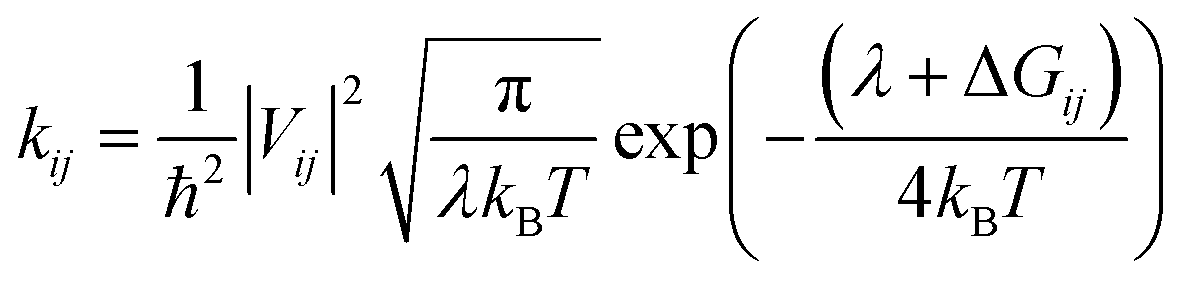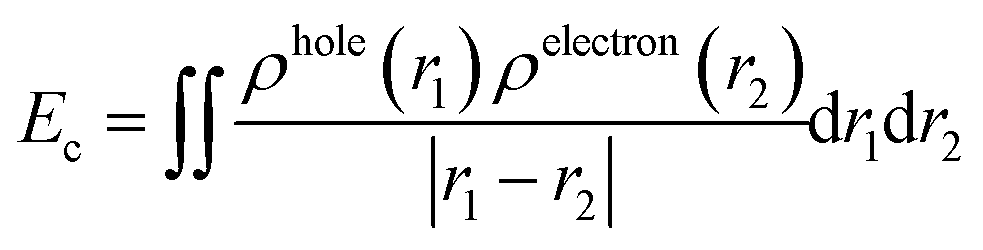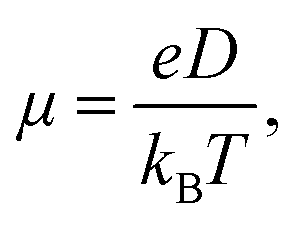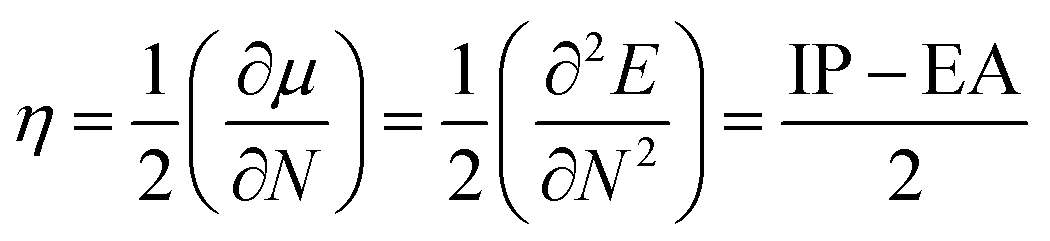DOI:
10.1039/D2TC02373C
(Paper)
J. Mater. Chem. C, 2022,
10, 11286-11295
Isomerization of two-dimensional non-fullerene electron acceptor materials for developing high-performance organic solar cells†
Received
7th June 2022
, Accepted 11th July 2022
First published on 21st July 2022
Abstract
Although the power conversion efficiency of organic solar cells has recently exceeded 19% benefiting from the development of non-fullerene acceptors (NFAs), the electron affinity and electron mobility of NFAs still lag behind those of fullerene acceptors. Therefore, it is necessary to investigate a design rule that can break the currently used A–D–A linear strategy. In our early study, we have proposed a kind of two-dimensional (2D) NFA containing four electron-withdrawing end groups, which exhibited better electron affinity and electron mobility than those of typical linear NFAs. As we know, isomerism is one of the major factors affecting the properties of materials. Herein, three isomeric 2D NFAs based on a 2D fused-ring core (naphthalene) and four electron-withdrawing end groups, 2D-ICH, d-2D-ICH and b-2D-ICH, are reported, which exhibited a similar chemical structure but different symmetries. The molecular structures, intramolecular interactions, energy level, electron affinity, electrostatic potential, absorption spectra, exciton binding energy, electron mobility, solubility and chemical stability of these isomers were investigated. The results indicated that the isomerism of the conjugated backbone is an efficient method to turn the optoelectronic properties of NFA materials. d-2D-ICH shows an appropriate LUMO energy of –3.99 eV, a high electron affinity of 3.34 eV, higher integrated absorption intensity and smaller exciton binding energy than those of Y6, good solubility of 52 mg mL−1 in chlorobenzene, the highest chemical stability, and high electron mobility of 9.42 × 10−4 cm2 V−1 s−1. This demonstrated that numerous critical factors including absorption intensity, exciton separation, chemical stability, and charge mobility of d-2D-ICH are superior to those of widely used Y6. We propose that d-2D-ICH will be an excellent candidate for the development of next-generation NFAs for high-performance organic solar cells.
Introduction
In last decades, organic solar cells (OSCs) have gone through rapid development due to their advantages of lightweight, semi-transparency, solution-processability and low cost.1,2 Conventional OSCs consist of mixed electron donor materials and electron acceptor materials to form a bulk heterojunction, which can generate donor and acceptor interfaces to accelerate exciton dissociation. Various donor and acceptor materials have been developed to improve the performance of OSCs.3–7 Especially for the appearance of non-fullerene acceptors (NFAs) in recent years, the power conversion efficiency of single-junction OSCs has made a great improvement8–11 and the highest one has exceeded 19%.12 These achievements mainly benefit from the distinguished advantages of NFAs such as strong absorption of visible and near-infrared solar energy, tuning of absorption and electronic energy levels, and easy and flexible synthetic method. Although NFAs have so many advantages, they exhibit smaller electron affinity and lower electron mobility than those of fullerene-based electron acceptors.13–15 Therefore, to further improve the performance of OSCs, increasing the electron affinity and electron mobility of NFAs will be an important path.
Currently, widely used NFAs consist of two strongly electron-withdrawing end groups connected by a planar fused-ring core to form an A–D–A-type linear structure.2,16 Although various efforts have been made to investigate the effect of electron-withdrawing end groups,11,17 fused-ring core,18 and side chains,4,19 the linear structure is still the most commonly used. In our previous study, we have proposed a quad-rotor-shaped two-dimensional (2D) NFA, BFT-TN, which exhibits larger electron affinity (3.45 eV) than that of ITIC (2.90 eV) and much higher electron mobility (5.32 × 10−3 cm2 V−1 s−1) than that of ITIC (8.20 × 10−4 cm2 V−1 s−1).20 We also demonstrated that as the degree of conjugation of these 2D NFAs increased, the spectrum red-shifted, the integrated absorption intensity enhanced, and the hole–electron coulombic attraction decreased.21 Therefore, further investigation of this kind of 2D NFA should be made.
Isomerism is the phenomenon in which more than one compounds have the same chemical formula but different chemical structures and properties, which is very important in chemistry. Isomers can be divided into two broad classes. Substances that differ in their connectivity are constitutional isomers. Isomers that have the same connectivity but different arrangements of the atoms in space are stereoisomers.22 The constitutional isomerization strategy has been used in many works to develop high-performance acceptor materials. Yang and co-workers investigated the performance of constitutional isomers of ITIC with para-alkyl-phenyl substitution and demonstrated that the side chain isomerization is an easy and convenient way to further improve the performance of OSCs.7 Recently, Lai and co-workers studied two isomeric acceptors based on acetoxy and methyl ester end group substituents.23 The results indicated that isomerism can fine tune the energy levels, adjust the absorption spectra and regulate the energy loss. Therefore, constitutional isomerization strategy is an effective way to fine tune the optoelectronic properties of materials. Although, as discussed above, 2D NFA materials are a new type of electron acceptors for developing high-performance NFA materials, the isomerization of these materials has not been paid enough attention.
In this work, three isomeric 2D NFA materials constructed by one 2D core (naphthalene) and four 1,1-dicya-nomethylene-3-indanone end groups, 2D-ICH, d-2D-ICH, and b-2D-ICH, (Fig. 1) were investigated. The effects of isomerization on their optoelectronic properties (containing energy levels, electron affinity, absorption spectra, exciton binding energy and electron mobility) and solubility in chloroform were studied by theoretical calculations and machine learning. The isomer d-2D-ICH exhibited larger electron affinity, stronger absorption intensity, smaller exciton binding energy, higher chemical stability and higher electron mobility than those of Y6. At the same time, linked with 4-ethyloctane as the side chain, it shows good solubility in chlorobenzene, which will facilitate the preparation of OSCs by a solution-processed method. These results imply that the isomerism of conjugated backbone is an efficient method to tune the optoelectronic properties of NFA materials. Simultaneously, these results indicate that d-2D-ICH is suitable as an important candidate for the development of next-generation NFAs for high-performance OSCs.
