
A graphdiyne oxide composite membrane for active electrolyte enhanced supercapacitors with super long self-discharge time†
Na
Liang
a,
Xueyan
Wu
a,
Yan
Lv
a,
Jixi
Guo
 *a,
Xiuli
Zhang
a,
Yingfu
Zhu
a,
Huibiao
Liu
*ab and
Dianzeng
Jia
*a
*a,
Xiuli
Zhang
a,
Yingfu
Zhu
a,
Huibiao
Liu
*ab and
Dianzeng
Jia
*a
aState Key Laboratory of Chemistry and Utilization of Carbon Based Energy Resources; Key Laboratory of Advanced Functional Materials, Autonomous Region, Institute of Applied Chemistry, College of Chemistry, Xinjiang University, Urumqi, 830046, Xinjiang, P. R. China. E-mail: jxguo1012@163.com; jdz@xju.edu.cn
bCAS Key Laboratory of Organic Solids, Beijing National Laboratory for Molecular Sciences, CAS Research/Education Center for Excellence in Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, P. R. China. E-mail: liuhb@iccas.ac.cn
First published on 11th January 2022
Abstract
Active electrolyte enhanced supercapacitors (AEESCs) are considered as promising tools for enhancing power capacity due to their high specific capacitance and simple construction. However, there are many challenges, such as severe self-discharge (SDC) and rapid capacity decay. Herein, we develop a novel strategy to fabricate AEESCs with the suppression of rapid SDC, in which a self-assembled composite membrane of graphdiyne oxide/polyvinyl alcohol (GDYO/PVA) is used as a separator. The new AEESCs show a SDC time of 37160 s (1.0 V–0.3 V), which is 153 times higher than that of commercial separators, as well as an excellent capacitance retention of 95.1% after 8000 cycles. The outstanding electrochemical performance of the GDYO/PVA composite membrane can be attributed to the inherent nano-network structure, excellent proton conductivity, and high selectivity of GDYO. This new strategy of a self-assembled composite membrane based on GDYO will provide new opportunities for constructing AEESCs with super long self-discharge time and high performance.
Introduction
Electrochemical supercapacitors (SCs) have become a hot research topic due to their excellent high power density and superior cycling stability.1–3 However, their low energy density greatly limits their development and practical applications.4 Since the electrochemical window of aqueous electrolytes is limited to ∼1 V, increasing the specific capacitance of SCs based on carbon materials and aqueous electrolytes has become a primary goal for researchers. Compared to the research studies on the modification of electrode materials, active electrolyte enhanced supercapacitors (AEESCs),5 prepared by adding redox active substances into the capacitor to store additional energy through electrochemical reactions, is simple, safe, and favorable for large-scale preparation.6,7 Currently, redox active species are usually divided into organic and inorganic additives. Organic additives mainly include p-phenylenediamine,8,9 hydroquinone,10 and methylene blue,11 while inorganic additives are KI,12 CuS,13 CuSO4,5 and FeSO4.14 AEESCs based on KI have recently dominated.15 Grzegorz Lota et al.16 firstly reported the application of KI in carbon/carbon electrochemical capacitors. In the two-electrode system, the capacitance in 1 mol L−1 KI (235 F g−1) was almost twice that of SC prepared with the same carbon electrodes in 1 mol L−1 H2SO4. Senthilkumar et al.12 prepared activated carbon from Eichhornia crassipes by the ZnCl2 activation method. By adding KI into the electrolyte, the specific capacitance also increased nearly twice, but they found that SC had a high severe self-discharge (SDC) after adding KI (the voltage decreased from 0.8 V to 0.3 V after 1800 s). As we all know, the problem of redox substances affects their stability.7 According to previous reports, there are two causes of this phenomenon: the first is due to the extensive redox reactions caused by the redox electrolyte;17 the second is that the redox reaction is not completed within the operating voltage window of the cell.18 The commonly used commercial separator for aqueous supercapacitors is polypropylene (PP) membranes, which have a large pore size, non-uniformity, no ion selectivity, and limited ionic conductivity in many electrolytes.19–21 The development of AEESC separators with ion selectivity, minimal resistance to ion movement, greater safety and stability is urgently needed for next-generation devices with better safety and performance characteristics. Chen et al.5 described the specific SDC phenomenon of the hybrid capacitor based on the electrolyte containing a quinone/hydroquinone redox pair. The reason for this phenomenon was that the active species migrate between the two electrodes. The SDC could be reduced by introducing an ion exchange membrane, as the potential of the device using a Nafion membrane decreased from 0.8 V to 0.3 V within 4686 s. Although the SDC has been suppressed, high performance is still a challenge.Graphdiyne (GDY), as a new two-dimensional carbon material, has attracted wide attention due to its unique physical and chemical properties, after it has been synthesized by our group.22 GDY is an emerging electrochemical energy material that has shown excellent potential in areas such as energy storage, catalysis, photonics, etc.23–28 GDY shows unique advantages in terms of selective nanopores, dimensional stability, and facile preparation. The uniform porous layer structure (its theoretically calculated pore size is about 3.8 Å) makes GDY an ideal membrane material.29,30 As a promising derivative of GDY, graphdiyne oxide (GDYO) retains the attractive structural features of GDY with additional hydrophilic carboxyl groups that facilitate proton transport and increase the dispersibility,31–33 making it a promising candidate as a separator for SCs.
Herein, we develop a novel strategy for constructing AEESCs using self-assembled graphdiyne oxide/polyvinyl alcohol (GDYO/PVA) composite membranes as separators. Since GDYO/PVA membranes can prevent the migration of the active electrolyte, SDC is significantly inhibited and specific capacitance retention is greatly improved compared with the commercially available membrane (PP membrane) in KI/H2SO4 electrolytes. The SDC time for AEESCs with a GDYO/PVA separator is up to 37160 s (1 V–0.3 V), which is over 153 times that of the commercial PP membrane (241.5 s). Remarkably, after 8000 charge–discharge cycles, the specific capacitance retention rate is 95.1%. The novel strategy for fabricating separator based GDYO opens a door to achieve ultra-long cycling stability and high performance for AEESCs.
