
Photoelectrochemical C–H activation of methane to methyl radical at room temperature†
Fumiaki
Amano
 *a,
Ayami
Shintani
b,
Tatsuya
Sakakura
c,
Yoshiyuki
Takatsuji
c and
Tetsuya
Haruyama
c
*a,
Ayami
Shintani
b,
Tatsuya
Sakakura
c,
Yoshiyuki
Takatsuji
c and
Tetsuya
Haruyama
c
aDepartment of Applied Chemistry for Environment, Tokyo Metropolitan University, 1-1 Minami-Osawa, Hachioji, Tokyo 192-0397, Japan. E-mail: f.amano@tmu.ac.jp
bDepartment of Chemical and Environmental Engineering, The University of Kitakyushu, 1-1 Hibikino, Wakamatsu-ku, Kitakyushu, Fukuoka 808-0135, Japan
cDepartment of Biological Functions and Engineering, Kyushu Institute of Technology, 2-4 Hibikino, Wakamatsu-ku, Kitakyushu, Fukuoka 808-0196, Japan
First published on 13th June 2023
Abstract
Herein, we report a continuous gas-fed photoelectrochemical (PEC) system with a proton exchange membrane for CH4 activation at ambient temperature and pressure. We found that both water splitting and steam reforming of CH4 were induced over oxide photoanodes. When the CH4 concentration was low, O2 and CO2 were formed on titanium oxide (TiO2) and tungsten trioxide (WO3) photoanodes under ultraviolet light irradiation. We also found that visible light enhanced CH4 activation and ethane (C2H6) formation over the WO3 photoanode. When the CH4 concentration increased, O2 formation was suppressed, with increasing production rates of CO2, C2H6, and CO. Under optimised conditions, the selectivity of C2H6 reached 57% on a carbon basis over the WO3 photoanode under visible-light irradiation. The production of C2H6 implies the formation of methyl radicals during the CH4 gas-fed PEC process. We also demonstrated the PEC coupling of ethane to n-butane and the visible-light-induced oxidation of CH4 without external bias.
Introduction
The catalytic conversion of CH4 into value-added products is challenging because of its high stability and a large energy gap between its highest occupied and lowest unoccupied molecular orbitals.1,2 Moreover, its electron affinity is low (−1.9 eV),3 the ionization potential (12.6 eV) and the C–H bond dissociation energy (439 kJ mol−1) are high,4 and the acidity is very weak (pKα = 56).5 The dipole moment of CH4 is zero because of its symmetric structure. Therefore, high-temperature processes are typically used for catalytic CH4 conversion. However, a low-temperature catalytic process may be promising for achieving high selectivity. Photoelectrochemical (PEC) reactions at room temperature differ from conventional catalytic processes.6–10There are three pathways that convert CH4 into methyl radicals (˙CH3), as shown in Fig. 1: electron transfer (ET), proton transfer (PT), and proton-coupled electron transfer (PCET). Among these reactions, ET (CH4 = ˙CH4+ + e−, ΔrG = 1163 kJ mol−1) and PT (CH4 = CH3− + H+, ΔrG = 270 kJ mol−1) are extremely difficult to achieve. In contrast, PCET is more advantageous than stepwise transfer because the transfer of H+ and e− together can avoid the formation of high-energy chemical intermediates.11–13 The ΔrG of PCET (CH4 = ˙CH3 + H+ + e−) is 198.7 kJ mol−1. Thus, the potential of the ˙CH3/CH4 couple is +2.06 V versus the standard hydrogen electrode (SHE).7
