
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVersatile methods for improving the mechanical properties of fullerene and non-fullerene bulk heterojunction layers to enable stretchable organic solar cells†
Emilie
Dauzon
 *a,
Xavier
Sallenave
b,
Cedric
Plesse
b,
Fabrice
Goubard
*b,
Aram
Amassian
*c and
Thomas D.
Anthopoulos
*a
*a,
Xavier
Sallenave
b,
Cedric
Plesse
b,
Fabrice
Goubard
*b,
Aram
Amassian
*c and
Thomas D.
Anthopoulos
*a
aKing Abdullah University of Science and Technology (KAUST), KAUST Solar Centre, Thuwal 23955-6900, Saudi Arabia. E-mail: emilie.dauzon@kaust.edu.sa; thomas.anthopoulos@kaust.edu.sa
bLaboratoire de Physicochimie des Polymères et des Interfaces (LPPI), CY Paris Cergy Université, 95000 Cergy, France. E-mail: fabrice.goubard@cyu.fr
cDepartment of Materials Science and Engineering, and Organic and Carbon Electronics Laboratories (ORaCEL), North Carolina State University, Raleigh, N.C. 27695, USA. E-mail: aamassi@ncsu.edu
First published on 27th January 2022
Abstract
We develop and evaluate two methods aimed at enhancing the mechanical resistance and stretchability of bulk heterojunction active layers in organic solar cells while maintaining photovoltaic performance intact. These techniques consist of adding a cross-linker or an elastomer into the active layer. First, we study P3HT:PC61BM blend as a reference polymer:fullerene acceptor system for our conceptual demonstration. The diazido cross-linker selectively reacts with PC61BM molecules and the resulting blend shows an overall softening effect of the mix, while the power conversion efficiency (PCE) increases by up to 20%, which we attribute to increased aggregation of P3HT. Addition of elastomers affects the blend differently: the crystallinity of the donor decreases, which can have a detrimental effect on the PCE of certain materials. However, the elastomer method is found to be considerably more universal by acting as a mechanical softener in modern fullerene-free blend systems as well. In particular, SEBS elastomer displayed a tailored elasticity with various fullerene and non-fullerene blends: P3HT:PC61BM, PCE10:PC71BM, and PCE13:IT-4F. The potential of our SEBS process is demonstrated in PCE13:IT-4F:SEBS solar cells with a PCE of 11.5%. This versatile approach highlights the ease of manufacturing and scalability achieved by the solution casting processes, along with the high compatibility of acceptor and donor blends.
Introduction
Organic photovoltaic (OPV) devices have received much attention in the last two decades due to their promise of low-cost and environmentally friendly photovoltaics fabricated using scalable solution processing methods such as roll-to-roll manufacturing. Research in this area has mainly focused on improving the power conversion efficiency (PCE), which has recently approached 19.6%.1 Besides this promising performance, OPVs certainly offer other advantages such as portability, lightweight, and flexibility. One aspect of material design, which is rarely considered, but essential is mechanical compliance. Several other interesting potential applications require elasticity, like foldable smartphones, human skin, and wearable electronic textiles.2 Extensive work has also focused on stabilizing OPV materials and devices, a key step towards their commercial viability.3–9 In this regard, Krebs et al. found that one of the significant problems of OPV prototypes tested in the field was the fragility of OPV modules in the hands of children due to mechanical failure.5 Material and device stability along with good mechanical compliance and resistance to failure are therefore important to achieving long-term durability of OPV-based products.2 In contrast to the common opinion that organic materials are ductile, archetypal materials such as P3HT:PC61BM are stiff (Young's modulus: 2–3 GPa)10–13 and therefore not the most suitable for plastic electronics. Mechanical compliance and stretchability can be induced by applying architectural modifications to a device extrinsically, or intrinsically by manipulating material chemistry and blending.14–17 However, with the fast progress in the organic bulk heterojunction (BHJ) systems in terms of materials development, an adaptable method is now required to satisfy non-fullerene acceptor blends as well. To minimize costs and avoid the time-consuming and complexity associated with the chemical alteration of new polymers, the development of a general method applicable to achieve stretchability in a wide range of BHJ material systems would be particularly useful.Cross-linking is a multipurpose tool to link molecules together by chemical bonds or bridges under a stimulus (ultraviolet irradiation or heat treatment). This strategy has been demonstrated to be a viable solution in OPVs for thermal and long-term stability.18–20 Furthermore, the three-dimensional network caused by cross-linking could provide opportunities to prevent mechanical failure and improve the overall elasticity. The benefit of cross-linking under strain has been demonstrated as an effective method.21–23 The decrease of the elastic modulus of 3,6-di-2-thienyl-pyrrolo[3,4-c]pyrrole-1,4-dione (DPP)-based conjugated polymer using siloxane as a cross-linker was confirmed by Wang et al.21 While there are many studies on the modification of the BHJ polymer chemistry to enhance the deformation,13,14,24,25 less work has explored the effect of the cross-linkable molecule on the elasticity of BHJ blends. Derue et al. developed cross-linking molecules by click chemistry having two functional azides, 4,4′-bis(azidomethyl)-1,1′-biphenyl (BABP), to limit the diffusion of PC61BM and hinder its crystallization.18,19,26
Another straightforward approach to enhance the stretchability is blending electronic materials with soft elastomers. Conjugated polymers dispersed in a stretchable matrix allow extreme mechanical deformations without compromising the electronic functionality. Materials embedded homogeneously in a rubber matrix have shown sufficient mechanical improvement to promote intrinsic stretchability of semiconductor polymers. For example, poly(3-hexylthiophene) (P3HT) blended with poly(dimethylsiloxane) (PDMS) leads to a stretchable material maintaining good charge mobility.27–29 Similarly, P3HT mixed with SEBS (Styrene Ethylene Butylene Styrene Block Copolymer) show high deformation.30 However, few reports demonstrate their mechanical benefits of blending in OPV devices.31,32
We investigate two different approaches to allow the photoactive BHJ layer to achieve mechanical compliance, namely the cross-linking or blending pathways. First, we investigate the effect of cross-linking on the elastic deformation of conjugated polymers. This is conceptually illustrated in Fig. 1a for a photoactive layer sandwiched between charge extracting layers and electrodes. In a first approach, we utilize three cross-linkable molecules (Fig. 1b) to tune the mechanical properties of the photoactive layer, namely 1,12-diazidododecane (C12N3), 1,11-diazido-3,6,9-trioxaundecane (PEG3N3), and poly(ethylene glycol)bisazide (PEGnN3). The chemistry of the cross-linker chains may play a role in the elasticity as suggested in some studies where organic polymers show a correlation between molecular weight and mechanical properties.33–35 Consequently, we decided to investigate different cross-linker chain natures: C12N3 has a short non-polar chain, PEG3N3 has a short polar chain, and PEGnN3 is long and polar.
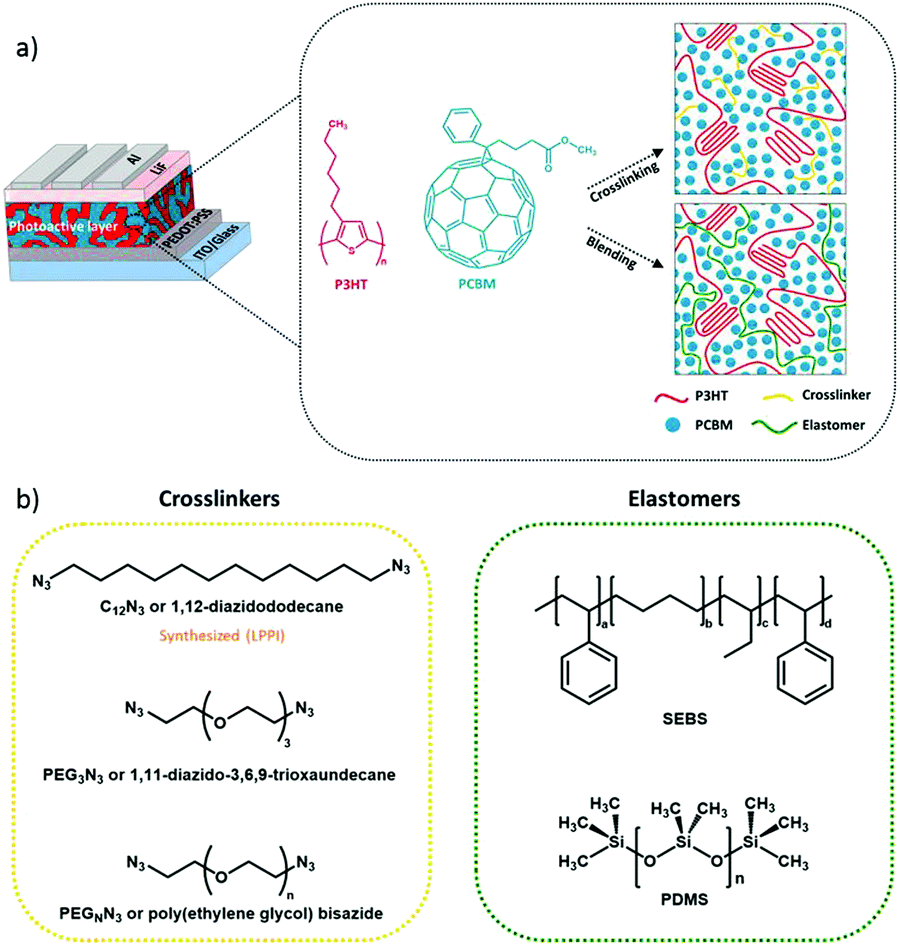 | ||
| Fig. 1 (a) Illustration of the strategies to improve the mechanical reliability of blend solar cells via fullerene cross-linking or elastomer blending. (b) Chemical structures of the cross-linker molecules and elastomers. | ||
In a second approach, we utilize a physical blending approach whereby an elastomer is composited with the BHJ as an effective approach to mechanically soften the photoactive layer.27,29,30,36,37 For the first time to our knowledge, we successfully implement elastomers as plasticizer components into photoactive BHJ layer of OPV devices. Unlike bis-azide molecules, which covalently bond to two PC61BM molecules, requires chemical specificity and treatment such as thermal annealing, the elastomer does not covalently bond with any BHJ component. Hence, this strategy can be extended in principle for thermally sensitive state-of-the-art non-fullerene OPV materials. We select a physical network, SEBS (Styrene Ethylene Butylene Styrene Block Copolymer) and a chemical network, PDMS (Polydimethylsiloxane) as elastomer components for this study (Fig. 1b). SEBS does not require heat treatment, which presents a major advantage over PDMS and cross-linkable molecules in solar cell fabrication. Heat-less method can increase the manufacturing speed and cuts the process complexity. Both of these strategies will be first evaluated on a reference BHJ photoactive layer consisting of P3HT:PC61BM, then extended to more modern donor:acceptor blends where small-molecule acceptors other than fullerene are commonly employed.