Results and discussion
A GDYO/PVA membrane has been fabricated by a self-assembly method at a liquid/air interface. Fig. 1a illustrates the manufacturing process of the GDYO/PVA membrane. GDY has been synthesized as previously reported.34 And GDYO has been synthesized by the oxidation of GDY using piranha solution (a mixture of H2SO4 and H2O2, v/v = 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).35 The addition of PVA formed a stable suspension. As a polymer soluble in water, PVA should exist in a long chain state in water. GDYO is rich in oxygen-containing functional groups (–OH and –COOH), which form GDYO/PVA conjugates by hydrogen bonding with the polymer matrix. Subsequently, these conjugates continuously gel due to the heat flow and bubbles reaching the liquid/air interface through the heating solution, forming a GDYO/PVA composite membrane. Finally, it has been assembled with the prepared electrode for the electrochemical test. Compared with the traditional vacuum filtration method, this method can be easily popularized, and is less time consuming, and easily recyclable. Fig. 1b and c show the morphology of GDY and GDYO powders, characterized by SEM. Compared with GDY, the GDYO powder is finer and easier to combine to form a porous structure. Moreover, it has a flat and continuous shape with occasional folds (Fig. S1a and b, ESI†). The surface composition and the nature of the chemical bonds in the GDY and GDYO have been investigated by XPS. Fig. S1c and d (ESI†) show two characteristic peaks corresponding to C 1s and O 1s at 284.8 and 531.2 eV, respectively, for both GDY and GDYO. As shown in Fig. S9 (ESI†), for GDY, peaks at around 531.8 and 533.2 eV correspond to C
:1).35 The addition of PVA formed a stable suspension. As a polymer soluble in water, PVA should exist in a long chain state in water. GDYO is rich in oxygen-containing functional groups (–OH and –COOH), which form GDYO/PVA conjugates by hydrogen bonding with the polymer matrix. Subsequently, these conjugates continuously gel due to the heat flow and bubbles reaching the liquid/air interface through the heating solution, forming a GDYO/PVA composite membrane. Finally, it has been assembled with the prepared electrode for the electrochemical test. Compared with the traditional vacuum filtration method, this method can be easily popularized, and is less time consuming, and easily recyclable. Fig. 1b and c show the morphology of GDY and GDYO powders, characterized by SEM. Compared with GDY, the GDYO powder is finer and easier to combine to form a porous structure. Moreover, it has a flat and continuous shape with occasional folds (Fig. S1a and b, ESI†). The surface composition and the nature of the chemical bonds in the GDY and GDYO have been investigated by XPS. Fig. S1c and d (ESI†) show two characteristic peaks corresponding to C 1s and O 1s at 284.8 and 531.2 eV, respectively, for both GDY and GDYO. As shown in Fig. S9 (ESI†), for GDY, peaks at around 531.8 and 533.2 eV correspond to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O, suggesting the existence of oxygen-functional groups like carbonyl and lactones. With the oxidation of GDY to GDYO, a new carboxylate peak appeared at around 529.7 eV and the O/C ratio of GDY became higher after the oxidation to GDYO, indicating that the oxidation treatment virtually leads to the generation of oxygen-containing species in the as-formed GDYO. In the high-resolution XPS spectra of C 1s for GDY and GDYO (Fig. 1e and f), the integral ratio of C–C (sp2) to C–C (sp) is 1:2, indicating that the chemical structure of GDYO remains unchanged. In the FT-IR spectra, GDYO shows the CO stretching vibration peak at 1654 cm−1, except for the three peaks of GDY, also proving the successful synthesis of GDYO (Fig. 1d). In the Raman spectra (Fig. 1g), the spectral intensity ratios ID/IG of GDY and GDYO are 0.82 and 0.78, respectively, indicating that the prepared GDYO has lower defects,36 which is conducive to inhibiting the diffusion of electrolyte.
O and C–O, suggesting the existence of oxygen-functional groups like carbonyl and lactones. With the oxidation of GDY to GDYO, a new carboxylate peak appeared at around 529.7 eV and the O/C ratio of GDY became higher after the oxidation to GDYO, indicating that the oxidation treatment virtually leads to the generation of oxygen-containing species in the as-formed GDYO. In the high-resolution XPS spectra of C 1s for GDY and GDYO (Fig. 1e and f), the integral ratio of C–C (sp2) to C–C (sp) is 1:2, indicating that the chemical structure of GDYO remains unchanged. In the FT-IR spectra, GDYO shows the CO stretching vibration peak at 1654 cm−1, except for the three peaks of GDY, also proving the successful synthesis of GDYO (Fig. 1d). In the Raman spectra (Fig. 1g), the spectral intensity ratios ID/IG of GDY and GDYO are 0.82 and 0.78, respectively, indicating that the prepared GDYO has lower defects,36 which is conducive to inhibiting the diffusion of electrolyte.
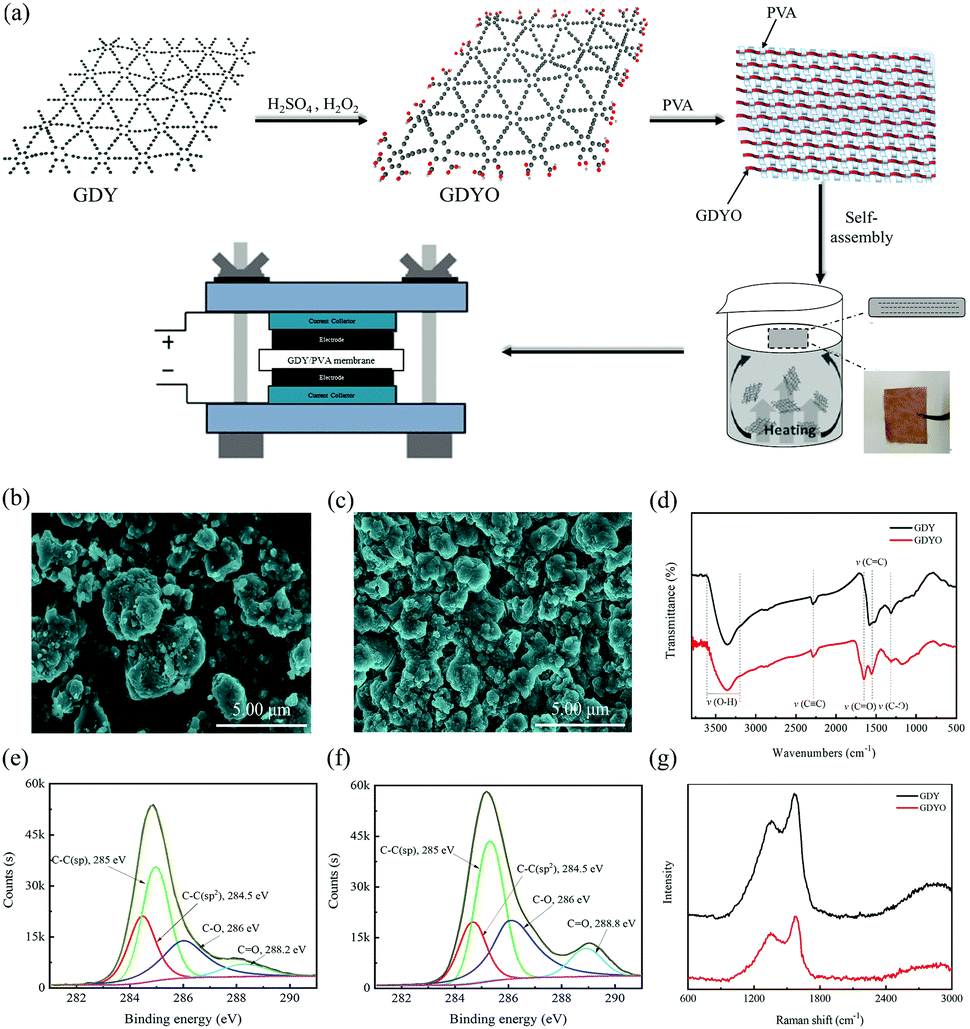 | ||
| Fig. 1 (a) Schematic illustration of the fabrication of the GDYO/PVA membrane. (b and c) SEM images of GDY and GDYO powder. (d) FT-IR spectra of GDY and GDYO powder. (e and f) C 1s spectra of GDY and GDYO powder. (g) Raman spectra of GDY and GDYO powder. | ||
The GDYO membrane has been prepared by the vacuum filtration method (Fig. S2, ESI†). The SEM images (Fig. S3a, ESI†) show that the GDYO membrane is uniform and porous, and the membrane thickness is about 23 μm. The SEM image of the GDYO/PVA membrane presents a direct observation of these nanoparticles (Fig. S3b, ESI†). And a well-developed network for ion transfer will be constructed along with these particles when the GDYO/PVA membrane is fully humidified with the electrolyte.