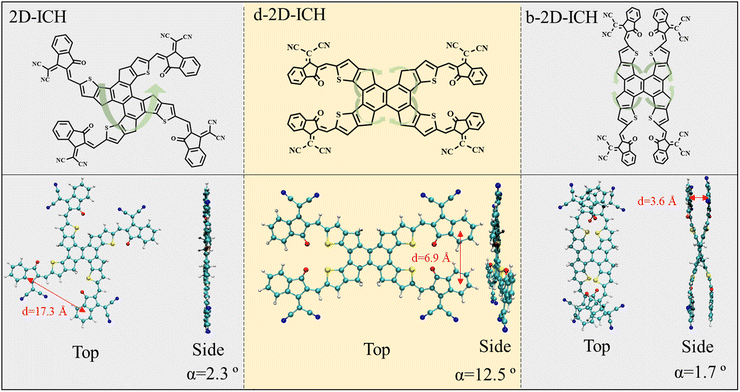 |
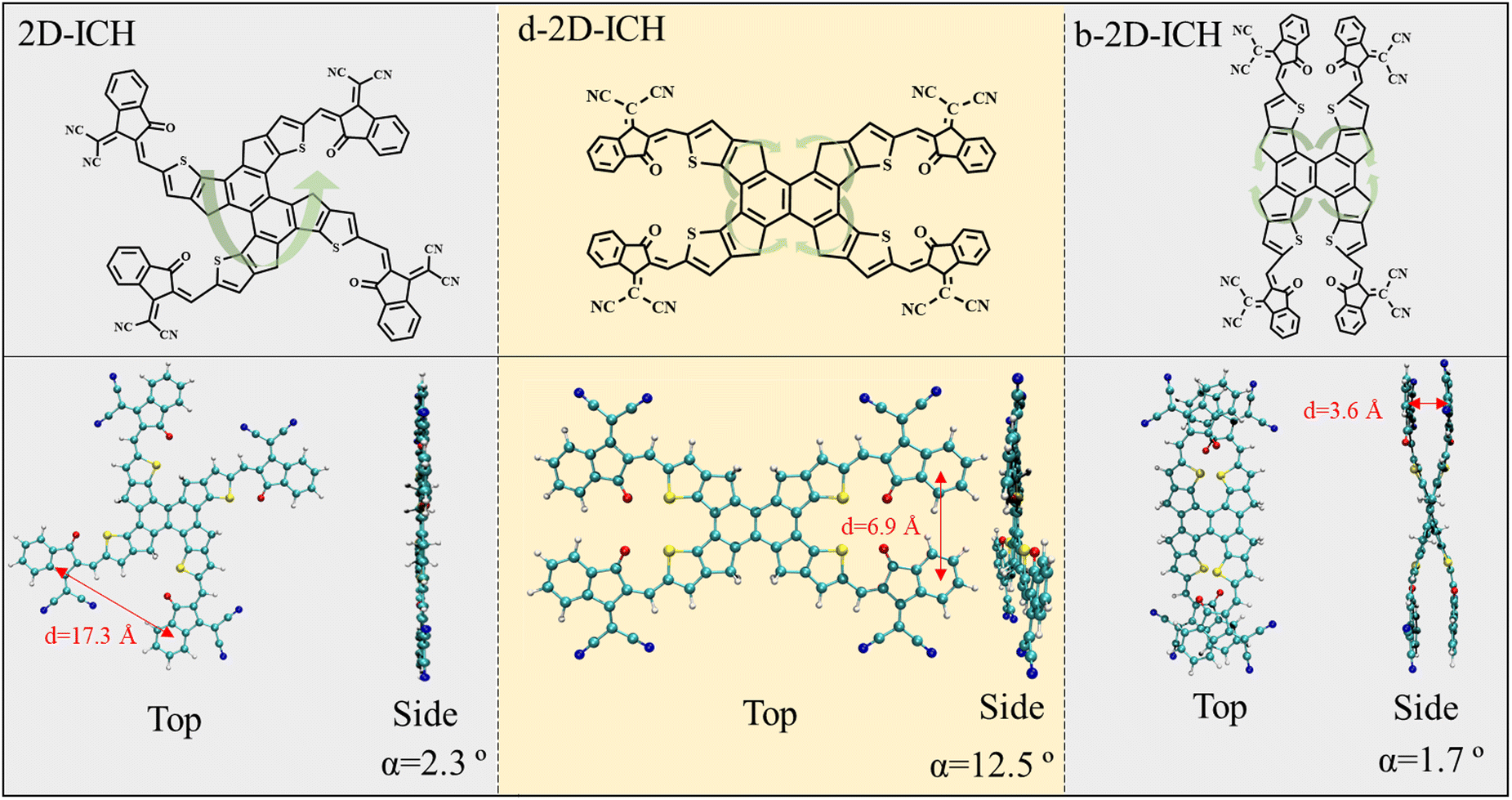
| | Fig. 1 Chemical structures and top/side view for the optimized structures of 2D-ICH, d-2D-ICH, and b-2D-ICH. Dihedral angle (α) and center distance (d) of the nearest adjacent end groups are listed at the bottom. | |
Computational method
Quantum chemical calculations
The ground-state geometry optimization of 2D-ICH, d-2D-ICH and b-2D-ICH was calculated at the B3LYP/def2-SVP level with the atom-pairwise dispersion correction23 to the DFT energy with Becke–Johnson (D3BJ).24 The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels, adiabatic electron affinities (AEA) and adiabatic ionization energy (AIP), electron reorganization energies (λ−) and spatial distribution of the frontier molecular orbitals were calculated at the B3LYP/def2-TZVP level. PBE0/def2-SVPD was used to investigate the absorption spectra of these materials. The atom-pairwise dispersion correction (D3ZERO) was used to account for the major parts of dispersion force contribution to the energy.25 The noncovalent interaction (NCI) method known as reduced density gradient (RDG) method,26 electrostatic potential (ESP), and excited state analysis were obtained using the Multiwfn 3.8 package.27 All quantum chemical calculations were carried out based on the ORCA Revision 5.0.1 program.28
Machine learning
The solubility of Y6, 2D-ICH, d-2D-ICH, and b-2D-ICH in 42 common solvents at 298 K was predicted by a machine learning model built in our previous work.29 In this model, four 4-ethyloctane molecules were linked to each molecule of 2D-ICH, d-2D-ICH, and b-2D-ICH as side chains. This model is based on Random Forest regression algorithm with a small set of descriptors (7 bits), which consist of area (molecular surface area), σ2+ (positive ESP variance when analyzing the distribution of ESP on vdW surface) and σ2- (negative ESP variance when analyzing the distribution of ESP on the vdW surface) of solvent and solute calculated by quantum chemistry and temperature. The geometries of these molecules with side chains were first optimized using the DS-PAW software30 integrated in the Device Studio Program.31 Further geometry optimization and single-point energy calculation were carried out at the B3LYP/def2-SVP and B3LYP/def2-TZVPD level respectively by ORCA 5.0.1. Then the surface descriptors of these materials were calculated by Multiwfn based on the wavefunction generated by the single-point energy calculations.
Multiscale simulations
The electron mobilities of Y6, 2D-ICH, d-2D-ICH, and b-2D-ICH amorphous thin films were evaluated by a multiscale simulation based on quantum chemical calculations, molecular dynamics (MD) simulations and Monte Carlo (MC) calculations.
MD simulations.
As the side chains can efficiently affect the packing of molecules during the MD simulations, four 4-ethyloctane side chains were introduced when processing the MD simulations of 2D-ICH, d-2D-ICH, and b-2D-ICH amorphous thin films. MD simulations were carried out using the GROMACS program with general amber force field (GAFF).32 The restrained electrostatic potential (RESP) atomic charge calculated by Multiwfn, was used for the GAFF. The MD simulation processes were built and imitated using the following procedures: (1) 520 and 420 molecules of Y6 and these 2D NFAs (2D-ICH, d-2D-ICH or b-2D-ICH) were randomly inserted in a large box (20 × 20 × 20 nm3) to generate the initial geometry; (2) 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps of the steepest descent were conducted on the system to remove the undesired forces and structural distortion; (3) NVT models were carried out at 300 K for 100 ps to naturally close the gap between molecules; (4) NPT ensemble were used to compress the box size at 300 K for 50 ps; (5) models in the NPT ensemble were annealed between 420 and 300 K for six times in 2.0 ns; and (6) the system was kept at a temperature of 300 K for 10 ns to achieve a completely converged system.
000 steps of the steepest descent were conducted on the system to remove the undesired forces and structural distortion; (3) NVT models were carried out at 300 K for 100 ps to naturally close the gap between molecules; (4) NPT ensemble were used to compress the box size at 300 K for 50 ps; (5) models in the NPT ensemble were annealed between 420 and 300 K for six times in 2.0 ns; and (6) the system was kept at a temperature of 300 K for 10 ns to achieve a completely converged system.
MC simulations.
As the internal reorganization energies are large and the electronic couplings are not strong for these systems, we employed the hopping model to calculate the electron mobility of Y6, 2D-ICH, d-2D-ICH, and b-2D-ICH. The hopping rate from one molecule to the adjacent molecules can be described by the Marcus formula,33 as follows:| | 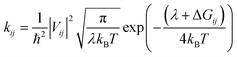 | (1) |
where ħ is reduced Planck's constant, and Vij is the transfer integral between the ith and jth molecules calculated using Zerner's intermediate neglect of a differential overlap (ZINDO) method.,34kB is the Boltzmann constant, T is the temperature and is set as 300 K in this work, λ denotes the reorganization energy and ΔGij is the free energy estimated to be the LUMO energy difference of ith and jth molecules.