Experimental section
Materials
Diazido-3,6,9-trioxaundecane (PEG3N3), poly(ethylene glycol) bisazide (PEGnN3), 1,12-dibromododecane, regioregular poly(3-hexylthiophene-2,5-diyl) (P3HT), [6,6]-phenyl-C61-butyric acid methyl ester (PC61BM) and [6,6]-phenyl-C71-butyric acid methyl ester (PC71BM) were purchased from Sigma. PEGnN3 has a molecular weight (MW) of 1100 g mol−1, compared to 244 g mol−1 for PEG3N3. PDMS was obtained from Dow Corning Sylgard 186. PEDOT:PSS low grade AI 4083 was purchased from Heraeus. Styrene ethylene butylene styrene block copolymer (SEBS) H1221 was purchased from Asahi Kasei. The following polymers were from Ossila: poly[4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)benzo[1,2-b;4,5-b′]dithiophene-2,6-diyl-alt-(4-(2-ethylhexyl)-3-fluorothieno[3,4-b]thiophene)-2-carboxylate-2-6-diyl)] named PTB7-th or PCE10, 3,9-bis(2-methylene-((3-(1,1-dicyanomethylene)-6,7-difluoro)-indanone))-5,5,11,11-tetrakis(4-hexylphenyl)-dithieno[2,3-d:2′,3′-d′]-s-indaceno[1,2-b:5,6-b′]di-thiophene (IT-4F), poly[(2,6-(4,8-bis(5-(2-ethylhexylthio)-4-fluorothiophen-2-yl)-benzo[1,2-b:4,5-b′]dithiophene))-alt-(5,5-(1′,3′-di-2-thienyl-5′,7′-bis(2-ethylhexyl)-benzo[1′,2′-c:4′,5′-c′]dithiophene-4,8-dione)] (PBDB-TSF or PCE13).C12N3 compound was synthesized by reacting NaN3 with 1,12-dibromododecane (see details in ESI†). P3HT:PC61BM blend (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.8 ratio) was dissolved in chlorobenzene at 60 °C for 3 hours. The final concentrations were 15 mg mL−1 of P3HT and 12 mg mL−1 of PC61BM. The solution was cooled down at room temperature, and different amounts of additive were incorporated in chlorobenzene into P3HT:PC61BM solution. The ratio of additive was determined in weight percent (wt%) compared to the acceptor (PC61BM) content. These solutions were either spin-coated or drop-casted, depending on the aimed characterization.
:0.8 ratio) was dissolved in chlorobenzene at 60 °C for 3 hours. The final concentrations were 15 mg mL−1 of P3HT and 12 mg mL−1 of PC61BM. The solution was cooled down at room temperature, and different amounts of additive were incorporated in chlorobenzene into P3HT:PC61BM solution. The ratio of additive was determined in weight percent (wt%) compared to the acceptor (PC61BM) content. These solutions were either spin-coated or drop-casted, depending on the aimed characterization.
Mechanical properties
Young's modulus was measured by traction on Q800 Dynamic Mechanical Analysis (DMA) from TA Instruments. The self-standing thin film (≈10 μm thick) was clamped, and a preload force of 0.05 N was applied until 0.05% strain. We used the mode strain rate at 20% min−1 to failure. The tensile test has its limitations.Young's modulus can be determined by buckling technique initially described by Stafford et al.38 The method is based on the formation of wrinkles in a relatively rigid film on a relatively compliant substrate under compressive strain.39 The tensile modulus of the film (Ef) is correlated to the modulus of the substrate (Es), the thickness of the coated film (tf), the wavelength of the buckles (λb), and the Poisson's ratios of the film and substrate (νf and νs):38
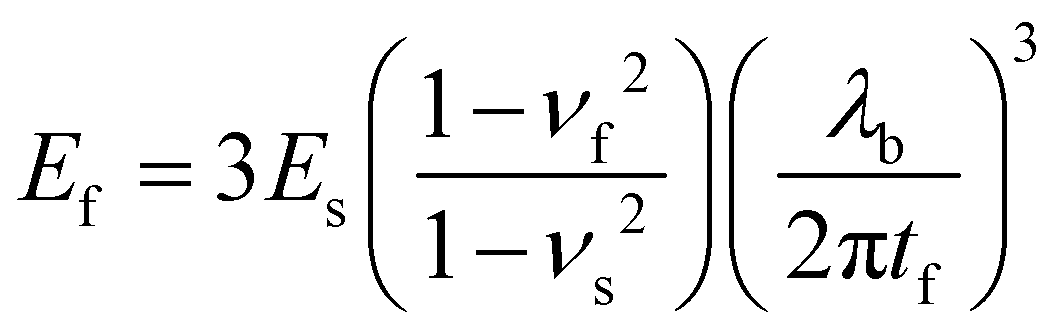 | (1) |
Thermal analysis
The variation of storage modulus and the loss tangent (tanδ) measured with Q800 Dynamic Mechanical Analysis (DMA) from TA Instruments, as a function of the temperature. We used infrared spectroscopy FTIR Cary 680 from Agilent, thermal gravimetric analysis (TGA) TG 209 from Netzsch and differential scanning calorimetry (DSC) TA Discovery 250.
Surface analysis and imaging
Optical microscope (Nikon eclipse LV100POL) mounted with a camera was used to observe the surface. Atomic force microscopy (AFM) from Bruker® was used to image the surface topography of a sample by analyzing point by point with a local probe scan. The images were taken in ScanAsyst mode (constant force, frequency of 2 kHz). The UV-Visible absorption spectra was measured using a UV-vis-NIR spectrophotometer (JASCO V-570).GIWAXS characterization
Grazing-Incidence Wide-Angle X-ray Scattering (GIWAXS) is a versatile technique for characterizing an object at the nanoscale structure. 2D GIWAXS experiments were performed at D1 beamline at the Cornell High Energy Synchrotron Source. The beamline energy was 1.17 Å, and the sample detector distance was 167.5 mm. An exposure time of 5 s was used for ex situ experiments. X-ray incidence angle was fixed at an optimal angle of 0.12° probing P3HT:PC61BM film and minimizing the signal from the substrate. 2D GIWAXS image is reshaped with GIXSGUI software.42 The reshaping is a simple two-dimensional image transformation taking account of some geometric and intensity correction resulting to a missing wedge image.OPV devices
Glass patterned with 140 nm of indium tin oxide (ITO) substrates were cleaned by an ultrasonic bath with sodium dodecyl sulfate solution (SDS), deionized water, acetone, and isopropanol. UV-Ozone cleaner was used for 15 min to remove all organic residues and to improve the wettability of PEDOT:PSS. We used PEDOT:PSS AI 4083 from Heraeus Clevios as a hole transport layer (HTL). After a pre-filtration (0.25 μm disc), the PEDOT:PSS mixture was deposited by spin coating (4000 rpm, 50 s, 2000 rpm s−1). Thermal treatment was performed at 140 °C for 10 min. The thickness was measured by a profilometer with average values of 60 ± 5 nm.P3HT:PC61BM mixture with additives, was deposited by spin coating at 500 rpm during 20 s. We kept the cells in a Petri dish overnight. Then, annealing was done at 140 °C for 10 min. PCE10:PC71BM (1:1.5, w/w, 25 mg mL−1) was mixed in chlorobenzene for 2 hours at 70 °C prior spin coating at 2000 rpm for 60 s. The samples were rinsed with 60 μL of methanol at 4000 rpm for 30 s. Then, they were kept under vacuum for 30 min prior further deposition. 0.6 nm of lithium fluoride (LiF) was evaporated on top of P3HT:PC61BM and PCE10:PC71BM.
PCE13:IT-4F (1:1, w/w, 20 mg mL−1) mixture was spin-coated (2000 rpm for 60 s) on top of AI4083, no annealing was required. (2-(1,10-phenanthrolin-3-yl)naphth-6-yl)diphenylphosphine (Phen-NaDPO) as cathode interfacial material (0.5 mg mL−1 in isopropanol) was spin-coated at 2000 rpm for 30 s on top of this active layer. Finally, we evaporated aluminum metal (100 nm) on all the cells. The area was 10 mm2. Organic solar cells were tested under AM 1.5G solar irradiation, in the nitrogen glove box, by solar simulator from Newport.
Results and discussion
Preamble – cross-linking characterization
Due to the thermodynamic instability of P3HT:PC61BM active layer, the diazido additives have been successfully used to stabilize the structure over time by decreasing the diffusion of the PC61BM,18,19,26 and here, we explore the possible improvement of the stretchability of the BHJ through this cross-linking approach. The reaction of fullerene with organic diazides produces “azafulleroids”. The mechanism of the reaction between diazide with PC61BM is shown in Fig. 2a. Under heat treatment, bis-azide molecules react selectively on the fullerene by a 1,3-cycloaddition and release two dinitrogen molecules.43 Differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), and infrared spectroscopy have been carried out to prove the selective reaction of cross-linker molecules on the fullerene.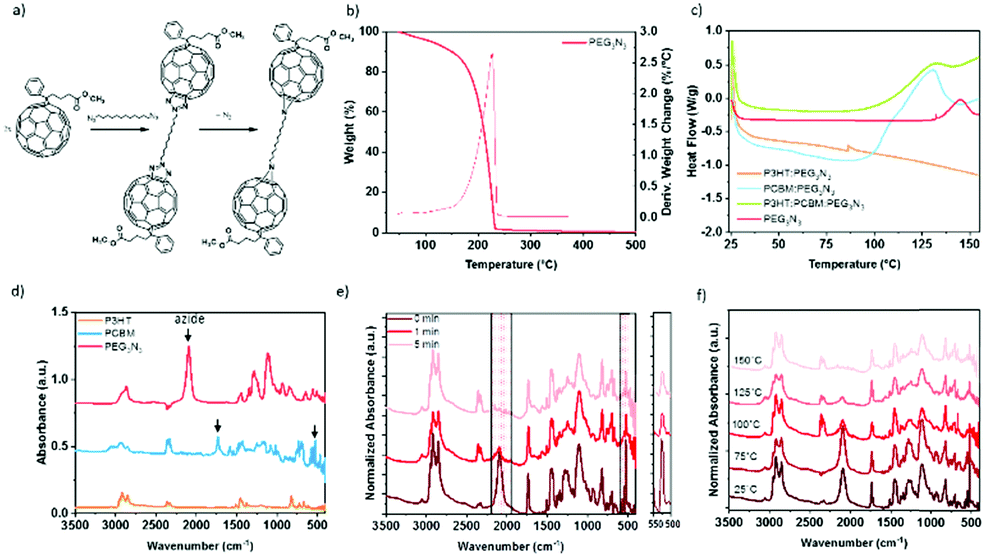 | ||
| Fig. 2 (a) Scheme of reaction between bis-azide and PC61BM molecule under heat treatment. (b) Thermal gravimetric analysis of cross-linkers PEG3N3. (c) DSC analysis of PEG3N3 mixed with either P3HT (1:1) or PC61BM (1:1), or P3HT:PC61BM (1:1:1). (d) FTIR spectra of the precursors before cross-linking. (e) Kinetic of cross-linking via monitoring P3HT:PC61BM:PEG3N3 after thermal annealing at 150 °C. (f) Temperature effect on P3HT:PC61BM:PEG3N3, for 5 min. | ||
Fig. 2b–f demonstrates the thermal change of PEG3N3 as cross-linker. The thermal characterizations of all other cross-linkers P3HT, and PC61BM are shown in the ESI.† The acceptor PC61BM presents two endothermic peaks at 247 and 280 °C (Fig. S2, ESI†), corresponding to the crystallization and the melting transition, respectively.44 The donor P3HT exhibits a Tg between 100 and 140 °C and a melting point at 220 °C (Fig. S2, ESI†). For PEG3N3 cross-linker, the TGA displays a rapid weight loss after 150 °C (Fig. 2b). The decomposition of azide function into dinitrogen is not seen as the decomposition of the entire molecule hides it. For PEGnN3, the thermal analysis reveals a weight loss of 6.5% between 140 and 180 °C, revealing the azide decomposition. Firstly, the molecule decomposes theoretically, by liberating four nitrogen atoms from the two azide groups, corresponding to an expected 5% weight loss (4 × MN/Mn = 56/1100). Then a second weight loss is observed from 180 to 220 °C, indicating the decomposition of the molecules. DSC was performed at several thermal ranges: from −90 °C to 250 °C (Fig. S2, ESI†) and from room temperature to 160 °C (Fig. 2c) to determine the thermal properties accurately. Below 160 °C, PEG3N3 exhibits an exothermic peak onset at 132 °C, corresponding to the decomposition of the azide function. Similarly, PEGnN3 displays a transition peak that starts at 29 °C, which is the melting of the molecule, while the decomposition of the bis-azide is found at the onset of 143 °C (see Fig. S2, ESI†). Thermal annealing up to 200 °C reveals the decomposition of both components above 170 °C, in agreement with TGA.