The interaction of GDYO/PVA composite membranes with different ratios was characterized by FT-IR, as shown in Fig. 2a. The peak shape of these membranes shows no change after three tests, indicating that the prepared membrane has good uniformity. There are mainly four peaks, namely, the O–H stretching vibration peak (3200–3700 cm−1), C–H stretching vibration peak (2653 cm−1), C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C stretching vibration peak (2152 cm−1), and CO stretching vibration peak (1708 cm−1). It is well known that the –OH stretching band and the –C–OH stretching band are sensitive to hydrogen bonding. After adding GDYO to PVA, the band near 3200–3700 cm−1 moves 2–3 cm−1 to the low wavenumber (blue shift), involving the strong hydroxyl band of free alcohols and hydrogen bonded alcohols. These results confirm the existence of hydrogen bonding between PVA and GDYO nanosheets.37–40 Besides, the stretching vibration peak of CC appears after the addition of GDYO nanosheets, reaffirming the successful preparation of the GDYO/PVA membrane.
C stretching vibration peak (2152 cm−1), and CO stretching vibration peak (1708 cm−1). It is well known that the –OH stretching band and the –C–OH stretching band are sensitive to hydrogen bonding. After adding GDYO to PVA, the band near 3200–3700 cm−1 moves 2–3 cm−1 to the low wavenumber (blue shift), involving the strong hydroxyl band of free alcohols and hydrogen bonded alcohols. These results confirm the existence of hydrogen bonding between PVA and GDYO nanosheets.37–40 Besides, the stretching vibration peak of CC appears after the addition of GDYO nanosheets, reaffirming the successful preparation of the GDYO/PVA membrane.
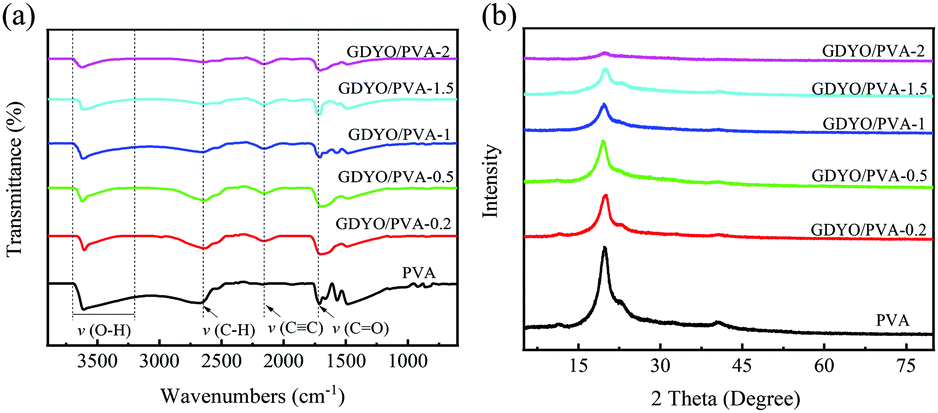 | ||
| Fig. 2 (a) FT-IR spectra of pure PVA and hybrid membranes. (b) XRD patterns of pure PVA and hybrid membranes. | ||
In the XRD patterns, since the crystalline planes of PVA are (101) and (200), all membranes have characteristic peaks at 2θ = 20° (Fig. 2b).41 After three tests, the peak shapes of these membranes did not change. The diffraction intensity of the peaks related to the PVA crystal region in the composite membrane decreases with the increase of the GDYO content, because the crystallinity of PVA is generally affected by the strong intermolecular hydrogen bonding interaction between PVA molecular chains.42 It is also confirmed that there are hydrogen bonds between GDYO nanosheets and PVA. The majority of scenarios for the application of proton exchange membranes in energy devices are in aqueous environments at room temperature and are carried out via the Grotthuss mechanism, which explains the unusually high proton mobility in water by the sequential transfer of protons along with a hydrogen bond (h-bond) network.29,43–45 The formation of hydrogen bonds in GDYO/PVA membranes facilitates the conduction of protons and improves the electrochemical performance.