The MC calculations were carried out as described in our previous work.35–37 The adjacent molecular pairs (the shortest interatomic distance is less than 0.5 nm) for each molecule under periodic conditions based on the model of amorphous films were obtained from the MD simulations. The center-of-mass (COM) of each molecular backbone was utilized as the hopping point in the MC simulations. During the hopping process, we first randomly selected one molecule (i) as the initial charge center. Then, the charge hops between the center molecule i and its adjacent molecule (j) with a probability of Pij = kij/Σkij. The hopping time was defined as 1/kij, and the hopping distance was taken to be the molecular COM(i)-COM(j) distance. Finally, we used the hopping coordinate and time to build the relationship between square diffusion displacement and the diffusion time to estimate the charge diffusion coefficient.
Results and discussion
Isomerism is one of the major factors affecting the properties of materials. Here, we first investigated three isomeric 2D NFA materials by isomerization of the conjugated backbone. The optoelectronic properties (containing energy levels, electron affinity, absorption spectra, exciton binding energy and electron mobility) and solubility in chloroform were studied by theoretical calculations and machine learning.
Geometry and intramolecular interactions
Three isomers constructed by one 2D core (naphthalene) and four 1,1-dicya-nomethylene-3-indanone end groups, namely, 2D-ICH, d-2D-ICH, and b-2D-ICH were designed, as shown in Fig. 1. These isomers are isomerized by changing the connection between naphthalene and fluorene. This is a new isomerization strategy by isomerizing the conjugated backbone, which is different from the commonly used side chain isomerization.
The top and side views for the optimized structures of these isomers are depicted at the bottom of Fig. 1. The center distance and dihedral angle of the nearest adjacent end groups of 2D-ICH are 17.3 Å and 2.3°, respectively, indicating that it presents an expended coplanar geometry. In d-2D-ICH, due to the existence of steric effect, the coplanar geometry is destroyed and the dihedral angle of the adjacent end groups is 12.5°. The center distance of the nearest adjacent end groups decreased to 6.9 Å. Compared with 2D-ICH and d-2D-ICH, b-2D-ICH shows a completely different geometry and exhibits an overlapping structure of the nearest adjacent end groups. These overlapped end groups are approximately parallel with a dihedral angle of 1.7°. At the same time, their center distance is only 3.6 Å, which is within the range of a typical π–π interaction distance (3.3–3.8 Å).38 These results indicate the existence of strong intramolecular interactions between the end groups in b-2D-ICH.
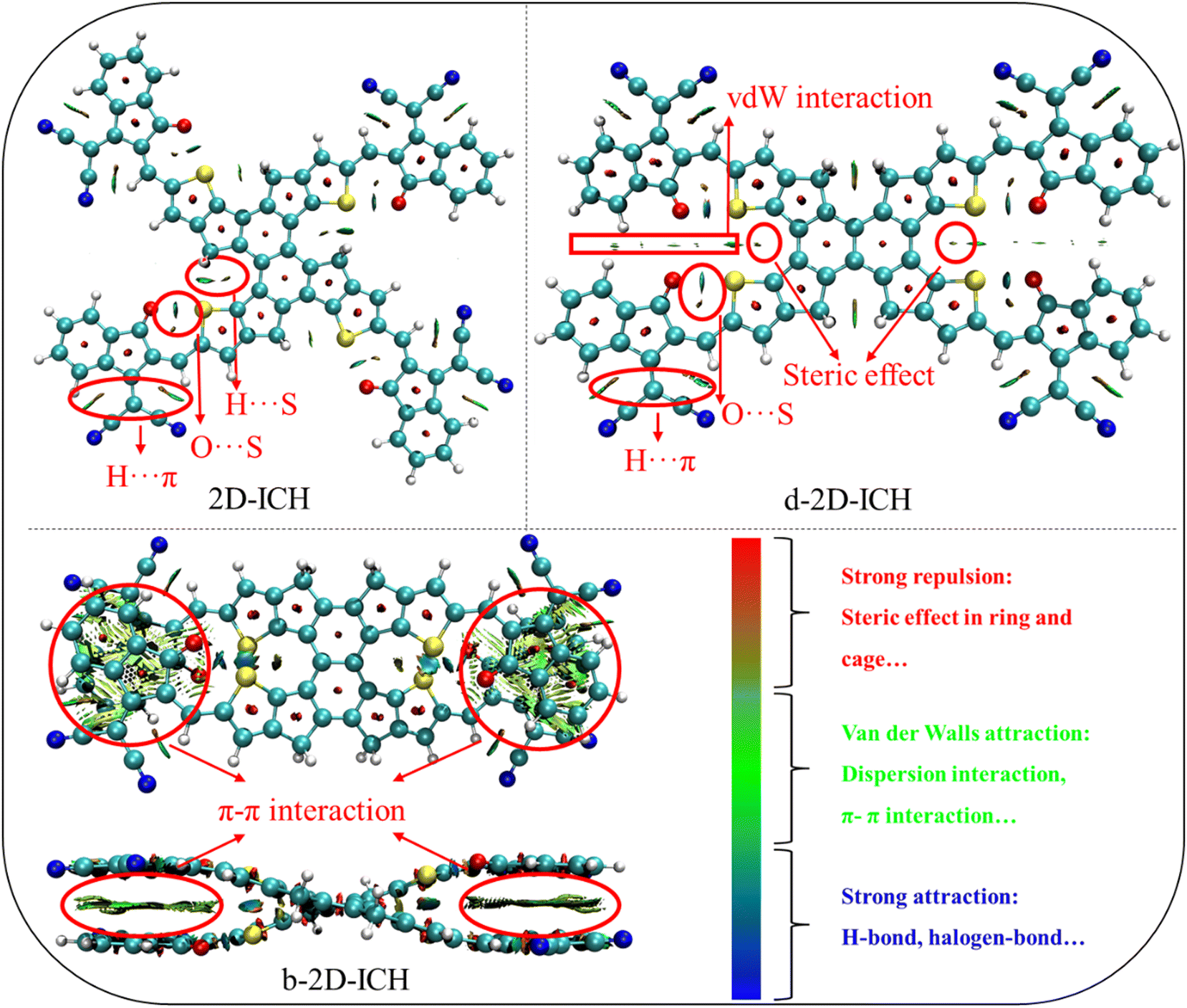
To better investigate the intramolecular interactions, the noncovalent interaction (NCI) method known as reduced density gradient (RDG) method26 was used to study the weak interaction in these isomers. The color filling maps to the RDG isosurfaces of these isomers obtained from Multiwfn27 are shown in Fig. 2. From the color-filled RDG isosurface, we identified different types of noncovalent interaction in them, as shown in Fig. 2. As shown by the scar bar, the bluer implies stronger attractive interaction, the redder indicates stronger repulsion interaction and the region marked by green can be identified as vdW interaction region. In 2D-ICH, there are three kinds of noncovalent intramolecular interactions (H⋯π, S⋯O and H⋯S), which have been demonstrated by the topological analysis of a similar structure in our previous work.20 In addition to H⋯π and S⋯O intramolecular interactions, d-2D-ICH has obvious vdW interaction and steric effect, which distorted the molecular structure, as shown in Fig. 1. There is a green isosurface between the end groups of b-2D-ICH, which can be identified as π–π interaction region combined with the above-discussed result. It is due to the existence of π–π interaction that the molecule of b-2D-ICH shows a completely overlapping structure between the adjacent end groups. Obviously, there are red RDG isosurfaces at the center of all rings in these isomers, which indicate strong steric effects in the ring.