DSC was carried out with cross-linkers mixed with P3HT or PC61BM, to prove the selectivity of the cross-linking and the temperature range, as plotted in Fig. 2c. For PEG3N3, the DSC curve in the presence of P3HT does not show any change, while an exothermic peak with a maximum at 125 °C is observed in the presence of PC61BM. This confirms the selectivity of the reaction between PC61BM and the bis-azide molecules.
Kinetics of reaction the bis-azide function can be detected by attenuated total-reflectance Fourier transform infrared (ATR-FTIR) spectroscopy (Fig. 2d). The infrared spectra of each precursor are presented in Fig. 2d–f and Fig. S3 (ESI†). The peak at 2090 cm−1 is attributed to azide functions, for all the cross-linkers: PEG3N3, C12N3, and PEGnN3. PC61BM shows several FTIR C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C characteristics peaks at 1464 and 526 cm−1, due to the C60 cage, and 1730 cm−1 corresponding to the CO bond of the butyric acid methyl ester, in agreement with the literature.45 IR spectra of P3HT:PC61BM:PEG3N3 with weight ratio (1:0.8:0.4) after thermal annealing at 150 °C during 0, 1, and 5 min is shown in Fig. 2e. The intensity of the azide peak (νN3 = 2090 cm−1) was reduced by 70% after only 1 min at 150 °C and disappeared totally in 5 min of heating. At the same time, the heat treatment decreases the vibrational bands of the C60 cage of the PC61BM (526 cm−1). This decrease is attributed to side group attachment of fullerenes to additive molecules, as described by the mechanism in Fig. 2a. This behavior is observed for the other cross-linkers in Fig. S3 (ESI†). In Fig. 2f, all materials were annealing for 5 min at various temperatures before the FTIR analysis. At room temperature and 75 °C, no change is detected, while above 100 °C, the intensity of azide peak decreases, in agreement with the DSC data. The kinetic of curing depends on the temperature, indicating a complete reaction at 150 °C of the azide group with the fullerene. Elevated temperature accelerates the cross-linking rate, as observed in Fig. 2f. According to these results, a 15 min curing at 150 °C was proceeded for each component to assure the complete selective reaction of azide and PC61BM, unless stated otherwise.
C characteristics peaks at 1464 and 526 cm−1, due to the C60 cage, and 1730 cm−1 corresponding to the CO bond of the butyric acid methyl ester, in agreement with the literature.45 IR spectra of P3HT:PC61BM:PEG3N3 with weight ratio (1:0.8:0.4) after thermal annealing at 150 °C during 0, 1, and 5 min is shown in Fig. 2e. The intensity of the azide peak (νN3 = 2090 cm−1) was reduced by 70% after only 1 min at 150 °C and disappeared totally in 5 min of heating. At the same time, the heat treatment decreases the vibrational bands of the C60 cage of the PC61BM (526 cm−1). This decrease is attributed to side group attachment of fullerenes to additive molecules, as described by the mechanism in Fig. 2a. This behavior is observed for the other cross-linkers in Fig. S3 (ESI†). In Fig. 2f, all materials were annealing for 5 min at various temperatures before the FTIR analysis. At room temperature and 75 °C, no change is detected, while above 100 °C, the intensity of azide peak decreases, in agreement with the DSC data. The kinetic of curing depends on the temperature, indicating a complete reaction at 150 °C of the azide group with the fullerene. Elevated temperature accelerates the cross-linking rate, as observed in Fig. 2f. According to these results, a 15 min curing at 150 °C was proceeded for each component to assure the complete selective reaction of azide and PC61BM, unless stated otherwise.
Film morphology in the presence of additives
In the previous section, the cross-linking process was studied and validated. The diazide cross-linkers react selectively with PC61BM molecules. From now, the preparation of P3HT:PC61BM (1:0.8 ratio) film involves blending fixed amounts of reactive cross-linkers or elastomers with the organic semiconductors. Solutions were mixed to achieve a target additive content in weight percent (wt%) compared to the acceptor weight. The blend solution was spin-coated and annealed at 150 °C for 15 min before testing. In this section, the impact of the addition of cross-linkers and rubbers into the active layer on the morphology, mechanical properties along photovoltaic characteristics are studied.
Optical microscopy allows us to understand, to some extent, phase separation. Thus, Fig. 3a and Fig. S4 (ESI†) show the micro-texture change in the presence of additives into P3HT:PC61BM blends after thermal annealing at 150 °C for 15 min. The phase segregation seems unchanged on the investigated scale with 10 wt% additives (Fig. 3a). With the increase of the additive content, phase separation is denoted. In particular, for SEBS and PDMS at 50 wt%, the elastomer and the blend appear highly segregated (Fig. 3a). The size of domains reaches up to 10 μm for 50 wt% SEBS and about 5 μm for 50 wt% PDMS. Furthermore, the morphology of P3HT:PC61BM in the presence of SEBS was analyzed by atomic force microscopy (AFM) (Fig. 3b). A phase-separated blend is visibly observed for different content, even as low as 10 wt% of SEBS (about 5 μm long). The poor miscibility of an elastomer with the blend could explain the phase separation.
 | ||
| Fig. 3 (a) Optical micrographs of P3HT:PC61BM in the presence of additives as indicated in the picture after annealing. (b) AFM images of P3HT:PC61BM blended with and without SEBS. | ||
P3HT:PC61BM active layer is thermodynamically unstable.46 The morphology changes with temperature. This problem is well known in the literature and ascribed to the dissemination and PC61BM crystallization molecules under thermal annealing.47,48 Crystallization could demonstrate dramatic effects on photovoltaic performances,46 but also on mechanical properties.21,49,50 While mechanical stability is the main concern in this work, the prevention of PC61BM crystallization remains essential to ensure elasticity. After 60 hours thermal annealing at 150 °C, PC61BM molecules diffuse into the active layer and form microcrystals, as shown in Fig. S5 and S6 (ESI†). Without additive, after heat treatment, PC61BM molecules form microscopic size crystals (about 20 μm2). When C12N3 is added, the cross-linking of the PC61BM molecules by –N3 groups prevents the growth of PC61BM microcrystals in the photoactive layer after 60 hours at 150 °C. The same effect is observed with PEG3N3 even after 60 h at 150 °C. In the presence of 10 wt% PEGnN3, the morphology after aging is not stabilized. This shows that 10 wt% PEGnN3 cross-linkers do not have the desired effect. This trend may be explained with the weight ratio of PEGnN3; there is only one azide function for about 24 molecules of PC61BM, while the two other cross-linkers are in a 1:1.4 ratio azide compared to PC61BM. Consequently, only a few of the fullerene derivatives are cross-linked with PEGnN3 and may explain the difference in cross-linker stability behavior compared to C12N3 and PEG3N3. This cross-linking prevents the crystallization of PC61BM during heat treatment at 150 °C. Several reports confirm the favorable effect of cross-linking on thermal aging.18,30,51 The embedding of P3HT:PC61BM with elastomeric SEBS slows down the diffusion of PC61BM molecules as a net reduction of the crystal is seen compared to the neat P3HT:PC61BM sample (Fig. S6, ESI†). This behavior can be beneficial for long-term stability.
UV-Visible absorption has been used for measuring the optical properties of semiconductors in films. The UV-vis spectra for as-casted films of PC61BM, P3HT, and P3HT:PC61BM blend is shown in Fig. S7a, (ESI†). PC61BM shows a low optical absorbance between 400 to 800 nm and a maximum at 335 nm. P3HT exhibits a broad absorption peak centered at 550 nm with three significant vibronic polymer bands at 520, 553, and 602 nm due to the relatively good organization of the polymer chains in the film.52,53 The additive effect on P3HT:PC61BM film morphology was investigated on the annealed thin films. Fig. S7b (ESI†) shows the absorption spectra normalized to the PC61BM maximum (335 nm). An increase of the absorbance intensity at 520 nm is observed for the cross-linked film with azide-based molecules, indicating an increase in the degree of P3HT aggregation. On the contrary, the elastomer addition decreases the absorption intensity with a blue shift at 510 nm. This indicates that the degree of P3HT aggregation decreases in the presence of elastomeric additives.
Understanding the effect of the additives on the morphology is critical to ensuring good charge transport in the OPV device. Fig. 4a corresponds to the GIWAXS 2D image obtained for P3HT:PC61BM thin film. The scattering patterns along with qz correspond to crystallized P3HT lamellar stacking with a primary edge-on orientation. The Bragg peaks rising at qz = 3.8, 7.5 and 11.5 nm−1 correspond to P3HT lamellae layer plans (100), (200), and (300), respectively. The (010) peak at q ∼ 16.5 nm−1, along qxy, is typical for P3HT π–π stacking, with a spacing of 0.37 nm. The ring positioned at q = 14.2 nm−1 is due to scattering from disordered PC61BM molecules. This is in agreement with previous studies.54–57
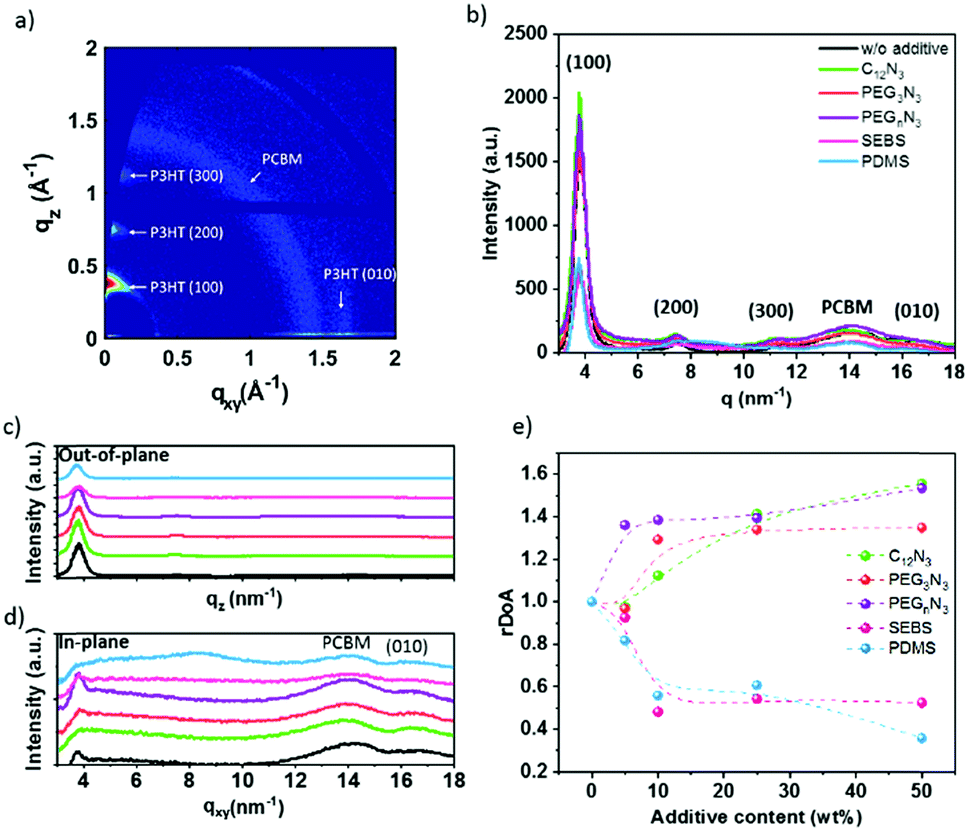 | ||
| Fig. 4 (a) Two-dimensional GIWAXS image of P3HT:PC61BM thin film. (b) Effect of the additives on 2D GIWAXS diffraction pattern: (b) fully integrated, (c) out-of-plane integrated and (d) in-plane integrated signal, and (e) the relative degree of aggregation to neat P3HT:PC61BM of (100) peak. | ||
Fig. 4b–e and Fig. S9 (ESI†) demonstrate the impact of the additives on the GIWAXS pattern. In agreement with absorption data, an increase in the intensity of the P3HT lamellae peak is observed in the presence of cross-linker molecules (C12N3, PEG3N3, PEGnN3). Fig. 4c shows strong out-of-plane alkyl diffraction for the latest molecules, while the blending with SEBS and PDMS indicates a decrease of intensity and broadening of the peak. Table 1 presents the calculated crystalline parameters of alkyl chain stacking and π–π stacking extracted from films with different additives. Table 1 exhibits a negligible change in d-spacing of (100) lamellar and (010) aromatic peaks for all additives. This indicates that there are no interactions between the additives and P3HT. No dramatic change in correlation length (CCL) is observed in the presence of cross-linkers. This suggests that the cross-linking does not disorder P3HT polymer, which is not surprising. In the presence of cross-linkers, the intensity of the PC61BM ring increases, and a slight shift to a lower q value is perceived from 14.0 to 13.7 nm−1 for C12N3 and 13.8 nm−1 for PEG3N3. This could be due to the cross-linking effect on PC61BM.
