
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransport of double-stranded ribonucleic acids (dsRNA) and deoxyribonucleic acids (DNA) in sand and iron oxide-coated sand columns under varying solution chemistries†
Katharina
Sodnikar
 a,
Ralf
Kaegi
b,
Iso
Christl
a,
Martin Herbert
Schroth
*a and
Michael
Sander
*a
a,
Ralf
Kaegi
b,
Iso
Christl
a,
Martin Herbert
Schroth
*a and
Michael
Sander
*a
aInstitute of Biogeochemistry and Pollutant Dynamics, ETH Zurich, 8092 Zurich, Switzerland. E-mail: michael.sander@env.ethz.ch; Tel: +41-44 632 8314
bEawag, Swiss Federal Institute of Aquatic Science and Technology, Überlandstrasse 133, 8600 Dübendorf, Switzerland
First published on 23rd October 2023
Abstract
Assessing ecological risks associated with the use of genetically modified RNA interference crops demands an understanding of the fate of crop-released insecticidal double-stranded RNA (dsRNA) molecules in soils. We studied the adsorption of one dsRNA and two double-stranded DNA as model nucleic acids (NAs) during transport through sand- and iron oxide-coated sand (IOCS)-filled columns over a range of solution pH and ionic compositions. Consistent with NA-sand electrostatic repulsion, we observed only slight retention of NAs in sand columns. Conversely, pronounced NA retention in IOCS columns is consistent with strong and irreversible NA adsorption involving electrostatic attraction to and inner-sphere complex formation of NAs with iron oxide coatings. Adsorption of NAs to iron oxides revealed a fast and a slow kinetic adsorption regime, possibly caused by the excluded-area effect. Adsorption of NAs to sand and IOCS increased in the presence of dissolved Mg2+ and with increasing ionic strength, reflecting cation-bridging and screening of repulsive electrostatics, respectively. The co-solute phosphate and a pre-adsorbed dissolved organic matter isolate competitively suppressed dsRNA adsorption to IOCS. Similar adsorption characteristics of dsRNA and similarly sized DNA suggest that existing information on DNA adsorption to soil particles helps in predicting adsorption and fate of dsRNA molecules in soils.
Environmental significanceDouble-stranded RNA (dsRNA) molecules are novel plant-incorporated protectants expressed in genetically modified crops and released into agricultural soils upon use. Ecological risk assessment (ERA) of dsRNA demands understanding dsRNA adsorption to soil particles, which affects its fate. We systematically studied adsorption of model dsRNA and DNA molecules during transport through sand and iron oxide-coated sand (IOCS) columns over a range of solution pH, ionic strengths and compositions. We show that adsorption is governed by nucleic acid (NA)-sorbent electrostatic interactions and NA-IOCS inner-sphere complex formation, and has a slow kinetic component. The presence of phosphate and dissolved organic matter suppress NA adsorption to IOCS. Our findings indicate that existing knowledge on DNA adsorption in soils applies to dsRNA and thereby informs ERA of dsRNA. |
Introduction
Modern agriculture relies on using genetically modified RNA interference (RNAi) crops that express insecticidal double-stranded RNA (dsRNA) molecules as plant-incorporated protectants targeting specific pest insects.1,2 When feeding on RNAi crops, the targeted insects co-ingest the dsRNA molecules, which subsequently trigger the breakdown of sequence-complementary insect mRNA3–6 that codes for the biosynthesis of a protein essential to the insects. As a consequence, the insects suffer from retarded development or die.4,7–9 Planting RNAi crops is expected to result in the release of dsRNA molecules to agricultural soils,10 therefore requiring an assessment of potential release-associated ecological risks, including possible dsRNA effects on non-targeted soil-dwelling organisms.11 Adsorption of dsRNA to soil mineral surfaces is a key process determining exposure of such organisms to dsRNA as well as overall dsRNA fate. On the one hand, dsRNA adsorption would decrease its mobility and exposure of soil organisms. On the other hand, dsRNA adsorption may protect it from hydrolytic enzymes in the soil pore water and, thereby, increase dsRNA stability in agricultural soils, as demonstrated previously for DNA molecules.12–15 However, a recent study suggests that adsorbed dsRNA molecules may also be destabilized due to mineral-catalyzed dsRNA hydrolysis over the timescale of days.16Adsorption of dsRNA molecules to mineral soil particle surfaces has been assessed only in a few previous studies.16–19 By comparison, DNA adsorption to soil particles and soil organic matter has been extensively studied and is well understood.12,13,15,20–47 This raises the question if the extensive information on DNA adsorption in soils is transferrable to dsRNA adsorption. In an effort to answer this question, we recently demonstrated comparable adsorption characteristics of similarly sized dsRNA and DNA molecules to iron oxide surfaces in a batch equilibration study.18 Similarities in the adsorption characteristics can be rationalized based on a central structural resemblance of these NAs: both are polyelectrolytes with negatively charged phosphodiester backbones. The same interaction forces thus likely control dsRNA and DNA adsorption to soil particle surfaces: electrostatic attraction and repulsion between the negatively charged NA and net positively and negatively charged soil particle surfaces, respectively. Similarly, inner-sphere complex formation between the phosphodiester groups in both dsRNA and DNA backbones with surface hydroxyl group on specific minerals, such as iron oxides,18,20,21,26,27,48–53 may lead to irreversible adsorption. Finally, polyelectrolyte adsorption theory may explain effects of solution chemistry on both dsRNA and DNA adsorption:54–62 increased NA adsorption with increasing solution ionic strength and in the presence of dissolved divalent cations likely resulted from NAs adopting more compact conformations and thus smaller footprints in adsorbed state, allowing for a larger number of adsorbed NA molecules at sorbent surface saturation.18,54,63–68
While our batch equilibration study supports similar adsorption characteristics of dsRNA and DNA,18 additional experimental verification is required given the nature of batch experiments. First and foremost, NA amounts ‘adsorbed’ to iron oxide particles in agitated batch reactors may be affected by NA-particle hetero-coagulation, a process not occurring to the same extent in soils. One possibility to exclude potential hetero-coagulation effects is to immobilize the iron oxide on a larger carrier material that is not itself suspended in solution. Second, continuous stirring of iron oxide-NA suspensions in batch reactors18 potentially results in iron oxide-particle abrasion69 and thus higher available surface area, leading to increased adsorbed NA concentrations. Third, batch reactors are well mixed, ensuring rapid transfer of NAs to particle surface and thereby fast adsorption kinetics. Conversely, NAs transported through soils may show more complex kinetics of adsorption to particle surfaces, including slow NA diffusion to iron oxide surfaces that are in contact with immobile water, e.g., in dead-end pores without advective water movement. These considerations jointly call for systematic transport studies on dsRNA and DNA through packed columns to evaluate if these NAs behave similar also under flow conditions commonly encountered in soils. To link to the results of batch experiments18 and to allow for a mechanistic interpretation of transport data, experiments in columns packed with minerals of defined surfaces are desirable before advancing to whole soil experiments.
The goal of this work was to systematically assess similarities in the transport (and hence adsorption) characteristics of dsRNA and DNA in columns filled with silica sand (hereafter referred to as sand) and iron oxide-coated (silica) sand (IOCS) over a range of solution conditions (i.e., solution pH, ionic strength, and composition) under steady-state flow. We selected a model dsRNA and two model DNA molecules of different sizes to also assess the size dependence of NA adsorption.18 Both dsRNA and DNA are negatively charged across the entire tested pH range from pH 5 to 9 as inferred from the low reported acidity constant of the phosphodiester group in the backbone of DNA of pKa ∼ 1.70 Sand as well as iron oxides are ubiquitous mineral phases in agricultural soils. Adsorption to IOCS surfaces in column transport experiments has routinely been used as a model system for transport studies in soils and sediments for bacteria,71–74 parasites,75,76 viruses,76 dissolved organic matter,77,78 low molecular weight organic acids,79–81 and DNA.82 For a quantitative understanding of NA adsorption to IOCS, we described the experimental column breakthrough data by a kinetic adsorption–desorption model83,84 implemented in a one-dimensional advection–dispersion transport model. Finally, to further approach conditions typically encountered in whole soils, we assessed potential competition of dsRNA with both dissolved organic matter (DOM) and phosphate for adsorption to IOCS. The obtained insights on NA adsorption during transport in sand and IOCS columns provide a better understanding of dsRNA adsorption in soils and further elucidate the applicability of existing information on DNA adsorption in soils to dsRNA.
Materials and methods
Chemicals
All chemicals were of high purity (>98%) and used as received. Detailed information is provided in the ESI, Section S1.†Solutions
All solutions were prepared using DI water (resistivity > 18 MΩ cm; Milli-Q IQ 7000, Millipore, USA) unless specified differently. Lyophilized, synthetic dsRNA (polyadenylic–polyuridylic acid) (hereafter dsRNA) from InvivoGen (USA) was dissolved in the supplied physiological water (containing 154 mM NaCl) to yield a 1 mg dsRNA mL−1 stock solution. UltraPure™ DNA stock solution (hereafter upDNA, 1 mg DNA mL−1) from salmon sperm was purchased from Invitrogen (USA). Stock solutions of dsRNA and upDNA were stored at −21 °C until use. The DNA sodium salt from salmon testes (hereafter genomic DNA (gDNA)) was purchased from Sigma-Aldrich (USA). Stock solutions were prepared by dissolving gDNA in buffer (3 mM of respective pH buffer, described below) containing 10 mM NaCl to prevent denaturing of gDNA.18 Stock solutions of gDNA were stored at 4 °C. The median size of dsRNA was 1367 base pairs (bp) (approximately equal to an extended length of 0.46 μm, estimated assuming that 1 bp has a length of 0.34 nm;85 and a molecular weight of approximately MW = 960![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1), 588 bp (0.22 μm length; MW = 380000 g mol−1) for upDNA, and 21.37 kbp (7.27 μm length; MW = 14000000 g mol−1) for gDNA, as previously reported (Section S2, ESI†).18 We calculated MW from the number of bp as described in Section S2, ESI.†
000 g mol−1), 588 bp (0.22 μm length; MW = 380000 g mol−1) for upDNA, and 21.37 kbp (7.27 μm length; MW = 14000000 g mol−1) for gDNA, as previously reported (Section S2, ESI†).18 We calculated MW from the number of bp as described in Section S2, ESI.†
Phosphate stock solutions (1 mM) from NaH2PO4 and Na2HPO4 were buffered to pH 7 and contained 10 mM NaCl as background electrolyte and 3 mM N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (HEPES). DOM stock solutions were prepared by dissolving 10 mg Pahokee peat humic acid (PPHA, used as model DOM) (International Humic Substances Society, USA) in 100 mL pH 7 buffer (3 mM HEPES and 10 mM NaCl) under vigorous stirring for at least 12 hours. Both phosphate and DOM stock solutions were filtered through 0.2 μm nominal cutoff polyethersulfone membranes (syringe filters, Pall Life Sciences) and stored at 4 °C. Stock solutions were used to prepare experimental solutions as detailed below.
