
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic hydrogen storage in liquid hydrogen carriers
Yuwen
Ni
,
Zhe
Han
*,
Yuchao
Chai
,
Guangjun
Wu
 and
Landong
Li
*
and
Landong
Li
*
Key Laboratory of Advanced Energy Materials Chemistry of Ministry of Education, College of Chemistry, Nankai University, Tianjin 300071, P. R. China. E-mail: zhehan@nankai.edu.cn; lild@nankai.edu.cn
First published on 22nd May 2023
Abstract
Hydrogen energy, often dubbed the “ultimate energy source”, boasts zero carbon emissions and no harmful by-products. Nevertheless, the storage and transportation of hydrogen remain significant hurdles for its commercialization and large-scale implementation. Liquid hydrogen carriers (LHC), such as cyclohexane, methylcyclohexane, N-heterocycles, methanol, and ammonia, have emerged as promising solutions in hydrogen energy conversion systems. The storage and release of hydrogen rely on molecular hydrogenation and dehydrogenation processes, which are heavily influenced by the presence of catalysts. As such, a thorough understanding of catalyst design and mechanism is essential to facilitate (de)hydrogenation reactions under milder conditions. In this review, we explore three prevalent LHC systems and the catalysts employed during (de)hydrogenation processes. While noble metal catalysts exhibit superior performance in catalytic hydrogen storage, non-noble metal catalysts have also made considerable advancements. Furthermore, some liquid organic molecules are close to commercialization, potentially providing new options for energy storage and transportation. This article aims to trigger interest in LHC research and inspire the development of innovative catalytic systems for the catalytic hydrogen storage process.’
 Yuwen Ni | Yuwen Ni received her Bachelor's degree from Nankai University in 2021. She then joined Prof. Landong Li's group as a MS student at the School of Materials Science and Engineering, Nankai University. Her current research focuses on the design and synthesis of catalysts for (de)hydrogenation. |
 Zhe Han | Zhe Han obtained his BSc in Chemistry in 2014 at Nankai University. Afterward, he pursued a PhD study with Prof. Can Li in CO2 hydrogenation with heterogeneous catalysts. He is currently a postdoctoral researcher with Prof. Landong Li at Nankai University. His research interests focus on the conversion of C1 molecules by heterogeneous catalysis. |
 Yuchao Chai | Yuchao Chai received his BS degree in chemistry from Hunan University in 2014 and PhD degree in materials physics and chemistry from Nankai University in 2019. Now he is a professor in the College of Chemistry of Nankai University. His research interest focuses on the construction and application of single-site catalysts. |
 Guangjun Wu | Guangjun Wu received his PhD degree in materials physics and chemistry from Nankai University in 2009. Now he is an associate professor in the College of Chemistry, Nankai University. His research interest focuses on the synthesis and catalytic applications of zeolites. |
 Landong Li | Landong Li received his PhD degree in physical chemistry from Nankai University in 2006. Now he is a distinguished professor in the College of Chemistry, Nankai University. Prof. Li directs comprehensive research on energy-related heterogeneous catalysis. |
Broader contextHydrogen is currently considered as the most promising renewable energy source, however the physical properties of dihydrogen molecule present significant challenges for its large-scale storage and transportation. In order to overcome these challenges, various liquid compounds, namely aromatic hydrocarbons, N-heterocycles, methanol, and ammonia-related compounds, have been explored as potential hydrogen carriers. Ideally, these liquid hydrogen carriers can absorb and release dihydrogen through reversible hydrogenation and dehydrogenation reactions under practical conditions, making them suitable for large-scale storage and transportation applications. Catalysts are indispensable in the processes of hydrogen storage and release processes. In the past decades, significant progress has been made in developing catalysts for the dehydrogenation and hydrogenation of liquid hydrogen carriers, while some key issues are still to be addressed. Herein, a comprehensive overview of current state of catalytic studies related to liquid hydrogen carriers is provided. A thorough understanding of catalyst design and mechanism can facilitate the progress of liquid hydrogen carriers, ultimately contributing to the broader applications of hydrogen as a renewable energy source. |
1. Introduction
Fossil fuels have been acting as the indispensable driving force in human development. However, since the Industrial Revolution, the widespread use of fossil fuels has led to a steady increase in atmospheric carbon dioxide concentrations. By 2022, the global average atmospheric carbon dioxide concentration has reached a record 416 parts per million (ppm) and has continued to rise. The increasing carbon dioxide concentration significantly affects the global environment and biodiversity.1 Sustainable and carbon-free energy systems need to be developed and deployed as soon as possible to achieve the goal of carbon neutrality.2 Moreover, the energy shortages exposed by regional conflicts have emphasized the need to replace fossil fuels with renewable energy sources. Overall, it is pressing to find alternatives to fossil energy.Hydrogen, one of the most promising renewable energy carriers, has the advantages of zero carbon emissions and no harmful by-products. Often referred to as the “ultimate energy source,” hydrogen has the potential to address global environmental issues and solve the energy crisis of the 21st century. To harness the full potential of hydrogen, challenges in production, storage, transportation, and utilization must be addressed. Hydrogen storage and transportation, in particular, are crucial due to the highly flammable and explosive nature of hydrogen at ambient conditions. Hydrogen storage technology faces difficulties related to storage density (including mass and volume storage density), energy consumption, and safety.3–5
There are two primary categories of hydrogen storage methods: physical and chemical. Physical methods, such as high-pressure gas storage, low-temperature liquid storage, and adsorptive storage, each have benefits and drawbacks. For instance, high-pressure gaseous hydrogen storage is low-cost, fast, and operates at room temperature, but it has low safety levels, requires significant compression work, and demands pressure-resistant vessels.5 Similarly, cryogenic liquid hydrogen storage has high energy density, but the high costs and energy consumption during liquefaction are significant drawbacks.4 In order to encourage the development of materials for on-board hydrogen storage in light-duty automobiles, the US Department of Energy (DOE) sets system-level technical hydrogen storage targets of 5.5 wt% and 40 g L−1 for 2025 and an ultimate target of 6.5 wt% and 50 g L−1. In this field, emerging research trends include metal hydride hydrogen storage and porous material adsorptive hydrogen storage, both offering potential solutions to current limitations. Metal hydride hydrogen storage refers to hydrogen storage in the form of metal hydrides. The hydrogen release process is realized by the decomposition of metal hydride upon heating. The hydrogen storage reversibility of metal hydride still needs to be improved.6 Porous material adsorptive hydrogen storage shows promise due to its safety, reliability, and efficiency. However, the volumetric storage capacity of porous materials is generally low.7
In contrast, hydrogen storage using specific molecules through reversible hydrogenation–dehydrogenation reactions can achieve high storage capacity. These molecules, typically liquid at room temperature, can be transported using existing infrastructure, ensuring safety and convenience.4 Liquid hydrogen carriers (LHC) offer high hydrogen storage capacity, high energy density, and safe, convenient liquid storage and transportation. This technology is considered an alternative to liquefaction or high-pressure hydrogen storage and is expected to play a significant role in future hydrogen energy storage and transportation. For example, 1,3-diaminopropane has a theoretical hydrogen content of up to 10.8 wt%, far exceeding the US DOE (2020) target of about 5.5 wt%.8 Research on utilizing these chemicals for hydrogen storage shows great potential for practical applications.9,10
The concept of LHC can be dated back to the 1980s.11 Several systems have been proposed and studied to assess their viability for practical application.12 Hydrogen carriers might be classified into two types: (1) hydrogen–lean organic liquids that achieve fully reversible hydrogenation and dehydrogenation storage cycles, such as aromatic hydrocarbons, and (2) hydrogen–lean inorganic small molecules such as CO2 or N2, which can form molecules containing elemental hydrogen after catalytic hydrogenation processes, like methanol and ammonia (Table 1). These systems show differences in their development timeline, hydrogen storage capacity, and energy density.13
| Hydrogen carrier | Dehydrogenated carrier | H2 density | Dehydrogenation enthalpy (kJ per mol H2) | |
|---|---|---|---|---|
| wt% | g L−1 | |||
| Cyclohexane (C6H12) | Benzene (C6H6) | 7.1 | 56.3 | 68.6 |
| Methylcyclohexane (C7H14) | Toluene (C7H8) | 6.1 | 47.1 | 68.3 |
| Decalin (C10H18) | Naphthalene (C10H8) | 7.3 | 64.9 | 63.9 |
| Cyclohexylbenzene (C12H16) | Biphenyl (C12H10) | 3.8 | 35.6 | 65.9 |
| Tercyclohexane (C18H32) | Terphenyl (C18H14) | 7.2 | 67.5 | — |
| Perhydro-dibenzyltoluene (C21H38) | Dibenzyltoluene (C21H20) | 6.2 | 56.4 | 62, 65, 71 |
| Dodecahydro-N-ethylcarbazole (C14H25N) | N-Ethylcarbazole (C14H13N) | 5.8 | — | 50.6 |
| Octahydro-1H-indole (C8H15N) | Indole (C8H7N) | 6.4 | 58.5 | 51.9 |
| Methanol (CH3OH) | Carbon dioxide (CO2) | 12.6 | 99.8 | 16.5 |
| Ammonia (NH3) | Nitrogen (N2) | 17.7 | 108 | 30.6 |
| Ammonia borane (NH3BH3) | Hydrolysis: NH4BO2 | 19.6 | 145 | −52 |
| Hydrous hydrazine (N2H4·H2O) | Nitrogen (N2) | 8.0 | 82.6 | −25.3 |
Cycloalkanes such as cyclohexane and methylcyclohexane are widely used in industry and can be obtained at a relatively low cost. However, the cyclohexane/benzene and methylcyclohexane/toluene systems have their drawbacks. These reactants and products have low boiling points, making them gaseous during the dehydrogenation processes. As a result, condensation and purification steps are needed to separate pure hydrogen from mixed gases. Additionally, the toxicity of benzene must be considered. In comparison, the perhydrodibenzyltoluene system has a lower vapor pressure and volatility, allowing it to release hydrogen in the liquid phase. The stability of the DBT system is also relatively high, making it advantageous. However, the viscosity of this system is quite high, posing challenges for reactor design. N-Heterocyclic molecules have a lower dehydrogenation enthalpy than alkanes, allowing dehydrogenation to occur at lower temperatures. Nevertheless, side reactions like disproportion and alkyl transfer can negatively impact long-term cycling performance and should be avoided. Methanol, which has high hydrogen content and is widely used in industry, has its limitations as well. Methanol decomposition produces carbon monoxide (CO), which is harmful to hydrogen fuel cell applications. Thus, removing CO from the effluent is necessary. Ammonia, on the other hand, produces only nitrogen and hydrogen when decomposed, without any carbon-containing gases. However, ammonia production is energy-intensive, and its storage presents challenges due to its low boiling point and toxic nature. Ammonia borane and hydrous hydrazine can be stored and transported more easily, but they come at a high cost.
The hydrogen storage process using liquid hydrogen carriers includes hydrogenation and dehydrogenation. The hydrogenation process of organic molecules is generally thermodynamically favourable, enabling high selectivity and conversion. In contrast, the dehydrogenation process is a strongly endothermic reaction that is thermodynamically unfavourable. Besides, dehydrogenation catalysts are prone to deactivation due to sintering and coking at high temperatures.
Thus, the main challenge in achieving hydrogen storage using liquid organic hydrogen carriers is to develop dehydrogenation catalysts that are low-cost, highly stable, and offer high activities. This review highlights the development of three types of substrates in hydrogen storage applications and introduces catalysts used in the reaction process, aiming to inspire further research in this hot field.
2. Cycloalkanes
When the concept of liquid hydrogen carriers (LHC) was initially proposed, the primary focus of research was on the hydrogen storage cycles of benzene-based hydrocarbons, such as benzene/cyclohexane and toluene/methylcyclohexane (MCH). In addition to these two classical systems, researchers have developed the hydrogen storage cycles of benzyltoluene (H0-BT)/perhydrobenzyltoluene (H12-BT), dibenzyltoluene (H0-DBT)/perhydro-dibenzyltoluene (H18-DBT), and naphthalene/hydrides of naphthalene, including tetralin and decalin over the past twenty years.3 In 2003, Air Products and Chemicals pioneered the use of carbazole-based molecules for hydrogen storage. A new class of heteroaromatic compounds, N-ethylcarbazole (NEC)/dodecahydro-N-ethylcarbazole (DNEC), was proposed, which can be used for reversible hydrogen storage under moderate conditions without generating unwanted impurities. The dehydrogenation enthalpy of this system is 51 kJ per mol H2, allowing the dehydrogenation process to occur at a much lower temperature compared to naphthenic hydrocarbons.14 Similar systems include N-propylcarbazole (NPCZ)/perhydro-N-propylcarbazole (12H-NPCZ),15 indole/indoline, 2-methylindole (2-MID)/8H-2-methylindole (8H-2-MID),16N-ethylindole (NEID)/octahydro-N-ethylindole (8H-NEID),17 and tetrahydroquinoline/quinoline.18The hydrogenation process of aromatic hydrocarbons (the term aromatic hydrocarbons in this context refers to a broader definition and includes non-benzene aromatics) results in a reduction of Gibbs free energy, often with a large absolute value, which is thermodynamically advantageous. Consequently, the conversion and selectivity of the hydrogenation process of aromatic hydrocarbons are very high, with conversion even approaching 100%. The aromatic hydrogenation process is well-established and has been industrialized. However, the dehydrogenation reaction is strongly endothermic that requires low-pressure and high-temperature conditions while also being limited by reaction equilibrium constraints.8 Early studies on chemical hydrides as hydrogen storage media indicated that chemical hydrides could not be recycled for these reasons. Moreover, dehydrogenation catalysts are susceptible to pore structure destruction, coking, and deactivation under high-temperature conditions. The dehydrogenation process might also involve side reactions, such as hydrogenolysis. Consequently, research on these systems primarily concentrates on the dehydrogenation process.
The activation and breaking of multiple C–H bonds are required during dehydrogenation, both for the traditional benzene-cycloalkane and the newly developed carbazole-based system. Therefore, the selectivity of catalysts is crucial, and the challenge lies in reducing the cost of dehydrogenation catalysts while ensuring high conversion, high selectivity, high stability, and good resistance to coking deactivation. Many studies indicate that noble metals, such as Pt and Pd, show excellent performance as the active component of catalysts. The activity of the catalyst largely depends on the presence of a suitable structure for the active sites, allowing the adsorption and activation of reactant molecules. Pt-Based and Pd-based catalysts continue to be the focus of current research. Although noble metal catalysts have shown exceptional catalytic activity, the scarcity of Pt, Pd, and other noble metals in nature, combined with high industrial costs, limit the practical application of these catalysts. From an industrial application standpoint, non-noble metal-based aromatic hydrocarbon dehydrogenation catalysts, such as those utilizing Ni, Cu, and other active metals, are more practical. Among these, Ni-based catalysts show great promise.
To further enhance the dehydrogenation activity of catalysts, it is common practice to add a second metal component to the catalyst. Bimetallic catalysts perform well in dehydrogenation reactions because the second active metal component can inhibit the migration and agglomeration of the original active metal on the catalyst surface, thus improving the stability and specific activity of the catalyst.19 In addition, the performance of the catalyst can be improved by modifying factors such as the catalyst support, the morphology of the catalyst, and the interaction between the active components and the support.
2.1 Benzene/cyclohexane
Benzene/cyclohexane is one of the most well-known and classical LHC systems and has been studied extensively. Dehydrogenation is the most challenging part of this cycle because the C–H bonds in cyclohexane are thermodynamically stable and kinetically inert. In most cases, catalysts are required to accelerate the dehydrogenation of cyclohexane to hydrogen and benzene.20 Transition metals are commonly used as catalysts for the activation of C–H bonds because they can hybridize their s- or d-orbitals with C–H s* anti-bonding orbitals so they can weaken the C–H bonds of hydrocarbons.21 Transition metal-based catalysts for this process mainly include Pt, Pd, and Co as the active component.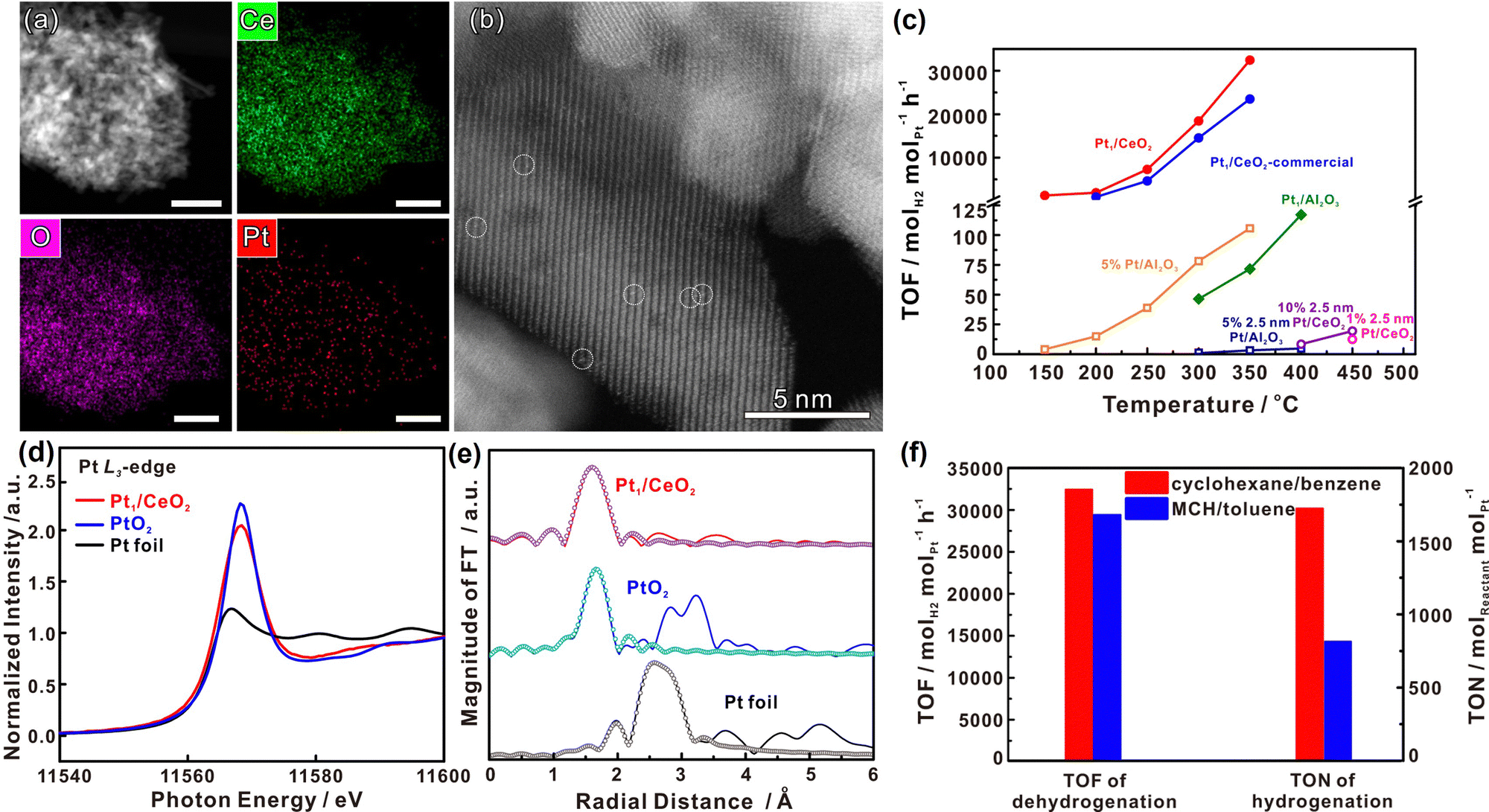 | ||
| Fig. 1 (a) HAADF-STEM images with corresponding elemental distribution images and (b) Cs-corrected HAADF-STEM images of Pt1/CeO2. (c) Comparison of the performance of Pt1/CeO2 with other catalysts in cyclohexane dehydrogenation. (d) Normalized Pt L3-edge XANES spectra and (e) k3-weighted Fourier transform EXAFS spectra (L3-edge) of Pt in Pt1/CeO2, PtO2, and bulk Pt foil. (f) Comparisons of the performance in dehydrogenation and hydrogenation of different molecules.24 Modified with permission from ref. 24. Copyright (2022) Springer Nature. | ||
| Catalyst | Temp. (°C) | Space velocity | Conv. (%) | Sel. (%) | Ref. |
|---|---|---|---|---|---|
| 3 wt% Pt/C | 280 | 1 h−1 (WHSV) | ∼58 | 35 | |
| Pt/CN (N-doped carbon) | 210 | 96.03 | 100 | 36 | |
| Pt1/CeO2 | 350 | 24 h−1 (WHSV) | ∼30 | ∼100 | 24 |
| Pt/MgAl(Sn)Ox | 550 | 360 h−1 (GHSV) | ∼11 | ∼48 | 29 |
| Pt/Al2O3 | 200 | 72.1 | 30 | ||
| Pt–Ca/Al2O3 | 350 | 10 h−1 (WHSV) | 97.0 | 31 | |
| Pt3Sn/SiO2 | 600 | ∼35 | ∼85 | 33 |
The dispersion of Pt and hydrogen spillover capability of the catalyst is also improved by CaO addition with a decreased product adsorption on Pt sites by electron transfer from Ca to Pt. The catalyst with Ca/Pt of 5 shows the best activity and stability. At low Ca loading, the hydrogen spillover from the Pt–Al interface is not enhanced efficiently. While at high Ca loading, The CaO covers the support surface. The strong interaction between CaO and Pt suppresses the reduction of Pt and reduces the initial activity. Komatsu et al. tested SiO2 supported Pt-based intermetallic compounds, including Co, Ge, Sn, Tl, and Zn, in this reaction.33 Pt3Zn/SiO2, Pt3Tl2/SiO2, Pt3Co/SiO2 were prepared by impregnation while Pt3Sn/SiO2, PtSn/SiO2, and PtGe/SiO2 were prepared by chemical vapor deposition. Pt3Sn/SiO2 and PtGe/SiO2 show higher activity and benzene selectivity than Pt/SiO2, while other catalysts show lower activity and poor benzene selectivity. The difference is ascribed to the different chemical properties of the second metal as well as geometric factors. Ali et al. studied the performance of Pt, Rh, Re, U, PtIr, PtRh, PtRe, and PtU on γ-Al2O3 using a pulsed micro-reactor.34 For the monometallic catalyst, the activity follows the order of Pt/Al2O3 > Rh/Al2O3 > Ir/Al2O3 > Re/Al2O3 ≥ U/Al2O3. For the bimetallic catalyst, the PtRh/Al2O3 shows the best activity, which is higher than the Pt/Al2O3. The addition of U in Pt/Al2O3 leads to a decreased activity, possibly because of the low electronegativity of U than Pt.
In addition to the thermal strategy, the dehydrogenation of cyclohexane using other energy sources, such as renewable solar energy, has been studied. This process can be performed under mild conditions. Recently, Zhang et al. synthesized Pt/black TiO2 photocatalysts via reduction–oxidation–reconstruction protocol.37 In this method, TiO2 nanoparticles were first reduced by NaBH4, then subjected to surface oxidation treatment with aqueous H2O2. Pt was finally introduced via a conventional wet impregnation. The catalyst obtained is designated as Pt@BT-O. The 0.2 wt% Pt@BT-O can achieve cyclohexane conversion of 99% under 1 h of visible-light irradiation at 43 °C with a ratio of produced benzene to H2 close to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.
:3.
| Catalyst | Temp. (°C) | Space velocity | Conv. (%) | Sel. (%) | Ref. |
|---|---|---|---|---|---|
| 10 wt% Ag−1 wt% Pt/ACC | 300 | 100 | 52 | ||
| Ni–Cu/SBA-15 | 350 | 12000 mL g−1 h−1 (GHSV) |
99.4 | 98.7 | 46 |
| Ni2P/Al2O3 | 340 | 1 h−1 (WHSV) | 80 | 99.9 | 47 |
| Pd/CeO2-HT | 450 | 12000 mL g−1 h−1 (GHSV) |
65.3 | 100 | 53 |
| Ni/Al2O3 | 340 | 1 h−1 (WHSV) | 65 | 90 | 47 |
| 1:4 Ag-Rh/Y2O3 |
300 | 35.8 | 54 | ||
| Co/SiO2 | 550 | 2.3 h−1 (WHSV) | 9.1 | 74.4 | 48 |
| Ni/20%CeO2–Al2O3 | 350 | 3 h−1 (WHSV) | ∼50 | 100 | 55 |
| RANEY®-Ni | 320 | 72.7 | 100 | 56 |
Some non-noble metal catalysts, such as Ni40 and Zn,41 also show good catalytic activity. Besides, bimetallic catalysts play an important role in enhancing activity in various reactions compared to monometallic catalysts, including the dehydrogenation of cyclohexane. Improving the performance of non-noble metal-based catalysts has practical significance for promoting the application of catalysts. Liu et al. studied the influence of Ni particle size on cyclohexane dehydrogenation using Ni/SiO2/Mo(110) model catalysts.42 Ni was deposited on the substrate by vapor deposition and the size was controlled by changing the deposition time. The results show that the TOF increases with the decrease of Ni particle size for the particles smaller than 2.5 nm due to the increase portions of low coordinated Ni atoms favoring H2 desorption, while the TOF is not sensitive to the particle size for the particles larger than 2.5 nm. Viveros et al. used Al2O3–TiO2 as support for Ni in this reaction.43 The effect of TiO2 on the performance of Ni/Al2O3 is influenced by the TiO2 content, Ni loading, and reduction temperature. At low TiO2 content, the dispersion and intrinsic activity of Ni can be promoted by TiO2. However, at high TiO2 content, the metallic Ni surface is partially covered by reduced TiOx species due to the strong metal support interaction, resulting in reduced activity. Ichikawa et al. used Pt to promote the performance of Ni supported on activated carbon cloth (ACC).44 The activity was tested using a spray-pulsed mode reactor. The addition of only 0.5 wt% Pt into 20 wt% Ni/ACC increases the hydrogenation production rate by ∼1.5 times. The promotion effect of Pt is ascribed to the easy formation and desorption of H2 on the Pt surface and a synergistic effect of Ni–Pt for C–H bond activation. Chen. et al. studied the performance of Ni–Cu/SiO2 in cyclohexane dehydrogenation.45 The catalysts were prepared by the one-step sol–gel method. Ni/SiO2 shows low dehydrogenation selectivity of <50% at 350 °C due to the small Ni particle size, which has high hydrogenolysis activity. The addition of Cu increases the selectivity to benzene, possibly due to the formation of NiCu alloy. The Ni0.85Cu0.15/SiO2 exhibits 94.9% conversion of cyclohexane with selectivity to benzene of 99.5% at 350 °C. SBA-15 was also used as support for Ni–Cu catalyst.46 The NiCu/SBA-15 with Ni and Cu of 4.9 wt% and 3.5 wt% shows a cyclohexane conversion of 99.4% and benzene selectivity of 98.7% at 350 °C. In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFT) shows that vinyl species are present on the catalyst surface during dehydrogenation. Liu et al. studied the performance of Ni2P/γ-Al2O3 in this reaction.47 The catalyst was prepared by the decomposition of nickel hypophosphite supported on Al2O3 under N2 atmosphere. The Ni2P/γ-Al2O3 shows higher activity, selectivity, and stability compared to Ni/Al2O3 and achieves cyclohexane conversion of 80% with benzene selectivity of 79.9% at 340 °C. The good performance of Ni2P is ascribed to the positive charge of Ni which enhances the interaction between reactant and active sites and reduces the adsorption of benzene as well as the ensemble effect of P in N2P that inhibits the sintering of Ni2P under reaction conditions. H2 temperature-programmed desorption (TPD) profiles show that the Ni2P has a stronger H2 spillover effect, which may contribute to the good performance. In situ DRIFT shows that the P-covered Ni sites of Ni2P(0001) should be the active sites.
Sooknoi et al. tested Co/SiO2 in cyclohexane dehydrogenation.48 A series of Co/SiO2 was prepared by electrostatic adsorption of Co salts on SiO2 using [Co(bipy)3](NO3)2, [Co(NH3)5Cl]Cl2, [Co(NH3)6]Cl3, and [Co(en)2Cl2]Cl. The activity of the catalyst prepared using [Co(bipy)3](NO3)2 is the highest among these catalysts, which is ascribed to the highest Co2+ content in the catalyst. A comparison of the activity for reduced and unreduced catalysts shows that metallic Co is much less active than the Co2+ species. It is proposed that the dehydrogenation on cationic Co2+ species has two steps. The first one is the C–H dissociation with the formation of cobalt–alkyl species and Si–OH, followed by β-H elimination of the cobalt–alkyl species. Since the activity of this catalyst under H2 is higher than N2 atmosphere, it is concluded that a cobalt hydride intermediate formed under H2 can facilitate the C–H activation of reactant without the necessity for dissociation of the Co–O bond. Pinard et al. used Ga containing MFI zeolite in cyclohexane dehydrogenation.49 Three catalysts were prepared by direct hydrothermal synthesis, ion exchange, and physical mixture. Ga incorporates into the zeolite framework by direct hydrothermal synthesis while deposits on the outer surface by ion exchange. For the physical mixture, Ga2O3 and zeolite are physically separated. The Ga species located on the outer surface of zeolite can be reduced and migrate into the zeolite crystal after high-temperature reducing treatment. The dehydrogenation reaction was conducted at 530 °C. Reaction results show that the catalysts prepared by hydrothermal synthesis show higher selectivity to benzene than the other two catalysts due to its weak acid strength. Du et al. reported that the C-Mo2C composite catalyst could catalyze cyclohexane dehydrogenation.50 Catalysts with different Mo contents were prepared by hydrothermal synthesis followed by carbonized at high temperatures. The catalyst with 25 wt% Mo content shows the best activity with conversion of ∼11% and selectivity to benzene of ∼100% at 315 °C. Chen et al. studied the performance of Cu/SBA-15 in this reaction.51 A series of Cu/SBA-15 with different Cu loading of 1.9–7.1 wt% was prepared by impregnation. For all the catalysts tested, the selectivity to benzene is 100%. The 3.7 wt% Cu/SBA-15 shows the highest turnover frequency (TOF) of 5578 mol h−1 molCu−1 at 350 °C due to the small Cu particle size under reaction conditions. This catalyst also shows good stability with only slight activity loss after 20 h on stream. Although the 1.9 wt% Cu/SBA-15 with lower Cu content should have a smaller Cu particle size at the initial state, the sintering of Cu nanoparticles after reduction makes the TOF of this catalyst lower than the 3.7 wt% Cu/SBA-15.