:0.8:0.4. Lamellar stacking was taken from the (100) peak, and the π–π stacking was extracted from the (010) peak
| Additive | MW (g mol−1) | Lamellar stacking | π–π stacking | ||
|---|---|---|---|---|---|
| d-Spacing (nm) | CCL (nm) | d-Spacing (nm) | CCL (nm) | ||
| w/o additive | — | 1.65 | 11.8 | 0.37 | 3.7 |
| C12N3 | 252 | 1.66 | 11.5 | 0.38 | 3.8 |
| PEG3N3 | 244 | 1.65 | 11.3 | 0.38 | 4.1 |
| PEGnN3 | 1100 | 1.65 | 11.5 | 0.38 | 3.6 |
| SEBS | — | 1.65 | 11.7 | 0.38 | 4.9 |
| PDMS | — | 1.67 | 14.3 | 0.38 | 4.9 |
On the other hand, the elastomer blending tends to increase the CCL for the π–π stacking, promoting phase separation of P3HT:PC61BM, as confirmed by the optical image. PDMS presents a significant increase of the CCL in both lamellar and π–π stacking and PC61BM scattering position center does not shift in the presence of elastomers. However, the crystallinity of PC61BM decreases in the embedded film with SEBS and PDMS.
The previous result revealed no change in the correlation length with cross-linkers. This effect does not take into account the volume distribution of the material. The quantification of the material blend could play a crucial role in understanding the morphology of film formation. The relative degree of crystalline aggregation (rDoA), also called the relative degree of crystallinity, is given in eqn (2).58 It is proportional to the azimuthal integration of a Bragg peak (in our case, it will concern (100) P3HT lamella peak), as seen in Fig. S10 (ESI†), taking account of a geometrical correction factor sin(χ).
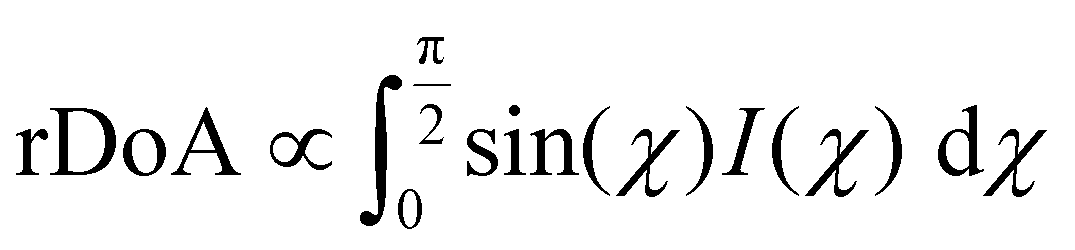 | (2) |
Mechanical properties
The Young's moduli of P3HT:PC61BM mixed with additive are plotted in Fig. 5b and were deduced by strain–stress curves in Fig. 5a. The pristine P3HT:PC61BM shows Young's modulus of 2.1 GPa. This is comparable to the literature.10–13 The diazide cross-linker components decrease the elastic modulus of the P3HT:PC61BM. The addition of 50 wt% cross-linker reduces P3HT:PC61BM modulus by 2.4-fold for C12N3, 2.9-fold for PEG3N3, and 4.6-fold for PEGnN3. Similarly, with elastomer additives, the elastic modulus of P3HT:PC61BM film drops from 2.1 GPa to 0.3 GPa in the presence of 50 wt% SEBS, and to 0.7 GPa for 50 wt% PDMS. Two effects due to the incorporation of additives are distinguishable: (1) the tensile modulus of a P3HT:PC61BM is drastically reduced with the additive concentration, and (2) the increase of the chain length of the cross-linker increases the softening effect in the semiconductors blend. The addition of small molecules has a plasticizing effect, which softens rigid polymers, while it is not perturbing significantly the crystallinity of P3HT as shown earlier. The Young's moduli of films decrease with elastomer blending, which agrees with the incorporation of soft rubber in the film concomitantly with the decrease of relative crystallinity of P3HT aggregates, deduced by UV-visible spectra and GIWAXS.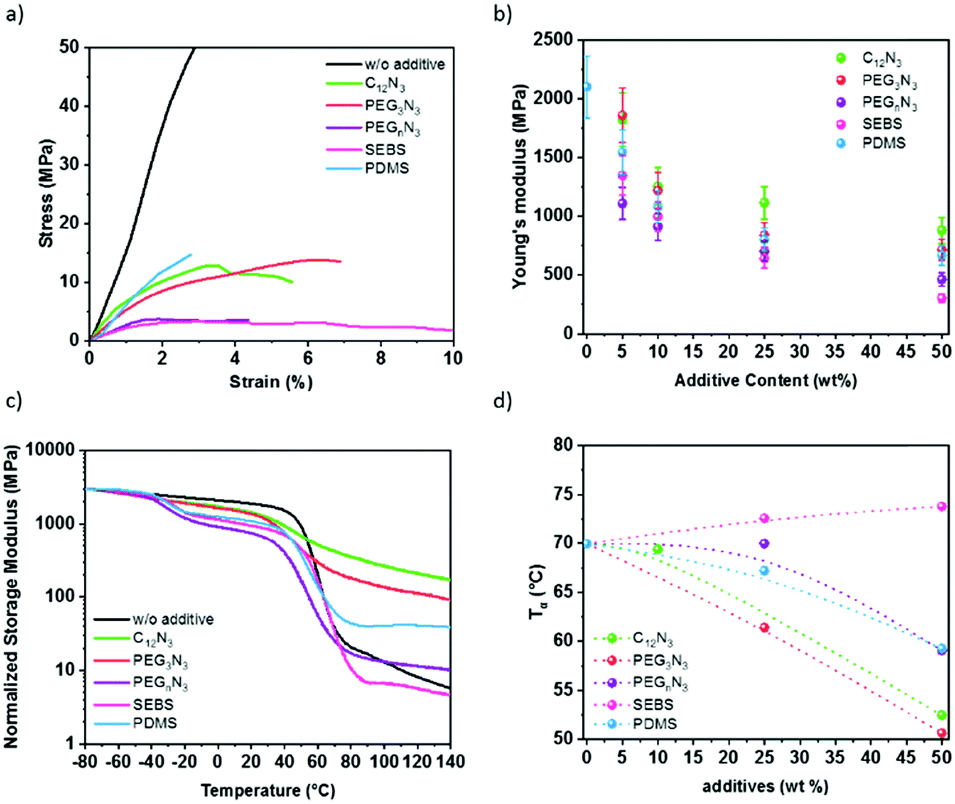 | ||
| Fig. 5 Effect of the additives on the mechanical properties i.e. (a) stress–strain curves with 50 wt% additive content. (b) Average Young's modulus according to the weight percent of additives added into P3HT:PC61BM blend prior to annealing. (c) Temperature dependence of storage modulus for the P3HT:PC61BM:additive composites (1:0.8:0.4), i.e., in the presence of 50 wt% additives. (d) Summary of the alpha temperature. | ||
Furthermore, Young's modulus determines the ability to resist stress, i.e., the material's stiffness. The material softness is not automatically related to the ability to be stretched. The elasticity of the material needs to be determined. For that reason, the fracture strain was extracted from a tensile test for an average of three samples. Table 2 shows a summary of the mechanical properties deduced from the measurements on 10–20 μm thick films. While the elastic modulus decreases exponentially with the additive content due to a plasticizer effect, the fracture strain does not increase as much. The fracture strain increases by ∼50% for all cross-linked films. Surprisingly, PDMS does not contribute to the elasticity more than the cross-linkers. In comparison, some studies show high deformability of P3HT nanowires blended with PDMS elastomer keeping the mobility constant under 100% strain.27,28 In our work, the presence of PC61BM as nano-charges might stiffen the resulting blends compared to only polymer/elastomer blends. On the other hand, the blending of P3HT:PC61BM with elastomers promotes the elasticity to 54% strain with 50 wt% SEBS. This suggests an excellent softening effect of SEBS, as previously seen for P3HT:SEBS composites.30
| Additive | E (MPa) | Fracture strain (%) | T α (°C) |
|---|---|---|---|
| w/o additive | 2103 | 4 | 70 |
| C12N3 | 880 | 6 | 53 |
| PEG3N3 | 715 | 7 | 51 |
| PEGnN3 | 462 | 5 | 59 |
| SEBS | 303 | 54 | 74 |
| PDMS | 667 | 6 | 59 |
Fig. 5c shows the variation of storage modulus and the loss tangent (tanδ) measured with dynamic mechanical analysis instrument (DMA), as a function of the temperature of P3HT:PC61BM thick film with different additives at 50 wt%. The typical behavior of P3HT:PC61BM blend without additive presents a sharp decrease of the storage modulus at 60 °C from the glassy to the rubbery state, corresponding to the α relaxation of the layer59,60 (Fig. 5c). The glass transition temperature of the blend increases with increasing PC61BM content. This trend is in agreement with the result reported by Zhao et al.61 After this transition, the material keeps softening with no stable rubbery plateau observed, indicating a creep behavior. As far as films with additives are concerned, two types of behavior can be observed. For minor molecular weight additives (C12N3 and PEG3N3), only one transition occurs in the same temperature range as neat P3HT:PC61BM but with a less pronounced modulus drop. For macromolecular additives (PEGnN3, SEBS, and PDMS), an additional transition can be observed at a lower temperature (−40 and −20 °C), corresponding to the α relaxation of the macromolecular additives themselves. The glass transition is −20 °C for SEBS and −40 °C for PDMS (Fig. S11, ESI†) which fits the first transition perceived for the P3HT:PC61BM:additives blend. Additionally, another small drop above 100 °C is observed for SEBS corresponding probably to the Tg of the polystyrene blocks responsible of the physical crosslinking of the SEBS network.