Sorbents
Silica sand (silicon dioxide, acid washed and calcined, ≥99.7%; Sigma Aldrich (Switzerland), hereafter referred to as ‘sand’) was sieved using stainless steel sieves (Retsch GmbH, Germany) to obtain particle sizes from 0.2 to 0.4 mm and acid-cleaned74 (Section S3, ESI†). The sand was net negatively charged at all studied solution pH based on published point of zero net charge (pHPZC) values of silica sand of about 2.86 We coated iron oxides onto the sand using the method of Mills et al.74 with slight modifications (Section S3, ESI†).Specific surface areas of sand and IOCS were 0.043 ± 0.002 and 0.312 ± 0.001 m2 g−1, respectively, determined by duplicate N2-BET analyses (Nova 3200e instrument, Quantachrome, USA) after 18–20 h of drying the materials under vacuum at 40 °C. Scanning electron microscopy (Magellan m400, FEI, USA) showed acicular particles on IOCS surfaces, typical for goethite (Section S4, ESI†). The coated mass of iron oxide on the sand was 0.86 ± 0.04 wt%, as determined from triplicate iron measurements using the phenanthroline assay87 and assuming that goethite had formed (Section S4, ESI†). Using a computer-controlled potentiometric acid–base titration system,88 we determined the point of zero charge (pHPZC) at 7.8 ± 0.1 for IOCS at different ionic strengths (I = 0.01 M and 0.1 M NaCl) (Section S4, ESI†).
Column experiments
Experimental NA, phosphate and PPHA solutions were prepared by diluting the respective stock solution in buffers of matching I and pH to desired concentrations (e.g., 5.1–7.4 μg NA mL−1 for the NAs). Experimental solutions were stored in pre-cleaned glassware at 4 °C and, before use, were sterile filtered through 0.2 μm nominal-cutoff filters.
The columns were wet-packed (Section S5, ESI†), and the pore volume (PV) of each packing was calculated by subtracting the volume of packed sorbent (calculated from packed sorbent mass and assuming a solid-phase density (ρs) of 2.65 g cm−3 for sand and IOCS) from the internal volume of the empty column (Vi,7cm = 2.40 cm3, Vi,2.2cm = 0.75 cm3). The calculated averaged porosity θ = PV/Vi over all columns was θ7cm = 0.488 ± 0.008 (average ± 1 SD, n = 18 packings) and θ2.2cm = 0.532 ± 0.013 (n = 46 packings). Sand or IOCS were initially equilibrated to experimental conditions by pumping at least 5 PVs of buffer solution through each packed column prior to collecting breakthrough curves.
Each experiment in sets 1–3 was run in duplicate in freshly packed columns. In each column, we first determined nitrate breakthrough curves (i.e., injection of 0.1 mM nitrate for 2.4 PVs followed by rinsing with nitrate-free solution for >2 PVs). The resulting breakthrough curve was subsequently modeled to determine the hydrodynamic dispersion coefficient (D) for each column packing (see details in Numerical model section below).
We subsequently delivered between 24 and 77 PVs of NA-containing solution to each column. At the end of this NA ‘loading’ step, we quantified the NA inflow concentration (Cinflow) by activating a column bypass to deliver NA solution from the syringe pumps directly to the spectrophotometric flow-through cell. The resulting short flow interruption inside the column led to a small decrease in NA column effluent concentration (Coutflow) when we re-initiated flow through the column (see Fig. 1c–e as an example). The slight concentration decreases likely reflected kinetic NA adsorption during the short time of interrupted flow in the column. Finally, we rinsed each column with NA-free buffer solution for >3 PVs. Between all experiments, we extensively cleaned the column system (Section S5, ESI†).
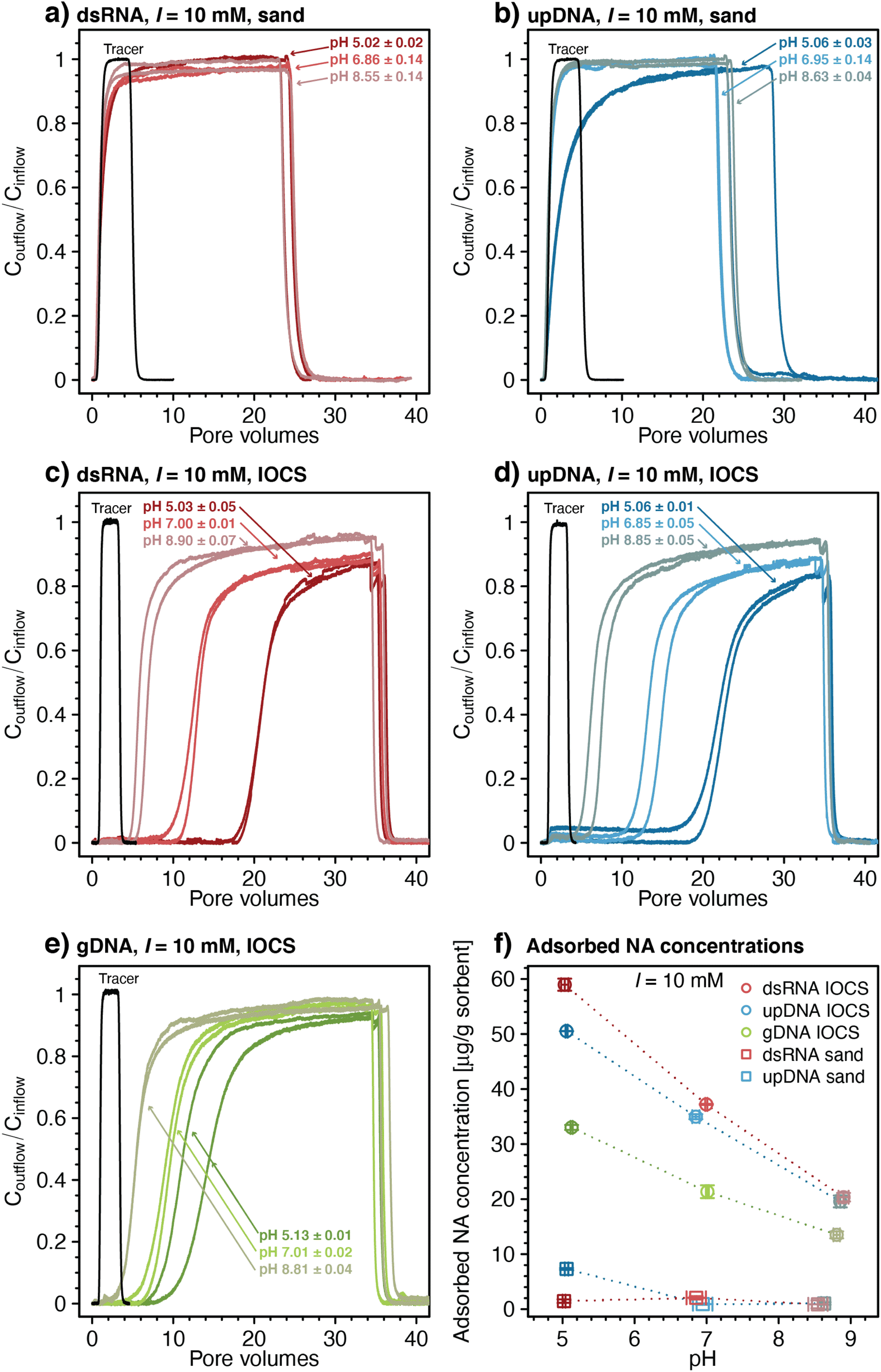 | ||
| Fig. 1 Effect of solution pH on transport of double-stranded RNA (dsRNA) and ultrapure DNA (upDNA) through sand columns (panels a and b) and of dsRNA, upDNA, and genomic DNA (gDNA) through iron oxide-coated sand (IOCS) columns (panels c–e). Panels a–e also show a selected breakthrough curve of the conservative tracer nitrate at pH 7 (only one representative breakthrough curve is shown in each panel). (f) Effect of solution pH on final adsorbed concentrations of dsRNA and upDNA to sand surfaces, and of dsRNA, upDNA, and gDNA to IOCS surfaces determined by mass balance calculations (see Materials and methods). Data points and error bars in panel f represent the mean and absolute deviation from the mean of the results of duplicate breakthrough experiments. | ||
We quantified NA retention in each column by determining the NA retardation factor (R), which is defined as the PV during the column loading step at which Coutflow/Cinflow reached 0.5. Moreover, we calculated final adsorbed NA concentrations [μg NA (g sorbent)−1] at the end of the loading step by subtracting the cumulative mass of NA measured in the column outflow from the total NA mass delivered to the column, divided by the mass of either sand or IOCS packed into the respective column.
We studied competitive adsorption between dsRNA and phosphate to IOCS in column experiments as described above except for co-delivering phosphate to the columns at varying inflow concentrations (0.025, 0.05, 0.15, and 0.5 mM phosphate; set 4; run in single experiments each) while maintaining approximately the same dsRNA inflow concentration (i.e., 5.0–6.3 μg dsRNA mL−1).
Competition between dsRNA and PPHA was studied as follows: prior to packing the columns, we pre-adsorbed varying amounts of PPHA onto IOCS in stirred batch-equilibration reactors (1.5 g IOCS in 20 mL solutions with initial PPHA concentrations of 0.88, 1.76, 3.52, and 7.02 μg mL−1; equilibration time: 18 hours; set 4; run in single experiments each). We estimated that these PPHA concentrations resulted in IOCS surface coverages of approximately 12.5, 25, 50, and 100% (i.e., 11.7, 23.4, 46.8, and 93.6 μg PPHA (g IOCS)−1), assuming a maximum adsorbed PPHA concentration of 30 ng cm−2 as previously reported for PPHA on a positively charged model surface.89 We subsequently washed the PPHA pre-adsorbed IOCS twice with 10 mL of pH 7 buffer solution (3 mM HEPES, 10 mM NaCl), individually packed them into columns, followed by determination of nitrate and dsRNA breakthrough curves, as described above. No PPHA desorption was detected during the experiments based on stable baseline absorbance readings at 425 nm (data not shown).
Numerical model
For pH-dependent experiments in IOCS columns, we modeled nitrate and NA breakthrough curves using an expanded form of the classical advection–dispersion equation (eqn (1)), which describes one-dimensional solute transport and adsorption and desorption under steady-state flow in a saturated, packed column: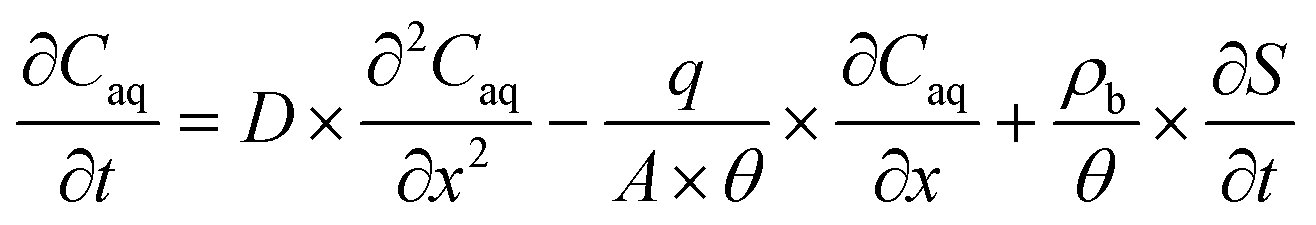 | (1) |
First, we fitted eqn (1) to breakthrough curves of nitrate with S set to zero (Aquasim software package; 2.1g, Eawag)90 (Section S6, ESI†) to estimate the hydrodynamic dispersion coefficient (D) for each column (D = 0.047 ± 0.006 cm2 min−1 for n = 18 columns; average ± 1 SD). We used the D value for each column as a constant when fitting the NA breakthrough curve subsequently collected in that column.
