
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bi-functional and mono-component organocatalysts for the ring-opening alternating co-polymerisation of anhydride and epoxide†
Max
Hirschmann‡
 *a,
Rachele
Zunino
bc,
Sara
Meninno
d,
Laura
Falivene
*d and
Tiziana
Fuoco
a
*a,
Rachele
Zunino
bc,
Sara
Meninno
d,
Laura
Falivene
*d and
Tiziana
Fuoco
a
aKTH Royal Institute of Technology, Department of Fibre and Polymer Technology, 100 44 Stockholm, Sweden. E-mail: max.hirschmann@kemi.uu.se
bScuola Superiore Meridionale, 80138 Napoli, Italy
cUniversity of Naples Federico II, Department of Chemical Sciences, 80126 Napoli, Italy
dUniversity of Salerno, Department of Chemistry and Biology, 84084 Fisciano, Italy. E-mail: lafalivene@unisa.it
First published on 7th November 2023
Abstract
Bi-functional organocatalysts constituted by a (thio-)urea moiety and an iminophosphorane moiety were synthesized and optimised for the ring-opening alternating co-polymerisation of phthalic anhydride with three different epoxides: cyclohexene oxide, propylene oxide and butylene oxide. The most effective catalyst featured a cyclohexyl urea moiety, an iminophosphorane moiety with three 2,4-dimethyl-3-methoxy phenyl substituents, and a short spacer length of two carbon atoms between them. All tested epoxides reached quantitative conversion within 24 hours with ester-selectivities up to >97%. NMR and DFT experiments reveal that the catalysts exist in solution as dimers that dissociate during the initiation of the polymerisation. During the polymerisation, the catalyst is coordinated to the growing chain and further modulates its reactivity through reversible protonation/deprotonation suppressing transesterification side reactions even at prolonged polymerisation times without the need for a co-catalyst. The rate-determining step of the polymerisation is the ring-opening of the epoxide by the carboxylate chain end, and accordingly, higher temperatures (up to 150 °C) and higher concentrations of epoxide and catalyst increase polymerisation rates.
Polyesters conform to the principles of a circular plastic economy.1,2 They can be synthesised from renewable feedstock and are recyclable and (bio-)degradable.3 Currently, polyesters are typically synthesised either via polycondensation, a step-growth polymerisation of alcohols and carboxylic acids (or their derivatives) or via ring-opening polymerisation (ROP) of lactones that follows a chain-growth mechanism.4 A third polymerisation technique, the ring-opening alternating co-polymerisation (ROAC)§ of anhydrides and epoxides, promises to combine the advantages of the aforementioned polymerisation strategies (cf.Scheme 1): plant oils,5 terpenes,6 and dicarboxylic acids7 from renewable sources and recycled polymers8 build a structurally-diverse monomer feedstock (an advantage shared with SGP) for the 100% atom-economic (an advantage shared with ROP) ROAC.9
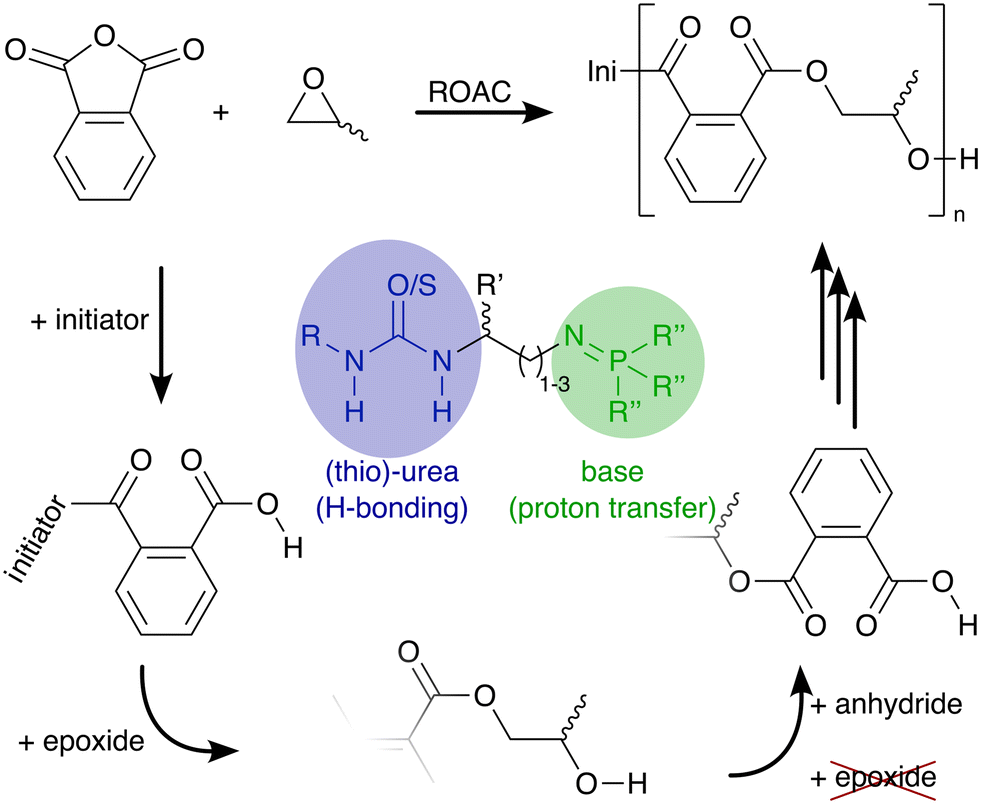 | ||
| Scheme 1 Ring-opening alternating co-polymerisation (ROAC) of phthalic anhydride and propylene oxide catalysed by the herein proposed bi-functional and mono-component organocatalyst. Hydrogen bonding and proton transfer are expected to facilitate the epoxide ring-opening by the carboxylic chain end and simultaneously attenuate the reactivity of the alcohol chain end (suppress side reactions). | ||
In the presence of an appropriate catalyst, the molar mass and macromolecular structure of the ROAC-derived polyesters can be further controlled.10 This catalyst needs to suppress side reactions such as (i) chain transfer caused by nucleophilic species, (ii) transesterification, and (iii) consecutive epoxide ring-openings. Chain transfers can be partly suppressed by employing highly pure monomers and solvents to avoid the presence of nucleophilic impurities such as water, diol (formed by the reaction between water and epoxide), and diacid (formed by the reaction between water and anhydride) that can promote such reactions. The other two side reactions are promoted in the presence of alkoxide chain ends that are too nucleophilic. Thus, they can only be suppressed in the presence of a catalyst that is able to attenuate the reactivity of the alkoxide chain-end such to prevent nucleophilic attacks of the chain end on ester groups (inter- and intra-molecularly, causing transesterification) and epoxides (causing ether-bonds in the backbone). The ideal catalyst achieves this chemo-selectivity by shielding the reaction centre (the growing polymer chain) from unwanted reaction partners such as impurities and functional groups present in the monomers.11
Besides the chemo-selectivity, a catalyst that promotes a regioselective12 and stereocontrolled13 ring-opening of the monomers is desirable to achieve further control of the macromolecular structure and polyester properties.14 A regioselective ring-opening of unsymmetrical monomers leads to either head–tail or head–head and tail–tail connections in the backbone and it is ideally imposed by the catalyst independent of the monomer's preferred side of attack. Stereocontrol occurs by a stereoregular or a stereoselective ring-opening of monomers. A stereoregular ring-opening preserves (or inverts) the chirality of the monomers, which is achievable by achiral catalysts including organocatalysts.15 On the other hand, a stereoselective ring-opening requires a chiral catalyst capable of enantio-differentiation (e.g. chiral resolution of racemic monomers) or stereoselective bond-formation. In the ROAC, such a stereoselective process has been reported in the presence of homo-chiral bimetallic catalysts.16,17
However, exchange of the potentially toxic metal catalysts commonly used in ROAC with less harmful organocatalysts18,19 is desirable, especially if the synthesised polyesters should be used in biomedical applications or for food packaging.15 Many organocatalytic systems20 including non-hazardous ones21 showed the ability to catalyse the ROAC of anhydrides and epoxides. Among these, bi-component mixtures of a Lewis acid (LA) and a Lewis base (LB) (referred to as dual catalysts or Lewis pairs) with balanced acidity, basicity and steric demand showed good performances in the ROAC.22 Dual catalysts made of boranes (LA) and phosphazene bases [Brønsted base (BB)] or onium salts (LB) enable regioselective, stereoregular23,24 and sequence-controlled ROAC.25 Among the others, (thio-)ureas are economically-favourable and sustainable LA that have shown to be effective in the ROAC together with bis(triphenylphosphine)iminium chloride (PPNCl)26 or phosphazene bases.27
More recently, mono-component and bi-functional organocatalysts were also proposed. These systems are constituted by a borane (LB) covalently linked to either an ammonium halide,28 a phosphonium halide,29 or a (thio-)urea30 (LA). The former two organocatalysts activate the epoxides through borane coordination, while their onium cations modulate the reactivity of the negatively charged chain end through loose coordination.31 In combination with [PPN]+ cation-containing co-catalysts, the latter bi-functional (thio-)urea/borane organocatalyst showed to improve the polymerisation rate of phthalic anhydride (PA) and propene oxide (PO) while maintaining good chemo-selectivity and preventing ether-bond formation. The increase in the catalytic activity, as compared to mono-functional catalysts, is explained by synergistic effects due to the catalyst's bi-functionality.32 However, the addition of a co-catalyst leads again to a bi-component catalytic system that is negatively affected by dilution and prevents low catalyst loadings.33 Furthermore, [PPN]+ cations (such as PPNCl) alone are efficient ROAC catalysts34 and no results on the performance of the mentioned bi-functional catalyst alone (without a [PPN]+-containing co-catalyst) are reported so far.30
Therefore, we decided to explore other bi-functional and mono-component organocatalysts and to test them in ROAC of PA and epoxides in the absence of any co-catalysts. We think that working in this direction towards a catalyst with a strong affinity to the growing polyester chain and the ability to confine the reaction centre has many advantages. Firstly, the reactivity of the growing chain can be modulated to suppress side reactions while maintaining polymerisability (chemo-selectivity). Secondly, a confined and potentially chiral surrounding of the growing chain could pave the way for catalyst-induced regio- and stereoselectivity. Thirdly, the need for highly purified monomers to obtain high molar mass polyesters would be eliminated; in fact, if the catalyst is non-detachable from the reaction centre, the system will be unaffected by nucleophilic impurities, thus preventing chain transfer reactions.
