
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Operando studies of Mn oxide based electrocatalysts for the oxygen evolution reaction
Andreas
Erbe
*a,
Marc Frederic
Tesch
 *b,
Olaf
Rüdiger
b,
Bernhard
Kaiser
c,
Serena
DeBeer
b and
Martin
Rabe
*d
*b,
Olaf
Rüdiger
b,
Bernhard
Kaiser
c,
Serena
DeBeer
b and
Martin
Rabe
*d
aDepartment of Materials Science and Engineering, NTNU, Norwegian University of Science and Technology, 7491 Trondheim, Norway
bMax Planck Institute for Chemical Energy Conversion, Stiftstr. 34-36, 45470 Mülheim an der Ruhr, Germany. E-mail: marc.tesch@cec.mpg.de
cSurface Science Laboratory, Department of Materials- and Earth Sciences, Technical University Darmstadt, Otto-Berndt-Str. 3, 64287 Darmstadt, Germany
dMax-Planck-Institut für Eisenforschung GmbH, Max-Planck-Str. 1, 40237 Düsseldorf, Germany. E-mail: m.rabe@mpie.de
First published on 7th August 2023
Abstract
Inspired by photosystem II (PS II), Mn oxide based electrocatalysts have been repeatedly investigated as catalysts for the electrochemical oxygen evolution reaction (OER), the anodic reaction in water electrolysis. However, a comparison of the conditions in biological OER catalysed by the water splitting complex CaMn4Ox with the requirements for an electrocatalyst for industrially relevant applications reveals fundamental differences. Thus, a systematic development of artificial Mn-based OER catalysts requires both a fundamental understanding of the catalytic mechanisms as well as an evaluation of the practicality of the system for industrial scale applications. Experimentally, both aspects can be approached using in situ and operando methods including spectroscopy. This paper highlights some of the major challenges common to different operando investigation methods and recent insights gained with them. To this end, vibrational spectroscopy, especially Raman spectroscopy, absorption techniques in the bandgap region and operando X-ray spectroelectrochemistry (SEC), both in the hard and soft X-ray regime are particularly focused on here. Technical challenges specific to each method are discussed first, followed by challenges that are specific to Mn oxide based systems. Finally, recent in situ and operando studies are reviewed. This analysis shows that despite the technical and Mn specific challenges, three specific key features are common to most of the studied systems with significant OER activity: structural disorder, Mn oxidation states between III and IV, and the appearance of layered birnessite phases in the active regime.
1. Introduction
The electrochemical oxygen evolution reaction (OER),| 2H2O → O2 + 4H+ + 4e− | (1) |
| 4OH− → O2 + 2H2O + 4e−, | (2) |
Biological oxygen evolution occurring in photosystem (PS) II is catalysed in an active center containing a CaMn4Ox cluster as the oxygen evolving complex (OEC).3–5 Computational and spectroscopic investigations have contributed significantly to the understanding of the reaction mechanisms leading to the production of O2 during photosynthesis.6–10 Researchers have been tempted to ask whether simple synthetic Mn-based systems can take over the function of the PS II active complex in an electrochemical system, leading to a lot of interest in Mn-based OER electrocatalysts. These could act as alternatives to highly performing, but also rare and expensive systems based, e.g., on Ir or Ru.11
For biological oxygen evolution, manganese was considered the “natural” choice for a catalyst, due to the combination of several properties such as its availability as soluble MnII, its versatile redox chemistry with reachable oxidation states and its aqueous coordination chemistry.5 For industrial applications, its high abundance on Earth makes it attractive, as Mn is available at a low price and with relatively low criticality.12–14 However, a fundamental difference between the requirements for OER catalysts in a biological system and in scenarios for the large-scale production of green hydrogen are the vastly varying current requirements. Biological O2 production in the respiratory chain involves several transport steps. Thus, transport limitation will at some point limit the currents. At these rather low currents, a catalytic cycle has evolved that is made possible only by the high electronic and structural control of PSII as a supramolecular system.6,15 Several technological approaches based on water splitting have been or are currently being developed, with very different requirements for the working conditions in which the required OER catalysts must persist.16–19 For instance, moderate currents in the range of 10–20 mA cm−2 would be appropriate for solar water splitting applications, which however in most cases disallow extreme pH values and require specific optical or morphological properties.17,20 On the other side, industrial zero gap alkaline water electrolyzers can operate at currents up to 1 A cm−2, in a 20–30 wt% KOH electrolyte and at temperatures of up to 100 °C,18,19 which differ significantly from the conditions in biological OERs. Yet, any economically useful catalyst should provide lifetimes in the range of years. Other aspects that require consideration for industrial applications are the costs and scalability of the catalyst preparation and electrode fabrication as well as robustness with respect to varying electrolyte compositions. Nevertheless, the high demand for affordable green energy storage technologies has triggered new research efforts towards Mn-based OER electrocatalysts.11
The mechanism of the dioxygen formation via heterogenous catalysis has been studied for decades (for a review see ref. 2). General schemes for metal oxide catalysts start from adsorbed OH at active surface (metal) sites, followed by single electron charge transfer steps to form M–O (terminal-oxo) species. The O–O bond formation may occur from adjacent M–O or M–OH sites or from a single site via peroxo intermediates.2 However, the nature of the intermediates and the intermediate steps remain controversial and likely depend on the exact conditions and the catalyst. Beside such atomic understanding, a knowledge driven catalyst development requires a detailed understanding of the processes occurring at application-relevant electrodes including the support and an evaluation of the practicality of the system in applications. Experimentally, these insights are obtained by characterisation of the catalyst using ‘in situ’ (i.e. in electrolyte “at the location” of the electrochemical process) and “operando” (i.e. “during the [electrochemical] process” itself) experiments.
The goal of electrochemical in situ and operando studies is the understanding of the chemical transformations taking place on the electrode surface (substrate and catalyst) as a function of potential or current. In spectroelectrochemical in situ experiments, the spectroscopic signature of a system is directly measured in a controlled environment, e.g., at an electrode in the electrolyte under potential control. Operando spectrolectrochemistry is a sub-type of the in situ experiment, which – for electrocatalytic applications – requires that a catalytic activity/selectivity is measured simultaneously to the spectroscopy.21,22 In OER electrocatalysis, the current density provided by the potentiostat is often used to monitor the catalytic activity. This article will discuss the potential of such operando studies to foster the understanding of Mn catalysed OERs. To that end, first, general and Mn specific challenges for such operando spectroelectrochemical methods are discussed, followed by a short review of selected recent in situ and operando studies.
2. Mn oxide-based systems pose special analytical challenges
2.1 General challenges for operando methods
Computational models can help to overcome the lack of reference data and also help to solve the inverse problem. However, for electrocatalytic systems, approaches to correctly describe the electrode potentials in computations with a relevant system size in interfacial systems are just emerging.23,24 The fact that Mn-based systems likely require an appropriate description of the correct spin state make calculations computationally expensive. Thus, such systems are not necessarily the best early adoption systems for novel calculation techniques.
(1) the metal (oxide) based active center, which can be inhomogeneous,
(2) possible coexistence of inactive phases,
(3) the support,
(4) the intermediates and precursors for O2 formation,
(5) the O2 gas bubbles which form, grow and detach from the electrode,
(6) the solvent, here water, which is also a reactant,
(7) the ions in solution,
(8) possible dopants in the solid, and
(9) impurities.
All of them are involved in the reaction to some extent, and several processes happen in parallel, but on different length and time scales.
Electron transfer itself occurs on the sub ps time scale. Solvent reorganisation and relaxation via vibrational modes occurs typically on a time scale of several hundred picoseconds. In electrochemical systems, charging and discharging of the double layer capacitance makes the intrinsic time scale externally accessible via electrochemical experiments, typically μs and above. Time scales for transport processes to and from the electrode depend on convection. Bubble detachment occurs on the s time scale, and affects convection. Catalyst or support degradation is typically the slowest process for an industrially relevant catalyst, where it happens on the time scale of years. A well designed operando experiment must try to maximize the impact of the process of interest on the measured signal, while diminishing the influence of the other processes. Unfortunately, it is not always obvious which process is the easiest to capture in a particular experiment. Particularly challenging are usually the slowest and the fastest processes. Depending on the turnover frequencies, the lifetime and the rate limiting step, the active center of the catalyst may be ‘invisible’ due to the relatively long time frame necessary for in situ data collection.25 Thus the dominating observed state is not necessarily the active state.