Second, we fitted eqn (1) to NA breakthrough curves using two kinetic versions of the Langmuir sorption model,83,84 featuring either single (n = 1) or dual (n = 2) adsorption kinetics:
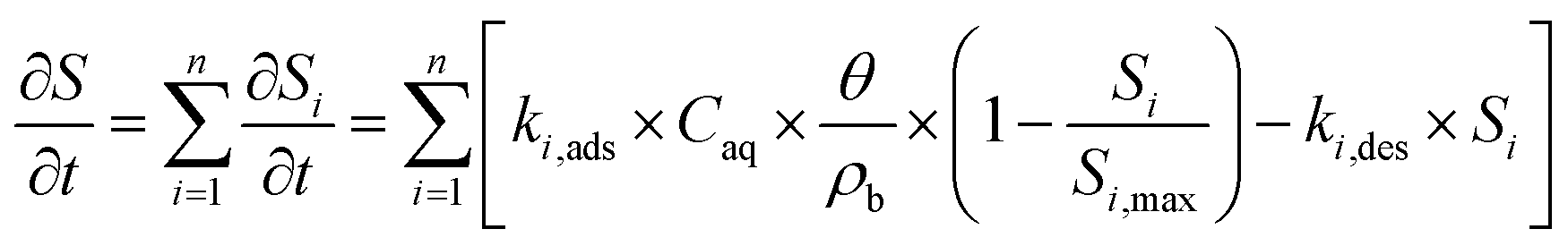 | (2) |
Results and discussion
Transport of NAs through sand and IOCS columns
The breakthrough of upDNA in sand columns at pH 6.95 and 8.63 occurred over similar PVs as those of dsRNA with negligible upDNA adsorption to the sand (i.e., Coutflow/Cinflow ≈ 0.98 after 3.5 PVs) (Fig. 1b). By comparison, Coutflow/Cinflow of upDNA at pH 5.06 increased slowly and showed slight retardation (retardation factor R = 2.13 ± 0.07), indicative of minor upDNA adsorption at this pH. Yet, like dsRNA, upDNA adsorption to sand over the tested pH range was only weak. Based on the weak adsorption of both dsRNA and upDNA, we did not assess adsorption of larger gDNA in the sand columns.
In contrast to sand columns, dsRNA transport was substantially retarded in IOCS columns (Fig. 1c). With decreasing pH, retardation factors increased from R = 6.42 ± 0.55 at pH 8.90 to 13.01 ± 0.15 at pH 7.0, and 21.31 ± 0.01 at pH 5.03. At all pH, dsRNA breakthrough occurred in two distinct phases: in the first phase, dsRNA breakthrough was initially delayed relative to nitrate, followed by dsRNA outflow concentrations increasing steadily to values of Coutflow/Cinflow ≈ 0.70–0.80. In the subsequent second phase, dsRNA breakthrough curves flattened with slower increases in Coutflow/Cinflow. The final Coutflow/Cinflow were ≈0.90 at pH 5.03 and pH 7.00, and ≈0.95 at pH 8.90 after about 34 PVs, when we terminated the loading step and initiated rinsing. Breakthrough of dsRNA was therefore incomplete (i.e., Coutflow/Cinflow remained below 1). Retarded and incomplete breakthrough implied significant dsRNA adsorption to IOCS surfaces. After initiating rinsing with dsRNA-free buffer solution, Coutflow/Cinflow decreased to zero within approximately two PVs, akin to sand columns, suggesting little if any dsRNA desorption from the iron oxide coatings. The breakthrough curves of upDNA (Fig. 1d) and gDNA (Fig. 1e) in IOCS columns showed very similar features as those for dsRNA.
Fig. 1f shows pH-dependent calculated final adsorbed concentrations of all NAs on IOCS and of dsRNA and upDNA on sand at the end of the loading steps (see Materials and methods for calculation). Overall low NA adsorption to sand is consistent with electrostatic repulsion of negatively charged NAs from likewise-charged sand surfaces (pHPZC of silica ≈2 86), in agreement with previous findings for DNA and silica.24,45,91
Substantial retention of all NAs in IOCS columns implies that NAs adsorbed largely to the iron oxide coatings on the sand. The IOCS had a pHPZC of 7.8 ± 0.1, which we determined by acid–base titrations (Section S4, ESI†). The decrease in adsorbed NA concentrations on IOCS with increasing pH (Fig. 1f) is therefore consistent with weakening of electrostatic attraction of NAs to the iron oxide given that its net positive surface charge – and hence the number of adsorption sites – decreased with increasing pH. In addition, decreasing NA adsorption with pH can also be explained by a decrease in the number of inner-sphere complexes formed between the phosphodiester groups of NAs and surface-hydroxyl groups of the iron oxide coating.18,20,21,48–52 However, the relative importance of inner-sphere complex formation to overall NA adsorption may have increased with increasing solution pH, particularly considering that NA adsorbed to IOCS surfaces even at pH 8.90 at which the IOCS was net negatively charged. Inner-sphere complexation of NAs with surface-hydroxyl groups through ligand exchange releases hydroxide ions. We separately tested for the latter in batch reactors in which we added dsRNA to suspended IOCS particles. We indeed observed solution pH increases from pH 6.1 to approximately pH 6.4 upon dsRNA addition, supporting inner-sphere complex formation of dsRNA with IOCS (Section S7, ESI†). Adding dsRNA to control batch reactors without IOCS did not alter solution pH.
Rinsing of IOCS columns yielded no noticeable NA desorption, implying that NA adsorption to iron oxide-coatings was irreversible under the applied experimental conditions. Irreversible polyelectrolyte adsorption is well established and reflects the low probability that all favorable NA-IOCS interactions are broken simultaneously, a prerequisite for complete detachment of NA molecules from the surface.54,92
Final adsorbed concentrations on IOCS at the end of the loading steps were comparable for similarly sized dsRNA and upDNA, but lower for the larger-sized gDNA (Fig. 1f). Higher final adsorbed concentrations of the shorter NAs likely reflected their smaller footprints in adsorbed states, allowing for a more efficient packing on the iron oxide surface as compared to the larger gDNA.18,24,34
We fitted all breakthrough curves of pH-dependent NA transport experiments in IOCS columns using a one-dimensional solute transport model that accounted for advective-dispersive transport coupled to kinetic, Langmuir-type sorption83,84 (eqn (1) and (2)). All NA breakthrough curves were described only poorly by models with a single kinetic adsorption regime83,84 (n = 1, eqn (2)). As shown exemplarily for one replicate of dsRNA breakthrough curves for each of the tested pH in Fig. 2a, dsRNA adsorption was underestimated (i.e., Coutflow/Cinflow was overestimated) both at the onset of dsRNA breakthrough and towards the end of the loading step when experimental Coutflow/Cinflow values increased more slowly than values obtained by fitting. An analysis of residuals between model fit and measured data for dsRNA breakthrough curves at pH 5 to 9 is provided in Section S6, ESI.† Poor fitting by a single kinetic adsorption model is consistent with breakthrough curves showing two distinct phases, suggesting that there were at least two kinetic adsorption regimes.
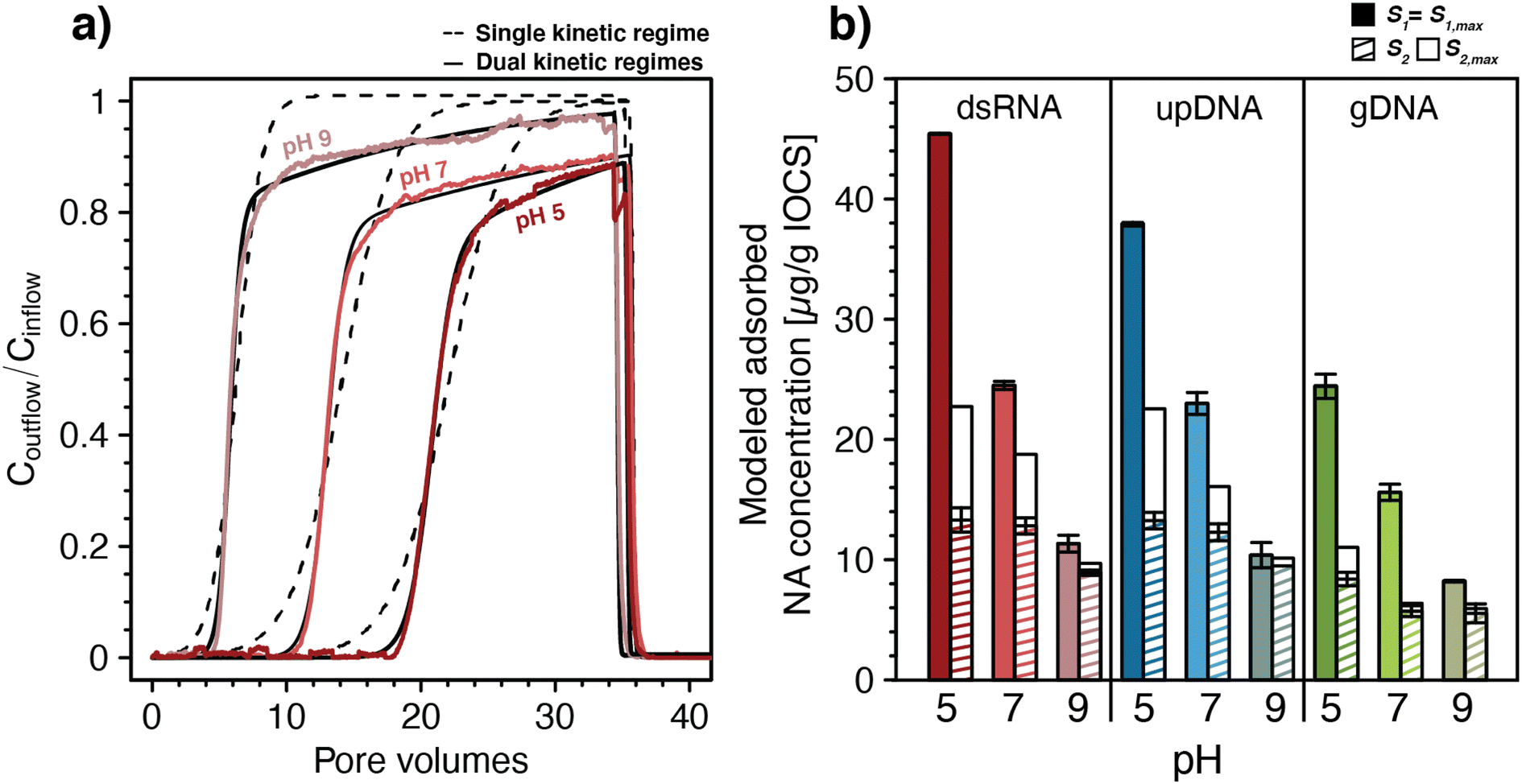 | ||
| Fig. 2 (a) Fitting of a one-dimensional solute transport model coupled to double-stranded RNA (dsRNA) adsorption with either single or dual kinetic adsorption regimes to the breakthrough curves of dsRNA in iron-oxide coated sand (IOCS) columns at nominal pH 5, 7, and 9 (actual solution pH of shown data was 4.98, 6.98, and 8.97, respectively). The dsRNA breakthrough curves in panel a were replotted from Fig. 1c. (b) Modeled final adsorbed concentrations S1 and S2 at the end of the loading steps and modeled maximum adsorption capacities S1,max and S2,max for dsRNA, ultrapure DNA (upDNA), and genomic DNA (gDNA) in IOCS columns at nominal pH values of 5, 7, and 9. Values were obtained by fitting with a model allowing for dual kinetic adsorption regimes (n = 2) with fixed pH-averaged rate constants for fast and slow adsorption kinetics. For exact solution pH values please refer to Fig. 1c–e. Bars and error bars in panel b represent the means and absolutes deviations from the means of fitted parameters from duplicate breakthrough experiments. Error bars for S1 for dsRNA at pH 5 and gDNA at pH 9 are too small to be seen in the figure. | ||
Fitting with two kinetic adsorption regimes83,84 (n = 2, eqn (2)) substantially improved the quality of fits for dsRNA and DNA breakthrough curves, again shown exemplarily for dsRNA in Fig. 2a and in Section S6, ESI.† For all NAs, fitted adsorption rate constants for the fast kinetic regime, k1,ads, ranged from 1.06–4.94 min−1 and were more than one order of magnitude higher than rate constants for the slow kinetic regime, k2,ads, ranging from 0.034–0.147 min−1. Maximum adsorption capacities associated with the fast regime, S1,max, ranged from 8.5–44.6 μg NA (g IOCS)−1 and were also higher than those of the slow kinetic regime, S2,max, which ranged from 4.7–25.6 μg NA (g IOCS)−1. All model parameters are summarized in Section S6, ESI.† Rate constants for both kinetic adsorption regimes were comparable for dsRNA and upDNA, which were higher than for gDNA. This finding supports similar adsorption characteristics of dsRNA and upDNA in terms of adsorption rates, in addition to similar final adsorbed amounts (Fig. 1d). Desorption rate constants for all NAs were small from S1 (k1,des ranged from 0.32 × 10−4 to 4.8 × 10−4 min−1) and zero from S2, consistent with the above qualitative assessment that NA adsorption to IOCS was largely irreversible under the tested experimental conditions.