We synthesised nine bi-functional and mono-component organocatalysts composed of a (thio-)urea moiety (LA) and an iminophosphorane (BB) moiety (cf.Chart 1). The selected systems differ in the Lewis acidity, Brønsted basicity, steric demand, and distance between the functional groups to evaluate the influence of these features on the ROAC. Moreover, the effect of the monomer structure was also explored by performing the co-polymerisation of PA with 3 different epoxides [cyclohexene oxide (CHO), butylene oxide (BO) and PO]. Special attention was focused on the mechanisms of initiation and chain propagation through a synergic experimental and theoretical approach. DFT calculations were performed to rationalise the experimental results and to achieve insights at the molecular level into the key interplay between the catalyst and growing chain/monomers. We think that this study can guide future ROAC catalyst development.
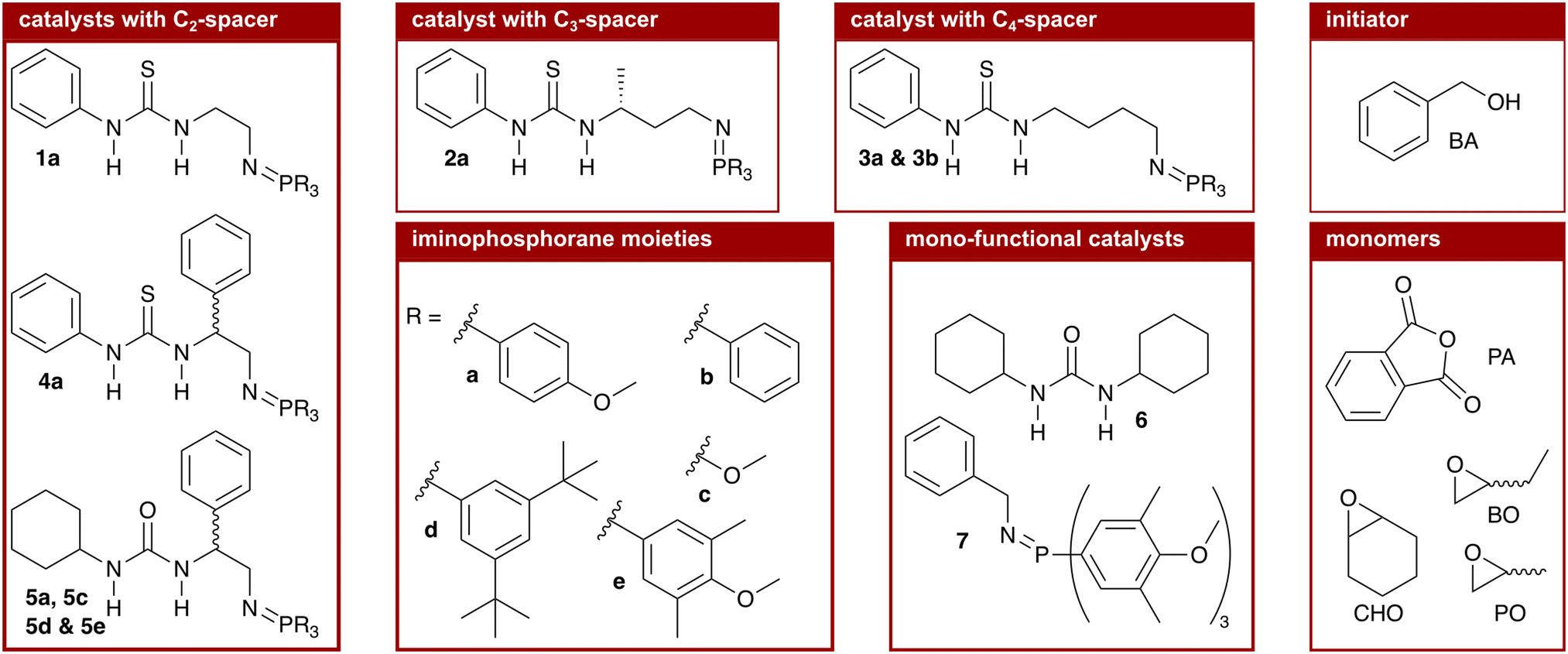 | ||
| Chart 1 Chemical structures of the catalysts, monomers and initiator. | ||
1 Results and discussion
As mentioned above, the catalysts we chose to test have the well-known scaffold of a (thio-)urea (LA) and an iminophosphorane (BB, cf. structure 1 in Chart 1) developed by the group of Dixon.35 Different (thio-)urea and iminophosphorane moieties are readily accessible36 and are covalently attached via a C2-linker obtainable from amino acids allowing the introduction and screening of chiral moieties at the linker.37 In addition to these C2-linkers, we extended the catalyst's modularity by synthesising catalysts with C3- (cat. 2a) and C4-linkers (cat. 3a and 3b). The catalysts were synthesised following literature-known procedures that are described in detail in the ESI† (cf. p. S23ff.).1.1 Characterisation of the catalytic species
The herein presented bi-functional catalysts are often generated in situ without characterisation of the catalyst itself.37 We, thus, started our investigation by identifying the catalyst species present in solution and assigning them to their 31P NMR chemical shift (cf.Scheme 2) in toluene, the same solvent which has been used in the ROAC. We performed the study on catalyst 5a as a model.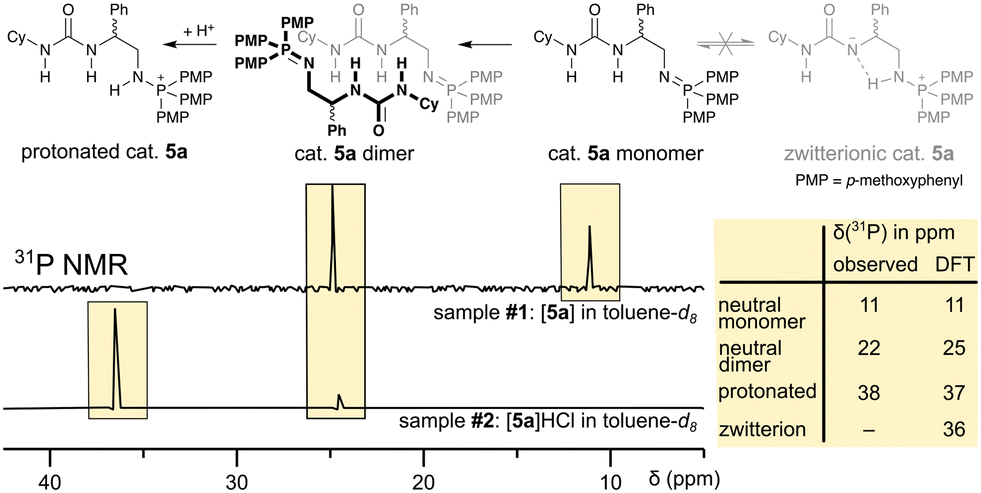 | ||
| Scheme 2 31P NMR spectra of catalyst 5a in toluene-d8 (top) and with additional HCl (bottom) are assigned to a neutral monomeric, a neutral dimeric, and a protonated species. A zwitterionic species of the catalyst is not observed (cf. ESI† for sample preparation and DFT calculation). | ||
Two different chemical shifts of the as-synthesised catalyst were observed at δ31P = 11 ppm and δ31P = 22 ppm (cf. sample #1 in Scheme 2). DFT NMR calculations have been performed with the ADF package to assign unambiguously the experimental signals observed (cf. ESI,† p. S78). As reported in the table in Scheme 2, the 31P chemical shifts are calculated to be 11 ppm for the neutral monomeric species and 25 ppm for a neutral dimeric species reproducing nicely the experimental findings. The 31P chemical shift of the neutral monomeric species is higher than the 31P chemical shift of a mono-functional neutral iminophosphorane (δ31P = 3 ppm),38 which indicates hydrogen bonding39 between the iminophosphorane's nitrogen and the urea moiety, as confirmed by the optimized DFT structure (cf. ESI,† Fig. S29). However, the urea is not deprotonated by the iminophosphorane, which would result in a zwitterionic species. Such a zwitterionic species is energetically unfavoured40 (with respect to the neutral monomer: ΔG = 9.0 kcal mol−1) and would significantly differ in its 31P chemical shift (δ31P = 36 ppm) from the neutral monomeric species. On the other hand, the dimerisation of the neutral monomeric species is energetically favoured (with respect to the neutral monomer: ΔG = −28.2 kcal mol−1) thanks to the formation of multiple hydrogen bonds (cf. ESI,† Fig. S29). The preference to form dimers is also known for similar bi-functional catalysts (thiourea + tertiary amine)41 and stems from the catalyst's bi-functionality. As such, the neutral catalyst 5a exists predominantly as dimers in solution, which need to break down into monomers to become the active catalytic species.42 The reported 31P chemical shift of a catalyst similar to 5a but with a more Lewis acidic thiourea moiety (δ31P = 27.4 ppm in CDCl3)37 suggests further that the dimer remains the predominant species even for catalysts with increased Lewis acidity.
Upon the addition of HCl to the solution containing catalyst 5a, the catalyst gets protonated and a new 31P signal at δ31P = 38 ppm appears (cf. sample #2 in Scheme 2). This 31P chemical shift coincides with the calculated chemical shift of δ31P = 37 ppm and it is in agreement with reported chemical shifts of protonated mono-functional iminophosphoranes.37,38 Both neutral species and the protonated species also differ in their diffusion behaviour, which allows the separation of their 1H NMR signals by 1H DOSY NMR¶ and full assignment of their 1H and 13C NMR signals (cf. ESI,† p. S72ff.).
