjustable structures. However, the understanding of absorption mechanisms especially in terms of theoretical insights is still not very clear, which is crucial for designing targeted ILs. In this work, a universal method that integrates density functional theory and molecular dynamic simulations was proposed to study the mechanisms of NH3 absorption by protic ionic liquids (PILs). The results showed that the NH3 absorption performance of the imidazolium-based PILs ([BIm][X], X= Tf2N, SCN and NO3) is determined by not only the hydrogen bonding between the N atom in NH3 and the protic site (H–N3) on the cation but also the cation–anion interaction. With the increase in NH3 absorption capacity, the hydrogen bonding between [BIm][Tf2N] and NH3 changed from orbital dominated to electrostatic dominated, so 3.0 mol NH3 per mol IL at 313.15 K and 0.10 MPa was further proved as a threshold for NH3 capacity of [BIm][Tf2N] by the Gibbs free energy results, which agrees well with the experimental results. Furthermore, the anions of [BIm][X] could also compete with NH3 for interaction with H-N3 of the cation, which weakens the interaction between the cation and NH3 and then decreases the NH3 absorption ability of PILs. This study provides further understanding on NH3 absorption mechanisms with ILs, which will guide the design of novel functionalized ILs for NH3 separation and recovery.
Keywords: Protic ionic liquids; NH3 absorption; Interaction mechanisms; Simulation calculations.
1. Introduction
Ammonia (NH3) mainly comes from various industrial emissions, such as in synthetic ammonia plants, melamine tail gas, urea granulating tower tail gas, metallurgical tail gas, and so on. The emission of NH3 into the atmosphere causes serious environmental issues.1–4 Therefore, the removal and recovery of NH3 from these gases is significant to satisfy environmental emission regulations and for resource reuse. Recently, ionic liquids (ILs) have attracted considerable attention as environmentally benign solvents for NH3 absorption and recovery due to their peculiar properties, such as negligible vapor pressures, tunable properties and high thermal stability.5–9 Various functionalized ILs, such as hydroxyl ILs,10–12 metal ILs,13–16 and protic ILs (PILs)17,18 were reported for efficient NH3 absorption. For instance, Shang et al. designed a kind of PIL with a strong hydrogen bond donating ability on cations, such as [BIm][Tf2N] with a high NH3 absorption capacity of 2.69 mol NH3 (mol IL)−1 at 313.15 K and 0.10 MPa, along with great selectivity and excellent recyclability.19 Luo et al. also designed an efficient PIL, [2PyH][Tf2N], by varying the cooperative hydrogen bonding, and the NH3 absorption capacity was high—up to 3.80 mol NH3 (mol IL)−1 at 303.15 K and 0.10 MPa.18 In addition, the elucidation of the mechanism of gas absorption is crucial for the design of targeted ILs, especially in terms of theoretical perspectives.20,21
In 2009, Shi et al. published the first work on molecular dynamic (MD) simulations of NH3 absorption by the IL [EMIm][Tf2N]. According to the analysis of the interaction and radial distribution function of NH3⋯cation and NH3⋯anion, they concluded that the NH3 absorption capacity of [EMIm][Tf2N] is governed by the hydrogen bonding between NH3 and cations.22 Inspired by their conclusions, a series of promising ILs, such as hydroxyl ILs and PILs, were designed to absorb NH3 efficiently by introducing effective sites that interact with NH3, and theoretical studies were performed to verify their ideas. For example, Li et al. performed density functional theory (DFT) simulations to confirm that the higher NH3 absorption capacity of a hydroxyl-functionalized IL than that of the analogous IL without the introduction of a hydroxyl group is mainly due to the hydrogen bonding between the hydroxyl group of the cation and the basic N atom of NH3.10 Cai et al. also conducted DFT simulations to verify the mechanism of NH3 absorption in lithium (Li)-triethylene glycol (TEG)-chelated ILs and their results indicated that the high NH3 capacity of ILs is due to the strong interaction between hydroxyl sites of the cation and NH3.23 Similarly, the DFT calculation results of our previous work19 also indicated that NH3 absorption of [BIm][Tf2N] may be mainly affected by the protic hydrogen of the cation. Further, MD simulations were conducted to confirm that NH3 can compete with [Tf2N]− for interaction with the protic hydrogen of the cation, forming a strong hydrogen bonding.24 So far, theoretical methods have attracted increasing attention to study the mechanism of NH3 absorption with ILs.
Our previous work revealed that PILs not only show impressively high NH3 absorption capacities but also their capacities are considerably affected by anions.17,19 For instance, the capacities for NH3 absorption of [BIm][Tf2N], [BIm][SCN] and [BIm][NO3] were 2.69, 1.96 and 1.30 mol NH3 (mol IL)−1 at 313.15 K and 0.10 MPa, respectively, as shown in Table 1. However, the mechanism is not clear. Here, DFT and MD simulations were conducted to further study the NH3 absorption mechanism by [BIm]+-based PILs to reveal the essence of the interaction between ILs and NH3, as well as the effect of anions on NH3 capacity. The comprehensive theoretical studies not only complement deeper understanding on NH3 absorption mechanisms by the PILs but also provide a guideline to design new efficient ILs.