For different KI doping levels in the mechanisms of SC and SDC, the electrochemical performance of the electrolytes is shown in Fig. 3. Fig. 3a and b show the GCD and CV curves of 1.0 M H2SO4 mixed with a certain mass of KI aqueous electrolyte in the SC, respectively. As shown in Fig. 3a, the GCD curves show linear contours and symmetric triangles in the electrolyte without KI. SC exhibits nonlinear charge–discharge behavior with increasing KI content. When the electrolyte is the mixture solution of 1.0 M H2SO4 and 0.5 M KI, the specific capacitance is the largest (327 F g−1). The specific capacitance decreases with the increase of KI concentration when the concentration exceeds 0.5 M. This is mainly because the high concentration of KI will lead to the aggregation of free ions and crystallization of KI in the H2SO4 system, hindering the separation and combination of free ions, and affecting the ion transport of the electrolyte. The addition of KI introduces a new reversible redox process for SC, resulting in additional Faraday pseudocapacitance. Therefore, the addition of KI incredibly improves the electrochemical performance of SC. In Fig. 3b, the CV curve is nearly rectangular without KI in the electrolytes, with the energy storage mechanism of a double-layer capacitor. In the process of oxidation, the redox peak appears between 0.1 and 0.3 V with the increase of the KI amount. At 0.5 M KI, the closed area and peak current of the curve are the maximum, corresponding to the maximum specific capacitance of SC. The redox reactions at the electrode–electrolyte interface region are as follows:46
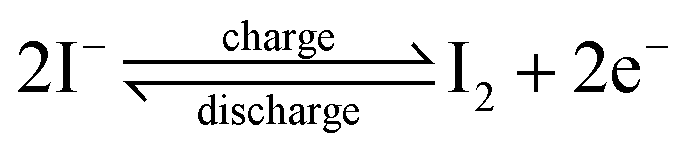 | (1) |
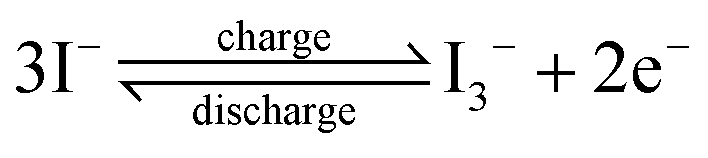 | (2) |
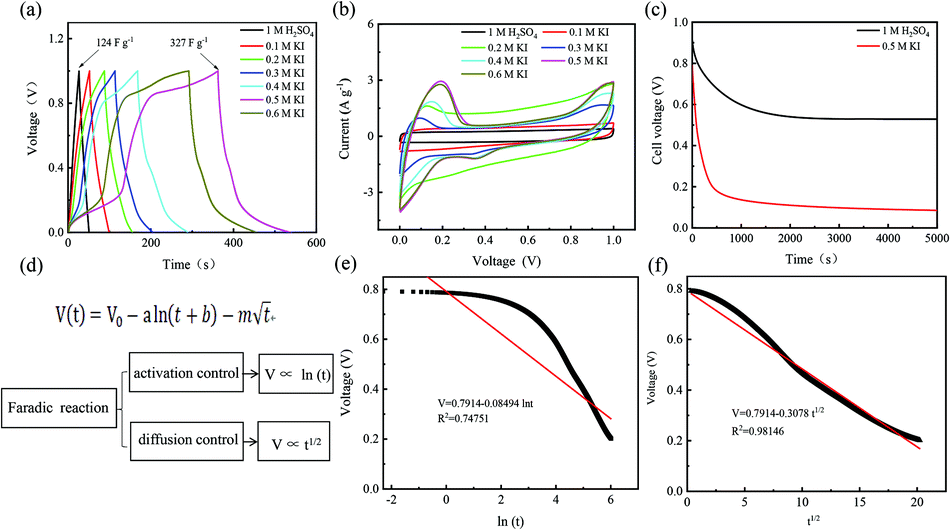 | ||
| Fig. 3 Supercapacitors based on different KI content electrolytes (a) charge and discharge curves at various constant current densities of 1 A g−1. (b) Cyclic voltammetry curves at 10 mV s−1 scan speed. (c) SDC curves were recorded after the device was charged to 1 V at 1 A g−1 constant current density. (d) The relationship between potential and time, independent variable ln(t) or t1/2. Supercapacitor SDC curve with 1.0 M H2SO4 and 0.5 M KI mixture electrolyte. (e) Fitting result of V and ln(t). (f) Fitting result of V and t1/2. | ||
So far, the Conway team has provided the most instructive work in the study of Faraday SDC, where the shape prediction of SDC is based on activation or diffusion-controlled SDC reactions47 (Fig. 3d). In order to determine the mechanism of SDC of AEESCs, several established models are used to analyze the SDC curve. The data conform to the following equation:48,49
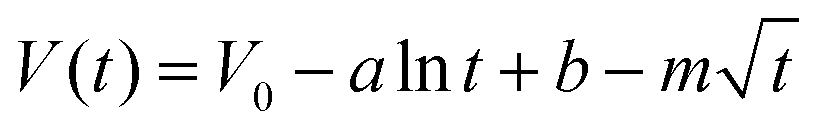 | (3) |
lnt + b is the overpotential activation of the decomposition reaction of the discharge electrode, and the term 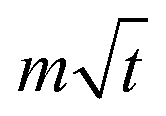 is the diffusion-limited side reaction caused by low concentration redox impurities.
is the diffusion-limited side reaction caused by low concentration redox impurities.
Based on eqn (3), if there is a linear relationship between V and ln(t), then the origin of SDC is mainly an activation-controlled reaction. If the relationship curve between V and t1/2 is linear, then the SDC mechanism is diffusion-controlled. As shown in Fig. 3e and f, there is no straight line between ln(t) and V (R2 = 0.74751), and V is proportional to t1/2 (R2 = 0.99). Therefore, the SDC of AEESCs may be controlled by ion diffusion.
The ionic sizes of I− and I3− are 3.9 Å and 6.3 Å, respectively.12 And the effective pore size of graphdiyne can be determined by the van der Waals diameter of the acetylene carbon chain, resulting in a triangular pore with a length of 3.8 Å (as previously reported),18,19 which can effectively prevent the diffusion of the active electrolyte. GDYO retains the structure of GDY, and GDYO has good hydrophilicity and solubility, which are conducive to the preparation of uniform separators. Therefore, the porous structure of the GDYO membrane material is proposed to prevent the diffusion of electrolyte ions, inhibit SDC and fundamentally improve its cycle performance.