1.2 Catalyst screening
All bi-functional catalysts were tested in the polymerisation of PA with an excess of either of the three epoxides CHO, BO and PO in toluene at 90 °C for 24 h (cf.Chart 1). The molar ratios of the catalyst to the benzyl alcohol (BA) initiator, PA and epoxide were kept constant at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:100:500 (cf. ESI,† p. S4). The catalytic activities of the catalysts are measured as the conversion of PA monomer into PA polymer units and the chemo-selectivity is expressed as the percentage of ester linkages in the polymer backbone. The results of the screening for the ROAC of PA/CHO, PA/BO and PA/PO are reported in Table 1.
:1:100:500 (cf. ESI,† p. S4). The catalytic activities of the catalysts are measured as the conversion of PA monomer into PA polymer units and the chemo-selectivity is expressed as the percentage of ester linkages in the polymer backbone. The results of the screening for the ROAC of PA/CHO, PA/BO and PA/PO are reported in Table 1.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| # | Catalyst | CHO | BO | PO | |||
| Conv.a | Sel.b | Conv.a | Sel.b | Conv.a | Sel.b | ||
| a Conversion of PA monomer into PA polymer units in mol%. b Selectivity to polyester over polyether in mol%. | |||||||
| 01 | 1a | 63 | 70 | 20 | 87 | 45 | 89 |
| 02 | 2a | 31 | 49 | 17 | 83 | 64 | 89 |
| 03 | 3a | 23 | 55 | 10 | 71 | 31 | 83 |
| 04 | 3b | 9 | 44 | 25 | 65 | 19 | 88 |
| 05 | 4a | 68 | 61 | 30 | 89 | 58 | >97 |
| 06 | 5a | 100 | 76 | 40 | 93 | 51 | >97 |
| 07 | 5c | 44 | 61 | 87 | 85 | 42 | 89 |
| 08 | 5d | 100 | 74 | 100 | >97 | 85 | >97 |
| 09 | 5e | 100 | 76 | 100 | >97 | 100 | >97 |
We started the screening with catalyst 1a, which is constituted by a phenyl thiourea and an iminophosphorane with three p-methoxy phenyl (PMP) substituents at the phosphorous atom that are covalently linked via a C2-spacer. The PMP iminophosphorane moiety was selected as it exhibits a similar basicity (pKBH+ = 25.0) to phosphazene base t-BuP1 (pKBH+ = 27.0) already known to be catalytically active in the ROAC.43 Catalyst 1a is found to promote the ROAC of all three epoxides (cf. entry 01, Table 1). The highest conversion is observed for the ROAC of PA/CHO with 63% and the best chemo-selectivity is observed for the ROAC of PA/PO with 89%.
Starting from catalyst 1a, we modified the spacer length between the two functional groups, cf. catalyst 2a with a C3- and catalyst 3a with a C4-spacer length. Comparing the results in Table 1 it emerges that, by increasing the spacer length, the catalytic activity drops down for the ROAC of PA with CHO and BO (1a > 2a > 3a), whereas the highest conversion of PA in the ROAC of PA/PO is observed for catalyst 2a (2a > 1a > 3a). A reduction in chemo-selectivity accompanies the drop in catalytic activity.
Next, we investigated the influence of a substituent at the spacer unit on the catalytic performance. It is known that adding a substituent to the C2-spacer improves the solubility of the catalyst.44 Compared to catalyst 1a, racemic catalyst 4a has a phenyl substituent at the C2-spacer unit. Although a small drop in the chemo-selectivity is observed in the ROAC of PA/CHO (61% vs. 70%), the addition of a phenyl substituent at the spacer unit leads to overall improved catalytic activity and chemo-selectivity.
We continued the screening by varying the features of the (thio-)urea moiety. A lower Lewis acidity was described to improve the catalytic activity of bi-component mixtures of PPNCl and (thio-)ureas in the ROAC and the highest activity was found for dicyclohexyl urea (6).26 We, thus, compared phenyl thiourea catalyst 4a with the analogous cyclohexyl urea catalyst 5a. The latter catalyst 5a has, overall, the highest catalytic activity and chemo-selectivity among the considered systems, reaching 100% conversion of PA in the ROAC of PA/CHO.
Finally, the influence of the changes at the iminophosphorane moiety on the ROAC was evaluated, starting from catalyst 5a. Changing the substituents at the phosphorous atom affects both the electronics and the sterics of the catalyst, but the two effects on the catalytic activity cannot be discerned experimentally. Firstly, more electron-withdrawing groups increase the basicity and secondly, bulkier groups increase the steric demand of the iminophosphorane moiety. Compared to catalyst 5a with PMP substituents, three catalysts were successfully synthesised:|| catalyst 5c with methoxy substituents, which is less basic and has a significantly lower steric demand; catalyst 5d with (m-tBu)2 phenyl substituents, which is less basic and has a higher steric demand and catalyst 5e with (m-Me)2–(p-OMe) phenyl substituents, which is similarly basic and has a higher steric demand.** The combination of a higher basicity and an increased steric demand (catalyst 5e) yields the best catalytic activity, resulting in the complete conversion of PA for the ROAC of all three epoxides tested.