2.2 Specific challenges of Mn oxides
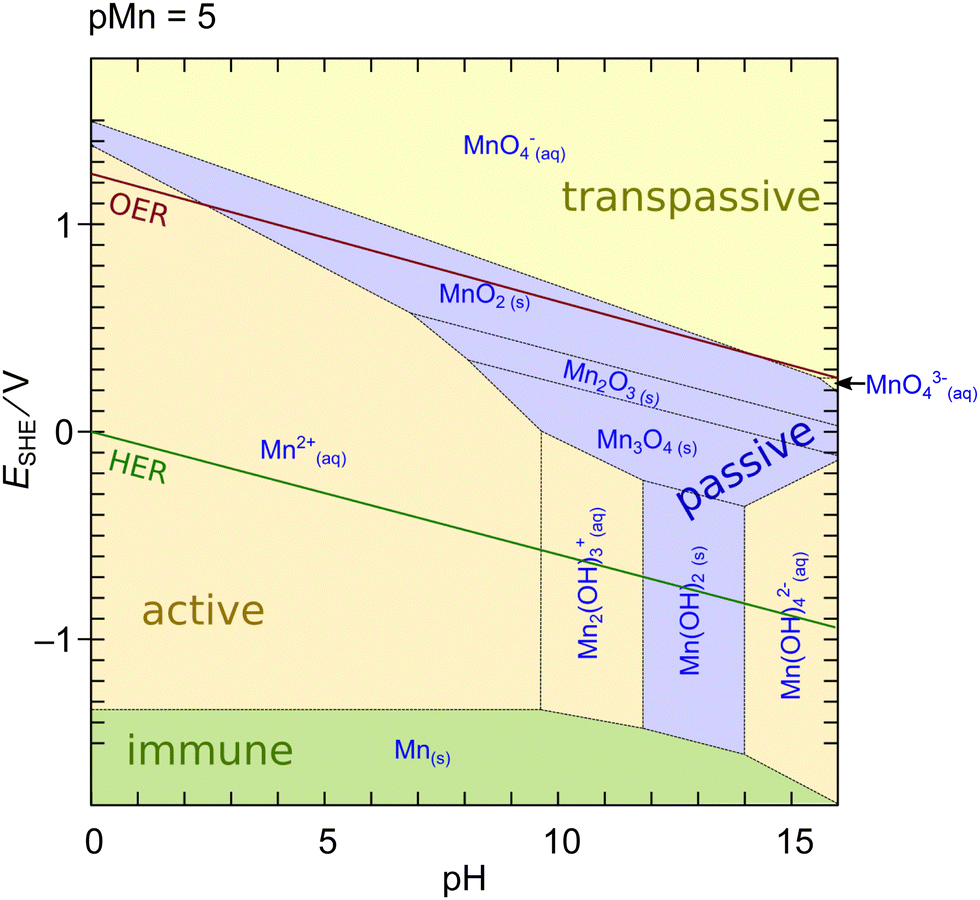 | ||
| Fig. 1 Potential-pH (Pourbaix) diagram of Mn calculated using MEDUSA27 at an activity of dissolved species of 10−5. Shading color of the areas refers to the water solubility of the respective phase: green – insoluble metal; blue – insoluble oxide/hydroxide, yellow – soluble ion. Red and green lines indicate oxidation/reduction potentials of water to molecular oxygen and hydrogen, respectively. | ||
An interesting feature of MnOx is that a region of limited stability exists at lower pH above the OER potential. This region makes MnOx particularly interesting for applications as an OER catalyst in photoelectrochemical cell devices (aka ‘artificial leaves’) at low currents in acidic to neutral pH.17,29,30 However, the dissolution of Mn oxides at elevated pH and currents is a major challenge for the applicability of such systems in industrial alkaline water electrolyzers or PEM electrolysis cells.11,31–33 The dissolution issue on the other hand implies that experimentally measured currents at elevated potentials often do not exclusively originate from oxygen evolution. In contrast, a purely thermodynamical reasoning is insufficient because the actual rate of dissolution is defined by the kinetics of the reactions which can be sluggish.26,33 Furthermore, the diagram in Fig. 1 may change in the presence of further ions or compounds which can significantly change the thermodynamics or kinetics of the dissolution.
| Mineral (compound) | Ideal chemical formula | Tunnel/layer structure |
|---|---|---|
| Hausmannite | MnIIMnIII2O4 | Spinel-like |
| Hollandite group (α-MnO2) | R0.8–1.5(MnIV, MnIII)8O16 R = Ba, Pb, K or Na | 2 × 2 tunnel |
| Pyrolusite (β-MnO2) | MnIVO2 | 1 × 1 tunnel |
| Romanechite | Ba(MnIV, MnIII)5O10·2H2O | 2 × 3 tunnel |
| Birnessite group (δ-MnO2) | (Na, Ca, K)(MnIV, MnIII)2O4·1.5H2O | Layered |
| Bixbyite | (MnIII)2O3 |
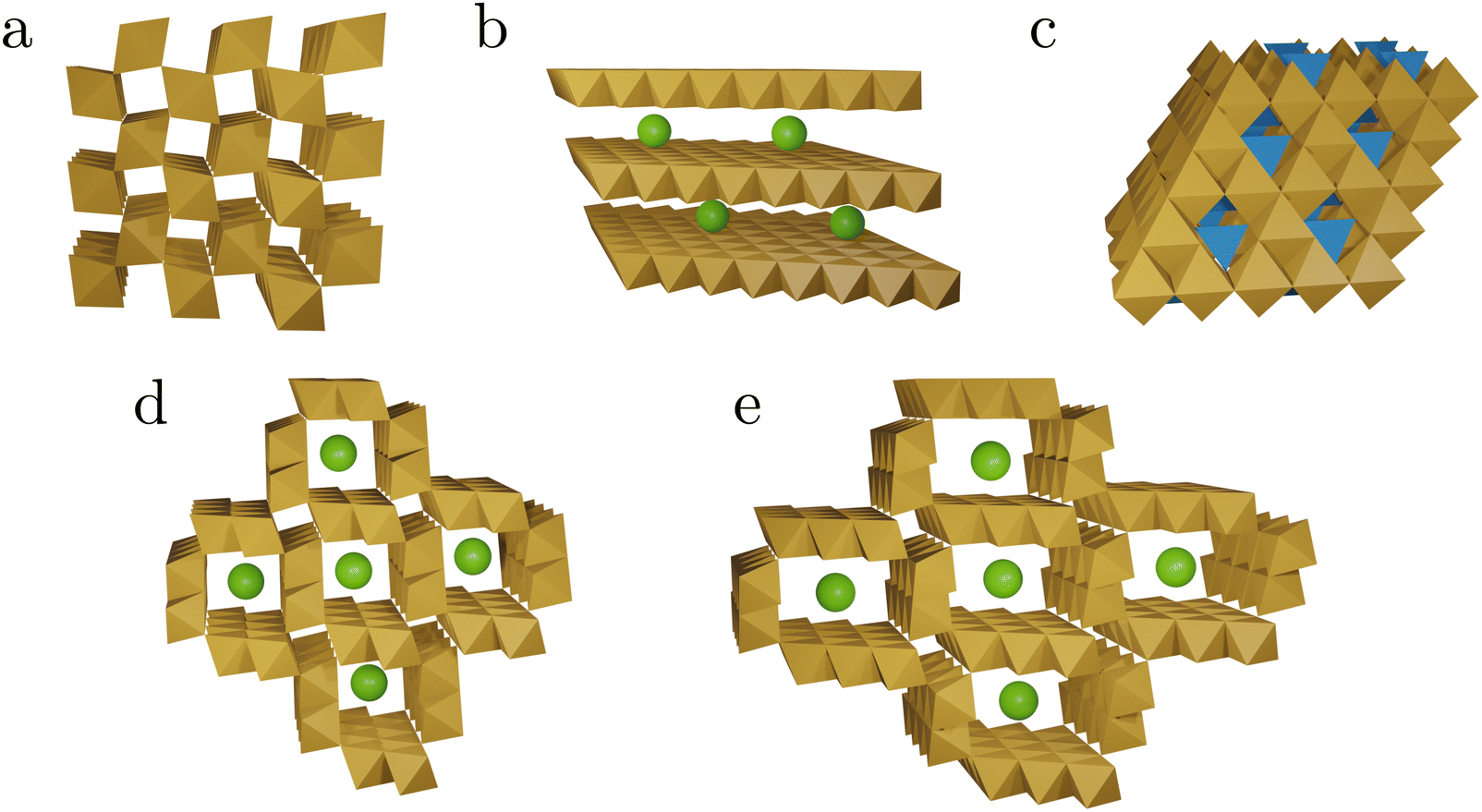 | ||
| Fig. 2 Schematic representations of the tunnel and layer structures in MnOx discussed in this work. The structures shown consist of MnO6 octahedra (brown), MnO4 tetrahedra (blue), and spheres representing other cations or water (green). Structural details are listed in Table 1. Pyrolusite, β-MnO2 (a), birnessite δ-MnO2 (b), spinel structure (c), hollandite α-MnO2 (d), and romanechite (e). | ||
On the one hand, this diversity makes MnOx interesting as potential catalysts.5 On the other hand, this heterogeneity makes fundamental studies, isolating individual aspects of a more complex problem, extremely difficult. An electrocatalyst prepared under certain conditions is likely to transform at elevated electrode potentials so that the actual catalytically active phase differs from the initially prepared material.
While all the features of the Mn oxide systems described above make a full interpretation of operando spectroscopic data challenging, they also highlight the strong need for studying such systems both “in situ” and “operando”. As one electrochemistry textbook puts it: “It cannot be over-emphasised how important it is to study any electrode process by as wide a variety of techniques as possible; only by this means can a reliable picture of the reaction be built up”.37
In the following, the specific basics for several important experimental methods shall briefly be highlighted and their recent application to Mn-based electrocatalyst systems shall be described, together with a critical analysis of the obtained results.