2.2 Toluene/methylcyclohexane
The toluene/methylcyclohexane system is another attractive system of LHC. This system can achieve gravimetric and volumetric hydrogen storage densities of up to 6.1 wt% and 47.1 g L−1, respectively.13 The enthalpy change for this reaction is 205 kJ mol−1 as shown in the equation below.57| C7H14 → C7H8 + 3H2 ΔHθ = 205 kJ mol−1 |
The methylcyclohexane–toluene–hydrogen (MTH) system, first proposed by Taube et al., shows great potential for application.11 Compared to benzene in the cyclohexane–benzene–hydrogen (CBH) cycle, toluene in the MTH cycle is less toxic, aligning with the current green energy development goals. While the hydrogenation of toluene has already been commercialized, the main challenge lies in resolving the issue of catalytic dehydrogenation.58 From an energy standpoint, the MCH system can form a relatively closed loop if the heat generated from the hydrogenation reaction is utilized for the dehydrogenation reaction and gasification of reactants.
Catalytic dehydrogenation of MCH is typically carried out using heterogeneous catalysts with metal and acid functionalities. A crucial aspect of developing catalysts for this system is optimizing the metal and acid functions of heterogeneous catalysts to prevent unwanted byproduct generation and enhance toluene formation.59,60 Dehydrogenation of MCH is an endothermic reaction accompanied by an increase in gas volume, making it more favorable at high temperatures and low pressures. However, most catalysts do not perform well under high-temperature conditions, primarily due to coke formation during dehydrogenation. Coke deposition can significantly reduce catalyst performance and even deactivate it, leading to an increase in byproducts, decreased selectivity, and reduced reactant conversion. Therefore, developing catalysts with good stability, product selectivity, and catalytic activity at relatively low temperatures is of great importance. The dehydrogenation of MCH is recognized as a structure-sensitive reaction, and in this context, the support structure greatly influences catalytic performance. Common supports, such as molecular sieves, metal oxides, composite metal oxides, and activated carbon, are generally suitable for MCH dehydrogenation catalysts. Several representative catalysts for this reaction are listed in Table 4.
| Catalyst | Temp. (°C) | Space velocity | Conv. (%) | Sel. (%) | Ref. |
|---|---|---|---|---|---|
| Mo10–SiO2 | 400 | 92.4 h−1 (WHSV) | 90 | 78 | |
| 8 wt% Ni-2 wt% Cu/ACC | 350 | 25.78 | 76 | ||
| Pt–Sn/MgAlOx | 300 | 9.2 h−1 (WHSV) | 90.5 | ∼100 | 69 |
| Ir/USY | 300 | 92.4 h−1 (WHSV) | 13.1 | 89 | 65 |
| Pt-B1/Al2O3-600 | 350 | 23 h−1 (WHSV) | 81.5 | >99.9 | 84 |
| Pt/Al2O3–TiO2 | 400 | 5 h−1 (WHSV) | 93 | 99 | 67 |
| Pt–Sn/Al2O3 | 300 | 600000 mL g−1 h−1 (GHSV) |
∼23.6 | ∼99.99 | 85 |
| Pt3(Fe0.75Zn0.25)/SiO2 | 320 | 99 | >99.8 | 86 | |
| Ni20AlOx | 450 | 3 h−1 (WHSV) | 77.4 | 85.6 | 87 |
| Ga52Pt/SiO2 | 450 | 1730 h−1 (GHSV) | 15 | 85 | 19 |
| Pt/GAC-S | 300 | 5 h−1 (WHSV) | 63 | 88 | |
| Zn/Ni–SiO2 (Ni:Zn = 80:20) |
350 | 18 h−1 (WHSV) | ∼76 | 97 | 89 |
| Pt/Y2O3 | 350 | 98 | ∼100 | 62 | |
| Pt/activated carbon | 300 | 28 h−1 (WHSV) | 42 | >99 | 63 |
| Pt/pyrolytic waste tire char | 300 | 3 h−1 (WHSV) | >95 | ∼100 | 64 |
| Cu–Pt/S-1 | 350 | 4.6 h−1 (WHSV) | 59.35 | 99.94 | 66 |
| Pt/TiO2–Al2O3 | 400 | 5 h−1 (WHSV) | 93.2 | 99.1 | 67 |
| Pt/Ce–Mg–Al–O | 350 | 9 h−1 (WHSV) | 98.5 | >99.9 | 70 |
For the catalytic dehydrogenation of MCH, similar to cyclohexane, Pt-based catalysts have been widely studied as Pt can effectively activate C–H bonds without splitting C–C bonds.58 Different supports have been used for Pt-based catalysts in MCH dehydrogenation. Biniwale et al. used single metal oxides and perovskites, including La2O3, ZrO2, TiO2, CeO2, Fe2O3, Al2O3, MnO2, LaNiO3, and La0.7Y0.3NiO3 as supports for Pt in MCH dehydrogenation in a spray-pulsed reactor.61 For the single metal oxide, the Pt/La2O3 shows the best activity of 21.1 mmol gmetal−1 min−1 at 350 °C. The performance can be further improved by using LaNiO3 and La0.7Y0.3NiO3 as supports, achieving hydrogenation formation rate of 30.3 mmol gmetal−1 min−1 and 45.3 mmol gmetal−1 min−1 at 350 °C, respectively. The Pt/La0.7Y0.3NiO3 also shows good selectivity, as no methane can be detected in the product. In a later work, V2O5 and Y2O3 were also used as supports for Pt.62 The Pt/Y2O3 shows a high hydrogen formation rate of 703 mmol gmetal−1 min−1 at 350 °C, which is stable up to 150 min. The good activity is ascribed to the high dispersion of Pt on Y2O3 (31.5%) as determined by CO adsorption. Bao et al. used activated carbon as support for Pt in MCH dehydrogenation.63 The surface of activated carbon was modified by oxidation with HNO3 and reduction in H2. Oxygen-containing groups, such as carboxylic acid groups, can be created on the surface of activated carbon during HNO3 oxidation. After H2 reduction, carboxylic acid and anhydride groups are removed while the thermal stable groups, such as lactone, phenol, and carbonyl, are preserved. The two-step modified catalysts show better activity than the unmodified or only oxidized catalysts. The difference in activity is ascribed to the different Pt dispersion caused by different surface groups. Liu et al. used pyrolytic waste tire char as support for Pt.64 The waste tire char was purified in multi-steps before being used as the support. The organic impurities in the waste tire char were extracted by toluene refluxing, while the inorganic metal oxides were removed by H2SO4 washing. Then the support was activated in steam to create the porous structure. The supported catalysts with Pt loading 0.4–1 wt% show better activity than the catalyst with low Pt content, achieving MCH conversion over 95% with selectivity to toluene of nearly 100%.
Besides Pt, other noble-metal can catalyze MCH dehydrogenation. Vasudevan et al. compared the performance of USY-supported Ir, Pt, Pd, and Ni.65 The activity follows the order of Ir ≫ Pt > Pd > Ni. The good activity of Ir/USY compared to Pt/USY is ascribed to the larger Ir nanoparticles formed due to the moderate interaction between Ir and USY. Larger particles have fewer kink sites which are active for the hydrogenolysis reaction and responsible for reduced selectivity. The deactivation of catalysts due to coke formation is related to the acidity of the catalyst. High total acidity of Pd/USY leads to severe coke formation and deactivation for this catalyst. The activity and selectivity of the noble-metal-based catalysts can be modified by adding promoters. Guo et al. improved the performance of Pt/S-1 in MCH dehydrogenation by doping Cu.66 The catalysts were prepared by adsorption of metal precursors using silicalite-1 as support, followed by calcination. The Cu–Pt/S-1 exhibits better activity, selectivity, and stability than Pt/S-1 and Cu/S-1, showing an average H2 formation rate of 288.9 mmol gmetal−1 min−1 with a MCH conversion of 59.35% at 350 °C. Formation of Pt–Cu alloy increases the dispersion of Cu, facilitates the charge transfer from Pt to Cu, decreases the electron density of Pt, and reduces the over-dehydrogenation and hydrogenolysis activity of the catalyst.
Lin et al. used TiO2 to modify Al2O3 support for Pt/Al2O3 catalysts.67 TiO2 was deposited on Al2O3 by chemical vapor deposition. The Pt/TiO2–Al2O3 with 11.5 wt% TiO2 shows the best activity with an average MCH conversion of 93.2% and toluene selectivity of 99.1% at 400 °C. The TiO2-coated Al2O3 combines the advantage of the high specific surface area of Al2O3 and strong interaction between Pt and TiO2, which results in a good Pt dispersion of 57.5%. The surface density of weak acid sites is reduced by TiO2 modification while the density of strong acid sites increases. At the same time, the electron density and H spillover on Pt are promoted. All these effects contribute to the increase in activity. Sekine et al. also studied the promotion effect of TiO2 on Pt/Al2O3.68 The repulsion between the π-coordination of toluene and electron-rich Pt on TiO2–Al2O3 promotes the desorption of toluene and suppresses the further demethylation to benzene and methane or coke formation. Yang et al. used Pt–Sn/MgAlOx in MCH dehydrogenation.69 The MgAlOx support was prepared from Mg–Al layered double hydroxides by calcination, and Pt and Sn were introduced into MgAlOx by impregnation. The activity and stability of Pt/MgAlOx were improved by the addition of Sn. X-ray photoelectron spectroscopy (XPS) results reveal the existence of electron transfer from Sn to Pt, which may be related to the improved activity by increasing the interaction between electron donor and Pt and thereby enhancing the reactant adsorption and product desorption. The good stability is ascribed to the absence of acid sites in support, large pore size that reduces the diffusion resistance of the product, and a drain-off effect of Sn in assisting the migration of coke precursors to the support. In a later work, Pt supported on CeMgAlOx derived from layered double hydroxide precursors was employed in this reaction.70 The catalysts with different Ce content were prepared by one-step co-precipitation and the introduction of Ce could promote the activity. The catalyst with Ce content of 14% shows the best activity with a hydrogen formation rate of 1358.6 mmol gmetal−1 min−1 due to the smallest Pt particle size of 2.28 nm and the largest specific surface area of 163.1 m2 g−1. This catalyst also shows a good stability as no coke formation and deactivation could be observed after 10 h on stream. The performance of Pt-based catalyst can also be improved by Mo, as reported by Boufaden et al.71 Mo was introduced into SiO2 support via sol–gel route before the introduction of Pt by impregnation. The catalyst with 8 wt% Mo shows the best activity. At higher Mo content (10.6 wt% and 12.7 wt%), MoOx–Pt core–shell nanoparticles will form with a decline in activity. There is a linear correlation between the activity and the surface Pt0/Mo4+ ratio, indicating that both MoO2 and Pt0 sites participate in the reaction and the catalyst shows a bifunctional character of both metallic and acidic character. Sekine et al. reported that Mn could act as a promoter to improve the performance of Pt/Al2O3.72,73 The MnOx selectively covers the unsaturated step facets on Pt which have high activity for demethylation, thus improving the dehydrogenation selectivity. Wasserscheid et al. studied the performance of Ga-rich, supported catalytically active liquid metal solution represented by Ga52Pt/SiO2 in MCH dehydrogenation.19 Pt was dissolved in the Ga matrix, which is supported on SiO2. Due to the low melting temperature of Ga, the alloy presents in the liquid state under reaction conditions and Pt in the atomically dispersed state. This catalyst shows higher activity and better stability than Pt/SiO2 and achieves conversion of 15% with toluene selectivity of 85% at 450 °C. No significant deactivation can be observed for 75 h on stream, possibly due to the atomically dispersed and dynamic state of Pt in the liquid Ga matrix under reaction conditions.
The non-noble metals such as Ni and Mo have also been used in MCH dehydrogenation. Unsupported RANEY®-Ni shows some activity in this reaction.74 Yolcular et al. studied the performance of Ni/Al2O3 with Ni content between 5–20 wt% in this reaction.75 MCH conversion increases with the Ni content in the range studied and 20 wt% Ni/Al2O3 achieves MCH conversion of 92% at 440 °C. Biniwale et al. used NiCu supported on activated carbon cloth with different Ni/Cu ratios in this reaction.76 The catalyst with 8 wt% Ni and 2 wt% Cu shows the best activity with a MCH conversion of 25.78%. The improved dehydrogenation activity and decreased hydrogenolysis activity are ascribed to decreases adsorption strength of hydrocarbon intermediates due to the formation of NiCu alloy. Takanabe et al. studied the effect of Zn on Ni/Al2O3 in MCH dehydrogenation.77 The Zn in NiZn/Al2O3 occupies the low coordination corner and edge sites which are active for C–C dissociation and thereby improves the toluene selectivity. The rate of MCH dehydrogenation on NiZn/Al2O3 shows a ∼0.5 order dependence on the H2 partial pressure, indicating that the rate-determining step involves H-derived species. Density functional theory (DFT) calculations suggest that the exothermic toluene dissociative adsorption by losing one H atom from the methyl group occurs on the catalyst surface during MCH dehydrogenation. The adsorption of this species is so strong that the reverse hydrogenation and desorption to form toluene needs to overcome an energy barrier of 1.29 eV and becomes the rate-determining step. Therefore, the rate of dehydrogenation reaction shows a positive order dependence on the H2 partial pressure. Boufaden et al. prepared Mo–SiO2 catalysts with different Mo content via sol–gel route and tested them in MCH dehydrogenation.78 MoO2 formed after the reduction is ascribed to be the active phase for dehydrogenation. The catalyst with Mo/Si of 0.1 shows the best activity at 400 °C with toluene selectivity of 90%. Further increase in the Mo content causes the decrease of activity due to the decreased MoO2 dispersion and formation of MoO3. The acidity of the catalysts also increases with the increased Mo content, resulting in coke formation.
It is worth mentioning that in the cycle of the MTH system, in addition to the conventional fixed-bed reactor, some other promising reaction systems have been studied. Takise et al. reported low-temperature catalytic dehydrogenation of MCH by applying an electric field in the fixed-bed flow reactor.57 Typically, 3 wt% Pt/CeO2 catalyst demonstrates the highest MCH conversion under the electric field which exceeds the thermodynamic equilibrium at 150 °C. The enhanced conversion under the electric field is ascribed to the proton hopping on the catalyst and the suppressed reverse hydrogenation reaction in the presence of electric field.
2.3 Polycyclic aromatic hydrocarbons
Decalin/naphthalene is one of the widely studied systems with a theoretical hydrogen capacity of 7.3 wt% and dehydrogenation heat of 63.9 kJ per mol H2. Naphthalene exists as a solid at ambient conditions. These factors complicate the implementation of the storage system and hinder practical applications to some extent.9 Despite these disadvantages, decalin is considered an excellent hydrogen source for fuel cells due to its high energy and power densities, low energy consumption, safety, economy, and ease of operation. Consequently, it holds promise for applications such as hydrogen sources in fuel-cell vehicles.79 Catalysts based on Pt-group metals exhibit exceptional performance in decalin dehydrogenation, with size-dependent activity. This underscores the importance of controlling Pt particle size and achieving ideal dispersion by adjusting the catalyst support for the dehydrogenation process.80 Studies have shown that Pt atoms with a low coordination number are more efficient than those with a high coordination number, as they have a lower energy barrier.81 Nevertheless, the dehydrogenation product of decalin, naphthalene, strongly adsorbs on the Pt surface. A few studies show that the kinetics of decalin dehydrogenation is limited by naphthalene desorption.79,82 Changing the adsorption behaviour of the reactant is crucial, and adjusting the electronic structure of Pt by modifying the support is an effective strategy. Commonly used catalyst supports include activated carbon and metal oxides. Activated carbon is particularly attractive due to its low cost and good performance. Sebastián et al. reported that 4 wt% Pt/C could effectively catalyze decalin dehydrogenation.83Tuo et al. improved the dehydrogenation performance of Pt/CNT by modifying the surface of CNT with nitrogen and oxygen functional groups.90 Recently, Tuo et al. used MgAl2O4 to modulate the electronic structure of Pt nanoparticles. The prepared catalyst, Pt/MgAl2O4, exhibited record-high dehydrogenation activity, which is almost as twice as the activity of Pt/CNF91 (Fig. 2). Some bimetallic catalysts have also been studied as they not only show distinctly different electronic and chemical properties from Pt itself, but also explore ways to reduce catalyst costs. Qi et al. designed Pt–Ni bimetallic catalysts supported on active carbon, with the bimetallic catalyst 1Ni–1Pt/C displaying higher dehydrogenation activities than the monometallic catalysts.92 Apart from Pt-based catalysts, other metal-based catalysts have shown promise for the reaction. Kim et al. found that decalin is more easily dehydrogenated on Pt/C, while tetrahydronaphthalene is more easily dehydrogenated on Pd/C.93 The difference in catalytic activity and selectivity can be attributed to the distinct structural properties and adsorption mode preferences between Pd and Pt catalysts during dehydrogenation. Non-precious metal-based catalysts have also been studied. For example, Al-Muntaser et al. reported that a Ni-based catalyst, nickel(II) stearate, reached the maximum decalin conversion rate of 21.95%.94 It is worth mentioning that some studies have found that the microwave effect appears in the process of tetrahydronaphthalene, but in the dehydrogenation of decalin, microwave heating is of little use to improve the catalytic performance.95
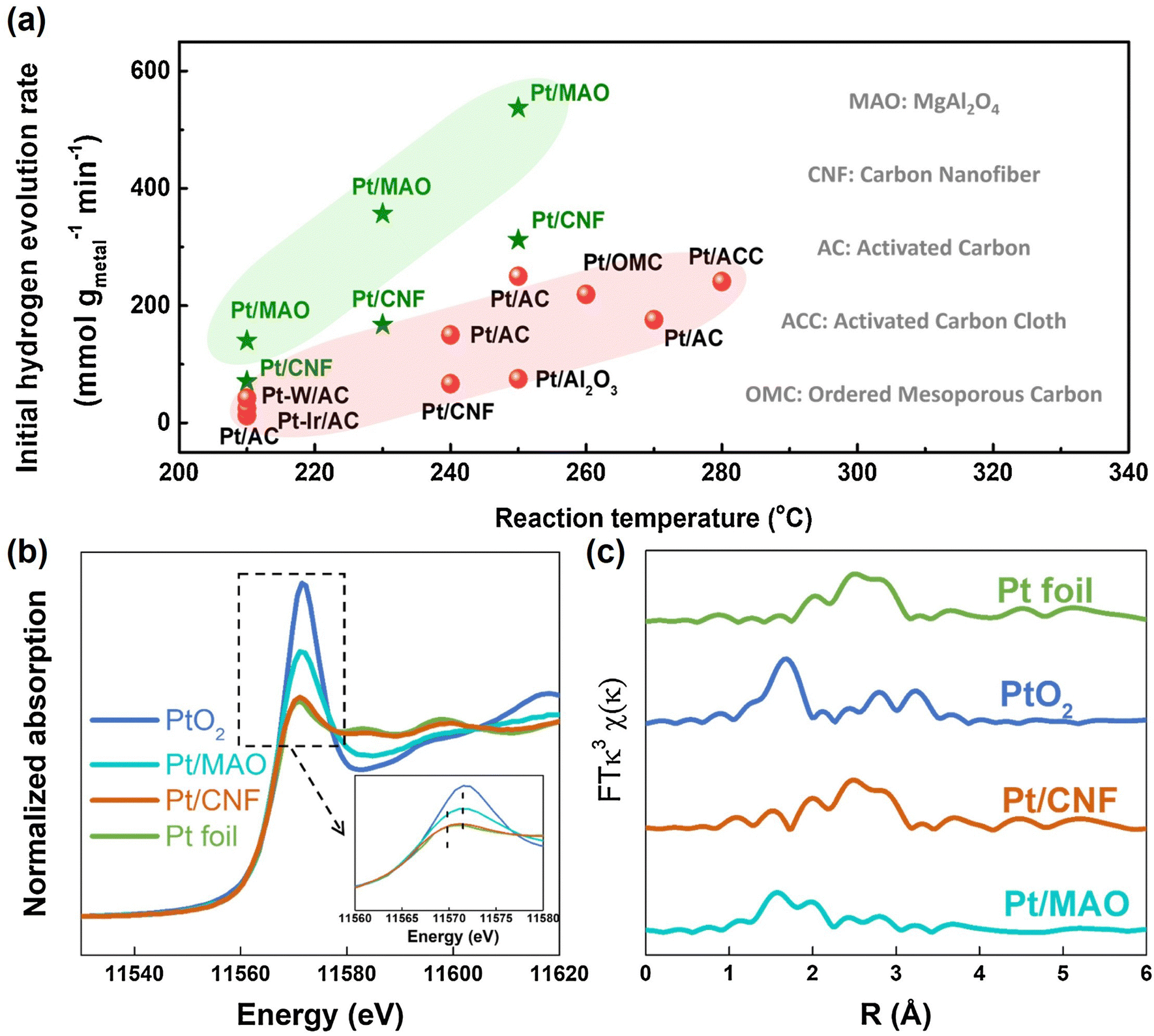 | ||
| Fig. 2 (a) Comparison of dehydrogenation activity of Pt/MgAl2O4 and Pt/CNF with other catalysts at different temperatures. (b) Normalized Pt L3-edge XANES spectra and (c) k3-weighted Fourier transform EXAFS spectra (L3-edge) of Pt for Pt foil, PtO2, Pt/CNF, and Pt/MgAl2O4.91 Modified with permission from ref. 91. Copyright (2021) Elsevier Inc. | ||
Perhydrodibenzyltoluene (H18-DBT) is another promising LOHC with a hydrogen storage content of 6.2 wt%. Mixtures of isometric benzyltoluenes and dibenzyltoluenes are used as heat-transfer oil in industry and produced on a large scale. The dehydrogenation of H18-DBT undergoes different intermediates, including H12-DBT, H6-DBT, to dibenzyltoluene (H0-DBT). This reaction can be catalyzed by Pt and Pd-based catalysts. Wasserscheid et al. compared the performance of Pt/C, Pt/Al2O3, Pt/SiO2, Pd/C, and Pd/Al2O3 in H18-DBT dehydrogenation.96 Pt-Based catalysts show higher activity than Pd-based catalysts and Pt/C exhibits the best activity among the catalysts tested, achieving 71% degree of dehydrogenation at 270 °C at catalysts loading of 0.15 mol%. In a later work, The reversible hydrogen storage in a hot pressure swing reactor using H18-DBT and dibenzyltoluene cycle catalyzed by Pt/Al2O3 was demonstrated by Wasserscheid et al.97 The reactor was operated at 290–310 °C. At this temperature, both hydrogenation and dehydrogenation can take place catalyzed by Pt/Al2O3. The reaction direction is changed by the hydrogen pressure. Hydrogenation is achieved at high pressures, while dehydrogenation is achieved at low pressures. Between the change of reaction direction, the reactor does not need to be cooled, making this system suitable for the fast start-up. Since the exothermic hydrogenation reaction can operate at a temperature slightly higher than the endothermic dehydrogenation, a heat storage system can be applied to utilize the hydrogenation heat for dehydrogenation. Qi et al. studied the influence of surface hydroxyl groups and oxygen vacancies of Al2O3 on the performance of Pt/Al2O3.98 The surface of Al2O3 was modified by H2 or O2 plasma treatment. Surface treatment by H2 increases the surface oxygen vacancies, while treatment by O2 increases the surface hydroxyl groups.
The catalysts treated by O2 plasma show improved hydrogenation and dehydrogenation activity, selectivity, and long-term performance. The high side reaction activity and poor stability for the catalyst pretreated by H2 plasma are due to the high proportion of low coordinated Pt species. Bessarabov et al. reported that the activity and selectivity of Pt/Al2O3 could be promoted by adding Mg dopants in Al2O3 support.99 The addition of Mg decreases the acidic property of the Al2O3 support, improves the stability of the catalyst, and promotes the desorption of H0-DBT. Kang et al. studied the dehydrogenation of H18-DBT on the Pt-based subsurface alloys (Pt/M/Pt(111), where M = Pd, Cu, or Ni) using DFT calculation.100 The results show that the rate-determined step is the first step of dehydrogenation in the middle ring of H18-DBT for all the surfaces studied. It is proposed that the hydrogen adsorption strength might be a descriptor for the dehydrogenation activity since the d-band centers and reaction energies for the rate-determining step correlate to the hydrogen adsorption energy. The Pt/Pd/Pt(111), tensile-strain-applied Pt/Pd/Pt(111), and tensile-strain-applied Pt/Cu/Pt(111) might be promising catalysts due to the low hydrogen adsorption energy of less than or equal to −0.6 eV.
2.4 N-Heterocyclic molecules
The lower endothermicity of heterocyclic organic liquids compounds is conducive to the dehydrogenation process, which means that heterocycles have the potential as hydrogen storage carriers. However, their disproportion, alkyl transfer, degradation by C–N cleavage, and other side reactions affect their application in industry.101 The introduction of nitrogen atoms into the conjugated structure of aromatic hydrocarbons has been shown to effectively lower the temperature required for dehydrogenation reactions.16,102–104 Researchers have expanded the LHC system with a variety of N-heterocycles, including carbazole,105 benzpyrole,106 pyridine,107 benzopyridine,108 and so on, among which the most studied is N-ethylcarbazole. Noble metal-based catalysts, such as Pd109 and Pt,110 have garnered the most attention for their role in the dehydrogenation of N-heterocycles. To assess the effectiveness of these N-heterocycles within the LHC system, researches rely on various performance indicators, including hydrogen storage capacity, operating conditions, conversions, and turnover frequency (TOF).| Reaction | Catalyst | Conditions | Time (h) | Conv./yield (%) | Cycles | H2 stored (wt%) | Ref. |
|---|---|---|---|---|---|---|---|
| Hydrogenation | LaNi5.5 | 453 K, 7 MPa | 8 | 96.8 | 9 | 5.5 | 145 |
| Ni70/AlSiO | 423 K, 7 MPa | 1.5 | 100 | ∼5.7 | 112 | ||
| Pd/Al2O3-YH3 | 453 K, 10 MPa | 2.0 | 94.8 | 3 | 5.5 | 114 | |
| Co-B/Al2O3-YH3−x | 453 K, 10 MPa | 2.0 | >94 | 3 | 5.5 | 135 | |
| RuPd/LDH | 393 K, 6 MPa | ∼1.3 | 99.3 | 8 | 5.75 | 147 | |
| Ru/YH3 | 363 K, 1 MPa | 100 | 146 | ||||
| Dehydrogenation | LaNi5.5 | 473 K, 0.1 MPa | 6 | 91.7 | 9 | 5.5 | 145 |
| h-BNNS | 393 K | 74 | 111 | ||||
| Pd/KIT-6 | 453 K, 0.1 MPa | 6.0 | 97.4 | 10 | 5.59 | 113 | |
| Pd/Al2O3-YH3 | 473 K, 0.1 MPa | 4.0 | 94.8 | 3 | 5.5 | 114 | |
| Pt/TiO2 | 453 K, 0.1 MPa | 7 | 100 | 5.38 | 117 | ||
| Pd/TiO2 | 453 K, 0.1 MPa | 7 | 100 | 5.25 | 117 | ||
| Rh/TiO2 | 453 K, 0.1 MPa | 7 | 100 | 3.72 | 117 | ||
| Au/TiO2 | 453 K, 0.1 MPa | 7 | 75.7 | 1.59 | 117 | ||
| Ru/TiO2 | 453 K, 0.1 MPa | 7 | 14.1 | 0.42 | 117 | ||
| Pd/Al2O3 | 453 K, 0.1 MPa | 7 | 100 | 4.64 | 117 | ||
| Pd/rGO-EG | 443 K, 0.1 MPa | 12.0 | 100 | 5 | 5.49 | 120 | |
| Co-B/Al2O3-YH3−x | 473 K, 0.1 MPa | 7.0 | >94 | 3 | 5.5 | 135 | |
| Pd3Au1/SiO2 | 453 K, 0.1 MPa | 8.0 | 100 | 5.7 | 148 | ||
| Pd3Ni1/SiO2 | 453 K, 0.1 MPa | 8.0 | 100 | 5.63 | 105 | ||
| Pd3Cu1/SiO2 | 453 K, 0.1 MPa | 8.0 | 100 | 5.47 | 105 | ||
| Pd/MgAl2O4 | 453 K, 0.1 MPa | 98.3 | 149 | ||||
| Pd/MoO3 | 473 K, 0.1 MPa | 1.5 | 5.8 | 136 | |||
| Pd1Co1/Al2O3 | 473 K, 0.1 MPa | 8.0 | 100 | 5.52 | 150 |
Pt catalysts are another kind of catalyst widely used in dehydrogenation reactions, with overall performance close to Pd catalysts. The reaction mechanism of Pt(111) is very similar to Pd(111). From 173 K, DNEC undergoes molecular adsorption at Pt(111) and stepwise dehydrogenation to NEC while heating to 380 K.130 Meanwhile, C–N bond will be broken above 390 K, ∼60 K higher than Pd(111). Al2O3 is the commonly used support for Pt-based catalysts.131 Wasserscheid et al. first applied egg-shell catalysts in the dehydrogenation of DNEC.132 An inert non-porous α-alumina core and an active, porous γ-alumina shell of defined thickness were impregnated with Pt by a hybrid sol–gel suspension process. Their results confirm that pore diffusion strongly affects the dehydrogenation reaction in almost all commercial catalysts. Recently, TiO2 has been applied in Pt-based catalysts.117,133 The catalytic activity of the noble metal catalysts on TiO2 followed the trend of Pt > Pd > Rh > Au > Ru.117 Jiang et al. proved the introduction of the Si–O–Ti species could strengthen the metal-supporting interaction and increase the oxygen vacancy concentration for efficient dedrogenation.134
At present, most catalysts used in the dehydrogenation reaction are noble metal-based catalysts. Non-noble metal catalysts have also been explored due to the scarcity of noble metals. Fu et al. reported that high crystallinity hexagonal boron nitride nanosheets (h-BNNS) performed high catalytic efficiency in the dehydrogenation of DNEC due to the synergistic effect of the nanoporous structure and highly ordered crystalline pattern.111 Zheng et al. prepared a two-direction catalyst, namely Co-B/Al2O3-YH3-x, for reversible hydrogen storage in NEC/DNEC,135 which is the first non-noble metal catalyst used for both NEC hydrogenation and DNEC dehydrogenation reaction. Synthesis of bimetallic catalysts is also an important strategy for dehydrogenation catalysts, which usually have catalytic activity for both hydrogenation and dehydrogenation.114,136,137 You et al. investigated the effect of interheteromolecular hyperconjugation on heterogeneous (de)hydrogenation catalyzed by Rh–Pd loaded silicon–aluminium oxide supports (Rh–Pd/SAO).138 Sun et al. synthesized reusable bimetallic Pd–Rh nanoparticle catalysts.139 The optimized catalyst performs a hydrogen release of 5.48 wt% in 4 h and the maximum hydrogen uptake of 5.43 wt% in 1 h. The good activity is ascribed to the synergistic effect between Pd and Rh nanoclusters.