The optimised catalyst 5e is highly chemo-selective (>97%) in the ROAC of PA/BO and PA/PO. MALDI analysis further supports a strictly alternating macromolecular structure of poly(PA-alt-BO) and poly(PA-alt-PO) with alcohol end groups that stem from a single ring-opening of epoxide after depletion of PA (cf. ESI† Fig. S7 and S10). In comparison, poly(PA-co-CHO) is constituted by species with one or more ether bonds, which is in agreement with the ether signals observed by and the lower chemo-selectivity of 76% concluded from 1H NMR analyses. Despite the imperfect chemo-selectivity, no further addition of CHO was observed after the complete conversion of PA (cf. ESI† Table S2, entries 04-4, 04-5 and 04-6). The inability to ring-open CHO in the absence of PA monomers indicates that PA promotes the ROAC, potentially by activating the epoxide.45 Based on the same experiment, no signs of transesterification are observed after consumption of PA as the SEC traces remain unchanged. Thus, catalyst 5e seems to suppress the transesterification, although a small shoulder in the SEC trace suggests that some transesterification occurs during the ROAC (cf.Scheme 3d).
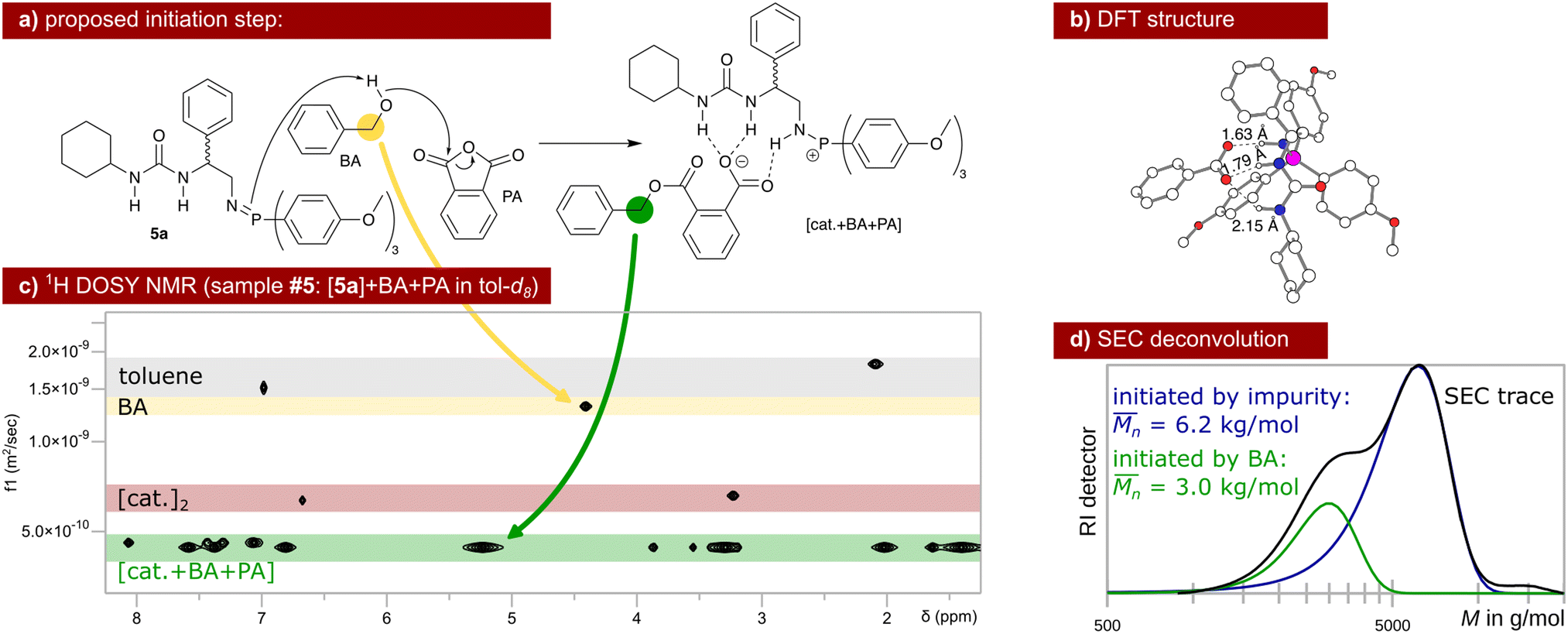 | ||
| Scheme 3 Proposed initiation of the ROAC: BA ring-opens PA assisted by the catalyst that gets protonated in the process (a). Hydrogen bond interaction between the catalyst and the carboxylic acid is energetically favoured (b) leading to aggregates observed via DOSY NMR (c, single diffusion coefficient in green). Unreacted dimeric species of catalyst 5a (red) and unreacted BA (yellow) are also observed at different diffusion coefficients via DOSY NMR. The initiation by BA leads to the lower molar mass fraction observed in the SEC trace of polymer poly(PA-co-CHO) (d, entry 09 in Table 1). The higher molar mass fraction stems from polymer chains initiated by water, diol (reaction between CHO and water) and/or diacid (reaction between PA and water). | ||
Further we have tested mono-functional cyclohexyl urea (6) and iminophosphorane 7 catalysts having each only one functional moiety of the optimised catalyst 5e. Both reference catalysts 6 and 7 are less catalytically active than catalyst 5e in the ROAC of PA/CHO, both alone and combined in a bi-component mixture (cf. Table S2† entries 08, 09, and 10). Further addition of 1 equivalent of either urea 6 or iminophosphorane 7 to catalyst 5e improves the catalytic activity slightly but leaves the chemo-selectivity unchanged (cf. Table S2† entries 11 and 12). Finally, the tested protonated catalyst showed to be incapable of catalysing the ROAC (cf. Table S2† entry 13) demonstrating that the catalyst's ability to undergo protonation/deprotonation cycles is responsible for its catalytic activity.
1.3 ROAC mechanism
The ROAC is initiated by BA deliberately added to the solution and by nucleophilic impurities. As a consequence, the synthesised polyesters have a bimodal molar mass distribution (cf. SEC results in Scheme 3d). The lower molar mass fraction constitutes BA-initiated polyesters (end group analysis by MALDI, cf. ESI† p. S14ff.). Their number average molar mass (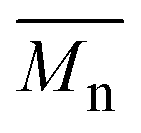 ) is halved compared to the higher molar mass fraction (Gaussian deconvolution of the SEC traces, cf. tabulated values in ESI† p. S5ff.). The higher molar mass fraction constitutes impurity-initiated polyesters. These nucleophilic impurities are water, diacid (ring-opened PA by water) and diol (ring-opened epoxide by water) leading to polyesters that are indistinguishable among themselves by MALDI analysis.34 In the absence of BA, the ROAC still occurs at a similar polymerisation rate because the impurities compensate for the lack of BA initiators (cf. ESI† Fig. S1).
) is halved compared to the higher molar mass fraction (Gaussian deconvolution of the SEC traces, cf. tabulated values in ESI† p. S5ff.). The higher molar mass fraction constitutes impurity-initiated polyesters. These nucleophilic impurities are water, diacid (ring-opened PA by water) and diol (ring-opened epoxide by water) leading to polyesters that are indistinguishable among themselves by MALDI analysis.34 In the absence of BA, the ROAC still occurs at a similar polymerisation rate because the impurities compensate for the lack of BA initiators (cf. ESI† Fig. S1).
The initiation of the ROAC was unveiled by NMR spectroscopy analysis performed on samples of catalyst 5a in various combinations with the initiator BA and the monomers PA and CHO (cf. sample composition in ESI† Table S4). As discussed in the previous section, the predominant species of catalyst 5a in toluene is a neutral dimer. This dimer resists the addition of BA, implicating that catalyst 5a is incapable of deprotonating BA (cf.31P NMR spectra in ESI† Fig. S23). Upon the addition of BA and PA, the dimer breaks down and catalyst 5a gets protonated. Simultaneously, a reaction between PA and BA occurs yielding a carboxylate (cf. the downfield shift of the benzyl protons) that intermolecularly binds to the protonated catalyst (single diffusion behaviour observed by 1H DOSY NMR, cf.Scheme 3c). This catalyst–carboxylate adduct is stabilised by multiple intermolecular hydrogen bonds between the protonated catalyst 5a and the carboxylate (cf. DFT structure in Scheme 3b). A similar catalyst–acid adduct has been described for a thiourea amine bi-functional catalyst.41,46 Thus, BA initiates the ROAC by ring-opening of PA in the presence of the bi-functional catalyst (cf.Scheme 3a) even at room temperature at the lower concentration used for the NMR study.