3. Operando studies of Mn oxide based systems during the OER
3.1 Vibrational spectroscopy
Vibrational spectroscopies, especially operando Raman spectroscopy, can yield information on the chemical identity of catalytically active materials. General introductions into the basic principles of these methods are available elsewhere.38–40 For an interpretation of the obtained spectra, thorough knowledge of the important vibrational modes of the bulk system is often required. To that end, several systematic studies of different common Mn oxides with X-ray diffraction based structural characterisation are available.41–48 Despite the many efforts to bring order into the vast variety of Mn oxide Raman spectra, the reference spectra in the literature remain contradictory and confusing. A recent study provides an extensive and systematic collection of natural Mn oxides and addressed several contradictions and conflicts in the literature of recent years.41 Especially, the thermal instability of some MnOx may have led to contradicting results in the study of reference materials (Challenge 6).The dominating Mn oxide vibrational modes are between 200 and 750 cm−1 (Fig. 3), which is easily accessible using standard lab-based Raman spectroscopy. Many MnII,III,IV oxides that may be of interest for OER catalysis, give rise to pronounced Raman modes above 500 cm−1, which result from the symmetry of the structure. A mode around 510 cm−1 has strong contributions from Mn–O–Mn deformation, while the typically most intense modes around 580 and 630 cm−1 have strong contributions from Mn–O bond stretching.41,44 For the latter it was shown that the position of these prominent ‘marker modes’ depends on the average Mn–O distance and the polymerisation of the framework, i.e., the number of shared edges between the MnO6 octahedra. This useful structural correlation was employed in the interpretation of Raman spectra of MnOx OER catalysts.32,49–51
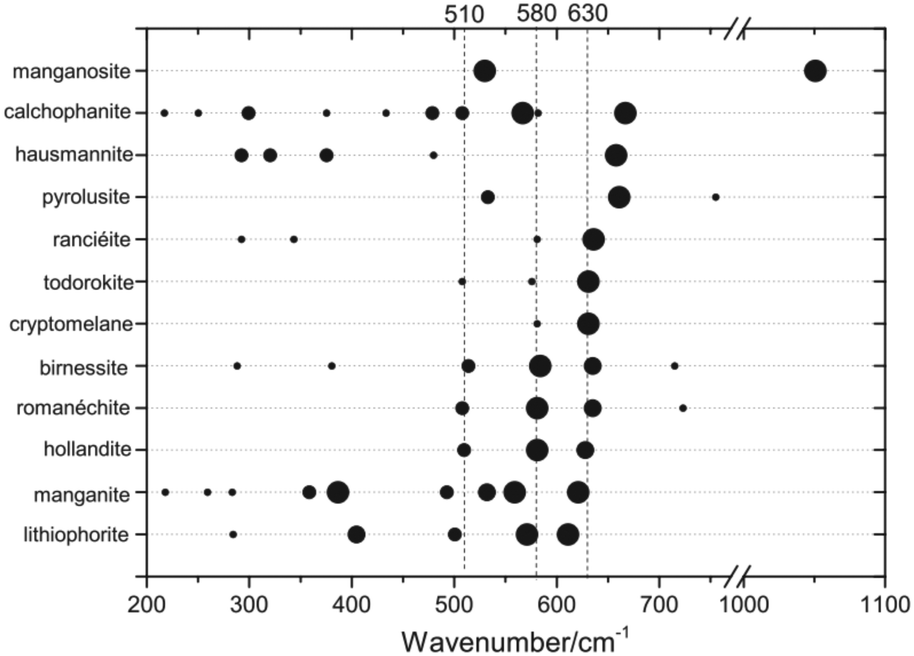 | ||
| Fig. 3 Raman band positions of selected MnOx. The dot size is proportional to the observed band intensities. Figure reprinted from ref. 41 with permission. Copyright 2019 John Wiley & Sons, Ltd. | ||
On the other hand, the strong similarities between spectra of different MnOx phases above 500 cm−1 can make a unique identification of a particular oxide extremely difficult, especially in mixed and dynamic systems as generally found for OER electrocatalysts. For instance, the patterns observed in situ during electrochemical oxidation of metallic Mn have been identified as α-MnO2,32 based on the similarity to reference spectra.44 Very similar spectral features have otherwise been assigned to the birnessite like phase prepared by electrochemical deposition50 based on previous XAS results.52 Indeed, from a recent extensive analysis of spectra of well-defined compounds, it was concluded that it is virtually impossible to make a clear distinction between hollandite (α-MnO2), birnessite (δ-MnO2) and romanechite on the basis of Raman data alone.41
The OER performance of differently prepared hollandite α-MnO2 materials (aka cryptomelane) was studied.53 Differences in the electrochemical OER performances of the different materials were found to mainly be governed by differences in conductivity originating from morphological differences. However, the initially good electrochemical OER performance of the materials degraded quickly over time. Operando Raman spectroscopy yielded remarkably stable spectra during the OER revealing no significant transformations in the materials, thus the reasons for the activity loss remained indistinct. It appears unlikely that the materials do not change when simultaneously a deactivation is observed. This illustrates a specific challenge for Raman spectroscopy, which is that the Raman cross-sections of different species differ significantly. Consequently, it is hard to get a representative picture of the surface species. Hence, quantitative information about the relative concentration of the species cannot be obtained and some species might even be virtually invisible in the Raman spectra. Furthermore, Raman spectroscopy is not per se surface specific (Challenge 7), however several strategies can be used to circumvent this problem.
Thin oxide layers of a few nm thickness on a Raman inactive substrate can be a workaround for the lack of surface specificity. To that end, electrochemically oxidized metals have been studied during the OER.32 The thicknesses of the formed oxide layers were below 10 nm, but Raman in situ measurements showed that the oxide layers remained intrinsically stable during voltammetric sweeps, down to relatively low potentials. During oxygen evolution the Raman spectra did not show similarities to any isolated stable oxide. Also, the spectrum shared some similarities with β-MnO2, but also distinct differences. Very broad bands around 600 cm−1, assigned to the Mn–O stretching vibrations showed that a strongly disordered MnO2 was present during the OER. The occurrence of the stretching vibration at this position indicates that the disordered oxide is structurally based on the octahedral coordination of Mn.
For well characterised materials the specific synthesis of reference compounds for the assignment of Raman bands can be very helpful. For instance, operando Raman spectroscopy was used to study a selection of MnOx based catalysts, prepared by electrodeposition on ITO working electrodes.50 The oxide layers featured thicknesses up to 300 nm and the two (out of three) catalysts with the higher onset potentials underwent structural changes in the pre-OER region, attributed to a charging process. Comparison of the observed spectral changes with spectra of well defined birnessite structures led to the conclusion that charging includes a transformation from triclinic to hexagonal Birnessite and the formation of point defects. It was concluded that the formation of an amorphized birnessite structure and bixbyite-like MnIII centers facilitated the OER.50
Alkali-metal manganese borophosphates were prepared using a mild-hydrothermal method and showed promising OER performances and stabilities.54 A thorough ex situ characterisation in combination with in situ Raman spectroscopy showed that these materials just served as pre-catalysts. Under OER conditions another active phase was formed which was termed a-δ-MnO2. It was described as being amorphous with a short range order similar to birnessite which was concluded from comparison of the Raman spectra with literature. It was concluded that during the oxygen evolution, corrosion of the pre-catalysts took place, forming a-δ-MnO2 while dissolving boron and phosphate species and incorporating potassium from the electrolyte. The leaching led to an increasing porosity of the materials which was assigned to the improved OER performance.
Using surface or tip enhanced Raman spectroscopy (SERS or TERS, respectively) can make Raman spectroscopy sensitive to sub-monolayer coverages. For instance the reduction of MnO2 and reoxidation of Mn(OH)2 was studied in an early application of in situ SERS.55
Thin MnOx OER catalyst films were prepared on SERS active substrates by RF-magnetron sputtering.51,56 The variation in the oxygen content in the sputter gas allowed for control of the Mn/O ratio in the final film, yielding samples, which were mainly made up of MnOx in the main oxidation states II, III and IV. Here, only the samples in the main oxidation states II and IV are discussed, since they are the most representative. In the atmosphere, films with low O content (termed Mn(II)) were easily oxidized and readily decomposed under laser irradiation, while the stable, higher oxidized MnOx phases (termed Mn(IV)) consisted of mixtures of different Mn oxides with an average oxidation number of 3.7–3.8 which was also supported by XPS studies. The operando Raman spectra indicated that Mn(II) samples (Fig. 4a and b) were oxidized when approaching the OER, showing spectra similar to the Mn(IV) samples (Fig. 4c and d) which also indicated the formation of catalytically active MnIII. For the Mn(IV) phase a slight blue shift of the Ag(2) mode (from 667 to 681 cm−1) and a red shift of the main Mn–O stretching band (from 599 to 568 cm−1) was found during the OER as well as a signal appearing at 503 cm−1 and shifting to 479 cm−1 at higher potentials. This was interpreted as being due to the formation of additional MnIII species and to an increase in the interplanar spacing between MnO6 octahedra, which could allow additional inclusion of potassium ions and water in the formed tunnel and/or layer structures. The shape of the signals and their potential dependent behaviour was found to be very similar to the Raman results of mixed Birnessite/Bixbiyte phases in ref. 50. The origins of different peaks in the region close to and above 850 cm−1 that were observed in the OER active MnOx ref. 32 and 51 (cf.Fig. 4) remain a matter of speculation. In fact, spectra of natural reference materials scarcely show peaks in this region, for instance a mode around 840–850 cm−1 was found only occasionally in reference spectra of β-MnO2.41,42,45,47 The formation of peroxo species maybe a possible origin of such peaks.57 An alternative explanation may be the excitation of multi-phonon modes or combination modes which significantly increase in intensity because of symmetry breaking in the presence of certain defects included in the defect rich films during the OER.
Note added in proof: During peer review, the question arose if the Raman spectra show any dependence on the excitation laser wavelength used. Such effects are indeed an important further aspect which could complicate operando Raman analysis and data interpretation. In this system excitation wavelength dependence could arise from (a) heat-induced or photochemical damage, (b) selective resonance enhancement of certain features, or (c) complex solid state phenomena. Manganese oxides are very well known to show transformations upon intense illumination, as documented systematically, e.g., in ref. 41. Some earlier studies may have been affected by such transformation. As transformations are typically induced by local heating upon, heating is affected by light absorption, which is strongly wavelength dependent. Hence illumination thresholds are very likely different for different excitation wavelengths. Resonance enhancement of certain features may or may not account for differences observed when comparing spectra with UV excitation to spectra with 633 nm excitation.41 However, a systematic understanding of such effects is according to our knowledge lacking. Specific phenomena in solids can also lead, e.g., to wavelength dependence of peak positions. A prominent example is the D band in sp2 carbon systems, which shifts systematically with excitation wavelength because of a Kohn anomaly.58 Such shifts have to our knowledge not been reported in literature for relevant manganese oxides. The conditions during OER and the nature of transient species make it difficult to rule out such phenomena, and to study them systematically.