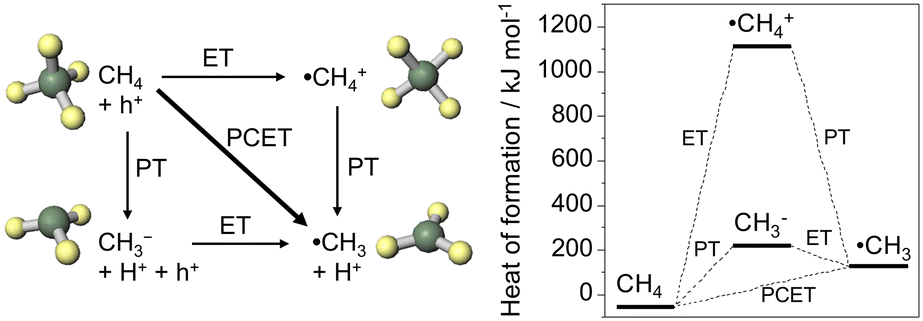 | ||
| Fig. 1 Activation of CH4 to methyl radical through electron transfer (ET), proton transfer (PT), and proton-coupled electron transfer (PCET). The heat of formation was calculated by MOPAC PM3. | ||
The potential to form ˙CH3 is more negative than that for ˙OH/H2O (+2.38 V vs. SHE). These potentials are suitable for titanium oxide (TiO2) photocatalysts. Fig. 2a shows the band diagram of anatase TiO2, in which the valence band maximum (VBM) is located at approximately 3.0 V vs. SHE. Oxide semiconductors without partially filled d levels also exhibit a similar VBM.14 Therefore, many semiconductor photocatalysts can activate CH4via the PCET mechanism. The nonoxidative coupling of methane (NOCM) is a well-known photocatalytic reaction (2CH4 → C2H6 + H2, ΔG298K = 68.6 kJ mol−1).15,16
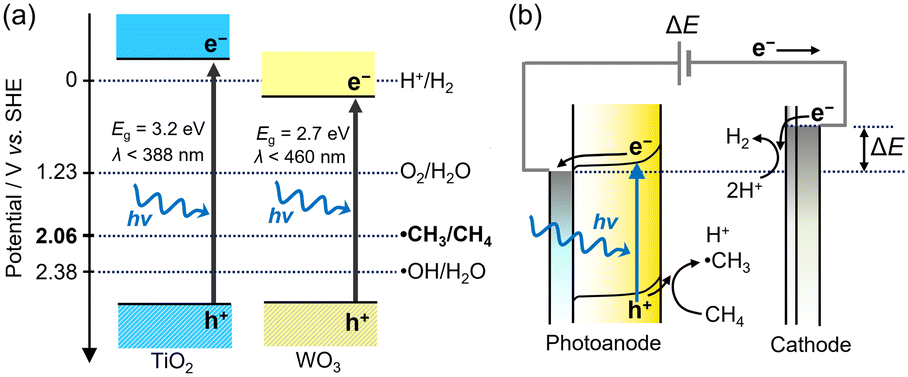 | ||
| Fig. 2 (a) Energy band diagram of TiO2 and WO3. (b) PEC process using photoanode and cathode under applied bias (ΔE). | ||
However, the reported photocatalytic activities for NOCM are quite low.17–24 In addition, TiO2 can only use UV light as the band gap (Eg) of anatase TiO2 is 3.2 eV.25
The PEC process can overcome the following limitations of TiO2 photocatalysis: low activity and lack of visible-light sensitivity (Fig. 2b). The external potential applied to the semiconductor electrodes improves the charge separation of the photoexcited carriers. Moreover, the applied voltage enables the use of visible light-responsive oxide semiconductors with a narrow Eg even though the conduction band minimum is too positive to induce the hydrogen evolution reaction (HER). Tungsten oxide (WO3) photocatalysts are theoretically inactive for NOCM accompanied by HER, but the PEC process enables the HER on the cathode by applying voltages between the two electrodes.
Herein, we investigate the PEC process for CH4 activation using TiO2 and WO3 photoanodes.7–10,26,27 We developed a continuous gas-flow PEC reactor using a proton exchange membrane (PEM) as a solid electrolyte.7,28–30 The all-solid-state PEM–PEC cell is suitable for hydrophobic CH4, which is insoluble in water. The maximum concentration of CH4 is only 0.0016 M in water while that of water is 55.5 M in liquid (Fig. 3).4 However, in the case of humidified CH4 gas, the saturated concentration of H2O(g) is 0.0013 M (3.16 kPa) at 25 °C, but the concentration of CH4 is 0.041 M (101 kPa). We studied the probability of a gas-fed PEC process for CH4 activation under both UV and visible-light irradiation.
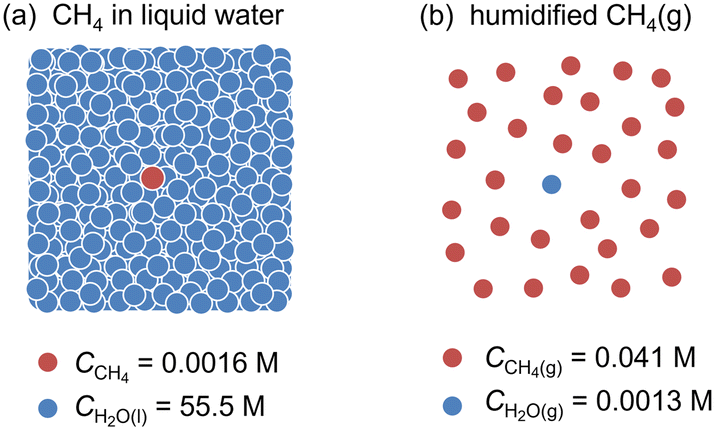 | ||
| Fig. 3 (a) Molar concentrations of (a) dissolved CH4 and H2O in liquid water, and (b) gaseous CH4 and water vapor in humidified condition at 25 °C. | ||
Results and discussion
Photoanode materials and light wavelengths
CH4 activation through PEC was tested using the PEM–PEC cell (Fig. S1 in ESI†) in a two-electrode system at 25 °C under atmospheric pressure. Fig. 4 shows the time course of the PEC process using the TiO2 and WO3 photoanodes under a continuous flow of 10 vol% CH4 and 3 vol% H2O(g) balanced with Ar. The cathode, which was separated from the photoanode using PEM, was a Pt/carbon electrocatalyst maintained under a humidified Ar flow. Humidification of the fed gases enhanced the proton conductivity of the PEM. The photoanode was irradiated with UV (365 nm) or blue light (453 nm). Before photoirradiation, the PEC system was kept in the dark to establish an adsorption–desorption equilibrium. Under irradiation, a good photocurrent response was observed for each condition when 1.2 V was applied between the photoanode and cathode catalysts (Fig. 4a). The incident photon-to-current conversion efficiencies (IPCE) at steady state were 19.8% and 11.3% for the WO3 and the TiO2 photoanodes, respectively, at 365 nm. The IPCE at 453 nm was 8.4% for the WO3 photoanode. This high quantum efficiency suggests efficient charge separation in the space-charge layer formed by the applied potential.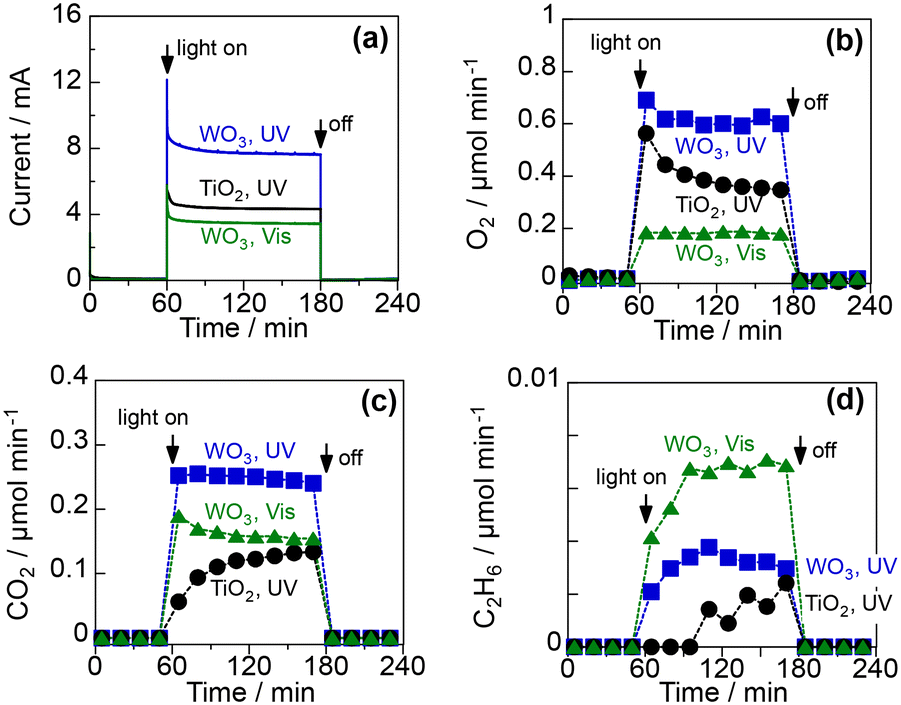 | ||
| Fig. 4 PEC activation of 10 vol% CH4 using WO3 and TiO2 photoanodes under UV light (6.5 mW cm−2 at 365 nm) and visible light (6.8 mW cm−2 at 453 nm): (a) the overall current at 1.2 V, (b) the rate of O2 evolution, (c) the rate of CO2 formation, and (d) the rate of C2H6 formation on the photoanode side. | ||
O2, CO2, and a small amount of C2H6 were obtained as products on the photoanodes (Fig. 4b–d). Carbon monoxide could not be analysed under these conditions because of its interference with the Ar diluent. The production of O2 and CO2 suggests that both water vapour and CH4 were oxidised on the photoanodes. The Faraday efficiencies (FE) of O2 and CO2 were approximately 50% and 40%, respectively, under UV irradiation for both photoanodes (Table S1 in ESI†). The FE of O2 decreased to 34% and that of CO2 increased to 59% under visible-light irradiation of the WO3 photoanode. The production rate of C2H6 also increased when 453 nm visible light was used instead of 365 nm UV light. These results suggest that CH4 oxidation to CO2 and C2H6 is more plausible than water oxidation under visible-light irradiation. The production of C2H6 implies that ˙CH3 is generated by the PEC process; this is because the homocoupling of ˙CH3 is involved in the formation mechanism of C2H6 in photo-Kolbe electrolysis.31–33
Methane concentration and light intensity
Fig. 5 shows the effect of the concentration of CH4 fed into the WO3 photoanode under visible-light irradiation. The O2 evolution rate decreased significantly when the CH4 concentration increased to 50 vol%. The IPCE was 10.5%, and the FEs for O2, CO2, and C2H6 were 5.1%, 81.7%, and 4.9%, respectively (Table S2 in the ESI†). For the cathode catalyst, stoichiometric H2 evolution (FE of ∼100%) was observed, indicating that the steam reforming of methane (CH4 + 2H2O(g) → CO2 + 4H2) was mainly promoted in this PEM–PEC reactor. To the best of our knowledge, this is the first report of the gas-fed PEC steam reforming of methane, which is an uphill reaction (ΔG298K = 114 kJ mol−1).15,16 Notably, the purity of the evolved H2 can be sustained by the membrane separation from CH4 and the oxidised products. The H2 production rate, and thus the photocurrent, did not depend on the CH4 concentration, suggesting that the charge-separation efficiency was determined by the applied potential rather than the surface reactions. | ||
| Fig. 5 Effect of CH4 concentration on the PEC reactions over the WO3 photoanode: (a) the overall current at 1.2 V, (b) the rate of H2 evolution on the cathode side, and (c) the rate of products formation on the photoanode side under visible-light irradiation (6.8 mW cm−2 at 453 nm). | ||
Fig. 6 shows the PEC properties of the WO3 photoanode under the flow of 97 vol% CH4 and 3 vol% H2O vapour.7 When the light intensity is 3.6 mW cm−2, the FEs of O2, CO2, CO, and C2H6 were 1.5%, 72.3%, 8.3%, and 14.0%, respectively. The sum of the FE values was 96%, suggesting that unidentified products were limited, and no methanol was formed. The FE of C2H6 was not very high because only two electrons were donated from CH4 to produce a C2H6 molecule and eight electrons produced a CO2 molecule. From the viewpoint of selectivity, the production rate of C2H6 was comparable to that of CO2 at high CH4 concentrations and low light intensities. The C2H6 selectivity on a carbon basis reached 57.4%, whereas the selectivities for CO2 and CO were 37.0% and 5.6%, respectively (Table S3 in the ESI†). This indicates that more than half of the PEC process can be explained by the dehydrogenative coupling of methane, similar to photocatalytic NOCM.7
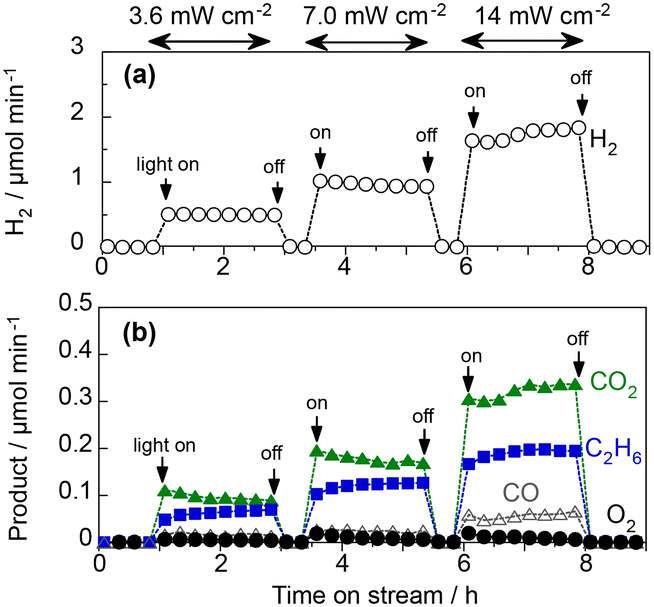 | ||
| Fig. 6 Effect of light intensity on the activation of humidified CH4 using the WO3 photoanode under 453 nm irradiation at 1.2 V; (a) the H2 evolution rate on the cathode side and (b) the rates of product formation on the photoanode side. | ||
The high C2H6 selectivity implies the efficient formation of ˙CH3 by the photogenerated holes of WO3. When the concentration of the generated ˙CH3 is high, homocoupling should easily occur to produce C2H6. In contrast, other side reactions of ˙CH3 are promoted, thereby decreasing C2H6 selectivity at low CH4 concentrations.
The production rates of CO2, CO, and C2H6 increased with the incident light intensity (Fig. 6). This indicated that the products were formed via the photoexcitation mechanism. The C2H6 selectivity gradually decreased from 57.4% to 49.1% when the irradiance intensity was changed from 3.6 to 14 mW cm−2, implying that overoxidation is promoted when the concentration of holes is high at the semiconductor surface. Although the photocurrent fluctuated over time, which may have been affected by humidity, the WO3 photoanode repeatedly exhibited sufficient stability for several hours. We also did not confirm the degradation of the crystallinity of WO3 or the structure of the ionomer coated on the photoanode surface, as shown in X-ray diffraction patterns (Fig. S2†) and Fourier transform infrared spectra (Fig. S3†).
PEC reaction mechanism
To further investigate the radical mechanism of the PEC process, we tested the PEC oxidation of C2H6.34Fig. 7 shows the time course of the C2H6 activation on the WO3 photoanode. We detected n-butane as the product. The formation of n-butane suggests the occurrence of the homocoupling of ethyl radicals (˙C2H5 + ˙C2H5 → CH3–CH2–CH2–CH3). We also detected the formation of ethylene, which could have been generated through intermolecular dehydrogenation (C2H6 + 2 h+ → CH2 = CH2 + 2H+). The FE of n-butane was 4.4%, and its selectivity was 39.7% (C-basis), as shown in Table S4 in the ESI.†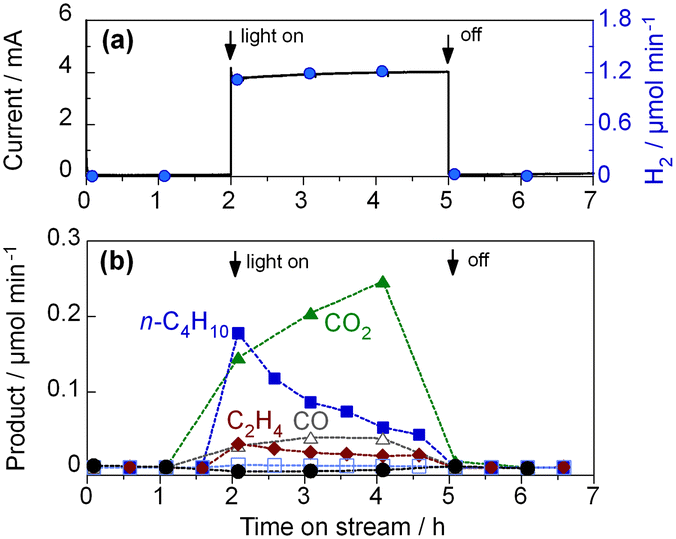 | ||
| Fig. 7 PEC activation of C2H6 using WO3 photoanode under visible-light irradiation (6.8 mW cm−2 at 453 nm) at 1.2 V; (a) the overall current and H2 evolution rate on the cathode side, and (b) the rates of product formation on the photoanode side. | ||
To investigate the radical intermediates involved in the activation of CH4, we conducted electron paramagnetic resonance (EPR) experiments using 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as a spin-trapping agent.35 Photoirradiation was performed for WO3 powder dispersed in a 50 mM AgNO3 aqueous solution with CH4 gas. The silver cation acted as an electron acceptor for the photoexcited WO3.36 We observed four-line EPR signals (g = 2.0056, AN = 1.49 mT, AH = 1.49 mT), which were consistent with the ˙DMPO–OH spin adduct, in the WO3 suspension after irradiation (Fig. 8). This implied that hydroxyl radical (˙OH) could form on the WO3 photoanode in the presence of water vapour. Therefore, ˙OH would be the active species for CH4 activation in the PEM–PEC system. In contrast, we could not detect a signal corresponding to the ˙DMPO–CH3 spin adduct, even in the presence of acetic acid and dimethyl sulfoxide. This suggests that spin trapping of ˙CH3 with DMPO is difficult in liquid water under our experimental conditions.
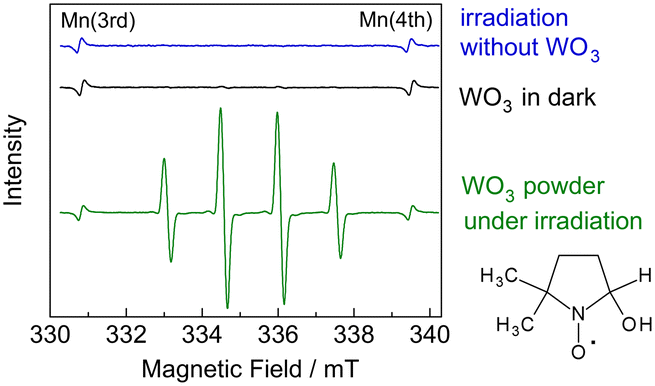 | ||
| Fig. 8 EPR spectra of an aqueous solution of DMPO and AgNO3 after 405 nm irradiation for 5 min without WO3 powder, aging in the dark with WO3 powder, and 405 nm irradiation for 5 min with WO3 powder. The standard Mn2+ marker shows signals at g = 2.0337 (third line) and g = 1.9803 (fourth line). | ||
We also attempted a photo-Kolbe reaction using WO3 powder, 5 vol% acetic acid, and 50 mM AgNO3.33,36 The gaseous products obtained were CO2, CH4, and O2 (Fig. S4 in ESI†). A trace amount of methanol was also formed in the aqueous solution. However, C2H6 was not formed in the aqueous system, indicating the importance of the vapour-fed conditions in C2H6 production.
The advantage of the gas-fed type reactor was also revealed by the gas flow rate dependence of C2H6 formation (Fig. S5 in ESI†). The production rate of C2H6 was very low at a gas flow rate of 1 mL min−1. We realised that gas diffusion plays an important role in C2H6 production in the PEM–PEC system because productivity increased at higher flow rates. The proposed reaction mechanism for the catalytic oxidative coupling of methane involves the coupling of ˙CH3 in the gas phase to form C2H6.2,37 Similar to this mechanism, continuous flow facilitates the desorption of ˙CH3 from the surface of the WO3 photoanode and the formation of C2H6.
Zero-bias PEC oxidation of methane
The photocatalytic oxidation of atmospheric CH4 to CO2 is another attractive reaction because the greenhouse gas effect of CH4 is over 30 times greater than that of CO2. The complete oxidation of methane is an exergonic reaction (CH4 + 2O2 → CO2 + 2H2O(g), ΔG298K = −801 kJ mol−1),15,16 but its high activation energy prevents the catalytic reaction at room temperature.Fig. 9 shows the PEC oxidation of CH4 in the PEM–PEC reactor using the WO3 photoanode under blue-light irradiation at 25 °C. CO2 formation by CH4 oxidation was confirmed without an external bias voltage. During the exergonic reaction, the oxygen reduction reaction (O2 + 4H+ + 4e− = H2O, 1.23 V vs. SHE) was promoted over the Pt/carbon catalyst in humidified air (Fig. 2a). The IPCE at zero bias was 2.5% at 453 nm, which was much higher than the quantum efficiency previously reported for photocatalytic systems.38,39
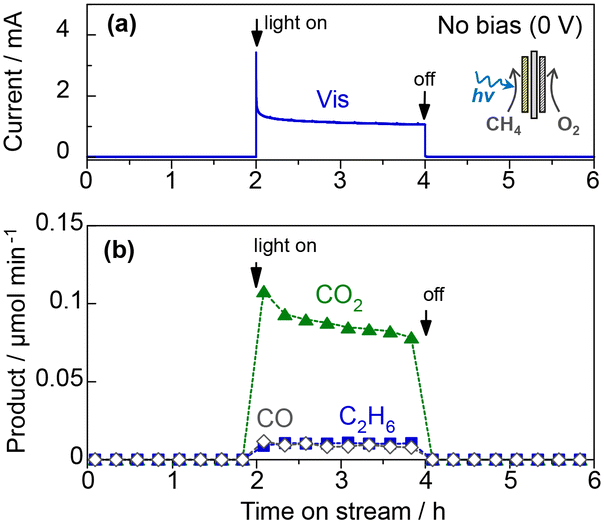 | ||
| Fig. 9 PEC oxidation of CH4 by the PEM–PEC reactor using the WO3 photoanode and Pt/carbon catalyst: (a) the overall current at zero bias, and (b) the rate of products formation on the photoanode side under 453 nm light irradiation (6.8 mW cm−2). | ||
Experimental
Preparation of photoanodes
Ti felt was used as the conductive substrate for the gas-diffusion photoanodes. The WO3 electrode was prepared by dip coating with an aqueous solution of (NH4)6H2W12O40 and polyethylene glycol; the electrode was then calcined at 923 K for 2 h.7,28,40 The TiO2 electrode was prepared by the anodization of Ti felt in ethylene glycol with 0.25 wt% NH4F and 10 vol% H2O at 50 V for 3 h.29,41,42 The anodized Ti felt was calcined at 823 K for 1 h to crystallize into anatase TiO2. The photoanodes were modified using a Nafion ionomer dispersion (Sigma-Aldrich).PEM–PEC reaction
The PEC measurements were performed at 25 °C and 1 bar using an AMETEK VersaSTAT3 workstation. For the proposed all-solid-state cells, a Nafion N117 film (DuPont) was sandwiched between the photoanode and the cathode, which was composed of Pt/carbon (Tanaka Kikinzoku Kogyo) and Toray carbon paper (Fuel Cell Store). The photoanode side was supplied with 20 mL min−1 of humidified CH4/Ar or humidified CH4 gas. The relative humidity was approximately 90% without liquid condensation. The cathode side was supplied with 20 mL min−1 of humidified Ar gas. The geometric surface areas of the two electrodes were 25 cm2, whereas the irradiation area of the photoanode was 16 cm2. Photoirradiation was performed through a glass window using light-emitting diodes (LED). The peak wavelengths were 365 and 453 nm for the UV (Nitride Semiconductor) and blue LED (OptoSupply), respectively. The IPCE was calculated as follows:Here, jphoto is the photocurrent density, λ is the wavelength (nm), and I0 is the intensity of incident light.
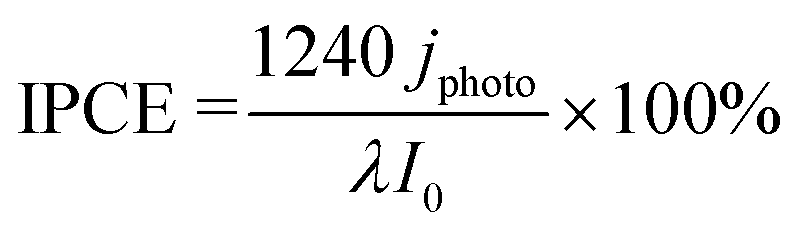
Product analysis
The gas products of the PEC reaction were analysed using gas chromatography (Shimadzu GC-8A and GC-2014). A thermal conductivity detector (TCD) equipped with a molecular sieve 5A column in an Ar carrier was used to quantify H2 and O2. A TCD with a Shincarbon ST column in an He carrier was used to quantify CO and CO2. A flame ionisation detector with a GS-CarbonPLOT was used to detect alkane species.The C2H6 selectivity in carbon basis was calculated using the below equation:
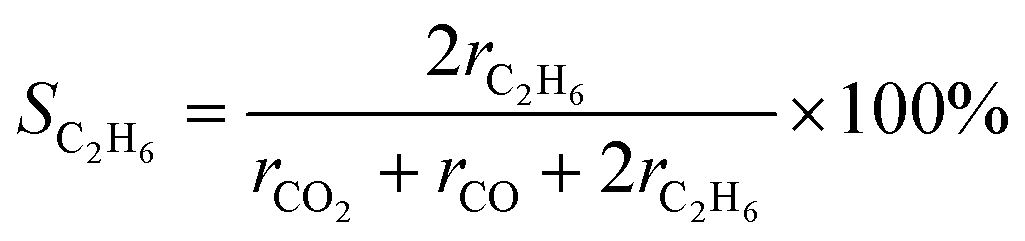
Here, ni is the number of electrons involved, and F is the Faradaic constant. The ni values for C2H6, O2, CO, and CO2 are 2, 4, 6, and 8, respectively.
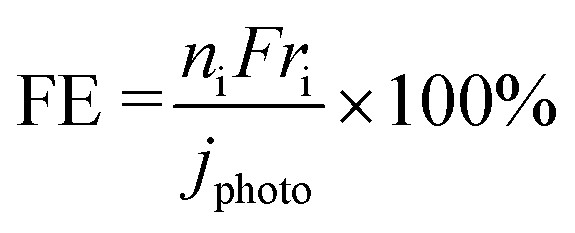
Characterization
X-ray diffraction patterns were recorded by a Rigaku SmartLab diffractometer using Cu Kα radiation. Fourier transform infrared spectroscopy was performed on a Shimadzu IR Affinity-1 spectrometer in the attenuated total reflection mode. Electron paramagnetic resonance (EPR) spectra were recorded using a JES-X310 spectrometer (JEOL, Japan) at room temperature. The sample suspension was taken out by a quartz capillary tube with the two ends sealed by sealing compound for the EPR measurement.Conclusions
We studied the PEC activation of CH4 over TiO2 and WO3 photoanodes in a gas-flow PEM–PEC system. We demonstrated that a WO3 photoanode excited by visible light converts CH4 into CO2 and C2H6. At high CH4 concentrations, the C2H6 selectivity was above 50% on a carbon basis. Moreover, we found that WO3 and visible light were more suitable than TiO2 and UV light to form C2H6. When C2H6 was used as the reactant, n-butane was formed, suggesting a radical coupling mechanism. The WO3 photoanode showed an IPCE of 7.6% at 453 nm with an applied voltage of 1.2 V. The current efficiency of H2 was nearly 100% in the cathode compartment, demonstrating that the PEM–PEC system is useful for steam reforming of methane and dehydrogenative methane coupling. Surprisingly, visible-light induced CH4 oxidation was also efficiently promoted in the PEM–PEC system using humidified air, even under zero bias.Author contributions
Fumiaki Amano: conceptualisation, methodology, validation, visualisation, writing – review & editing, and supervision. Ayami Shintani: investigation visualisation, and writing – original draft. Tatsuya Sakakura and Yoshiyuki Takatsuji: investigation. Tetsuya Haruyama: validation and resources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Japan Science and Technology Agency (JST) PRESTO [grant number JPMJPR18T1] and the Japan Society for the Promotion of Science (JSPS) KAKENHI [Grant No. JP20H02525].Notes and references
- K. Yoshizawa, Acc. Chem. Res., 2006, 39, 375–382 CrossRef CAS PubMed
.
- P. Schwach, X. Pan and X. Bao, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS PubMed
- C.-G. Zhan, J. A. Nichols and D. A. Dixon, J. Phys. Chem. A, 2003, 107, 4184–4195 CrossRef CAS
-
J. R. Rumble, CRC handbook of chemistry and physics, CRC Press, Boca Raton, 100th edn, 2019 Search PubMed
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS
- J. Baltrusaitis, I. Jansen and J. D. Schuttlefield Christus, Catal. Sci. Technol., 2014, 4, 2397–2411 RSC
- F. Amano, A. Shintani, K. Tsurui, H. Mukohara, T. Ohno and S. Takenaka, ACS Energy Lett., 2019, 4, 502–507 CrossRef CAS
- J. Ma, K. K. Mao, J. X. Low, Z. H. Wang, D. W. Xi, W. Q. Zhang, H. X. Ju, Z. M. Qi, R. Long, X. J. Wu, L. Song and Y. J. Xiong, Angew. Chem., Int. Ed., 2021, 60, 9357–9361 CrossRef CAS PubMed
- A. Mehmood, S. Y. Chae and E. D. Park, Catalysts, 2021, 11, 1387 CrossRef CAS
- H. Tateno, S. Iguchi, Y. Miseki and K. Sayama, Angew. Chem., Int. Ed., 2018, 57, 11238–11241 CrossRef CAS PubMed
- J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS PubMed
- J. N. Schrauben, R. Hayoun, C. N. Valdez, M. Braten, L. Fridley and J. M. Mayer, Science, 2012, 336, 1298–1301 CrossRef CAS PubMed
- H. Schwarz, S. Shaik and J. L. Li, J. Am. Chem. Soc., 2017, 139, 17201–17212 CrossRef CAS PubMed
- D. E. Scaife, J. Sol. Energy, 1980, 25, 41–54 CrossRef CAS
- H. Yokokawa, S. Yamauchi and T. Matsumoto, Calphad, 1999, 23, 357–364 CrossRef CAS
- D. D. Wagman, W. H. Evans, V. B. Parker, R. H. Schumm, I. Halow, S. M. Bailey, K. L. Churney and R. L. Nuttall, J. Phys. Chem. Ref. Data, 1982, 11, 2 Search PubMed
- L. Yuliati, T. Hattori, H. Itoh and H. Yoshida, J. Catal., 2008, 257, 396–402 CrossRef CAS
- K. Shimura and H. Yoshida, Catal. Surv. Asia, 2014, 18, 24–33 CrossRef CAS
- L. Yu and D. Li, Catal. Sci. Technol., 2017, 7, 635–640 RSC
- S. Q. Wu, X. J. Tan, J. Y. Lei, H. J. Chen, L. Z. Wang and J. L. Zhang, J. Am. Chem. Soc., 2019, 141, 6592–6600 CrossRef CAS PubMed
- S. Q. Wu, L. Z. Wang and J. L. Zhang, J. Photochem. Photobiol., C, 2021, 46, 100400 CrossRef CAS
- S. P. Singh, A. Anzai, S. Kawaharasaki, A. Yamamoto and H. Yoshida, Catal. Today, 2021, 375, 264–272 CrossRef CAS
- S. P. Singh, A. Yamamoto, E. Fudo, A. Tanaka, H. Kominami and H. Yoshida, ACS Catal., 2021, 11, 13768–13781 CrossRef CAS
- L. Li, G.-D. Li, C. Yan, X.-Y. Mu, X.-L. Pan, X.-X. Zou, K.-X. Wang and J.-S. Chen, Angew. Chem., Int. Ed., 2011, 50, 8299–8303 CrossRef CAS PubMed
- W. Q. Zhang, C. F. Fu, J. X. Low, D. L. Duan, J. Ma, W. B. Jiang, Y. H. Chen, H. J. Liu, Z. M. Qi, R. Long, Y. F. Yao, X. B. Li, H. Zhang, Z. Liu, J. L. Yang, Z. G. Zou and Y. J. Xiong, Nat. Commun., 2022, 13, 2806 CrossRef CAS PubMed
- W. Li, D. He, G. Hu, X. Li, G. Banerjee, J. Li, S. H. Lee, Q. Dong, T. Gao, G. W. Brudvig, M. M. Waegele, D.-E. Jiang and D. Wang, ACS Cent. Sci., 2018, 4, 631–637 CrossRef CAS PubMed
- S. J. Xie, Z. B. Shen, J. Deng, P. Guo, Q. H. Zhang, H. K. Zhang, C. Ma, Z. Jiang, J. Cheng, D. H. Deng and Y. Wang, Nat. Commun., 2018, 9, 1181 CrossRef PubMed
- F. Amano, A. Shintani, H. Mukohara, Y. M. Hwang and K. Tsurui, Front. Chem., 2018, 6, 598 CrossRef CAS PubMed
- F. Amano, H. Mukohara, A. Shintani and K. Tsurui, ChemSusChem, 2019, 12, 1925–1930 CrossRef CAS PubMed
- F. Amano, H. Mukohara, H. Sato, C. Tateishi, H. Sato and T. Sugimoto, Sustainable Energy Fuels, 2020, 4, 1443–1453 RSC
- B. Kraeutler and A. J. Bard, J. Am. Chem. Soc., 1977, 99, 7729–7731 CrossRef CAS
- B. Kraeutler, C. D. Jaeger and A. J. Bard, J. Am. Chem. Soc., 1978, 100, 4903–4905 CrossRef CAS
- S. Sato, J. Phys. Chem., 1983, 87, 3531–3537 CrossRef CAS
- S. P. Singh, A. Yamamoto and H. Yoshida, Catal. Sci. Technol., 2022, 12, 1551–1561 RSC
- M. H. Ab Rahim, M. M. Forde, R. L. Jenkins, C. Hammond, Q. He, N. Dimitratos, J. A. Lopez-Sanchez, A. F. Carley, S. H. Taylor, D. J. Willock, D. M. Murphy, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2013, 52, 1280–1284 CrossRef CAS PubMed
- T. Sakata, T. Kawai and K. Hashimoto, J. Phys. Chem., 1984, 88, 2344–2350 CrossRef CAS
- B. L. Farrell, V. O. Igenegbai and S. Linic, ACS Catal., 2016, 6, 4340–4346 CrossRef CAS
- J. Zhang, Y. Wang, Y. Wang, Y. Bai, X. Feng, J. Zhu, X. Lu, L. Mu, T. Ming, R. de Richter and W. Li, Chem. – Eur. J., 2022, 28, e202201984 CAS
- X. Chen, Y. Li, X. Pan, D. Cortie, X. Huang and Z. Yi, Nat. Commun., 2016, 7, 12273 CrossRef CAS PubMed
- F. Amano, A. Shintani, K. Tsurui and Y.-M. Hwang, Mater. Lett., 2017, 199, 68–71 CrossRef CAS
- M. V. Makarova, F. Amano, S. Nomura, C. Tateishi, T. Fukuma, Y. Takahashi and Y. E. Korchev, ACS Catal., 2022, 12, 1201–1208 CrossRef CAS
- F. Amano, S. Nomura, C. Tateishi and S. Nakayama, J. Electrochem. Soc., 2023, 170, 026501 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy00632h |
| This journal is © The Royal Society of Chemistry 2023 |