Table 1 The viscosities, densities and NH3 capacities at 313.15 K and 0.10 MPa of the PILs17
| ILs |
Viscosity (mPa s) |
Density (g L−1) |
NH3 capacity (mol NH3 (mol IL)−1) |
| [BIm][Tf2N] |
47.14 |
1460.24 |
2.69 |
| [BIm][SCN] |
84.30 |
1078.66 |
1.96 |
| [BIm][NO3] |
136.95 |
1165.50 |
1.30 |
2. Results and discussion
2.1 The interaction between NH3 and [BIm][Tf2N]
The optimized structures that illustrate [BIm][Tf2N] interacting with one, two, three and four NH3 molecules are demonstrated in Fig. 2. The results showed that the NH3 molecule mainly interacts with the site of protic hydrogen (H–N3, the atom labels are provided in Fig. 1) of [BIm][Tf2N] via hydrogen bonding. For example, the distance between N of the 1st NH3 and H of H–N3 was 1.7 Å (Fig. 2a), which is less than the sum of the van der Walls radii of N and H atoms.25 On the other hand, hydrogen bonding was also discovered between NH3 and the anion [Tf2N]−, as shown in Fig. 2a–d. For [BIm][Tf2N]⋯NH3, the interaction energy (ΔE) between the PIL and NH3 was −17.30 kcal mol−1 when the NH3 was located around H–N3 (Fig. 2a), which is almost twice as those of NH3 interacting with H–C2/4/5 (see Fig. S1†). For [BIm][Tf2N]⋯2NH3, the most stable structure was obtained when the 2nd NH3 molecule connected to the 1st NH3 molecule (see Fig. 2b). The ΔE between the IL and two NH3 molecules was −27.30 kcal mol−1, implying that the ΔE for the 2nd NH3 molecule was −10.00 kcal mol−1. Interestingly, the distance between the N atom of the 1st NH3 molecule and H–N3 decreased from 1.7 Å to 1.6 Å while the 2nd NH3 molecule was absorbed. In other words, the hydrogen bonding formed between the 1st NH3 molecule and H–N3 was enhanced. A similar phenomenon was discovered from the interactions between H2O molecules and [EIm][Tf2N], which means that the distance between the O atom of the 1st H2O molecule and H–N3 of [EIm]+ is shortened when the 2nd H2O molecule interacts with the 1st H2O molecule.26
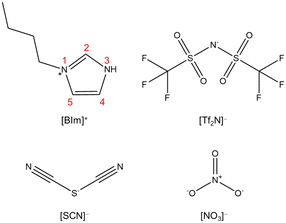 |
| | Fig. 1 Structures of the cation and anions studied in this work. | |
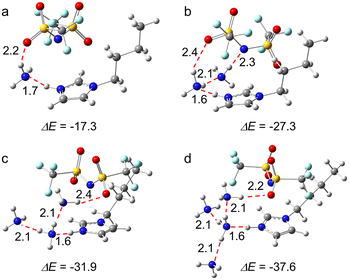 |
| | Fig. 2 The optimized structures of [BIm][Tf2N] with (a) one, (b) two, (c) three, and (d) four NH3 molecules. (Color of ligand: N, blue; H, gray; C, dark gray; S, yellow; O, red; F, pale turquoise. All the distances are given in Å. ΔE = E(IL⋯nNH3) − E(IL) – n × E(NH3)). | |
The structures of [BIm][Tf2N] interacting with three and four NH3 molecules are demonstrated in Fig. 2c and d, respectively. It was found that the rest of NH3 molecules can interact with the 1st absorbed NH3 molecule. It is worth noting that the calculated ΔE of absorption of the 1st and 2nd NH3 molecules is much larger than that of the 3rd and 4th NH3 molecules, i.e., −17.30 and −10.00 kcal mol−1versus −4.60 and −5.70 kcal mol−1. The findings revealed that the 1st and 2nd NH3 molecules are absorbed via different types of interactions compared to the 3rd and 4th NH3 molecules. The former two are through strong hydrogen bonding while the latter two are dominated by VdW interactions. The essence of these interactions was further analyzed via energy decomposition analysis (EDA), and detailed discussions are given in the next sections.
2.2 Energy decomposition analysis of NH3 absorption with [BIm][Tf2N]
In order to present a quantitative interpretation and more details of the interactions between [BIm][Tf2N] and NH3, EDA was performed. Within the EDA scheme, the total interaction energy consists of three meaningful physical terms, namely, the repulsive exchange (Pauli) interaction ΔEPauli, the electrostatic interaction ΔEelstat and the orbital interaction ΔEorb, which includes the orbital relaxation and orbital mixing between the fragments.27,28 In addition, the summation of Pauli repulsion and electrostatic interactions is often defined as the steric interaction ΔEsteric, which arises from the fact that each atom in a molecule occupies a certain amount of space. The total and the individual interactions of the process for each NH3 absorbed by [BIm][Tf2N] are given in Table 2 and Fig. 3. The results showed that the interactions between the 1st and 2nd NH3 molecules with [BIm][Tf2N] are considerable, especially for the 1st NH3 molecule, and its total interaction energy is as high as −16.79 kcal mol−1. However, the total interaction energies of the 3rd and 4th NH3 molecules are much lower than those of the 1st and 2nd NH3 molecules, which are around −3.50 kcal mol−1. The 3rd NH3 molecule is favored by electrostatic interactions of 8.16 kcal mol−1 compared to the 4th NH3 molecule. This is most likely because the 3rd NH3 molecule interacts with the anion through hydrogen bonding (see Fig. 2c).
Table 2 EDA at the M062X/TZP level for each step ([BIm][Tf2N]⋯(i-1) NH3 + NH3 →[BIm][Tf2N]⋯iNH3, i = 1–4) of [BIm][Tf2N] to capture one NH3 molecule, and all the energies are expressed in kcal mol−1
| Interaction energiesa |
1st NH3 |
2nd NH3 |
3rd NH3 |
4th NH3 |
|
ΔE = E(IL⋯nNH3) − E(IL⋯(n-1)NH3) – E(NH3).