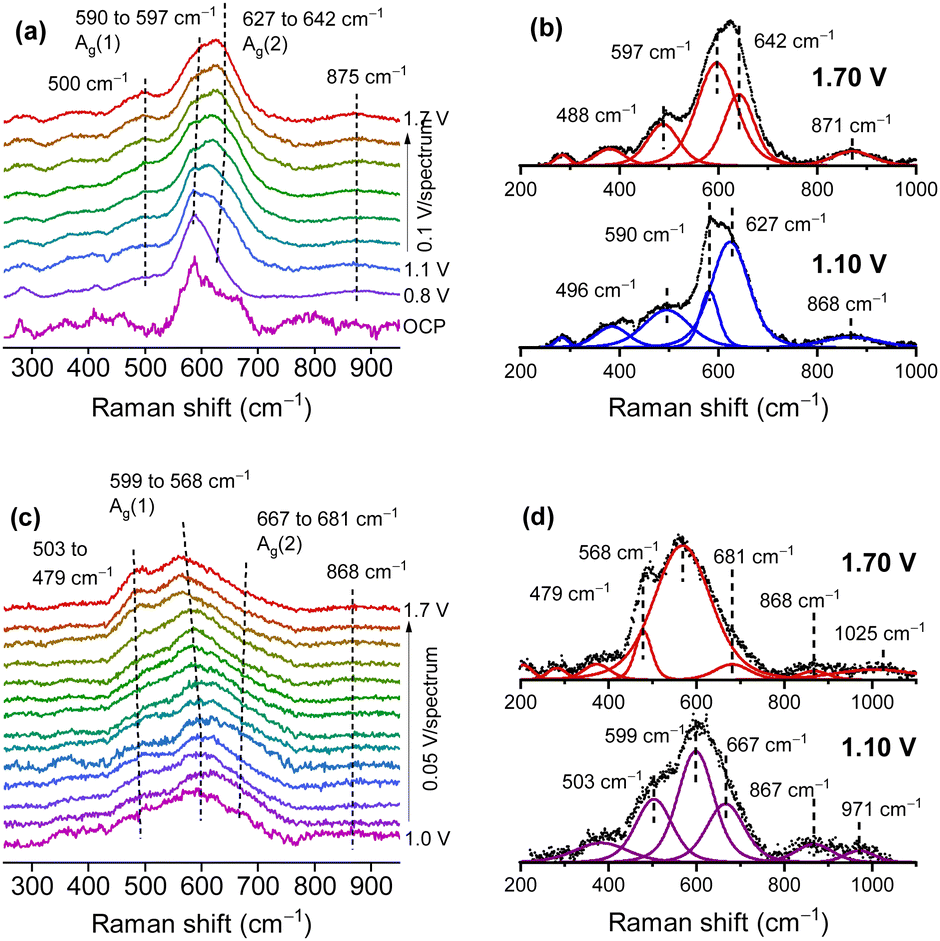 | ||
| Fig. 4 Operando Raman spectra of the Mn(II) (a) and (b) and Mn(IV) (c) and (d) phase in 0.1 M KOH. Fitted spectra (b) and (d) at two selected potentials below and above the oxygen evolution potential. Potentials are vs. the reversible hydrogen electrode (RHE). Figure adapted from ref. 56 under CC BY 4.0 license. | ||
3.2 UV/Vis absorption and photoluminescence
MnIII and MnIV containing oxides are typically brown to black in colour, i.e. they have a strong light absorption in the visible spectrum. While on the one hand, black substances are difficult to characterise by absorption spectroscopy, their strong optical absorption also implies that absorption spectroscopy and possible photoluminescence (PL) as a follow up process is a sensitive tool to detect them. The use of UV/Vis absorption, but also PL spectroscopy, for operando characterisation has been described by some of us recently.39MnIV oxides typically have a small bandgap of 1–2 eV.59 Consequently, UV/Vis absorption spectroscopy can be used to analyse the electronic structure around the band gap, and the presence of intragap states indicative of defects. For instance the local environment of MnIII in a reduced, activated MnO2 catalyst was modeled by oxidizing MnII,III in a hausmannite like (α-Mn3O4) structure to MnIII,IV.49 The authors found significantly lowered energy gaps between the highest occupied molecule orbital (HOMO) dominated by O 2p and the lowest unoccupied MO (LUMO) originating from antibonding states from tetrahedrally coordinated MnIII. The reduced gap can facilitate the formation of oxygen holes, i.e. reactive oxyl radicals as intermediates. Reduced HOMO–LUMO gaps should manifest in a measurable red shift in the UV/Vis absorbance.
Spectroscopic ellipsometry was employed to obtain spectra of MnOx thin films, which showed reversible red shifts in potential regions before the OER sets in and during the OER (Fig. 5).32 However, the observed spectral shapes are not well understood and could not be easily interpreted on the basis of the spectra of known oxides (Challenge 2). The observed peaks cannot be explained by the commonly reported absorption spectra of crystalline MnOx. Hence, the occurrence of broad peaks in the region around where the band gap is expected implied a complex defect chemistry inducing states in the gap, but also modifying significantly the density of states just below the valence band level or just above the conduction band level. Thus, it was concluded that the active MnOx consists of a defect rich, disordered MnO2.
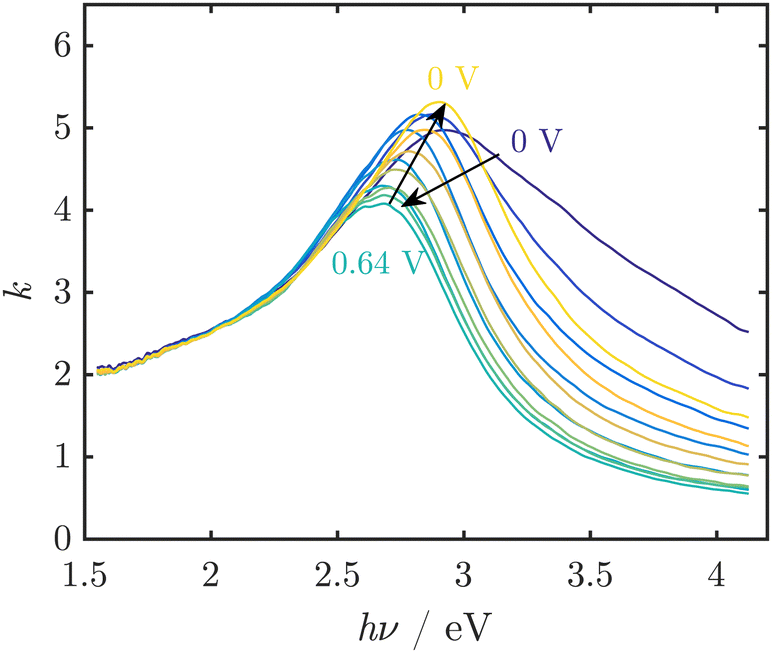 | ||
| Fig. 5 UV/Vis spectra of thin MnOx films, represented by the imaginary part k of the complex refractive index, at pH 13 during electrochemical cycling show reversible peak shifts. In this example, the OER onset was observed at ≈0.55 V vs. Ag/AgCl/3 M KCl. Figure adapted from ref. 32 under CC BY 3.0 license. | ||
Also the bifunctionality, i.e. the ability to catalyse both the OER and ORR, of MnOx catalysts has been studied. These catalysts are specifically interesting for reversible applications such as rechargeable metal–air batteries or regenerative fuel cells.60 An Mn2O3 bifunctional catalyst was studied by in situ UV/Vis/nIR spectroscopy and ex situ PL spectroscopy.61 To that end, characteristic emissions from electronic transitions of various cationic defects were assigned to the observed PL spectra in the nIR, which was further supported by reference spectra. The bifunctional catalyst featured a reversible polarity switch from p-type to n-type between OER and ORR potentials as determined by electrochemical impedance spectroscopy and Mott–Schottky analysis. The spectroscopic results indicated that the two different states were caused by formation of MnIV, MnV high valence and MnII low valence states, respectively. Interestingly, ex situ PL recorded with a confocal micro Raman setup indicated that the changes occurred in the material surface only, not in the bulk. The surface-only nature of the modification was explained by the structure of bixbyite α-Mn2O3 which, unlike many other MnOx with tunnel or layer structures, has no major voids allowing –OH ion insertion in the bulk material during the OER and ORR. The high and low valence states were proposed to act as the catalytic centers during the OER and ORR, enabled by the low defect formation potentials.
The pH dependency of the OER activity of δ-MnO2 nanoparticles (potassium birnessite) was examined by operando UV/Vis absorption spectroscopy.62 In particular the OER onset with this catalyst had an increasing overpotential η in the pH range 4–8 and a decreasing η for pH >9, indicating different mechanisms in these regions. A reversible absorption peak that showed a similar pH dependency was found at 510 nm but could not be assigned based on existing literature (cf. Challenge 2). Therefore, an assignment to MnIII was based on comparison experiments using pyrophosphate as a probe. The MnIII is assumed to be a precursor for O2 evolution and the observed higher η at pH <9 is thus a result of MnIII instability because of disproportionation. However at pH >9, MnIII can be generated by conproportionation leading to a decreasing η. Following up on these results the authors were able to significantly reduce η at pH 8 by means of an amine containing polymer.63 Also here in situ UV/Vis spectroscopy substantiated that stabilisation of surface MnIII caused this effect. For the same catalyst an additional blue shifted absorption peak at 470 nm appeared under OER conditions at elevated pH was reported later.64 The shift was explained by deprotonation of surface specific MnIII species. Accumulation of protons was shown to vary the ratio of the two absorbance peaks and was correlated with the OER current. Due to missing reference spectra (Challenge 2) the exact MnIII species could not be determined unequivocally, but the authors argued for the formation of MnIII–O− at basic and of MnIII–OH at neutral to acidic pH.
3.3 X-Ray spectroscopy
At higher photon energies, X-ray spectroscopy is a powerful tool to elucidate the core level electronic structure of a material on an atomic level. The theory of these methods and their applications to water oxidation catalysts in general have been extensively reviewed recently.65,66 Both X-ray absorption spectroscopy (XAS) and X-ray emission spectroscopy (XES) are frequently used techniques. Absorbance of X-ray photons leads to edge features in the spectra. Their position depends on the energy level of the excited core electrons. Therefore, 3d transition metal K-edges are located in the hard X-ray regime (5–10 keV), while oxygen K-edges and metal L-edges are observed in the soft X-ray regime (400–1200 eV).XES experiments monitor the fluorescence relaxation process after ionization. They are greatly limited by the effective solid angle of detection imposed by the spectrometer. Therefore, these experiments remain very challenging for electrochemical operando studies and to the best of our knowledge to date there have been only a few examples of operando spectroelectochemical XES on Mn, both in the soft and hard X-ray regime.67–70 For a non-ionizing excitation into an intermediate (previously unoccupied) electronic state, the emission experiment becomes resonant and is referred to as resonant XES (RXES) or resonant inelastic X-ray scattering (RIXS).