 |
| | Fig. 2 Color filling maps to the reduced density gradient isosurfaces of 2D-ICH, d-2D-ICH, and b-2D-ICH. | |
Electronic properties
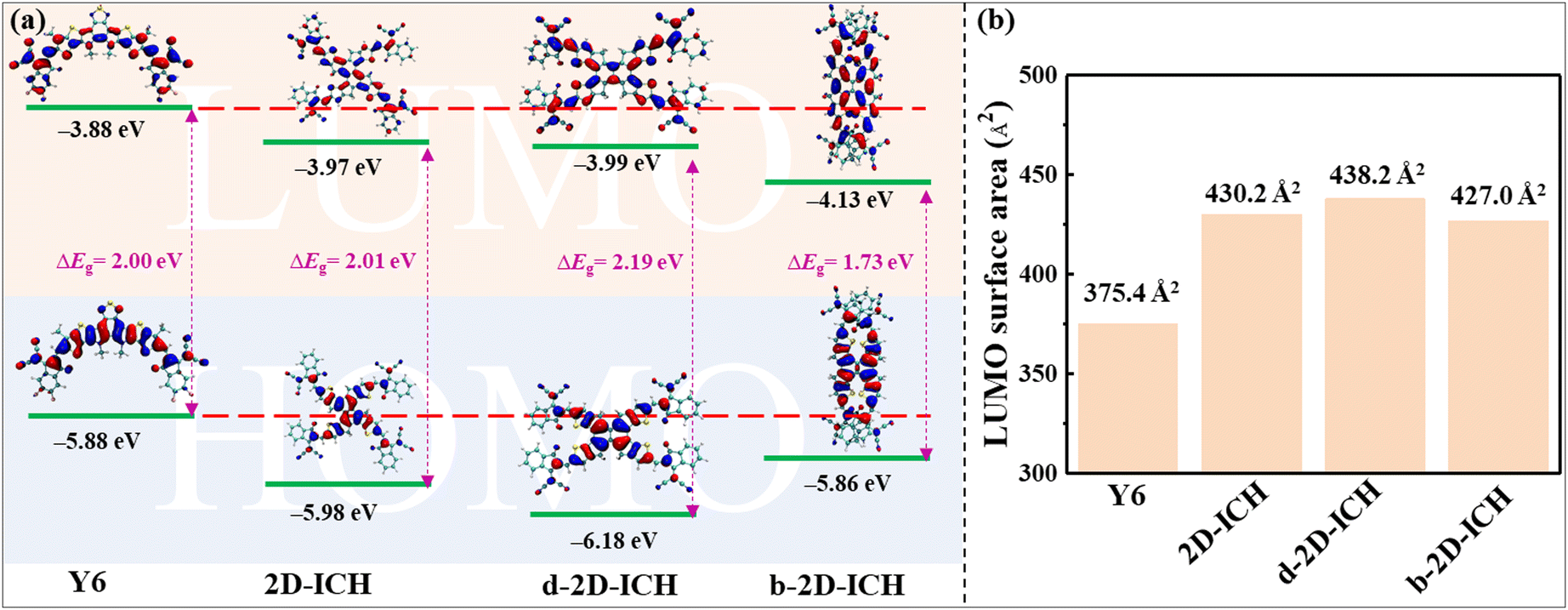
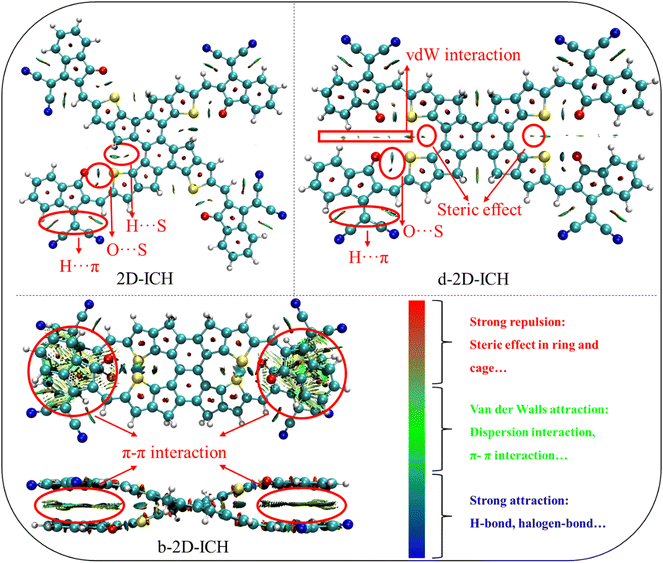
As HOMO and LUMO energy levels dominate the exciton separation, charge transportation and open circuit voltage of OSCs, the HOMO and LUMO energy levels of 2D-ICH, d-2D-ICH and b-2D-ICH compared with the widely used electron acceptor Y6 are shown in Fig. 3(a). 2D-ICH, d-2D-ICH and b-2D-ICH exhibit lower-lying LUMOs than that of Y6, which will result in a larger current density when combined with a large-bandgap donor material.39 The LUMO energy levels 2D-ICH, d-2D-ICH and b-2D-ICH are 0.09, 0.11 and 0.25 eV, respectively, lower than that of Y6, which implied that 2D-ICH, d-2D-ICH and b-2D-ICH will have stronger electronic attraction than that of Y6. It is confirmed by the calculated adiabatic electron affinities (AEA) of Y6 (3.12 eV), 2D-ICH (3.33 eV), d-2D-ICH (3.34) and b-2D-ICH (3.45 eV), as presented in Table 1. Although b-2D-ICH exhibits stronger electronic attraction, the LUMO energy level is much lower, which will dramatically reduce the open circulate voltage of OSCs. Therefore, when focusing on the energy levels, 2D-ICH and d-2D-ICH will have more advantages. The HOMO and LUMO energy band gaps of Y6, 2D-ICH, d-2D-ICH, and b-2D-ICH are 2.00, 2.00, 2.20 and 1.73 eV, respectively. This result indicates that compared with Y6, 2D-ICH will exhibit similar absorption peaks, and d-2D-ICH and b-2D-ICH will show blue- and red-shifted absorption peaks, respectively. It is obvious that the spatial distribution of LUMO delocalized on the whole conjugation backbone of Y6 and these 2D isomers (Fig. 3(a)). To quantitatively describe the distribution of LUMOs, the surface area of LUMO with an isosurface of 0.02 for Y6 and these isomers was calculated using Multiwfn, as shown in Fig. 3(b). The LUMO surface area of 2D-ICH (430.2 Å2), d-2D-ICH (438.2 Å2) and b-2D-ICH (427.0 Å2) is larger than that of Y6 (375.4 Å2), which indicates that when an electron is obtained in these 2D isomers, the delocalization region of this electron will be larger than Y6.
 |
| | Fig. 3 HOMO and LUMO energy levels and spatial distribution (a), and surface area (b) of LUMO with an isosurface of 0.02 a.u. for Y6, 2D-ICH, d-2D-ICH, and b-2D-ICH. | |
Table 1 HOMO and LUMO energy levels, HOMO and LUMO band gap (ΔEg), adiabatic electron affinities (AEA), adiabatic ionization energy (AIP), and electron reorganization energies (λ−) of Y6, 2D-ICH, d-2D-ICH, and b-2D-ICH
|
|
HOMO (eV) |
LUMO (eV) |
ΔEg (eV) |
AEA (eV) |
AIP (eV) |
λ
− (eV) |
|
Y6
|
−5.88 |
−3.88 |
2.00 |
3.12 |
6.60 |
0.14 |
|
2D-ICH
|
−5.98 |
−3.97 |
2.01 |
3.33 |
6.67 |
0.15 |
|
d-2D-ICH
|
−6.18 |
−3.99 |
2.19 |
3.34 |
6.84 |
0.13 |
|
b-2D-ICH
|
−5.86 |
−4.13 |
1.73 |
3.45 |
6.54 |
0.20 |
The electron reorganization energies (λ−) of Y6, 2D-ICH, d-2D-ICH and b-2D-ICH are 0.14, 0.15, 0.13 and 0.20 eV, respectively. This is a key parameter affecting the electron mobility of materials, which is inversely proportional to the hopping rate, as shown in eqn (1).
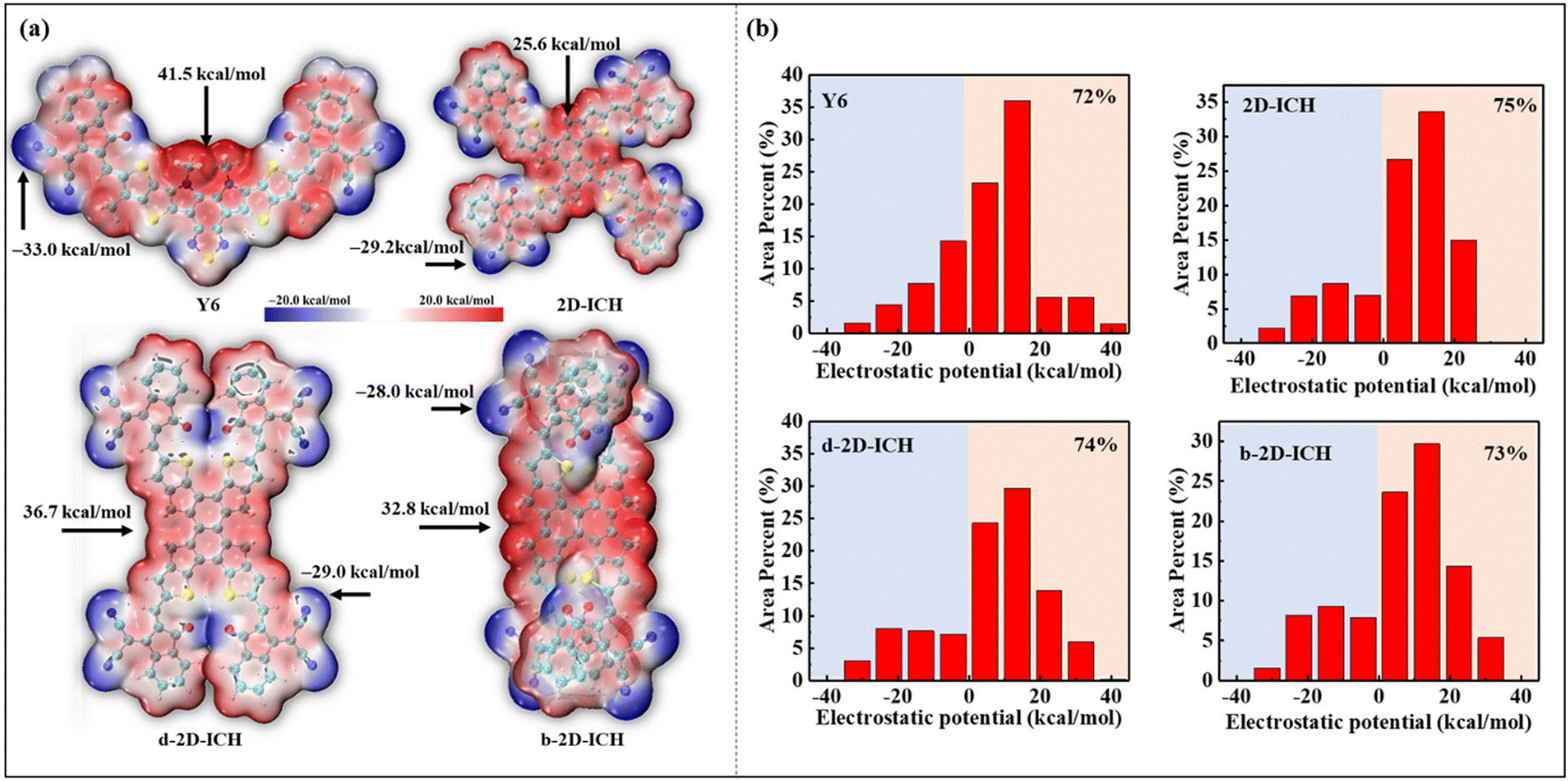
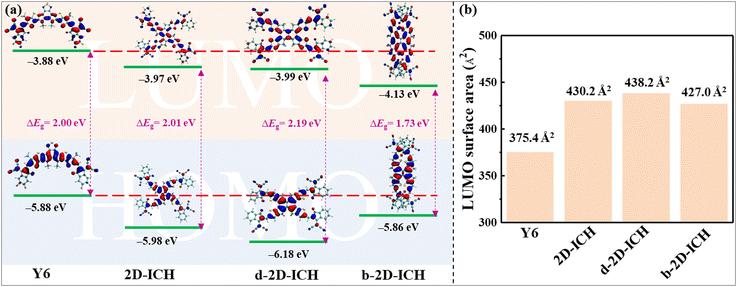