|
| ΔEPauli |
+92.02 |
+72.05 |
+61.64 |
+55.71 |
| ΔEelstat |
−29.76 |
−16.79 |
−15.87 |
−7.71 |
| ΔEsteric (ΔEPauli + ΔEelstat) |
+62.26 |
+55.26 |
+45.77 |
+48.00 |
| ΔEorb |
−79.05 |
−64.40 |
−49.58 |
−51.54 |
| ΔEtotal |
−16.79 |
−9.14 |
−3.81 |
−3.54 |
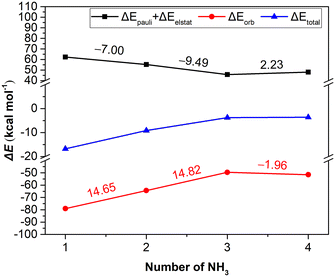 |
| | Fig. 3 The EDA results at the M062X/TZP level of theory for each step of [BIm][Tf2N] to capture one NH3. The numbers on the lines indicate the changes of values of adjacent two points, and their colours are consistent with the lines. | |
The plots in Fig. 3 demonstrate that the orbital interaction is more likely to decay than the steric interaction. For example, the orbital interaction decreased by 14.65 kcal mol−1 from the 1st to the 2nd NH3 molecule, while the corresponding steric interactions only decreased by 7.00 kcal mol−1. The results indicated that the orbital interaction makes a great contribution to the total interaction. However, in the case of the 4th NH3 molecule, the changes of ΔEsteric and ΔEorb were small compared to those of the 3rd NH3 molecule, which is most likely due to physical interaction of the 4th NH3 molecule. In this case, the electrostatic interaction plays the most important role.
2.3 Gibbs free energy of NH3 absorption with [BIm][Tf2N]
Our previous experimental results16 showed that the NH3 absorption capacity of [BIm][Tf2N] is 2.69 mol NH3 (mol IL)−1, which is close to 3.0 mol NH3 (mol IL)−1 at 313.15 K and 0.10 MPa. The high capacity for NH3 of [BIm][Tf2N] was probably attributed to the strong hydrogen bonding site H–N3 in the [BIm]+ cation. Here, we revisited this open question by computing the Gibbs free energy (ΔG) for [BIm][Tf2N] absorption of different moles of NH3 molecules. The calculated ΔG values are shown in Fig. 4, and their values are highlighted in red. The ΔG values for [BIm][Tf2N] absorbing one and two moles NH3 were −3.6 and −3.1 kcal mol−1, respectively. These two negative values indicated that the process of [BIm][Tf2N] absorbing one or two moles NH3 takes place spontaneously and easily.
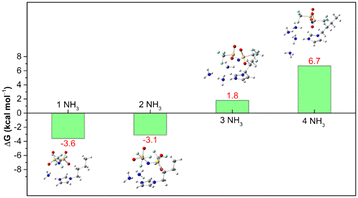 |
| | Fig. 4 ΔG of [BIm][Tf2N] + iNH3 → [BIm][Tf2N]⋯iNH3, (i = 1–4) calculated at the M062X/def2TZVP level, including the B3LYP/6-31 + G* thermodynamic correction. | |
The ΔG value for [BIm][Tf2N] absorbing three moles NH3 was 1.8 kcal mol−1. This slightly positive value reveals that the absorption of three moles NH3 by [BIm][Tf2N] is thermodynamically feasible. Moreover, the other conformers of [BIm][Tf2N]⋯3NH3 are shown in Fig. S3.† The relative energies (Er), ΔG values and Boltzmann distribution weighting factors (wi) are summarized in Table S1.† The results indicated that most of the conformers have comparable Er values and their ΔG values are close to zero as well. It should be noted that the most stable structure of [BIm][Tf2N]⋯3NH3 (Fig. S3a†) is slightly more stable than the structure in Fig. 2c (equal to Fig. S3d†). The energy of the former was 1.1 kcal mol−1 lower than the latter. Moreover, the most stable structure was dominant among the displayed conformers with a wi value of 0.432, and its corresponding ΔG value was −0.05 kcal mol−1. The result indicated that [BIm][Tf2N] is capable of absorbing close to 3 moles NH3 in consideration of thermodynamics. It is also consistent with the experimental fact that the NH3 capacity of H–C2 is less than 1.0 mol NH3 (mol IL)−1.5,6,17
We also calculated the ΔG value for the [BIm][Tf2N]⋯4NH3 system. The DFT calculations yielded a large positive value of 6.7 kcal mol−1 for the hypothetical process, indicating that [BIm][Tf2N] failed to absorb 4 moles NH3 spontaneously at 313.15 K and 0.10 MPa. Furthermore, the energy of the most stable structure of [BIm][Tf2N]⋯4NH3 (Fig. S4a†) was 3.8 kcal mol−1 lower than that of the structure shown in Fig. 2d (equal to Fig. S4t†), but the ΔG value of [BIm][Tf2N] absorbed 4 moles NH3 molecules based on the most stable structure of [BIm][Tf2N]⋯4NH3 (2.5 kcal mol−1), which indicated that this structure is also unfavorable from a thermodynamic point of view.
2.4 The interaction between anions and NH3
Based on the above analysis, it was concluded that H–N3 plays an important role in NH3 absorption of [BIm][Tf2N]. However, the experimental results (see Table 1) also indicated that the anions have an observable influence on NH3 capacity of PILs. To reveal the possible mechanisms, the interactions between anions and NH3 were also studied by DFT calculations. As seen in Fig. 5, all the anions interacted with NH3via hydrogen bonding. The interaction energy between [NO3]− and NH3 was the largest one among them, while the difference of interaction energies was less than 3.0 kcal mol−1. Although the anion [Tf2N]− offered more hydrogen bond acceptors than the other two anions, it seemed impossible to cause such a great difference in NH3 capacity of PILs (NH3 capacity of [BIm][Tf2N] is almost twice that of [BIm][NO3] in Table 1). Furthermore, in terms of interaction energies, [BIm][NO3] should show a more likely higher NH3 capacity than [BIm][SCN], which conflicts with experimental results. In sum, the interaction between NH3 and anions is not the direct reason that affects NH3 absorption capacity of PILs.
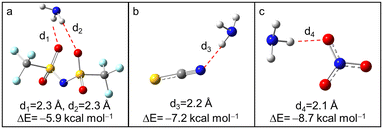 |
| | Fig. 5 The optimized structures of NH3 interact with (a) [Tf2N]−, (b) [SCN]− and (c) [NO3]− (color of ligand: N, blue; H, gray; C, dark gray; S, yellow; O, red; F, pale turquoise). | |
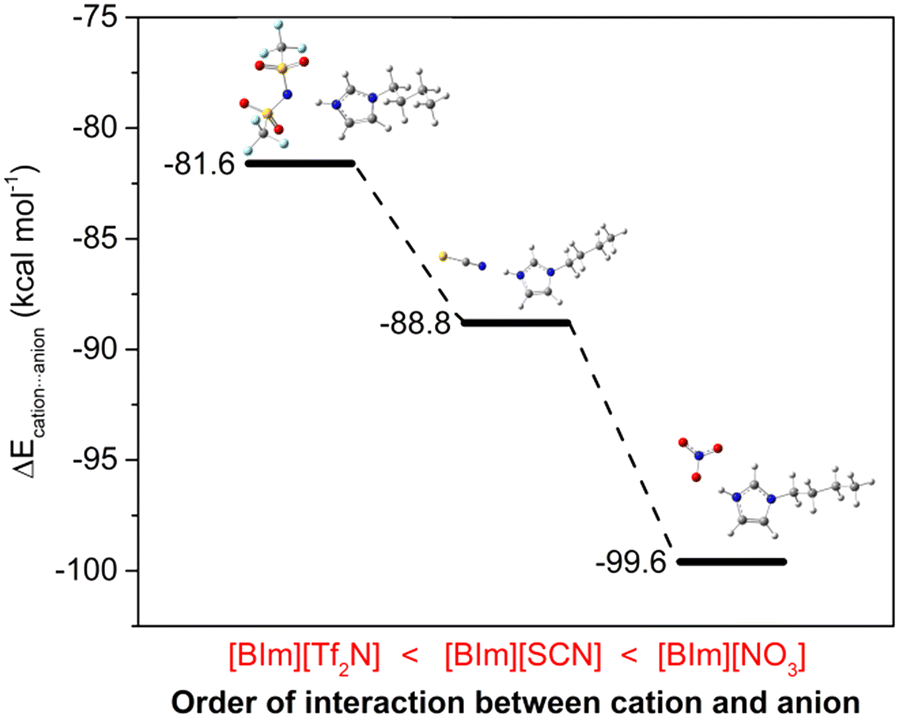
2.5 The interaction between cations and anions
The H–N3 site is a main hydrogen bond donor that interacts with NH3, and then the anions may also provide hydrogen bond acceptors to form hydrogen bonding with the protic site, therefore influencing the NH3 absorption capacity of PILs. In order to verify this assumption, DFT calculations were conducted to study the interactions between cations and anions. The most stable structures for these three “ion pairs” of [BIm][Tf2N], [BIm][SCN] and [BIm][NO3] are depicted in Fig. 6. In addition, the interaction energies between cations and anions are indicated as well (the detailed values are in Fig. S10†). It was found that all the anions prefer to interact with H–N3, meaning that the anions compete against NH3 for H–N3. It was implied that the stronger interaction between cations and anions results in a lower NH3 absorption capacity. As seen in Fig. 6, the interaction energies between the cations and anions of these PILs followed the order [BIm][Tf2N] < [BIm][SCN] < [BIm][NO3]. Moreover, the increasing interaction between cations and anions would decrease the free volume in PILs that accommodates NH3, decreasing the NH3 capacity of PILs.29 Therefore, the reason for the anions influencing NH3 capacity of PILs originates from the strength of interaction between anions and cations.
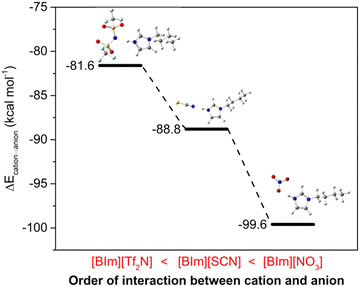 |
| | Fig. 6 The interaction energies between cations and anions of PILs. A more negative value indicates a stronger interaction (color of ligand: N, blue; H, gray; C, dark gray; S, yellow; O, red; F, pale turquoise). | |
2.6 The distribution of hydrogen bonding around [BIm]+
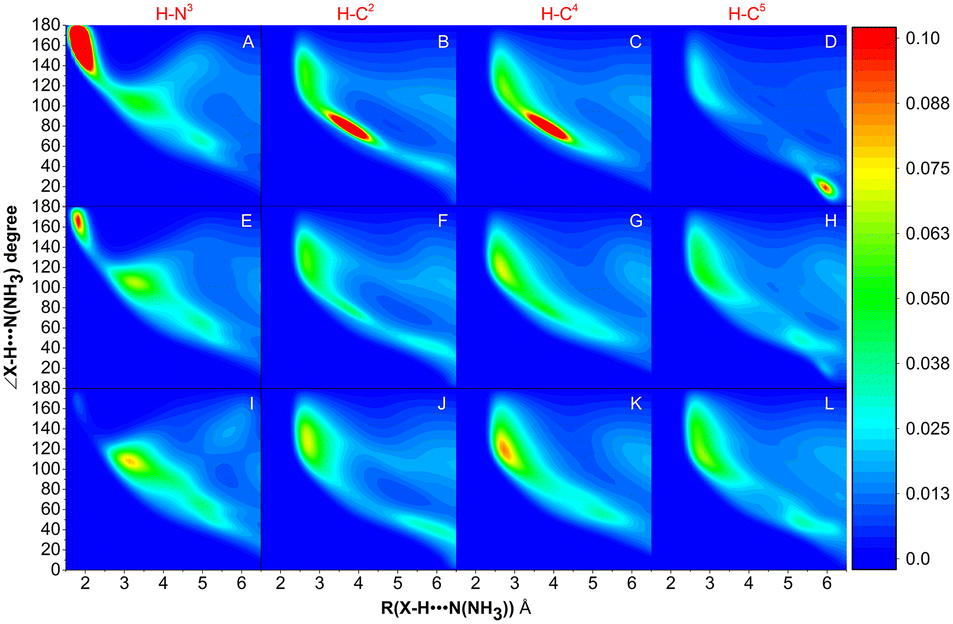
The MD simulations of NH3 absorption of ILs were further carried out to study the distribution of NH3 and anions in PILs. Fig. 7 shows the combined distribution functions of ∠X–H⋯N(NH3) versus the distance between the H atom of the cation and the N atom of NH3 (RH⋯N(NH3)). X refers to C (i.e., C2-site, C4-site, and C5-site, indicated as C2, C4, and C5, respectively) and N (i.e., N3-site, indicated as N3) atoms on the imidazolium ring of [BIm]+. It was found that NH3 molecules are mainly distributed around H–N3 in [BIm][Tf2N] (with a large region in red color, panel A). In panel A, the red region represented the range where 130° < ∠N–H⋯N(NH3) < 180° and 1.5 Å < RH⋯N(NH3) < 2.0 Å, which indicated that most of the NH3 molecules interact with H–N3 by strong hydrogen bonding. A similar red region was also found in panel E, but its area was much less than that of [BIm][Tf2N]. It was demonstrated that part of NH3 molecules distribute around H–N3 in [BIm][SCN] by strong hydrogen bonding. However, it was hard to find any red region in panel I, which means that few NH3 molecules distribute around H–N3 in [BIm][NO3] by strong hydrogen bonding. In addition, regions with red color were also found in panels B–D. These are possible indications that H–C2, H–C4, and H–C5 are sharing part of NH3 molecules around H–N3.
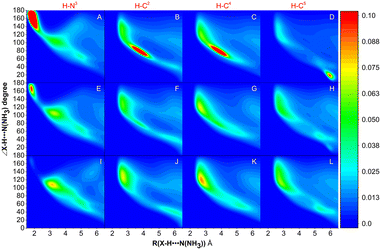 |
| | Fig. 7 Combined distribution functions of ∠N/C–H⋯N(NH3) versus the distance of H⋯N(NH3) between the cation and NH3. The first column of panels A, F, and H reflects the distribution of NH3 around H–N3. The 2nd to 4th columns of the panels reflect the distribution of NH3 around H–C2, H–C4, and H–C5, respectively. Panels A–D, E–H and I–L are combined distribution functions for NH3 absorption by [BIm][Tf2N], [BIm][SCN] and [BIm][NO3], respectively. | |
2.7 The distribution of anions around the [BIm]+ cation
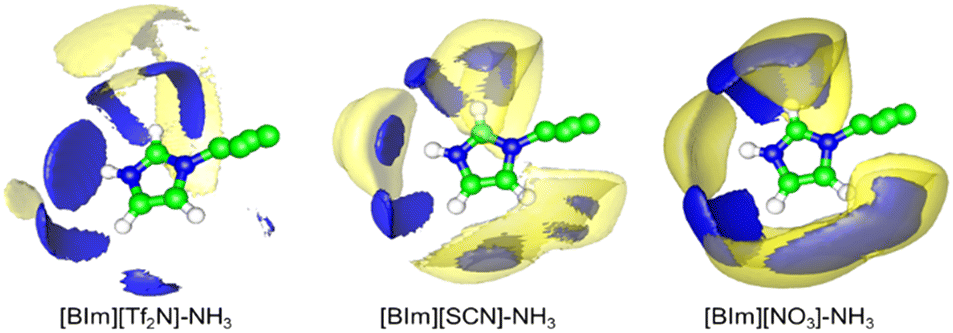
To study the distribution of anions around H–N3 of the cation, the radial distribution functions (RDFs) before and after NH3 absorption of H–N3(cation)⋯N/C(anion) and the three-dimensional probability distribution of NH3, and the anions around the [BIm]+ cation in PILs after NH3 absorption were calculated as shown in Fig. 8 and 9, respectively. The results showed that the distance of the first evident peak of RDFs for the three PILs follows the order [BIm][NO3] < [BIm][SCN] < [BIm][Tf2N]. The [NO3]− anions were organized closer, and more [SCN]− anions were around H–N3, which leads to a smaller probability for NH3 molecules interacting with H–N3 in [BIm][NO3] and a lower NH3 capacity. However, for the [Tf2N]− anion, it was the most far away from H–N3 among the three anions. Consequently, [BIm][Tf2N] provided more opportunities for H–N3 to attract NH3 and more free volume to accommodate NH3.30,31 Interestingly, the RDFs of [BIm][Tf2N] before and after NH3 absorption exhibited more evident changes than those of the other two PILs. This is likely because the [Tf2N]− anion has the largest size among these three anions, so it is vulnerable to be attacked by NH3 resulting in considerable spatial displacement. Fig. 8 also shows that the anion [Tf2N]− keeps away from H–N3 in the bulk phase of [BIm][Tf2N] with NH3 absorption compared to the bulk phase without NH3 absorption.
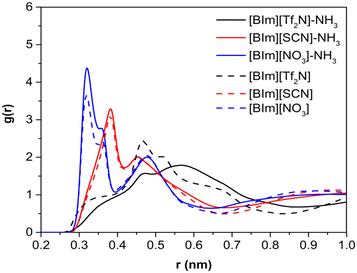 |
| | Fig. 8 RDF presentation between the H atom of H–N3 in [BIm]+ and the central atom in anions (the N atom is assigned as the central atom for [Tf2N]− and [NO3]−, and the C atom is assigned as the central atom for [SCN]−). | |
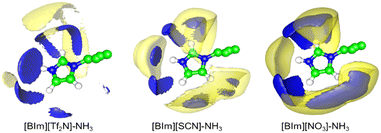 |
| | Fig. 9 Three-dimensional probability distribution of NH3 (blue) and anions (yellow) around [BIm]+ in the bulk liquid of ILs after NH3 absorption. In each case, the blue and yellow surfaces are drawn at 4 times the average density of the corresponding NH3 or anions in the bulk region. | |
3. Conclusions
In this work, both DFT calculations and MD simulations were conducted to deeply understand the mechanisms of NH3 absorption with PILs, including [BIm][Tf2N], [BIm][SCN] and [BIm][NO3], in terms of theoretical insights. The interactions between [BIm][Tf2N] and NH3, anions and NH3, cations and anions, and the NH3 distribution around H–N3 and H–C2/4/5 in [BIm]+, as well as the anion organization around H–N3 before and after NH3 absorption for the three PILs, were systematically investigated. The results indicated that both the strong hydrogen bonding between NH3 and H–N3 of the cation and the weak interaction between cations and anions lead to a high NH3 capacity of the PILs. Moreover, the orbital moiety of the total interaction between NH3 and [BIm][Tf2N] decays dramatically with the increase of NH3 absorption, and the major part changed from orbital to electrostatic, which further proved that 3.0 mol NH3 (mol IL)−1 is a threshold for the NH3 capacity of [BIm][Tf2N]. On the other hand, the anions of the PILs could also compete with NH3 for interaction with H–N3 of the cations, when the interaction between the cations and anions became stronger, the corresponding anions would prefer to gather near or around the H–N3 of the cation and decreased the probability of H–N3 to interact with NH3, thus reducing the NH3 absorption capacity of the PILs. This study provides a deep understanding of the mechanism of NH3 absorption with ILs, which will guide the design of novel functionalized ILs for NH3 separation and recovery applications.
4. Computational details
4.1 Density functional theory (DFT) calculations
The structures of PILs and PIL-NH3 were first optimized at the B3LYP/6-31 + G* theoretical level in consideration of solvation via polarizable continuum mode (PCM),32 together with the employment of the DFT-D3 approach to consider the interaction of dispersion.33 Frequency analyses were performed at the same level to confirm the optimized structures: no imaginary frequencies were found for the local minima. Then single point calculations were conducted for optimized structures at the M062X/def2-TZVP level to obtain the energy in the ideal gas phase. The zero point energies (ZPE) were obtained from the frequency calculation at the B3LYP-D3/6-31 + G* level. When applying PCM, the solvent parameters of ILs are necessary. The dielectric constant and refractive index of the ILs studied were estimated to be 15.0 and 1.43, respectively, based on published works.34–36 All the above calculations were performed by employing the Gaussian09 package.37 The Amsterdam density functional (ADF) suite of programs was employed to perform energy decomposition analysis (EDA).38,39 EDA was conducted via single point calculations at the M062X level with an all-electron basis set of TZP quality.40
The interaction energy (ΔE), which is the change in energy between the complex and its monomers, was calculated using eqn (1):
| | | ΔE = E(A⋯B) − E(A) − E(B) | (1) |
where
E(
A),
E(
B) and
E(
A⋯
B) are the ideal gas phase electronic energies of
A,
B and
A⋯
B, respectively.
The final Gibbs energy (G) in solution included two parts, i.e., the ideal gas phase electronic energy (E) and the gas phase thermostatistical contribution (GRROH) (see eqn (2)).
The change in Gibbs energy (Δ
G) for a process can be calculated by
eqn (3):
The thermostatistical contribution is calculated as follows:
| | | GRROH = ZPE + Etherm + PV − TS | (4) |
where ZPE is the zero point vibrational energy,
Etherm is the thermal energy, and
P,
V,
T, and
S denote the pressure, volume, temperature and entropy, respectively.
T and
P were set to 313.15 K and 0.10 MPa, respectively.
4.2 Molecular dynamic (MD) calculations
All the MD simulations were performed using the GROMACS simulation package41 at 313.15 K and 0.10 MPa. The time step was set to 2 fs and the velocity-Verlet algorithm was used to integrate the equations of motion. All the covalent bonds were constrained using the LINCS algorithm. A cutoff of 1.2 nm was set for both the LJ and Coulombic interactions. The LJ tail correction was added to the energy and pressure, and long-range electrostatic interactions were calculated using the particle-mesh Ewald (PME) method.42 Cross interactions between different atom types were computed using standard Lorentz–Berthelot rules. The temperature was controlled using a Nosé–Hoover thermostat with a coupling time constant of 0.2 ps and the pressure was maintained using a Parrinello–Rahman barostat with a coupling time of 2.0 ps. Periodic boundary conditions were applied in all directions.
The initial configurations were built with the PACKMOL package43 by randomly disposing 1000 ILs for pure IL systems and 1000 ILs + m NH3 (m = 3000, 2000, 1500 for [BIm][Tf2N], [BIm][SCN] and [BIm][NO3], respectively) molecules. The force field values of the ILs were obtained from the literature,44–47 and the NH3 molecule was modelled using the OPLS-AA force field.48 Due to the low-diffusion of ILs, the system was heated up to 500 K for 10 ns to keep the ion mobility. The temperature was gradually decreased to 313.15 K. The production run of 100 ns was carried out in an NPT ensemble, and structure analysis was conducted based on the last 50 ns.
Abbreviation
| [BIm][Tf2N] | 1-Butyl-imidazolium bis(trifluoromethylsulfonyl)imide |
| [2PyH][Tf2N] | 2-Aminopyridinium bis(trifluoromethylsulfonyl)imide |
| [EMIm][Tf2N] | 1-Ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide |
| [BMIm][Tf2N] | 1-Butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide |
| [BMIm][SCN] | 1-Butyl-3-methylimidazolium thiocyanide |
| [BMIm][DCA] | 1-Butyl-3-methylimidazolium dicyandiamide |
| [EIm][Tf2N] | 1-Ethyl-imidazolium bis(trifluoromethylsulfonyl)imide |
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This study was financially supported by the National Key R&D Program of China (2020YFA0710200), the National Natural Science Foundation of China (22122814, 21890764 and 21838010), the Youth Innovation Promotion Association of the Chinese Academy of Sciences (2018064), and the Major Scientific and Technological Innovation Project of Shandong Province of China (2019JZZY010518).
References
- J. J. Gao, K. Wang, Y. Wang, S. H. Liu, C. Y. Zhu, J. M. Hao, H. J. Liu, S. B. Hua and H. Z. Tian, Temporal-spatial characteristics and source apportionment of PM2.5 as well as its associated chemical species in the Beijing-Tianjin-Hebei region of China, Environ. Pollut., 2018, 233, 714–724 CrossRef CAS PubMed.
- Z. X. Wang, L. W. Wang, P. Gao, Y. Yu and R. Z. Wang, Analysis of composite sorbents for ammonia storage to eliminate NOx emission at low temperatures, Appl. Therm. Eng., 2018, 128, 1382–1390 CrossRef CAS.
- P. J. He, S. Y. Wei, L. M. Shao and F. Lu, Emission potential of volatile sulfur compounds (VSCs) and ammonia from sludge compost with different bio-stability under various oxygen levels, Waste Manage., 2018, 73, 113–122 CrossRef CAS PubMed.
- S. Zeng, Y. Cao, P. Li, X. Liu and X. Zhang, Ionic liquid–based green processes for ammonia separation and recovery, Curr. Opin. Green Sustainable Chem., 2020, 25, 100354 CrossRef.
- A. Yokozeki and M. B. Shiflett, Vapor-liquid equilibria of ammonia plus ionic liquid mixtures, Appl. Energy, 2007, 84, 1258–1273 CrossRef CAS.
- A. Yokozeki and M. B. Shiflett, Ammonia solubilities in room-temperature ionic liquids, Ind. Eng. Chem. Res., 2007, 46, 1605–1610 CrossRef CAS.
- Z. Lei, C. Dai and B. Chen, Gas solubility in ionic liquids, Chem. Rev., 2014, 114, 1289–1326 CrossRef CAS PubMed.
- H. Qin, X. Hu, J. Wang, H. Cheng, L. Chen and Z. Qi, Overview of acidic deep eutectic solvents on synthesis, properties and applications, Green Energy Environ., 2020, 5, 8–21 CrossRef.
- H. Qin, Z. Song, H. Cheng, L. Deng and Z. Qi, Physical absorption of carbon dioxide in imidazole-PTSA based deep eutectic solvents, J. Mol. Liq., 2021, 326, 115292 CrossRef CAS.
- Z. Li, X. Zhang, H. Dong, X. Zhang, H. Gao, S. Zhang, J. Li and C. Wang, Efficient absorption of ammonia with hydroxyl-functionalized ionic liquids, RSC Adv., 2015, 5, 81362–81370 RSC.
- J. Palomar, M. Gonzalez-Miquel, J. Bedia, F. Rodriguez and J. J. Rodriguez, Task-specific ionic liquids for efficient ammonia absorption, Sep. Purif. Technol., 2011, 82, 43–52 CrossRef CAS.
- J. Lemus, J. Bedia, C. Moya, N. Alonso-Morales, M. A. Gilarranz, J. Palomar and J. J. Rodriguez, Ammonia capture from the gas phase by encapsulated ionic liquids (ENILs), RSC Adv., 2016, 6, 61650–61660 RSC.
- S. Zeng, L. Liu, D. Shang, J. Feng, H. Dong, Q. Xu, X. Zhang and S. Zhang, Efficient and reversible absorption of ammonia by cobalt ionic liquids through Lewis acid–base and cooperative hydrogen bond interactions, Green Chem., 2018, 20, 2075–2083 RSC.
- F. T. Kohler, S. Popp, H. Klefer, I. Eckle, C. Schrage, B. Böhringer, D. Roth, M. Haumann and P. Wasserscheid, Supported ionic liquid phase (SILP) materials for removal of hazardous gas compounds–efficient and irreversible NH3 adsorption, Green Chem., 2014, 16, 3560–3568 RSC.
- W. Chen, S. Liang, Y. Guo, X. Gui and D. Tang, Investigation on vapor–liquid equilibria for binary systems of metal ion-containing ionic liquid [bmim]Zn2Cl5/NH3 by experiment and modified UNIFAC model, Fluid Phase Equilib., 2013, 360, 1–6 CrossRef CAS.
- A. Kaftan, H. Klefer, M. Haumann, M. Laurin, P. Wasserscheid and J. Libuda, An operando DRIFTS-MS study of NH3 removal by supported ionic liquid
phase (SILP) materials, Sep. Purif. Technol., 2017, 174, 245–250 CrossRef CAS.
- D. W. Shang, L. Bai, S. J. Zeng, H. F. Dong, H. S. Gao, X. P. Zhang and S. J. Zhang, Enhanced NH3 capture by imidazolium-based protic ionic liquids with different anions and cation substituents, J. Chem. Technol. Biotechnol., 2018, 93, 1228–1236 CrossRef CAS.
- X. Y. Luo, R. X. Diu, X. Y. Chen, B. Y. Pei, J. Q. Li and C. M. Wang, Reversible construction of ionic networks through cooperative hydrogen bonds for efficient ammonia absorption, ACS Sustainable Chem. Eng., 2019, 7, 9888–9895 CrossRef CAS.
- D. W. Shang, X. P. Zhang, S. J. Zeng, K. Jiang, H. S. Gao, H. F. Dong, Q. Y. Yang and S. J. Zhang, Protic ionic liquid [Bim][NTf2] with strong hydrogen bond donating ability for highly efficient ammonia absorption, Green Chem., 2017, 19, 937–945 RSC.
- J. Ruan, X. Ye, R. Wang, L. Chen, L. Deng and Z. Qi, Experimental and theoretical study on efficient CO2 absorption coordinated by molecules and ions of DBN and 1, 2, 4-triazole formed deep eutectic solvents, Fuel, 2023, 334, 126709 CrossRef CAS.
- J. Tian and B. Liu, Ammonia capture with ionic liquid systems: A review, Crit. Rev. Environ. Sci. Technol., 2022, 52, 767–809 CrossRef CAS.
- W. Shi and E. J. Maginn, Molecular simulation of ammonia absorption in the ionic liquid 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([emim][Tf2N]), AIChE J., 2009, 55, 2414–2421 CrossRef CAS.
- Z. Cai, J. Zhang, Y. Ma, W. Wu, Y. Cao, K. Huang and L. Jiang, Chelation-activated multiple-site reversible chemical absorption of ammonia in ionic liquids, AIChE J., 2022, 68, e17632 CrossRef CAS.
- T. Zhao, S. Zeng, Y. Li, Y. Bai, L. Bai, W. Li, X. Zhang and S. Zhang, Molecular insight into the effect of ion structure and interface behavior on the ammonia absorption by ionic liquids, AIChE J., 2022, 68, e17860 CAS.
- A. V. Bondi, van der Waals volumes and radii, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- H. E. Bailey, Y. L. Wang and M. D. Fayer, Impact of hydrogen bonding on the dynamics and structure of protic ionic liquid/water binary mixtures, J. Phys. Chem. B, 2017, 121, 8564–8576 CrossRef CAS PubMed.
- M. von Hopffgarten and G. Frenking, Energy decomposition analysis, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 43–62 CAS.
- L. Liu, E. Osorio and T. Heine, The importance of dynamics studies on the design of sandwich structures: a CrB24 case, Phys. Chem. Chem. Phys., 2016, 18, 18336–18341 RSC.
- G. B. Damas, A. B. A. Dias and L. T. Costa, A quantum chemistry study for ionic liquids applied to gas capture and separation, J. Phys. Chem. B, 2014, 118, 9046–9064 CrossRef CAS PubMed.
- A. R. Shaikh, H. Karkhanechi, E. Kamio, T. Yoshioka and H. Matsuyama, Quantum mechanical and molecular dynamics simulations of dual-amino-acid ionic liquids for CO2 capture, J. Phys. Chem. C, 2016, 120, 27734–27745 CrossRef CAS.
- F. J. Liu, W. Chen, J. X. Mi, J. Y. Zhang, X. Kan, F. Y. Zhong, K. Huang, A. M. Zheng and L. L. Jiang, Thermodynamic and molecular insights into the absorption of H2S, CO2, and CH4 in choline chloride plus urea mixtures, AIChE J., 2019, 65, e16574 CrossRef.
- S. Miertus, E. Scrocco and J. Tomasi, Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects, Chem. Phys., 1981, 55, 117–129 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for
the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- V. S. Bernales, A. V. Marenich, R. Contreras, C. J. Cramer and D. G. Truhlar, Quantum mechanical continuum solvation models for ionic liquids, J. Phys. Chem. B, 2012, 116, 9122–9129 CrossRef CAS PubMed.
- X. Wang, S. Zhang, J. Yao and H. Li, The polarity of ionic liquids: relationship between relative permittivity and spectroscopic parameters of probe, Ind. Eng. Chem. Res., 2019, 58, 7352–7361 CrossRef CAS.
- M. M. Huang, Y. P. Jiang, P. Sasisanker, G. W. Driver and H. Weingartner, Static relative dielectric permittivities of ionic liquids at 25 °C, J. Chem. Eng. Data, 2011, 56, 1494–1499 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision D.01, Gaussian Inc., Wallingford CT, 2013 Search PubMed.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. F. Guerra, S. J. A. Van Gisbergen, J. G. Snijders and T. Ziegler, Chemistry with ADF, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- M. Du, C. Dai, A. Chen, X. Wu, Y. Li, Y. Liu, W. Li and M. Zhao, Investigation on the aggregation behavior of photo-responsive system composed of 1-hexadecyl-3-methylimidazolium bromide and 2-methoxycinnamic acid, RSC Adv., 2015, 5, 68369–68377 RSC.
- E. Van Lenthe and E. J. Baerends, Optimized slater-type basis sets for the elements 1-118, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS PubMed.
- D. Van der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, GROMACS: Fast, flexible, and free, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- T. Darden, D. York and L. Pedersen, Particle mesh Ewald: An N.Log(N) method for Ewald sums in large systems, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- L. Martinez, R. Andrade, E. G. Birgin and J. M. Martinez, PACKMOL: A package for building initial configurations for molecular dynamics simulations, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef CAS PubMed.
- J. N. C. Lopes, J. Deschamps and A. A. H. Padua, Modeling ionic liquids using a systematic all-atom force field, J. Phys. Chem. B, 2004, 108, 2038–2047 CrossRef CAS.
- A. S. L. Gouveia, C. E. S. Bernardes, L. C. Tome, E. I. Lozinskaya, Y. S. Vygodskii, A. S. Shaplov, J. N. C. Lopes and I. M. Marrucho, Ionic liquids with anions based on fluorosulfonyl derivatives: from asymmetrical substitutions to a consistent force field model, Phys. Chem. Chem. Phys., 2017, 19, 29617–29624 RSC.
- J. N. C. Lopes and A. A. H. Padua, Molecular force field for ionic liquids III: Imidazolium, pyridinium, and phosphonium cations; Chloride, bromide, and dicyanamide anions, J. Phys. Chem. B, 2006, 110, 19586–19592 CrossRef PubMed.
- K. I. Oh, J. H. Choi, J. H. Lee, J. B. Han, H. Lee and M. Cho, Nitrile and thiocyanate IR probes: Molecular dynamics simulation studies, J. Chem. Phys., 2008, 128, 154504–154514 CrossRef PubMed.
- W. L. Jorgensen, D. S. Maxwell and J. TiradoRives, Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The structures and coordinates of each conformer of the ILs studied, NH3, ILs combined with NH3, isolated cations or anions, and anions combined with NH3 are provided. See DOI: https://doi.org/10.1039/d2im00041e |
|
| This journal is © Institute of Process Engineering of CAS 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *abc
*abc