Instead, in the presence of CHO (cf. sample #6, ESI† Table S4), the same catalyst–carboxylate adduct forms but the expected ring-opening of CHO does not occur as confirmed by the absence of ester signals in the 1H NMR spectrum even after heating the sample to 100 °C for 60 min. The absence of the ring-opening reaction of CHO, which is the rate-determining step (rds)47 of the ROAC (vide infra), is partly reasoned by the low concentration of reactants48 and further indication that excess PA promotes the ROAC.
To get better insight into the operative mechanism and disentangle the effects of the different structural components (e.g. spacer lengths and steric demand of the iminophosphorane moiety) on the reactivity of these catalysts, DFT calculations were also performed. Computational investigations [using the dispersion-corrected PBE0-D3BJ method with the 6-31G(d) basis set followed by single point energy calculations with the TZVP basis set in toluene (cf. ESI† p. S78, ‘Computational methodology’)] reveal that the catalytic performance of these bi-functional and mono-component organocatalysts relies on the two H-bond donor sites that activate the monomers and enhance the nucleophilicity of the propagation intermediates. In Fig. 1, the Gibbs-free energy profile for the organocatalysed ROAC of PO and PA promoted by catalyst 5e is reported as an example. For the sake of calculation efficiency and clarity, the propagating polymer chain was simplified to a benzoate.
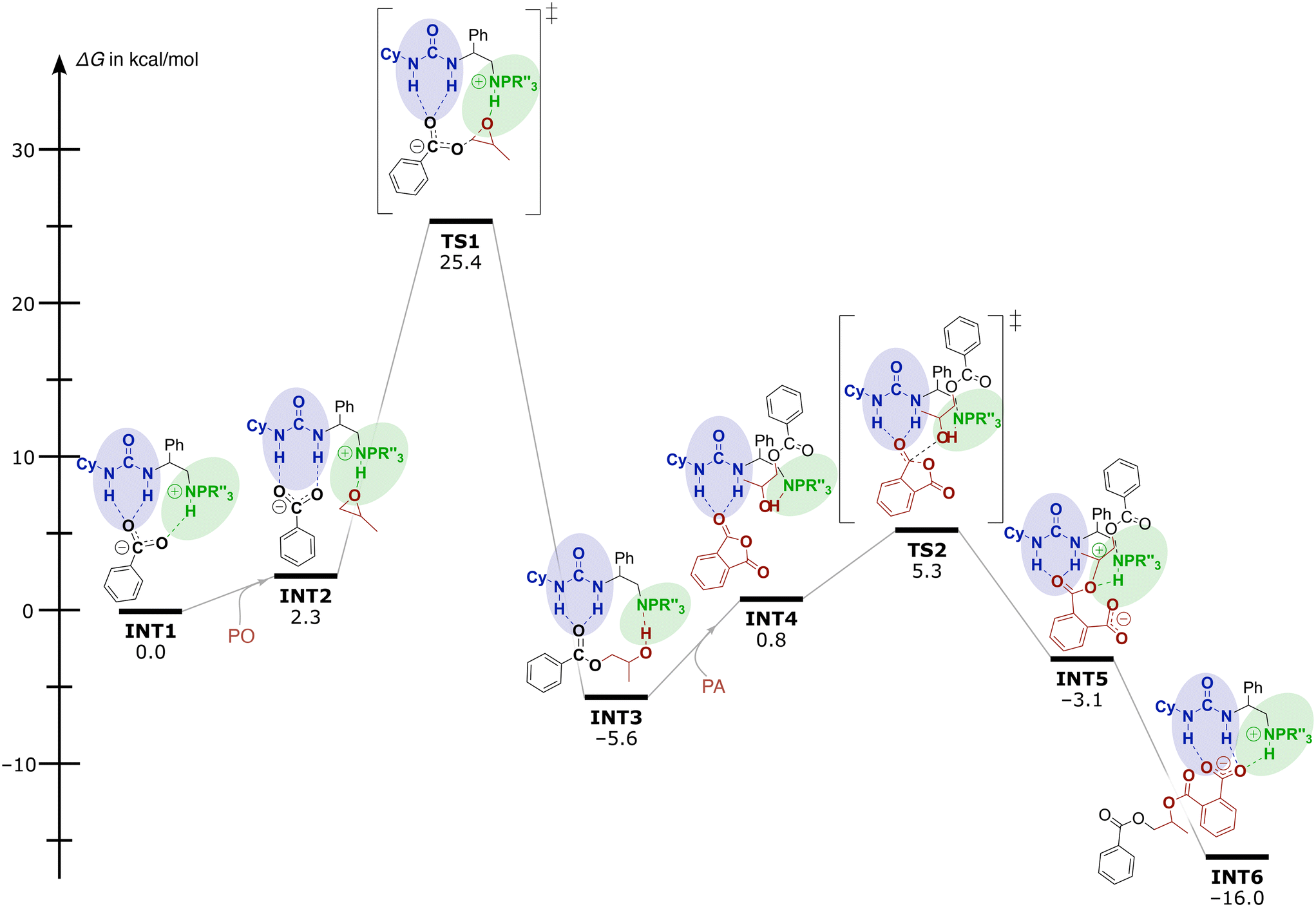 | ||
| Fig. 1 Gibbs free energy profile and mechanism for the organocatalysed ROAC of PO and PA promoted by catalyst 5e. | ||
Initially the mechanism involves a carboxylate chain-end bound to both urea and iminophosphorane moieties through H-bonding (INT1 in Fig. 1). Then, the LA pulls off the propagating chain-end from the BB residue facilitating PO coordination and activation at the iminophosphorane centre affording the intermediate INT2. The subsequent nucleophilic attack of the bound carboxylate on PO proceeds via the transition state TS1 with an activation barrier ΔG‡ of 25.4 kcal mol−1. During this process, the carboxyl group remains bound to the urea moiety and the proton switches position from the catalyst onto the chain end resulting in an alcohol and a neutral catalyst (INT3). The following addition of PA starts with the release of the growing chain from the urea moiety allowing the freed urea to intermolecularly bind to a PA monomer in INT4. Then, a nucleophilic attack of the alkoxide on the activated PA occurs with a barrier of 5.3 kcal mol−1, resulting in PA ring-opening to form the intermediates stabilized by both LA and BB residues (INT5 and INT6). Proton transfer between INT5 and INT6 leads again to a carboxylate end group and the ROAC cycle starts again.
The calculations suggest the epoxide ring-opening by carboxylate to form INT3 as the rds on the reaction coordinate. Analysis of the polymerisation rate in dependence on the feed ratio and the polymerisation temperature supports the proposed mechanism (cf. ESI† Tables S2 and S3). A reduction of either the catalyst or the epoxide feed ratio decreases the polymerisation rate as they are involved in the rds. The initial turnover frequency (TOF) of the catalyst is less dependent on the PA feed ratio (cf. ESI† Fig. S2), which agrees with the proposed non-involvement of PA in the rds. Slightly higher initial TOFs obtained for higher PA feed ratios could indicate an indirect involvement of PA in the rds by e.g. epoxide activation.45 On the other hand, the TOFs gradually increase over time for the higher PA feed ratios. Interestingly, the chemo-selectivity is unaffected by the polymerisation temperature and the feed ratio of PA and CHO (cf. ESI† Table S2, entries 02–07) and remains at 75–80%.
Comparison of the activation energies of the ROAC (ΔG‡ = INT1 → TS1) allows the influence of the different catalysts' moieties on the ROAC to be individually assessed. These activation energies should further qualitatively correlate with the obtained catalytic activities (measured as PA conversion, cf.Table 1), which allows a comparison between computational and experimental results: a lower ΔG‡ should correspond to a greater PA conversion. The observed catalytic activity and chemo-selectivity of the ROAC are similar for BO and PO but differed for CHO (using the same catalyst). We, thus, narrowed down the DFT analysis to the ROAC of PA/CHO and PA/PO.