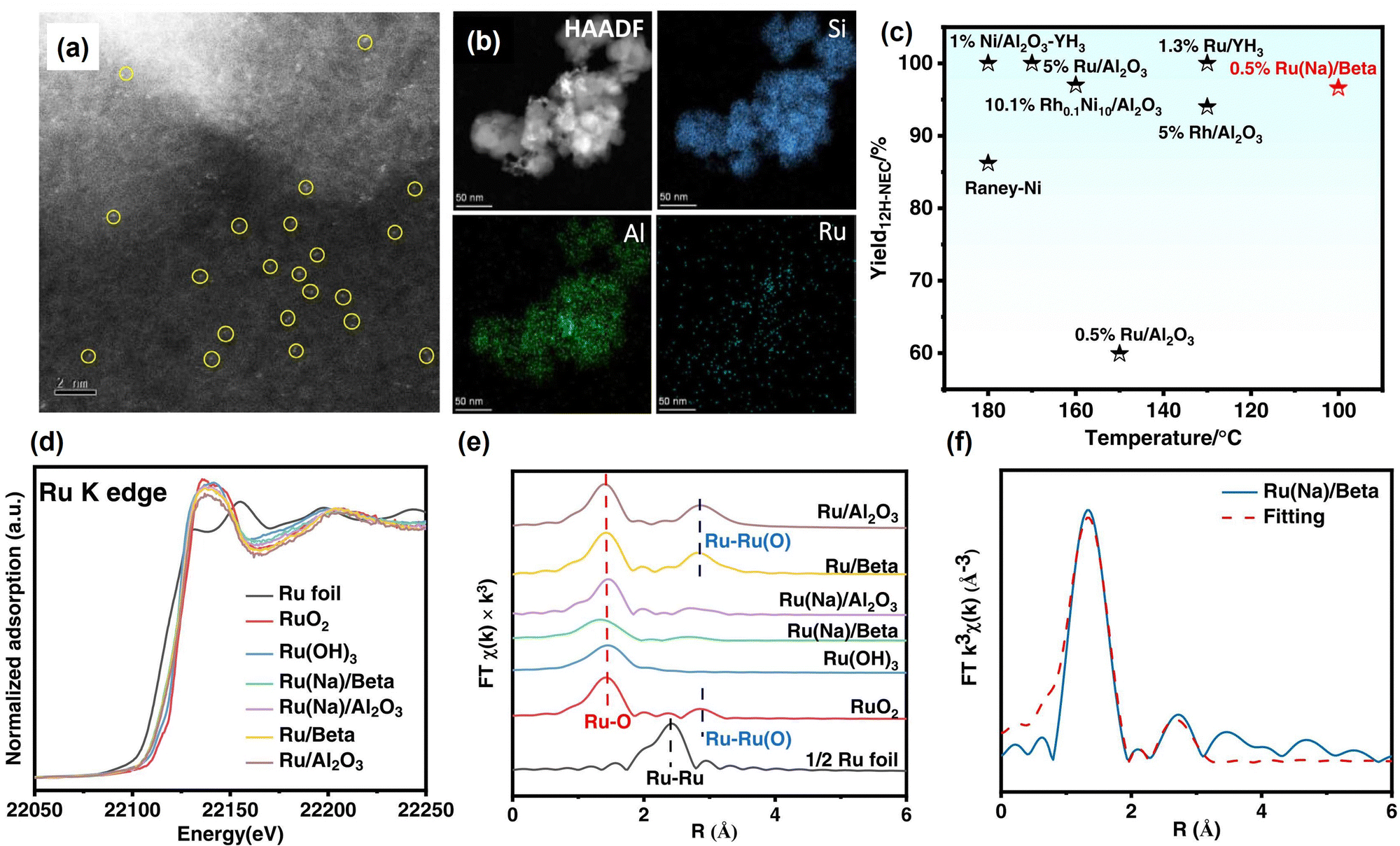 | ||
| Fig. 3 (a) Aberration-corrected HAADF-STEM image and (b) corresponding element mapping images of Ru(Na)/Beta. (c) Comparison of the performance of different catalysts in the hydrogenation of N-ethylcarbazole. (d) Ru K-edge XANES spectra, (e) k3-weighted Fourier transform Ru K-edge EXAFS spectra, and (f) EXAFS fitting curve of Ru(Na)/Beta.144 Modified with permission from ref. 144. Copyright (2022) Elsevier Inc. | ||
3. Methanol
The consumption of fossil fuels increases the emission of carbon dioxide (CO2). Carbon capture and utilization (CCU) is an effective way to reduce the excessive CO2 concentration in the atmosphere.155 Furthermore, the synthesis of methanol from CO2 and hydrogen is a feasible way to reduce the concentration of CO2 in the atmosphere.156 Methanol has been proposed to replace fossil fuels partially.157 From another perspective, it is possible to use methanol as LHC. Methanol can store 12.6 wt% H2 in each molecule, which is higher than cyclohexane and other LHC substances. Methanol is a stable liquid substance at room temperature that can be easily transported. Hydrogen can be generated from methanol through steam reforming as shown in the following equation.| CH3OH + H2O = CO2 + 3H2 ΔHθ289K = +49.7 kJ mol−1 |
MSR reaction is endothermic and heat from an external source is required. On the other hand, due to the absence of C–C bond in methanol molecule, its steam reforming can be achieved at a relatively low temperature (150–350 °C) compared to other alcohols and hydrocarbons. During MSR, the reaction of methanol decomposition and water–gas shift reaction158 might also occur.
| CH3OH = CO2 + 2H2 ΔHθ289K = +90.7 kJ mol−1 |
| CO + H2O = CO2 + H2 ΔHθ289K = +41.2 kJ mol−1 |
The main drawback of methanol decomposition is the byproduct of this process, CO, which can poison some catalysts. Therefore, reducing the CO content in the effluent is one of the targets of concern. In recent years, there have been many reports on the dehydrogenation of methanol to hydrogen. However, in many catalytic reactions, less than three molecules of hydrogen can be obtained from a single molecule of methanol. The by-products, CH4 and CO, will reduce the purity of the hydrogen product, which brings additional problems of hydrogen purification.
3.1 Heterogeneous catalysis for MSR
Catalysts play a crucial role in methanol steam reforming by increasing the reaction rate. Heterogeneous catalysts, in particular, have garnered attention in this area due to their benefits in large-scale applications, such as good stability, simple separation, and reusability. A wide range of catalysts, including Cu, Pt, Pd, and metal oxide-based catalysts, have been explored for their potential use in MSR (Table 6).| Catalyst | Temp. (°C) | Pressure (MPa) | Space velocity | Methanol conv. (%) | CO2 sel. (%) | CO sel. (%) | Ref. |
|---|---|---|---|---|---|---|---|
| 15 wt% Cu/ZnO | 300 | 0.1 | 1.4 h−1 (WHSV) | 82.2 | ∼2 | 172 | |
| Cu/ZnO/Al2O3 | 225 | 0.1 | 6 h−1 (WHSV) | 67.0 | 0.07 | 173 | |
| Cu/ZnO/Al2O3 | 263 | 0.1 | 25714 h−1 (GHSV) |
58 | 2.5 | 204 | |
| 10 wt% Cu/ZnO–Al | 250 | 0.1 | 15.3 h−1 (WHSV) | 57 | ∼98.7 | 235 | |
| 5% MgO–Cu/ZnO/Al2O3 | 200 | 0.1 | 3.84 h−1 (WHSV) | ∼70 | ∼1 | 174 | |
| 30% Ni–Cu/Al2O4 | 300 | 0.1 | 0.9 h−1 (WHSV) | 96.89 | 33.5 | 181 | |
| Ru–Cu/ZnO/Al2O3 | 240 | 0.1 | 4 h−1 (WHSV) | 95 | 1.97 | 175 | |
| CuNiAlOx spinel oxide | 255 | 1 | 1.09 h−1 (WHSV) | ∼92 | <0.8 | 180 | |
| CuFeMg/γ-Al2O3/Al mesh | 250 | 0.1 | 3000 mL gcat−1 h−1 (GHSV) | 93.5 | ∼4 | 199 | |
| CuTi1.9/γ-Al2O3/Al mesh | 275 | 0.1 | 4000 mL gcat−1 h−1 (GHSV) | ∼100 | ∼3 | 236 | |
| 20 wt% Cu/ZrO2–Al2O3 | 240 | 0.1 | 3.8 h−1 (WHSV) | 95 | 0.37 | 185 | |
| Cu/ZrO2-10 wt% SiO2 | 260 | 0.1 | 5 h−1 (WHSV) | 73 | <0.1 | 186 | |
| Cu/ZnO/ZrO2 | 300 | 0.1 | 24.9 h−1 (WHSV) | 84 | 1.6 | 203 | |
| Cu/ZnO/ZrO2/Al2O3 | 305 | 0.1 | 0.77 h−1 (WHSV) | 80 | 3.6 | 205 | |
| CuO/ZnO/ZrO2/Al2O3 | 270 | 0.1 | 8.7 h−1 (WHSV) | ∼80 | ∼0.7 | 237 | |
| CuO/ZnO/CeO2/ZrO2/Al2O3 | 270 | 0.1 | 8.7 h−1 (WHSV) | 90.5 | 0.91 | 238 | |
| CuO/ZnO/ZrO2/Al2O3 | 270 | 0.1 | 8.7 h−1 (WHSV) | 92.4 | 0.97 | 238 | |
| 2 wt% In2O3–Cu/ZnO/ZrO2 | 400 | 0.1 | 29.1 h−1 (WHSV) | 79 | 1.7 | 239 | |
| CuPd/ZrO2 | 260 | 0.1 | 1.4 h−1 (WHSV) | ∼86 | 5 | 187 | |
| CuZnGaOx | 150 | 0.1 | 0.12 h−1 (WHSV) | 36 | <1 ppm (Conc.) | 189 | |
| Cu/CeO2 | 200 | 0.1 | 6.2 h−1 (WHSV) | 7.4 | <0.5 | 240 | |
| Ce0.8Cu0.2Ox | 300 | 0.1 | 36000 mL gcat−1 h−1 (GHSV) |
∼99 | ∼1 | 241 | |
| Ce–Cu/KIT-6 | 325 | 0.1 | 2 h−1 (WHSV) | 96.8 | 3.7 | 198 | |
| Cu–Ce/SBA-15 | 250 | 0.1 | 6 h−1 (WHSV) | ∼82 | ∼1.2 | 197 | |
| ZrO2–CeO2–Cu/KIT-6 | 300 | 0.1 | 2 h−1 (WHSV) | 96 | 0.7 | 242 | |
| Cu-MCM-41 | 300 | 0.1 | 2838 h−1 (GHSV) | 69.9 | 16.4 | 243 | |
| 10% Cu/γ-Al@ZnAlOx | 300 | 0.1 | 800 h−1 (GHSV) | 99.98 | 0.92 (Conc.) | 176 | |
| CeCuZn/CNTs | 300 | 0.1 | 7.5 h−1 (WHSV) | 94.2 | 2.6 | 244 | |
| CuIn/SiO2 | 260 | 0.1 | 18 h−1 (WHSV) | 71.20 | 0.08 | 195 | |
| CuZn/MCM-41 | 300 | 0.1 | 2.85 h−1 (WHSV) | 88 | <1.8 | 196 | |
| Ru1/CeO2 | 350 | 0.1 | 6 h−1 (WHSV) | 25.6 | 97.8 | 206 | |
| Pt/NiAl2O4 | 210 | 2.9 | 2.94 h−1 (WHSV) | >99.9 | 99.72 | 211 | |
| 0.5 wt% Zn–Pt/MoC | 160 | 0.1 | 1.8 h−1 (WHSV) | 65.9 | 0 | 210 | |
| Pt–SnO/MIL-101(Cr) | 300 | 0.1 | 1.8 h−1 (WHSV) | 92 | 3.4 | 245 | |
| KOH-Pt/Al2O3 | 230 | 0.5 | 360 h−1 (WHSV) | <1 | 214 | ||
| Pt/In2O3/Al2O3 | 350 | 0.1 | 3 h−1 (WHSV) | 100 | 3.2 | 246 | |
| In2Pt | 400 | 0.1 | 7 h−1 (WHSV) | 0.2 | 247 | ||
| Pt/In2O3/CeO2 | 350 | 0.1 | 3 h−1 (WHSV) | ∼100 | <4.5 | 248 | |
| Pt/In2O3 | 300 | 0.1 | 11 h−1 (WHSV) | 0.6 | 249 | ||
| PtNi/CeO2 | 400 | 0.1 | 31 h−1 (WHSV) | 100 | ∼20 | 207 | |
| K-Pt@Silicalite-1 | 400 | 0.1 | 1.0 h−1 (WHSV) | 100 | 1.9 | 212 | |
| Pd/ZnO | 250 | 0.1 | 2.03 mL gcat−1 s−1 (WHSV) | ∼67 | ∼15 | 218 | |
| 10 wt% Pd/ZnO | 220 | 0.1 | 0.47 s−1 (WHSV) | ∼58 | 2 | 216 | |
| 15 wt% Pd/ZnO | 250 | 0.1 | 3.4 h−1 (WHSV) | ∼65 | 4 | 250 | |
| 0.1 wt% Pd/ZnAl2O4 | 250 | 0.1 | 3.3 h−1 (WHSV) | <20 | 3 | 220 | |
| 4 wt% Pd/ZnO (002) | 330 | 0.1 | 30130 mL gcat−1 h−1 (GHSV) |
97.3 | 2.7 | 219 | |
| Fe/Mo2C | 200 | 0.1 | 9000 mL gcat−1 h−1 (GHSV) | ∼8 | ∼4 | 251 | |
| Co/Mo2C | 200 | 0.1 | ∼12 | ∼30 | 251 | ||
| Ni/Mo2C | 200 | 0.1 | ∼26 | ∼30 | 251 | ||
| Pt/Mo2C | 200 | 0.1 | 100 | ∼8 | 251 | ||
| Cu/Mo2C | 400 | 0.1 | ∼100 | 8 | 252 | ||
| 1% Au/CeO2 | 225 | 0.1 | 42000 h−1 (GHSV) |
∼50 | 253 | ||
| Au/CuO–CeO2 | 300 | 0.1 | 14.8 h−1 (WHSV) | ∼79 | 0.43 | 254 | |
| Au–Cu/CeZrOx | 350 | 0.1 | 21000 mL gcat−1 h−1 (GHSV) |
100 | ∼3 | 229 | |
| Au/ZnO | ∼300 | 0.1 | 50 | 20 | 228 | ||
| ZnO/ZnZrOx | 380 | 0.1 | 3.3 h−1 (WHSV) | 99.7 | 2.4 | 233 | |
| ZnCeZrOx | 400 | 0.1 | 4.5 h−1 (WHSV) | 99.8 | 6.0 | 234 | |
| ZnO | 300 | 0.1 | 0.18 h−1 (WHSV) | 3.6 | 99.6 | 231 | |
| ZnO–Cr2O3/CeO2–ZrO2/Al2O3 | 460 | 0.1 | 22594 mL gcat−1 h−1 (GHSV) |
∼100 | ∼9 | 232 | |
| ZnO–Al2O3 | 420 | 0.1 | 13275 mL gcat−1 h−1 (GHSV) |
∼100 | ∼6 | 255 |
Cu/ZnO and Cu/Al2O3 catalysts. Among copper catalysts, the most-studied catalysts are Cu/ZnO and Cu/Al2O3 based catalysts. Ressler et al. studied the structure change of Cu/ZnO during reduction and MSR using in situ X-ray diffraction (XRD) and X-ray absorption spectroscopy (XAS).167 Reaction results show that catalysts reduced with the mixture of H2 (2.5%) and H2O (3%) exhibit lower CO selectivity than catalysts reduced by H2 alone (2%) at similar methanol conversion levels. The effect of reduction followed by an oxidation cycle was also studied. The addition of oxygen in the reactant stream transfers the metallic Cu into Cu+ and Cu2+ with the loss of catalytic activity. Besides, the methanol conversion and CO2 selectivity increase with repeated oxidation/reduction cycle. In situ XRD shows that the lattice parameters of Cu and ZnO increase after the oxidation/reduction cycle, indicating an expansion of the unit cell. XAS shows that the medium-range structure disorder of Cu increases with a reduced Zn concentration in Cu clusters after repeated oxidation and reduction. Zn migrated out of Cu clusters and formed ZnO on the Cu surface with increased interface interaction between Cu and ZnO during this process. The influence of aging during preparation on the structure and activity for the Cu/ZnO catalyst was studied by Ressler et al.170,171 Four Cu/ZnO catalysts with different aging times (0, 15, 30, and 120 min) were investigated. An amorphous precursor was obtained with short (15 min) or without aging, while crystalline hydroxycarbonate was obtained after a long aging time. For the catalysts after calcination, the longer aging time results in a smaller CuO crystallite size and a lower reduction temperature. In situ XRD and XAS show that the microstrain in Cu increases with the aging time. The high Cu surface area and increased microstrain both contribute to the high reaction activity for the catalysts with long aging time. Mendes et al. studied the influence of surface area and polarity of ZnO on the activity of Cu/ZnO.172 Different calcination temperatures (300–400 °C) were used to control the ZnO surface area, while the polarity (002) surface ratio was adjusted by using different Zn precursors. A higher calcination temperature results in a lower surface area of ZnO and Cu, thus a decreased activity. Cu species on the polar surface of ZnO can be reduced more easily, as caused by the strong interaction between Cu and polar facets. The results suggest that the CO2 selectivity increase with the polar surface ratio of ZnO, indicating that the Cu–ZnO polar interface is more selective for MSR. Xu et al. studied the influence of the activation process on the catalytic activity of commercial Cu/ZnO/Al2O3 catalyst.173 The catalyst reduced by H2 first followed by a 10 min reduction under H2/H2O/CH3OH/N2 showed the best activity and stability among various pretreatment conditions. CuO could be reduced to Cu under all activation conditions as confirmed by in situ XRD. Moreover, scanning transmission electron microscopy (STEM) results of the catalysts after pretreatment show that activation in H2 only results in discrete ZnO and Cu nanoparticles. In contrast, activation in H2/H2O/CH3OH/N2 mixture induces the formation of thin layers of ZnO covering the Cu nanoparticles due to the strong metal support interaction (Fig. 4). The formation of ZnOx overlayers outside of the Cu nanoparticle after treatment under H2/H2O/CH3OH/N2 was also confirmed by quasi in situ XPS and IR spectroscopy using CO as a probe molecule. Therefore, the better catalytic performance is ascribed to the formation of abundant ZnOx–Cu interfacial sites after activation in H2/H2O/CH3OH/N2 mixture for a proper period. DFT calculations of the migration energy of small ZnO clusters over the Cu surface indicate that adsorbed CH3OH molecules on the ZnO clusters can accelerate the migration of ZnOx over the Cu nanoparticles, explaining the formation of plenty of Cu–ZnOx interfacial sites for the catalysts activated under H2/H2O/CH3OH/N2 mixture.
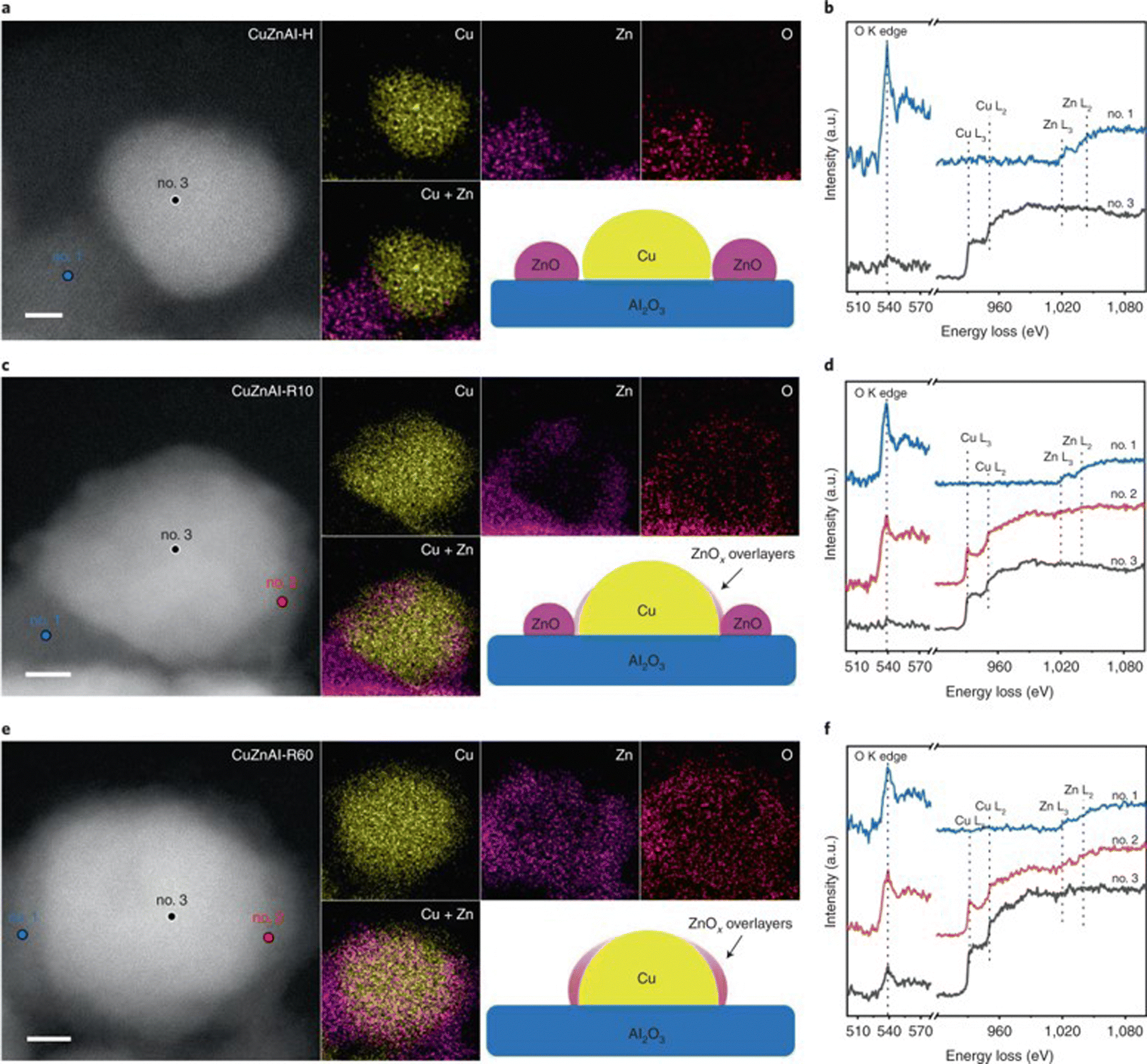 | ||
| Fig. 4 HAADF-STEM images and corresponding EDS elemental maps of CuZnAl-H (reduced in H2/N2), (a), CuZnAl-R10 (reduced in H2/N2) followed by H2/H2O/CH3OH/N2, (c) and CuZnAl-R60 (reduced in H2/H2O/CH3OH/N2), (e). Scale bars, 2 nm. Insets give schematic illustrations of the catalyst structures. EELS spectra of marked regions for CuZnAl-H (b), CuZnAl-R10 (d), and CuZnAl-R60 (f) in (a), (c) and (e), respectively.173 Reproduced with permission from ref. 173. Copyright (2022) Springer Nature. | ||
Li et al. introduced Mg into Cu/ZnO/Al2O3 by co-precipitation.174 The H2 formation rate increases with the increasing Mg content in Cu/ZnO/Al2O3 up to 5% with the decrease of CO2 selectivity. The highest H2 formation rate was 172 mol kgcat−1 h−1 at 200 °C. However, a further high Mg content of 7% leads to a slightly low H2 formation rate and high CO2 selectivity. The inverse trend for H2 formation rate and CO2 selectivity can be explained by the increased reverse water–gas shift reaction at high H2 and CO2 concentrations. Mg2+ can incorporate into the malachite structure, replace the Cu2+ partially and improve the dispersion of Cu and ZnO after calcination. The Cu+/Cu0 ratio increased with the Mg content up to 5%, indicating that Mg can stabilize the Cu+ and promotes the Cu–ZnO synergy, leading to enhanced performance. Hu et al. studied the influence of Pt, Pd, Rh, Au, Ag, Ru, Ni, and Co on CuO/ZnO/Al2O3 derived from precursors with hydrotalcite structure in MSR.175 The catalyst modified with Ru shows the best activity with methanol conversion increase of 20% at 240 °C compared to CuO/ZnO/Al2O3. At the same time, the introduction of Ru does not have a significant influence on CO selectivity. In contrast, the introduction of Ni and Rh improves the CO formation significantly with a slight drop in methanol conversion. Other metals lead to a decrease in methanol conversion without a significant change in CO selectivity. The promotion effect of Ru is ascribed to the electron interaction between Ru and Cu, as confirmed by XPS. The decrease of activity for other metals is due to the partly destroyed lamellar structure and reduced active surface area after impregnation and calcination.
Zhang et al. prepared ZnAl layered double hydroxides (LDHs) on Al2O3 and used them as support precursors for Cu catalyst.176 The ZnAl-LDHs were prepared by precipitation of Zn ions in the presence of Al2O3, and the LDHs were calcinated and transferred into mixed metal oxides before supporting Cu with different loading by impregnation. XRD confirms the formation of the hydrotalcite phase on the support precursor. After calcination, the hydrotalcite phase transfers into metal oxide. The catalyst with 10% Cu loading shows the highest activity with methanol conversion of 99.98% at 300 °C. The high Cu surface area and the easy reduction of Cu due to the interaction with ZnO contributed to the good activity. Gao et al. reported that Cu–Al spinel oxide was active in MSR.177 The Cu–Al spinel catalyst with a high surface area was prepared by the solid-state reaction between Cu(OH)2 and pseudo boehmite. The sintering of Cu during pre-reduction can be avoided by eliminating the pre-reduction step before reaction. During MSR, Cu in the spinel structure was reduced by methanol and released slowly in order to form Cu active sites. The influence of calcination temperature on the activity was systemically studied.178 The catalyst calcinated at 950 °C shows the best activity, which can be ascribed to the modest release rate of Cu from the spinel structure under reaction conditions and the small copper nanoparticles stabilized by the defective spinel structure. A detailed study of the dynamic change of CuAl2O4 reveals that the releasing rate of Cu species increases first and then decreases with time on stream, in agreement with the activity at the initial state of reaction.179 The deactivation after a long time on stream is caused by the coke formation. Even after 300 h on stream, a small amount (5.8%) of Cu species still stays in the spinel lattice. Ni was introduced in the Cu–Al spinel oxide to improve its performance.180 The catalysts were prepared by the solid phase reaction between Cu(OH)2, nickel acetate, and pseudo-boehmite at 900 °C. H2-TPR profiles indicate that the introduction of Ni slows the reduction behaviour. The catalyst with Ni/Cu of 0.05 exhibits the best activity and good stability. A higher Ni content (Ni/Cu = 0.1 and 0.2) results in an increased CO selectivity, which is related to the formation of metallic Ni from the non-spinel NiO at high Ni loading. However, the Ni2+ species in the spinel lattice are stable against reduction under reaction conditions, as proved by the low and stable CO selectivity of the catalyst with low Ni content (Ni/Cu = 0.01 and 0.05). The improvement effect of Ni on Cu–Al spinel oxide catalyst is ascribed to the stabilization effect of Ni containing defective spinel for the small Cu nanoparticles. Hsu et al. also studied the performance of Ni–Cu/Al2O4 in MSR.181 The catalysts with Cu/Ni ratios of 10:1, 5:1, 10:3 and a fixed Cu/Al ratio of 1:10 were prepared by a precipitation–adsorption method.
Characterization results show that the as-synthesis catalysts possess Ni–Cu/Al2O4 spinel structure. For the catalyst with the highest Ni content, NiCu alloy can also be observed by XRD. The stability test shows only a small amount of coke (∼1.0 wt%) formed on the catalyst after 20 h on stream, which can be removed by calcination in air. However, the selectivity to CO increases after regeneration.