To investigate the ESP distribution of these 2D isomers, the ESP distribution of Y6, 2D-ICH, d-2D-ICH and b-2D-ICH is depicted in Fig. 4(a). Obviously, in all these molecules, the ESP values on the conjugated backbone are almost positive. The maximum ESP values of Y6, 2D-ICH, d-2D-ICH and b-2D-ICH are 41.5, 25.6, 36.7 and 32.8 kcal mol−1, respectively. The results indicate that these 2D isomers show similar electron-accepting ability to that of Y6. The negative ESP values are mainly distributed around −C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N and ketone groups. The minimum values are −33.0, −29.2, −29.0 and −28.0 kcal mol−1, respectively, distributed around −CN groups of Y6 and these 2D isomers. This is mainly caused by the strong electron-withdrawing property of −CN groups. To quantitatively study the distribution of ESP, we mapped the ESP area distribution, as shown in Fig. 4(b). The area percentages of 2D-ICH, d-2D-ICH and b-2D-ICH with ESP values larger than 0 are 75%, 74% and 73%, respectively, which are larger than that of Y6 (72%). Combined with the results of AEA as shown in Table 1, we suggest that these 2D isomers will have sufficient electron-affinity, even larger than that of Y6.
N and ketone groups. The minimum values are −33.0, −29.2, −29.0 and −28.0 kcal mol−1, respectively, distributed around −CN groups of Y6 and these 2D isomers. This is mainly caused by the strong electron-withdrawing property of −CN groups. To quantitatively study the distribution of ESP, we mapped the ESP area distribution, as shown in Fig. 4(b). The area percentages of 2D-ICH, d-2D-ICH and b-2D-ICH with ESP values larger than 0 are 75%, 74% and 73%, respectively, which are larger than that of Y6 (72%). Combined with the results of AEA as shown in Table 1, we suggest that these 2D isomers will have sufficient electron-affinity, even larger than that of Y6.
 |
| | Fig. 4 ESP distribution (a) and area percentage (b) of Y6, 2D-ICH, d-2D-ICH, and b-2D-ICH. Maximum and minimum values of the ESP distribution are added in (a) and percentage with an ESP value larger than 0 of these molecules is added in (b). | |
Electronic absorption spectra analysis
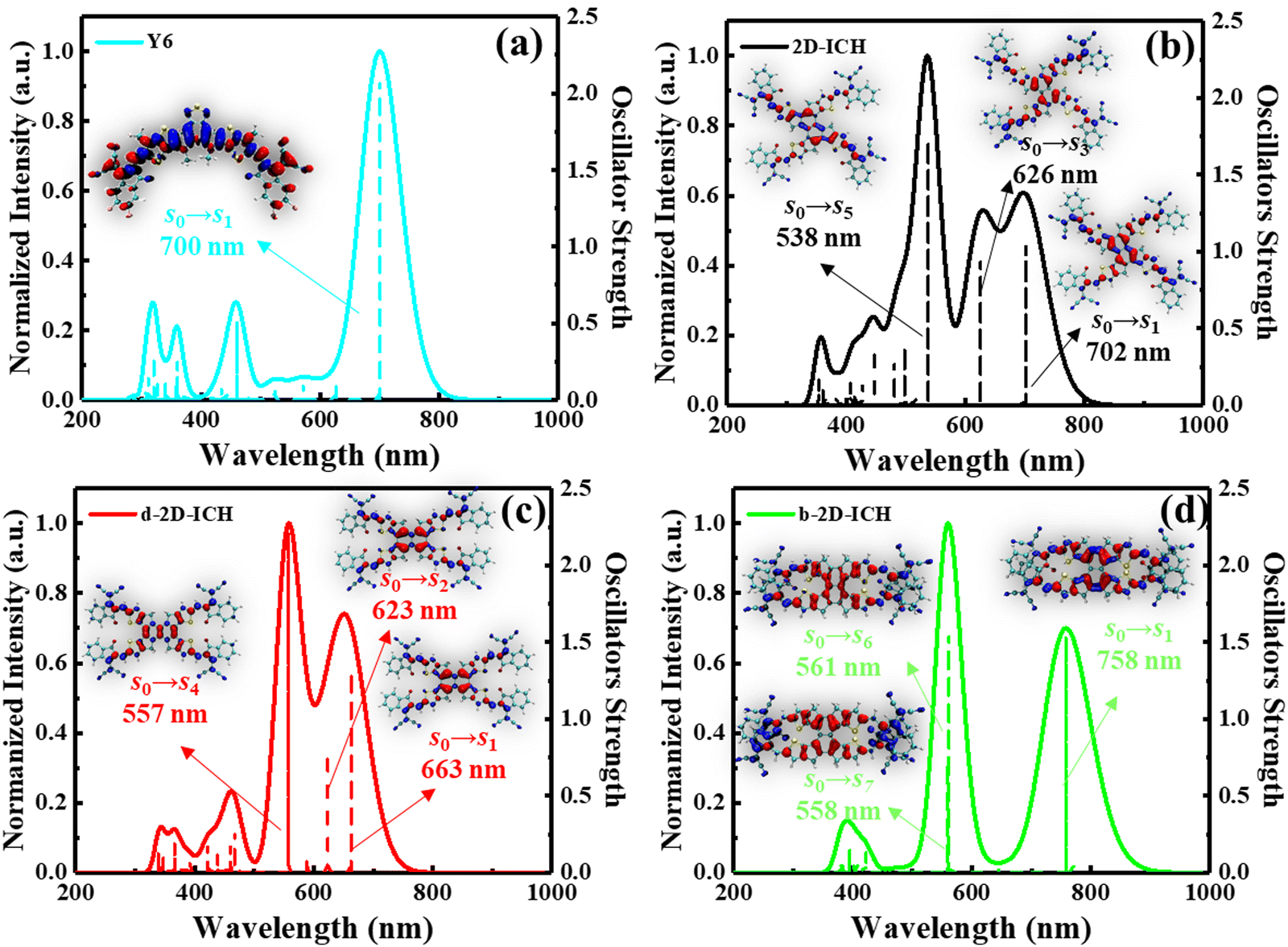
Strong absorption in visible and NIR spectrum regions is an essential characteristic of excellent FREAs, which will generate more excitons under the sunlight to increase the power conversion efficiency. Here, we investigated the electronic absorption spectra of Y6, 2D-ICH, d-2D-ICH and b-2D-ICH in chloroform by time-dependent density functional theory (TDDFT). The spectrum broadened by Multiwfn with the molar absorption coefficient as the ordinate is shown in Fig. S1(a) (ESI†). Compared with Y6, the absorption spectrum of 2D-ICH shows a similar absorption peak around 700 nm and strong absorption peaks in the blue-shifted region at 626 and 538 nm. d-2D-ICH exhibits blue-shifted and broadened absorption bands around 500–600 nm. Two significant absorption peaks appeared in the absorption spectrum of b-2D-ICH, which are located in the red and green regions, respectively. To quantitatively investigate the absorption intensity, we calculated the integrated intensity of these spectra, as shown in Fig. S1(b), (ESI†) which are 3.40 × 107, 4.12 × 107, 4.36 × 107 and 4.14 × 107 for Y6, 2D-ICH, d-2D-ICH and b-2D-ICH, respectively. This result indicates that these 2D isomers will have stronger absorption of sunlight than that of Y6, which will contribute to improve the performance of the corresponding OSCs. It is obvious that d-2D-ICH has the strongest absorption intensity in these 2D isomers, which indicates that it has significant advantages in absorbing sunlight.