First, the influence of a substituent at the spacer was evaluated and found to be negligible as observed experimentally. In fact, adding a phenyl substituent to the C2-spacer of the catalyst 1a, resulting in catalyst 4a, leads to a ΔΔG‡ of 0.3 kcal mol−1 for PA/CHO and 0.2 kcal mol−1 for PA/PO, respectively. Conversely, by changing the length of the spacer between BB and LA residues going from the C4-spacer of catalyst 3a to the C2-spacer of catalyst 1a, the ΔG‡ is reduced by 1.7 kcal mol−1 for PA/CHO and 1.0 kcal mol−1 for PA/PO, respectively. These results are in line with the higher catalytic activity found experimentally for catalyst 1a having the shortest spacer length.
Next, the effect of the different LA moieties was evaluated. Experimentally, a higher catalytic activity was found for the less acidic cyclohexyl urea catalyst 5a compared to the phenyl thiourea catalyst 4a for the ROAC of PA/CHO (100% vs. 68% PA conversion) while the opposite effect was found for the ROAC of PA/PO (58% vs. 51% PA conversion). Based on the DFT results, ΔΔG‡ is 0.1 kcal mol−1 for the ROAC of both PA/PO and PA/CHO which confirms that the experimentally found catalytic activities are inconclusive. Since catalysts 4a and 5a differ not only for the LA nature but also for the N-linked substituent, two new catalysts were modelled to evaluate these effects separately. In this regard, the phenyl substituent of thiourea catalyst 4a has been replaced with a cyclohexyl substituent (catalyst 4a-Cy) and the cyclohexyl substituent of urea catalyst 5a has been replaced with a phenyl substituent (catalyst 5a-Ph). The presence of a cyclohexyl substituent leads to a decrease of the activation energy barrier; ΔΔG‡ of 2.2 kcal mol−1 (PA/CHO) and 1.0 kcal mol−1 (PA/PO) was found for the thiourea catalysts (4avs.4a-Cy) whereas ΔΔG‡ of 1.6 kcal mol−1 (PA/CHO) and 0.8 kcal mol−1 (PA/PO) was found for the urea catalysts (5a-Phvs.5a). Inspection of the corresponding transition states (TS1, cf. ESI† Fig. S26 and S27) reveals the presence of unfavourable steric interactions between the phenyl ring of the (thio-)urea and the iminophosphorane substituent, which increases the energy of TS1. On the other hand, ΔΔG‡ of 1.5 kcal mol−1 for PA/CHO and 0.9 kcal mol−1 for PA/PO was found for phenyl-substituted catalysts 4a and 5a-Ph, whereas ΔΔG‡ of 2.1 kcal mol−1 (PA/CHO) and 1.1 kcal mol−1 (PA/PO) was found for the cyclohexyl-substituted catalysts 4a-Cy and 5a. The higher Lewis acidity of the thiourea moiety increases the hydrogen bonding strength and favours the ring-opening of the epoxide, which decreases the energy of TS1. Based on these results, the ideal LA moiety is more acidic and small/flexible.
The iminophosphorane moiety can influence the catalytic activity through its steric demand and basicity. The basicity of the systems affects the degree of elongation of the N–H bond of the iminophosphorane residue (cf. ESI,† Table S5). To have a further measure of the electronic effect of the different phosphines, we have considered the energy of the N protonation for the catalyst alone. Obtained data clearly indicate system 5c to be by far the least basic, while systems 5a and 5e are more similar. Concerning the steric demand, catalyst 5e is bulkier than systems 5a and 5c as evidenced by steric maps49–51 reported in Fig. 2. It is clear that the basicity of the phosphine moiety does not explain the relative reactivity order of the three catalysts, instead, we observe that the catalytic activity improves with higher steric demand. The increase of the steric demand of the phosphine moiety is clearly highlighted by the %VBur in the NE and SE quadrants in Fig. 2. The increases of these %VBur contribute to the creation of a tighter catalytic pocket that forces the two substrates to be closer to each other. In fact, if in the TS1 of system 5c the nucleophilic attack distance is 2.20 Å, in systems 5a and 5e with similar steric footprint, the same distance is 1.96 Å and 1.98 Å, respectively. Additionally, the presence of the –CH3 substituents on the rings of the phosphine moiety of system 5e causes the –OCH3 groups to face inward into the catalytic pocket promoting its closure and overall allowing for a better hydrogen bond interaction between the substrates and the catalyst. Catalyst 5e, thus, leads to the lowest ΔG‡ of 24.3 kcal mol−1 (PA/CHO) and 25.4 kcal mol−1 (PA/PO) in agreement with its superior catalytic activity found experimentally.
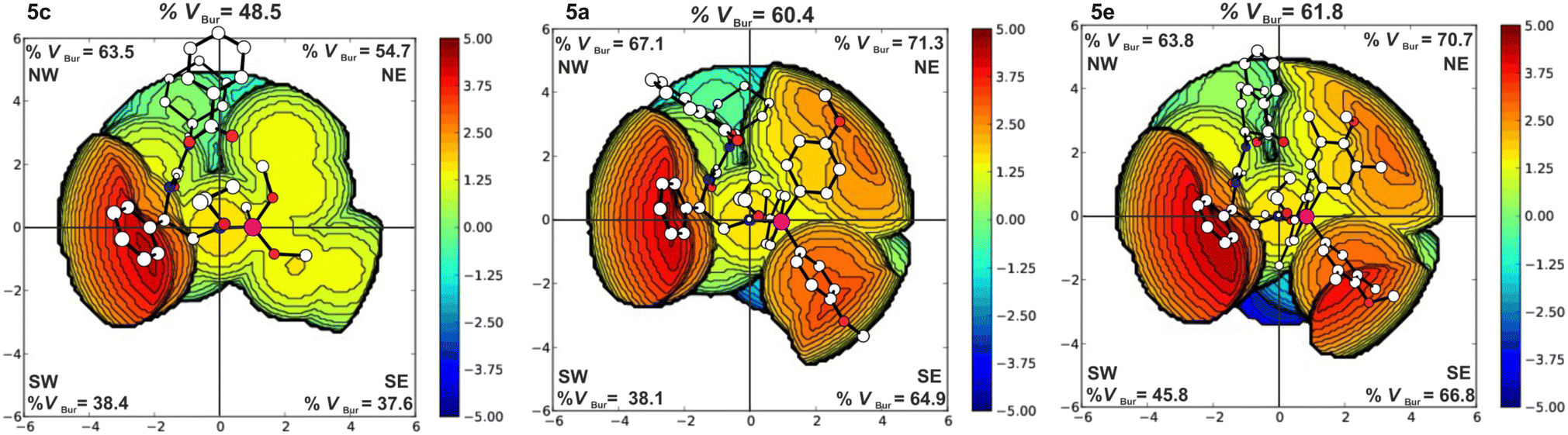 | ||
| Fig. 2 Steric maps of catalysts 5c (left), 5a (middle), and 5e (right). | ||
1.4 Comparison to bi-functional organocatalysts
The ROAC mechanisms, as investigated by DFT, of two other bi-functional organocatalysts (8 and 9) were previously published.28,30 The chemical structures of these systems (catalyst in black, initiating species in blue) and the activation energies of the rds (ring-opening of the epoxide, TS1) are displayed and compared to catalyst 5e in Table 2.|
|
|||||
|---|---|---|---|---|---|
| #: | Catalyst | Ini.a | ΔG‡b | ROAC | Ref. |
| a Initiating species. b Gibbs free energy difference in kcal mol−1 (equal to energy of TS1). c Initiating species is independent of the catalyst (extra component). | |||||
| 01: | [Urea/base] (5e) | BAc | 24.3 | PA/CHO | — |
| 02: | [Urea/base] (5e) | BAc | 25.4 | PA/PO | — |
| 03: | [Borane/ammonium] (8) | Cl− | 24.3 | PA/CHO | 28 |
| 04: | [Borane/thiourea] (9) + PPN+ | BDC2− | 23.0 | PA/PO | 30 |

These previously published bi-functional organocatalysts are constituted by a borane moiety as one functional group and either an ammonium cation (8) or a thiourea (9) moiety as the other functional group. The initiating species of the ROAC is in both cases the counter-anion to the system, being a chloride for catalyst 8 and 1,2-benzene dicarboxylate (BDC) added as part of the [PPN]+-containing co-catalyst for catalyst 9. As a result, the ratio between the catalytically active and initiating species is fixed and cannot be changed independently.