Given that the electrodes and electrolyte are the same across the devices, the differences in cell performance are mainly attributed to the separators. From Fig. S4 and Table S2 (ESI†), GDYO/PVA-1 has the best electrochemical performance. In order to demonstrate the role of hydrogen bonding in GDYO/PVA membranes, GDY/PVA-1 was prepared by the same method. Since GDY powder has no oxygen-containing functional groups, the non-uniform dispersion in the PVA solution leads to the non-uniform distribution of GDY in the prepared membrane, which does not prevent the electrolyte diffusion well (Fig. S5, ESI†). In Fig. S6a (ESI†), the specific capacitance of AEESCs with GDY/PVA-1 and GDYO/PVA-1 membrane assemblies are 210.2 F g−1 and 325.6 F g−1. And in the SDC curve (Fig. S6c, ESI†), it can be seen that the discharge times of GDY/PVA-1 and GDYO/PVA-1 are 12490 and 37160 s (1 V–0.3 V), respectively. The electrochemical properties show that the hydrogen bonding network in GDYO/PVA is more favorable for inhibiting the diffusion of the electrolyte. Fig. 4 shows the electrochemical properties of the commodity membrane (PP), GDYO membrane, and GDYO/PVA-1 membrane. Fig. 4a shows the GCD curves of AEESCs with the different membranes between 0 and 1 V at 1 A g−1. The specific capacitance of PP, GDYO, and GDYO/PVA-1 calculated by the galvanostatic charge/discharge is 327 F g−1, 348 F g−1, and 326 F g−1, respectively. And a faradaic plateau (non-ideal triangle) is observed due to the voltage dependent redox reactions.10 This is further substantiated by the CV technique (Fig. 4b), where these systems show non-ideal capacitive behavior due to the larger number of possible redox reactions between the pairs of 3I−/I3− and 2I−/I2. The GDYO membrane has the largest area. EIS measurements have been carried out for different membranes (Fig. 4c). A semicircle is observed at a high frequency for all AEESCs, which corresponds to the internal resistance of the AEESCs. Subsequently, all AEESCs exhibit a 45° inclined line at medium frequencies which confirms the occurrence of a redox reaction.12 Two resistances can be immediately obtained from the Nyquist plot, namely the solution resistance Rs and the charge transfer resistance Rct. Rs comes from the bulk solution resistance, and its size depends on the electrolyte conductivity, material, and separator.50 Considering that AEESC uses the same electrode and electrolyte, the difference of Rs is attributed to the separator. In the high frequency region, the resistances of AEESC Rs from small to large are GDYO, PP, and GDYO/PVA, possibly due to the proton conductivity of the separator (Table S1, ESI†). At low frequencies, the diffusion resistance of the solution has a great influence on the curve. The vertical shape at lower frequencies indicates a pure capacitive behavior, representative of the ion diffusion in the structure.51 The more vertical the curve, the more closely the supercapacitor behaves like an ideal capacitor.52 At a low frequency, the angles between the curve and Z′ from small to large are PP, GDYO, GDYO/PVA, respectively. This indicates that GDYO and GDYO/PVA membranes effectively inhibit electrolyte diffusion, and the GDYO/PVA membrane has the best effect. For practical applications, AEESCs should have extensive cycling stability. The cycling stability of these devices has been evaluated by the galvanostatic charge–discharge test at 1 A g−1 and the results are shown in Fig. 4d, which suggests that the GDYO/PVA membrane has the best cycling stability. And the capacitance retention is 96.2% compared to the original values after 1000 charge–discharge cycles and 95.1% after 8000 charge–discharge cycles (Fig. S4f, ESI†). The specific capacitance retentions of the GDYO membrane and PP membrane are 75% and 65%, after 1000 cycles. Compared with previous reports (Table S3, ESI†), GDYO and GDYO/PVA membranes greatly improve the cycling stability. To elucidate the morphological changes of the septum after long-term cycling, we have performed SEM imaging (Fig. S7, ESI†). The GDYO/PVA continues to exhibit a uniform and stable structure even after the charge–discharge cycle (Fig. S7b, ESI†). This is also the reason for the high cycling stability of the GDYO/PVA membrane.
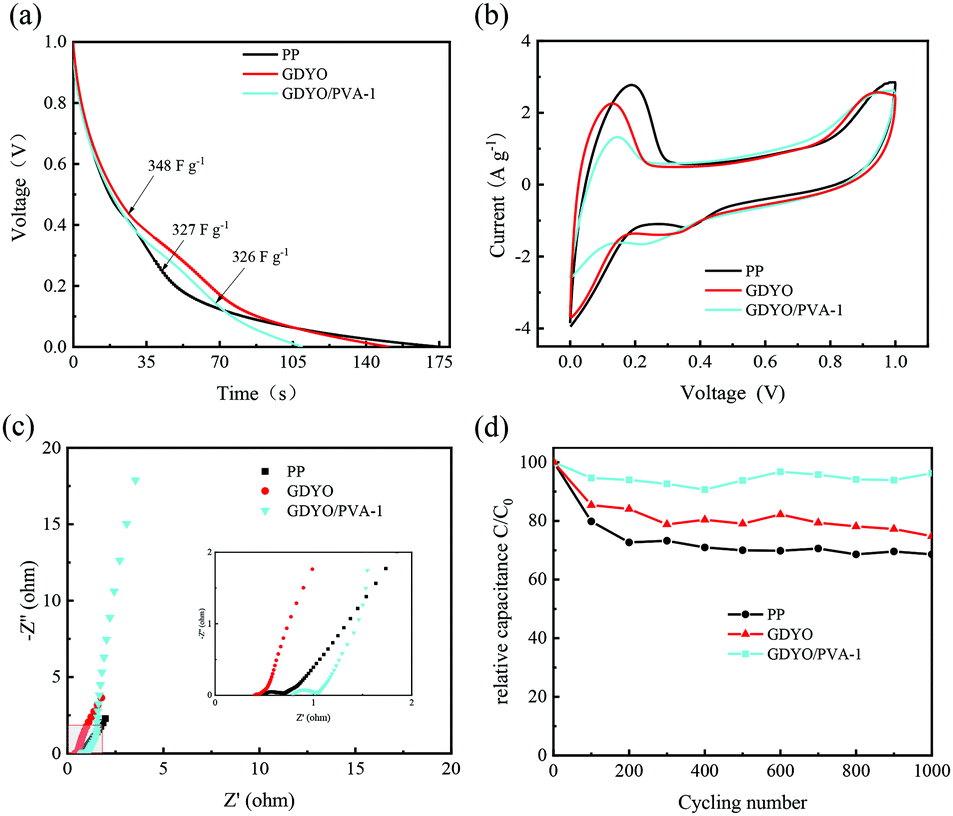 | ||
| Fig. 4 AEESEs based on the different membranes (commodity membrane: PP; GDYO membrane and GDYO/PVA-1 membrane) when the electrolyte is a 1 M H2SO4 and 0.5 M KI mixture (a) Charge–discharge curves at a current density of 1 A g−1. (b) CVs at a scan rate of 10 mV s−1. (c) EIS analysis. (d) Cycling stability at a current density of 1 A g−1. | ||
The SDC behavior of AEESCs has been studied by charging it to 1 V. Then, the voltage has been recorded as open circuit voltage concerning time. In the SDC curve (Fig. 5), when SDC of the capacitor device is taken into account, an obvious drawback of PP is observed, indicating the fast self-discharge rate. The discharge times of SDC are from fast to slow for PP, GDYO, and GDYO/PVA, corresponding to 241 s, 27150 s, and 37160 s (1 V–0.3 V), respectively.