For each of the NAs, adsorption rate constants showed no systematic variation with pH (post hoc Tukey test, p < 0.05). Conversely, S1,max decreased significantly for all NAs and S2,max decreased significantly only for dsRNA and upDNA (post hoc Tukey test, p < 0.05) with increasing solution pH. The decrease in NA adsorption with increasing pH (Fig. 1) therefore reflected decreases in adsorption capacities and not in adsorption rate constants with pH.
To separately assess the effect of pH on S1,max and S2,max without them being also affected by slight differences in ki,ads and ki,des between the pH values, we re-fitted NA breakthrough curves using pH-averaged adsorption and desorption rate constants for all pH values as fixed model input parameters (see Section S6, ESI† for values). Fixing the rate constants to values averaged across the three experimental pH had little effect on the quality of the fit as compared to fitting also the rate constants, as shown by residual analysis in Section S6, ESI.† This modeling revealed that increases in solution pH from 5.1 to 8.9 decreased S1,max almost fourfold for dsRNA and upDNA, and threefold for gDNA (Fig. 2b), whereas S2,max decreased approximately twofold for all NAs. The modeling further showed that adsorption sites with fast kinetics were completely filled at the end of the loading step (i.e., S1 attained S1,max), whereas adsorption sites with slow kinetics only reached saturation (i.e., S2 attained S2,max) for gDNA at pH 7 and 9. Therefore, the later phases of the breakthrough were dominated by slow kinetic NA adsorption to S2, as expected by the slow increase in Coutflow/Cinflow values towards unity.
Slow kinetic NA adsorption may have had several causes. First, it may have resulted from adsorption to a subset of adsorption sites in IOCS columns that were not in direct contact with mobile pore water. In this case, slow diffusion of NAs from mobile into the immobile pore water in the columns controlled adsorption rates to these sites. Second, it is possible that slow and fast adsorption occurred to the same iron oxide surfaces but that the slow adsorption started only after fast adsorption was completed. For instance, dissolved NAs may have experienced increasing electrostatic repulsion from the IOCS surface as its surface became increasingly filled adsorbed NA molecules, thereby causing remaining unoccupied adsorption sites to fill more slowly (i.e., an effect also referred to as the ‘excluded-area effect’93,94). It is also conceivable that beyond a certain surface loading, a slow rearrangement of adsorbed NAs on IOCS surfaces was needed to allow for additional NA adsorption. To allow for the possibility that fast and slow adsorption did not co-occur to two distinct sites but instead occurred to the same surfaces, we modified the kinetic sorption model to allow for slow kinetic adsorption only after a significant fraction of IOCS surface was occupied by NAs (Section S6, ESI†). This modified model also well-described the breakthrough curves and resulted in only slight changes in fitted parameters compared with the original dual kinetic adsorption model (eqn (2)).
While the experimental design does not allow to elucidate the exact causation of the slow kinetic adsorption, our results highlight the need to consider non-equilibrium adsorption of dsRNA (and DNA) during advective-dispersive transport in porous media. Furthermore, the modeling revealed similar adsorption kinetics of dsRNA and upDNA to IOCS. This finding supports that existing information on DNA adsorption to mineral surfaces can be leveraged to predict adsorption of similarly sized dsRNA molecules to the same mineral surfaces in soils.
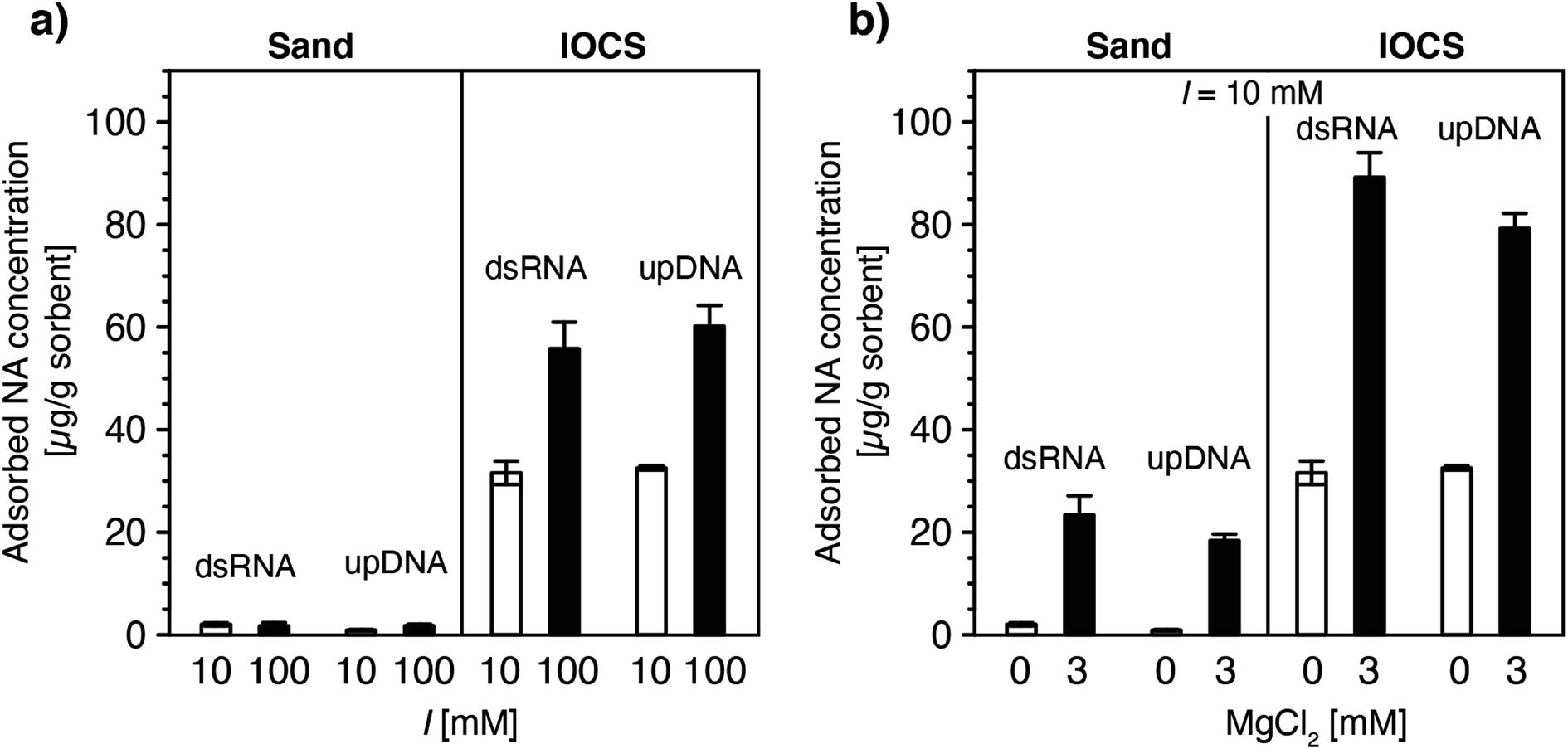 | ||
| Fig. 3 Effect of (a) solution ionic strength (10 and 100 mM NaCl) and (b) solution ionic composition (0 and 3 mM MgCl2 at a constant ionic strength of 10 mM) on final adsorbed concentrations of double-stranded RNA (dsRNA) and ultrapure DNA (upDNA) to sand and iron oxide-coated sand (IOCS) at the end of the loading steps, determined by mass balance calculations (see Materials and methods). The solution pH was pH 6.89 ± 0.10 (averaged over all experiments shown). Data at I = 10 mM in panels a and b were replotted from Fig. 1f. Each bar and error bars represent the mean and absolute deviation from the mean of the results of duplicate breakthrough experiments. | ||
In IOCS columns, increasing I from 10 to 100 mM approximately doubled final adsorbed dsRNA and upDNA concentrations at the end of the loading steps (Fig. 3a). By comparison, 3 mM Mg2+ (at a total I = 10 mM) resulted in 2.8- and 2.4-fold increases in dsRNA and upDNA adsorption, respectively (Fig. 3b). These findings can be rationalized by three factors. First, the findings of increased adsorption with increasing I and Mg2+ likely reflected increasing charge screening of intra-NA electrostatic repulsion with increasing I100–105 and, for Mg2+, intra-NA cation-bridging18,54,63–67 between negatively charged phosphodiester groups in the NA backbone. Both of these effects are expected to result in NAs adopting more compact conformations when adsorbed on the IOCS surfaces, allowing for higher maximum sorbent surface loadings at saturation. Consistent with our findings, previous studies have reported that divalent cations (here: Mg2+) are more effective in compacting DNA conformations than monovalent cations (here: Na+).18,54,63–67 Second, the higher I and presence of Mg2+ likely also resulted in increased charge screening and cation bridging between neighboring adsorbed NA molecules, respectively, and, thereby in denser packings of NA molecules on the iron oxides.93,94 Third, we expect that Mg2+-cation bridging also resulted in NA adsorption to potentially non-coated sand surface between iron oxide coatings, as inferred from the observation of increased NA adsorption in the sand columns in the presence of Mg2+ (Fig. 3b). Since we could not delineate the relative contributions of the non-coated sand and of iron oxide surfaces to the total increased adsorption of NAs at higher I and in the presence of Mg2+, we refrained from modeling NA breakthrough in IOCS columns under these solution conditions.