The mechanism of methanol steam reforming on Ni–Cu based catalysts (Ni@Cu(111) and Ni@Cu(110) models) was studied using DFT calculations by Fajín et al.182 The possible reaction routes include methanol decomposition, water–gas shift reaction, direct CO2 production from reforming, and formation of methane. Coke can form from the dissociation of adsorbed species like COH* or from the dehydrogenation of 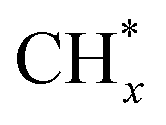 species. According to the results, methanol decomposition followed by the water–gas shift reaction is the main reaction route on the Ni–Cu alloy surface, while CO2 formation from methanol direct reforming is a minor route. The desorption of CO, formation of methane, and coke on the Ni–Cu surface are unfavorable due to the high energy barriers.
species. According to the results, methanol decomposition followed by the water–gas shift reaction is the main reaction route on the Ni–Cu alloy surface, while CO2 formation from methanol direct reforming is a minor route. The desorption of CO, formation of methane, and coke on the Ni–Cu surface are unfavorable due to the high energy barriers.
Cu/ZrO2 catalysts. Penner et al. compared the performance of Cu/ZrO2 catalysts prepared with different ZrO2 phases (m-ZrO2 and t-ZrO2), synthesis methods, and Cu precursors in MSR.183 Cu/t-ZrO2 exhibited a higher activity with higher CO selectivity compared to Cu/m-ZrO2. The difference is explained by the more defective and hydroxylated surface of t-ZrO2 with plenty of reactive Lewis acidic and Brønsted basic sites. Different binding sites for methanol and water on the surface of t-ZrO2 promote the decarbonylation and splitting of C–O bond, leading to the formation of CO and CH4. On Cu/m-ZrO2, the CO formation comes from the spillover of formate to the support exclusively, while the Cu–ZrO2 interface sites on Cu/t-ZrO2 also contribute to the CO formation. Meanwhile, the Cu precursors used in synthesis show some influence on the activity and product selectivity. The performance of Cu/ZrO2 with different Cu loadings was also studied by Penner et al.184 Cu dispersion decreased from 18% to 0.09% with increasing Cu content from 0.2 wt% to 80 wt%. The Cu morphology was also influenced by the Cu loading, changing from a highly dispersed state at low loading over nanoparticles to bulk Cu grains wetting and covering ZrO2 at 80 wt%, with the change of fraction of exposed ZrO2 simultaneously. It is interesting that the TOF value increases with increasing Cu loading despite the reduced Cu dispersion. Moreover, 80 wt% Cu/ZrO2 showed the highest H2 TOF of ∼0.015 s−1 at 220 °C. Covering of exposed ZrO2 surface by Cu reduced the exposure of CO generation sites on ZrO2. Mateos-Pedrero et al. used ZrO2–Al2O3 with different Zr/Al ratios prepared by hydrothermal route as supports for Cu and tested in MSR.185 The increase of support Zr/Al ratio up to 1 results in a decrease of CuO crystallite size for the as-prepared catalysts. However, a further increase of Zr/Al ratio higher than 1 leads to an increase in CuO crystallite size as evidenced by XRD. The dispersion of Cu, influenced by the Zr/Al, is revealed by N2O chemisorption. The catalyst with Zr/Al ratio of 0.4 shows the highest Cu dispersion of 19.7% and the best activity, suggesting the activity is closely related to the dispersion of Cu.
Dal Santo et al. investigated the influence of Si on Cu/ZrO2 for MSR.186 Characterization results show that the introduction of Si results in more amorphous content in the final catalyst and changes the reduction behaviour as well as the final size of Cu species. The Cu/ZrO2–SiO2 with 10 wt% SiO2 content shows the best methanol conversion of 73% and hydrogen productivity of 370 mmol h−1 gcat−1 (with the lowest apparent activation energy of 54.9 kJ mol−1), which is higher than Cu/ZrO2 and Cu/ZrO2–SiO2 with other SiO2 contents. The improved activity is ascribed to the Lewis acidity from the electron-deficient small Cu nanoparticles. XPS results clearly demonstrate the introduction of Si results in a lower electron density for Zr and Cu. Mendes et al. investigated the influence of the impregnation sequence of Cu and Pd for CuPd/ZrO2 in MSR.187 The results show that the impregnation sequence has a significant impact on the property and activity of catalysts. Segregated Pd nanoparticles form on the catalyst prepared by impregnating Cu first, while Cu and Pd are well dispersed and homogeneous mixed with the formation of CuPd alloy on the catalyst prepared by adding Pd first. The catalyst with Pd loaded first shows a higher activity, achieving methanol conversion of ∼86% with CO2 selectivity of ∼95% at 260 °C. Xiao et al. used CuZn containing MOFs as precursors for the construction of CuO/ZnO/CeO2/ZrO2 catalysts for MSR.188 Typically, Cu/Zn/CuZn-BTC were impregnated with other metal precursors and calcinated in air at 500 °C to form CuO/ZnO/CeO2/ZrO2 mixed oxides. Then the oxides were mixed with PVA and deposited on the cordierite honeycomb ceramic support to get the final catalyst. The catalyst derived from CuZn-BTC shows better activity than the catalyst derived from Cu-BTC or Zn-BTC. Characterization results show that the addition of Cu2+ during the formation of CuZn-BTC MOF introduces lattice distortions and increases the specific surface area. Meanwhile, the ZnO formed during calcination can act as a spacer to prevent the sintering of CuO, resulting in the formation of Cu active sites with fine and uniform dispersion and high activity. The catalyst also shows low CO selectivity, which is related to oxygen vacancies on the CexZr1−xO2.
Other Cu-based catalysts. According to thermodynamic calculation, low reaction temperature (100–150 °C) is favorable for MSR while equilibrium shifts to methanol decomposition at high temperature and results in the formation of CO. Therefore, it is important to develop catalysts active at low temperatures. Tsang et al. reported that CuZnGaOx could achieve MSR with a very low CO content (<1 ppm) at 150 °C.189 The incorporation of Ga3+ into Cu/ZnO promotes the reduction of Cu species at lower temperatures, as proved by H2-TPR. Through reduction, small Cu clusters of 0.4–0.8 nm are formed from trapped Cu ions, leading to the high activity at low reaction temperature. Kinetic analysis suggests that the selectivity to CO can be lowered by increasing contact time. Chiu et al. studied a series of Cu-containing delafossite materials, including CuCrO2,190,191 CuCrxFe1−xO2,192 CuFeO2–CeO2,193 and CuYO2194 in MSR. The promotion effect of In2O3 on Cu/SiO2 was studied by Santo et al.195 The CO selectivity drops from 0.37% to 0.08% at 260 °C while the H2 formation rate increases from 237 mmol h−1 gcat−1 to 301 mmol h−1 gcat−1 with the addition of 1 wt% In. The stronger reduction peak for Cu–1In/SiO2 than Cu/SiO2 in CO-TPR indicates that In promoted the reduction of Cu. H2O-TPD further reveals that InOx can promote the activation of water.
Mesoporous silica with ordered structure and carbon nanotubes have also been used for catalysts construction. Jibril et al. compared the performance of CuZn/MCM-41 prepared by impregnation and hydrothermal routes.196 The best activity is achieved by co-impregnation, offering methanol conversion of 88% and CO selectivity of less than 1.8%. Huang et al. studied the influence of Ce at Cu/SBA-15 on the catalytic performance.197 The finely dispersed Ce species show strong interaction with Cu as confirmed by CO-adsorbed FTIR. Reaction results indicate that Cu–Ce/SBA-15 with Ce ion exchange for one time exhibits the best CO2 selectivity and stability. The improved CO2 selectivity is explained by the redox properties of Ce species, which facilitate H2O activation. Taghizadeh et al. studied the performance of Ce–Cu/KIT-6 in MSR.198 Enhanced activation of water by CeO2 results in methanol conversion of 96.8% and CO selectivity of 3.7% at 325 °C. Zhang et al. prepared mesh-type structured CuFeMg/γ-Al2O3/Al catalysts and used in MSR.199,200 A commercial Al mesh was used as substrate and Al2O3 was fabricated by anodic oxidation and calcination. The Cu, Fe, and Mg species were introduced by sequential impregnation. The introduction of Mg decreases the formation of dimethyl ether, while Fe acts as both electronic and structure promoters.200 The comparison of the two catalysts in the same size range (20–50 mesh) indicates that the commercial granular catalyst shows a better intrinsic activity as its activity at low temperatures is higher. However, when used in the shaped form, the mesh-type catalyst shows better performance than the granular catalyst. This should be ascribed to the better mass transportation for the mesh-type catalyst. Further, the arrangement of mesh-type catalysts in the reactor can influence the activity due to the difference in the effective contact area. The hole-to-edge arrangement can improve the methanol conversion by 14% compared to the hole-to-hole arrangement.
Cu-Based catalysts tend to deactivate due to different reasons such as sintering, change of oxidation states, and coke deposition. The poisons such as sulphur and chloride can also lead to deactivation.169,201 Valdés-Solís et al. concluded that coke formation and sintering lead to the deactivation of Cu/ZnO/ZrO2 and Cu/MnOx.202 Adding s small amount of oxygen in the feed may be a solution to coke formation and also change the reaction partially to oxidative methanol steam reforming. However, the amount of H2 produced will be reduced. Matsumura et al. studied the deactivation of Cu/ZnO/ZrO2 in MSR.203 It is found that the growth of Cu nanoparticles is not significant during the reaction. The deactivation is ascribed to the growth of ZnO, which changes the interaction between Cu and ZnO. Hagelin-Weaver et al. studied the deactivation of nanoparticle alumina supported Cu/ZnO.204 XPS shows that the surface Zn/Cu ratio increases after the reaction, indicating that the Cu surface is covered by ZnO due to the migration of ZnO under reaction conditions. It is concluded that the deactivation is related to the reduced Cu surface area due to ZnO cover. No carbon build-up can be observed, indicating that coking is not the deactivation reason for this catalyst. In another work, the deactivation of Cu/ZnO based catalysts is ascribed to the sintering of Cu nanoparticles.205 Pettersson et al. studied the poison effect of sulphur and chlorine on Cu/ZnO/Al2O3.169 It is found that sulphur is more detrimental to the catalysts than chlorine. The poison effect of sulphur may be due to the formation of ZnS or ZnSO4 from ZnO and sulphur, while the poison effect of chlorine may occur from the formation of volatile copper chloride compounds. The high reaction temperature is also detrimental due to the sintering. Adding promoters such as ZrO2, Al2O3, CeO2, and In2O3 can improve the stability of Cu-based catalysts by modifying the Cu support interaction and inhibiting sintering. Using zeolite may also be a promising method to improve stability by spatial confinement.
Ru, Pt, and Ni based catalysts. Su et al. investigated the MSR on single-site Me/CeO2 catalysts where Me includes Pt, Pd, Rh, and Ru.206 The catalysts were prepared by an ascorbic acid-assisted reduction method. Ru1/CeO2 shows the highest methanol conversion and CO2 selectivity compared to other catalysts, which can be explained by the strong adsorption of CO on the Ru1/CeO2 as confirmed by CO-TPD. It is suggested that MSR is a tandem reaction, consisting of methanol decomposition to CO and the subsequent wafer–gas shift reaction. On the single metal site, the intermediate CO comes from methanol dehydrogenation. CO can either desorb or react with water adsorbed on the neighbouring Ce3+ sites to produce CO2. The strong adsorption of CO on Ru site can promote the water–gas shift reaction and the formation of CO2, leading to low CO selectivity. On the Ru/CeO2 surface, the single Ru sites and neighbouring Ce3+ sites construct an ensemble reaction pool, catalyzing the methanol decomposition and water–gas shift reaction efficiently (Fig. 5).
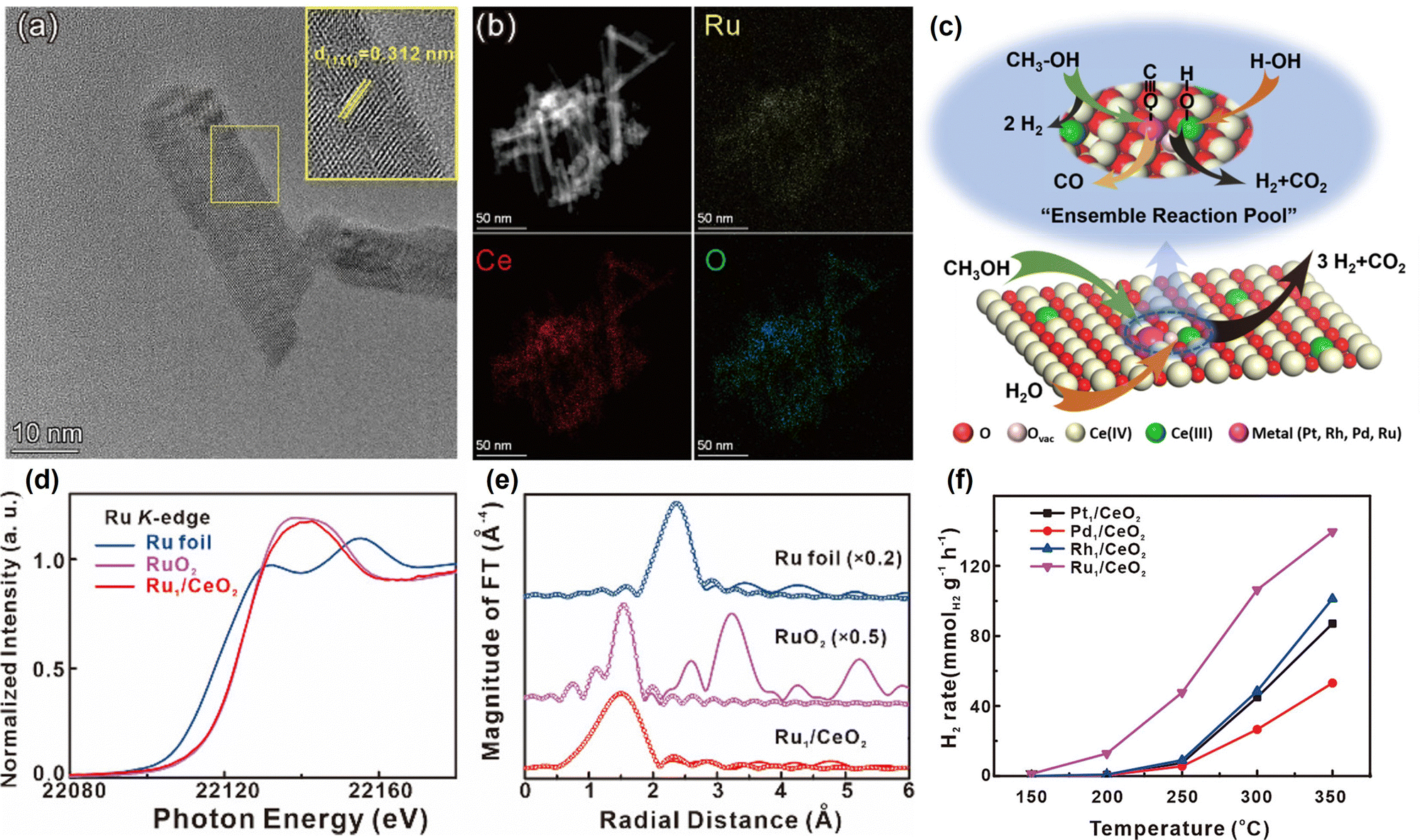 | ||
| Fig. 5 (a) HRTEM image, (b) HAADF-STEM image, and elemental mapping images of Ru1/CeO2. (c) Proposed mechanism for methanol steam reforming on M1/CeO2 single-site catalysts. (d) Normalized Ru K-edge XANES spectra and (e) k3-weighted Fourier transform EXAFS spectra for Ru K-edge of Ru1/CeO2, RuO2, and Ru foil. (f) H2 formation rate for different catalysts.206 Modified with permission from ref. 206. Copyright (2021) American Chemical Society. | ||
The performance of PtNi/CeO2 in MSR was studied by Hernández et al.207 PtNi/CeO2 shows higher activity and H2 yield than Pt/CeO2 and Ni/CeO2. Unfortunately, a high methane yield of ∼10% and a CO yield of ∼10% are achieved with PtNi/CeO2. The CO-adsorbed FTIR indicates that the surface of PtNi/CeO2 is terminated by Pt atoms. Mechanism studies show that methoxy species from methanol adsorption react with the surface oxygen and transform into formate species during the reaction. Ma et al. reported that single atom Pt supported on α-MoC enabled MSR at 150–190 °C with a very high average TOF of 18046 h−1 calculated based on Pt.208 The Pt/α-MoC catalysts were prepared by temperature-programmed carburization of Pt/MoO3 under a flowing mixture of H2 and CH4. The support, α-MoC, shows strong interaction with Pt and facilitates the atomic dispersion of Pt, as proved by STEM and XAS. DFT calculations suggest that Pt/α-MoC provides bifunctional sites for MSR. The electron transfer from Pt to α-MoC makes Pt1 sites electron deficient. Methanol is activated on the Pt site with an energy barrier of 0.67 eV and decomposes to adsorbed CO. Water is adsorbed and activated on α-MoC, forming hydroxy groups, which react with the adsorbed CO to produce CO2 and finish the reforming reaction. The excellent activity is ascribed to the synergy effects between Pt1 sites and α-MoC (Fig. 6).
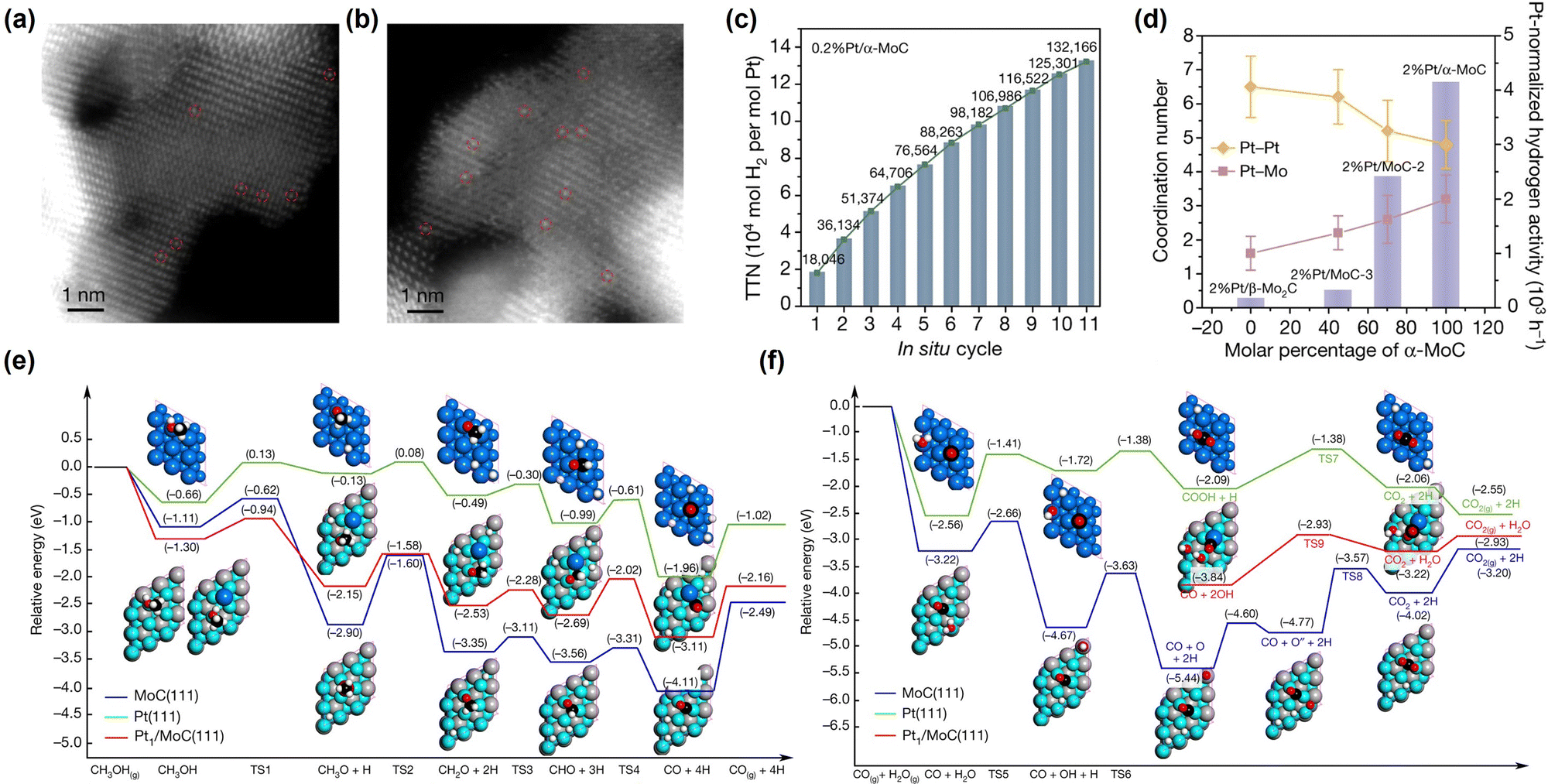 | ||
| Fig. 6 (a and b) HAADF-STEM images of (a) fresh and (b) used 0.2% Pt/α-MoC. (c) Cycling performance of 0.2% Pt/α-MoC in aqueous-phase methanol reforming. (d) The coordination numbers of Pt–Pt and Pt–Mo shells and the activity change with the change in the molar ratio of α-MoC in Pt/MoC. (e and f) Energy profiles for CH3OH dissociation and CO2 formation on α-MoC(111), Pt(111), and Pt1/α-MoC(111) surfaces.208 Modified with permission from ref. 208. Copyright (2017) Springer Nature. | ||
In a later work, Ma et al. reported that atomically dispersed Ni on α-MoC could also catalyze MSR efficiently.209 A series of Ni catalysts with different Ni loadings were prepared by impregnation under inert gas protection. The best activity is achieved with 2% Ni/α-MoC, which offers an average TOF of 1805 h−1 at 240 °C. Although this catalyst shows low CO selectivity of 0.7%, the methane selectivity was very high (∼20%). XPS results reveal the charge transfer from Ni to support and the strong electronic interaction between Ni and α-MoC. The local coordination study shows that Ni is anchored on the surface of α-MoC through carbon bridge bonds. The MSR mechanism on this catalyst is studied by DFT calculations and the decomposition of adsorbed methoxy is established as the rate-determining step. The atomically dispersed Ni sites are responsible for the C–H bond activation and CO reforming, while H2O activation is more easily on the Mo terminated α-MoC site. Therefore, the Ni–C–Mo interfacial sites play an important role in MSR reaction. Sun et al. investigated the promotion effect of Zn on the Pt/MoC catalyst.210 A series of Zn–Pt/MoC with different Zn loadings were prepared by temperature-programmed carburization of mixtures of ammonium molybdate, chloroplatinic acid, and ZnO. Characterization results show that the introduction of a suitable amount of Zn can promote the formation of the α-MoC1−x phase and improve the dispersion of Pt species through the electronic interaction between Zn and Pt/Mo. The best performance is achieved with Zn–Pt/MoC with Zn content of 0.5%, which shows methanol conversion of 65.9% without the detection of CO at 160 °C. The activity decreases when Zn content is higher than 0.5%, possibly because of the formation of PtZn alloy. The effect of H2O/CH3OH ratio on the stability of the catalyst is also studied. High H2O/CH3OH ratios will accelerate the deactivation of the catalysts, possibly due to the oxidation of α-MoCx by water. Mechanism study suggests that aldehyde is the intermediate for MSR, which can react with adsorbed methoxy species to produce methyl formate and finally decomposes into CO2 and hydrogen through formic acid. The aldehyde species can also convert into formic acid directly via the nucleophilic attack of water (Fig. 7).
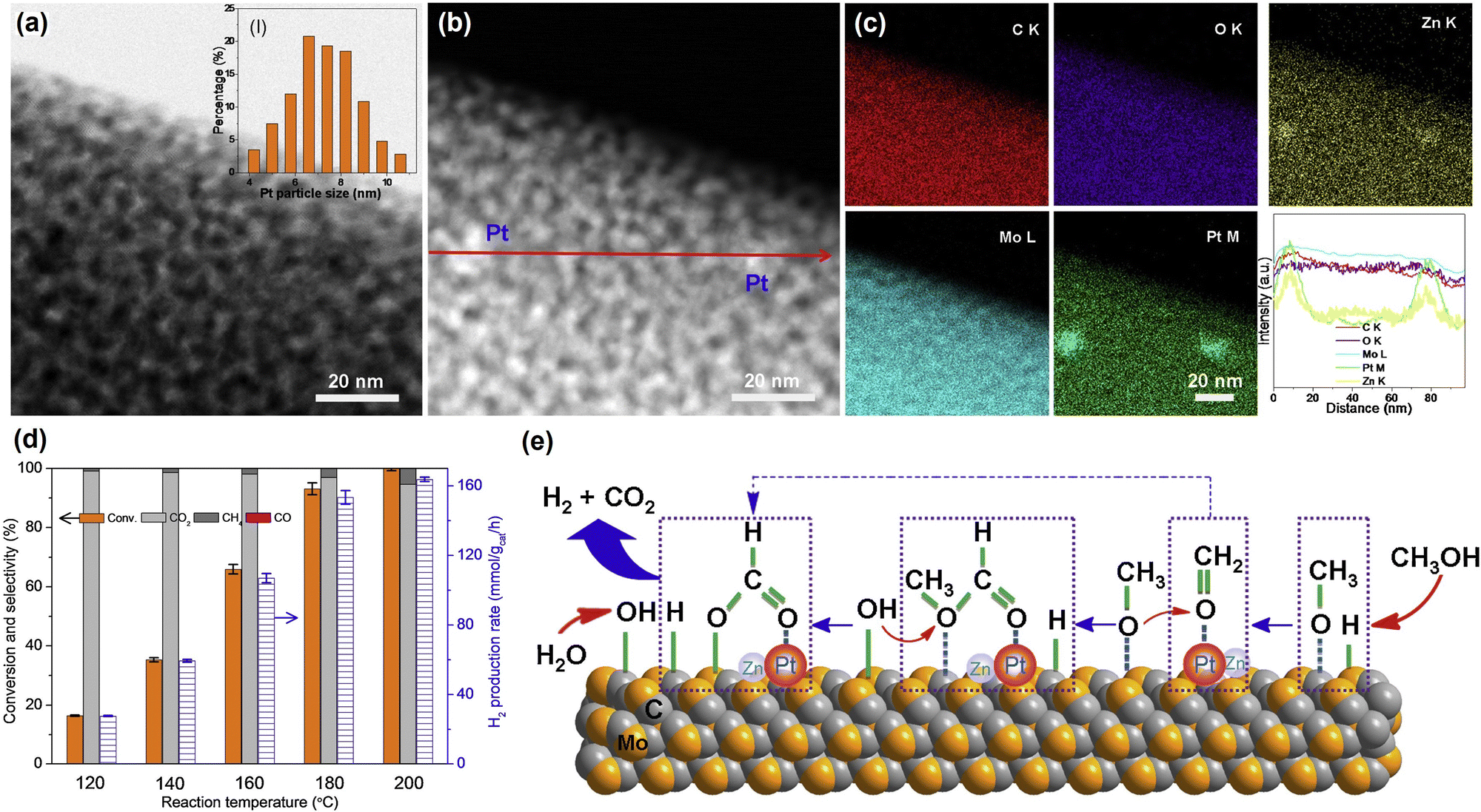 | ||
| Fig. 7 (a) HRTEM image, (b) HAADF-STEM image, and (c) EDX element mapping and line spectra along the red arrow in (b) of 0.5% Zn–Pt/MoC. (d) Catalytic performance of 0.5% Zn–Pt/MoC for methanol steam reforming at different temperatures. (e) Proposed mechanism for methanol steam reforming on 0.5% Zn–Pt/MoC.210 Modified with permission from ref. 210. Copyright (2020) Elsevier Inc. | ||
Guo et al. compared the activity of Pt/NiAl2O4 and Pt/γ-Al2O3 for methanol aqueous phase reforming.211 The Pt/NiAl2O4 catalyst shows a methanol conversion of 99.9% and a H2 yield of 95.7%, much higher than the Pt/γ-Al2O3. This catalyst also exhibits good stability with 90% of the original activity being preserved for 600 h on stream. Mechanism study suggests two tandem reactions, namely methanol decomposition and water–gas shift reaction, occur sequentially on the Pt catalyst. For the first reaction, adsorbed methoxy species form and then dehydrogenate to formaldehyde species and finally to absorbed CO step by step. The Pt species on Pt/NiAl2O4 catalyst under working conditions are closer to the metallic state compared with those on Pt/γ-Al2O3, thus exhibiting higher activity for methanol decomposition. For the water–gas shift reaction, the IR study suggests that the mechanisms for these two catalysts are different. On Pt/γ-Al2O3, adsorbed CO converts to CO2via formate intermediate, while a redox pathway happens on Pt/NiAl2O4. The redox pathway on Pt/NiAl2O4 is faster than the formate pathway on Pt/γ-Al2O3, leading to higher water–gas shift activity for the former catalyst. Sun et al. investigated the performance of K-promoted Pt/silicalite-1 catalyst in MSR.212 The Pt species were confined in the silicalite-1 via a ligand protection one-pot synthesis route and K was introduced by adding KOH in the synthesis gel. The final Pt content in the catalysts is ∼0.3 wt% while the K content varies from 0.1 wt% to 0.8 wt%. The size of Pt nanoparticles is influenced by K loading, possibly due to the Pt–O–Kx interaction. The catalyst with 0.2 wt% K shows the highest Pt dispersion (66.5%) and smallest Pt particle size (1.7 nm), which offers the best activity and lowest CO selectivity. A detailed analysis of the Pt state indicates that Pt0 and Ptδ+ co-exist on the K-modified catalysts. The Pt-0.2 K@S-1 has the highest specific surface area of both Pt0 and Ptδ+, contributing to its high activity. The different roles of Pt0 and Ptδ+ sites are studied by DFT calculations. Pt0 sites are suitable for methanol adsorption and activation, forming HCHO and HCOOCH3. CO is formed more easily on Pt0 sites through the decomposition of HCHO. On the other hand, HCOOH is formed on the Ptδ+ sites from the nucleophilic attack of HCOOCH3 by water, while the decomposition of HCOOH on Ptδ+ sites produces CO2 and H2. The synergy effect between Pt0 and Ptδ+ sites is responsible for the high activity and low CO selectivity. Xiao et al. used titanosilicalite-1 supported Pt catalyst in MSR, in comparison with Pt catalysts supported on ZSM-5, SiO2, and TiO2.213 The Pt nanoparticles on TS-1 show an average diameter of ∼2.6 nm, smaller than those on other supports, indicating that TS-1 can stabilize the Pt nanoparticles more efficiently. CO adsorption results show that Pt0 and Ptδ+ coexist on Pt/SiO2 and Pt/ZSM-5 while the major species on Pt/TS-1 and Pt/TiO2 are Pt0, as also confirmed by XPS and XAS, indicating the enhanced electron transferred from Ti containing support to Pt. Reaction results show that the activity of Pt/TS-1 is highest among all these catalysts, offering a H2 formation rate of 6.6 mmol h−1 gcat−1 at 250 °C. The good activity is ascribed to the small Pt nanoparticle size and the metallic state of Pt. Moreover, this catalyst shows good stability as Pt nanoparticles only sinter slightly to ∼2.8 nm after reaction at 300 °C.