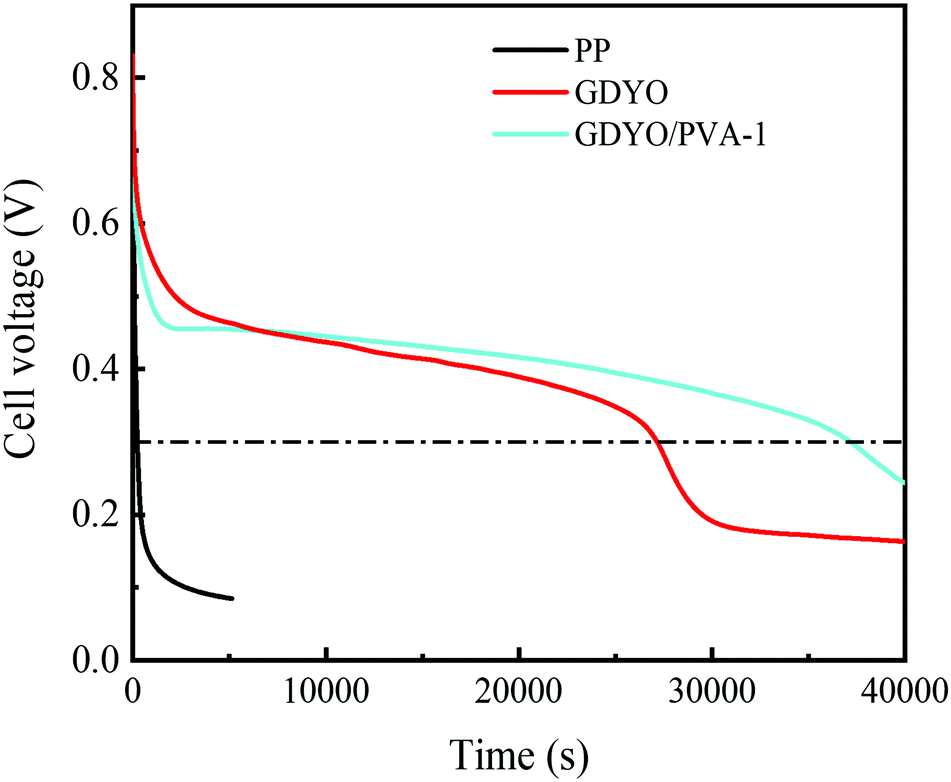 | ||
| Fig. 5 SDC of the supercapacitors with different membranes when the electrolyte is a 1 M H2SO4 and 0.5 M KI mixture. | ||
We find that when the GDYO is present, the SDC of AEESCs is longer than that previously reported (Table S3, ESI†). Thus, the comparison of the SDC time of the devices distinctly reveals that GDYO and GDYO/PVA membranes effectively inhibit the SDC of supercapacitors.
Conclusions
In summary, we have developed a novel strategy for fabricating AEESCs using self-assembled GDYO/PVA composite membranes as separators. The GDYO/PVA composite membrane is prepared by a self-assembly process at the liquid/air interface, with controllable size and thickness by adjusting the time of treatment. When GDYO/PVA is used as the separator of AEESCs with KI/H2SO4 electrolytes, SDC is significantly inhibited and the specific capacitance retention is obviously improved compared with the commercial PP membranes since the GDYO/PVA composite membrane effectively prevents the migration of the active electrolyte. The SDC time with the GDYO/PVA separator is 37160 s (1 V–0.3 V), which is 153 times that of commercial PP seperators (241.5 s). The long-term cycling behavior of the GDYO/PVA separator shows that the capacitance retention rate is 96.2% after 1000 cycles, and 95.1% after 8000 cycles, much higher than those of other films reported in the literature. The results demonstrate that GDYO as a separator has potential application in energy storage.Experimental
Chemicals and reagents
The polypropylene (PP) membrane (NKK-MPF30AC-100) was purchased from Nippon Kodoshi Corporation. Polyvinyl alcohol (PVA 98.0–99.0 mol%) and tetrabutylammonium fluoride (TBAF) were purchased from Aladdin Chemical Reagent, Inc. N,N,N′,N′-Tetramethylethylenediamine (TMEDA) was purchased from Alfa Aesar Chemical Reagents, Inc. Unless otherwise stated, other analytical chemicals were used as received.Synthesis of GDYO
Hexaethynylbenzene (HEB) was synthesized according to a reported synthesis method.22 HEB (100 mg) was dissolved in pyridine solution, and then, TMEDA (10 mL) and CuI (8 mg) were added. The mixture was heated in nitrogen at 50 °C for 12 hours while avoiding light. Excess CuI was removed by ammonia and the obtained powder was washed to neutral with distilled water. After vacuum drying, GDY powder was successfully synthesized. GDYO was obtained by acid oxidation of GDY. Briefly, GDY powder (15 mg) was first mixed carefully with concentrated H2SO4 (4 mL) in an ice bath for 10 min, H2O2 (30 wt%, 1.5 mL) was slowly added and stirred violently. The mixed solution reacted at room temperature for 4 h to obtain a brown suspension, denoted as graphdiyne oxide solution. Finally, GDYO powder was obtained by freeze-drying the graphdiyne oxide solution.Synthesis of the GDYO membrane
The graphdiyne oxide solution was vacuum filtered using the hydrophilic PTFE membrane (pore diameter 0.22 μm) to form a homogeneous film, which was washed repeatedly with ethanol and water, and characterized after natural drying.Synthesis of the GDYO/PVA membrane
The GDYO/PVA membrane was synthesized by a self-assembly method at a liquid/air interface. First, PVA (4 g) was dissolved in deionized water (50 mL). The blend was heated at 90 °C for 3 h with continuous stirring until the solution became transparent. After that, 8% (w/v) of the PVA solution was prepared. Then, a certain proportion of GDYO powder (0 mg, 5.0 mg, 12.5 mg, 25.0 mg, 37.5 mg, and 50.0 mg) was added to 25 mL of PVA aqueous solution, and the cell comminution apparatus was used for ultrasound treatment for 30 min at a high power of 200 W. The GDYO/PVA solutions with different concentrations and proportions were uniformly dispersed. The obtained solution was uniform and stable, and it could still exhibit stability without precipitation after several weeks of storage. Finally, the uniformly dispersed GDYO/PVA solution was heated by water bath heating (80 °C, 1 h). Different proportions of GDYO/PVA composite membranes were obtained at the liquid/air interface. The membranes were denoted as PVA, GDYO/PVA-0.2, GDYO/PVA-0.5, GDYO/PVA-1, GDYO/PVA-1.5, and GDYO/PVA-2, respectively.Structure characterization
The FTIR spectra of the samples were recorded using a VERTEX70 spectrometer. Scanning electron microscopy (SEM, Japan Hitachi SU-4800) and transmission electron microscopy (TEM, Japan JEM2100F, Japan JEOL) were used to observe the microstructure of the material. X-ray photoelectron spectroscopy (XPS, USA Escalab250) was used to determine the surface chemical properties of the material. Structure information of the samples was studied by X-ray powder diffraction (XRD, Bruker D8, filtered Cu Kα radiation) and Raman spectroscopy (Raman; Bruker Senterra R200-L spectrometer).Proton conductivity
The ionic conductivity (σ) of the membrane was measured by electrochemical impedance spectroscopy (CHI 770, Shanghai Chenhua Co., Ltd) in the frequency range of 1 Hz–500 kHz. The 1 cm × 1 cm sample was sandwiched between two stainless steel electrodes at room temperature. The sample was soaked in deionized water before measurement. The transverse σ value of the membrane was calculated from the impedance data using the following formula.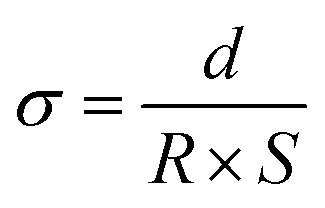 | (4) |
Water uptake
All membranes were vacuum dried in a vacuum at 120 °C, and then the swelling ratio was tested. The sample film was immersed in deionized water at room temperature for 24 h to measure the weight of the dry film and the wet film. Water absorption content is the average of the two measured values, and the error is within ±3.0%.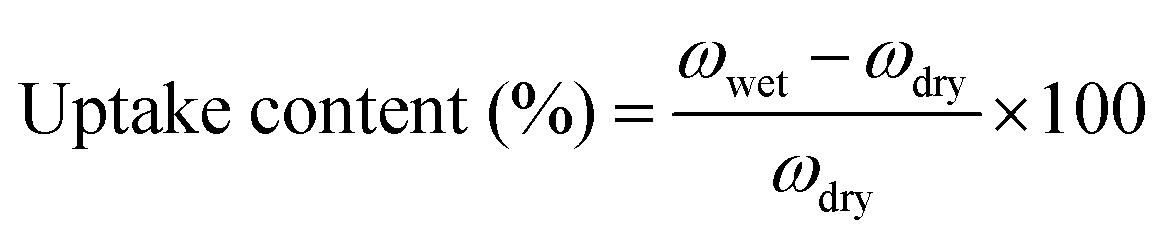 | (5) |
Electrochemical characterization
The electrodes were prepared on a stainless steel mesh using activated carbon (AC, 80 wt%), polytetrafluoroethylene (5 wt%), and carbon black (15 wt%). The area loading amount of stainless steel mesh was 5–7 mg cm−2. The supercapacitor consisted of two identical electrodes, with the 1 M H2SO4 and KI solution of different concentrations as the electrolyte, the separator is the PP membrane, GDYO membrane and GDYO/PVA membrane, respectively. The illustration of such a device is shown in Fig. 1a.The capacitance performance of SC was tested on the dual-electrode battery on a CHI 760E electrochemical workstation. 1 M H2SO4 mixed with KI aqueous solution was used as the electrolyte. The cell capacitance (CCell) and electrode specific capacitance (Csp) were determined using the following equation.12,53
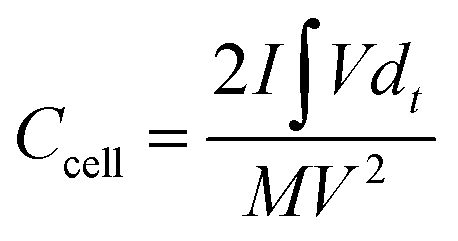 | (6) |
| Csp = 4Ccell | (7) |
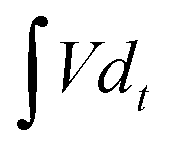 is the integral current area, and V is the potential window.
is the integral current area, and V is the potential window.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work is supported by the National Natural Science Foundation of China (22071251, 21875258, 201972123, U1703251, and 21861037), the Program for Tianshan Innovative Research Team of Xinjiang Uygur Autonomous Region (2018D14002), the Open Fund of the Key Laboratory of Xinjiang Uygur Autonomous Region (2017D040414), and the Scientific Research Program of the Higher Education Institution of Xinjiang (XJEDU2017A001).References
- J. Chen and P. S. Lee, Adv. Energy Mater., 2020, 11, 2003311 CrossRef.
- A. Borenstein, O. Hanna, R. Attias, S. Luski, T. Brousse and D. Aurbach, J. Mater. Chem. A, 2017, 5, 12653–12672 RSC.
- J. Yan, S. Li, B. Lan, Y. Wu and P. S. Lee, Adv. Funct. Mater., 2019, 30, 1902564 CrossRef.
- T. Wu, X. Wu, L. Li, M. Hao, G. Wu, T. Zhang and S. Chen, Angew. Chem., Int. Ed., 2020, 59, 23800–23809 CrossRef CAS PubMed.
- L. Chen, H. Bai, Z. Huang and L. Li, Energy Environ. Sci., 2014, 7, 1750–1759 RSC.
- K. Nasrin, S. Gokulnath, M. Karnan, K. Subramani and M. Sathish, Energy Fuels, 2021, 35, 6465–6482 CrossRef CAS.
- S. T. Senthilkumar, R. K. Selvan and J. S. Melo, J. Mater. Chem. A, 2013, 1, 12386 RSC.
- Y. Yan, T. Kuila, N. H. Kim, S. H. Lee and J. H. Lee, Carbon, 2015, 85, 60–71 CrossRef CAS.
- J. Wu, H. Yu, L. Fan, G. Luo, J. Lin and M. Huang, J. Mater. Chem., 2012, 22, 19025–19030 RSC.
- H. Yu, J. Wu, L. Fan, Y. Lin, K. Xu, Z. Tang, C. Cheng, S. Tang, J. Lin, M. Huang and Z. Lan, J. Power Sources, 2012, 198, 402–407 CrossRef CAS.
- S. Roldán, M. Granda, R. Menéndez, R. Santamaría and C. Blanco, Electrochim. Acta, 2012, 83, 241–246 CrossRef.
- S. T. Senthilkumar, R. K. Selvan, Y. S. Lee and J. S. Melo, J. Mater. Chem. A, 2013, 1, 1086–1095 RSC.
- L. Qian, X. Tian, J. Mao, H. Yuan and D. Xiao, RSC Adv., 2013, 3, 1703–1708 RSC.