Interestingly, with increasing the temperature above the transition of P3HT:PC61BM phase, a rubbery plateau is reached for all three samples indicating that no creep is observed in these conditions, on the opposition of neat films and films modified with low molecular weight additives. From the variation of loss tangent depicted in Fig. 5d, it can be seen that no significant modification of the α temperature is observed for the sample with SEBS, demonstrating poor interaction between SEBS and P3HT:PC61BM phase, due to the heterogeneity of the composite in accordance with the micrographs (Fig. 3). On the other side, a single glass transition is observed with all different compositions with a shift to lower temperature and a broader transition range, attesting to the miscibility of the additives with semiconductor polymer. Plasticizer addition results in a progressive decrease of the α relaxation, acting as a lubricant in polymer chains.62 Accordingly, the glass temperature depression occurring (Fig. 5d) attests to the softening effect, as suggested by the Young's modulus.
Photovoltaic performances
Photovoltaic performances were measured to investigate the influence of additives on photovoltaic efficiency. Fig. 6 shows the photovoltaic characteristics, namely the short-circuit current density Jsc, the open-circuit voltage Voc, the fill factor FF, and the power conversion efficiency (PCE) in the function of the content of the additive. The J–V characteristics curves are plotted in Fig. S12 (ESI†). P3HT:PC61BM was blended with different amounts of additives (0, 5, 10, 25 and 50 wt%) and spin-coated on top of ITO coated with PEDOT:PSS.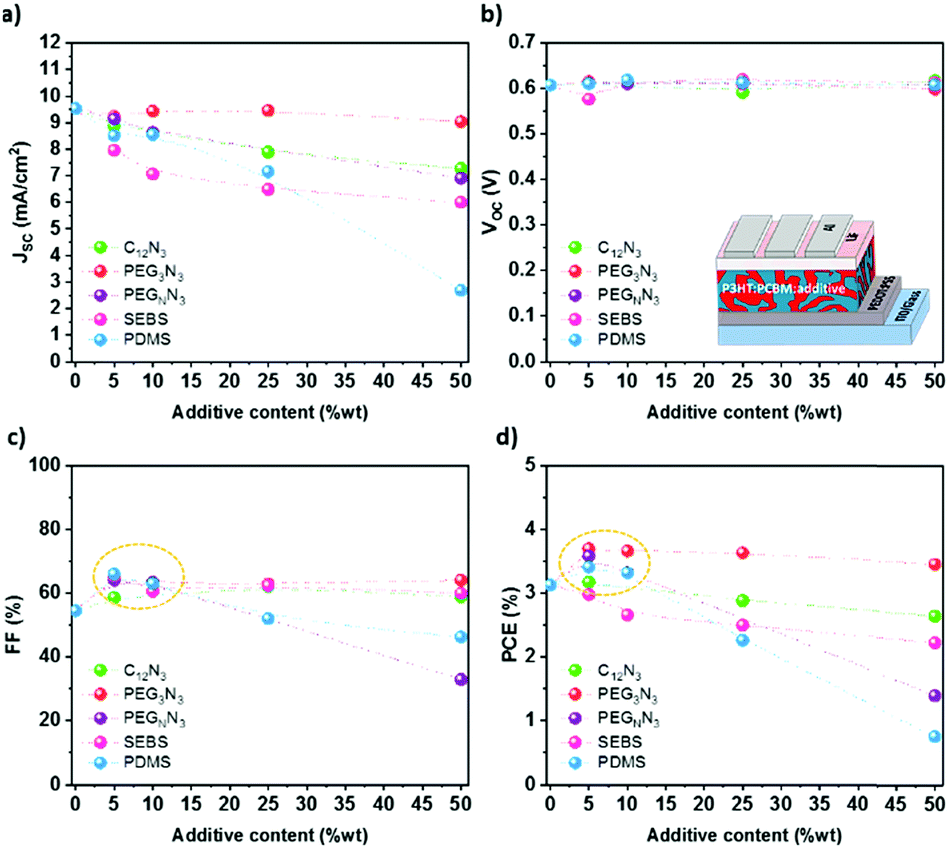 | ||
| Fig. 6 Photovoltaic performances of P3HT:PC61BM-based OPV in the function of the additive content. | ||
The addition of an insulating ternary component into the blend does not significantly influence the Voc, as observed in Fig. 6b. On the other hand, the Jsc decreases with the additive content, even if not significantly for PEG3N3 (Fig. 6a). Below 10 wt%, the increase of the photovoltaic efficiency (Fig. 6d) results from the balance between the increase in the fill factor and the decrease in the short-circuit current. Beyond 25 wt%, the additions of high molecular weight polymer sharply decrease the photovoltaic operational characteristics, especially the short circuit current. At the same time, C12N3 and PEG3N3 based OPVs maintain excellent efficiency with more than 90% of their initial PCE. At 25 wt% SEBS, the PCE drops up to 20% compared to the initial PCE. In analogy with the mechanical measurements, a maximum additive content of 25 wt% allows up to 50% decrease of the Young Modulus and less than 25% decrease of the PCE.
Diazide components have demonstrated their ability to improve the softness and to maintain good electrical properties for OPVs. It is worth noting that the apolar C12N3 cross-linker and the polar PEG3N3 cross-linker have relatively similar lengths and behave similarly as a stretchable additive for OPV. Thus, the polarity of the cross-linkers does not appear to play a significant role in the mechanical and electronics properties of the material systems studied here. While these cross-linkers are suitable for fullerene acceptors only, they require thermal curing, as shown by TGA and DSC analyses. However, solar cells fabrication without thermal annealing is beneficial for several reasons, including reducing process complexity, manufacturing speed, increasing embedded energy and limiting the exposition of thermally sensitive active layer to thermal treatment. The recent progress of our group in the OPV devices shows an efficiency of up to 17% without thermal annealing.63 This solar cell was made from a ternary PBDB-T-2F:Y6:PC71BM bulk-heterojunction. Recently, the chlorination or fluorination of conjugated polymer has also demonstrated annealing-free OPVs with high crystallization and aggregation due to the intermolecular interactions between the halogens and carbon/hydrogen.64 The morphology is therefore optimized and ordered without requiring thermal annealing. While some new systems do not require thermal treatment, any strategy involving thermal annealings like diazide cross-linking or PDMS curing is highly undesirable to avoid a drastic drop in performance. Instead, the second strategy, i.e., the blending with a thermoplastic elastomer, does not require any additional step for curing since physical crosslinks will occur during the casting and drying process. It appears as the ideal pathway to be explored for high-efficiency and more thermally sensitive polymers. This method seems to have excellent versatility and is a straightforward technique to creating stretchability, and may be suitable for other acceptor/donor blends. Our study shows that physically cross-linked SEBS is a great candidate for improving the mechanical properties and keeping excellent photovoltaic performances when it is blended with P3HT:PC61BM at 25 wt% loadings or less. In the next section, we broaden our investigation to modern high-efficiency BHJ systems blended with SEBS.
Other bulk heterojunction blends for high power conversion efficiency
The SEBS additive was introduced to poly[4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)benzo[1,2-b:4,5-b′]dithiophene-alt-3-fluorothieno[3,4-b]thiophene-2-carboxylate] (PTB7-Th or PCE10) and [6,6]-phenyl-C71-butyric acid methyl ester (PC71BM) blends, also referred to PCE10:PC71BM. Fig. 7a shows the chemical structure, the energy band diagram, the absorption spectra, and the J–V curve associated with the PCE10:PC71BM blend at a weight ratio of 1:1.5. PCE10 polymer has a low bandgap (1.6 eV), with a broad absorption between 500 and 800 nm with two peaks at 645 and 700 nm. The solar spectrum between 300 and 800 nm is covered in a complementary manner by PC71BM blended with PCE10 (Fig. 7b). The solar cell layout is glass/ITO/PEDOT:PSS/PCE10:PC71BM/LiF/Al. This BHJ blend yields a PCE of up to 10% (Jsc = 17.3 mA cm−2, Voc = 0.8 V, FF = 60.0%), in agreement with several reports.65–67
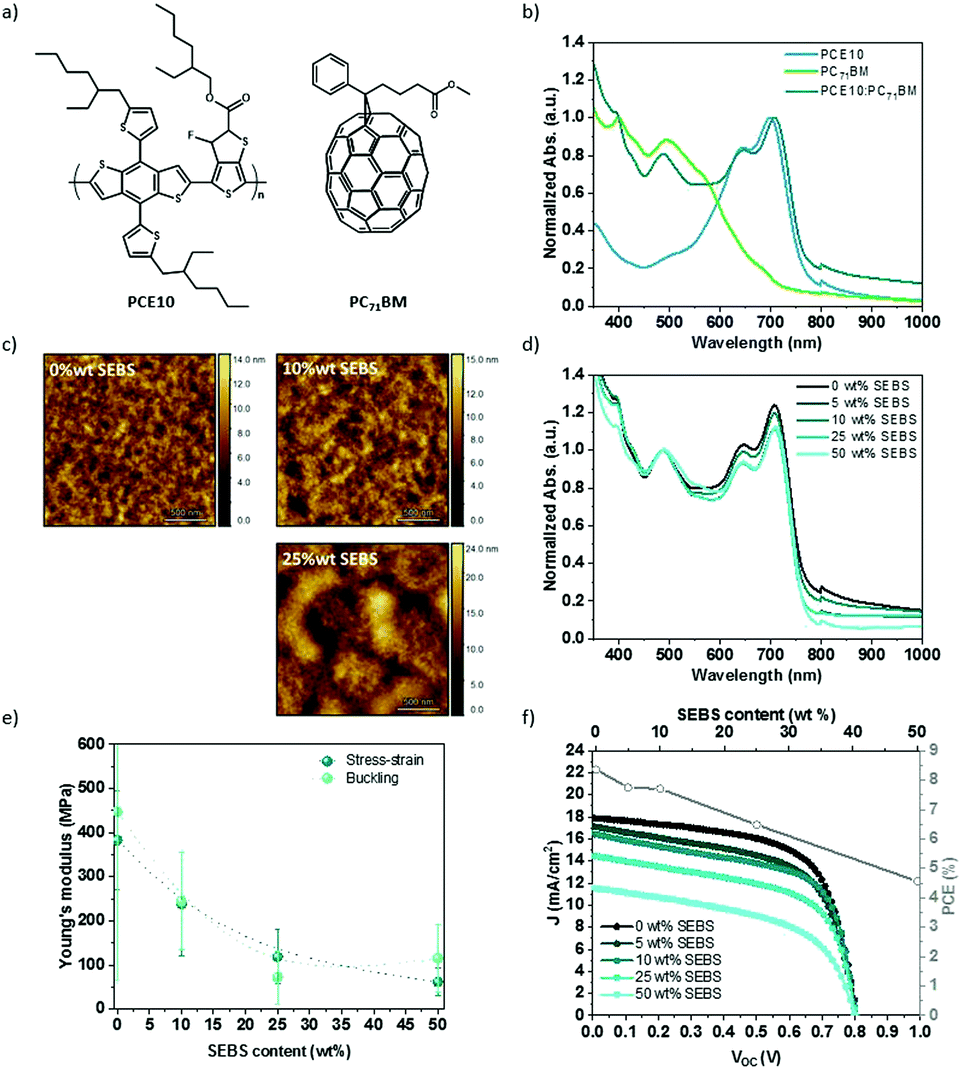 | ||
| Fig. 7 PCE10:PC71BM (1:1.5) solar cell: (a) chemical structures, and (b) absorption spectra. (c) PCE10:PC71BM:SEBS morphology characterized by atomic force microscopy. (d) Absorption spectra of PCE10:PC71BM film mixed with SEBS elastomer. (e) Young's moduli by stress–strain or buckling methods with different amounts of SEBS (wt/wt% PC71BM). (f) J–V characteristics curves of PCE10:PC71BM with SEBS elastomer and associated PCE. | ||
SEBS elastomer was diluted in chlorobenzene at 90 °C, then mixed with PCE10:PC71BM solution (1:1.5) with 3 vol% 1,8-diiodooctane (DIO), and cast. SEBS elastomer content is determined by the amount relative to PC71BM weight. DIO is used to improve morphology.66Fig. 7c and Fig. S13 (ESI†) show the resulting morphology after spin-coating observed by atomic force microscopy (AFM) and optical microscope, respectively. Between 0 to 25 wt% SEBS, no drastic change is seen at the microscale. The addition of 50 wt% reveals a micro-phase separation of the polymers blend. AFM additionally characterized the morphology of the BHJ thin film. The roughness and the domain size increase with SEBS content. The phase-separated thin film shows nano-domains at 10 wt% to micro-domains at 25 wt%. This suggests that the introduction of SEBS increases the phase separation of the blend. In comparison, the phase-separation in PCE10:PC71BM mixture is less noticeable than P3HT:PC61BM for the same content of SEBS (Fig. S13, ESI†). On the contrary, the topography indicates relatively good miscibility between SEBS and PCE10:PC71BM blend.