Wasserscheid et al. used molten salt to modify the Pt/Al2O3 catalysts and tested them in MSR.214,215 Methanol conversion and CO2 selectivity both increase with the addition of promoters. The best loading is determined to be 7.5 wt% and 5.35 wt% for KOH and NaOH, respectively. Phase behaviour study showed that under reaction conditions, a liquid film containing alkali hydroxide/alkali carbonate solution forms on the catalyst surface, which increases the local water concentration and contributes to the improved performance. Besides, the electronic interaction between alkali ions and Pt promotes the intrinsic activity of Pt active sites.
Pd-Based catalysts. The most used Pd catalysts are Pd/ZnO. In an early work, Takezawa et al. compared the performance of Pd/SiO2, Pd/ZrO2, and Pd/ZnO in MSR.216 The Pd/ZnO shows higher CO2 selectivity than the other two catalysts. The CO2 selectivity of Pd/ZnO is influenced by the reduction temperature. Characterization results show that PdZn alloy is formed at high reduction temperature and results in the high CO2 selectivity. The morphology of ZnO for Pd/ZnO catalyst has a significant impact on the catalytic activity as reported by Datye et al.217 In a later work, Wang et al. studied the effect of ZnO crystallite faceting in detail.218 The needle-like ZnO predominantly exposed (10
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) nonpolar facets were prepared by a PVP-assisted alcohol thermal method and a commercial ZnO prism without preferential exposed facets was used for comparison. Reaction results show that the selectivity to CO decreases with the increase of Pd loading for both supports. This is ascribed to the increased particle size of PdZn at high Pd loadings. Meanwhile, the needle-like ZnO support shows higher CO selectivity compared to ZnO prism, which is explained by the difficulty in the formation of β-PdZn on the nonpolar (100) surface compared to the (0001) polar facet. On the nonpolar (100) surface, Pd dominated PdxZny (x > y) phase and Pd nanoparticles are easily formed and show higher selective to CO. Huang et al. also studied the facet effect of ZnO. Typically, Pd nanoparticles with narrow size distribution were deposited on the surface of ZnO with different major exposed facets of (002) or (100).219 The results demonstrate that the (002) face of ZnO has a stronger interaction with Pd species, which promotes the reduction of Pd species and the formation of PdZn alloy. Thus, the Pd/ZnO (002) catalysts show better MSR activity and CO2 selectivity. The strong interaction also contributes to the stability of formed PdZn alloy against oxidative decomposition under oxidizing atmosphere. Wang et al. studied the effect of Pd loading (0.1 to 15 wt%) of Pd/ZnAl2O4 in MSR.220 Characterization results show that α-PdZn alloy phase forms on the catalyst with high Pd contents of ≥7.5 wt%, while β-PdZn alloy phase forms on the catalysts with low Pd content of ≤2.5 wt%. The Pd loading has a significant influence on CO2 selectivity. The catalysts with low Pd loadings show high selectivity to CO2 and the highest CO2 selectivity of 97% is achieved with 0.1 wt% Pd/ZnAl2O4. When the Pd content increases to 7.5 wt%, the CO2 selectivity drops to ∼50.5%. Therefore, the β-PdZn alloy phase is more selective for MSR. It is proposed that the polar face of the ZnAl2O4 promotes the formation of the β-PdZn alloy at low Pd loadings while the formation of α-PdZn at high Pd loadings is due to the limited Zn atoms on the ZnAl2O4 surface.
0) nonpolar facets were prepared by a PVP-assisted alcohol thermal method and a commercial ZnO prism without preferential exposed facets was used for comparison. Reaction results show that the selectivity to CO decreases with the increase of Pd loading for both supports. This is ascribed to the increased particle size of PdZn at high Pd loadings. Meanwhile, the needle-like ZnO support shows higher CO selectivity compared to ZnO prism, which is explained by the difficulty in the formation of β-PdZn on the nonpolar (100) surface compared to the (0001) polar facet. On the nonpolar (100) surface, Pd dominated PdxZny (x > y) phase and Pd nanoparticles are easily formed and show higher selective to CO. Huang et al. also studied the facet effect of ZnO. Typically, Pd nanoparticles with narrow size distribution were deposited on the surface of ZnO with different major exposed facets of (002) or (100).219 The results demonstrate that the (002) face of ZnO has a stronger interaction with Pd species, which promotes the reduction of Pd species and the formation of PdZn alloy. Thus, the Pd/ZnO (002) catalysts show better MSR activity and CO2 selectivity. The strong interaction also contributes to the stability of formed PdZn alloy against oxidative decomposition under oxidizing atmosphere. Wang et al. studied the effect of Pd loading (0.1 to 15 wt%) of Pd/ZnAl2O4 in MSR.220 Characterization results show that α-PdZn alloy phase forms on the catalyst with high Pd contents of ≥7.5 wt%, while β-PdZn alloy phase forms on the catalysts with low Pd content of ≤2.5 wt%. The Pd loading has a significant influence on CO2 selectivity. The catalysts with low Pd loadings show high selectivity to CO2 and the highest CO2 selectivity of 97% is achieved with 0.1 wt% Pd/ZnAl2O4. When the Pd content increases to 7.5 wt%, the CO2 selectivity drops to ∼50.5%. Therefore, the β-PdZn alloy phase is more selective for MSR. It is proposed that the polar face of the ZnAl2O4 promotes the formation of the β-PdZn alloy at low Pd loadings while the formation of α-PdZn at high Pd loadings is due to the limited Zn atoms on the ZnAl2O4 surface.
Armbrüster et al. prepared Pd/In2O3 aerogel catalysts by gelation of InCl3 and PdCl2 using propylene oxide in water and ethanol mixture and applied them in MSR.221 InPd bimetallic nanoparticles are formed after mild reduction at 230 °C, which show a high H2 formation rate of 50 mmol mmolPd−1 h−1 and a CO2 selectivity of 99% at 300 °C. The apparent activation energy is 142 kJ mol−1 at 225–300 °C and 54 kJ mol−1 at 300–400 °C. The high activation energy at low temperatures is similar to the performance of In2O3 while the low activation energy at high temperatures resembles InPd, indicating the existence of In2O3 on the catalyst surface at below 300 °C and a dynamic change of surface structure with temperatures. Characterization results show that In3Pd2 formed from the reduction of In2O3 and incorporation of In into InPd at temperatures equal to or higher than 300 °C. Isotope-labelling experiments confirm that the reaction involves the Mars–van-Krevelen mechanism, and oxygen atoms in the phase boundary of In2O3 and PdIn participate in the reaction.
Compared to the Cu-based catalyst, the Pd-based catalysts show better thermal stability. Penner et al. studied the thermal stability of PdZn alloy using TEM.222 PdZn alloy is thermally and structurally stable on reduction between 200 °C and 600 °C from the TEM observation. Iwasa et al. compared the stability of Cu/ZnO and Pd/Zn/CeO2 at 350 °C. The Pd/Zn/CeO2 is quite stable and no deactivation was observed for 3 h on stream.223 In contrast, the Cu/ZnO shows a significant deactivation with activity loss by 20% after 3 h on stream. Datye et al. compared the stability of Pd/ZnO/Al2O3 with commercial Cu/ZnO/Al2O3.224 The Pd/ZnO/Al2O3 is tested at 250 °C, slightly higher than 230 °C for Cu/ZnO/Al2O3. The Cu-based catalysts show a 40% activity loss after 60 h on stream, while Pd/ZnO/Al2O3 only shows a 17% loss in activity. The deactivation of Pd-based catalysts is not due to sintering, as the average diameter only increases slightly. A simple oxidation–reduction cycle can regenerate the Pd/ZnO/Al2O3 and fully recover the activity. Tomishige et al. found that zinc carbonate hydroxide forms for the used Pd/ZnO and may be related to the deactiviation.225 Datye et al. proposed that the deactivation of PdZn is caused by coke formation.226 Pérez-Hernández et al. studied the performance of Pd supported on TiO2, ZrO2, and ZrO2–TiO2 mixed oxide and ascribed the deactivation to sintering of active phase and coke deposition.227
Au-Based catalysts. Wang et al. studied the performance of Au/ZnO in methanol steam reforming, decomposition, partial oxidation, and oxidative stream refroming.228 Au/ZnO with Au content of 4.3 wt% and a mean particle size of ∼3.2 nm was prepared by deposition precipitation. The catalytic activity for methanol decomposition and steam reforming is lower than partial oxidation and oxidative steam reforming, as the initiation temperatures are 300 °C, 250 °C, 150 °C, and 150 °C respectively. A comparison with Cu-based catalysts shows that the methanol decomposition and steam reforming activity of Au/ZnO is lower. However, the activity can be greatly enhanced by adding oxygen to the reactant to induce partial oxidation and oxidative steam reforming. This is ascribed to the good oxygen activation ability of the Au catalyst in the Au-support interface. The oxidative steam reforming shows a good methanol conversion of 95%, high H2/CO2 yield ratio of 2.4, and low CO selectivity of 1% at 200 °C. Luengnaruemitchai et al. tested Au/Ce1−xZrxO2 and Au–Cu/Ce1−xZrxO2 in MSR.229 Au and Cu were deposited on the supports by deposition precipitation. The metal loading is influenced by the point of zero charge of the support, which is controlled by the Ce/Zr ratio and preparation method. XRD patterns indicate the formation of AuCu alloy on the Au–Cu/Ce1−xZrxO2. The particle size of AuCu alloy is influenced by the deposition pH value. For the Au/Ce1−xZrxO2, the catalyst with Ce/Zr of 3
:1 shows the best activity with methanol conversion of ∼95% at 350 °C. For the Au–Cu/Ce0.75Zr0.25O2, the catalyst prepared at pH = 7 exhibits the best performance with 100% methanol conversion at 350 °C.
Penner et al. explored the potential of In2O3 catalysts in MSR.230 In2O3 thin films deposited on NaCl(001) surfaces by thermal evaporation and commercial In2O3 powders were investigated. Pretreatment in H2 or O2 at 400 °C or 450 °C does not show significant impacts on the bulk structure of In2O3. Temperature-programmed reaction results show that MSR starts at 177 °C and 277 °C on In2O3 film and powders, respectively. The highest CO selectivity is less than 5% in the temperature range studied (<400 °C), indicating that In2O3 is very selective for MSR. Later, the performance of ZnO in MSR was studied.231 Commercial ZnO with a specific surface area of 14 m2 g−1 was employed. In the batch reactor, ZnO shows activity starting from 267 °C and increases strongly above 327 °C. In the flow reactor, ZnO achieves methanol conversion of 3.6% with high CO2 selectivity of 99.6% at 300 °C. Chen et al. applied ZnO–Cr2O3/CeO2–ZrO2/Al2O3 prepared by impregnation in MSR.232 The optimized catalyst achieves methanol conversion of 100% and CO selectivity of ∼8% at 460 °C. Reaction results of methanol steam reforming, water–gas shift, and reverse water–gas shift reaction reveal that water–gas shift and reverse water–gas shift reaction can be neglected during MSR on this catalyst. The by-product CO mainly comes from methanol decomposition. The catalyst shows good stability during MSR without significant deactivation for 6 h on stream at 440 °C. However, the activity declines significantly when subjected to methanol decomposition reaction. H2 and CO do not show significant impacts on MSR, but H2 inhibits methanol decomposition. CO2 suppresses MSR, and H2O suppresses both methanol steam reforming and decomposition. Li et al. studied the performance of ZnO/ZnZrOx in MSR reaction.233 Reaction results at 400 °C show that methanol conversion increases with Zn content and reaches the maximum of 99% at Zn content of 9–13%. The good performance of catalysts with 9–13% Zn content is ascribed to the synergy effect between small ZnO clusters and ZnZrOx solid solution support. Moreover, ZnO/ZnZrOx-9% shows good stability during 230 h on stream with a slight decrease of methanol conversion from 90.4% to 80.7% and an almost unchanged CO selectivity of ca. 2%. The mechanism of MSR on ZnO/ZnZrOx-9% was studied by temperature-programmed DRIFTS and TPSR. From the FTIR study, methoxy species are observed at 100 °C and transferred to formate species at 300–400 °C. The surface OH species participate in the reaction as the negative bands corresponding to OH species appear at high temperatures. HCHO is detected by TPSR ahead of HCOOH, which comes from methanol dehydrogenation. HCOOH is produced from the reaction of HCHO with OH species and decomposes into CO2 and H2 subsequently. In the later work, the promotion effect of Ce on ZnZrOx was studied by Li et al.234 Characterization results from XRD and Raman show that Zn and Ce incorporate in the lattice of t-ZrO2, forming ternary solid solutions in the composition range studied. Zn1Ce0.5Zr9Ox appears to be more active than the Zn1Zr10Ox and Zn1Ce10Ox, achieving methanol conversion of 99.8% at 400 °C. The CO selectivity of Zn1Ce0.5Zr9Ox (6.0%) is lower than Zn1Ce10Ox (11.6%) but higher than Zn1Zr10Ox (2.2%). The participation of oxygen vacancy in MSR is confirmed by in situ Raman. TPSR profiles indicate that the reaction follows the formate pathway on Zn1Ce0.5Zr9Ox, similar to that on unmodified ZnZrOx. The enhanced activity is due to the improved H2O activation ability on oxygen vacancy sites stabilized by Ce3+. Ce doping can also stabilize the phase structure of ZnCeZrOx during the reaction and inhibit carbon deposition, leading to the improved stability.
3.2 Homogeneous catalysis for MSR
Homogeneous catalysis refers to a homogeneous phase in which the catalytic reaction system is indistinguishable from the reaction medium and other components. Compared with heterogeneous catalysts, homogeneous catalysts show the characteristics of high activity, high selectivity, and catalyze specific reactions quickly with definite mechanisms. The most used homogeneous catalysts in dehydrogenation reactions are organometallic complexes, such as Ru-based catalysts.256 Some representative results are summarised in Table 7.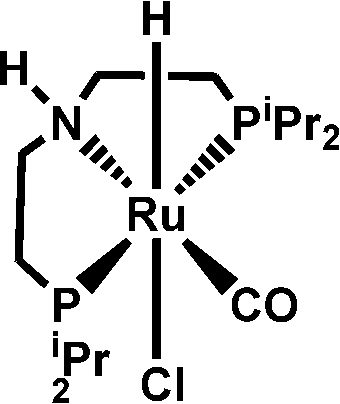
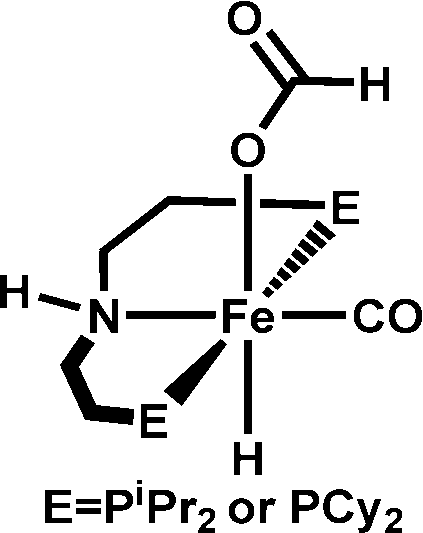
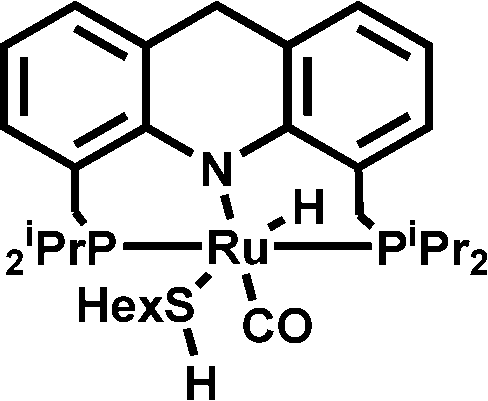
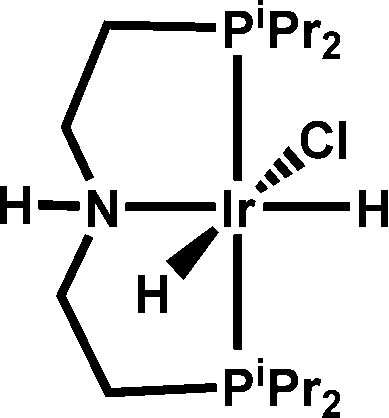
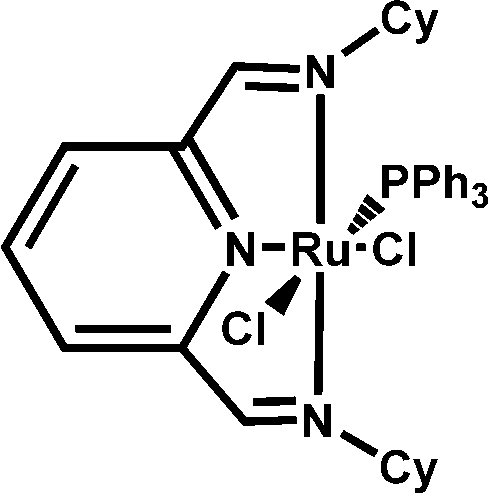
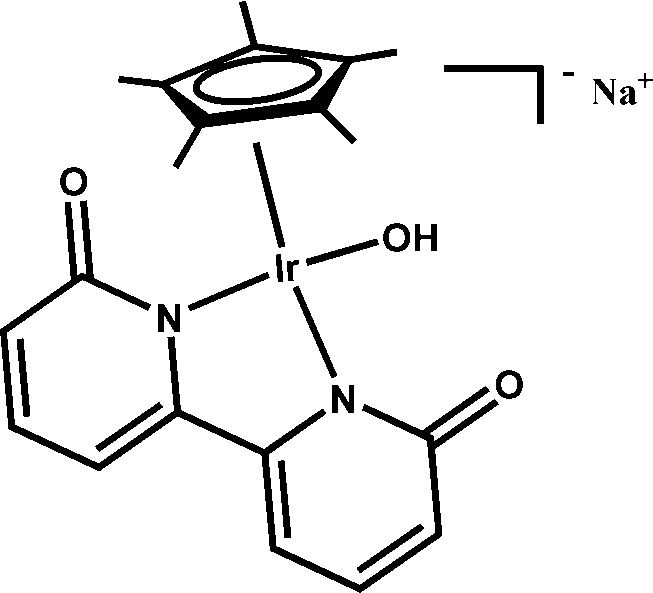
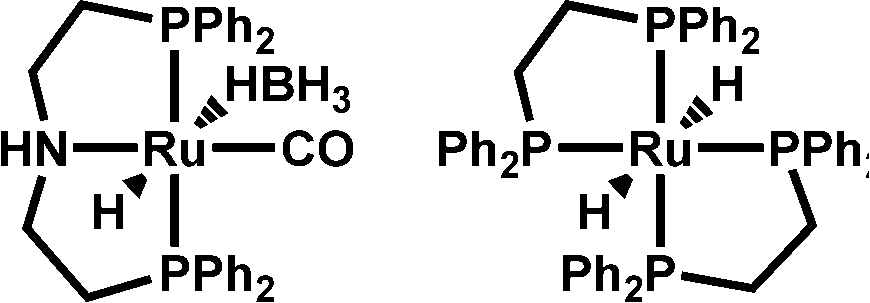
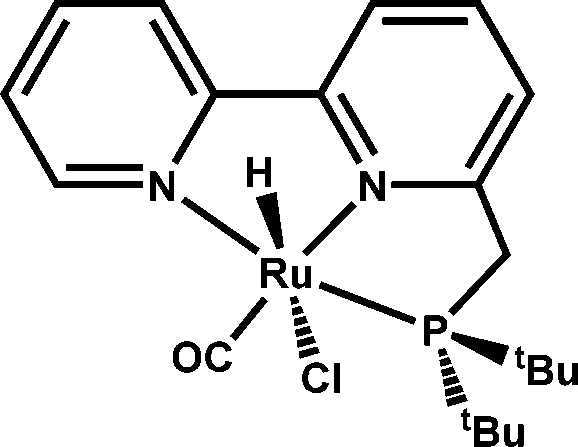
From the perspective of reducing energy consumption and side reactions, lowering the reaction temperature is a key objective for MSR. In 1987, Cole-Hamilton et al. reported that MSR could be catalyzed by [Rh(bipy)2]Cl (bipy = 2,2′-bipyridyl).257 This catalyst achieved a TOF of 7 h−1 at 120 °C in the presence of NaOH. Beller et al. reported a process facilitated by Ru pincer complexes [RuHCl(CO)(HN(C2H4PiPr2)2)], offering a TOF of 4700 h−1 and a turnover number (TON) of 35000 at reaction temperature of 93 °C.258 The reaction mechanism was studied in the later work, as shown in Fig. 8. Ru–Amido complex is sorted and considered as the key reaction intermediate. This intermediate is highly reactive with methanol, formic acid, and water, providing mono and dihydride Ru complexes, which can be deprotonated at the nitrogen atom of the pincer ligand in the presence of base. The deprotonated dihydride complex releases hydrogen and formaldehyde during the protonation to generate monohydride complex. Beller et al. also reported a bi-catalytic system for methanol steam reforming in the absence of base.259 This system contains Ru-MACHO-BH and Ru(H)2(dppe)2 that work synergistically. A TON of higher than 4200 was achieved at ∼94 °C and only a trace amount of CO (<8 ppm) was detected in the products. Grützmacher et al. developed a Ru-bases complex with a chelating bis(olefin) diazadiene ligand.260 The complex, [K(dme)2][Ru(H)(trop2dad)], can intramolecularly store up two equivalents of hydrogen reversibly. This catalyst can achieve 80% conversion after 10 h at the temperature of 90 °C, producing CO2/H2 from methanol aqueous solution under neutral conditions. Milstein et al. reported a Ru-PNN pincer complex in this reaction.261 This catalyst achieves a H2 yield of up to 82% in 9 days at ∼100 °C and is very stable. No deactivation can be observed after ∼1 month, achieving a TON of ∼29000. Reek et al. reported a ruthenium complex Ru(salbinapht)(CO)(Pi-Pr3) could also catalyze this reaction.262 A TOF of 55 h−1 is achieved at 82 °C.
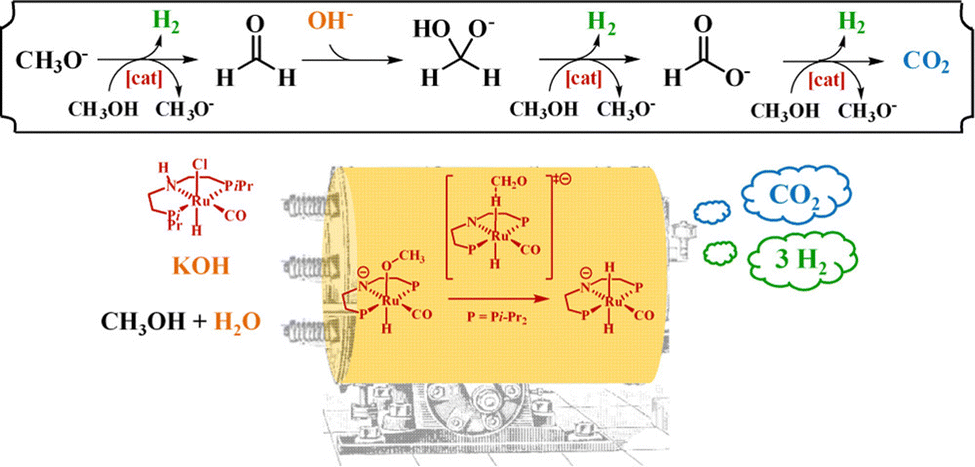 | ||
| Fig. 8 The mechanism of RuHCl(CO)(HN(C2H4PiPr2)2) catalyzed methanol dehydrogenation.268 Reproduced with permission from ref. 268. Copyright (2016) American Chemical Society. | ||
Yamaguchi et al. reported that an anionic Ir complex with a functional bipyridonate ligand was active for MSR.263 A continuous H2 production is achieved for 150 h by adding methanol, water, and NaOH continuously at refluxing conditions, achieving a TON of 10510 with a H2 yield of 64%. Beller et al. reported a Ir-PNP pincer complex in this reaction.264 A TOF of 326 is achieved at 70 °C. During the reaction, the formation of an iridium dihydride carbonyl complex results in the deactivation of this catalyst in low basic media. Using strongly basic conditions can inhibit the deactivation.
It is worth noting that non-noble metal-based complex catalysts for MSR have been explored in recent years. Beller et al. reposted an iron pincer complex with a TON close to 10000 obtained with a TOF of 644 h−1 at 91 °C.265 Deactivation is observed for this catalyst and adding additional ligands in the reaction mixture can improve the lifetime as the decomposition of the catalyst is hindered. Later, a Mn pincer catalyst was reported for this reaction.266 This catalyst shows good stability and a TON of more than 20000 is reached. However, the PNPiPr ligand is sensitive to light and the experiments need to be carried out under the exclusion of light. Holthausen et al. applied a pincer-supported Fe compound in the methanol dehydrogenation reaction with TON up to 51000.267 A co-catalytic amount of a Lewis acid, LiBF4, plays an important role in facilitating the decarboxylation of the Fe formate species.
3.3 Heterogeneous catalysis for methanol synthesis from CO2 hydrogenation
Methanol can be produced from CO hydrogenation and CO2 hydrogenation. Methanol synthesis from syngas in the presence of a small amount of CO2 is a well-established industry process while direct CO2 hydrogenation has been a hot research topic in recent years. A brief introduction of heterogeneous catalysts for CO2 hydrogenation to methanol will be given below.Cu/ZnO based catalysts show good performance in both methanol synthesis from CO and CO2.272,273 Urakawa et al. studied the performance of Cu/ZnO/Al2O3 in CO2 hydrogenation under very high pressure.274 The one-pass CO2 conversion can achieve 95% with a methanol selectivity of 98% at 36 MPa and 260 °C. Other Cu-based catalysts such as Cu/ZrO2 and Cu/CeO2 also show good performance for CO2 hydrogenation to methanol. Rodriguez et al. compared the performance of CeOx/Cu(111) and Cu(111) using model catalysts.275 The TOF of CeOx/Cu(111) is 1.3 s−1 at 302 °C, much higher than Cu(111) (6.3 × 10−3 s−1). The activation energy of CeOx/Cu(111) (12 kcal mol−1) is also lower than Cu(111) (25 kcal mol−1). Satokawa et al. compared the performance of Cu supported on ZrO2 with different crystal structures, including amorphous ZrO2 (a-ZrO2), monoclinic ZrO2 (m-ZrO2), and amorphous/tetragonal ZrO2 supported on KIT-6 (a-ZrO2/KIT-6 and t-ZrO2/KIT-6).276 Reaction results show that the catalysts using supports including amorphous ZrO2 show higher methanol selectivity and formation rate. The difference is related to the different existence states of Cu species on the as-prepared catalysts. XRD and XAS show that CuO exists on the Cu/m-ZrO2 and Cu/t-ZrO2/KIT-6 while CuxZryOz exists on the a-ZrO2 containing catalyst. After reduction, the Cu size of a-ZrO2 containing catalyst is smaller, leading to the higher activity. At the same time, methanol vapor adsorption indicates that the adsorption of methanol on a-ZrO2 is weaker. The weak adsorption of methanol can inhibit the decomposition of methanol into CO and improve the methanol selectivity. Recently, Li et al. reported a faujasite-encaged mononuclear Cu catalyst (Cu@FAU) that showed good performance in CO2 hydrogenation to methanol.277 A methanol formation rate of 12.8 mmol gcat−1 h−1 and methanol selectivity of 89.5% can be achieved at 240 °C. No deactivation is observed over 200 h on stream, showing that this catalyst is very stable under reaction conditions and may be a promising catalyst for large-scale industry applications. H2–D2 exchange experiment results show that the activation of H2 on this catalyst is assisted by CO2. Surface species study and DFT calculations prove that CO2 hydrogenation to methanol follows the formate pathway. The mononuclear Cu and neighbouring O sites act as classical Lewis pairs and activates the H2 with the assistance from adsorbed CO2 species.