One of the first operando SEC XAS studies at the Mn K-edge was performed on MnOx catalysts that have been developed to perform both ORRs and OERs at moderate overpotentials.71,72 XANES and EXAFS identified Mn3II,III,IIIO4 (hausmannite) as the phase present during the ORR, and a mixture of a birnessite-like phase and a more reduced phase that probably corresponded to unchanged Mn3II,III,IIIO4 under OER conditions (cf.Fig. 6). Since the reduced phase represented a small fraction of the sample, it was assumed that the porous structure of the electrocatalyst enabled good electrochemical control and electrolyte access to most of the sample and therefore increased activity.72
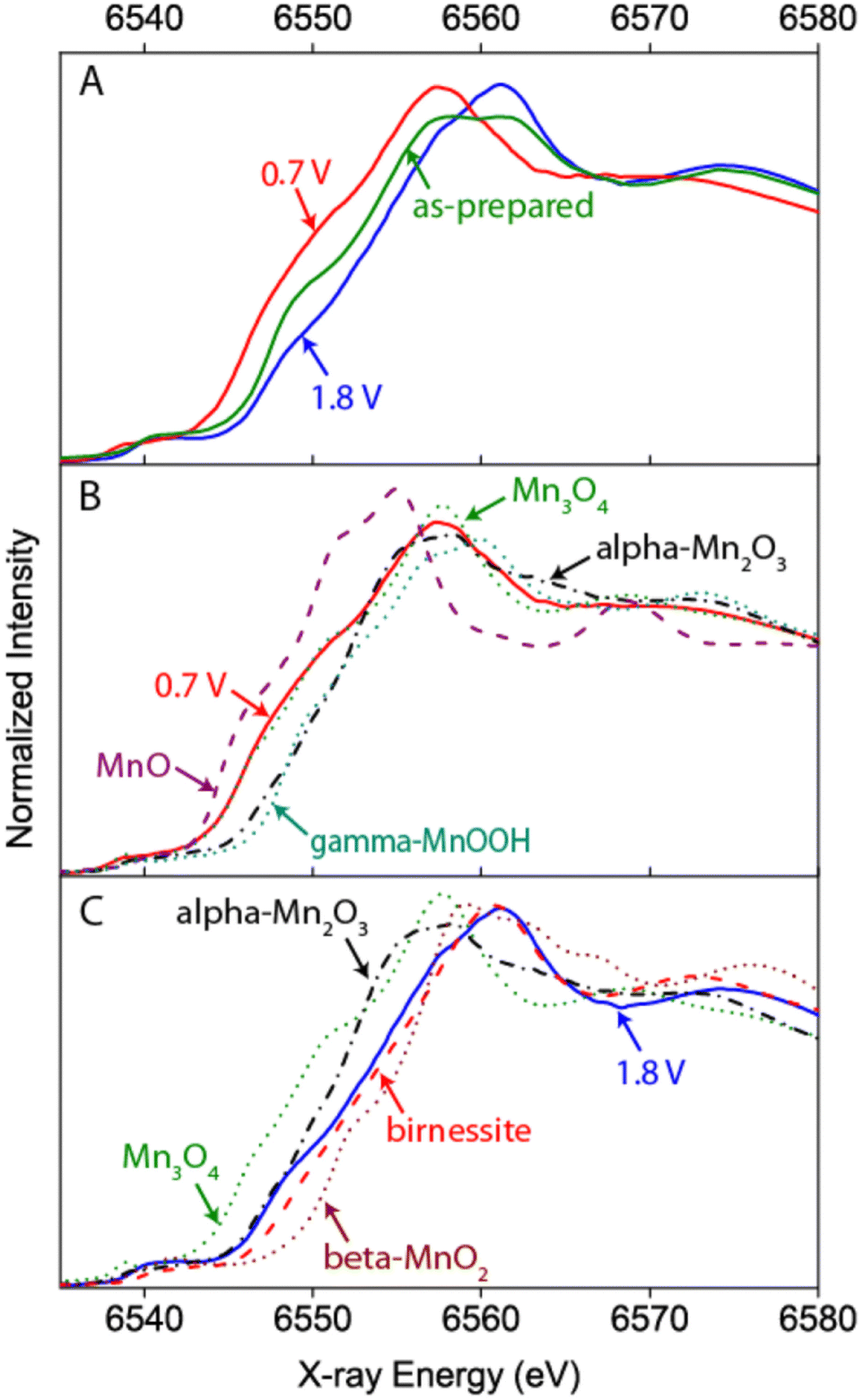 | ||
| Fig. 6 Example of a hard X-ray study undertaken by Gorlin et al. on a MnOx film prepared on a Au coated Si3N4 membrane.72 The Mn K-edge XANES undergoes clear changes with respect to the “as-prepared” state when ORR or OER conditions are applied indicating a reduction or oxidation of the catalyst, respectively (A). A comparison to spectra of reference samples additionally allows an estimation of the oxidation state and phase composition under the respective conditions (B and C). Reprinted with permission from ref. 72. Copyright 2013 American Chemical Society. | ||
Dissociation of a tetranuclear Mn cluster into MnII compounds upon immobilization in a Nafion matrix has been investigated in detail.73 These compounds became electrochemicaly re-oxidized to form nanoparticles of a disordered MnIII/IV (birnessite resembling) phase, which is catalytically active. Although the study was not exactly performed under operando conditions its results demonstrated that structures resembling that of the PSII water oxidation complex are not a requirement for catalytic performance and that complex structures in a non-biological environment may act only as precatalysts.73
The effect of Ni addition to a bifunctional MnNiOx catalyst was studied by in situ XAS complemented by an XES method that allowed for simultaneous detection of signals from multiple metal centers, thereby disentangling the redox behaviour of Mn and Ni.67 The catalyst as prepared consisted of nano-phase separated crystalline Ni rich and Mn rich oxide phases. The presence of Ni resulted in a lower oxidation potential for Mn, facilitating the oxidation towards MnIV during the OER as compared to the Ni-free catalyst. The MnNiOx was found to be bifunctional for the ORR and OER. While the oxidation states of Ni and Mn changed upon transition from the ORR to the OER regime, no major structural changes where observed.
Thin films of an MnOx co-catalyst on a SrTiO3 photoelectrode were studied using in situ XAS.74,75 The results implied UV irradiation and potential dependent slow oxidation of the co-catalyst, from an average Mn oxidation number of +3.1 to +4 at an applied potential of 1.0 V (RHE) and to +3.3 at 0.5 V (RHE). The irradiation strongly influenced the rate of the oxidation, indicating that oxidation occurred by holes forming as a result of photoexcitation. The correlation found between hole transfer and OER activity was explained by a potential induced upward band bending in the n-type semiconductor that facilitates the charge separation and the hole transfer to the MnOx co-catalyst. Interestingly, the OER rate did not show strong dependence on the Mn oxidation state, thus it was concluded that both MnIII and MnIV rich phases can be photoelectrochemically active to catalyse the OER.
In a combined quasi in situ and time-resolved in situ XAS study the redox behavior of the biological Mn4Ca oxide cluster and three different birnessite type Mn oxides (with and without Ca) were analyzed.76 For the quasi in situ technique the samples were freeze-quenched after a potential was applied for 3 minutes and subsequently XAS was measured at 20 K. The low measuring temperature can help to minimize photoreduction by the beam and decreases thermal contributions to the EXAFS, which can greatly increase experimental resolution. For the time-resolved experiment, samples were studied at room temperature and the absorption signal was tracked over time at a fixed excitation energy when changing the potential. The results of catalytically active birnessite type MnOx and Ca containing MnOx were compared to an inactive MnOx and the biological OEC of PSII. It was found that the catalytically active birnessite type MnOx undergoes oxidation and structural changes that share certain features with the OEC. The catalysts comprised metastable MnIII centers at OER potentials which can accumulate oxidizing equivalents (holes) by MnIII → MnIV oxidation coupled to changes in the μ-oxo bridging. Kinetic experiments implied that metal oxidation precedes the O2− oxidation. The inactive MnOx showed a significantly higher ordered structure than the active one, implying that non-crystalline materials promote the OER because they allow dynamic and flexible oxidation states and structural changes. This dynamic picture of the MnOx OER catalyst instead of that of a static active center is quite akin to the mechanism of the biological oxygen evolution in the OEC.76
Another study at room temperature, also reported shifts in the Mn K-edge position as a function of applied potential. It was observed that electrodeposited δ-MnO2 birnessite films initially displayed very low electrocatalytic activity that increased upon cycling between −0.4 and +1.1 V vs. NHE (at pH 8.0).49 A continuous shift of the K-edge position towards lower energies with increasing number of cycles indicating an accumulation of MnIII centers, and EXAFS analysis suggested that the activated material comprised a mixture of α-Mn3O4 hausmannite-like and δ-MnO2 (birnessite) structures in a mixed oxidation state containing MnIII and MnIV. The authors proposed that all octahedral (Oh) sites contain MnIV, while MnIII centers remain metastable, kinetically locked in tetrahedral (Td) sites, unable to disproportionate. A computational model, describing the resulting electronic structure around these MnIII(Td), features a HOMO dominated by oxygen 2p valence states and a metal-based LUMO with a reduced energy gap as compared to pure δ-MnO2. Oxidation would create oxygen holes leading to the formation of a very reactive oxyl radical that would explain the enhanced activity of these MnIII containing materials.49
The co-deposition of gold particles (2–3 nm diameter) on MnOx OER catalysis, studied by operando XAS, resulted in an increased oxidation state of the MnOx catalyst under OER conditions compared to a MnOx without Au particles.77 The increased oxidation state may have been related to the observed increase in OER activity caused by the gold particles.
An operando hard, tender, and soft XAS study was performed on a layered MnOx catalyst with intercalated potassium ions in an KOH containing aqueous phosphate buffer solution (pH 8).78 In agreement with the previously discussed studies, the Mn K-edge position shifted to higher energies with increasing potential, indicating a reversible increase of the average oxidation state from ∼+3.1 to ∼+3.9. The shape of the Mn K-edge spectra and soft X-ray O K-edge spectra resembled δ-MnO2. The catalytic activity of the MnOx film was attributed to the remaining MnIII species at high potentials. Complementary XAS with tender X-rays at the K K-edge suggested the formation of buserite – the hydrated form of birnessite – under in situ conditions.
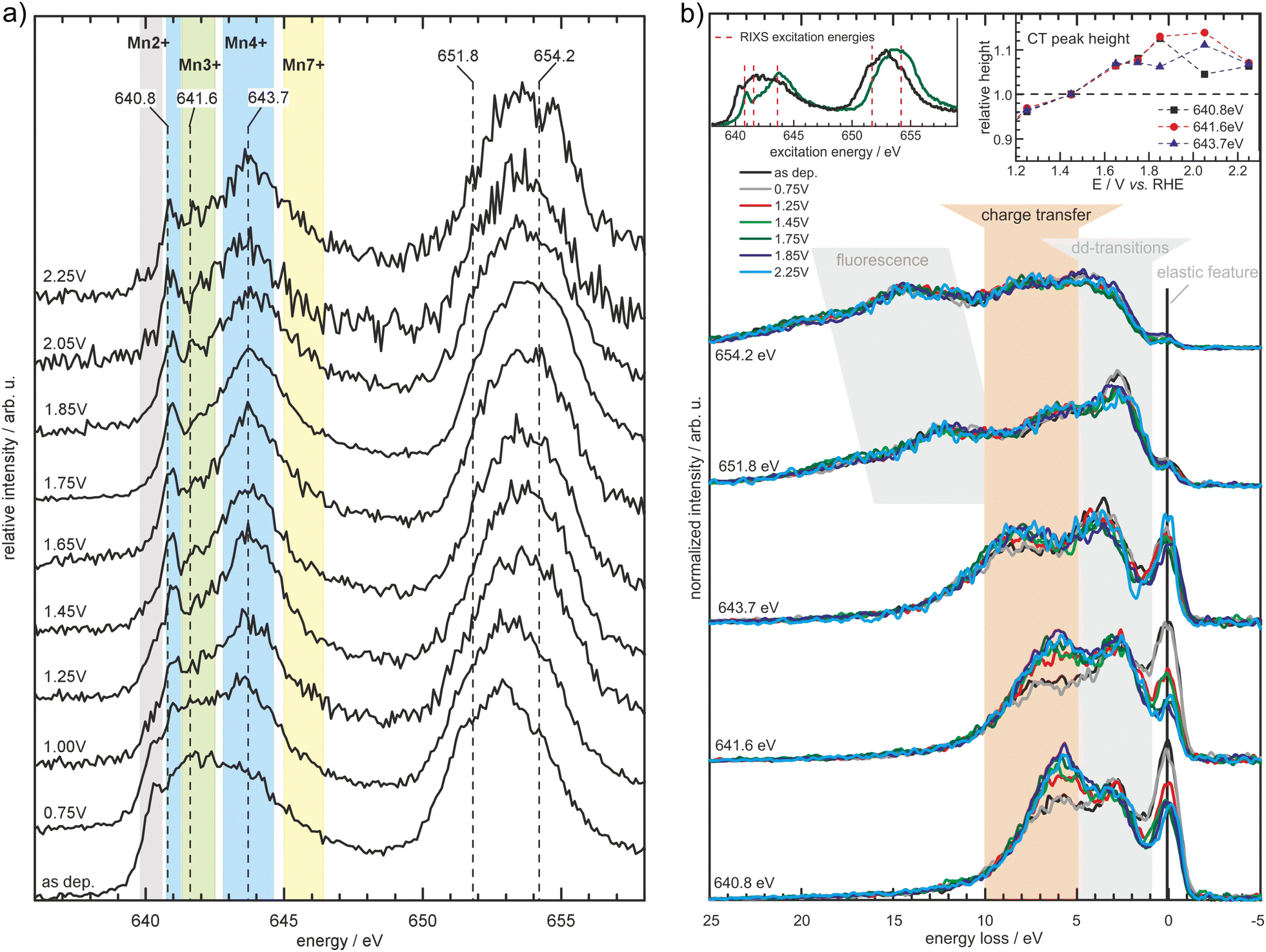 | ||
| Fig. 7 Partial fluorescence yield spectra measured at the Mn L2,3-edges (a) for MnOx in borate buffer (0.1 M, pH 9.2) at the specified potentials (vs. RHE). The spectral shape changes depending on the applied potential with the background shading indicating spectral regions where particular Mn oxidation states typically exhibit prominent features. Further results obtained by inelastic X-ray scattering (b) measured at the excitation energies depicted in (a) and the left inset of (b) reveal further potential induced changes in the electronic structure of the MnOx-catalyst, manifesting itself as a decrease of the elastic feature and increase in the charge transfer feature (highlighted by the orange background shading and the right inset). Reprinted with permission from ref. 68. Copyright 2019 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
However, the operando application of soft X-ray spectroscopy comes with some additional challenges.79–81 In contrast to experiments in the visible or hard X-ray regime, the propagation of soft X-rays requires vacuum conditions and the penetration depth of the X-ray photons is limited to a few hundred nanometres. Its limited penetration makes the application of this technique rather challenging for operando experiments, in particular because the catalyst needs to be in contact with both the liquid electrolyte and an electrode support to apply a potential. This experimental challenge might be one reason why operando soft X-ray spectroscopy studies are still exceptional compared to hard X-ray experiments. In addition, the emission intensity of soft X-ray photons during the 3d2d-relaxation processes is rather low and photon yield experiments have to be conducted very thoroughly to avoid radiation induced changes of oxidation states or radiolysis of the electrolyte (Challenge 6). Nevertheless, during the past few years several in situ/operando studies with soft X-rays have demonstrated their value for the interpretation of Mn based water oxidation catalysts.
A combined absorption/emission study, complemented by operando total electron yield and combined with modeling, was undertaken on an electrodeposited MnOx thin film for the OER applied in 0.1 M borate buffer (pH 9.2).68 It was shown that δ-MnO2 (birnessite) is the main stable phase at OER conditions and no evidence for Mn in a higher oxidation state than +4 was found. Furthermore, RIXS experiments revealed an increase of a feature that is related to metal–ligand charge transfer (Fig. 7). This was associated with the evolution of the hybridization of Mn–O bonds with increasing potential leading to facilitation of metal–ligand electron transfer and the formation of O− species.
Mn valency and the role of Mn–O hybridization was studied in detail with a layered Mn oxide catalyst for the ORR and OER, by (inverse) partial fluorescence yield experiments at the Mn L-edges and the O K-edge.82 The catalyst was an electrodeposited birnessite type film applied in 0.1 M KOH. Upon cycling between ORR and OER conditions, the Mn valence varied between 2.4–2.6 for the ORR with tetrahedral MnII identified in the sample, and 3.1–3.4 under OER conditions. Analysis of the O K-edge allowed an estimation of the maximum Mn–O hybridization for a Mn valence close to +3 (and an eg occupancy of nearly one) and the presence of MnIII was suggested to be critical for the OER as well as ORR.
Soft XAS in transmission mode was carried out on an electrodeposited MnOx film, as a proof of principle to validate the function of a soft X-ray transmission flow cell.83 Although the authors point out that some of their data may have suffered from radiation-induced damage (Challenge 6) a clear transformation of the Mn L-edge absorption spectrum was observed when increasing the potential from 0.3 to 1.5 V vs. NHE. By using a linear combination fitting approach based on spectra taken on reference materials, the authors disentangle the spectra into contributions of individual MnOx phases. Decreasing the potential back to 0.3 V showed the hysteretic behavior of the phase transitions. Furthermore, the effect of radiation damage was demonstrated and related to the actual radiation dose during data collection.
The same setup was used to study the oxidation and reduction behavior of a MnOx thin film electrodeposited in a methylphospate solution and applied for the OER at pH 7 in a KNO3 containing 0.1 M phosphate buffer solution.84 Using a linear fitting approach it was found that the freshly prepared films mostly contained MnO2 (in an oxidation state of +4) with a small amount of birnessite, transforming into a birnessite dominating phase with minor contributions of MnO2 at 1.5 V vs. NHE. A continuous transformation into birnessite was also observed when activating the film at 1.2 V for 1 h. Subsequent reduction at 0.3 V lead to a decrease of birnessite and an increase of MnO2 as well as to the formation of Mn2O3. No other Mn phases were identified; however, a small mismatch of the fitted data to the recorded spectra was reported (Challenge 2).
A porous layer of MnOx deposited on a BiVO4 photoanode following a similar deposition and analysis approach, was shown to also transform into a birnessite phase with increasing potential up to 1.91 V vs. RHE.85 The transformation was facilitated by illumination, attributed to the creation of photogenerated holes in the BiVO4 and subsequent electron migration. In addition it was found that oxidation of Mn also occurred in this sample system with illumination only.
The influence of Mn loading on Ni catalysts was studied in a Ni L-edge XAS/2p3d-RIXS spectroelectrochemical study.70 The Mn loaded oxide, Ni3MnO4, was compared to pure NiO, and the structure–activity correlations were studied by tracking the changes of the Ni centers before and during the OER. It was found that Mn did not improve the catalytic activity of a pure NiO. Additionally, no significant changes in the electronic structure of the Ni could be identified as a result of adding Mn to the oxide. The study could identify γ-NiOOH as the active phase.
4. Summary and outlook
While the results from the different studies discussed are not free of contradictions, some important common interdependent key features have been extracted for several MnOx.I. Disordered structures and structural flexibility. Under working conditions MnOx are observed that are disordered, amorphous or mixed phases. Furthermore, phases based on octahedral Mn coordination are frequently observed.32,49,54,72,76,78 In this regard the operando results are consistent with many ex situ studies.52,73 Several positive influences of the disorder on the OER performance have been discussed. For instance structural defects may kinetically trap metastable intermediates or precusors as proposed for MnIII.49,62,76 Also, it was argued that non-crystalline materials comprise structural flexibility, allowing structural as well as oxidation state changes during the catalysis,76 and facilitate charge separation through electron–hole pair formation by reduced energy gaps.49 Also, the positive influence of flexible μ-oxo bridges in amorphous oxides has been proposed to facilitate redox-potential leveling by coupling μ-OH deprotonation to oxidation steps.2,76
II. Flexibility in the oxidation number in the range III–IV. MnOx found under OER working conditions mostly featured oxidation numbers between +3 and +4. In this regard the MnOx resemble the biological OEC, where oxidation equivalents are accumulated by redox steps between MnIII and MnIV. An enhancement of OER activity by the presence of MnIII in MnOx was also repeatedly reported.50,54,62,63,76,78 Accordingly, it is assumed that these MnIII act as precursors62 or constitute the catalytic center.76 One mechanism was proposed that involved resting states of MnIII centers in acidic environments and mixed MnIII,IV resting states in the alkaline regime.30 However, OER potentials largely exceed the oxidation of MnIII (cf.Fig. 1). Thus, the structural properties of the catalysts are thought to keep the MnIII centers locally in a metastable state.
III. Activity of birnessite like phases. MnOx catalysts with as prepared birnessite-like structure have been identified to have good OER activities and thus have been the focus of many studies that used operando methods.49,50,52,68,76,78,82 Moreover, the formation of additional birnessite phases from other materials under OER conditions or upon catalyst activation has also been observed by operando methods at various pH.54,68,72,73 Thus it is possible that other Mn-based materials also convert (partially) into birnessite like structures under OER conditions which can only be probed by operando studies. Birnessites, intrinsically unite the properties of the above points I & II. They comprise disordered, layered structures based on edge sharing MnO6 octahedra, including MnIII and MnIV centers (Fig. 2b).35,36 The structural flexibility in birnessites is given by several features. In the interlayer space (distance ca. 7 Å) H2O as well as metal ions can be incorporated and the latter can be easily exchanged.36 For instance the incorporation of Ca2+, which is essential in biological OER, may be beneficial here. Defects in the MnO6 layers may act as H2O coordination sites and missing oxo- or hydroxo-ligands can enable additional μ-O bridges.86 In contrast, recently the intrinsic OER activity of layered MnOx has been challenged by the discovery that traces of Ni and Fe ions significantly increase their activity and in the absence of these ions, birnessite's intrinsic OER activity appears to be low.87
Despite the challenges outlined in Section 2, in situ and operando spectroscopy has contributed a great deal to the understanding of reaction mechanisms of Mn-containing electrocatalysts for the OER. A wealth of studies has revealed key features that seem to determine whether a certain Mn material features efficient OER catalysis. Even though the challenges these methods face will not be overcome in the short term, it is hard to imagine progressing our understanding without operando spectroscopic analysis. Thus, there is plenty of room for ongoing method development with the existing methods, and also for the application of less widely available techniques. A number of questions remain open, and operando spectroscopy may be able to contribute to answering them. For example, the role of and need for the calcium in the biocatalyst, remains at least partly in the dark. Also, the stability issues generally observed at high current densities require further understanding. Stability is without any doubt important for their application, and solving stability issues is crucial for the application of Mn-based electrocatalysts in large scale water splitting applications.
List of abbreviations
| EXAFS | Extended X-ray absorption fine structure |
| HER | Hydrogen evolution reaction |
| IR | Infrared |
| nIR | Near infrared |
| OEC | Oxygen evolving center |
| OER | Oxygen evolution reaction |
| ORR | Oxygen reduction reaction |
| PL | Photo luminescence |
| PS II | Photo system II |
| RF | Radio frequency |
| RHE | Reversible hydrogen electrode |
| RIXS | Resonant inelastic X-ray scattering |
| RXES | Resonant X-ray emission spectroscopy |
| SEC | Spectroelectrochemistry |
| SERS | Surface enhanced Raman spectroscopy |
| TERS | Tip enhanced Raman spectroscopy |
| UV | Ultraviolet |
| Vis | Visible |
| XANES | X-Ray absorption near edge spectroscopy |
| XAS | X-Ray absorbance spectroscopy |
| XES | X-Ray emission spectroscopy |
| XPS | X-Ray photoelectron spectroscopy |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from the German Federal Ministry of Education and Research (Bundesministeriums für Bildung und Forschung, BMBF) within the Framework of the MANGAN project (grant numbers 03EK3545, 03SF0504, 03SF0507, and 03SF0513). MR, MFT, OR and SDB acknowledge the Max Planck Society for funding. M. R.'s current work is funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2033 – 390677874 – RESOLV. AE acknowledges Siri Marie Skaftun and Andrew Gibson for inspiring discussions. BK acknowledges financial support by the german science foundation DFG under project numbers GSC 1070 and PAK 981 (JA 859/35-1), and H. Radinger for valuable contributions. Open Access funding provided by the Max Planck Society.Notes and references
- K. Kinoshita, Electrochemical Oxygen Technology, Wiley, New York, 1992, vol. 1117 Search PubMed.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS.
- A. Tanaka, Y. Fukushima and N. Kamiya, J. Am. Chem. Soc., 2017, 139, 1718–1721 CrossRef CAS PubMed.
- Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS PubMed.
- F. A. Armstrong, Philos. Trans. R. Soc. Lond., B, Biol. Sci., 2008, 363, 1263–1270 CrossRef CAS PubMed.
- D. A. Pantazis, ACS Catal., 2018, 8, 9477–9507 CrossRef CAS.
- V. Krewald, M. Retegan, N. Cox, J. Messinger, W. Lubitz, S. DeBeer, F. Neese and D. A. Pantazis, Chem. Sci., 2015, 6, 1676–1695 RSC.
- J. Yano and V. Yachandra, Chem. Rev., 2014, 114, 4175–4205 CrossRef CAS PubMed.
- N. Cox, D. A. Pantazis, F. Neese and W. Lubitz, Acc. Chem. Res., 2013, 46, 1588–1596 CrossRef CAS PubMed.
- P. E. M. Siegbahn, Acc. Chem. Res., 2009, 42, 1871–1880 CrossRef CAS PubMed.
- J. Melder, P. Bogdanoff, I. Zaharieva, S. Fiechter, H. Dau and P. Kurz, Z. Phys. Chem., 2020, 234, 925–978 CrossRef CAS.
- D. B. Wellbeloved, P. M. Craven and J. W. Waudby, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, Germany, 2000, pp. 175–221 Search PubMed.
- L. Erdmann, S. Behrendt and M. Feil, Kritische Rohstoffe für Deutschland, Institut für Zukunftsstudien und Technologiebewertung/adelphi technical report, 2011.
- G. Blengini, C. El Latunussa, U. Eynard, C. Torres De Matos, D. Wittmer, K. Georgitzikis, C. Pavel, S. Carrara, L. Mancini, M. Unguru, D. Blagoeva, F. Mathieux and D. Pennington, Study on the EU's list of critical raw materials (2020): final report, European Commission and Directorate-General for Internal Market, Industry, Entrepreneurship and SMEs, 2020.
- B. Kok, B. Forbush and M. McGloin, Photochem. Photobiol., 1970, 11, 457–475 CrossRef CAS PubMed.
- X. Li, L. Zhao, J. Yu, X. Liu, X. Zhang, H. Liu and W. Zhou, Nano-Micro Lett., 2020, 12, 131 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- D. Pletcher and X. Li, Int. J. Hydrogen Energy, 2011, 36, 15089–15104 CrossRef CAS.
- J. Brauns and T. Turek, Processes, 2020, 8, 248 CrossRef CAS.
- C. Ros, T. Andreu and J. R. Morante, J. Mater. Chem. A, 2020, 8, 10625–10669 RSC.
- B. M. Weckhuysen, Chem. Commun., 2002, 97–110 RSC.
- M. A. Bañares and I. E. Wachs, J. Raman Spectrosc., 2002, 33, 359–380 CrossRef.
- F. Deißenbeck, C. Freysoldt, M. Todorova, J. Neugebauer and S. Wippermann, Phys. Rev. Lett., 2021, 126, 136803 CrossRef PubMed.
- J.-B. Le, X.-H. Yang, Y.-B. Zhuang, M. Jia and J. Cheng, J. Phys. Chem. Lett., 2021, 12, 8924–8931 CrossRef CAS PubMed.
- H. J. King, M. Fournier, S. A. Bonke, E. Seeman, M. Chatti, A. N. Jumabekov, B. Johannessen, P. Kappen, A. N. Simonov and R. K. Hocking, J. Phys. Chem. C, 2019, 123, 28533–28549 CrossRef CAS.
- A. R. Akbashev, Curr. Opin. Electrochem., 2022, 35, 101095 CrossRef CAS.
- I. Puigdomenech, Chemical Equilibrium Diagrams, https://www.kth.se/che/medusa/, https://sites.google.com/site/chemdiagr/, https://github.com/ignasi-p/eq-diagr/, 2018.
- M. Pourbaix, in Atlas of electrochemical equilibria in aqueous solutions, ed. M. Pourbaix, National Association of Corrosion Engineers/Centre Belge d'Etude de la Corrosion CEBELCOR, Houston/Bruxelles, 1974, ch. 4-Corrosion, pp. 70–83 Search PubMed.
- M. Huynh, C. Shi, S. J. L. Billinge and D. G. Nocera, J. Am. Chem. Soc., 2015, 137, 14887–14904 CrossRef CAS PubMed.
- M. Huynh, D. K. Bediako and D. G. Nocera, J. Am. Chem. Soc., 2014, 136, 6002–6010 CrossRef CAS PubMed.
- F. D. Speck, P. G. Santori, F. Jaouen and S. Cherevko, J. Phys. Chem. C, 2019, 123, 25267–25277 CrossRef CAS.
- M. Rabe, C. Toparli, Y.-H. Chen, O. Kasian, K. J. J. Mayrhofer and A. Erbe, Phys. Chem. Chem. Phys., 2019, 21, 10457–10469 RSC.
- J. Peng, L. Giordano, T. C. Davenport and Y. Shao-Horn, Chem. Mater., 2022, 34, 7774–7787 CrossRef CAS.
- L. I. Simandi, M. Jaky, C. R. Savage and Z. A. Schelly, J. Am. Chem. Soc., 1985, 107, 4220–4224 CrossRef CAS.
- J. E. Post, Proc. Nat. Acad. Sci. U. S. A., 1999, 96, 3447–3454 CrossRef CAS PubMed.
- Q. Feng, H. Kanoh and K. Ooi, J. Mater. Chem., 1999, 9, 319–333 RSC.
- C. H. Hamann, A. Hamnett and W. Vielstich, Electrochemistry, Wiley-VCH, Weinheim, Germany, 2nd edn, 2007, p. 251 Search PubMed.
- A. Erbe, A. Sarfraz, C. Toparli, K. Schwenzfeier and F. Niu, in Soft Matter at Aqueous Interfaces, ed. P. R. Lang and Y. Liu, Springer, Cham, Switzerland, 2016, pp. 459–490 Search PubMed.
- A. Erbe, S. Nayak, Y.-H. Chen, F. Niu, M. Pander, S. Tecklenburg and C. Toparli, in Encyclopedia of Interfacial Chemistry, ed. K. Wandelt, Elsevier, Oxford, 2018, pp. 199–219 Search PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds: Part A: Theory and Applications in Inorganic Chemistry, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2008 Search PubMed.
- S. Bernardini, F. Bellatreccia, A. Casanova Municchia, G. Della Ventura and A. Sodo, J. Raman Spectrosc., 2019, 50, 873–888 CAS.
- C. Julien, M. Massot, S. Rangan, M. Lemal and D. Guyomard, J. Raman Spectrosc., 2002, 33, 223–228 CrossRef CAS.
- C. Julien, M. Massot, R. Baddour-Hadjean, S. Franger, S. Bach and J. P. Pereira-Ramos, Solid State Ionics, 2003, 159, 345–356 CrossRef CAS.
- C. M. Julien, M. Massot and C. Poinsignon, Spectrochim. Acta A, 2004, 60, 689–700 CrossRef CAS PubMed.
- C. Julien, Solid State Ionics, 2006, 177, 11–19 CrossRef CAS.
- T. Gao, H. Fjellvåg and P. Norby, Nanotechnology, 2009, 20, 055610 CrossRef PubMed.
- T. Gao, H. Fjellvåg and P. Norby, Anal. Chim. Acta, 2009, 648, 235–239 CrossRef CAS PubMed.
- B. Lafuente, R. T. Downs, H. Yang and N. Stone, in Highlights in Mineralogical Crystallography, ed. T. Armbruster and R. Danisi, De Gruyter, Berlin, Germany, 2015, ch. The power of databases: the RRUFF project, pp. 1–30 Search PubMed.
- Z. Morgan Chan, D. A. Kitchaev, J. Nelson Weker, C. Schnedermann, K. Lim, G. Ceder, W. Tumas, M. F. Toney and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E5261–E5268 CrossRef PubMed.
- H. An, Z. Chen, J. Yang, Z. Feng, X. Wang, F. Fan and C. Li, J. Catal., 2018, 367, 53–61 CrossRef CAS.
- H. Radinger, P. Connor, R. Stark, W. Jaegermann and B. Kaiser, ChemCatChem, 2021, 13, 1175–1185 CrossRef CAS.
- I. Zaharieva, P. Chernev, M. Risch, K. Klingan, M. Kohlhoff, A. Fischer and H. Dau, Energy Environ. Sci., 2012, 5, 7081–7089 RSC.
- J. Heese-Gärtlein, D. M. Morales, A. Rabe, T. Bredow, W. Schuhmann and M. Behrens, Chem. – Eur. J., 2020, 26, 12256–12267 CrossRef PubMed.
- P. W. Menezes, C. Walter, B. Chakraborty, J. N. Hausmann, I. Zaharieva, A. Frick, E. Hauff, H. Dau and M. Driess, Adv. Mater., 2021, 33, 2004098 CrossRef CAS PubMed.
- D. Gosztola and M. J. Weaver, J. Electroanal. Chem. Interfacial Electrochem., 1989, 271, 141–154 CrossRef CAS.
- H. Radinger, MSc thesis, Technische Universität Darmstadt, Darmstadt, 2023.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds: Part B: Applications in Coordination, Organometallic, and Bioinorganic Chemistry, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2008, ch. 1.21, pp. 161–175 Search PubMed.
- A. C. Ferrari, Solid State Commun., 2007, 143, 47–57 CrossRef CAS.
- D. M. Sherman, Geochim. Cosmochim. Acta, 2005, 69, 3249–3255 CrossRef CAS.
- J. Pan, X. L. Tian, S. Zaman, Z. Dong, H. Liu, H. S. Park and B. Y. Xia, Batteries Supercaps, 2019, 2, 336–347 CrossRef CAS.
- I. Roy, Q. Wang and V. Chakrapani, J. Phys. Chem. C, 2020, 124, 5286–5299 CrossRef CAS.
- T. Takashima, K. Hashimoto and R. Nakamura, J. Am. Chem. Soc., 2012, 134, 1519–1527 CrossRef CAS PubMed.
- T. Takashima, K. Hashimoto and R. Nakamura, J. Am. Chem. Soc., 2012, 134, 18153–18156 CrossRef CAS PubMed.
- T. Takashima, A. Yamaguchi, K. Hashimoto, H. Irie and R. Nakamura, Electrochemistry, 2014, 82, 325–327 CrossRef CAS.
- C. H. M. van Oversteeg, H. Q. Doan, F. M. F. de Groot and T. Cuk, Chem. Soc. Rev., 2017, 46, 102–125 RSC.
- J. Timoshenko and B. Roldan Cuenya, Chem. Rev., 2021, 121, 882–961 CrossRef CAS PubMed.
- S. Gul, J. W. D. Ng, R. Alonso-Mori, J. Kern, D. Sokaras, E. Anzenberg, B. Lassalle-Kaiser, Y. Gorlin, T.-C. Weng, P. H. Zwart, J. Z. Zhang, U. Bergmann, V. K. Yachandra, T. F. Jaramillo and J. Yano, Phys. Chem. Chem. Phys., 2015, 17, 8901–8912 RSC.
- M. F. Tesch, S. A. Bonke, T. E. Jones, M. N. Shaker, J. Xiao, K. Skorupska, R. Mom, J. Melder, P. Kurz, A. Knop-Gericke, R. Schlögl, R. K. Hocking and A. N. Simonov, Angew. Chem., Int. Ed., 2019, 58, 3426–3432 CrossRef CAS PubMed.
- V. Celorrio, A. S. Leach, H. Huang, S. Hayama, A. Freeman, D. W. Inwood, D. J. Fermin and A. E. Russell, ACS Catal., 2021, 11, 6431–6439 CrossRef CAS.
- M. Al Samarai, A. W. Hahn, A. Beheshti Askari, Y.-T. Cui, K. Yamazoe, J. Miyawaki, Y. Harada, O. Rüdiger and S. DeBeer, ACS Appl. Mater. Interfaces, 2019, 11, 38595–38605 CrossRef CAS PubMed.
- Y. Gorlin and T. F. Jaramillo, J. Am. Chem. Soc., 2010, 132, 13612–13614 CrossRef CAS PubMed.
- Y. Gorlin, B. Lassalle-Kaiser, J. D. Benck, S. Gul, S. M. Webb, V. K. Yachandra, J. Yano and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 8525–8534 CrossRef CAS PubMed.
- R. K. Hocking, R. Brimblecombe, L.-Y. Chang, A. Singh, M. H. Cheah, C. Glover, W. H. Casey and L. Spiccia, Nat. Chem., 2011, 3, 461–466 CrossRef CAS PubMed.
- M. Yoshida, T. Yomogida, T. Mineo, K. Nitta, K. Kato, T. Masuda, H. Nitani, H. Abe, S. Takakusagi, T. Uruga, K. Asakura, K. Uosaki and H. Kondoh, Chem. Commun., 2013, 49, 7848–7850 RSC.
- M. Yoshida, T. Yomogida, T. Mineo, K. Nitta, K. Kato, T. Masuda, H. Nitani, H. Abe, S. Takakusagi, T. Uruga, K. Asakura, K. Uosaki and H. Kondoh, J. Phys. Chem. C, 2014, 118, 24302–24309 CrossRef CAS.
- I. Zaharieva, D. González-Flores, B. Asfari, C. Pasquini, M. R. Mohammadi, K. Klingan, I. Zizak, S. Loos, P. Chernev and H. Dau, Energy Environ. Sci., 2016, 9, 2433–2443 RSC.
- R. Frydendal, L. C. Seitz, D. Sokaras, T.-C. Weng, D. Nordlund, I. Chorkendorff, I. E. Stephens and T. F. Jaramillo, Electrochim. Acta, 2017, 230, 22–28 CrossRef CAS.
- S. Tsunekawa, F. Yamamoto, K.-H. Wang, M. Nagasaka, H. Yuzawa, S. Takakusagi, H. Kondoh, K. Asakura, T. Kawai and M. Yoshida, J. Phys. Chem. C, 2020, 124, 23611–23618 CrossRef CAS.
- M. F. Tesch, S. A. Bonke, R. Golnak, J. Xiao, A. N. Simonov and R. Schlögl, Electrochem. Sci. Adv., 2022, 2, e2100141 CAS.
- E. A. Carbonio, J.-J. Velasco-Velez, R. Schlögl and A. Knop-Gericke, J. Electrochem. Soc., 2020, 167, 054509 CrossRef CAS.
- J.-J. Velasco-Vélez, L. J. Falling, D. Bernsmeier, M. J. Sear, P. C. J. Clark, T.-S. Chan, E. Stotz, M. Hävecker, R. Kraehnert, A. Knop-Gericke, C.-H. Chuang, D. E. Starr, M. Favaro and R. V. Mom, J. Phys. D: Appl. Phys., 2021, 54, 124003 CrossRef.
- M. Risch, K. A. Stoerzinger, B. Han, T. Z. Regier, D. Peak, S. Y. Sayed, C. Wei, Z. Xu and Y. Shao-Horn, J. Phys. Chem. C, 2017, 121, 17682–17692 CrossRef CAS.
- C. Schwanke, L. Xi and K. M. Lange, J. Synchrotron Radiat., 2016, 23, 1390–1394 CrossRef CAS PubMed.
- L. Xi, C. Schwanke, J. Xiao, F. F. Abdi, I. Zaharieva and K. M. Lange, J. Phys. Chem. C, 2017, 121, 12003–12009 CrossRef CAS.
- L. Xi, F. Wang, C. Schwanke, F. F. Abdi, R. Golnak, S. Fiechter, K. Ellmer, R. van de Krol and K. M. Lange, J. Phys. Chem. C, 2017, 121, 19668–19676 CrossRef CAS.
- M. Wiechen, H.-M. Berends and P. Kurz, Dalton Trans., 2012, 41, 21–31 RSC.
- M. Salmanion, I. Kondov, M. Vandichel, P. Aleshkevych and M. M. Najafpour, Inorg. Chem., 2022, 61, 2292–2306 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2023 |