Fig. 7d displays the normalized absorption spectra of PCE10:PC71BM thin film with SEBS at a weight ratio between 0 and 50 wt% relative to PC71BM. All spectra were normalized at 486 nm corresponding to PC71BM peak. The addition of SEBS decreases the absorption intensity of PCE10 peaks at 645 and 708 nm. SEBS does not absorb in the visible range; its addition leads to the dilution of PCE10 and PC71BM, as expected.
To test the elastic modulus and stretchability of the BHJ films, thick films were made by the solution drop-casting of PCE10:PC71BM:SEBS with 3% DIO on glass, annealed at 50 °C for two hours and washed for 30 min in a methanol bath to remove any residue of DIO co-solvent. While DIO induces a better morphology,66,68 it may act as a lubricant and decrease Young's modulus in some cases13,69 and increase the stiffness of polymers blend in others.70 The drying time is critical to avoid any contribution of the solvent and co-solvent to the mechanical properties. We measured the loss of mass under vacuum at 50 °C, at a fixed time to make sure that the elasticity is not due to an excess of solvent in PCE10:PC71BM films (Fig. S14, ESI†). Our observation suggests that thick PCE10:PC71BM film requires at least an hour of drying before the tensile test. After applying 90 min annealing under vacuum, stress–strain characteristics were recorded at a fixed weight percent of SEBS. The Young's modulus deduced by the slope at the origin of the stress–strain curve is plotted in Fig. 7e, and compared to the buckling method. The two techniques show similar values. At 0 wt% SEBS, the film displays an elastic modulus of around 350 MPa compared to 450 MPa by the buckling method. Our results are in accordance with the elastic modulus reported between 0.1–0.4 GPa for neat PCE10.71 Elastic moduli decrease up to 60 MPa by stress–strain and 77 MPa by buckling with 50 wt% SEBS. This decrease represents between 80 to 85% reduction of initial Young's modulus. The buckling method leads to Young's modulus of the same order of magnitude compared to the stress–strain way. SEBS blending offers an excellent softening effect. The strain at break increases by 4-fold with 50 wt% SEBS compared to the pristine, demonstrating an increase of the stretchability. These results are coherent compared to the previous study with P3HT:PC61BM in presence of SEBS.
Fig. 7f sows the evolution of the photovoltaic characteristics in function of SEBS content. The addition of SEBS demonstrates a decrease in the short circuit current density from 17.3 for the neat polymers blend (1:1.5) to 11.1 mA cm−2 for the mixture with 50 wt% SEBS. FF decreases less than 15% with 50 wt% SEBS compared to neat blend, while Voc remains unchanged. An overall decay of PCE is noticed. PCE drops more than 20% at 25 wt% SEBS and 45% at 50 wt% SEBS. This suggests that SEBS has the same effect on the efficiency of PCE10:PC71BM that, as previously seen with P3HT:PC61BM.
With the recent development of the non-fullerene acceptors, the PCE has progressively increased by over 18%.72–75 Poly[(2,6-(4,8-bis(5-(2-ethylhexylthio)-4-fluorothiophen-2-yl)-benzo[1,2-b:4,5-b′]dithiophene))-alt-(5,5-(1′,3′-di-2-thienyl-5′,7′-bis(2-ethylhexyl)benzo[1′,2′-c:4′,5′-c′]dithiophene-4,8-dione)] (PCE13 or PBDB-T-SF) was blended with 3,9-bis(2-methylene-((3-(1,1-dicyanomethylene)-6,7-difluoro)-indanone))-5,5,11,11-tetrakis(4-hexylphenyl)-dithieno[2,3-d:2′,3′-d′]-s-indaceno[1,2-b:5,6-b′]dithiophene (IT-4F). This produces PCE13:IT-4F blend, whose chemical structures are given in Fig. 8a. Fig. 8b displays the absorption spectra of the polymers. The low bandgap donor polymer (PCE13) exhibits an absorption peak onset at 684 nm corresponding to an optical band gap of 1.8 eV, while IT-4F acceptor presents at peak onset at 786 nm. PCE13:IT-4F at a weight ratio of 1:1, covers the absorption spectra of the two component fully. The ITO/PEDOT:PSS/PCE13:IT-4F/DPO/Al solar cell demonstrates a power conversion efficiency of over 11%, in accordance with the literature.63 Next, we investigate the mechanical and electrical properties of PCE13:IT-4F bulk heterojunction in the presence of SEBS.
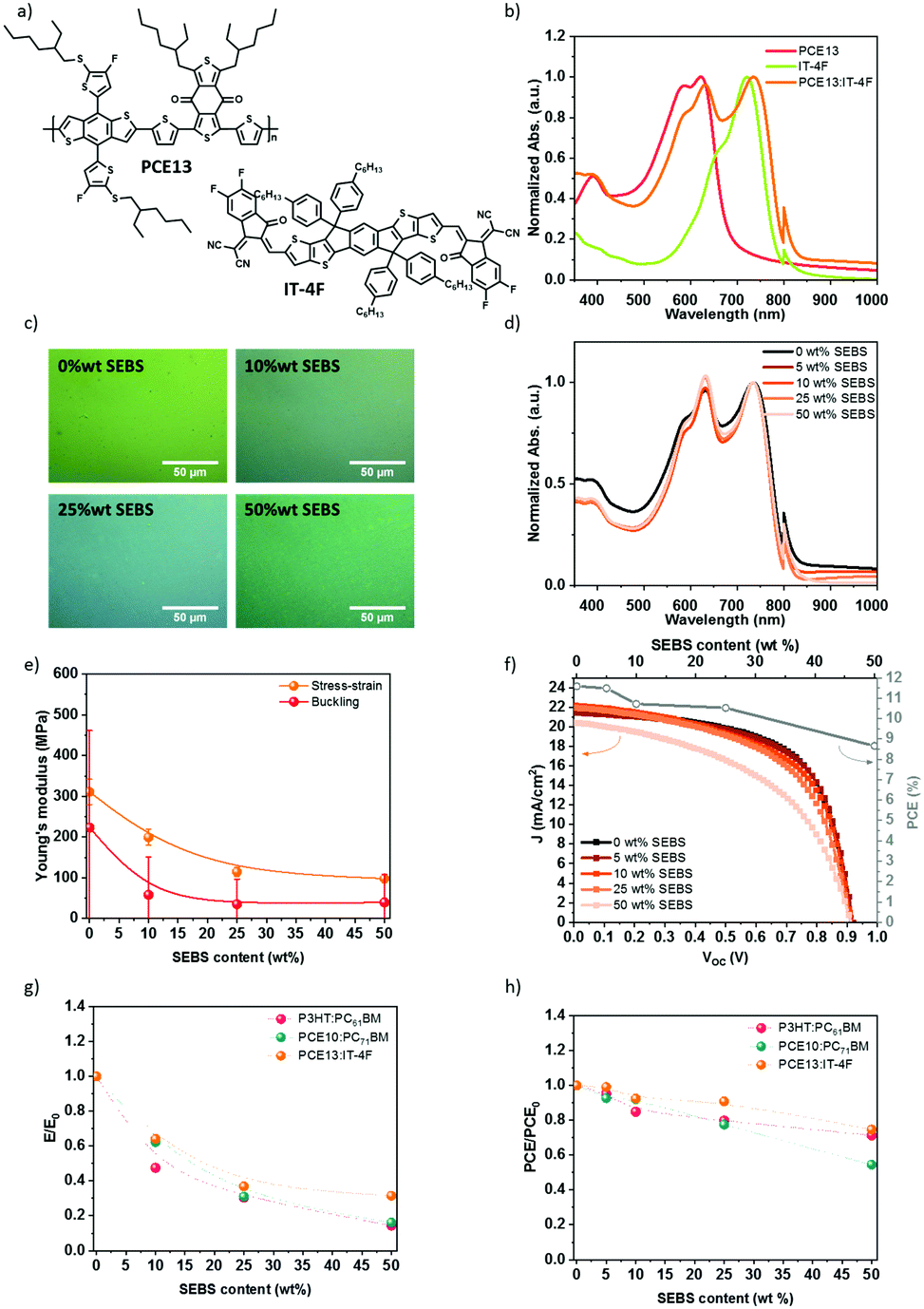 | ||
| Fig. 8 (a) chemical structures of PCE13 and IT-4F. (b) UV-visible absorption spectra of PCE13, IT-4F, and PCE13:IT-4F (1:1) blend. (c) Optical morphology and (d) UV-visible absorption spectra of PCE13:IT-4F mixed with SEBS elastomer. (e) Determination of Young's moduli of PCE13:IT-4F with 0, 10, 25, and 50 wt% SEBS by buckling method or by direct stress–strain measurements and (f) J–V characteristics curves of PCE13:IT-4F with SEBS elastomer and the associated PCE. Comparative effect of SEBS on the (g) initial elastic modulus and (h) initial power conversion efficiency. | ||
PCE13, IT-4F, and SEBS were mixed in solution before deposition. The SEBS content is described as relative to IT-4F acceptor. The morphology of the thin film was characterized by optical microscopy (Fig. 8c). A micro-phase separation is noticed at 50 wt% SEBS. The absorption spectra in Fig. 8d confirmed the phase-separated morphology. The spectra were normalized to IT-4F (786 nm), and PCE13 peak intensity increases with the progressive introduction of SEBS.