Pd-Based catalyst is another kind of catalyst used for CO2 hydrogenation to methanol. Different supports, such as ZnO, Ga2O3, and CNTs, have been used. For Pd/ZnO, the PdZn alloy formed after reduction is ascribed to the active sites.278 Mu et al. studied the influence of Al doping in ZnO support for Pd/ZnO on this reaction.279 The activity increases with Al doping up to 3.93 wt%. Al doping in ZnO can facilitate the adsorption and activation of CO2, leading to the improved performance. However, Al content greater than 3.93 wt% leads to a decrease in activity. This is caused by the formation of ZnAl2O4 spinel and amorphous Al2O3 on the ZnO surface. Tsang et al. studied the influence of the exposed surface of Ga2O3 for Pd/Ga2O3.280,281 β-Ga2O3 with plate and rod morphologies are used as supports. The plate type Pd/β-Ga2O3 shows better performance than the rod type catalyst. XRD and TEM show that the major exposed facets are different for rod and plate β-Ga2O3. The majority surface of rod Ga2O3 is terminated with (111) and (110) faces while it is (002) for plate Ga2O3. The (002) surface of Ga2O3 has polarity due to the unbalanced arrangement of cation and anion and a stronger interaction with Pd, which improve the metal dispersion and lead to the formation of PdGax, resulting in a better activity. Li et al. investigated the position effect of Pd on CNTs.282 Pd nanoparticles with high selectivity located inside or outside CNTs were prepared and tested in CO2 hydrogenation to methanol. The TOF of Pd inside of CNTs is 0.33 h−1, much higher than the Pd outside of CNTs (0.09 h−1). The activation energy of Pd inside of CNTs is also lower. The difference is caused by the different ratios of Pdδ+ species. The deviation from the plane structure of CNTs causes the hybridization of the p orbital of C to become intermediate between sp2 and sp3. Therefore, the π-electron density is shifted from the concave inner surface to the convex outer surface of CNTs. The inner surface thus becomes electron deficient and helps to stabilize the Pdδ+ species, forming more Pd0–Pdδ+ structures. During CO2 hydrogenation, H2 is activated on Pd0 sites while CO2 is activated on Pdδ+. More Pd0–Pdδ+ structures lead to the higher activity.
Besides metal-based catalysts, metal oxides and metal sulfides catalysts have also been used in CO2 hydrogenation, such as In2O3, In2O3/ZrO2, ZnO–ZrO2, GaZrOx, CdZrOx, and MoS2. Metal oxide-based catalysts show high methanol selectivity than Cu-based catalysts. However, the activity at low temperatures is unsatisfactory and should be improved. Using DFT calculations, Liu et al. predicted that In2O3 could catalyze CO2 hydrogenation to methanol.283,284 Later, the activity of commercial In2O3 is confirmed by experiments.285 Pérez-Ramírez et al. reported a very high selectivity to methanol catalyzed by In2O3 prepared from the decomposition of In(OH)3.286 The oxygen vacancies on the surface of In2O3 are crucial in this reaction. The easy reduction of In2O3 under reducing atmosphere at high temperature limit its practical application. The activity of In2O3 can be improved by supporting on monoclinic ZrO2.287 The lattice mismatch between ZrO2 and In2O3 causes pronounced tensile forces and more oxygen vacancies for In2O3. ZrO2 also contributes to CO2 activation. Both these factors contribute to the improved In-based specific activity. Metals, including Pd,288 Pt,289 Ni,290 Rh,291 Re,292 and Au,293 have been used as promoters for In2O3, mainly to improve the hydrogen activation ability of the catalyst at low reaction temperatures. Li et al. studied the performance of ZnO–ZrO2 solid solution catalyst in CO2 hydrogenation to methanol.294 This catalyst shows a high methanol selectivity of 86–91% with CO2 conversion higher than 10% at 320–315 °C. The resistance of ZnO–ZrO2 to SO2 and H2S is good. No deactivation is observed in the presence of 50 ppm SO2 or H2S, making this catalyst suitable for industrial application. DFT calculations show that the synergetic effect between Zn and Zr sites leads to the good performance. Later, the solid solution catalyst system is extended to GaZrOx and CdZrOx.295 Wang et al. reported that MoS2 nanosheet could catalyze CO2 hydrogenation to methanol at low temperatures.296 A methanol selectivity of 94.3% with a CO2 conversion of 12.5% is achieved at 180 °C. Theoretical study shows that the in-plane S vacancies of MoS2 are the active sites for methanol formation.
It should be noted that this part is only a brief introduction for CO2 hydrogenation to methanol. A comprehensive summary on this topic can be found in some nice review articles.297,298
4. Ammonia and related chemicals
Ammonia and its derivatives, including ammonia borane and hydrazine hydrate, also deserve attention as carbon-free energy carriers.299 Ammonia, a well-established industrial product, is produced and transported globally at a scale of approximately 150 million tons per year via trains, pipelines, and other means.300 In contrast to carbon-containing carriers, ammonia does not emit CO or CO2, and ammonia is not a greenhouse gas. In 1982, Green first proposed the use of ammonia as an energy carrier, suggesting that it could be an economically viable energy vector.301 Other ammonia-based compounds like ammonia borane and hydrazine are also carbon-free. Ammonia is a particularly versatile energy carrier because the hydrogen generated from ammonia can be utilized in hydrogen fuel cells, and meanwhile, ammonia itself and related compounds can be directly employed as fuels.3024.1 Ammonia
Ammonia (NH3) offers great potential as a liquid-phase hydrogen storage and transport medium due to its ability to be stored in liquid form under relatively mild conditions. Notably, its mass hydrogen density surpasses that of liquid H2, and NH3 is much easier to liquefy than H2, given its higher temperature (∼25 °C) and lower pressure (∼3 bar) requirements. NH3 has several advantages as a hydrogen source, as summarized below.High hydrogen capacity: with a hydrogen storage capacity of up to 17.7 wt% (108 g L−1) and an energy density of 3000 W h kg−1, NH3 outperforms other LHCs such as methanol in terms of capacity and density.302
Environment-friendly: NH3 decomposition results in the production of H2 and stoichiometric N2 without generating harmful CO or greenhouse gas CO2,303 thereby benefiting both the environment and the purity of H2 produced.302
Since the synthesis of NH3 from N2 and H2 (H2 storage process) is very mature in industry, using NH3 as LHC is mainly decided by H2 release process. There are two key challenges of NH3 decomposition and H2 separation, which will be discussed in the following sections.
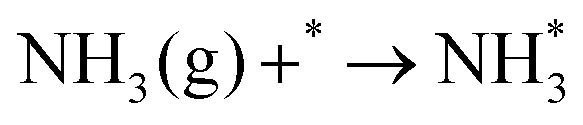
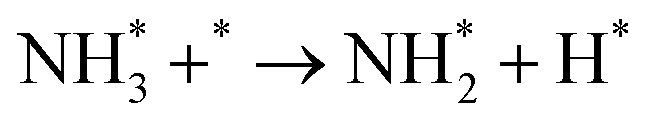
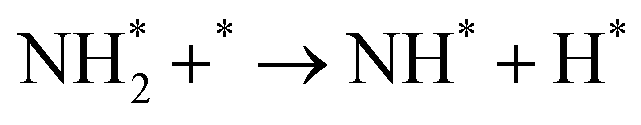
| NH* + * → N* + H* |
| 2N* → N2(g) |
| 2H* → H2(g) |
The binding strength of the metal site to N is the most dominant factor throughout the reaction. A strong M–H bond favours N–H bond scission but goes against N* desorption, while a weak M–H bond is not sufficient to cause N and H separation. Thus, a suitable strength of M–H bond is crucial for enhancing catalytic activity for NH3 decomposition.305 Among all single metal catalysts, Ru exhibits the highest catalytic activity due to their appropriate Ru–N binding energy.306 Currently, catalysts for NH3 decomposition can be divided into two categories, namely Ru-based catalysts and Ru-free catalysts (Table 8).
| Catalyst | Temp. (°C) | GHSV (mL gcat−1 h−1) | Conv. (%) | H2 production (mmol g−1 min−1) | Ref. |
|---|---|---|---|---|---|
| Ru/CP33 | 500 | 30000 |
99.5 | 33.3 | 308 |
| Ru/K2SiO3 | 450 | 30000 |
60.5 | 20.3 | 309 |
| Ru/CNTs | 550 | 30000 |
100 | 33.5 | 316 |
| Ru/Cr2O3 | 600 | 30000 |
100 | 30.7 | 317 |
| Ru-K/CaO | 400 | 9000 | 53.7 | 5.4 | 318 |
| Ru/CaAlOx | 500 | 6000 | 98.2 | 6.6 | 319 |
| Mo2N/SBA-15/rGO | 600 | 60000 |
68.5 | 184.6 | 312 |
| 20Ni/Al1Ce0.05Ox | 600 | 54000 |
81.8 | 49.3 | 313 |
| Ni7.5Co2.5/CeO2 | 650 | 30000 |
96.96 | 32.5 | 320 |
| 20Co-10Ni/Y2O3 | 550 | 9000 | 85.02 | 8.5 | 321 |
| Ni10Ce5Ox/Al2O3 | 525 | 30000 |
75 | 25.1 | 322 |
In recent years, significant advancements have been made in the synthesis and mechanism of ruthenium-based catalysts, aiming to achieve higher activity at lower temperatures.305,307 Yamazaki et al. prepared Ru/CeO2–PrOxvia a coprecipitation route for on-site NH3-decomposition H2 fuelling station in 2022, which showed the highest level of decomposition activity among all catalysts ever reported.308 Typically, the optimized catalyst Ru/CP33 shows NH3 conversion of >99.5% at 500 °C within 1800 h on stream. The benefits of H2 production from NH3 decomposition exceed the actual cost, which lays a solid foundation for its commercialization. Zhong et al. prepared a series of alkali metal silicates A2SiO3 (A = Li, Na, and K) supported Ru nanoparticles for NH3 decomposition, and the optimized catalyst exhibited the highest conversion of 60.5% with a TOF value of 2.03 s−1.309 The improved catalytic performance is ascribed to strong electronic metal support interaction (SMSI) between Ru particles and oxygen vacancies. Yun et al. confirmed that modulating the SMSI could efficiently enhance the catalytic properties of Ru/BCY-x catalysts, which provided an efficient way for catalyst modification.310 Chae et al. prepared a series of Ru doped LaxCe1−xOy composites for NH3 catalytic decomposition and confirmed that N2 desorption was the rate-determining step in the reaction311 To reduce costs, non-noble metal-based catalysts for NH3 decomposition have been explored. Various strategies have been employed to improve the catalytic activity, such as designing spatially confined metal nitrides, using one-pot cation–anion double hydrolysis synthesis route, and employing a sol–gel method with a second support material. Jia et al. designed spatially confined metal nitrides in order to weaken the associative desorption of adsorbed N effectively.312 The synthesized Mo2N/SBA-15/rGO exhibits the highest NH3 decomposition rate of 30.58 mmol g−1 min−1 among all Mo-based catalysts ever reported. Chae et al. prepared a series of Ni/Al1CeaOx composites with higher Ni dispersion and surface area via one-pot cation–anion double hydrolysis synthesis.313 The introduction of Ce can modulate the synergy of oxygen vacancies, NiO reducibility, Ni-support interaction, and basicity. Jiang et al. developed a CeO2 and BN hybrid-supported Ni catalyst by a sol–gel method, which showed high activity with H2 yield of approximately 516 mmol gNi−1 min−1 at 600 °C.314 The result demonstrates a simple but effective strategy of adding a second support material to improve catalytic activity for NH3 decomposition. Besides, Bertola et al. performed kinetic assessment of NH3 decomposition reaction based on experiments and computational fluid dynamics modeling.315 A flat-plate microreactor operating under kinetic-control conditions with negligible mass transfer resistance is designed, which facilitates the assemblage and disassemblage of components.
4.2 Ammonia borane
Ammonia borane (AB), a solid at room temperature, has emerged as one of the most promising materials for chemical hydrogen storage since its first synthesis in 1955 by Shore and Parry.328 Due to its high hydrogen content of 19.6 wt%, non-toxic nature, and sustainable stability under ambient conditions, AB has garnered significant interest as a potential hydrogen carrier. The structure and properties of AB can be found in a recent review paper by Demirci.329 AB dissolves well in water and methanol, allowing it to decompose and release H2 in the presence of catalysts in the liquid system. The chemical equations for AB hydrolysis and methanolysis are as follows.| Hydrolysis: NH3BH3 + 2H2O → NH4BO2 + 3H2 |
| Methanolysis: NH3BH3 + 4CH3OH → NH4B(OCH3)4 + 3H2 |
However, at high temperatures, AB decomposes to produce H2 and volatile toxic substances such as NH3 and diborane.330,331
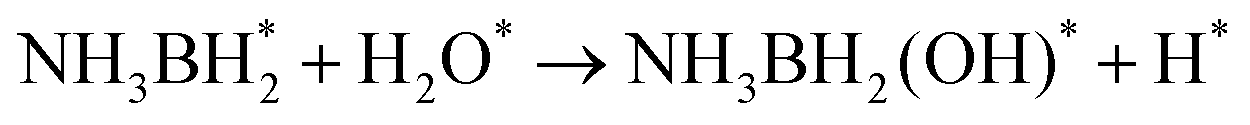 is the rate-determining step.333 The results suggest that the key to hydrolysis catalysis lies in O–H breaking in H2O. Some of the representative catalysts for AB hydrolysis are listed in Table 9.
is the rate-determining step.333 The results suggest that the key to hydrolysis catalysis lies in O–H breaking in H2O. Some of the representative catalysts for AB hydrolysis are listed in Table 9.
| Catalyst | Temp. (°C) | TOF (min−1) | E a (kJ mol−1) | Ref. |
|---|---|---|---|---|
| Ru/CS | 30 | 331.8 | 41.3 | 334 |
| N-hcp-Ni/C | 25 | 17.2 | 35.3 | 337 |
| PVP stabilized Co | 40 | 14 | 46 | 336 |
| Co/CTF | 25 | 42.3 | 42.7 | 338 |
| Fe–Ni–Ni3B | 30 | ∼293 | 39.95 | 339 |
| P2-Cu–Co3O4@CNF | 30 | 35.6 | 29.86 | 340 |
| hcp-CuNi/C | 25 | 22.64 | 29.92 | 351 |
| Co–P | 25 | 23.5 | 38.7 | 352 |
| Cu@Ni6-MOF | 25 | 69.1 | 31.6 | 341 |
| PdCo@NCHP | 30 | 881.91 | 36.9 | 353 |
| Pt/MoO3−x-500 | 25 | 331.4 | 13.97 | 354 |
Similar to the dehydrogenation reaction of toluene and NEC, noble metal catalysts used in the hydrolysis of AB have better performance than non-noble metal catalysts. In 2022, Liu et al. immobilized Ru nanoparticles on natural chitosan polymers, achieving a TOF value of 331.8 min−1 and an Ea value of 41.3 kJ mol−1.334 Xu et al. first applied metal–organic frameworks in hydrolysis with the help of Pt nanoparticles in MIL-101's nanopores, which showed an activation energy of 40.7 kJ mol−1.335 The present results bring light to new opportunities in the superiority of porous materials in hydrolysis.
Despite their superior performance, the high cost of noble metal catalysts has driven researchers to explore more affordable non-noble metal catalysts, such as Ni and Co catalysts. Özkar et al. prepared polymer-stabilized cobalt(0) nanoclusters from the reduction of cobalt(II) chloride, which was found to be active in the hydrolysis reaction of AB.336 Li et al. presented for the first time that N-hcp-Ni/C could perform admirable catalytic properties for AB dehydrogenation.337 N-hcp-Ni/C delivered good catalytic activity with a TOF value of 15.2 min−1, owing to the unusual hcp engineering phase of Ni with N doping. In 2016, Chen et al. synthesized Co/CTF and Ni/CTF through an impregnation method with a total turnover frequency of 42.3 min−1 at room temperature.338 The covalent triazine framework (CTF) was rich in N, which gave it an electron-donating effect to increase the electron density of the Co and Ni. Besides, the KIE measurements showed that the breaking of an O–H bond in H2O was the rate-determining step for AB hydrolysis.
Recently, bimetallic catalysts have gained attention for their potential synergistic effects in the hydrolysis of AB. Vernekar et al. reported that the addition of Fe in the Ni–Ni3B produced strongly positive cooperativity, similar to nature-designed hydrogenase metalloenzymes.339 The catalyst can effectively reduce the reaction activation energy and maintain a catalytic conversion rate of 100% in several catalytic cycles with a TOF of ∼293 min−1. Yu et al. prepared a ZIF-67 derived Co3O4 nanoparticles in a carbon-based nano framework with P and Cu co-doping strategy which exhibited an astonishing nearly 51-fold improvement.340 According to the experimental results and DFT calculations, the strong electronic coupling between Co cation and P anions brought electronic structure regulation and a downshift of d-band, similar to the Cu doping effect. This structural shift produced the apparent activation energy with 29.86 kJ mol−1 and a TOF of 35.6 min−1. Li et al. designed an out-of-plane CoRu nanoalloy axially coupling CosNC with superior intrinsic activity and cycle stability at room temperature.341 The result presented that the electron-enrichment-boosting effect could effectively reduce the reaction energy barrier according to DFT calculations.
Increasing studies have been focused on light-assisted photocatalytic AB hydrolysis to H2. With the help of visible light, dehydrogenation can proceed quickly without heat from external systems. Furthermore, the photocatalytic system is more environmentally friendly and promotes carbon neutrality. The photocatalysts are based on the semiconductor materials such as titanium oxide342 and carbon nitride.343 In 2022, Astruc and coworkers prepared AuNi@ZIF-8 alloys under visible-light illumination.344 The researchers designed a new strategy involving plasmon-induced visible light and maximum synergy among the nanocomponents to boost AB dehydrogenation performances. Filiz et al. reported a series of Co doped n-type semiconductors and presented that Co@TiO2–WO3 heterojunction structure had the best performance.345
4.3 Hydrous hydrazine
Hydrous hydrazine (N2H4·H2O) is considered to be used in the liquid chemical hydrogen storage system, and N2 is the only complete decomposition by-product. Hydrazine hydrate has the advantage of liquid phase at room temperature, high hydrogen content (8.0 wt%), and satisfactory stability.349,350 It is worth noting that pure hydrazine is prone to explode after contact with metal and hydrous hydrazine does not have this safety hazard. The decomposition of hydrazine includes complete decomposition and incomplete decomposition. The complete decomposition is the ideal reaction, while the incomplete decomposition will produce toxic NH3 and possibly poison catalysts.| N2H4 → N2 + 2H2 |
| 3N2H4 → N2 + 4NH3 |
To achieve hydrazine complete decomposition, reducing the energy barrier of N–H cleavage is crucial for the reaction, while N–N bond is easier to be broken than N–H bond.355 Thus, the metal catalysts are necessary for the reaction due to the stronger M–H bonds than M–N bond.356 In recent years, some progress has been made in the nanocatalysts for hydrazine dehydrogenation (Table 10). In an early study, Xu et al. applied Rh, Co, Ru, Ir, Cu, Ni, Fe, Pt, and Pd nanoparticles (NPs) in hydrazine decomposition.357 Among them, Rh NPs are confirmed to be the most active catalyst under aqueous conditions, while Fe, Ni, Cu, Pt, and Pd NPs exhibit lower activity. Khan et al. prepared tri-metallic nanocatalysts for hydrazine decomposition in a basic solution at low temperatures.358 Compared to the mono-metallic and bi-metallic catalysts, the tri-metallic catalysts perform good durability and 100% hydrogen selectivity. Lu et al. reported a series of Rh-MoOx NPs prepared via a one-step chemical reduction approach without any surfactant/support at room temperature.359 Typically, Rh0.5(MoOx)0.5 NPs exhibit good catalytic activity with a TOF of 750 h−1 and 100% H2 selectivity at 50 °C.
5. Concluding remarks
In this article, an overview of major existing LHC systems is provided and some relevant catalytic studies are involved. Currently, LHC technology is at a crucial stage, transitioning from laboratory settings to industrial applications, where it can address the challenges in H2 storage and transportation. As LHC technology advances toward commercialization, it has the potential to transform the energy landscape and create a more open market through the development of diverse hydrogen sources. However, LHC systems still face several key issues that need to be addressed. Hydrocarbons, although economically viable, may not be the optimal choice due to dehydrogenation challenges. Methanol can effectively utilize CO2 as the synthesis raw material; however, CO produced during dehydrogenation can lead to catalyst deactivation. Although ammonia decomposition has achieved profitability in gas station applications, its overall economic viability for large-scale commercialization is still uncertain. Moreover, the low-cost, large-scale production of NH3 remains a challenge. N-ethyl carbazole and ammonia borane have attracted considerable attention in recent years, but their costs remain high. Importantly, there is a pressing need to develop catalysts with good activity, selectivity, and stability, either by modifying existing catalysts or employing novel preparation methods like atomic layer deposition.For LOHC, methanol appears to be the optimized choice due to the high H2 mass and volumetric density as well as the low dehydrogenation enthalpy (Table 1). That is, methanol is the intrinsic better LOHC in comparison with other candidates. In practice, both the synthesis of methanol from CO2–H2 and the H2 release from methanol steam reforming can take place at relatively low temperatures of <250 °C in the presence of non-noble metal catalysts such as Cu-based catalysts. Overall, methanol shows unparalleled advantages as LOHC and is ready for industrial applications. Furthermore, using methanol as LOHC can be combined with the management and utilization of CO2 for local carbon neutrality. Ammonia and its derivatives are rising candidates as hydrogen carriers with very high H2 mass and volumetric density. However, many key issues should be addressed before their practical applications. Keep in mind ammonia synthesis from N2 and H2 remains one of the most energy-extensive processes in the chemical industry.
Last but not least, improvements beyond the materials themselves (LHC and catalysts) should be made, focusing on the development of more efficient and cost-effective systems for hydrogen storage and release. By integrating advanced control systems, heat management, and energy recovery strategies, the overall performance of these systems can be significantly enhanced, making them more practical for large-scale implementation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Key R&D Program of China (2021YFA1501202), National Natural Science Foundation of China (22025203), and the Fundamental Research Funds for the Central Universities (Nankai University).References
- H. J. Schellnhuber, S. Rahmstorf and R. Winkelmann, Nat. Clim. Change, 2016, 6, 649–653 CrossRef.
- C. F. Shih, T. Zhang, J. Li and C. Bai, Joule, 2018, 2, 1925–1949 CrossRef CAS.
- P. Preuster, C. Papp and P. Wasserscheid, Acc. Chem. Res., 2017, 50, 74–85 CrossRef CAS PubMed.
- M. Niermann, S. Drünert, M. Kaltschmitt and K. Bonhoff, Energy Environ. Sci., 2019, 12, 290–307 RSC.
- A. Züttel, Mater. Today, 2003, 6, 24–33 CrossRef.
- F. Marques, M. Balcerzak, F. Winkelmann, G. Zepon and M. Felderhoff, Energy Environ. Sci., 2021, 14, 5191–5227 RSC.
- M. D. Allendorf, Z. Hulvey, T. Gennett, A. Ahmed, T. Autrey, J. Camp, E. Seon Cho, H. Furukawa, M. Haranczyk, M. Head-Gordon, S. Jeong, A. Karkamkar, D.-J. Liu, J. R. Long, K. R. Meihaus, I. H. Nayyar, R. Nazarov, D. J. Siegel, V. Stavila, J. J. Urban, S. P. Veccham and B. C. Wood, Energy Environ. Sci., 2018, 11, 2784–2812 RSC.
- N. Rao, A. K. Lele and A. W. Patwardhan, Int. J. Hydrogen Energy, 2022, 47, 28530–28547 CrossRef CAS.
- E. Gianotti, M. Taillades-Jacquin, J. Rozière and D. J. Jones, ACS Catal., 2018, 8, 4660–4680 CrossRef CAS.
- G. Sievi, D. Geburtig, T. Skeledzic, A. Bösmann, P. Preuster, O. Brummel, F. Waidhas, M. A. Montero, P. Khanipour, I. Katsounaros, J. Libuda, K. J. J. Mayrhofer and P. Wasserscheid, Energy Environ. Sci., 2019, 12, 2305–2314 RSC.
- M. Taube, D. W. T. Rippin, D. L. Cresswell and W. Knecht, Int. J. Hydrogen Energy, 1983, 8, 213–225 CrossRef CAS.
- F. Valentini, A. Marrocchi and L. Vaccaro, Adv. Energy Mater., 2022, 12, 2103362 CrossRef CAS.
- A. Bourane, M. Elanany, T. V. Pham and S. P. Katikaneni, Int. J. Hydrogen Energy, 2016, 41, 23075–23091 CrossRef CAS.
- S. P. Verevkin, V. N. Emel’yanenko, A. Heintz, K. Stark and W. Arlt, Ind. Eng. Chem. Res., 2012, 51, 12150–12153 CrossRef CAS.
- Y. Dong, M. Yang, T. Zhu, X. Chen, G. Cheng, H. Ke and H. Cheng, ACS Appl. Energy Mater., 2018, 1, 4285–4292 CrossRef CAS.
- L. Li, M. Yang, Y. Dong, P. Mei and H. Cheng, Int. J. Hydrogen Energy, 2016, 41, 16129–16134 CrossRef CAS.
- Y. Dong, M. Yang, Z. Yang, H. Ke and H. Cheng, Int. J. Hydrogen Energy, 2015, 40, 10918–10922 CrossRef CAS.
- K. He, F. Tan, C. Zhou, G. Zhou, X. Yang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 3080–3084 CrossRef CAS PubMed.
- O. Sebastian, S. Nair, N. Taccardi, M. Wolf, A. Søgaard, M. Haumann and P. Wasserscheid, ChemCatChem, 2020, 12, 4533–4537 CrossRef CAS.
- D. Balcells, E. Clot and O. Eisenstein, Chem. Rev., 2010, 110, 749–823 CrossRef CAS PubMed.
- X. Lin, W. Wu and Y. Mo, Coord. Chem. Rev., 2020, 419, 213401 CrossRef CAS.
- M. Boudart, in Adv. Catal., ed. D. D. Eley, H. Pines and P. B. Weisz, Academic Press, 1969, vol. 20, pp. 153–166 Search PubMed.
- J. A. C. Rodriguez and C. T. Campbel, J. Phys. Chem., 1989, 93, 826–835 CrossRef CAS.
- L. Chen, P. Verma, K. Hou, Z. Qi, S. Zhang, Y.-S. Liu, J. Guo, V. Stavila, M. D. Allendorf, L. Zheng, M. Salmeron, D. Prendergast, G. A. Somorjai and J. Su, Nat. Commun., 2022, 13, 1092 CrossRef CAS PubMed.
- M. Peng, C. Dong, R. Gao, D. Xiao, H. Liu and D. Ma, ACS Cent. Sci., 2021, 7, 262–273 CrossRef CAS PubMed.
- Y. Deng, Y. Guo, Z. Jia, J. Liu, J. Guo, X. Cai, C. Dong, M. Wang, C. Li, J. Diao, Z. Jiang, J. Xie, N. Wang, H. Xiao, B. Xu, H. Zhang, H. Liu, J. Li and D. Ma, J. Am. Chem. Soc., 2022, 144, 3535–3542 CrossRef CAS PubMed.
- B. Wang, G. Froment and D. Goodman, J. Catal., 2008, 253, 239–243 CrossRef CAS.
- J. Du, C. Song, J. Song, J. Zhao and Z. Zhu, J. Fuel Chem. Technol., 2009, 37, 468–472 CrossRef CAS.
- N. Wang, J. Qiu, J. Wu, X. Yuan, K. You and H. Luo, Appl. Catal., A, 2016, 516, 9–16 CrossRef CAS.
- B.-H. Jeong, K.-I. Sotowa and K. Kusakabe, J. Membr. Sci., 2003, 224, 151–158 CrossRef CAS.
- J. Yu, Q. Ge, W. Fang and H. Xu, Int. J. Hydrogen Energy, 2011, 36, 11536–11544 CrossRef CAS.
- J. Yu, R. Wang, S. Ren, X. Sun, C. Chen, Q. Ge, W. Fang, J. Zhang, H. Xu and D. S. Su, ChemCatChem, 2012, 4, 1376–1381 CrossRef CAS.
- S. Furukawa, A. Tamura, K. Ozawa and T. Komatsu, Appl. Catal., A, 2014, 469, 300–305 CrossRef CAS.
- L. I. Ali, A.-G. A. Ali, S. M. Aboul-Fotouh and A. K. Aboul-Gheit, Appl. Catal., A, 1999, 177, 99–110 CrossRef CAS.
- A. N. Kalenchuk, V. I. Bogdan, S. F. Dunaev and L. M. Kustov, Fuel Process. Technol., 2018, 169, 94–100 CrossRef CAS.
- J. Wang, H. Liu, S. Fan, W. Li, Z. Li, H. Yun, X. Xu, A. Guo and Z. Wang, Energy Fuels, 2020, 34, 16542–16551 CrossRef CAS.
- L. Zhang, L. Liu, Z. Pan, R. Zhang, Z. Gao, G. Wang, K. Huang, X. Mu, F. Bai, Y. Wang, W. Zhang, Z. Cui and L. Li, Nat. Energy, 2022, 7, 1042–1051 CrossRef CAS.
- J. S. Kang, J. Y. Baek, H. Hwang, H. S. Shin, C. W. Yoon and H.-J. Shin, J. Mater. Chem. A, 2022, 10, 22701–22706 RSC.
- I. Rodríguez-Ramos and A. Guerrero-Ruiz, J. Catal., 1992, 135, 458–466 CrossRef.
- A. H. Al-ShaikhAli, A. Jedidi, L. Cavallo and K. Takanabe, Chem. Commun., 2015, 51, 12931–12934 RSC.
- D. Gao, Y. Zhi, L. Cao, L. Zhao, J. Gao, C. Xu, M. Ma and P. Hao, Chin. J. Chem. Eng., 2022, 43, 124–134 CrossRef CAS.
- Y. Yao, Z. Yan, L. Chen, Z. Zhou, L. Liu and D. W. Goodman, Catal. Lett., 2012, 142, 1437–1444 CrossRef CAS.
- J. Escobar, J. A. De Los Reyes, T. Viveros and M. C. Barrera, Ind. Eng. Chem. Res., 2006, 45, 5693–5700 CrossRef CAS.
- R. B. Biniwale, N. Kariya and M. Ichikawa, Catal. Lett., 2005, 105, 83–87 CrossRef CAS.