Increasing concentrations of phosphate co-delivered with dsRNA to IOCS columns at pH 7 and 10 mM NaCl increasingly shifted dsRNA breakthrough towards smaller PVs (Fig. 4a, inset). Corresponding calculated final adsorbed dsRNA concentrations at the end of the loading steps decreased with increasing phosphate co-solute concentrations (Fig. 4a). These findings show that phosphate competed with dsRNA for adsorption sites on iron oxides, likely due to phosphate adsorption inverting the iron oxide surface charge from net positive to net negative,107–110 resulting in electrostatic repulsion of dsRNA molecules from phosphate-coated IOCS surfaces. In addition, adsorbed phosphate is expected to also have blocked potential sites for dsRNA inner-sphere complexation with the IOCS surface (Section S7, ESI†). We note that the lowest tested phosphate concentration of 0.025 mM was in the range of reported phosphate concentrations in drainage waters of agricultural soils (0.002–0.032 mM),111 suggesting that such competition will also occur in agricultural soils. At the phosphate concentration of 0.025 mM, the phosphorous concentration approximately equaled the concentration of phosphorous in the diester groups in dsRNA at the used dsRNA concentration of ∼6 μg mL−1 (i.e., 0.018 mM phosphodiesters in the dsRNA backbone). This low phosphate concentration decreased the adsorbed dsRNA concentration by approximately 30% as compared with dsRNA adsorption in the absence of phosphate (i.e., from 32.2 ± 2.45 to 21.4 μg dsRNA (g IOCS)−1). Interestingly, suppression of dsRNA adsorption by phosphate remained incomplete even at the highest tested phosphate concentration of 0.5 mM. Consistently, separately run batch experiments showed that dsRNA adsorbed to IOCS to which we had pre-adsorbed phosphate at such high phosphate concentrations that phosphate binding sites were saturated (Section S9, ESI†). These findings suggest that either dsRNA molecules can displace a fraction of pre-adsorbed phosphate molecules from the IOCS surfaces or that a fraction of adsorption sites is not available to phosphate molecules. At the same time, increasing competition with phosphate decreased the slow kinetic component of dsRNA adsorption to IOCS: Coutflow/Cinflow of dsRNA at the end of the loading step increased to unity at all tested phosphate concentrations (i.e., dsRNA breakthrough was complete; Fig. 4a, inset).
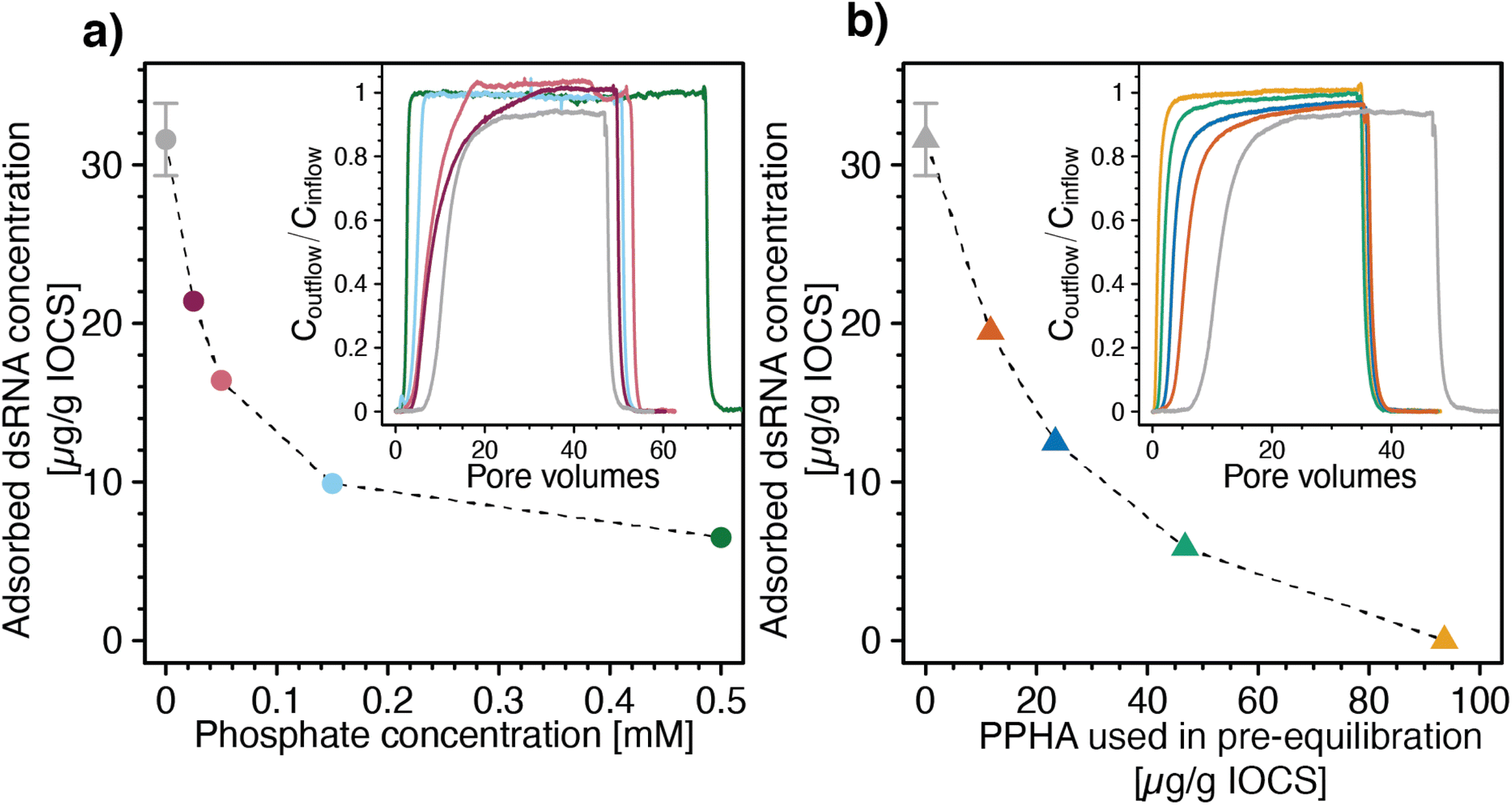 | ||
| Fig. 4 Effects of the adsorptive competitors phosphate and Pahokee Peat humic acid (PPHA, a model dissolved organic matter isolate) on adsorption of double-stranded RNA (dsRNA) to iron oxide-coated sand (IOCS). (a) Calculated final adsorbed dsRNA concentrations at the end of the loading steps as a function of phosphate concentrations co-delivered with dsRNA to IOCS columns. Inset of panel a: corresponding dsRNA breakthrough curves through IOCS columns at increasing concentrations of the co-solute phosphate (i.e., 0.025, 0.05, 0.15, and 0.5 mM). (b) Calculated final adsorbed dsRNA concentrations on IOCS at the end of the loading steps as a function of PPHA concentrations pre-adsorbed to IOCS in batch reactors. Adsorbed concentrations are expressed as μg PPHA added per g of suspended IOCS, corresponding to 11.7, 23.4, 46.8, 93.6 μg PPHA (g IOCS)−1, assuming complete adsorption of added PPHA in the batch pre-equilibration. Inset panel b: corresponding breakthrough curves of dsRNA through IOCS columns that contained different concentrations of pre-adsorbed PPHA. Calculated dsRNA adsorbed concentrations in panel a and b are determined by mass balance calculation, as detailed in the Materials and methods section. Adsorbed dsRNA concentrations in panel a and b in the absence of the competitor were replotted from Fig. 3a. Lines are linear interpolations between consecutive data points and serve to guide the eye. The dsRNA breakthrough curves without adsorptive competitor in the insets of panel a and b were replotted from Section S8, ESI.† | ||
We also assessed competitive suppression of dsRNA adsorption to IOCS by PPHA. With increasing amounts of PPHA pre-adsorbed to IOCS, dsRNA breakthrough shifted towards smaller PVs (Fig. 4b, inset) and the corresponding calculated final adsorbed dsRNA concentrations at the end of the loading steps decreased (Fig. 4b). At the highest tested PPHA concentration (i.e., 93.6 μg PPHA (g IOCS)−1), dsRNA showed no retardation in the column (R = 0.9) and breakthrough was complete (Coutflow/Cinflow ≈ 1) (Fig. 4b, inset). In fact, the slightly lower retardation factor for dsRNA than for nitrate (R = 1, data not shown) suggests that dsRNA molecules were excluded from a fraction of the column pore space, possibly due to electrostatic repulsion of dsRNA from negatively charged PPHA adsorbed onto the IOCS.