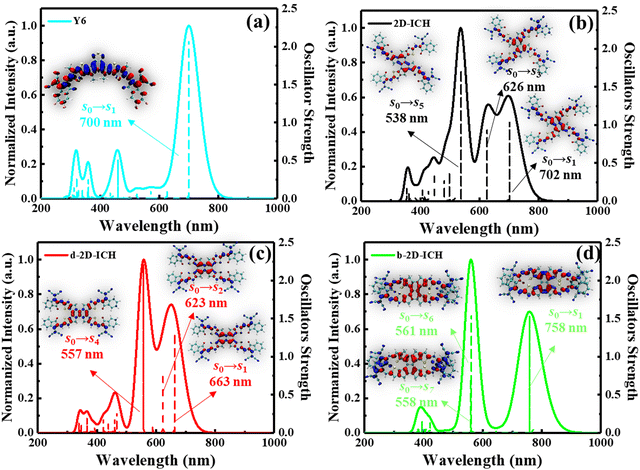
To further identify the contribution of each excited state to the absorption spectra, the detailed electron excited states calculated by TDDFT of Y6, 2D-ICH, d-2D-ICH and b-2D-ICH were analyzed, as shown in Fig. 5. The absorption spectrum of Y6 is mainly dominated by the S0 → S1 transition with a high oscillator strength (f) of 2.08 and an absorption wavelength (λ) at 700 nm. Analyzing the electron and hole distribution of this excitation, we noticed that during the excitation, the electron transformed from the center to the end groups. As shown in Fig. 5(b), the absorption spectrum of 2D-ICH consists of three main excitations with f larger than 0.5, which are S0 → S1 (λ: 702 nm, f: 1.04), S0 → S3 (λ: 626 nm, f: 0.95) and S0 → S5 (λ: 538 nm, f: 1.72). The absorption spectra of d-2D-ICH is dominated by the excitations of S0 → S1 (λ: 663 nm, f: 1.31), S0 → S2 (λ: 623 nm, f: 0.78) and S0 → S4 (λ: 557 nm, f: 2.21), as shown in Fig. 5(c). The electron and hole distributions of the excitations of 2D-ICH and d-2D-ICH were depicted in a related place, which demonstrated that during these excitations, the electrons and holes are delocalized around the center region and exhibit a degree of overlap. b-2D-ICH also exhibits three main excitations (Fig. 5(d)) with f larger than 0.5, which are S0 → S1 (λ: 758 nm, f: 1.54), S0 → S6 (λ: 561 nm, f: 1.55) and S0 → S7 (λ: 558 nm, f: 0.72). Herein, the electron and hole distribution of S0 → S1 exhibits a similar overlap, as shown in 2D-ICH and d-2D-ICH, while those of S0 → S6 and S0 → S7 exhibit obvious separation of the electron and hole. To compare the overlapping extent of holes and electrons quantitatively, we characterized the Sr index20 of each excited state, as summarized in Table 2. The Sr index of b-2D-ICH is between 0.006 and 0.023, which is much smaller than that of 2D-ICH (0.561–0.694), d-2D-ICH (0.610–0.710) and Y6 (0.650). This result also indicates less overlap between the hole and the electron of excitons in b-2D-ICH, which can result in a smaller exciton binding energy. Simultaneously, we studied the molecular orbital (MO) contribution for each transition process by Multiwfn, as summarised in Table 2. The absorption spectrum of Y6 is dominated by the transition between HOMO and LUMO, whereas HOMO−1, HOMO, LUMO, LUMO+1, and LUMO+4 contribute to the transition of 2D-ICH, d-2D-ICH and b-2D-ICH, which may be due to the increased symmetry of these 2D isomers.
 |
| | Fig. 5 TDDFT-calculated detailed electron excited states contribute to the absorption spectra of Y6 (a), 2D-ICH (b), d-2D-ICH (c) and b-2D-ICH (d), and the electron (blue) and hole (red) distribution of the excitations with an oscillator strength larger than 0.5 is inserted. | |
Table 2 Excited state analysis (λ, f, MO contribution, Dindex, Sr index, Ec, and Eb) of Y6, 2D-ICH, d-2D-ICH, and b-2D-ICH with an oscillator strength larger than 0.5
|
|
Excited states |
λ (nm) |
f
|
MO contributiona |
S
r index (a.u.) |
E
c (eV) |
E
b
(eV) |
|
H = HOMO, L = LUMO, H−1 = HOMO−1, L+1 = LUMO+1.
E
b = AIP-AEA-Es1.
|
|
Y6
|
S0 → S1 |
700 |
2.08 |
H → L (97%) |
0.65 |
2.55 |
1.72 |
|
2D-ICH
|
S0 → S1 |
702 |
1.04 |
H → L (98%) |
0.69 |
2.18 |
1.51 |
|
|
S0 → S3 |
626 |
0.95 |
H → L+2 (95%) |
0.56 |
2.08 |
|
|
|
S0 → S5 |
538 |
1.72 |
H−1 → L (93%) |
0.65 |
1.98 |
|
|
d-2D-ICH
|
S0 → S1 |
663 |
1.31 |
H → L (95%) |
0.63 |
2.12 |
1.63 |
|
|
S0 → S2 |
623 |
0.78 |
H−1 → L (95%) |
0.71 |
2.07 |
|
|
|
S0 → S4 |
557 |
2.21 |
H−1 → L+1 (93%) |
0.61 |
1.98 |
|
|
b-2D-ICH
|
S0 → S1 |
758 |
1.54 |
H → L (96%) |
0.02 |
0.77 |
1.45 |
|
|
S0 → S6 |
561 |
1.55 |
H−1 → L+1 (72%), H → L+4 (23%) |
0.01 |
0.69 |
|
|
|
S0 → S7 |
558 |
0.72 |
H−1 → L+1 (23%), H → L +4(23%) |
0.02 |
0.62 |
|
The difficulty of exciton separation will also affect the efficiency of OSCs. Here, we investigated the coulombic attraction (Ec) and exciton binding energy (Eb) of each electronic excited state. The definitions of Ec and Eb are shown as in eqn (2)20 and eqn (3):40
| | 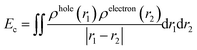 | (2) |
ρhole(
r1) and
ρelectron(
r2) represent the hole and electron density at
r1 and
r2, respectively.
Es1 represents the excitation energy of S
0 → S
1. The calculated
Ec values of each excited state are summarized in
Table 2.
Y6 (1.72 eV) exhibits a higher
Ec than that of
2D-ICH (1.98–2.18 eV),
d-2D-ICH (1.98–2.12 eV) and
b-2D-ICH (0.62–0.77 eV), which indicated that the excitons generated in these 2D isomers are easier to separate into free charges than those in
Y6, and those in
b-2D-ICH are the easiest one. It can be demonstrated by the estimated
Eb, which is 1.72, 1.51, 1.63 and 1.45 eV for
Y6,
2D-ICH,
d-2D-ICH, and
b-2D-ICH, respectively. These results indicate that the 2D isomers
2D-ICH,
d-2D-ICH and
b-2D-ICH can facilitate the separation of excitons to generate free charges.
Electron mobilities
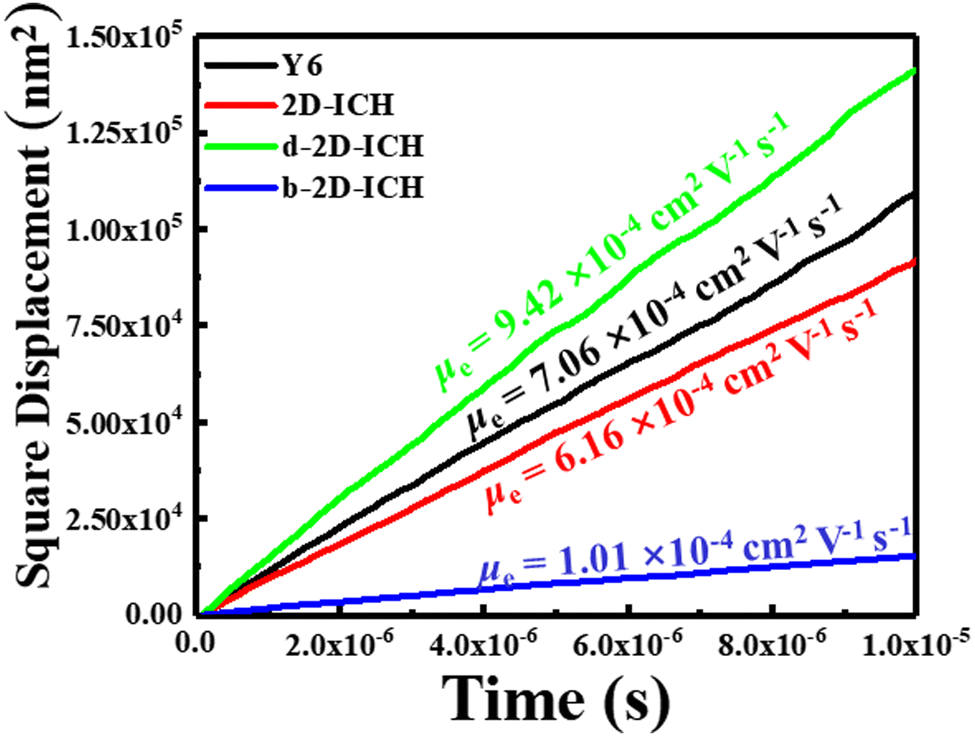
In addition to energy levels, absorption intensity and exciton separation, the hole and electron mobility of electron donor material and electron acceptor material plays a decisive role in the performance of OSCs. High electron mobility is an important property for excellent electron acceptor materials. A multiscale simulation method described in the computational method section was used to study the electron mobility of Y6, 2D-ICH, d-2D-ICH and b-2D-ICH in amorphous films. As the side chains can efficiently affect the packing of molecules during the MD simulations, four 4-ethyloctane side chains were introduced when processing the MD simulations of these 2D isomers to build up the amorphous thin films. The molecular structures are shown in Fig. S2 (ESI†). As described in Einstein's formula (eqn (4)), the electron mobility can be estimated when obtaining the electron diffusion constant:| | 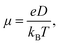 | (4) |
in which e is the electron charge, kB is the Boltzmann constant, T is the temperature, and D is the diffusion constant. Using the multiscale simulation method, we obtained the hopping coordinate and time. Then, we built up the relationship between square diffusion displacement and the diffusion time, as shown in Fig. 6. Combined with eqn (4), the electron mobilities were estimated to be 7.06 × 10−4 cm2 V−1 s−1, 6.16 × 10−4 cm2 V−1 s−1, 9.42 × 10−4 cm2 V−1 s−1 and 1.01 × 10−3 cm2 V−1 s−1 for Y6, 2D-ICH, d-2D-ICH and b-2D-ICH, respectively. The electron mobility of Y6 estimated here is in agreement with the experiment data (3.12 × 10−4 cm2 V−1),15 demonstrating the accuracy of this simulation method. The results indicate that the electron mobilities of 2D-ICH, d-2D-ICH and b-2D-ICH are in the same order of magnitude as Y6. This result indicates that isomerism of the backbone can effectively affect the molecular structure and molecular packing in amorphous films to turn the electron mobility. We note that the mobility of d-2D-ICH is approximately 1.5 times that of Y6, which indicates that d-2D-ICH has significant advantages in electron transport.