- L. Qiang, L. Kai-Xi, S. Guo-Hua, F. Hui and G. Jian-Yu, Acta Phys. Chim. Sin., 2006, 22, 1445–1450 Search PubMed.
- B. Gorska, E. Frackowiak and F. Beguin, Curr. Opin. Electrochem., 2018, 9, 95–105 CrossRef CAS.
- G. Lota and E. Frackowiak, Electrochem. Commun., 2009, 11, 87–90 CrossRef CAS.
- W. Chen, R. B. Rakhi and H. N. Alshareef, Nanoscale, 2013, 5, 4134–4138 RSC.
- S. Roldan, C. Blanco, M. Granda, R. Menendez and R. Santamaria, Angew. Chem., Int. Ed., 2011, 50, 1699–1701 CrossRef CAS PubMed.
- M. Liu, K. Turcheniuk, W. Fu, Y. Yang, M. Liu and G. Yushin, Nano Energy, 2020, 71, 104627 CrossRef CAS.
- H. Liu, J. Xu, B. Guo and X. He, J. Mater. Sci., 2014, 49, 6961–6966 CrossRef CAS.
- S. Janakiraman, M. Khalifa, R. Biswal, S. Ghosh, S. Anandhan and A. Venimadhav, J. Power Sources, 2020, 460, 228060 CrossRef CAS.
- G. Li, Y. Li, H. Liu, Y. Guo, Y. Li and D. Zhu, Chem. Commun., 2010, 46, 3256–3258 RSC.
- H. Changshui, L. Yongjun, W. Ning, X. Yurui, Z. Zicheng, L. Huibiao and L. Yuliang, Chem. Rev., 2018, 118(16), 7744–7803 CrossRef PubMed.
- Y. Li, L. Xu, H. Liu and Y. Li, Chem. Soc. Rev., 2014, 43, 2572–2586 RSC.
- R. Sakamoto, N. Fukui, H. Maeda, R. Matsuoka, R. Toyoda and H. Nishihara, Adv. Mater., 2019, 31, e1804211 CrossRef PubMed.
- Y. Du, W. Zhou, J. Gao, X. Pan and Y. Li, Acc. Chem. Res., 2020, 53, 459–469 CrossRef CAS PubMed.
- S. Thangavel, K. Krishnamoorthy, V. Krishnaswamy, N. Raju, S. J. Kim and G. Venugopal, J. Phys. Chem. C, 2015, 119, 22057–22065 CrossRef CAS.
- K. Krishnamoorthy, S. Thangavel, J. Chelora Veetil, N. Raju, G. Venugopal and S. J. Kim, Int. J. Hydrogen Energy, 2016, 41, 1672–1678 CrossRef CAS.
- L. Shi, A. Xu, D. Pan and T. Zhao, Nat. Commun., 2019, 10, 1165 CrossRef PubMed.
- J. Guo, M. Guo, F. Wang, W. Jin, C. Chen, H. Liu and Y. Li, Angew. Chem., Int. Ed., 2020, 59, 16712–16716 CrossRef CAS PubMed.
- H. Yan, S. Guo, F. Wu, P. Yu, H. Liu, Y. Li and L. Mao, Angew. Chem., Int. Ed., 2018, 57, 3922–3926 CrossRef CAS PubMed.
- J. Li, Y. Chen, J. Guo, F. Wang, H. Liu and Y. Li, Adv. Funct. Mater., 2020, 30(42), 2004115 CrossRef CAS.
- A. Donnadio, M. Nocchetti, F. Costantino, M. Taddei, M. Casciola, F. da Silva Lisboa and R. Vivani, Inorg. Chem., 2014, 53, 13220–13226 CrossRef CAS PubMed.
- J. Li, Y. Chen, J. Gao, Z. Zuo, Y. Li, H. Liu and Y. Li, ACS Appl. Mater. Interfaces, 2019, 11, 2591–2598 CrossRef CAS PubMed.
- Z. Zhu, Q. Bai, S. Li, S. Li, M. Liu, F. Du, N. Sui and W. W. Yu, Small, 2020, 16, e2001440 CrossRef PubMed.
- C. Huang, S. Zhang, H. Liu, Y. Li, G. Cui and Y. Li, Nano Energy, 2015, 11, 481–489 CrossRef CAS.
- J. Sun, X. Qian, Z. Wang, F. Zeng, H. Bai and N. Li, J. Membr. Sci., 2020, 599, 117838 CrossRef CAS.
- B. Torabi and E. Ameri, Chem. Eng. J., 2016, 288, 461–472 CrossRef CAS.
- A. Shameli and E. Ameri, Chem. Eng. J., 2017, 309, 381–396 CrossRef CAS.
- Y. Zhu, H. Wang, J. Zhu, L. Chang and L. Ye, Appl. Surf. Sci., 2015, 349, 27–34 CrossRef CAS.
- P. S. Rachipudi, M. Y. Kariduraganavar, A. A. Kittur and A. M. Sajjn, J. Membr. Sci., 2011, 383, 224–234 CrossRef CAS.
- K. E. Strawhecker and E. Manias, Chem. Mater., 2000, 12, 2943–2949 CrossRef CAS.
- A. Hassanali, F. Giberti, J. Cuny, T. D. Kühne and M. Parrinello, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 13723–13728 CrossRef CAS PubMed.
- Y. L. S. Tse, C. Knight and G. A. Voth, JCP, 2015, 142, 014104 Search PubMed.
- W. Conrad, Science, 2016, 6316 Search PubMed.
- H. Yu, J. Wu, L. Fan, K. Xu, X. Zhong, Y. Lin and J. Lin, Electrochim. Acta, 2011, 56, 6881–6886 CrossRef CAS.
- A. H. Andreas, J. Electrochem. Soc., 2015, 162, A5047–A5053 CrossRef.
- B. E. Conway, W. G. Pell and T. C. Liu, J. Power Sources, 1997, 65, 53–59 CrossRef CAS.
- K. Wang, L. Yao, M. Jahon, J. Liu and T. N. Ng, ACS Energy Lett., 2020, 5, 3276–3284 CrossRef CAS.
- C. Lei, F. Markoulidis and Z. Ashitaka, Electrochim. Acta, 2013, 92, 183–187 CrossRef CAS.
- W. C. Chen, T. C. Wen and H. Teng, Electrochim. Acta, 2003, 48, 641–649 CrossRef CAS.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed.
- L. Q. Mai, A. Minhas-Khan, X. Tian, K. M. Hercule, Y. L. Zhao, X. Lin and X. Xu, Nat. Commun., 2013, 4, 2923 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc04406k |
| This journal is © The Royal Society of Chemistry 2022 |