Environmental implications
This study demonstrates similar adsorption characteristics of dsRNA and similarly sized DNA molecules during transport through both sand and IOCS columns. Electrostatic repulsion of NAs from sand surfaces resulted in high NA mobility in sand columns and little retardation. Conversely, NA electrostatic attraction and inner-sphere complex formation of NAs with iron oxide coatings in IOCS columns resulted in significant adsorption and retarded transport. Similar transport characteristics, including kinetic adsorption rate constants and final adsorbed amounts, of dsRNA and upDNA strongly support that dsRNA and similarly sized DNA molecules have comparable adsorption characteristics also in soils. Based on existing DNA adsorption data, we therefore anticipate strong dsRNA adsorption to positively charged surfaces of minerals such as iron and aluminum (oxyhydr-)oxide as well as edge sites of clay minerals, but only weak adsorption to negatively charged surfaces of solids including silicon dioxides, basal planes of many clay minerals, and soil organic matter. At the same time, this work highlights that dsRNA adsorption may be strongly modulated in soils with elevated concentrations of dissolved divalent cations, which facilitate adsorption to negatively charged surfaces by cation bridging. Similarly, in soils with elevated ionic strengths, increased dsRNA intramolecular charge screening may result in adsorbed dsRNA molecules adopting compact conformations and thus higher adsorbed concentrations. Adsorption of dsRNA to positively charged mineral surfaces in soils may, however, also be strongly suppressed in the presence of dissolved organic matter and phosphate, resulting in enhanced mobility of dsRNA.Our work supports that fate and ecological risk assessment of dsRNA from RNAi crops in soils may leverage known information on DNA adsorption to soil particles. Future work is needed, however, to elucidate overall dsRNA stability in soils by assessing potential biotic and abiotic degradation pathways of dsRNA both in adsorbed states and in soil solution. Furthermore, irreversible dsRNA adsorption observed under the experimental conditions used herein points at potential analytical challenges in the development of extraction buffers for dsRNA from soils in efforts to monitor dsRNA concentrations in soils.19 Adsorption may largely contribute to measured decreases in dissolved dsRNA concentrations in soils, therefore calling for a clear delineation of dsRNA adsorption from dsRNA (bio)degradation in future studies reporting dsRNA dissipation in soils.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. S. and K. S. acknowledge support from an ETH Zurich Research Grand (ETH-14 17-1). We thank Kimberly M. Parker for her help in preliminary experiments that led to the work presented herein. We thank Numa Pfenninger for the determination of the specific surface area of the IOCS and the sand, the Genetic Diversity Centre (GDC) at ETH Zurich for access to the Tapestation, and the Scientific Center for Optical and Electron Microscopy (ScopeM) at ETH Zurich for access to the SEM.References
- K. H. J. Gordon and P. M. Waterhouse, RNAi for Insect-Proof Plants, Nat. Biotechnol., 2007, 25(11), 1231–1232, DOI:10.1038/nbt1107-1231.
- L. Shaffer, Inner Workings: RNA-Based Pesticides Aim to Get around Resistance Problems, Proc. Natl. Acad. Sci. U. S. A., 2020, 15, 202024033, DOI:10.1073/pnas.2024033117.
- D. R. G. Price and J. A. Gatehouse, RNAi-Mediated Crop Protection against Insects, Trends Biotechnol., 2008, 26(7), 393–400, DOI:10.1016/j.tibtech.2008.04.004.
- H. Huvenne and G. Smagghe, Mechanisms of DsRNA Uptake in Insects and Potential of RNAi for Pest Control: A Review, J. Insect Physiol., 2010, 56(3), 227–235, DOI:10.1016/j.jinsphys.2009.10.004.
- P. Ramaseshadri, G. Segers, R. Flannagan, E. Wiggins, W. Clinton, O. Ilagan, B. McNulty, T. Clark and R. Bolognesi, Physiological and Cellular Responses Caused by RNAi- Mediated Suppression of Snf7 Orthologue in Western Corn Rootworm (Diabrotica Virgifera Virgifera) Larvae, PLoS One, 2013, 8(1), e54270, DOI:10.1371/journal.pone.0054270.
- A. Fire, S. Xu, M. K. Montgomery, S. A. Kostas, S. E. Driver and C. C. Mello, Potent and Specific Genetic Interference by Double-Stranded RNA in Caenorhabditis Elegans, Nature, 1998, 391(6669), 806–811, DOI:10.1038/35888.
- R. Bolognesi, P. Ramaseshadri, J. Anderson, P. Bachman, W. Clinton, R. Flannagan, O. Ilagan, C. Lawrence, S. Levine, W. Moar, G. Mueller, J. Tan, J. Uffman, E. Wiggins, G. Heck and G. Segers, Characterizing the Mechanism of Action of Double-Stranded RNA Activity against Western Corn Rootworm (Diabrotica Virgifera Virgifera LeConte), PLoS One, 2012, 7(10), e47534, DOI:10.1371/journal.pone.0047534.
- J. G. Scott, K. Michel, L. C. Bartholomay, B. D. Siegfried, W. B. Hunter, G. Smagghe, K. Y. Zhu and A. E. Douglas, Towards the Elements of Successful Insect RNAi, J. Insect Physiol., 2013, 59(12), 1212–1221, DOI:10.1016/j.jinsphys.2013.08.014.
- V. S. R. Kola, P. Renuka, M. S. Madhav and S. K. Mangrauthia, Key Enzymes and Proteins of Crop Insects as Candidate for RNAi Based Gene Silencing, Front. Physiol., 2015, 6, 1, DOI:10.3389/fphys.2015.00119.
- K. M. Parker and M. Sander, Environmental Fate of Insecticidal Plant-Incorporated Protectants from Genetically Modified Crops: Knowledge Gaps and Research Opportunities, Environ. Sci. Technol., 2017, 51(21), 12049–12057, DOI:10.1021/acs.est.7b03456.
- U.S. Environmental Protection Agency, A Set of Scientific Issues Being Considered by the Environmental Protection Agency Regarding: RNAi Technology: Program Formulation for Human Health and Ecological Risk Assessment, Arlington, Virginia, 2014, https://www.epa.gov/sites/production/files/2015-06/documents/012814minutes.pdf Search PubMed.
- G. Stotzky, Persistence and Biological Activity in Soil of Insecticidal Proteins from Bacillus Thuringiensis and of Bacterial DNA Bound on Clays and Humic Acids, J. Environ. Qual., 2000, 29(3), 691–705, DOI:10.2134/jeq2000.00472425002900030003x.
- P. Cai, Q. Y. Huang and X. W. Zhang, Interactions of DNA with Clay Minerals and Soil Colloidal Particles and Protection against Degradation by DNase, Environ. Sci. Technol., 2006, 40(9), 2971–2976, DOI:10.1021/es0522985.
- B. W. Aardema, M. G. Lorenz and W. E. Krumbein, Protection of Sediment-Adsorbed Transforming DNA against Enzymatic Inactivation, Appl. Environ. Microbiol., 1983, 46(2), 417–420, DOI:10.1128/aem.46.2.417-420.1983.
- E. Paget, L. J. Monrozier and P. Simonet, Adsorption of DNA on Clay Minerals: Protection against DNase I and Influence on Gene Transfer, FEMS Microbiol. Lett., 1992, 97, 31–39, DOI:10.1111/j.1574-6968.1992.tb05435.x.
- K. Zhang, K.-P. Ho, A. Chatterjee, G. Park, Z. Li, J. G. Catalano and K. M. Parker, RNA Hydrolysis at Mineral–Water Interfaces, Environ. Sci. Technol., 2023, 57(22), 8280–8288, DOI:10.1021/acs.est.3c01407.
- K. Zhang, J. Wei, K. E. H. Hartz, M. J. Lydy, T. S. Moon, M. Sander and K. M. Parker, Analysis of RNA Interference (RNAi) Biopesticides: Double-Stranded RNA (DsRNA) Extraction from Agricultural Soils and Quantification by RT-QPCR, Environ. Sci. Technol., 2020, 54(8), 4893–4902, DOI:10.1021/acs.est.9b07781.
- K. Sodnikar, K. M. Parker, S. R. Stump, L. K. ThomasArrigo and M. Sander, Adsorption of Double-Stranded Ribonucleic Acids (DsRNA) to Iron (Oxyhydr-)Oxide Surfaces: Comparative Analysis of Model DsRNA Molecules and Deoxyribonucleic Acids (DNA), Environ. Sci.: Processes Impacts, 2021, 23, 605–620, 10.1039/d1em00010a.
- K. M. Parker, V. B. Borrero, D. M. van Leeuwen, M. A. Lever, B. Mateescu and M. Sander, Environmental Fate of RNA Interference Pesticides: Adsorption and Degradation of Double-Stranded RNA Molecules in Agricultural Soils, Environ. Sci. Technol., 2019, 53(6), 3027–3036, DOI:10.1021/acs.est.8b05576.
- M. P. Schmidt and C. E. Martinez, Ironing Out Genes in the Environment: An Experimental Study of the DNA-Goethite Interface, Langmuir, 2017, 33(34), 8525–8532, DOI:10.1021/acs.langmuir.7b01911.
- S. J. Parikh, F. N. D. Mukome and X. Zhang, ATR–FTIR Spectroscopic Evidence for Biomolecular Phosphorus and Carboxyl Groups Facilitating Bacterial Adhesion to Iron Oxides, Colloids Surf., B, 2014, 119, 38–46, DOI:10.1016/j.colsurfb.2014.04.022.
- S. Blum, M. G. Lorenz and W. Wackernagel, Mechanism of Retarded DNA Degradation and Prokaryotic Origin of DNases in Nonsterile Soils, Syst. Appl. Microbiol., 1997, 20(4), 513–521, DOI:10.1016/S0723-2020(97)80021-5.
- C. M. Gardner and C. K. Gunsch, Adsorption Capacity of Multiple DNA Sources to Clay Minerals and Environmental Soil Matrices Less than Previously Estimated, Chemosphere, 2017, 175, 45–51, DOI:10.1016/j.chemosphere.2017.02.030.
- A. Ogram, M. L. Mathot, J. B. Harsh, J. Boyle and C. A. Pettigrew, Effects of DNA Polymer Length on Its Adsorption to Soils, Appl. Environ. Microbiol., 1994, 60(2), 393–396, DOI:10.1128/AEM.60.2.393-396.1994.
- A. Ogram, G. S. Sayler, D. Gustin and R. J. Lewis, DNA Adsorption to Soils and Sediments, Environ. Sci. Technol., 1988, 22(8), 982–984, DOI:10.1021/es00173a020.
- P. Cai, Q. Huang, X. Zhang and H. Chen, Adsorption of DNA on Clay Minerals and Various Colloidal Particles from an Alfisol, Soil Biol. Biochem., 2006, 38(3), 471–476, DOI:10.1016/j.soilbio.2005.05.019.
- P. Cai, Q. Huang, J. Zhu, D. Jiang, X. Zhou, X. Rong and W. Liang, Effects of Low-Molecular-Weight Organic Ligands and Phosphate on DNA Adsorption by Soil Colloids and Minerals, Colloids Surf., B, 2007, 54(1), 53–59, DOI:10.1016/j.colsurfb.2006.07.013.
- P. Cai, Q. Y. Huang and X. W. Zhang, Microcalorimetric Studies of the Effects of MgCl2 Concentrations and PH on the Adsorption of DNA on Montmorillonite, Kaolinite and Goethite, Appl. Clay Sci., 2006, 32(1–2), 147–152, DOI:10.1016/j.clay.2005.11.004.
- M. Khanna and G. Stotzky, Transformation of Bacillus Subtilis by DNA Bound on Montmorillonite and Effect of DNase on the Transforming Ability of Bound DNA, Appl. Environ. Microbiol., 1992, 58(6), 1930–1939 CrossRef CAS PubMed.
- C. Crecchio, P. Ruggiero, M. Curci, C. Colombo, G. Palumbo and G. Stotzky, Binding of DNA from Bacillus Subtilis on Montmorillonite–Humic Acids–Aluminum or Iron Hydroxypolymers, Soil Sci. Soc. Am. J., 2005, 69(3), 834–841, DOI:10.2136/sssaj2004.0166.