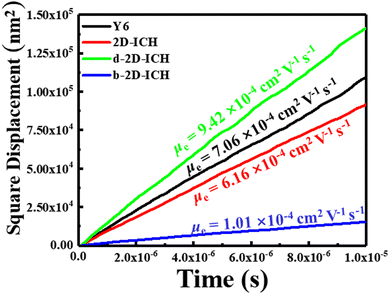 |
| | Fig. 6 Square displacement of averages over 2000 times versus the simulation of Y6, 2D-ICH, d-2D-ICH, and b-2D-ICH. | |
Solubility
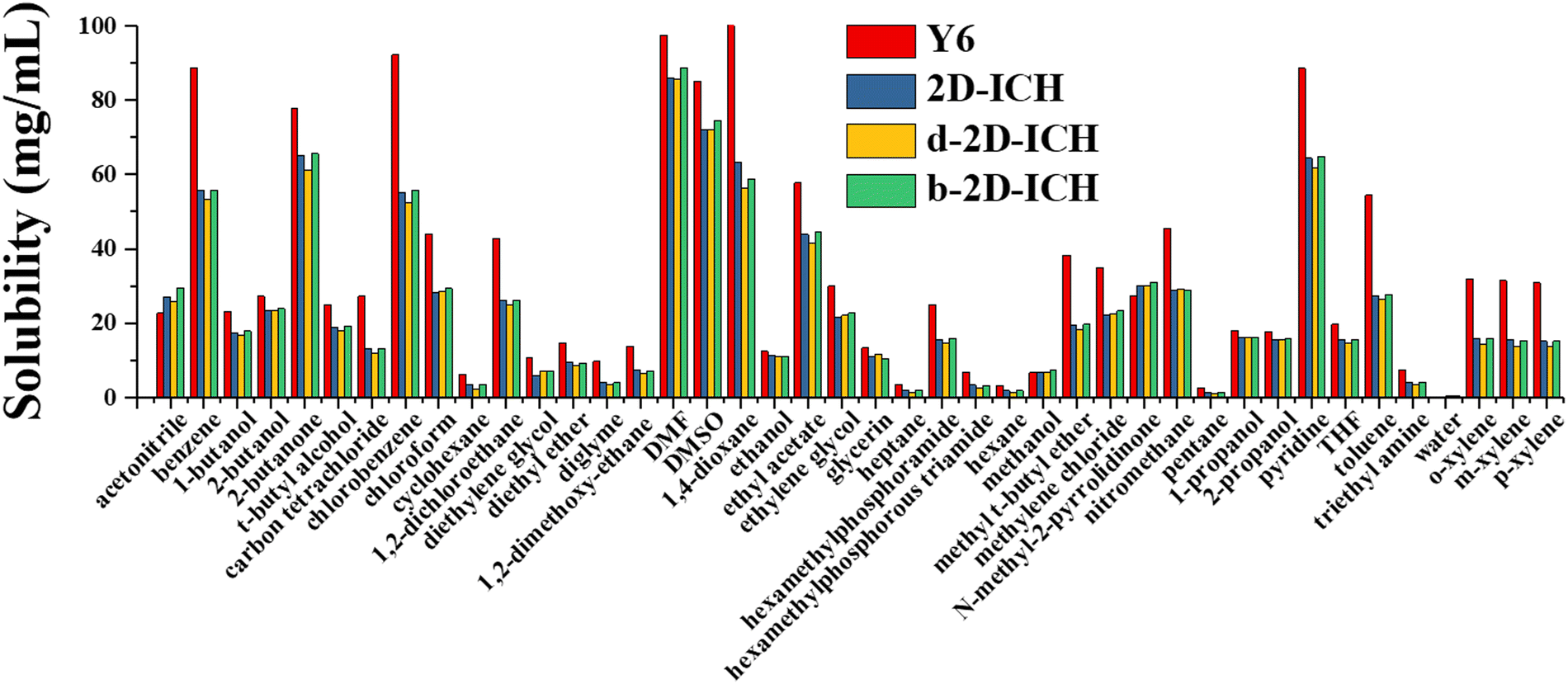
The solubility of electron acceptor materials is a critical physical property, which can profoundly affect the morphology of the photoactive layer and the tuning of the performance of OSCs. Therefore, the solubility of electron acceptor materials should be considered during the design stage. The solubility of Y6, 2D-ICH, d-2D-ICH, and b-2D-ICH in 42 common solvents at 298 K was predicted by a machine learning model built in our previous work.29 As side chains can effectively affect the solubility of molecules, four 4-ethyloctane molecules were linked to each molecule of 2D-ICH, d-2D-ICH and b-2D-ICH, as shown in Fig. S2 (ESI†). The molecular surface descriptors (area, σ2+ and σ2-) of these materials were calculated, as shown in Table S1 (ESI†). The solubility of Y6 in chloroform was predicted to be 44 mg mL−1, which is in good agreement with the experimental result (40 mg mL−1). This result demonstrates the accuracy of this model. As shown in Fig. 7, Y6 exhibits good solubility in some typical used solvents such as benzene (88 mg mL−1), chlorobenzene (92 mg mL−1), 2-butanone (78 mg mL−1), DMF (97 mg mL−1), DMSO (85 mg mL−1), 1,4-dioxane (100 mg mL−1) and pyridine (89 mg mL−1). Although these 2D isomers have smaller solubility than that of Y6 in almost all solvents, they also exhibit high solubility in chlorobenzene (>52 mg mL−1), benzene (>53 mg mL−1), DMF (>85 mg mL−1), DMSO (>72 mg mL−1), 1,4-dioxane (>56 mg mL−1), chloroform (>28 mg mL−1) and pyridine (>61 mg mL−1), which are larger than that commonly used to prepare the photoactive layer (total concentration of donor and acceptor <20 mg mL−1).12,41 Therefore, these 2D isomers not only have excellent optoelectronic properties, but also exhibit sufficient solubility in common solvents for preparing solution-processed OSCs.
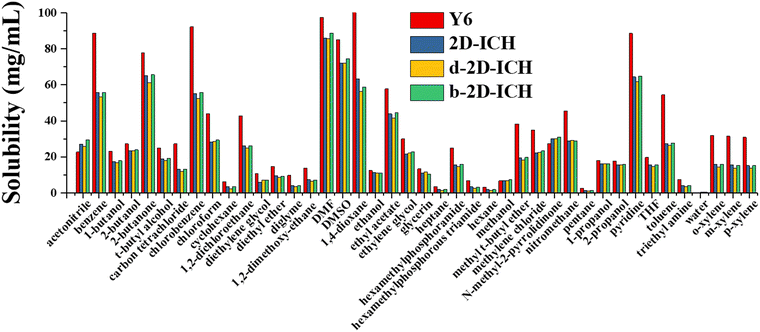 |
| | Fig. 7 Predicted solubility (mg mL−1) of Y6, 2D-ICH, d-2D-ICH and b-2D-ICH in 42 common solvents at 298 K. | |
Chemical stability
While the function and performance of optoelectronic materials were highly important, their stability also needs to be addressed if these materials have to find their way in industrial applications. Therefore, it is necessary to characterize the stability of these 2D isomers. The absolute chemical hardness (η) based on the molecular orbital theory was utilized to evaluate the chemical stability of these 2D isomers and calculated using operational definitions as follows:36| | 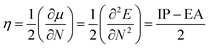 | (5) |
where μ, N, IP and EA represent the chemical potential, total electron number, electron ionization energy and electron affinities, respectively. Herein, we mainly used AIP and AEA, as shown in Table 1, to calculate η of these designed 2D isomers, which is shown in Table 3.