Compared to the borane-containing catalysts 8 (reported for the ROAC of PA/CHO) and 9 (reported for the ROAC of PA/PO), catalyst 5e enables the ROAC with similar activation energies (ΔΔG‡ of 0.0 kcal mol−1 for PA/CHO and ΔΔG‡ of 2.4 kcal mol−1 for PA/PO). Nonetheless, the TOF of catalyst 5e is reduced by more than one magnitude compared to those of catalysts 8 and 9, which cannot be explained by the difference in activation energies.
In an attempt to explain these findings, we first compared the ability of the borane and iminophosphorane moiety to coordinate to an epoxide, as this coordination initiates the epoxide ring-opening. To this extent, we have computed the two functionalities as two small models, i.e. 9-BBNB(CH3) for borane and CH3NP(CH3)3 for iminophosphorane. Calculations of the binding of propylene oxide to model borane (ΔG of 6.0 kcal mol−1) and iminophosphorane (ΔG of 0.1 kcal mol−1) reveal that iminophosphorane is the better coordination partner. Less effective coordination and activation of epoxide by the iminophosphorane compared to borane are, thus, ruled out, and differences in the TOF due to the presence of borane instead of iminophosphorane are unlikely.
The second evident difference between the catalysts is the ability of catalyst 5e to participate in protonation/deprotonation reactions (via its iminophosphorane group), whereas catalysts 8 (ammonium cation) and 9 ([PPN]+ cation) are constituted by permanent cations incapable of being deprotonated. We, thus, analysed the impact of this difference on the catalytic activity. As described in the previous section, the acidic adduct of anhydride and water (a diacid) is present in the reaction system. Based on the mass ratio of low (∼20%) and high (∼80%) molecular weight polymer fractions (SEC results, cf. ESI†), we estimate that the amount of diacid is two times the amount of benzyl alcohol/catalyst. This diacid can interact with the catalyst modifying the relative stability of the species involved in the reaction pathway. For instance, we have investigated the possible formation of INT1diacid and INT3diacid (cf. ESI† Fig. S28). Calculations revealed that INT1diacid and INT3diacid are −17.9 and −16.5 kcal mol−1 more stable than INT1 and INT3 in Fig. 1, respectively. We hypothesise that the formation of these species (INT1diacid and INT3diacid) can affect the performance of the catalyst as they (i) force the reaction to proceed along a pathway of higher energy and/or (ii) lower the concentrations of INT1 and INT3, which result in a lower concentration of catalytically active species. This hypothesis is supported by the fact that higher catalyst loadings lead to higher TOFs (cf. ESI† Fig. S1). Furthermore, when using catalyst 5e together with PPNCl (ratio 1:1, cf. Table S2† entry 14) in the ROAC of PA/CHO, both the catalytic activity (full conversion already after 2 h) and chemo-selectivity (>97%) are drastically improved compared to catalyst 5e alone (full conversion after 21 h with 75% chemo-selectivity). In combination, these findings provide evidence that the ability of iminophosphorane-containing bi-functional organocatalysts to participate in protonation/deprotonation reactions together with the good interaction between the catalyst and growing polymer chain makes this catalytic system prone to acidic species present in the ROAC system.
2 Conclusions
All tested bi-functional and mono-component catalysts composed of a (thio-)urea Lewis acidic moiety and an iminophosphorane Brønsted basic moiety catalyse the ROAC between PA and either CHO, BO and PO. In general, for the ROAC of one epoxide, catalysts that are more catalytically active are also more chemo-selective. The highest catalytic activity was found for catalyst 5e with a cyclohexyl urea LA and a sterically large iminophosphorane BB separated by a C2-spacer. Irrespective of the catalyst, the nature of the epoxide impacts both the polymerisation rate (CHO > PO ∼ BO) and the chemo-selectivity (PO > BO ≫ CHO) of the ROAC.A strong interaction between the catalyst and the growing chain is observed and results from the bi-functionality of the system. This interaction is enabled by multiple hydrogen bonds, which also lead to the dimerisation of the catalysts in the absence of PA. In the presence of PA, the dimers break down and get themselves protonated while keeping the carboxylate chain end deprotonated. After the addition of an epoxide, the proton transfers from the iminophosphorane moiety to the chain end, which attenuates its reactivity. Thereby, the bi-functional catalysts suppress transesterification side reactions, in contrast to the widely used PPNCl ROAC catalyst, which is unable to protonate the alkoxide chain end.52
Compared to other bi-functional catalyst systems,28–30 the herein presented (thio-)urea/iminophosphorane bi-functional catalysts are constituted by a Brønsted base (iminophosphorane) instead of a cation incapable of participating in protonation/deprotonation reactions. The iminophosphorane enables coordination of the catalysts with the growing polymer chain [together with the (thio-)urea moiety] and reversible protonation/deprotonation of the chain end (carboxylate vs. alcohol chain ends), which opens up an energetically favourable trajectory for the ROAC. In the presence of acidic species formed by secondary reactions, however, the ability of the iminophosphorane residue to strongly coordinate acid moieties slows down the polymerisation rate by partially trapping the catalytically active species. Future work will, thus, focus on maintaining the tight association of the catalyst with the polymer chain end, while eliminating possible side reactions with acidic impurities, e.g., by shielding the reactive site from these acidic species. If successful, such a catalyst will improve the polymerisation rate and promises to suppress the initiation of the ROAC by impurities, making the—currently necessary22—extensive monomer purification obsolete.
Author contributions
M. H.: conceptualization, formal analysis, investigation, project administration, visualization, writing – original draft, writing – review & editing. R. Z.: formal analysis, investigation, visualization, writing – review & editing. S. M.: conceptualization, writing – review & editing. L. F.: formal analysis, investigation, supervision, writing – review & editing. T. F.: conceptualization, funding acquisition, project administration, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. H. and T. F. acknowledge Formas – a Swedish Research Council for Sustainable Development (Early-career Researchers Grant no. 2020-00910) for the financial support.Notes and references
- The New Plastics Economy Catalysing Action, World Economic Forum and Ellen Macarthur Foundation Technical Report, 2017 Search PubMed.
- F. M. Haque, J. S. A. Ishibashi, C. A. L. Lidston, H. Shao, F. S. Bates, A. B. Chang, G. W. Coates, C. J. Cramer, P. J. Dauenhauer, W. R. Dichtel, C. J. Ellison, E. A. Gormong, L. S. Hamachi, T. R. Hoye, M. Jin, J. A. Kalow, H. J. Kim, G. Kumar, C. J. LaSalle, S. Liffland, B. M. Lipinski, Y. Pang, R. Parveen, X. Peng, Y. Popowski, E. A. Prebihalo, Y. Reddi, T. M. Reineke, D. T. Sheppard, J. L. Swartz, W. B. Tolman, B. Vlaisavljevich, J. Wissinger, S. Xu and M. A. Hillmyer, Chem. Rev., 2022, 122, 6322–6373 CrossRef CAS PubMed.
- Y. D. Y. L. Getzler and R. T. Mathers, Acc. Chem. Res., 2022, 55, 1869–1878 CrossRef CAS PubMed.
- J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167–15197 CrossRef CAS PubMed.
- K. Nomura and N. W. Binti Awang, ACS Sustainable Chem. Eng., 2021, 9, 5486–5505 CrossRef CAS.
- O. Santoro, L. Izzo and F. Della Monica, Sustainable Chem., 2022, 3, 259–285 CrossRef CAS.
- M. N. Rashed, S. M. A. H. Siddiki, M. A. Ali, S. K. Moromi, A. S. Touchy, K. Kon, T. Toyao and K.-I. Shimizu, Green Chem., 2017, 19, 3238–3242 RSC.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y.-X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS PubMed.
- X. Liang, F. Tan and Y. Zhu, Front. Chem., 2021, 9, 150 Search PubMed.
- X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef CAS PubMed.
- X.-L. Chen, B. Wang, L. Pan and Y.-S. Li, Macromolecules, 2022, 55, 3502–3512 CrossRef CAS.
- A. Kummari, S. Pappuru and D. Chakraborty, Polym. Chem., 2018, 9, 4052–4062 RSC.