- M. Franchi, E. Bramanti, L. M. Bonzi, P. L. Orioli, C. Vettori and E. Gallori, Clay-Nucleic Acid Complexes: Characteristics and Implications for the Preservation of Genetic Material in Primeval Habitats, Orig. Life Evol. Biosph., 1999, 29(3), 297–315, DOI:10.1023/A:1006557832574.
- M. P. Greaves and M. J. Wilson, The Adsorption of Nucleic Acids by Montmorillonite, Soil Biol. Biochem., 1969, 1(4), 317–323, DOI:10.1016/0038-0717(69)90014-5.
- F. Poly, C. Chenu, P. Simonet, J. Rouiller and L. J. Monrozier, Differences between Linear Chromosomal and Supercoiled Plasmid DNA in Their Mechanisms and Extent of Adsorption on Clay Minerals, Langmuir, 2000, 16(3), 1233–1238, DOI:10.1021/la990506z.
- G. Pietramellara, M. Franchi, E. Gallori and P. Nannipieri, Effect of Molecular Characteristics of DNA on Its Adsorption and Binding on Homoionic Montmorillonite and Kaolinite, Biol. Fertil. Soils, 2001, 33(5), 402–409, DOI:10.1007/s003740100341.
- C. A. Goring and W. V. Bartholomew, Adsorption of Mononucleotides, Nucleic Acids, and Nucleoproteins by Clays, Soil Sci., 1952, 74(2), 149–164 CrossRef CAS.
- G. W. Beall, D. S. Sowersby, R. D. Roberts, M. H. Robson and L. K. Lewis, Analysis of Oligonucleotide DNA Binding and Sedimentation Properties of Montmorillonite Clay Using Ultraviolet Light Spectroscopy, Biomacromolecules, 2009, 10(1), 105–112, DOI:10.1021/bm800970v.
- K. Saeki, T. Kunito and M. Sakai, Effects of PH, Ionic Strength, and Solutes on DNA Adsorption by Andosols, Biol. Fertil. Soils, 2010, 46(5), 531–535, DOI:10.1007/s00374-010-0447-y.
- K. Saeki, M. Morisaki and M. Sakai, The Contribution of Soil Constituents to Adsorption of Extracellular DNA by Soils, Microbes Environ., 2008, 23(4), 0809240008, DOI:10.1264/jsme2.ME08531.
- K. Saeki and T. Kunito, in Current Research, Technology and Education Topics in Applied Microbiology and Mikrobial Biotechnology, ed. A. Méndez-Vilas, Formatex Research Center, 2010, vol. 2, pp. 188−195 Search PubMed.
- X. Chen, Q. Huang and W. Chen, Adsorption of DNA by Bacteria and Their Composites with Minerals, Geomicrobiol. J., 2016, 33(9), 822–831, DOI:10.1080/01490451.2015.1117545.
- T. H. Nguyen and M. Elimelech, Plasmid DNA Adsorption on Silica: Kinetics and Conformational Changes in Monovalent and Divalent Salts, Biomacromolecules, 2007, 8(1), 24–32, DOI:10.1021/bm0603948.
- C. Crecchio and G. Stotzky, Binding of DNA on Humic Acids: Effect on Transformation of Bacillus Subtilis and Resistance to DNase, Soil Biol. Biochem., 1998, 30(8–9), 1061–1067, DOI:10.1016/S0038-0717(97)00248-4.
- M. G. Lorenz and W. Wackernagel, Adsorption of DNA to Sand and Variable Degradation Rates of Adsorbed DNA, Appl. Environ. Microbiol., 1987, 53(12), 2948–2952, DOI:10.4155/fso.15.32.
- B. Chamier, M. G. Lorenz and W. Wackernagel, Natural Transformation of Acinetobacter Calcoaceticus by Plasmid DNA Adsorbed on Sand and Groundwater Aquifer Material, Appl. Environ. Microbiol. , 1993, 59(5), 1662–1667 CrossRef CAS PubMed.
- G. Romanowski, M. G. Lorenz and W. Wackernagel, Adsorption of Plasmid DNA to Mineral Surfaces and Protection against DNase I, Appl. Environ. Microbiol., 1991, 57(4), 1057–1061, DOI:10.1128/aem.57.4.1057-1061.1991.
- K. A. Melzak, C. S. Sherwood, R. F. B. Turner and C. A. Haynes, Driving Forces for DNA Adsorption to Silica in Perchlorate Solutions, J. Colloid Interface Sci., 1996, 181(2), 635–644, DOI:10.1006/jcis.1996.0421.
- X. Sheng, C. Qin, B. Yang, X. Hu, C. Liu, M. G. Waigi, X. Li and W. Ling, Metal Cation Saturation on Montmorillonites Facilitates the Adsorption of DNA via Cation Bridging, Chemosphere, 2019, 235, 670–678, DOI:10.1016/j.chemosphere.2019.06.159.
- S. J. Parikh and J. Chorover, ATR-FTIR Spectroscopy Reveals Bond Formation During Bacterial Adhesion to Iron Oxide, Langmuir, 2006, 22(20), 8492–8500, DOI:10.1021/la061359p.
- A. Omoike, J. Chorover, K. D. Kwon and J. D. Kubicki, Adhesion of Bacterial Exopolymers to α-FeOOH: Inner-Sphere Complexation of Phosphodiester Groups, J. Phys. Chem. B, 2004, 20, 11108–11114, DOI:10.1021/la048597.
- A. Omoike and J. Chorover, Adsorption to Goethite of Extracellular Polymeric Substances from Bacillus Subtilis, Geochim. Cosmochim. Acta, 2006, 70(4), 827–838, DOI:10.1016/j.gca.2005.10.012.
- L. Fang, Y. Cao, Q. Huang, S. L. Walker and P. Cai, Reactions between Bacterial Exopolymers and Goethite: A Combined Macroscopic and Spectroscopic Investigation, Water Res., 2012, 46(17), 5613–5620, DOI:10.1016/j.watres.2012.07.046.
- Y. Cao, X. Wei, P. Cai, Q. Huang, X. Rong and W. Liang, Preferential Adsorption of Extracellular Polymeric Substances from Bacteria on Clay Minerals and Iron Oxide, Colloids Surf., B, 2011, 83(1), 122–127, DOI:10.1016/j.colsurfb.2010.11.018.
- A. Omoike and J. Chorover, Spectroscopic Study of Extracellular Polymeric Substances from Bacillus Subtilis: Aqueous Chemistry and Adsorption Effects, J. Phys. Chem. B, 2004, 5(4), 1219–1230, DOI:10.1021/bm034461z.
- F. T. Hesselink, Adsorption of Polyelectrolytes from Dilute Solution, in Adsorption from Solution at the Solid/Liquid Interface, ed. G. D. Parfitt and C. H. Rochester, London, 1983, pp. 377–412 Search PubMed.
- F. T. Hesselink, On the Adsorption of Polyelectrolyte Macromolecules on a Flat Interface. An Approximate Theory for Low Potentials, J. Electroanal. Chem. Interfacial Electrochem., 1972, 37(1), 317–325, DOI:10.1016/S0022-0728(72)80236-5.
- M. Kawaguchi and A. Takahashi, Polymer Adsorption at Solid-Liquid Interfaces, Adv. Colloid Interface Sci., 1992, 37(3–4), 219–317, DOI:10.1016/0001-8686(92)80085-C.
- V. Shubin and P. Linse, Effect of Electrolytes on Adsorption of Cationic Polyacrylamide on Silica: Ellipsometric Study and Theoretical Modeling, J. Phys. Chem. B, 1995, 99(4), 1285–1291, DOI:10.1021/j100004a031.
- J. de Reese and J. Plank, Adsorption of Polyelectrolytes on Calcium Carbonate – Which Thermodynamic Parameters Are Driving This Process?, J. Am. Ceram. Soc., 2011, 94(10), 3515–3522, DOI:10.1111/j.1551-2916.2011.04682.x.
- T. G. M. Van De Ven, Kinetic Aspects of Polymer and Polyelectrolyte Adsorption on Surfaces, Adv. Colloid Interface Sci., 1994, 48, 121–140, DOI:10.1016/0001-8686(94)80006-5.
- H. G. M. Van de Steeg, M. A. Cohen Stuart, A. De Keizer and B. H. Bijsterbosch, Polyelectrolyte Adsorption: A Subtle Balance of Forces, Langmuir, 1992, 8(10), 2538–2546, DOI:10.1021/la00046a030.
- M. A. Cohen Stuart, G. J. Fleer, J. Lyklema, W. Norde and J. M. Scheutjens, Adsorption of Ions, Polyelectrolytes and Proteins, Adv. Colloid Interface Sci., 1991, 34, 477–535, DOI:10.1016/0001-8686(91)80056-P.
- A. Dobrynin and M. Rubinstein, Theory of Polyelectrolytes in Solutions and at Surfaces, Prog. Polym. Sci., 2005, 30(11), 1049–1118, DOI:10.1016/j.progpolymsci.2005.07.006.
- X. Wang, H. Kweon, S. Lee, H. Shin, B. Chua, M. R. Liles, M. Lee and A. Son, Vulnerability of DNA Hybridization in Soils Is Due to Mg2+ Ion Induced DNA Aggregation, Soil Biol. Biochem., 2018, 125, 300–308, DOI:10.1016/j.soilbio.2018.08.003.
- D. H. Taylor and A. T. Wilson, The Adsorption of Yeast RNA by Allophane, Clays Clay Miner., 1979, 27(4), 261–268 CrossRef CAS.
- V. K. Misra and D. E. Draper, On the Role of Magnesium Ions in RNA Stability, Biopolymers, 1998, 48(2–3), 113–135, DOI:10.1002/(SICI)1097-0282(1998)48:2<113::AID-BIP3>3.0.CO;2-Y.
- J. Anastassopoulou, Metal–DNA Interactions, J. Mol. Struct., 2003, 651–653, 19–26, DOI:10.1016/S0022-2860(02)00625-7.
- G. Minasov, V. Tereshko and M. Egli, Atomic-Resolution Crystal Structures of B-DNA Reveal Specific Influences of Divalent Metal Ions on Conformation and Packing, J. Mol. Biol., 1999, 291(1), 83–99, DOI:10.1006/jmbi.1999.2934.
- B. K. G. Theng, Polymer Behaviour at Clay and Solid Surfaces, in Developments in Clay Science, Elsevier, 2012, pp. 47–75, DOI:10.1016/B978-0-444-53354-8.00002-5.
- C. S. Buergisser, M. Cernik, M. Borkovec and H. Sticher, Determination of Nonlinear Adsorption Isotherms from Column Experiments: An Alternative to Batch Studies, Environ. Sci. Technol., 1993, 27(5), 943–948, DOI:10.1021/es00042a018.
- P. Thaplyal and P. C. Bevilacqua, Experimental Approaches for Measuring pKa's in RNA and DNA, Methods Enzymol., 2014, 549, 189–219, DOI:10.1016/B978-0-12-801122-5.00009-X.