Table 3 Calculated η values in eV for investigated molecules at the B3LYP/def2-TZVP level of theory
|
|
Y6
|
2D-ICH
|
d-2D-ICH
|
b-2D-ICH
|
|
η
|
1.74 |
1.67 |
1.75 |
1.55 |
The results indicated that the η values of these 2D isomers are almost the same as Y6, which indicated that the chemical stability of 2D NFAs is similar to that of 1D linear NFAs. The η value of d-2D-ICH (1.75) is the largest one in these isomers, indicating that d-2D-ICH has the highest chemical stability, even larger than that of Y6.
Conclusions
In conclusion, we have investigated the isomerization stratagem of 2D NFAs by isomerizing the conjugated backbone. Three 2D isomers, namely, 2D-ICH, d-2D-ICH, and b-2D-ICH, were constructed by an isomerized 2D fused-ring core and four difluorinated 1,1-dicyanomethylene-3-indanone. Compared with the famous acceptor material Y6, these 2D isomers exhibit a lower LUMO energy level and a stronger electron affinity, which is conducive to the acceptance of electron. These 2D isomers exhibited broadened electron absorption spectrum and the integrated absorption intensity are larger than that of Y6 with an order of d-2D-ICH > b-2D-ICH > 2D-ICH > Y6. This indicates that these 2D isomers can effectively increase the absorption of solar radiation to generate more excitons. Simultaneously, they have smaller Ec and Eb, which can facilitate the separation of excitons to produce more free charges. In addition, these 2D isomers show good electron mobility in the same order of magnitude as Y6 and d-2D-ICH even exhibits higher mobility than that of Y6. This demonstrates that d-2D-ICH can transport the free electron more efficiently. Finally, these 2D isomers show good solubility in chlorobenzene, benzene, DMF and DMSO, which are sufficient to fabricate solution-processed OSCs. All these results indicate that in addition to the isomerization of side chains, the isomerism of conjugated backbone is also an efficient method to turn the optoelectronic properties of NFA materials. Simultaneously, considering the electron affinity, sunlight absorption, exciton separation, electron transportation, solubility and chemical stability, d-2D-ICH is suitable as an important candidate for the development of next-generation NFAs for high-performance OSCs.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Science and Technology Research Program of the Chongqing Education Commission of China (KJQN202001431 and KJQN202101412) and the Scientific Research Grants of Yangtze Normal University (2017XJQN04). We acknowledge the Hongzhiwei Technology (Shanghai) Co., Ltd for providing the computational resources.
References
- L. Dou, Y. Liu, Z. Hong, G. Li and Y. Yang, Chem. Rev., 2015, 115, 12633–12665 CrossRef CAS PubMed.
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K. Y. Jen, S. R. Marder and X. Zhan, Nat. Rev. Mater., 2018, 3, 18003 CrossRef CAS.
- H. Fu, Z. Wang and Y. Sun, Angew. Chem., Int. Ed., 2019, 58, 4442–4453 CrossRef CAS PubMed.
- B. Qiu, Z. Chen, S. Qin, J. Yao, W. Huang, L. Meng, H. Zhu, Y. Yang, Z.-G. Zhang and Y. Li, Adv. Mater., 2020, 32, 1908373 CrossRef CAS PubMed.
- G.-U. Kim, C. Sun, J. S. Park, H. G. Lee, D. Lee, J.-W. Lee, H. J. Kim, S. Cho, Y.-H. Kim, S.-K. Kwon and B. J. Kim, Adv. Funct. Mater., 2021, 31, 2100870 CrossRef CAS.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- Y. Yang, Z.-G. Zhang, H. Bin, S. Chen, L. Gao, L. Xue, C. Yang and Y. Li, J. Am. Chem. Soc., 2016, 138, 15011–15018 CrossRef CAS PubMed.
- B. Fan, D. Zhang, M. Li, W. Zhong, Z. Zeng, L. Ying, F. Huang and Y. Cao, Sci. China: Chem., 2019, 62, 746–752 CrossRef CAS.
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiao, K. Sun, S. Yang, X. Zhang and L. Ding, Sci. Bull., 2020, 65, 272–275 CrossRef CAS.
- Y. Cheng, B. Huang, X. Huang, L. Zhang, S. Kim, Q. Xie, C. Liu, T. Heumüller, Z. Liu, Y. Zhang, F. Wu, C. Yang, C. J. Brabec, Y. Chen and L. Chen, Angew. Chem., Int. Ed., 2022, e202200329 CAS.
- G. Han, Y. Guo, L. Ning and Y. Yi, Sol. RRL, 2018, 0, 1800251 Search PubMed.
- Y. Cui, Y. Xu, H. Yao, P. Bi, L. Hong, J. Zhang, Y. Zu, T. Zhang, J. Qin, J. Ren, Z. Chen, C. He, X. Hao, Z. Wei and J. Hou, Adv. Mater., 2021, 33, 2102420 CrossRef CAS PubMed.
- M. Liu, P. Fan, Q. Hu, T. P. Russell and Y. Liu, Angew. Chem., Int. Ed., 2020, 59, 18131–18135 CrossRef CAS PubMed.
- Y. Liu, C. e Zhang, D. Hao, Z. Zhang, L. Wu, M. Li, S. Feng, X. Xu, F. Liu, X. Chen and Z. Bo, Chem. Mater., 2018, 30, 4307–4312 CrossRef CAS.
- Z. Luo, R. Sun, C. Zhong, T. Liu, G. Zhang, Y. Zou, X. Jiao, J. Min and C. Yang, Sci. China: Chem., 2020, 63, 361–369 CrossRef CAS.
- Y. Wang, J. Lee, X. Hou, C. Labanti, J. Yan, E. Mazzolini, A. Parhar, J. Nelson, J.-S. Kim and Z. Li, Adv. Energy Mater., 2021, 11, 2003002 CrossRef CAS.
- A. Wadsworth, H. Bristow, Z. Hamid, M. Babics, N. Gasparini, C. W. Boyle, W. Zhang, Y. Dong, K. J. Thorley, M. Neophytou, R. S. Ashraf, J. R. Durrant, D. Baran and I. McCulloch, Adv. Funct. Mater., 2018, 0, 1808429 Search PubMed.
- Y. Lin, T. Li, F. Zhao, L. Han, Z. Wang, Y. Wu, Q. He, J. Wang, L. Huo, Y. Sun, C. Wang, W. Ma and X. Zhan, Adv. Energy Mater., 2016, 6, 1600854 CrossRef.
- H. Bristow, K. J. Thorley, A. J. P. White, A. Wadsworth, M. Babics, Z. Hamid, W. Zhang, A. F. Paterson, J. Kosco, J. Panidi, T. D. Anthopoulos and I. McCulloch, Adv. Electron. Mater., 2019, 5, 1900344 CrossRef CAS.
- C. Yao, Y. Yang, L. Li, M. Bo, C. Peng and J. Wang, J. Mater. Chem. A, 2019, 7, 18150–18157 RSC.
- Y. Yang, C. Yao, L. Li, M. Bo, J. Zhang, C. Peng, Z. Huang and J. Wang, Dyes Pigm., 2020, 181, 108542 CrossRef CAS.
-
R. J. Ouellette and J. D. Rawn, 2 - Functional Groups and Their Properties. in Organic Chemistry, ed., R. J. Ouellette, J. D. Rawn, Academic Press, 2nd edn, 2018, pp 31–49 Search PubMed.
- E. Caldeweyher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2017, 147, 034112 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- D. Aravena, M. Atanasov and F. Neese, Inorg. Chem., 2016, 55, 4457–4469 CrossRef CAS PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- C. Yao, L. Xin, Y. Yang, L. Li, M. Bo, C. Peng and J. Wang, J. Mater. Chem. A, 2022 10.1039/D2TA03728A.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
-
D. Studio, Device Studio, 2021A, Hongzhiwei Technology, China, 2021 Search PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- R. A. Marcus, Rev. Mod. Phys., 1993, 65, 599–610 CrossRef CAS.
- J. Ridley and M. Zerner, Theor. Chim. Acta, 1973, 32, 111–134 CrossRef CAS.
- C. Yao, C. Peng, Y. Yang, L. Li, M. Bo and J. Wang, J. Mater. Chem. C, 2018, 6, 4912–4918 RSC.
- C. Yao, C. Peng, Y. Yang, L. Li, M. Bo and J. Wang, J. Phys. Chem. C, 2018, 122, 22273–22279 CrossRef CAS.
- C. Yao, Y. Yang, L. Li, M. Bo, C. Peng and J. Wang, J. Mater. Chem. C, 2018, 6, 6146–6152 RSC.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148–7151 CrossRef CAS PubMed.
- J.-C. Lee, J.-D. Chai and S.-T. Lin, RSC Adv., 2015, 5, 101370–101376 RSC.
- T.-J. Wen, Z.-X. Liu, Z. Chen, J. Zhou, Z. Shen, Y. Xiao, X. Lu, Z. Xie, H. Zhu, C.-Z. Li and H. Chen, Angew. Chem., Int. Ed., 2021, 60, 12964–12970 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Lei
Li
a,
Maolin
Bo
*a,
Lei
Li
a,
Maolin
Bo