- Z.-Q. Wan, J. M. Longo, L.-X. Liang, H.-Y. Chen, G.-J. Hou, S. Yang, W.-P. Zhang, G. W. Coates and X.-B. Lu, J. Am. Chem. Soc., 2019, 141, 14780–14787 CrossRef CAS PubMed.
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC.
- A. Kummari, S. Pappuru, P. K. Gupta, D. Chakraborty and R. S. Verma, Mater. Today Commun., 2019, 19, 306–314 CrossRef CAS.
- J. Li, B.-H. Ren, Z.-Q. Wan, S.-Y. Chen, Y. Liu, W.-M. Ren and X.-B. Lu, J. Am. Chem. Soc., 2019, 141, 8937–8942 CrossRef CAS PubMed.
- G.-H. He, Y.-L. Liu, Y. Liu and X.-B. Lu, Macromolecules, 2022, 55, 3869–3876 CrossRef CAS.
- D. W. C. MacMillan, Nature, 2008, 455, 304–308 CrossRef CAS PubMed.
- H. C. Erythropel, J. B. Zimmerman, T. M. de Winter, L. Petitjean, F. Melnikov, C. H. Lam, A. W. Lounsbury, K. E. Mellor, N. Z. Janković, Q. Tu, L. N. Pincus, M. M. Falinski, W. Shi, P. Coish, D. L. Plata and P. T. Anastas, Green Chem., 2018, 20, 1929–1961 RSC.
- D. Ryzhakov, G. Printz, B. Jacques, S. Messaoudi, F. Dumas, S. Dagorne and F. Le Bideau, Polym. Chem., 2021, 12, 2932–2946 RSC.
- M. Sihtmäe, E. Silm, K. Kriis, A. Kahru and T. Kanger, ChemSusChem, 2022, 15, e202201045 CrossRef PubMed.
- H.-Y. Ji, B. Wang, L. Pan and Y.-S. Li, Green Chem., 2018, 20, 641–648 RSC.
- H.-Y. Ji, X.-L. Chen, B. Wang, L. Pan and Y.-S. Li, Green Chem., 2018, 20, 3963–3973 RSC.
- J. Zhang, L. Wang, S. Liu, X. Kang and Z. Li, Macromolecules, 2021, 54, 763–772 CrossRef CAS.
- H.-Y. Ji, D.-P. Song, B. Wang, L. Pan and Y.-S. Li, Green Chem., 2019, 21, 6123–6132 RSC.
- L. Lin, J. Liang, Y. Xu, S. Wang, M. Xiao, L. Sun and Y. Meng, Green Chem., 2019, 21, 2469–2477 RSC.
- X. Zhu, R. Wang, X. Kou, F. Liu and Y. Shen, Macromol. Chem. Phys., 2021, 222, 2100104 CrossRef CAS.
- R. Xie, Y. Zhang, G. Yang, X. Zhu, B. Li and G. Wu, Angew. Chem., Int. Ed., 2021, 60, 19253–19261 CrossRef CAS PubMed.
- Y.-Y. Zhang, C. Lu, G.-W. Yang, R. Xie, Y.-B. Fang, Y. Wang and G.-P. Wu, Macromolecules, 2022, 55, 6443–6452 CrossRef CAS.
- J. Wang, Y. Zhu, M. Li, Y. Wang, X. Wang and Y. Tao, Angew. Chem., Int. Ed., 2022, 61, e202208525 CrossRef CAS PubMed.
- J. Xu, P. Zhang, Y. Yuan and N. Hadjichristidis, Angew. Chem., Int. Ed., 2023, 62, e202218891 CrossRef CAS PubMed.
- X. Wang, J. Hui, M. Shi, X. Kou, X. Li, R. Zhong and Z. Li, ACS Catal., 2022, 12, 8434–8443 CrossRef CAS.
- C. A. L. Lidston, S. M. Severson, B. A. Abel and G. W. Coates, ACS Catal., 2022, 12, 11037–11070 CrossRef CAS.
- Z. Hošt'álek, O. Trhlíková, Z. Walterová, T. Martinez, F. Peruch, H. Cramail and J. Merna, Eur. Polym. J., 2017, 88, 433–447 CrossRef.
- M. Formica, D. Rozsar, G. Su, A. J. M. Farley and D. J. Dixon, Acc. Chem. Res., 2020, 53, 2235–2247 CrossRef CAS PubMed.
- D. Rozsar, M. Formica, K. Yamazaki, T. A. Hamlin and D. J. Dixon, J. Am. Chem. Soc., 2022, 144, 1006–1015 CrossRef CAS PubMed.
- M. G. Núñez, A. J. M. Farley and D. J. Dixon, J. Am. Chem. Soc., 2013, 135, 16348–16351 CrossRef PubMed.
- T. A. Albright, W. J. Freeman and E. E. Schweizer, J. Org. Chem., 1976, 41, 2716–2720 CrossRef CAS.
- A. L. Llamas-Saiz, C. Foces-Foces, J. Elguero, F. Aguilar-Parrilla, H.-H. Limbach, P. Molina, M. Alajarín, A. Vidal, R. M. Claramunt and C. López, J. Chem. Soc., Perkin Trans. 2, 1994, 209–212 RSC.
- J. C. Golec, E. M. Carter, J. W. Ward, W. G. Whittingham, L. Simón, R. S. Paton and D. J. Dixon, Angew. Chem., 2020, 132, 17570–17575 CrossRef.
- R. S. Klausen, C. R. Kennedy, A. M. Hyde and E. N. Jacobsen, J. Am. Chem. Soc., 2017, 139, 12299–12309 CrossRef CAS PubMed.
- R. P. Singh, B. M. Foxman and L. Deng, J. Am. Chem. Soc., 2010, 132, 9558–9560 CrossRef CAS PubMed.
- H. Li, J. Zhao and G. Zhang, ACS Macro Lett., 2017, 6, 1094–1098 CrossRef CAS PubMed.
- A. M. Goldys and D. J. Dixon, Macromolecules, 2014, 47, 1277–1284 CrossRef CAS.
- H. Li, H. Luo, J. Zhao and G. Zhang, ACS Macro Lett., 2018, 7, 1420–1425 CrossRef CAS PubMed.
- M. Žabka, A. Kocian, S. Bilka, S. Andreǰcák and R. Šebesta, Eur. J. Org. Chem., 2019, 35, 6077–6087 CrossRef.
- S. Pappuru and D. Chakraborty, Eur. Polym. J., 2019, 121, 109276 CrossRef CAS.
- M. E. Fieser, M. J. Sanford, L. A. Mitchell, C. R. Dunbar, M. Mandal, N. J. Van Zee, D. M. Urness, C. J. Cramer, G. W. Coates and W. B. Tolman, J. Am. Chem. Soc., 2017, 139, 15222–15231 CrossRef CAS PubMed.
- A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, Eur. J. Inorg. Chem., 2009, 13, 1759–1766 CrossRef.
- L. Falivene, R. Credendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, Organometallics, 2016, 35, 2286–2293 CrossRef CAS.
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed.
- B. Abel, C. A. L. Lidston and G. W. Coates, J. Am. Chem. Soc., 2019, 141, 12760–12769 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Catalyst syntheses and characterisation, polymerisation procedures, tabulated results and representative NMR, SEC & MALDI spectra, 31P NMR (including δ31P predictions by DFT) and 1H DOSY NMR spectra of the catalyst/catalyst mixtures, and DFT calculations (including optimised structures as xyz). See DOI: https://doi.org/10.1039/d3cy01424j |
| ‡ Present address: Uppsala University, Department of Chemistry – Ångström Laboratory, 752 37 Uppsala, Sweden. |
| § The ROAC is also referred to as ring-opening co-polymerisation (ROCOP) and ring-opening alternating polymerisation (ROAP). |
| ¶ Diffusion coefficients were only used qualitatively because of the different nature of species (ionic vs. neutral) and their different ability to form hydrogen bonds, which hampers the quantitative interpretation of diffusion coefficients (cf. ESI,† p. S2 for further information). |
| || The syntheses of bulkier catalysts with ortho-substituted phenyl rings (mono-methyl and di-methoxy) failed because no coupling reaction between azide 18 and the respective phosphine occurred. |
| ** A catalyst with phenyl substituents was tested as well: catalyst 3b is catalytically less active and less chemo-selective than the PMP-substituted catalyst 3a (cf.Table 1, entry 04) |
| This journal is © The Royal Society of Chemistry 2023 |