The mechanical properties of PCE13:IT-4F blend have been determined by the buckling method and stress–strain technique in Fig. 8e. PCE13:IT-4F was deposited by spin coating on pre-strained PDMS rectangles using different spin speeds to achieve at least three thicknesses for the buckling method. Buckling wavelengths were obtained from the optical microscopy image, and a profilometer measured the thickness. In Fig. 8e, the Young's moduli are plotted in function of the concentration. The stress–strain method gives higher tensile modulus values, but the same behavior is observed compared to the buckling technique. With no SEBS elastomer, Young's modulus is about 310 MPa by stress–strain or 220 MPa by buckling method. This is a bit less than PCE10:PC71BM. It decreases to 200 MPa with 10 wt% SEBS, 115 MPa for 25 wt% SEBS, and 98 MPa for 50 wt% SEBS by the stress–strain method. With the buckling method, the tensile modulus decreases up to 35 MPa with 50 wt% SEBS. This suggests that again SEBS plays a crucial role in the mechanical properties.
Fig. 8f illustrates the impact of SEBS on the photovoltaic performances of PCE13:IT-4F. For this system, Jsc decreases slightly from 21.7 to 19.4 mA cm−2 with a SEBS content of 0 and 50 wt%, respectively (Fig. 8f). Voc remains constant at 0.91 V, and the fill factor decays from 58.8% at 0 wt% SEBS to 49% at 50 wt% SEBS, which is more pronounced than for PCE10:PC71BM. Overall, the average PCE decreases from 11.6 to 8.6%, while 50 wt% is added, corresponding to a 25% loss of the initial PCE. At 25 wt% SEBS, PCE decreases only to 10.6%, i.e. less than 10% PCE loss. Fig. S17 (ESI†) shows a decrease in EQE when SEBS is added to the blend. This result suggests that the addition of SEBS increases the charge losses by lowering the exciton dissociation yield or increasing the charge recombination. The decrease in the integrated EQE is consistent with the decrease in Jsc.
Fig. 8g and h summarizes the effect of SEBS on Young's modulus and the PCE, depending on the studied BHJ. For all the acceptor:donor blends, the addition of SEBS as a ternary blend component improves the ductility of the film as suggested by a decrease of the elastic modulus over 70% at 50 wt% SEBS, while PCE remains within 60%. The introduction of SEBS is a versatile tool that can be adaptable to any BHJ system (providing that common solvent can be found), assuring good mechanical properties and a minimal loss of PCE. These results demonstrate the possibility of tailoring mechanical properties and photovoltaic performances. A concentration up to 25 wt% SEBS could be introduced into acceptor:donor blend with primarily tailored softness i.e., Young's modulus. Moreover, it seems that the chemical nature of SEBS is well adapted whatever the used acceptor (fullerene or non-fullerene) and donor. Until 25% wt SEBS, miscibility has minor importance in PCE results. Indeed, the GIWAXS analysis of PCE10:PC71BM and PCE13:IT-4F in the presence of SEBS would reveal similar results to P3HT:PC61BM. Furthermore, the crack-onset has to be studied to attest the ability to be stretched on PDMS. Besides the efficiency and the elasticity, nowadays the thermal stability remains an issue, and the addition of SEBS in acceptor:donor blend could be explored as a solution to limit the losses over time.
Conclusions
Different strategies have been used to improve the mechanical properties of the photoactive BHJ layer of organic photovoltaic devices to enable mechanically robust and stretchable devices. The addition of C12N3, PEG3N3, and SEBS into P3HT:PC61BM blend slows down the PC61BM diffusivity and crystallization, which are the source of degradation. Both elastomer blending and cross-linking strategies demonstrate a softening effect. However, the blending approach is shown to extend beyond fullerene-based solar cells to non-fullerene systems processed at low or room temperature. The alpha temperature decreases in the presence of the diazide cross-linker, suggesting that plasticization occurs and is responsible for the softening effect. In the case of elastomer addition, the softening seems more related to incorporating a soft rubber with low interaction with the photoactive components, which form an effective bulk heterojunction. While the elastomer blending leads to the softest films among both strategies studied, the cross-linking strategy appears to be better at not disturbing the BHJ phase-separation, maintaining high photovoltaic performance in fullerene-based solar cells. Despite this advantage, the elastomer blending approach, thermoplastic elastomer such as using SEBS, provides a more versatile strategy to enhance the mechanical properties (e.g., Young's modulus) of a wide range of non-fullerene BHJ systems making it widely applicable to modern OPV systems. With the fast progress in OPV, this kind of method would easily find its place due to its low cost, high adaptability, and high mechanical compliance. Besides some engineering issues to build a fully stretchable solar cell that needs to be solved, our investigation demonstrates the efficiency of elastomer blending in P3HT:PC61BM, PCE10:PC71BM, and PCE13:IT-4F, all of which appear to retain their photoactive functionality in OPV devices despite significant loading of the elastomer. Although this result gives a possible route for inherently stretchable solar cells, more progress is needed on the multi-layer integration front to construct fully stretchable solar cell device stacks and modules.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This publication is based upon work supported by the King Abdullah University of Science and Technology (KAUST) Office of Sponsored Research (OSR) under Award No. OSR-2018-CARF/CCF-3079, and No. OSR-2019-CRG8-4095.3.Notes and references
- J. Wang, Z. Zheng, Y. Zu, Y. Wang, X. Liu, S. Zhang, M. Zhang and J. Hou, Adv. Mater., 2021, 2102787 CrossRef CAS PubMed.
- E. Dauzon, X. Sallenave, C. Plesse, F. Goubard, A. Amassian and T. D. Anthopoulos, Adv. Mater., 2021, 2101469 CrossRef CAS PubMed.
- F. C. Krebs, M. Biancardo, B. Winther-Jensen, H. Spanggard and J. Alstrup, Sol. Energy Mater. Sol. Cells, 2006, 90, 1058–1067 CrossRef CAS.
- M. Jørgensen, K. Norrman and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2008, 92, 686–714 CrossRef.
- F. C. Krebs, T. D. Nielsen, J. Fyenbo, M. Wadstrøm and M. S. Pedersen, Energy Environ. Sci., 2010, 3, 512 RSC.
- F. C. Krebs, S. A. Gevorgyan and J. Alstrup, J. Mater. Chem., 2009, 19, 5442 RSC.
- M. Jørgensen, K. Norrman, S. A. Gevorgyan, T. Tromholt, B. Andreasen and F. C. Krebs, Adv. Mater., 2012, 24, 580–612 CrossRef PubMed.
- W. R. Mateker and M. D. McGehee, Adv. Mater., 2017, 29, 1603940 CrossRef PubMed.
- M. Karakawa, K. Suzuki, T. Kuwabara, T. Taima, K. Nagai, M. Nakano, T. Yamaguchi and K. Takahashi, Org. Electron., 2020, 76, 105448 CrossRef CAS.
- S. Savagatrup, A. D. Printz, T. F. O’Connor, A. V. Zaretski, D. Rodriquez, E. J. Sawyer, K. M. Rajan, R. I. Acosta, S. E. Root and D. J. Lipomi, Energy Environ. Sci., 2015, 8, 55–80 RSC.
- A. D. Printz, S. Savagatrup, D. Rodriquez and D. J. Lipomi, Sol. Energy Mater. Sol. Cells, 2015, 134, 64–72 CrossRef CAS.
- D. J. Lipomi, H. Chong, M. Vosgueritchian, J. Mei and Z. Bao, Sol. Energy Mater. Sol. Cells, 2012, 107, 355–365 CrossRef CAS.
- S. Savagatrup, A. S. Makaram, D. J. Burke and D. J. Lipomi, Adv. Funct. Mater., 2014, 24, 1169–1181 CrossRef CAS.
- S. Savagatrup, A. D. Printz, D. Rodriquez and D. J. Lipomi, Macromolecules, 2014, 47, 1981–1992 CrossRef CAS.
- D. J. Lipomi, B. C.-K. C.-K. Tee, M. Vosgueritchian and Z. Bao, Adv. Mater., 2011, 23, 1771–1775 CrossRef CAS PubMed.
- Y.-T. Hsieh, J.-Y. Chen, C.-C. Shih, C.-C. Chueh and W.-C. Chen, Org. Electron., 2018, 53, 339–345 CrossRef CAS.
- N. El-Atab, N. Qaiser, R. Bahabry and M. M. Hussain, Adv. Energy Mater., 2019, 1902883, 1–7 Search PubMed.
- L. Derue, O. Dautel, A. Tournebize, M. Drees, H. Pan, S. Berthumeyrie, B. Pavageau, E. Cloutet, S. Chambon, L. Hirsch, A. Rivaton, P. Hudhomme, A. Facchetti and G. Wantz, Adv. Mater., 2014, 26, 5831–5838 CrossRef CAS PubMed.
- G. Wantz, L. Derue, O. Dautel, A. Rivaton, P. Hudhomme and C. Dagron-Lartigau, Polym. Int., 2014, 63, 1346–1361 CrossRef CAS.
- J. W. Rumer and I. McCulloch, Mater. Today, 2015, 18, 425–435 CrossRef CAS.
- G.-J. N. Wang, L. Shaw, J. Xu, T. Kurosawa, B. C. Schroeder, J. Y. Oh, S. J. Benight and Z. Bao, Adv. Funct. Mater., 2016, 26, 7254–7262 CrossRef CAS.
- J. Y. Oh, S. Rondeau-Gagné, Y.-C. Chiu, A. Chortos, F. Lissel, G.-J. N. Wang, B. C. Schroeder, T. Kurosawa, J. Lopez, T. Katsumata, J. Xu, C. Zhu, X. Gu, W.-G. Bae, Y. Kim, L. Jin, J. W. Chung, J. B.-H. Tok and Z. Bao, Nature, 2016, 539, 411–415 CrossRef CAS PubMed.
- G.-J. N. Wang, Y. Zheng, S. Zhang, J. Kang, H.-C. Wu, A. Gasperini, H. Zhang, X. Gu and Z. Bao, Chem. Mater., 2019, 31, 6465–6475 CrossRef CAS.
- A. D. Printz, S. Savagatrup, D. J. Burke, T. N. Purdy and D. J. Lipomi, RSC Adv., 2014, 4, 13635–13643 RSC.
- F. Sugiyama, A. T. Kleinschmidt, L. V. Kayser, D. Rodriquez, M. Finn, M. A. Alkhadra, J. M.-H. Wan, J. Ramírez, A. S.-C. Chiang, S. E. Root, S. Savagatrup and D. J. Lipomi, Polym. Chem., 2018, 9, 4354–4363 RSC.
- M. T. Dang, L. Hirsch, G. Wantz and J. D. Wuest, Chem. Rev., 2013, 113, 3734–3765 CrossRef CAS PubMed.
- E. Song, B. Kang, H. H. Choi, D. H. Sin, H. Lee, W. H. Lee and K. Cho, Adv. Electron. Mater., 2016, 2, 1–8 Search PubMed.
- G. Zhang, M. McBride, N. Persson, S. Lee, T. J. Dunn, M. F. Toney, Z. Yuan, Y. H. Kwon, P. H. Chu, B. Risteen and E. Reichmanis, Chem. Mater., 2017, 29, 7645–7652 CrossRef CAS.
- D. Choi, H. Kim, N. Persson, P. H. Chu, M. Chang, J. H. Kang, S. Graham and E. Reichmanis, Chem. Mater., 2016, 28, 1196–1204 CrossRef CAS.
- M. Shin, J. Y. Oh, K.-E. Byun, Y.-J. Lee, B. Kim, H.-K. Baik, J.-J. Park and U. Jeong, Adv. Mater., 2015, 27, 1255–1261 CrossRef CAS PubMed.