- Z. Xia, H. Lu, H. Liu, Z. Zhang and Y. Chen, Catal. Commun., 2017, 90, 39–42 CrossRef CAS.
- Z. Xia, H. Liu, H. Lu, Z. Zhang and Y. Chen, Appl. Surf. Sci., 2017, 422, 905–912 CrossRef CAS.
- J. Li, Y. Chai, B. Liu, Y. Wu, X. Li, Z. Tang, Y. Liu and C. Liu, Appl. Catal., A, 2014, 469, 434–441 CrossRef CAS.
- K. Choojun, A. Worathanaseth, S. Kuhatasanadeekul, T. Kurato, S. Ketaniruj, P. Phichitsurathaworn, P. Promchana, K. Prakobtham, N. Numwong, Y. Poo-arporn and T. Sooknoi, Catal. Commun., 2019, 125, 108–113 CrossRef CAS.
- M. Raad, A. Astafan, S. Hamieh, J. Toufaily, T. Hamieh, J. D. Comparot, C. Canaff, T. J. Daou, J. Patarin and L. Pinard, J. Catal., 2018, 365, 376–390 CrossRef CAS.
- H. Wang, N. Zhang, R. Liu, R. Zhao, T. Guo, J. Li, T. Asefa and J. Du, ACS Omega, 2018, 3, 10773–10780 CrossRef CAS PubMed.
- Z. Xia, H. Liu, H. Lu, Z. Zhang and Y. Chen, Catal. Lett., 2017, 147, 1295–1302 CrossRef.
- J. V. Pande, A. Shukla and R. B. Biniwale, Int. J. Hydrogen Energy, 2012, 37, 6756–6763 CrossRef CAS.
- G. Zhou, T. Li, J. Chen, L. Deng and H. Xie, Int. J. Hydrogen Energy, 2021, 46, 14540–14555 CrossRef CAS.
- J. V. Pande, A. B. Bindwal, Y. B. Pakade and R. B. Biniwale, Int. J. Hydrogen Energy, 2018, 43, 7411–7423 CrossRef CAS.
- H. M. Gobara, R. S. Mohamed and W. A. Aboutaleb, Microporous Mesoporous Mater., 2021, 323, 111151 CrossRef CAS.
- Z. Kou, S. Shen, K. Liu, G. Xu, Y. An and C. He, Int. J. Hydrogen Energy, 2013, 38, 11930–11936 CrossRef CAS.
- K. Takise, A. Sato, K. Murakami, S. Ogo, J. G. Seo, K.-I. Imagawa, S. Kado and Y. Sekine, RSC Adv., 2019, 9, 5918–5924 RSC.
- J. Meng, F. Zhou, H. Ma, X. Yuan, Y. Wang and J. Zhang, Top. Catal., 2021, 64, 509–520 CrossRef CAS.
- Q. Zhu and Q. Xu, Energy Environ. Sci., 2015, 8, 478–512 RSC.
- F. Alhumaidan, D. Tsakiris, D. Cresswell and A. Garforth, Int. J. Hydrogen Energy, 2013, 38, 14010–14026 CrossRef CAS.
- A. A. Shukla, P. V. Gosavi, J. V. Pande, V. P. Kumar, K. V. R. Chary and R. B. Biniwale, Int. J. Hydrogen Energy, 2010, 35, 4020–4026 CrossRef CAS.
- A. Shukla, J. V. Pande and R. B. Biniwale, Int. J. Hydrogen Energy, 2012, 37, 3350–3357 CrossRef CAS.
- X. Li, D. Ma and B. Xinhe, Chin. J. Catal., 2008, 29, 259–263 CrossRef CAS.
- C. Zhang, X. Liang and S. Liu, Int. J. Hydrogen Energy, 2011, 36, 8902–8907 CrossRef CAS.
- D. K. Cromwell, P. T. Vasudevan, B. Pawelec and J. L. G. Fierro, Catal. Today, 2016, 259, 119–129 CrossRef CAS.
- X. Zhang, N. He, L. Lin, Q. Zhu, G. Wang and H. Guo, Catal. Sci. Technol., 2020, 10, 1171–1181 RSC.
- X. Yang, Y. Song, T. Cao, L. Wang, H. Song and W. Lin, Mol. Catal., 2020, 492, 110971 CrossRef CAS.
- Y. Sugiura, T. Nagatsuka, K. Kubo, Y. Hirano, A. Nakamura, K. Miyazawa, Y. Iizuka, S. Furuta, H. Iki, T. Higo and Y. Sekine, Chem. Lett., 2017, 46, 1601–1604 CrossRef CAS.
- J. Yan, W. Wang, L. Miao, K. Wu, G. Chen, Y. Huang and Y. Yang, Int. J. Hydrogen Energy, 2018, 43, 9343–9352 CrossRef CAS.
- W. Wang, L. Miao, K. Wu, G. Chen, Y. Huang and Y. Yang, Int. J. Hydrogen Energy, 2019, 44, 2918–2925 CrossRef CAS.
- N. Boufaden, R. Akkari, B. Pawelec, J. L. G. Fierro, M. S. Zina and A. Ghorbel, J. Mol. Catal. A: Chem., 2016, 420, 96–106 CrossRef CAS.
- S. Manabe, T. Yabe, A. Nakano, S. Nagatake, T. Higo, S. Ogo, H. Nakai and Y. Sekine, Chem. Phys. Lett., 2018, 711, 73–76 CrossRef CAS.
- A. Nakano, S. Manabe, T. Higo, H. Seki, S. Nagatake, T. Yabe, S. Ogo, T. Nagatsuka, Y. Sugiura, H. Iki and Y. Sekine, Appl. Catal., A, 2017, 543, 75–81 CrossRef CAS.
- L. Zhang, G. Xu, Y. An, C. Chen and Q. Wang, Int. J. Hydrogen Energy, 2006, 31, 2250–2255 CrossRef CAS.
- S. Yolcular and Ö. Olgun, Catal. Today, 2008, 138, 198–202 CrossRef CAS.
- S. P. Patil, J. V. Pande and R. B. Biniwale, Int. J. Hydrogen Energy, 2013, 38, 15233–15241 CrossRef CAS.
- A. H. Al-ShaikhAli, A. Jedidi, D. H. Anjum, L. Cavallo and K. Takanabe, ACS Catal., 2017, 7, 1592–1600 CrossRef CAS.
- N. Boufaden, R. Akkari, B. Pawelec, J. L. G. Fierro, M. Said Zina and A. Ghorbel, Appl. Catal., A, 2015, 502, 329–339 CrossRef CAS.
- S. Hodoshima, Int. J. Hydrogen Energy, 2003, 28, 1255–1262 CrossRef CAS.
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS PubMed.
- Y. Tuo, L. Yang, H. Cheng, M. Yang, Y. Zhu and P. Li, Int. J. Hydrogen Energy, 2018, 43, 19575–19588 CrossRef CAS.
- S. Hodoshima, S. Takaiwa, A. Shono, K. Satoh and Y. Saito, Appl. Catal., A, 2005, 283, 235–242 CrossRef CAS.
- D. Sebastián, E. G. Bordejé, L. Calvillo, M. J. Lázaro and R. Moliner, Int. J. Hydrogen Energy, 2008, 33, 1329–1334 CrossRef.
- X. Wu, H. Lu, Y. Xiao, H. Guo, L. Jia and D. Li, Int. J. Hydrogen Energy, 2022, 47, 34955–34962 CrossRef CAS.
- K. Murata, N. Kurimoto, Y. Yamamoto, A. Oda, J. Ohyama and A. Satsuma, ACS Appl. Nano Mater., 2021, 4, 4532–4541 CrossRef CAS.
- Y. Nakaya, M. Miyazaki, S. Yamazoe, K.-I. Shimizu and S. Furukawa, ACS Catal., 2020, 10, 5163–5172 CrossRef CAS.
- D. Wang, Q. Lei, H. Li, G. Li and Y. Zhao, Catalysts, 2022, 12, 958 CrossRef CAS.
- H. Ye, S. Liu, C. Zhang, Y. Cai and Y. Shi, RSC Adv., 2021, 11, 29287–29297 RSC.
- M. V. Alekseeva, Y. K. Gulyaeva, O. A. Bulavchenko, A. A. Saraev, A. M. Kremneva, S. A. Stepanenko, A. P. Koskin, V. V. Kaichev and V. A. Yakovlev, Dalton Trans., 2022, 51, 6068–6085 RSC.
- Y. Tuo, L. Yang, X. Ma, Z. Ma, S. Gong and P. Li, Int. J. Hydrogen Energy, 2021, 46, 930–942 CrossRef CAS.
- Y. Tuo, Y. Meng, C. Chen, D. Lin, X. Feng, Y. Pan, P. Li, D. Chen, Z. Liu, Y. Zhou and J. Zhang, Appl. Catal., B, 2021, 288, 119996 CrossRef CAS.
- S. Qi, Y. Li, J. Yue, H. Chen, C. Yi and B. Yang, Chin. J. Catal., 2014, 35, 1833–1839 CrossRef CAS.
- K. Kim, J. Oh, T. W. Kim, J. H. Park, J. W. Han and Y.-W. Suh, Catal. Sci. Technol., 2017, 7, 3728–3735 RSC.
- A. A. Al-Muntaser, M. A. Varfolomeev, M. A. Suwaid, M. M. Saleh, R. Djimasbe, C. Yuan, R. R. Zairov and J. Ancheyta, Fuel, 2022, 313, 122652 CrossRef CAS.
- Y. Suttisawat, H. Sakai, M. Abe, P. Rangsunvigit and S. Horikoshi, Int. J. Hydrogen Energy, 2012, 37, 3242–3250 CrossRef CAS.
- N. Brückner, K. Obesser, A. Bösmann, D. Teichmann, W. Arlt, J. Dungs and P. Wasserscheid, ChemSusChem, 2014, 7, 229–235 CrossRef PubMed.
- H. Jorschick, P. Preuster, S. Dürr, A. Seidel, K. Müller, A. Bösmann and P. Wasserscheid, Energy Environ. Sci., 2017, 10, 1652–1659 RSC.
- L. Shi, Y. Zhou, S. Qi, K. J. Smith, X. Tan, J. Yan and C. Yi, ACS Catal., 2020, 10, 10661–10671 CrossRef CAS.
- R. Garidzirai, P. Modisha, I. Shuro, J. Visagie, P. van Helden and D. Bessarabov, Catalysts, 2021, 11, 490 CrossRef CAS.
- J. Zhou, J. S. Chung and S. G. Kang, Appl. Surf. Sci., 2022, 579, 152142 CrossRef CAS.
- M. Niermann, S. Timmerberg, S. Drünert and M. Kaltschmitt, Renewable Sustainable Energy Rev., 2021, 135, 110171 CrossRef CAS.
- Z. Chen, M. Yang, T. Zhu, Z. Zhang, X. Chen, Z. Liu, Y. Dong, G. Cheng and H. Cheng, Int. J. Hydrogen Energy, 2018, 43, 12688–12696 CrossRef CAS.
- S. Lim, Y. Song, K. Jeong, J. H. Park and K. Na, ACS Sustainable Chem. Eng., 2022, 10, 3584–3594 CrossRef CAS.
- A. Søgaard, M. Scheuermeyer, A. Bösmann, P. Wasserscheid and A. Riisager, Chem. Commun., 2019, 55, 2046–2049 RSC.
- Z. Jiang, X. Gong, S. Guo, Y. Bai and T. Fang, Int. J. Hydrogen Energy, 2021, 46, 2376–2389 CrossRef CAS.
- J. Steinhauer, P. Bachmann, E. M. Freiberger, U. Bauer, H. P. Steinrück and C. Papp, J. Phys. Chem. C, 2020, 124, 22559–22567 CrossRef CAS.
- Y. Cui, S. Kwok, A. Bucholtz, B. Davis, R. A. Whitney and P. G. Jessop, New J. Chem., 2008, 32, 1027 RSC.
- P. Ryabchuk, A. Agapova, C. Kreyenschulte, H. Lund, H. Junge, K. Junge and M. Beller, Chem. Commun., 2019, 55, 4969–4972 RSC.
- Y. Dong, H. Zhao, Y. Zhao, M. Yang, H. Zhang and H. Cheng, RSC Adv., 2021, 11, 15729–15737 RSC.
- M. Schwarz, P. Bachmann, T. N. Silva, S. Mohr, M. Scheuermeyer, F. Späth, U. Bauer, F. Düll, J. Steinhauer, C. Hohner, T. Döpper, H. Noei, A. Stierle, C. Papp, H. P. Steinrück, P. Wasserscheid, A. Görling and J. Libuda, Chem. – Eur. J., 2017, 23, 14806–14818 CrossRef CAS PubMed.
- H. Chen, Z. Yang, Z. Zhang, Z. Chen, M. Chi, S. Wang, J. Fu and S. Dai, Angew. Chem., Int. Ed., 2019, 58, 10626–10630 CrossRef CAS PubMed.
- Y. Ding, Y. Dong, H. Zhang, Y. Zhao, M. Yang and H. Cheng, Int. J. Hydrogen Energy, 2021, 46, 27026–27036 CrossRef CAS.
- Z. Feng and X. Bai, Microporous Mesoporous Mater., 2022, 335, 111789 CrossRef CAS.
- X. Yang, Y. Wu, H. Yu, M. Sun, J. Zheng, X. Li, W. Lin and Y. Wu, Int. J. Hydrogen Energy, 2020, 45, 33657–33662 CrossRef CAS.
- F. Sotoodeh, B. J. M. Huber and K. J. Smith, Appl. Catal., A, 2012, 419–420, 67–72 CrossRef CAS.
- Y.-Q. Zou, N. von Wolff, A. Anaby, Y. Xie and D. Milstein, Nat. Catal., 2019, 2, 415–422 CrossRef CAS PubMed.
- Z. Jiang, X. Gong, B. Wang, Z. Wu and T. Fang, Int. J. Hydrogen Energy, 2019, 44, 2951–2959 CrossRef CAS.
- M. Yang, Y. Dong, S. Fei, H. Ke and H. Cheng, Int. J. Hydrogen Energy, 2014, 39, 18976–18983 CrossRef CAS.
- F. Sotoodeh, L. Zhao and K. J. Smith, Appl. Catal., A, 2009, 362, 155–162 CrossRef CAS.
- B. Wang, T. Yan, T. Chang, J. Wei, Q. Zhou, S. Yang and T. Fang, Carbon, 2017, 122, 9–18 CrossRef CAS.
- M. Zhu, L. Xu, L. Du, Y. An and C. Wan, Catalysts, 2018, 8, 638 CrossRef.
- B. Wang, Y. Chen, T. Chang, Z. Jiang, Z. Huang, S. Wang, C. Chang, Y. Chen, J. Wei, S. Yang and T. Fang, Appl. Catal., B, 2020, 266, 118658 CrossRef CAS.
- F. Sotoodeh and K. J. Smith, J. Catal., 2011, 279, 36–47 CrossRef CAS.
- Y. Dong, M. Yang, P. Mei, C. Li and L. Li, Int. J. Hydrogen Energy, 2016, 41, 8498–8505 CrossRef CAS.
- M. Sobota, I. Nikiforidis, M. Amende, B. S. Zanón, T. Staudt, O. Höfert, Y. Lykhach, C. Papp, W. Hieringer, M. Laurin, D. Assenbaum, P. Wasserscheid, H.-P. Steinrück, A. Görling and J. Libuda, Chem. – Eur. J., 2011, 17, 11542–11552 CrossRef CAS PubMed.
- H. Shuang, H. Chen, F. Wu, J. Li, C. Cheng, H. Li and J. Fu, Fuel, 2020, 275, 117896 CrossRef CAS.
- Z. Feng, Y. Wang and X. Bai, Environ. Sci. Pollut. Res., 2022, 29, 39266–39280 CrossRef CAS PubMed.
- C. Tang, Z. Feng and X. Bai, Fuel, 2021, 302, 121186 CrossRef CAS.
- C. Dong, Z. Gao, Y. Li, M. Peng, M. Wang, Y. Xu, C. Li, M. Xu, Y. Deng, X. Qin, F. Huang, X. Wei, Y. Wang, H. Liu, W. Zhou and D. Ma, Nat. Catal., 2022, 5, 485–493 CrossRef CAS.
- M. Amende, C. Gleichweit, K. Werner, S. Schernich, W. Zhao, M. P. A. Lorenz, O. Höfert, C. Papp, M. Koch, P. Wasserscheid, M. Laurin, H.-P. Steinrück and J. Libuda, ACS Catal., 2014, 4, 657–665 CrossRef CAS PubMed.
- M. Amende, C. Gleichweit, S. Schernich, O. Höfert, M. P. A. Lorenz, W. Zhao, M. Koch, K. Obesser, C. Papp, P. Wasserscheid, H.-P. Steinrück and J. Libuda, J. Phys. Chem. Lett., 2014, 5, 1498–1504 CrossRef CAS PubMed.
- W. Peters, A. Seidel, S. Herzog, A. Bösmann, W. Schwieger and P. Wasserscheid, Energy Environ. Sci., 2015, 8, 3013–3021 RSC.
- X. Gong, Z. Jiang and T. Fang, Int. J. Hydrogen Energy, 2020, 45, 6838–6847 CrossRef CAS.
- Z. Yang, X. Gong, L. Li, Z. Jiang, R. Zhang and T. Fang, Fuel, 2023, 334, 126733 CrossRef CAS.
- Y. Wu, Y. Guo, H. Yu, X. Jiang, Y. Zhang, Y. Qi, K. Fu, L. Xie, G. Li, J. Zheng and X. Li, CCS Chem., 2021, 3, 974–984 CrossRef CAS.
- S. Fei, Y. Wang, H. Wu, N. Zheng, X. Fang and D. Liu, ACS Appl. Energy Mater., 2022, 5, 10562–10571 CrossRef CAS.
- B. Wang, T. Chang, X. Gong, Z. Jiang, S. Yang, Y. Chen and T. Fang, ACS Sustainable Chem. Eng., 2018, 7, 1760–1768 CrossRef.
- W. Xue, H. Liu, B. Zhao, C. Tang, B. Y. Xia and B. You, ChemSusChem, 2023, 16, e202201512 CrossRef CAS PubMed.
- W. Xue, H. Liu, B. Mao, H. Liu, M. Qiu, C. Yang, X. Chen and Y. Sun, Chem. Eng. J., 2021, 421, 127781 CrossRef CAS.
- A. Ali, U. Kumar G and H. J. Lee, J. Mech. Sci. Technol., 2019, 33, 5561–5569 CrossRef.
- K. M. Eblagon, K. Tam and S. C. E. Tsang, Energy Environ. Sci., 2012, 5, 8621 RSC.
- M. Yang, C. Han, G. Ni, J. Wu and H. Cheng, Int. J. Hydrogen Energy, 2012, 37, 12839–12845 CrossRef CAS.
- K. Morawa Eblagon, K. Tam, K. M. K. Yu, S.-L. Zhao, X.-Q. Gong, H. He, L. Ye, L.-C. Wang, A. J. Ramirez-Cuesta and S. C. Tsang, J. Phys. Chem. C, 2010, 114, 9720–9730 CrossRef CAS.
- L. Ge, M. Qiu, Y. Zhu, S. Yang, W. Li, W. Li, Z. Jiang and X. Chen, Appl. Catal., B, 2022, 319, 121958 CrossRef CAS.
- H. Yu, X. Yang, X. Jiang, Y. Wu, S. Chen, W. Lin, Y. Wu, L. Xie, X. Li and J. Zheng, Nano Energy, 2021, 80, 105476 CrossRef CAS.
- Y. Wu, H. Yu, Y. Guo, X. Jiang, Y. Qi, B. Sun, H. Li, J. Zheng and X. Li, Chem. Sci., 2019, 10, 10459–10465 RSC.
- X. Liu, J. Shi, X. Bai and W. Wu, Environ. Sci. Pollut. Res., 2022, 29, 48558–48572 CrossRef CAS PubMed.
- Z. Jiang, S. Guo and T. Fang, ACS Appl. Energy Mater., 2019, 2, 7233–7243 CrossRef CAS.
- J. Li, F. Tong, Y. Li, X. Liu, Y. Guo and Y. Wang, Fuel, 2022, 321, 124034 CrossRef CAS.
- X. Gong, S. Guo, Z. Jiang, B. Yang and T. Fang, Int. J. Hydrogen Energy, 2021, 46, 33835–33848 CrossRef CAS.
- C. N. M. Ouma, P. M. Modisha and D. Bessarabov, Appl. Surf. Sci., 2019, 471, 1034–1040 CrossRef CAS.
- S. A. Stepanenko, D. M. Shivtsov, A. P. Koskin, I. P. Koskin, R. G. Kukushkin, P. M. Yeletsky and V. A. Yakovlev, Catalysts, 2022, 12, 1260 CrossRef CAS.
- P. Bachmann, J. Steinhauer, F. Späth, F. Düll, U. Bauer, R. Eschenbacher, F. Hemauer, M. Scheuermeyer, A. Bösmann, M. Büttner, C. Neiß, A. Görling, P. Wasserscheid, H.-P. Steinrück and C. Papp, J. Chem. Phys., 2019, 151, 144711 CrossRef PubMed.
- B. Brigljević, M. Byun and H. Lim, Appl. Energy, 2020, 274, 115314 CrossRef.
- R. M. Cuéllar-Franca and A. Azapagic, J. CO2 Util., 2015, 9, 82–102 CrossRef.
- F. Sha, Z. Han, S. Tang, J. Wang and C. Li, ChemSusChem, 2020, 13, 6160–6181 CAS.
- G. A. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636–2639 CrossRef CAS PubMed.
- A. Iulianelli, P. Ribeirinha, A. Mendes and A. Basile, Renewable Sustainable Energy Rev., 2014, 29, 355–368 CrossRef CAS.
- S. T. Yong, C. W. Ooi, S. P. Chai and X. S. Wu, Int. J. Hydrogen Energy, 2013, 38, 9541–9552 CrossRef CAS.
- C. Rameshan, W. Stadlmayr, S. Penner, H. Lorenz, N. Memmel, M. Hävecker, R. Blume, D. Teschner, T. Rocha, D. Zemlyanov, A. Knop-Gericke, R. Schlögl and B. Klötzer, Angew. Chem., Int. Ed., 2012, 51, 3002–3006 CrossRef CAS PubMed.
- I. Kasatkin, P. Kurr, B. Kniep, A. Trunschke and R. Schlögl, Angew. Chem., Int. Ed., 2007, 46, 7324–7327 CrossRef CAS PubMed.
- K. C. Waugh, Catal. Today, 1992, 15, 51–75 CrossRef CAS.
- K. Klier, in Adv. Catal., ed. D. D. Eley, H. Pines and P. B. Weisz, Academic Press, 1982, vol. 31, pp. 243–313 Search PubMed.
- V. Ponec, Surf. Sci., 1992, 272, 111–117 CrossRef CAS.
- N. Y. Topsøe and H. Topsøe, Top. Catal., 1999, 8, 267–270 CrossRef.
- Y. Choi, K. Futagami, T. Fujitani and J. Nakamura, Appl. Catal., A, 2001, 208, 163–167 CrossRef CAS.
- M. M. Günter, T. Ressler, R. E. Jentoft and B. Bems, J. Catal., 2001, 203, 133–149 CrossRef.
- J. C. Frost, Nature, 1988, 334, 577–580 CrossRef CAS.
- B. Lindström and L. J. Pettersson, Catal. Lett., 2001, 74, 27–30 CrossRef.
- B. Kniep, F. Girgsdies and T. Ressler, J. Catal., 2005, 236, 34–44 CrossRef CAS.
- B. L. Kniep, T. Ressler, A. Rabis, F. Girgsdies, M. Baenitz, F. Steglich and R. Schlögl, Angew. Chem., Int. Ed., 2004, 43, 112–115 CrossRef PubMed.
- C. Mateos-Pedrero, H. Silva, D. A. Pacheco Tanaka, S. Liguori, A. Iulianelli, A. Basile and A. Mendes, Appl. Catal., B, 2015, 174–175, 67–76 CrossRef CAS.
- D. Li, F. Xu, X. Tang, S. Dai, T. Pu, X. Liu, P. Tian, F. Xuan, Z. Xu, I. E. Wachs and M. Zhu, Nat. Catal., 2022, 5, 99–108 CrossRef CAS.
- Z. Cheng, W. Zhou, G. Lan, X. Sun, X. Wang, C. Jiang and Y. Li, J. Energy Chem., 2021, 63, 550–557 CrossRef CAS.
- J. Lin, C. Hu, X. Xu, M. Shao, Y. Hu and C. Ma, Chem. Eng. Technol., 2021, 44, 1121–1130 CrossRef CAS.
- J. He, Z. Yang, L. Zhang, Y. Li and L. Pan, Int. J. Hydrogen Energy, 2017, 42, 9930–9937 CrossRef CAS.
- H. Xi, X. Hou, Y. Liu, S. Qing and Z. Gao, Angew. Chem., Int. Ed., 2014, 53, 11886–11889 CrossRef CAS PubMed.
- Y. Liu, S. Qing, X. Hou, F. Qin, X. Wang, Z. Gao and H. Xiang, Catal. Sci. Technol., 2017, 7, 5069–5078 RSC.
- Y. Liu, H. Kang, X. Hou, S. Qing, L. Zhang, Z. Gao and H. Xiang, Appl. Catal., B, 2023, 323, 122043 CrossRef CAS.
- Y. Liu, S. Qing, X. Hou, F. Qin, X. Wang, Z. Gao and H. Xiang, ChemCatChem, 2018, 10, 5698–5706 CrossRef CAS.
- H.-K. Huang, Y.-K. Chih, W.-H. Chen, C.-Y. Hsu, K.-J. Lin, H.-P. Lin and C.-H. Hsu, Int. J. Hydrogen Energy, 2022, 47, 37542–37551 CrossRef CAS.
- J. L. C. Fajín and M. N. D. S. Cordeiro, ACS Catal., 2021, 12, 512–526 CrossRef.
- K. Ploner, M. Watschinger, P. D. Kheyrollahi Nezhad, T. Götsch, L. Schlicker, E.-M. Köck, A. Gurlo, A. Gili, A. Doran, L. Zhang, N. Köwitsch, M. Armbrüster, S. Vanicek, W. Wallisch, C. Thurner, B. Klötzer and S. Penner, J. Catal., 2020, 391, 497–512 CrossRef CAS.
- K. Ploner, P. Delir Kheyrollahi Nezhad, M. Watschinger, L. Schlicker, M. F. Bekheet, A. Gurlo, A. Gili, A. Doran, S. Schwarz, M. Stöger-Pollach, J. Bernardi, M. Armbrüster, B. Klötzer and S. Penner, Appl. Catal., A, 2021, 623, 118279 CrossRef CAS.
- C. Mateos-Pedrero, C. Azenha, D. A. Pacheco Tanaka, J. M. Sousa and A. Mendes, Appl. Catal., B, 2020, 277, 119243 CrossRef CAS.
- F. Bossola, N. Scotti, F. Somodi, M. Coduri, C. Evangelisti and V. Dal Santo, Appl. Catal., B, 2019, 258, 118016 CrossRef CAS.
- C. Azenha, T. Lagarteira, C. Mateos-Pedrero and A. Mendes, Int. J. Hydrogen Energy, 2021, 46, 17490–17499 CrossRef CAS.
- M. Liao, H. Qin, W. Guo, P. Gao, J. Liu and H. Xiao, Int. J. Hydrogen Energy, 2022, 47, 35136–35148 CrossRef CAS.
- K. M. K. Yu, W. Tong, A. West, K. Cheung, T. Li, G. Smith, Y. Guo and S. C. E. Tsang, Nat. Commun., 2012, 3, 1230 CrossRef PubMed.
- T.-W. Chiu, R.-T. Hong, B.-S. Yu, Y.-H. Huang, S. Kameoka and A.-P. Tsai, Int. J. Hydrogen Energy, 2014, 39, 14222–14226 CrossRef CAS.
- R.-J. Huang, S. Sakthinathan, T.-W. Chiu and C. Dong, RSC Adv., 2021, 11, 12607–12613 RSC.
- B.-Y. Hwang, S. Sakthinathan and T.-W. Chiu, Int. J. Hydrogen Energy, 2019, 44, 2848–2856 CrossRef CAS.
- C.-L. Yu, S. Sakthinathan, B.-Y. Hwang, S.-Y. Lin, T.-W. Chiu, B.-S. Yu, Y.-J. Fan and C. Chuang, Int. J. Hydrogen Energy, 2020, 45, 15752–15762 CrossRef CAS.
- C.-L. Yu, S. Sakthinathan, S.-Y. Chen, B.-S. Yu, T.-W. Chiu and C. Dong, Microporous Mesoporous Mater., 2021, 324, 111305 CrossRef CAS.
- F. Bossola, T. Roongcharoen, M. Coduri, C. Evangelisti, F. Somodi, L. Sementa, A. Fortunelli and V. Dal Santo, Appl. Catal., B, 2021, 297, 120398 CrossRef CAS.
- O. O. Fasanya, A. Y. Atta, M. T. Z. Myint, J. Dutta and B. Y. Jibril, Int. J. Hydrogen Energy, 2021, 46, 3539–3553 CrossRef CAS.
- Y. Chen and Y. Huang, Int. J. Hydrogen Energy, 2023, 48, 1323–1336 CrossRef CAS.
- M. Taghizadeh, H. Akhoundzadeh, A. Rezayan and M. Sadeghian, Int. J. Hydrogen Energy, 2018, 43, 10926–10937 CrossRef CAS.
- Q. Wang, G. Zhang, J. Zhao, L. Zhang and Q. Zhang, Int. J. Hydrogen Energy, 2022, 47, 25256–25265 CrossRef CAS.
- G. Zhang, J. Zhao, Q. Wang, T. Yang, Q. Zhang and L. Zhang, Chem. Eng. J., 2021, 426, 130644 CrossRef CAS.