- J. W. A. Foppen and J. F. Schijven, Transport of E. Coli in Columns of Geochemically Heterogeneous Sediment, Water Res., 2005, 39(13), 3082–3088, DOI:10.1016/j.watres.2005.05.023.
- S.-J. Park and S.-B. Kim, Influence of (Bi)Carbonate on Bacterial Interaction with Quartz and Metal Oxide-Coated Surfaces, Colloids Surf., B, 2010, 76(1), 57–62, DOI:10.1016/j.colsurfb.2009.10.010.
- S.-B. Kim, S.-J. Park, C.-G. Lee, N.-C. Choi and D.-J. Kim, Bacteria Transport through Goethite-Coated Sand: Effects of Solution PH and Coated Sand Content, Colloids Surf., B, 2008, 63(2), 236–242, DOI:10.1016/j.colsurfb.2007.12.003.
- A. L. Mills, J. S. Herman, G. M. Hornberger and T. H. Dejesús, Effect of Solution Ionic Strength and Iron Coatings on Mineral Grains on the Sorption of Bacterial Cells to Quartz Sand, Appl. Environ. Microbiol., 1994, 60(9), 3300–3306, DOI:10.1128/aem.60.9.3300-3306.1994.
- Y. Park, E. R. Atwill, L. Hou, A. I. Packman and T. Harter, Deposition of Cryptosporidium Parvum Oocysts in Porous Media: A Synthesis of Attachment Efficiencies Measured under Varying Environmental Conditions, Environ. Sci. Technol., 2012, 46(17), 9491–9500, DOI:10.1021/es300564w.
- F. A. Abudalo, Y. G. Bogatsu, J. N. Ryan, R. W. Harvey, D. W. Metge and M. Elimelech, Effect of Ferric Oxyhydroxide Grain Coatings on the Transport of Bacteriophage PRD1 and Cryptosporidium Parvum Oocysts in Saturated Porous, Environ. Sci. Technol., 2005, 39(17), 6412–6419, DOI:10.1021/es050159h.
- X. Qin, F. Liu, G. Wang, H. Hou, F. Li and L. Weng, Fractionation of Humic Acid upon Adsorption to Goethite: Batch and Column Studies, Chem. Eng. J., 2015, 269, 272–278, DOI:10.1016/j.cej.2015.01.124.
- H. Weigand and K. U. Totsche, Flow and Reactivity Effects on Dissolved Organic Matter Transport in Soil Columns, Soil Sci. Soc. Am. J., 1998, 62(5), 1268–1274, DOI:10.2136/sssaj1998.03615995006200050017x.
- Y. S. Hwang and J. J. Lenhart, Dicarboxylic Acid Transport through Hematite-Coated Sand, Chemosphere, 2010, 78(8), 1049–1055, DOI:10.1016/j.chemosphere.2009.11.028.
- J. D. Filius, J. C. L. Meeussen and W. H. van Riemsdijk, Transport of Malonate in a Goethite–Silica Sand System, Colloids Surf., A, 1999, 151(1–2), 245–253, DOI:10.1016/S0927-7757(98)00698-0.
- H. Ouachtak, S. Akhouairi, A. Ait Addi, R. Ait Akbour, A. Jada, J. Douch and M. Hamdani, Mobility and Retention of Phenolic Acids through a Goethite-Coated Quartz Sand Column, Colloids Surf., A, 2018, 546, 9–19, DOI:10.1016/j.colsurfa.2018.02.071.
- F. Chen, X. Yang, F. Zhao, S. Xu and Y. Yang, Transport of an Antibiotic Resistance Plasmid through Iron-Oxide-Coated Sand: Influence of the Anionic Hofmeister Effect and Coexisting Kaolinite Colloids, ACS ES T Water, 2021, 1(2), 388–395, DOI:10.1021/acsestwater.0c00148.
- Z. Adamczyk, B. Siwek, M. Zembala and P. Belouschek, Kinetics of Localized Adsorption of Colloid Particles, Adv. Colloid Interface Sci., 1994, 48, 151–280, DOI:10.1016/0001-8686(94)80008-1.
- C. W. Fetter, T. Boving and D. Kreamer, Contaminant Hydrogeology, Waveland Press, 3rd edn, 2017 Search PubMed.
- W. Saenger, DNA Structure, in Principles of Nucleic Acid Structure, Springer, New York, NY, 1984, pp. 253–282. DOI:10.1007/978-1-4612-5190-3.
- M. Kosmulski, Isoelectric Points and Points of Zero Charge of Metal (Hydr)Oxides: 50 Years after Parks' Review, Adv. Colloid Interface Sci., 2016, 238, 1–61, DOI:10.1016/j.cis.2016.10.005.
- W. B. Fortune and M. G. Mellon, Determination of Iron with O-Phenanthroline: A Spectrophotometric Study, J. Phys. Chem. B, 1938, 10(2), 60–64, DOI:10.1021/ac50118a004.
- D. G. Kinniburgh, C. J. Milne and P. Venema, Design and Construction of a Personal-Computer-Based Automatic Titrator, Soil Sci. Soc. Am. J., 1995, 59(2), 417–422, DOI:10.2136/sssaj1995.03615995005900020021x.
- J. E. Tomaszewski, R. P. Schwarzenbach and M. Sander, Protein Encapsulation by Humic Substances, Environ. Sci. Technol., 2011, 45(14), 6003–6010, DOI:10.1021/es200663h.
- P. Reichert, Aquasim - a Tool for Simulation and Data-Analysis of Aquatic Systems, Water Sci. Technol., 1994, 30(2), 21–30 CrossRef CAS.
- T. H. Nguyen and M. Elimelech, Plasmid DNA Adsorption on Silica: Kinetics and Conformational Changes in Monovalent and Divalent Salts, Biomacromolecules, 2006, 8(1), 24–32, DOI:10.1021/bm0603948.
- R. T. Podoll, K. C. Irwin and S. Brendlinger, Sorption of Water-Soluble Oligomers on Sediments, Environ. Sci. Technol., 1987, 21, 562–568, DOI:10.1021/es00160a006.
- R. Rajagopalan and R. Q. Chu, Dynamics of Adsorption of Colloidal Particles in Packed Beds, J. Colloid Interface Sci., 1982, 86(2), 299–317, DOI:10.1016/0021-9797(82)90076-5.
- P. R. Johnson and M. Elimelech, Dynamics of Colloid Deposition in Porous Media: Blocking Based on Random Sequential Adsorption, J. Phys. Chem. B, 1995, 11, 801–812, DOI:10.1021/la00003a023.
- K. Li, X. Zhao, B. K. Hammer, S. Du and Y. Chen, Nanoparticles Inhibit DNA Replication by Binding to DNA: Modeling and Experimental Validation, ACS Nano, 2013, 7(11), 9664–9674, DOI:10.1021/nn402472k.
- G. Romanowski, M. G. Lorenz and W. Wackernagel, Adsorption of Plasmid DNA to Mineral Surfaces and Protection against DNase I, Appl. Environ. Microbiol., 1991, 57(4), 1057–1061, DOI:10.1128/AEM.57.4.1057-1061.1991.
- B. K. G. Theng, Nucleic Acids, in Developments in Clay Science, Elsevier, 2012, pp. 319–337, DOI:10.1016/B978-0-444-53354-8.00009-8.
- T. H. Nguyen and K. L. Chen, Role of Divalent Cations in Plasmid DNA Adsorption to Natural Organic Matter-Coated Silica Surface, Environ. Sci. Technol., 2007, 41(15), 5370–5375, DOI:10.1021/es070425m.
- N. Lu, S. E. Mylon, R. Kong, R. Bhargava, J. L. Zilles and T. H. Nguyen, Interactions between Dissolved Natural Organic Matter and Adsorbed DNA and Their Effect on Natural Transformation of Azotobacter Vinelandii, Sci. Total Environ., 2012, 426, 430–435, DOI:10.1016/j.scitotenv.2012.03.063.
- J. Lipfert, S. Doniach, R. Das and D. Herschlag, Understanding Nucleic Acid–Ion Interactions, Annu. Rev. Biochem., 2014, 83(1), 813–841, DOI:10.1146/annurev-biochem-060409-092720.
- M. I. Zarudnaya, A. L. Potyahaylo and D. M. Hovorun, Dependence of DNA Persistence Length on Ionic Conditions, Biopolym. Cell, 2017, 33(2), 81–91, DOI:10.7124/bc.000946.
- J. Seils and R. Pecora, Dynamic Light Scattering Study of a Monodisperse 2311 Base Pair Circular DNA, J. Phys. Chem. B, 1992, 25, 354–362, DOI:10.1021/ma00027a055.
- M. Salomo, K. Kegler, C. Gutsche, M. Struhalla, J. Reinmuth, W. Skokow, U. Hahn and F. Kremer, The Elastic Properties of Single Double-Stranded DNA Chains of Different Lengths as Measured with Optical Tweezers, Environ. Chem. Lett., 2006, 284(11), 1325–1331, DOI:10.1007/s00396-006-1517-4.
- H. Liu, J. Gapinski, L. Skibinska, A. Patkowski and R. Pecora, Effect of Electrostatic Interactions on the Dynamics of Semiflexible Monodisperse DNA Fragments, J. Chem. Phys., 2000, 113(14), 6001–6010, DOI:10.1063/1.1290477.
- N. Borochov, H. Eisenberg and Z. Kam, Dependence of DNA Conformation on the Concentration of Salt, Biopolymers, 1981, 20(1), 231–235, DOI:10.1002/bip.1981.360200116.
- Y.-T. Huang, D. J. Lowe, G. J. Churchman, L. A. Schipper, R. Cursons, H. Zhang, T.-Y. Chen and A. Cooper, DNA Adsorption by Nanocrystalline Allophane Spherules and Nanoaggregates, and Implications for Carbon Sequestration in Andisols, Appl. Clay Sci., 2016, 120, 40–50, DOI:10.1016/j.clay.2015.11.009.
- F. J. Hingston, R. J. Atkinson, A. M. Posner and J. P. Quirk, Specific Adsorption of Anions, Nature, 1967, 215(5109), 1459–1461, DOI:10.1038/2151459a0.
- T. Hiemstra and W. H. Van Riemsdijk, A Surface Structural Approach to Ion Adsorption: The Charge Distribution (CD) Model, J. Colloid Interface Sci., 1996, 179(2), 488–508, DOI:10.1006/jcis.1996.0242.
- J. Beek and W. H. Van Riemsdijk, Interaction of Orthophosphate Ions with Soil, in Developments in Soil Science, Elsevier, 1979, pp. 259–284 Search PubMed.
- L. Sigg and W. Stumm, The Interaction of Anions and Weak Acids with the Hydrous Goethite (α-FeOOH) Surface, Colloids Surf., 1981, 2(2), 101–117, DOI:10.1016/0166-6622(81)80001-7.
- V. L. Snoeyink, Water Chemistry, J. Wiley, New York, 1980 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3em00294b |
| This journal is © The Royal Society of Chemistry 2023 |