- J. W. Mok, Z. Hu, C. Sun, I. Barth, R. Muñoz, J. Jackson, T. Terlier, K. G. Yager and R. Verduzco, Chem. Mater., 2018, 30, 8314–8321 CrossRef CAS.
- S. Chen, S. Jung, H. J. Cho, N.-H. Kim, S. Jung, J. Xu, J. Oh, Y. Cho, H. Kim, B. Lee, Y. An, C. Zhang, M. Xiao, H. Ki, Z.-G. Zhang, J.-Y. Kim, Y. Li, H. Park and C. Yang, Angew. Chem., Int. Ed., 2018, 57, 13277–13282 CrossRef CAS PubMed.
- J. T. Seitz, J. Appl. Polym. Sci., 1993, 49, 1331–1351 CrossRef CAS.
- F. P. V. Koch, J. Rivnay, S. Foster, C. Müller, J. M. Downing, E. Buchaca-Domingo, P. Westacott, L. Yu, M. Yuan, M. Baklar, Z. Fei, C. Luscombe, M. A. McLachlan, M. Heeney, G. Rumbles, C. Silva, A. Salleo, J. Nelson, P. Smith and N. Stingelin, Prog. Polym. Sci., 2013, 38, 1978–1989 CrossRef CAS.
- D. Rodriquez, J.-H. Kim, S. E. Root, Z. Fei, P. Boufflet, M. Heeney, T.-S. Kim and D. J. Lipomi, ACS Appl. Mater. Interfaces, 2017, 9, 8855–8862 CrossRef CAS PubMed.
- M. Selivanova, S. Zhang, B. Billet, A. Malik, N. Prine, E. Landry, X. Gu, P. Xiang and S. Rondeau-Gagné, Macromolecules, 2019, 52, 7870–7877 CrossRef CAS.
- J. Xu, S. Wang, G.-J. N. Wang, C. Zhu, S. Luo, L. Jin, X. Gu, S. Chen, V. R. Feig, J. W. F. To, S. Rondeau-Gagné, J. Park, B. C. Schroeder, C. Lu, J. Y. Oh, Y. Wang, Y.-H. Kim, H. Yan, R. Sinclair, D. Zhou, G. Xue, B. Murmann, C. Linder, W. Cai, J. B.-H. Tok, J. W. Chung and Z. Bao, Science, 2017, 355, 59–64 CrossRef CAS PubMed.
- C. M. Stafford, C. Harrison, K. L. Beers, A. Karim, E. J. Amis, M. R. VanLandingham, H.-C. Kim, W. Volksen, R. D. Miller and E. E. Simonyi, Nat. Mater., 2004, 3, 545–550 CrossRef CAS PubMed.
- N. Bowden, S. Brittain, A. G. Evans, J. W. Hutchinson and G. Whitesides, Nature, 1998, 393, 146–149 CrossRef CAS.
- V. Koncar, Smart Textiles for In Situ Monitoring of Composites, Elsevier, 2019, pp. 295–381 Search PubMed.
- D. Tahk, H. H. Lee and D.-Y. Khang, Macromolecules, 2009, 42, 7079–7083 CrossRef CAS.
- Z. Jiang, J. Appl. Crystallogr., 2015, 48, 917–926 CrossRef CAS.
- M. Prato, Q. C. Li, F. Wudl and V. Lucchini, J. Am. Chem. Soc., 1993, 115, 1148–1150 CrossRef CAS.
- E. F. V. Scriven and K. Turnbull, Chem. Rev., 1988, 88, 297–368 CrossRef CAS.
- J. C. Hummelen, B. W. Knight, F. LePeq, F. Wudl, J. Yao and C. L. Wilkins, J. Org. Chem., 1995, 60, 532–538 CrossRef CAS.
- M. Jørgensen, K. Norrman and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2008, 92, 686–714 CrossRef.
- A. Swinnen, I. Haeldermans, M. vandeVen, J. D’Haen, G. Vanhoyland, S. Aresu, M. D’Olieslaeger and J. Manca, Adv. Funct. Mater., 2006, 16, 760–765 CrossRef CAS.
- M. Campoy-Quiles, T. Ferenczi, T. Agostinelli, P. G. Etchegoin, Y. Kim, T. D. Anthopoulos, P. N. Stavrinou, D. D. C. Bradley and J. Nelson, Nat. Mater., 2008, 7, 158–164 CrossRef CAS PubMed.
- B. O’Connor, E. P. Chan, C. Chan, B. R. Conrad, L. J. Richter, R. J. Kline, M. Heeney, I. McCulloch, C. L. Soles and D. M. DeLongchamp, ACS Nano, 2010, 4, 7538–7544 CrossRef PubMed.
- J.-S. Kim, J.-H. Kim, W. Lee, H. Yu, H. J. Kim, I. Song, M. Shin, J. H. Oh, U. Jeong, T.-S. Kim and B. J. Kim, Macromolecules, 2015, 48, 4339–4346 CrossRef CAS.
- B. J. Kim, Y. Miyamoto, B. Ma and J. M. J. Fréchet, Adv. Funct. Mater., 2009, 19, 2273–2281 CrossRef CAS.
- H. Sirringhaus, P. J. Brown, R. H. Friend, M. M. Nielsen, K. Bechgaard, B. M. W. Langeveld-Voss, A. J. H. Spiering, R. A. J. Janssen, E. W. Meijer, P. Herwig and D. M. de Leeuw, Nature, 1999, 401, 685–688 CrossRef CAS.
- N. Kiriy, E. Jähne, H.-J. Adler, M. Schneider, A. Kiriy, G. Gorodyska, S. Minko, D. Jehnichen, P. Simon, A. A. Fokin and M. Stamm, Nano Lett., 2003, 3, 707–712 CrossRef CAS.
- P. Müller-Buschbaum, Adv. Mater., 2014, 26, 7692–7709 CrossRef PubMed.
- Y.-C. Huang, C.-S. Tsao, C.-M. Chuang, C.-H. Lee, F.-H. Hsu, H.-C. Cha, C.-Y. Chen, T.-H. Lin, C.-J. Su, U.-S. Jeng and W.-F. Su, J. Phys. Chem. C, 2012, 116, 10238–10244 CrossRef CAS.
- N. D. Treat, M. A. Brady, G. Smith, M. F. Toney, E. J. Kramer, C. J. Hawker and M. L. Chabinyc, Adv. Energy Mater., 2011, 1, 82–89 CrossRef CAS.
- B. Schmidt-Hansberg, M. Sanyal, M. F. G. Klein, M. Pfaff, N. Schnabel, S. Jaiser, A. Vorobiev, E. Müller, A. Colsmann, P. Scharfer, D. Gerthsen, U. Lemmer, E. Barrena and W. Schabel, ACS Nano, 2011, 5, 8579–8590 CrossRef PubMed.
- J. Rivnay, S. C. B. Mannsfeld, C. E. Miller, A. Salleo and M. F. Toney, Chem. Rev., 2012, 112, 5488–5519 CrossRef CAS PubMed.
- T. T. Ngo, D. N. Nguyen and V. T. Nguyen, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2012, 3, 045001 CAS.
- J. Zhao, A. Swinnen, G. Van Assche, J. Manca, D. Vanderzande and B. Van Mele, J. Phys. Chem. B, 2009, 113, 1587–1591 CrossRef CAS PubMed.
- J. Zhao, A. Swinnen, G. Van Assche, J. Manca, D. Vanderzande and B. Van Mele, J. Phys. Chem. B, 2009, 113, 1587–1591 CrossRef CAS PubMed.
- A. Rudin and P. Choi, The Elements of Polymer Science & Engineering, Elsevier, 2013, pp. 231–274 Search PubMed.
- Y. Lin, B. Adilbekova, Y. Firdaus, E. Yengel, H. Faber, M. Sajjad, X. Zheng, E. Yarali, A. Seitkhan, O. M. Bakr, A. El-Labban, U. Schwingenschlögl, V. Tung, I. McCulloch, F. Laquai and T. D. Anthopoulos, Adv. Mater., 2019, 31, 1902965 CrossRef CAS PubMed.
- Z. Liu, Y. Gao, J. Dong, M. Yang, M. Liu, Y. Zhang, J. Wen, H. Ma, X. Gao, W. Chen and M. Shao, J. Phys. Chem. Lett., 2018, 9, 6955–6962 CrossRef CAS PubMed.
- L. K. Jagadamma, H. Hu, T. Kim, G. O. N. Ndjawa, A. E. Mansour, A. El Labban, J. C. D. Faria, R. Munir, D. H. Anjum, M. A. McLachlan and A. Amassian, Nano Energy, 2016, 28, 277–287 CrossRef CAS.
- L. Song, W. Wang, E. Barabino, D. Yang, V. Körstgens, P. Zhang, S. V. Roth and P. Müller-Buschbaum, ACS Appl. Mater. Interfaces, 2019, 11, 3125–3135 CrossRef CAS PubMed.
- Z. He, B. Xiao, F. Liu, H. Wu, Y. Yang, S. Xiao, C. Wang, T. P. Russell and Y. Cao, Nat. Photonics, 2015, 9, 174–179 CrossRef CAS.
- J. J. van Franeker, M. Turbiez, W. Li, M. M. Wienk and R. A. J. Janssen, Nat. Commun., 2015, 6, 6229 CrossRef CAS PubMed.
- L. Li, J. Liang, H. Gao, Y. Li, X. Niu, X. Zhu, Y. Xiong and Q. Pei, ACS Appl. Mater. Interfaces, 2017, 9, 40523–40532 CrossRef CAS PubMed.
- J.-H. Kim, J. Noh, H. Choi, J.-Y. Lee and T.-S. Kim, Chem. Mater., 2017, 29, 3954–3961 CrossRef CAS.
- N. Balar and B. T. O’connor, Macromolecules, 2017, 50, 8611–8618 CrossRef CAS.
- Y. Lin, Y. Firdaus, F. H. Isikgor, M. I. Nugraha, E. Yengel, G. T. Harrison, R. Hallani, A. El-Labban, H. Faber, C. Ma, X. Zheng, A. Subbiah, C. T. Howells, O. M. Bakr, I. McCulloch, S. De Wolf, L. Tsetseris and T. D. Anthopoulos, ACS Energy Lett., 2020, 5, 2935–2944 CrossRef CAS.
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiao, K. Sun, S. Yang, X. Zhang and L. Ding, Sci. Bull., 2020, 65, 272–275 CrossRef CAS.
- Y. Lin, M. I. Nugraha, Y. Firdaus, A. D. Scaccabarozzi, F. Aniés, A.-H. Emwas, E. Yengel, X. Zheng, J. Liu, W. Wahyudi, E. Yarali, H. Faber, O. M. Bakr, L. Tsetseris, M. Heeney and T. D. Anthopoulos, ACS Energy Lett., 2020, 5, 3663–3671 CrossRef CAS.
- Y. Lin, A. Magomedov, Y. Firdaus, D. Kaltsas, A. El-Labban, H. Faber, D. R. Naphade, E. Yengel, X. Zheng, E. Yarali, N. Chaturvedi, K. Loganathan, D. Gkeka, S. H. AlShammari, O. M. Bakr, F. Laquai, L. Tsetseris, V. Getautis and T. D. Anthopoulos, ChemSusChem, 2021, 14(17), 3569–3578 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures, TGA curve, FTIR spectra, optical microscopic images, visible-ultraviolet spectra, GIWAXS patterns, storage modulus, photovoltaic curves, stress–strain characterization, EQE. See DOI: 10.1039/d1tc05263b |
| This journal is © The Royal Society of Chemistry 2022 |