- S. Sá, H. Silva, L. Brandão, J. M. Sousa and A. Mendes, Appl. Catal., B, 2010, 99, 43–57 CrossRef.
- T. Valdés-Solís, G. Marbán and A. B. Fuertes, Catal. Today, 2006, 116, 354–360 CrossRef.
- Y. Matsumura and H. Ishibe, Appl. Catal., B, 2009, 91, 524–532 CrossRef CAS.
- S. D. Jones, L. M. Neal and H. E. Hagelin-Weaver, Appl. Catal., B, 2008, 84, 631–642 CrossRef CAS.
- S. D. Jones and H. E. Hagelin-Weaver, Appl. Catal., B, 2009, 90, 195–204 CrossRef CAS.
- L. Chen, Z. Qi, X. Peng, J.-L. Chen, C.-W. Pao, X. Zhang, C. Dun, M. Young, D. Prendergast, J. J. Urban, J. Guo, G. A. Somorjai and J. Su, J. Am. Chem. Soc., 2021, 143, 12074–12081 CrossRef CAS PubMed.
- R. Pérez-Hernández, Int. J. Hydrogen Energy, 2021, 46, 25954–25964 CrossRef.
- L. Lin, W. Zhou, R. Gao, S. Yao, X. Zhang, W. Xu, S. Zheng, Z. Jiang, Q. Yu, Y. Li, C. Shi, X. Wen and D. Ma, Nature, 2017, 544, 80–83 CrossRef CAS PubMed.
- L. Lin, Q. Yu, M. Peng, A. Li, S. Yao, S. Tian, X. Liu, A. Li, Z. Jiang, R. Gao, X. Han, Y. Li, X. Wen, W. Zhou and D. Ma, J. Am. Chem. Soc., 2021, 143, 309–317 CrossRef CAS PubMed.
- F. Cai, J. J. Ibrahim, Y. Fu, W. Kong, J. Zhang and Y. Sun, Appl. Catal., B, 2020, 264, 118500 CrossRef.
- D. Li, Y. Li, X. Liu, Y. Guo, C.-W. Pao, J.-L. Chen, Y. Hu and Y. Wang, ACS Catal., 2019, 9, 9671–9682 CrossRef CAS.
- Z. Shao, S. Zhang, X. Liu, H. Luo, C. Huang, H. Zhou, Z. Wu, J. Li, H. Wang and Y. Sun, Appl. Catal., B, 2022, 316, 121669 CrossRef CAS.
- H. Wang, L. Wang, D. Lin, X. Feng, X. Chu, L. Li and F.-S. Xiao, Catal. Today, 2021, 382, 42–47 CrossRef CAS.
- M. Kusche, F. Agel, N. Ní Bhriain, A. Kaftan, M. Laurin, J. Libuda and P. Wasserscheid, ChemSusChem, 2014, 7, 2516–2526 CrossRef CAS PubMed.
- M. Kusche, F. Enzenberger, S. Bajus, H. Niedermeyer, A. Bösmann, A. Kaftan, M. Laurin, J. Libuda and P. Wasserscheid, Angew. Chem., Int. Ed., 2013, 52, 5028–5032 CrossRef CAS PubMed.
- N. Iwasa, S. Masuda, N. Ogawa and N. Takezawa, Appl. Catal., A, 1995, 125, 145–157 CrossRef CAS.
- A. M. Karim, T. Conant and A. K. Datye, Phys. Chem. Chem. Phys., 2008, 10, 5584–5590 RSC.
- H. Zhang, J. Sun, V. L. Dagle, B. Halevi, A. K. Datye and Y. Wang, ACS Catal., 2014, 4, 2379–2386 CrossRef CAS.
- Y. Chen, Q. Dai, Q. Zhang and Y. Huang, Int. J. Hydrogen Energy, 2022, 47, 14869–14883 CrossRef CAS.
- L. Liu, Y. Lin, Y. Hu, Z. Lin, S. Lin, M. Du, L. Zhang, X. Zhang, J. Lin, Z. Zhang, H. Xiong, S. Wang, B. Ge, S. Wan and Y. Wang, ACS Catal., 2022, 12, 2714–2721 CrossRef CAS.
- N. Köwitsch, L. Thoni, B. Klemmed, A. Benad, P. Paciok, M. Heggen, I. Köwitsch, M. Mehring, A. Eychmüller and M. Armbrüster, ACS Catal., 2021, 11, 304–312 CrossRef.
- S. Penner, B. Jenewein, H. Gabasch, B. Klötzer, D. Wang, A. Knop-Gericke, R. Schlögl and K. Hayek, J. Catal., 2006, 241, 14–19 CrossRef CAS.
- N. Iwasa, T. Mayanagi, W. Nomura, M. Arai and N. Takezawa, Appl. Catal., A, 2003, 248, 153–160 CrossRef CAS.
- T. Conant, A. M. Karim, V. Lebarbier, Y. Wang, F. Girgsdies, R. Schlögl and A. Datye, J. Catal., 2008, 257, 64–70 CrossRef CAS.
- Y. Suwa, S.-i Ito, S. Kameoka, K. Tomishige and K. Kunimori, Appl. Catal., A, 2004, 267, 9–16 CrossRef CAS.
- B. Halevi, E. J. Peterson, A. Roy, A. DeLariva, E. Jeroro, F. Gao, Y. Wang, J. M. Vohs, B. Kiefer, E. Kunkes, M. Hävecker, M. Behrens, R. Schlögl and A. K. Datye, J. Catal., 2012, 291, 44–54 CrossRef CAS.
- R. Pérez-Hernández, A. D. Avendaño, E. Rubio and V. Rodríguez-Lugo, Top. Catal., 2011, 54, 572–578 CrossRef.
- C.-W. Tang, Y.-J. Chen, C.-T. Yeh, R.-C. Wu, C.-C. Wang and C.-B. Wang, Int. J. Hydrogen Energy, 2021, 46, 80–88 CrossRef CAS.
- C. Pojanavaraphan, A. Luengnaruemitchai and E. Gulari, Int. J. Hydrogen Energy, 2013, 38, 1348–1362 CrossRef CAS.
- H. Lorenz, W. Jochum, B. Klötzer, M. Stöger-Pollach, S. Schwarz, K. Pfaller and S. Penner, Appl. Catal., A, 2008, 347, 34–42 CrossRef CAS.
- H. Lorenz, M. Friedrich, M. Armbrüster, B. Klötzer and S. Penner, J. Catal., 2013, 297, 151–154 CrossRef CAS PubMed.
- W. Cao, G. Chen, S. Li and Q. Yuan, Chem. Eng. J., 2006, 119, 93–98 CrossRef CAS.
- X. Chen, J. Wang, Z. Han, C. Li, C. Tang, D. Zhao, Q. Yang and C. Li, ChemCatChem, 2021, 14, e202101232 Search PubMed.
- X. Chen, Z. Feng, D. Zhao, Q. Yang and C. Li, Catal. Sci. Technol., 2022, 12, 7048–7056 RSC.
- Z. Cheng, C. Jiang, X. Sun, G. Lan, X. Wang, L. He, Y. Li, H. Tang and Y. Li, Ind. Eng. Chem. Res., 2022, 61, 11699–11707 CrossRef CAS.
- J. Zhao, G. Zhang, H. Liu, Q. Shu and Q. Zhang, Int. J. Hydrogen Energy, 2022, 47, 18294–18304 CrossRef CAS.
- C.-C. Chang, J.-W. Wang, C.-T. Chang, B.-J. Liaw and Y.-Z. Chen, Chem. Eng. J., 2012, 192, 350–356 CrossRef CAS.
- G. Huang, B.-J. Liaw, C.-J. Jhang and Y.-Z. Chen, Appl. Catal., A, 2009, 358, 7–12 CrossRef CAS.
- Y. Matsumura and H. Ishibe, J. Power Sources, 2012, 209, 72–80 CrossRef CAS.
- S. Jin, D. Li, Z. Wang, Y. Wang, L. Sun and M. Zhu, Catal. Sci. Technol., 2022, 12, 7003–7009 RSC.
- X. Liu, J. Xu, S. Li, Z. Chen, X. Xu, X. Fang and X. Wang, Catal. Today, 2022, 402, 228–240 CrossRef CAS.
- M. Taghizadeh and M. H. Abbandanak, Int. J. Hydrogen Energy, 2022, 47, 16362–16374 CrossRef CAS.
- R. Y. Abrokwah, V. G. Deshmane and D. Kuila, J. Mol. Catal. A: Chem., 2016, 425, 10–20 CrossRef CAS.
- H. Shahsavar, M. Taghizadeh and A. D. Kiadehi, Int. J. Hydrogen Energy, 2021, 46, 8906–8921 CrossRef CAS.
- N. Kamyar, Y. Khani, M. M. Amini, F. Bahadoran and N. Safari, ChemistrySelect, 2019, 4, 6113–6122 CrossRef CAS.
- D. Liu, Y. Men, J. Wang, G. Kolb, X. Liu, Y. Wang and Q. Sun, Int. J. Hydrogen Energy, 2016, 41, 21990–21999 CrossRef CAS.
- N. Köwitsch, S. Barth, K. Ploner, R. Blume, D. Teschner, S. Penner and M. Armbrüster, ChemPhysChem, 2022, 23, e202200074 CrossRef PubMed.
- X. Liu, Y. Men, J. Wang, R. He and Y. Wang, J. Power Sources, 2017, 364, 341–350 CrossRef CAS.
- N. Köwitsch, L. Thoni, B. Klemmed, A. Benad, P. Paciok, M. Heggen, A. Eychmüller and M. Armbrüster, J. Phys. Chem. C, 2021, 125, 9809–9817 CrossRef.
- A. Karim, T. Conant and A. Datye, J. Catal., 2006, 243, 420–427 CrossRef CAS.
- Y. Ma, G. Guan, C. Shi, A. Zhu, X. Hao, Z. Wang, K. Kusakabe and A. Abudula, Int. J. Hydrogen Energy, 2014, 39, 258–266 CrossRef CAS.
- Y. Ma, G. Guan, X. Hao, Z. Zuo, W. Huang, P. Phanthong, K. Kusakabe and A. Abudula, RSC Adv., 2014, 4, 44175–44184 RSC.
- N. Yi, R. Si, H. Saltsburg and M. Flytzani-Stephanopoulos, Energy Environ. Sci., 2010, 3, 831–837 RSC.
- S. Pongstabodee, S. Monyanon and A. Luengnaruemitchai, J. Ind. Eng. Chem., 2012, 18, 1272–1279 CrossRef CAS.
- M. Yang, S. Li and G. Chen, Appl. Catal., B, 2011, 101, 409–416 CrossRef CAS.
- N. Onishi and Y. Himeda, Coord. Chem. Rev., 2022, 472, 214767 CrossRef CAS.
- D. Morton and D. J. Cole-Hamilton, J. Chem. Soc., Chem. Commun., 1987, 248–249, 10.1039/C39870000248.
- M. Nielsen, E. Alberico, W. Baumann, H.-J. Drexler, H. Junge, S. Gladiali and M. Beller, Nature, 2013, 495, 85–89 CrossRef CAS PubMed.
- A. Monney, E. Barsch, P. Sponholz, H. Junge, R. Ludwig and M. Beller, Chem. Commun., 2014, 50, 707–709 RSC.
- R. E. Rodríguez-Lugo, M. Trincado, M. Vogt, F. Tewes, G. Santiso-Quinones and H. Grützmacher, Nat. Chem., 2013, 5, 342–347 CrossRef PubMed.
- P. Hu, Y. Diskin-Posner, Y. Ben-David and D. Milstein, ACS Catal., 2014, 4, 2649–2652 CrossRef CAS.
- F. F. van de Watering, M. Lutz, W. I. Dzik, B. de Bruin and J. N. H. Reek, ChemCatChem, 2016, 8, 2752–2756 CrossRef CAS PubMed.
- K.-I. Fujita, R. Kawahara, T. Aikawa and R. Yamaguchi, Angew. Chem., Int. Ed., 2015, 54, 9057–9060 CrossRef CAS PubMed.
- C. Prichatz, E. Alberico, W. Baumann, H. Junge and M. Beller, ChemCatChem, 2017, 9, 1891–1896 CrossRef CAS.
- E. Alberico, P. Sponholz, C. Cordes, M. Nielsen, H.-J. Drexler, W. Baumann, H. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 14162–14166 CrossRef CAS PubMed.
- M. Andérez-Fernández, L. K. Vogt, S. Fischer, W. Zhou, H. Jiao, M. Garbe, S. Elangovan, K. Junge, H. Junge, R. Ludwig and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 559–562 CrossRef PubMed.
- E. A. Bielinski, M. Förster, Y. Zhang, W. H. Bernskoetter, N. Hazari and M. C. Holthausen, ACS Catal., 2015, 5, 2404–2415 CrossRef CAS.
- E. Alberico, A. J. J. Lennox, L. K. Vogt, H. Jiao, W. Baumann, H.-J. Drexler, M. Nielsen, A. Spannenberg, M. P. Checinski, H. Junge and M. Beller, J. Am. Chem. Soc., 2016, 138, 14890–14904 CrossRef CAS PubMed.
- J. Luo, S. Kar, M. Rauch, M. Montag, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2021, 143, 17284–17291 CrossRef CAS PubMed.
- C. Prichatz, E. Alberico, W. Baumann, H. Junge and M. Beller, ChemCatChem, 2017, 9, 1891–1896 CrossRef CAS.
- V. Arora, E. Yasmin, N. Tanwar, V. R. Hathwar, T. Wagh, S. Dhole and A. Kumar, ACS Catal., 2023, 13, 3605–3617 CrossRef CAS.
- S. Kuld, M. Thorhauge, H. Falsig, C. F. Elkjær, S. Helveg, I. Chorkendorff and J. Sehested, Science, 2016, 352, 969–974 CrossRef CAS PubMed.
- F. Studt, M. Behrens, E. L. Kunkes, N. Thomas, S. Zander, A. Tarasov, J. Schumann, E. Frei, J. B. Varley, F. Abild-Pedersen, J. K. Nørskov and R. Schlögl, ChemCatChem, 2015, 7, 1105–1111 CrossRef CAS.
- A. Bansode and A. Urakawa, J. Catal., 2014, 309, 66–70 CrossRef CAS.
- J. Graciani, K. Mudiyanselage, F. Xu, A. E. Baber, J. Evans, S. D. Senanayake, D. J. Stacchiola, P. Liu, J. Hrbek, J. F. Sanz and J. A. Rodriguez, Science, 2014, 345, 546–550 CrossRef CAS PubMed.
- S. Tada, A. Katagiri, K. Kiyota, T. Honma, H. Kamei, A. Nariyuki, S. Uchida and S. Satokawa, J. Phys. Chem. C, 2018, 122, 5430–5442 CrossRef CAS.
- Y. Chai, B. Qin, B. Li, W. Dai, G. Wu, N. Guan and L. Li, Natl. Sci. Rev., 2023, nwad043, DOI:10.1093/nsr/nwad043.
- N. Iwasa, H. Suzuki, M. Terashita, M. Arai and N. Takezawa, Catal. Lett., 2004, 96, 75–78 CrossRef CAS.
- J. Song, S. Liu, C. Yang, G. Wang, H. Tian, Z.-J. Zhao, R. Mu and J. Gong, Appl. Catal., B, 2020, 263, 118367 CrossRef CAS.
- X. Zhou, J. Qu, F. Xu, J. Hu, J. S. Foord, Z. Zeng, X. Hong and S. C. Edman Tsang, Chem. Commun., 2013, 49, 1747–1749 RSC.
- J. Qu, X. Zhou, F. Xu, X.-Q. Gong and S. C. E. Tsang, J. Phys. Chem. C, 2014, 118, 24452–24466 CrossRef CAS.
- J. Wang, S.-m Lu, J. Li and C. Li, Chem. Commun., 2015, 51, 17615–17618 RSC.
- J. Ye, C. Liu and Q. Ge, J. Phys. Chem. C, 2012, 116, 7817–7825 CrossRef CAS.
- J. Ye, C. Liu, D. Mei and Q. Ge, ACS Catal., 2013, 3, 1296–1306 CrossRef CAS.
- K. Sun, Z. Fan, J. Ye, J. Yan, Q. Ge, Y. Li, W. He, W. Yang and C.-J. Liu, J. CO2 Util., 2015, 12, 1–6 CrossRef CAS.
- O. Martin, A. J. Martín, C. Mondelli, S. Mitchell, T. F. Segawa, R. Hauert, C. Drouilly, D. Curulla-Ferré and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2016, 55, 6261–6265 CrossRef CAS PubMed.
- M. S. Frei, C. Mondelli, A. Cesarini, F. Krumeich, R. Hauert, J. A. Stewart, D. Curulla Ferré and J. Pérez-Ramírez, ACS Catal., 2020, 10, 1133–1145 CrossRef CAS.
- N. Rui, Z. Wang, K. Sun, J. Ye, Q. Ge and C.-J. Liu, Appl. Catal., B, 2017, 218, 488–497 CrossRef CAS.
- Z. Han, C. Tang, J. Wang, L. Li and C. Li, J. Catal., 2021, 394, 236–244 CrossRef CAS.
- X. Jia, K. Sun, J. Wang, C. Shen and C.-J. Liu, J. Energy Chem., 2020, 50, 409–415 CrossRef.
- J. Wang, K. Sun, X. Jia and C.-J. Liu, Catal. Today, 2021, 365, 341–347 CrossRef CAS.
- S. Tang, Z. Feng, Z. Han, F. Sha, C. Tang, Y. Zhang, J. Wang and C. Li, J. Catal., 2023, 417, 462–472 CrossRef CAS.
- N. Rui, F. Zhang, K. Sun, Z. Liu, W. Xu, E. Stavitski, S. D. Senanayake, J. A. Rodriguez and C.-J. Liu, ACS Catal., 2020, 10, 11307–11317 CrossRef CAS.
- J. Wang, G. Li, Z. Li, C. Tang, Z. Feng, H. An, H. Liu, T. Liu and C. Li, Sci. Adv., 2017, 3, e1701290 CrossRef PubMed.
- J. Wang, C. Tang, G. Li, Z. Han, Z. Li, H. Liu, F. Cheng and C. Li, ACS Catal., 2019, 9, 10253–10259 CrossRef CAS.
- J. Hu, L. Yu, J. Deng, Y. Wang, K. Cheng, C. Ma, Q. Zhang, W. Wen, S. Yu, Y. Pan, J. Yang, H. Ma, F. Qi, Y. Wang, Y. Zheng, M. Chen, R. Huang, S. Zhang, Z. Zhao, J. Mao, X. Meng, Q. Ji, G. Hou, X. Han, X. Bao, Y. Wang and D. Deng, Nat. Catal., 2021, 4, 242–250 CrossRef CAS.
- X. Jiang, X. Nie, X. Guo, C. Song and J. G. Chen, Chem. Rev., 2020, 120, 7984–8034 CrossRef CAS PubMed.
- S.-T. Bai, G. De Smet, Y. Liao, R. Sun, C. Zhou, M. Beller, B. U. W. Maes and B. F. Sels, Chem. Soc. Rev., 2021, 50, 4259–4298 RSC.
- A. Klerke, C. H. Christensen, J. K. Nørskov and T. Vegge, J. Mater. Chem., 2008, 18, 2304 RSC.
- K. E. Lamb, M. D. Dolan and D. F. Kennedy, Int. J. Hydrogen Energy, 2019, 44, 3580–3593 CrossRef CAS.
- L. Green, Int. J. Hydrogen Energy, 1982, 7, 355–359 CrossRef CAS.
- R. Lan, J. T. S. Irvine and S. Tao, Int. J. Hydrogen Energy, 2012, 37, 1482–1494 CrossRef CAS.
- S. Lee, T. Kim, G. Han, S. Kang, Y.-S. Yoo, S.-Y. Jeon and J. Bae, Renewable Sustainable Energy Rev., 2021, 150, 111447 CrossRef CAS.
- X. Duan, J. Ji, G. Qian, C. Fan, Y. Zhu, X. Zhou, D. Chen and W. Yuan, J. Mol. Catal. A: Chem., 2012, 357, 81–86 CrossRef CAS.
- C. Chen, K. Wu, H. Ren, C. Zhou, Y. Luo, L. Lin, C. Au and L. Jiang, Energy Fuels, 2021, 35, 11693–11706 CrossRef CAS.
- A. Boretti and S. Castelletto, ACS Energy Lett., 2022, 7, 2557–2564 CrossRef CAS.
- S. F. Yin, B. Q. Xu, X. P. Zhou and C. T. Au, Appl. Catal., A, 2004, 277, 1–9 CrossRef CAS.
- K. Yamazaki, M. Matsumoto, M. Ishikawa and A. Sato, Appl. Catal., B, 2023, 325, 122352 CrossRef CAS.
- F. Zhiqiang, W. Ziqing, L. Dexing, L. Jianxin, Y. Lingzhi, W. Qin and W. Zhong, Fuel, 2022, 326, 125094 CrossRef.
- N. Jeon, S. Kim, A. Tayal, J. Oh, W. Yoon, W. B. Kim and Y. Yun, ACS Sustainable Chem. Eng., 2022, 10, 15564–15573 CrossRef CAS.
- T. A. Le, Y. Kim, H. W. Kim, S.-U. Lee, J.-R. Kim, T.-W. Kim, Y.-J. Lee and H.-J. Chae, Appl. Catal., B, 2021, 285, 119831 CrossRef CAS.
- L. Huo, X. Han, L. Zhang, B. Liu, R. Gao, B. Cao, W. Wang, C. Jia, K. Liu, J. Liu and J. Zhang, Appl. Catal., B, 2021, 294, 120254 CrossRef CAS.
- Q. C. Do, Y. Kim, T. A. Le, G. J. Kim, J.-R. Kim, T.-W. Kim, Y.-J. Lee and H.-J. Chae, Appl. Catal., B, 2022, 307, 121167 CrossRef CAS.
- C. Zhou, K. Wu, H. Huang, C. Cao, Y. Luo, C. Chen, L. Lin, C. Au and L. Jiang, ACS Catal., 2021, 11, 10345–10350 CrossRef CAS.
- H. Maleki, M. Fulton and V. Bertola, Chem. Eng. J., 2021, 411, 128595 CrossRef CAS.
- S.-F. Yin, B.-Q. Xu, C.-F. Ng and C.-T. Au, Appl. Catal., B, 2004, 48, 237–241 CrossRef CAS.
- L. Li, Y. Wang, Z. P. Xu and Z. Zhu, Appl. Catal., A, 2013, 467, 246–252 CrossRef CAS.
- S. Sayas, N. Morlanés, S. P. Katikaneni, A. Harale, B. Solami and J. Gascon, Catal. Sci. Technol., 2020, 10, 5027–5035 RSC.
- J. Zhao, S. Xu, H. Wu, Z. You, L. Deng and X. Qiu, Chem. Commun., 2019, 55, 14410–14413 RSC.
- H. He, H. Jiang, F. Yang, J. Liu, W. Zhang, M. Jin and Z. Li, Int. J. Hydrogen Energy, 2023, 48, 5030–5041 CrossRef CAS.
- H. Li, L. Guo, J. Qu, X. Fang, Y. Fu, J. Duan, W. Wang and C. Li, Int. J. Hydrogen Energy, 2023, 48, 8985–8996 CrossRef CAS.
- L. Zhou, X. Fu, Y. Xu, W. Wang and C. Jia, ACS Appl. Nano Mater., 2023, 6, 2952–2962 CrossRef CAS.
- S.-J. Park and B.-J. Kim, J. Colloid Interface Sci., 2005, 291, 597–599 CrossRef CAS PubMed.
- C. W. Skarstrom, Ann. N. Y. Acad. Sci., 1959, 72, 751–763 CrossRef CAS.
- K. P. Kostroski and P. C. Wankat, Ind. Eng. Chem. Res., 2006, 45, 8117–8133 CrossRef CAS.
- T. Inui, Y. Okugawa and M. Yasuda, Ind. Eng. Chem. Res., 1988, 27, 1103–1109 CrossRef CAS.
- G. Q. Lu, J. C. Diniz da Costa, M. Duke, S. Giessler, R. Socolow, R. H. Williams and T. Kreutz, J. Colloid Interface Sci., 2007, 314, 589–603 CrossRef CAS PubMed.
- S. G. Shore and R. W. Parry, J. Am. Chem. Soc., 1955, 77, 6084–6085 CrossRef CAS.
- U. B. Demirci, Int. J. Hydrogen Energy, 2017, 42, 9978–10013 CrossRef CAS.
- B. L. Conley, D. Guess and T. J. Williams, J. Am. Chem. Soc., 2011, 133, 14212–14215 CrossRef CAS PubMed.
- J. M. B. Richard, J. Keaton and R. Tom Baker, J. Am. Chem. Soc., 2007, 129, 1844–1845 CrossRef PubMed.
- M. Chandra and Q. Xu, J. Power Sources, 2006, 156, 190–194 CrossRef CAS.
- W. Chen, D. Li, Z. Wang, G. Qian, Z. Sui, X. Duan, X. Zhou, I. Yeboah and D. Chen, AlChE J., 2017, 63, 60–65 CrossRef CAS.
- J. Liu, P. Li, R. Jiang, X. Zheng and P. Liu, ChemCatChem, 2021, 13, 4142–4150 CrossRef CAS.
- A. Aijaz, A. Karkamkar, Y. J. Choi, N. Tsumori, E. Rönnebro, T. Autrey, H. Shioyama and Q. Xu, J. Am. Chem. Soc., 2012, 134, 13926–13929 CrossRef CAS PubMed.
- S. Ö. Önder Metin, Energy Fuels, 2009, 23, 3517–3526 CrossRef.
- P. Li, Y. Huang, Q. Huang, R. Chen, J. Li and S. Tian, J. Energy Chem., 2023, 79, 72–82 CrossRef CAS.
- Z. Li, T. He, L. Liu, W. Chen, M. Zhang, G. Wu and P. Chen, Chem. Sci., 2017, 8, 781–788 RSC.
- R. K. Borah, A. P. Fatrekar, P. Bakre, S. G. Tilve and A. A. Vernekar, J. Mater. Chem. A, 2022, 10, 25490–25499 RSC.
- C. Yuan, T. Xu, M. Guo, T. Zhang and X. Yu, Appl. Catal., B, 2023, 321, 122044 CrossRef CAS.
- W. Xu, M. Liu, S. Wang, Z. Peng, R. Shen and B. Li, Int. J. Hydrogen Energy, 2022, 47, 23213–23220 CrossRef CAS.
- B. Pant, H. R. Pant, M. Park, Y. Liu, J.-W. Choi, N. A. M. Barakat and H.-Y. Kim, Catal. Commun., 2014, 50, 63–68 CrossRef CAS.
- S. Hu, X. Qu, J. Bai, P. Li, Q. Li, F. Wang and L. Song, ACS Sustainable Chem. Eng., 2017, 5, 6863–6872 CrossRef CAS.
- N. Kang, X. Wei, R. Shen, B. Li, E. G. Cal, S. Moya, L. Salmon, C. Wang, E. Coy, M. Berlande, J.-L. Pozzo and D. Astruc, Appl. Catal., B, 2023, 320, 121957 CrossRef CAS.
- B. Coşkuner Filiz and A. Kantürk Figen, Int. J. Hydrogen Energy, 2023 DOI:10.1016/j.ijhydene.2022.12.176.
- X. Li, Q. Yao, Z. Li, H. Li, Q. Zhu and Z. Lu, J. Mater. Chem. A, 2022, 10, 326–336 RSC.
- J. Liao, Y. Shao, Y. Feng, J. Zhang, C. Song, W. Zeng, J. Tang, H. Dong, Q. Liu and H. Li, Appl. Catal., B, 2023, 320, 121973 CrossRef CAS.
- J. Liao, Y. Wu, Y. Shao, Y. Feng, X. Zhang, W. Zhang, J. Li, M. Wu, H. Dong, Q. Liu and H. Li, Chem. Eng. J., 2022, 449, 137755 CrossRef CAS.
- Y. Cheng, X. Wu and H. Xu, Sustainable Energy Fuels, 2019, 3, 343–365 RSC.
- X. Yang, D. A. Bulushev, J. Yang and Q. Zhang, Energies, 2022, 15, 6360 CrossRef CAS.
- P. Li, R. Chen, Y. Huang, W. Li, S. Zhao and S. Tian, Appl. Catal., B, 2022, 300, 120725 CrossRef CAS.
- P. Li, Y. Huang, Q. Huang, R. Chen, J. Li and S. Tian, Appl. Catal., B, 2022, 313, 121444 CrossRef CAS.
- J. Deng, X. Zhou, J. Zou, Y. Qin and P. Wang, ACS Appl. Energy Mater., 2022, 5, 7408–7419 CrossRef CAS.
- S. Zhou, Y. Yang, P. Yin, Z. Ren, L. Wang and M. Wei, ACS Appl. Mater. Interfaces, 2022, 14, 5275–5286 CrossRef CAS PubMed.
- J. Prasad and J. L. Gland, Langmuir, 1991, 7, 722–726 CrossRef CAS.
- D. J. Alberas, J. Kiss, Z. M. Liu and J. M. White, Surf. Sci., 1992, 278, 51–61 CrossRef CAS.
- S. K. Singh, X.-B. Zhang and Q. Xu, J. Am. Chem. Soc., 2009, 131, 9894–9895 CrossRef CAS PubMed.
- K. S. Al-Thubaiti and Z. Khan, Int. J. Hydrogen Energy, 2020, 45, 13960–13974 CrossRef CAS.
- Q. Yao, M. He, X. Hong, X. Chen, G. Feng and Z. Lu, Int. J. Hydrogen Energy, 2019, 44, 28430–28440 CrossRef CAS.
- S. K. Singh and Q. Xu, Inorg. Chem., 2010, 49, 6148–6152 CrossRef CAS PubMed.
- P. Zhao, N. Cao, J. Su, W. Luo and G. Cheng, ACS Sustainable Chem. Eng., 2015, 3, 1086–1093 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |