
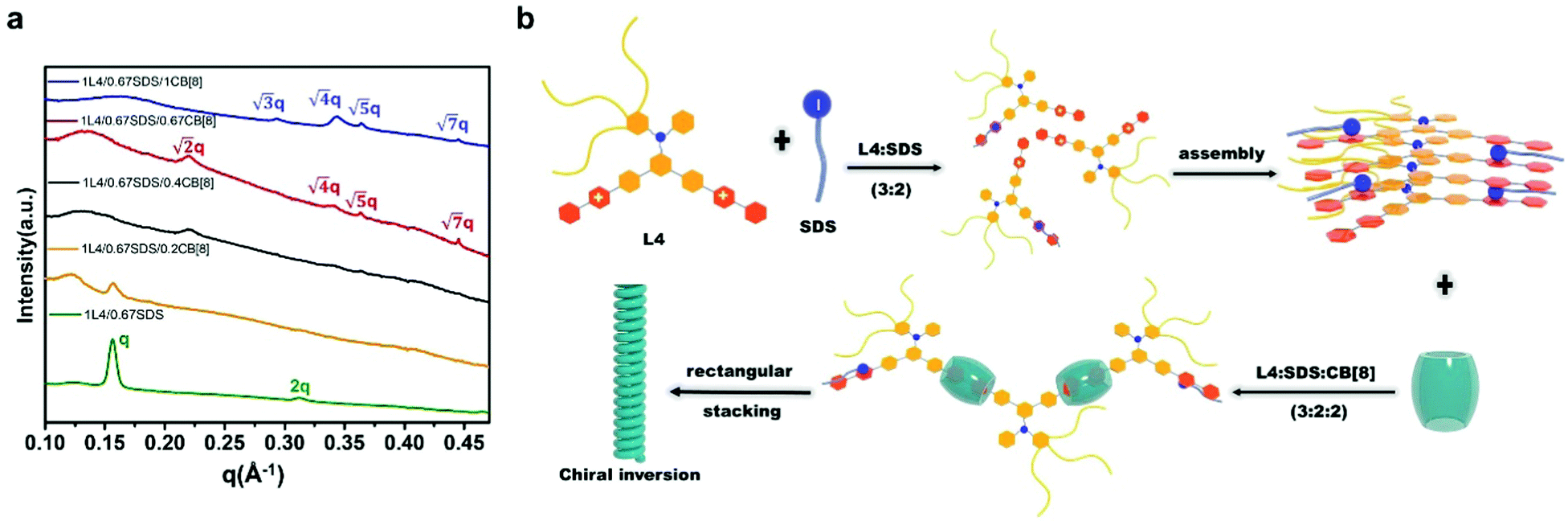
Surfactant-induced chirality transfer, amplification and inversion in a cucurbit[8]uril–viologen host–guest supramolecular system†
Hui
Xu
,
Huanhuan
Lu
,
Qi
Zhang
,
Meng
Chen
,
Yahan
Shan
,
Tian-Yi
Xu
,
Fei
Tong
 * and
Da-Hui
Qu
*
* and
Da-Hui
Qu
*
Key Laboratory for Advanced Materials and Joint International Research Laboratory of Precision Chemistry and Molecular Engineering, Feringa Nobel Prize Scientist Joint Research Center, Frontiers Science Center for Materiobilogy and Dynamic Chemistry, Institute of Fine Chemicals, School of Chemistry and Molecular Engineering, East China University of Science and Technology, Shanghai 200237, People's Republic of China. E-mail: feitong@ecust.edu.cn; dahui_qu@ecust.edu.cn
First published on 25th October 2021
Abstract
Chirality manipulation in supramolecules, especially in multicomponent supramolecular structures, is crucially desired. Herein, we report a feasible approach to achieve the chirality transfer, amplification, and inversion in a ternary supramolecular system consisting of new chiral molecules L4, which contain two positively charged viologen units and express an R configuration, cucurbit[8]uril (CB[8]) molecules, and surfactant molecules sodium dodecyl sulfate (SDS). A series of experimental techniques and measurements were used to investigate the mechanism of chirality changes, including circular dichroism (CD) spectroscopy, absorption spectroscopy, isothermal titration calorimetry (ITC), and small-angle X-ray scattering (SAXS). The achiral SDS molecules were found to facilitate the transformation of chirality from the molecular level to the supramolecular level. The CD signals could be regulated precisely as the coassembled ternary supramolecular structure of the L4/SDS/CB[8] system strongly depended on the quantity of each component. When the molar ratio of L4, SDS, and CB[8] reached 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2, a significant chirality inversion from the positive upward CD signals to the negative downward CD signals was observed. The combination effects of non-covalent interactions which involve electrostatic and host–guest interactions caused a supramolecular structure packing change from the lamellar to the rectangular stacking pattern and resulted in a large chirality inversion at the supramolecular level. Our results may provide a facile approach to regulate multiple chiral activities in multicomponent supramolecular systems.
:2:2, a significant chirality inversion from the positive upward CD signals to the negative downward CD signals was observed. The combination effects of non-covalent interactions which involve electrostatic and host–guest interactions caused a supramolecular structure packing change from the lamellar to the rectangular stacking pattern and resulted in a large chirality inversion at the supramolecular level. Our results may provide a facile approach to regulate multiple chiral activities in multicomponent supramolecular systems.
Introduction
Chiral structures are widely discovered in nature such as amino acids, monosaccharides, and double-helical DNA structures, and they are essential for most forms of life.1–10 These chiral molecules and assemblies possess unique symmetrical and asymmetrical features, involving replication, recognition, adhesion, and catalytic activities in many disciplines such as biology, chemistry, and pharmaceuticals.11–13 Chiral supramolecules14 that consist of chiral/achiral compounds and other building blocks held together by non-covalent interactions (e.g., hydrogen bonding,15,16 electrostatic interactions,17 π–π stacking interactions,18–20 and host–guest interactions21) offer a promising strategy to build ordered chiral assembly structures and achieve versatile chiral activities at the macroscopic scale. Consequently, chirality induction,22 transfer,23 amplification,24 and inversion25–28 have been studied extensively in order to elaborate on the chiral activities in different supramolecular systems.29–31 Nevertheless, these processes are usually investigated and reported separately, and it still remains a considerable challenge to obtain multiple chirality changes and chiral activities that occur in a single supramolecular system. This may be attributed to the difficulty in generating highly compatible and cooperative supramolecular architectures that can afford different chiral activities in one system. Besides, as the chiral supramolecular structures are vulnerable to the external environment, it would be challenging to characterize and manipulate multiple chiral behaviors in a supramolecular structure, especially for complicated supramolecular coassemblies that include two or more components.32–35One possible way to circumvent these issues is to predesign suitable molecular structures and to utilize these components as building blocks to construct designed supramolecular architectures. For instance, a well-defined macrocycle-based host–guest system36–39 with a specific host and guest molar ratio may provide a feasible approach to build such supramolecular structures. Cucurbit[8]uril (CB[8]) is widely used as a host molecule to produce various dynamic host–guest supramolecular systems. The versatility of its macrocycle structure and design based on CB[8] allows supramolecules to achieve multiple orthogonal properties and to be prepared in predictable fashions. Viologens,40,41N,N′ di-quaternized bipyridyl salts with cationic units, are well-studied species and commonly used as guest components to interact with different cucurbituril macrocycles.42–45 Due to the reversible redox states, cationic properties, and tunability of the nitrogen substituents, viologens are used as functional groups in many systems such as molecular machines, electrochemical devices, batteries, and stimuli-responsive materials.46–50 Zhang et al. recently developed a new light-powered dissipative supramolecular polymerization approach by taking advantage of a 2:1 host–guest complexation of viologen moieties and CB[8] in the aqueous solution, offering a rational way to regulate different supramolecular states using light.51 Qu et al. successfully prepared a dual-stimuli-responsive supramolecular system that contains water-soluble titanium dioxide nanoparticles for the recyclable catalytic activity using methyl viologen radical cations and CB[8] macrocyclic structures.52 The idea of combining inorganic semiconductor nanoparticles and organic supramolecules has provided a new strategy to make complicated hybrid supramolecules that embrace multifunctionalities.
The aforementioned research has opened up new ways of preparing appropriate and sophistical viologen-cucurbituril supramolecular structures that can exhibit multiple properties. A supramolecular system that can afford different chiral activities may also be manufactured. Herein, we synthesized a new chiral molecule L4 that consists of a hydrophilic glycol chain at one end and two positively charged viologen moieties at the other end. The hydrophilic part and hydrophobic part were connected by a chiral phenylalanine linker and the L4 molecule that exhibits R configuration (Scheme 1). Thermally stable ternary supramolecular structures of L4/CB[8]/SDS with different molar ratios were prepared in water by a coassembling process. Circular dichroism (CD) spectroscopy showed that the chirality of the L4 molecule could be effectively transferred, amplified, and inversed at the supramolecular scale by simply adjusting the molar amount of each component in the system.
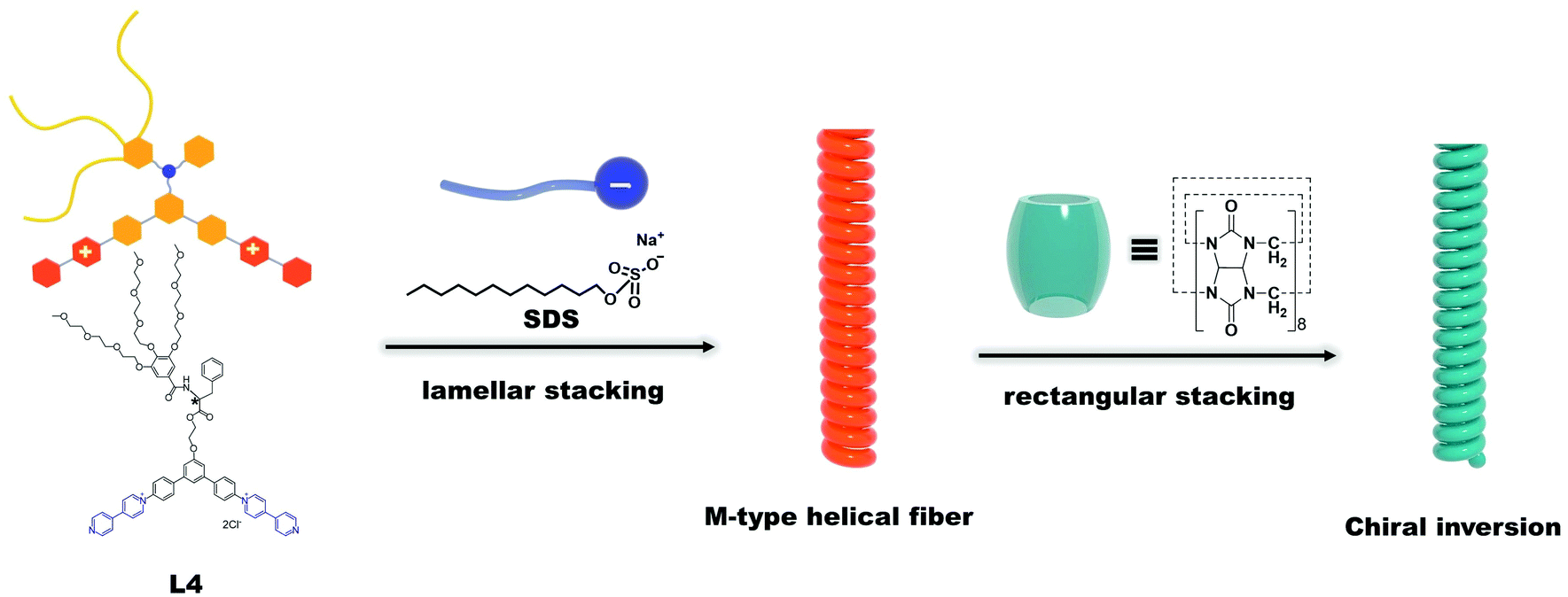 | ||
| Scheme 1 A schematic diagram of the possible supramolecular assembly route for L4/SDS/CB[8] systems. SDS molecules coassembled with L4 molecules to form a binary L4/SDS supramolecule and generated the M-type helical fibers via lamellar stacking, Adding CB[8] to the L4/SDS system resulted in a new ternary L4/SDS/CB[8] supramolecular structure possessing a rectangular stacking arrangement, which also caused a chirality inversion. | ||
The SDS was found to facilitate the chirality manipulation effectively by coassembling with L4 and CB[8]. First, SDS molecules generated anionic organosulfate groups in water and coassembled with L4 molecules via electrostatic interactions, which allowed L4 molecules to assemble towards a specific direction and to present the chirality at the supramolecular level. This coassembly process also resulted in a lamellar stacking pattern in the L4/SDS supramolecular structure. Second, by introducing CB[8] to the aqueous solution, the CD signals could be regulated precisely and exhibited a significant inversion from the original positive upward CD to the negative downward CD. The small-angle X-ray scattering (SAXS) measurements demonstrated that the original lamellar stacking pattern in the binary L4/SDS supramolecule was disrupted and gradually turned to a rectangular stacking arrangement with the increased amount of CB[8] (Scheme 1), which indicated a supramolecular morphology revolution. The isothermal titration calorimetry (ITC) measurements53,54 showed that two binding constants, k1 (6.17 × 105 M−1) of L4 with CB[8] and k2 (9.52 × 105 M−1) of L4 with SDS, were relatively close to each other, which would cause a cooperative effect of non-covalent interactions, involving the host–guest interactions of CB[8] and L4 molecules, the electrostatic interactions of L4 and SDS molecules, and the van der Waals interactions of each component. The comprehensive non-covalent interactions induced significant changes in both the supramolecular structure and the chirality. Our results may provide a new approach to manipulate multiple supramolecular chiral activities by using achiral surface species like ionic surfactants, and also demonstrate that the chiral properties at the molecular level can be dynamically regulated at the supramolecular level for complicated supramolecular systems that involve multicomponents.
Experimental
Materials
The synthesis procedures of L4 molecules can be found in the ESI.† All reagents and solvents, including SDS and CB[8] used in this experiment, were of reagent grade (TCI) and were used as received. The deionized water used was collected from a millipore ultrapure water machine (conductivity 18.2 mΩ cm−1, Labconco, USA).Coassembly
The formation process of L4/CB[8] co-assembly: the CB [8] aqueous solution (0.5 mM, 0.3 mL) was added to the L4 aqueous solution (0.05 mM, 3 mL); the formation process of L4/SDS co-assembly: different volumes of SDS aqueous solutions (0.1 M) were added to the L4 aqueous solution (0.05 mM, 3 mL); the formation process of L4/0.67SDS/bCB[8] ternary assembly: the SDS aqueous solution (0.1 M, 1 μL) was added to the L4 aqueous solution (0.05 mM, 3 mL), after 5 min, different volumes of CB[8] aqueous solutions (0.5 mM) were added to the L4/0.67SDS mixture. All mixtures containing the coassemblies were sealed, mixed evenly by manual shaking for 1 min, and then aged at room temperature for 24 h. The pH values of all the prepared solution samples were around 7.0 ± 0.12 (Table S1, ESI†).Characterization
Proton nuclear magnetic resonance (1H NMR) and 13C nuclear magnetic resonance (13C NMR) measurements were performed using tetramethyl silane (TMS) as the internal standard at room temperature (25 °C, 298 K). D2O was used as the deuterated solvent in the NMR titration measurements. Both 1H NMR and 13C NMR data were collected from a super conducting Fourier NMR spectrometer (Avance III 400, 400 MHz, Brucker). High-resolution time-of-flight mass spectrometry (HR-ESI) was performed using an LCT Premier XE mass spectrometer from the Water Company of the United States. The UV-Vis absorption spectrum was recorded using a Varian Cary 100 UV-Vis spectrophotometer with an optical path of 1 cm. TEM images were obtained using a transmission electron microscope JEM-1400 from the JEOL company (Japan). The aged solution was dropped onto a 230-mesh copper net, evaporated and dried under ambient conditions. The SEM images were obtained using a JSM-6360LV vacuum scanning electron microscope from the JEOL company (Japan). The aged solution was dripped on a silica plate coated with a thin layer of platinum to increase contrast, evaporated and dried under ambient conditions. The binding constant was measured with a nano-ITC instrument. The CD spectra were recorded using a ChirascanPlus CD spectrophotometer. The experiments were carried out at room temperature in a 1 cm path length quartz cell.Results and discussion
A chiral molecule L1 previously reported was chosen as the fundamental chiral unit for preparing the sequential chiral molecule L4.55 After combining two positively charged viologen phenyl diazonium salts, the L4 molecule exhibiting an R configuration was obtained (ESI,† and Scheme S1). The CB[8] molecule, as a good receptor and host of the cationic guest molecules, was used to encapsulate the viologen units of L4. UV-Vis absorption spectroscopy was first conducted in order to investigate the optical properties of this binary coassembly system of L4/CB[8]. As shown in Fig. 1a, the absorption spectrum of the L4 aqueous solution (0.02 mM) initially exhibited two main broad absorption bands, which peaked at about 326 nm associated with the viologen units and 263 nm corresponding to the phenyl groups. The absorption band at 263 nm decreased slowly while the absorption band at 326 nm showed a slight red shift to 365 nm after adding CB[8] (0.2 mM) to the L4 aqueous solution using a gradual titration method. The evolution of the absorption spectra suggested that the CB[8] molecules gradually coassembled with the L4 molecules as the positively charged viologen units were recognized and encapsulated by CB[8] cavities via host–guest interactions. When the amount of CB[8] reached 1.0 equivalent (eq.) relative to the L4, the absorption spectra remained almost unchanged. Excessive CB[8] would not influence the spectra, indicating that the L4/CB[8] coassembly system reached a steady equilibrium state when the molar ratio of L4 and CB[8] was 1:1. The binding ratio between L4 and CB[8] was also determined as 1:1 by Job's plot analysis56 (Fig. S1, ESI†), and by fitting the titration data to a 1:1 binding model, the binding constant of L4/CB[8] was 590751 M−1,56,57 which is close to the binding constants of other chirality regulation coassemblies that involved CB[8] in the literature.58,59 The isothermal titration calorimetry (ITC) measurements53,54 further confirmed that the L4 and CB[8] molecules established a stable host–guest supramolecular complex of 1:1 (Fig. 1b and Fig. S2, ESI†) with a binding constant of 6.17 × 105 M−1, which was close to the binding constant measured by fitting the UV-Vis titration data.
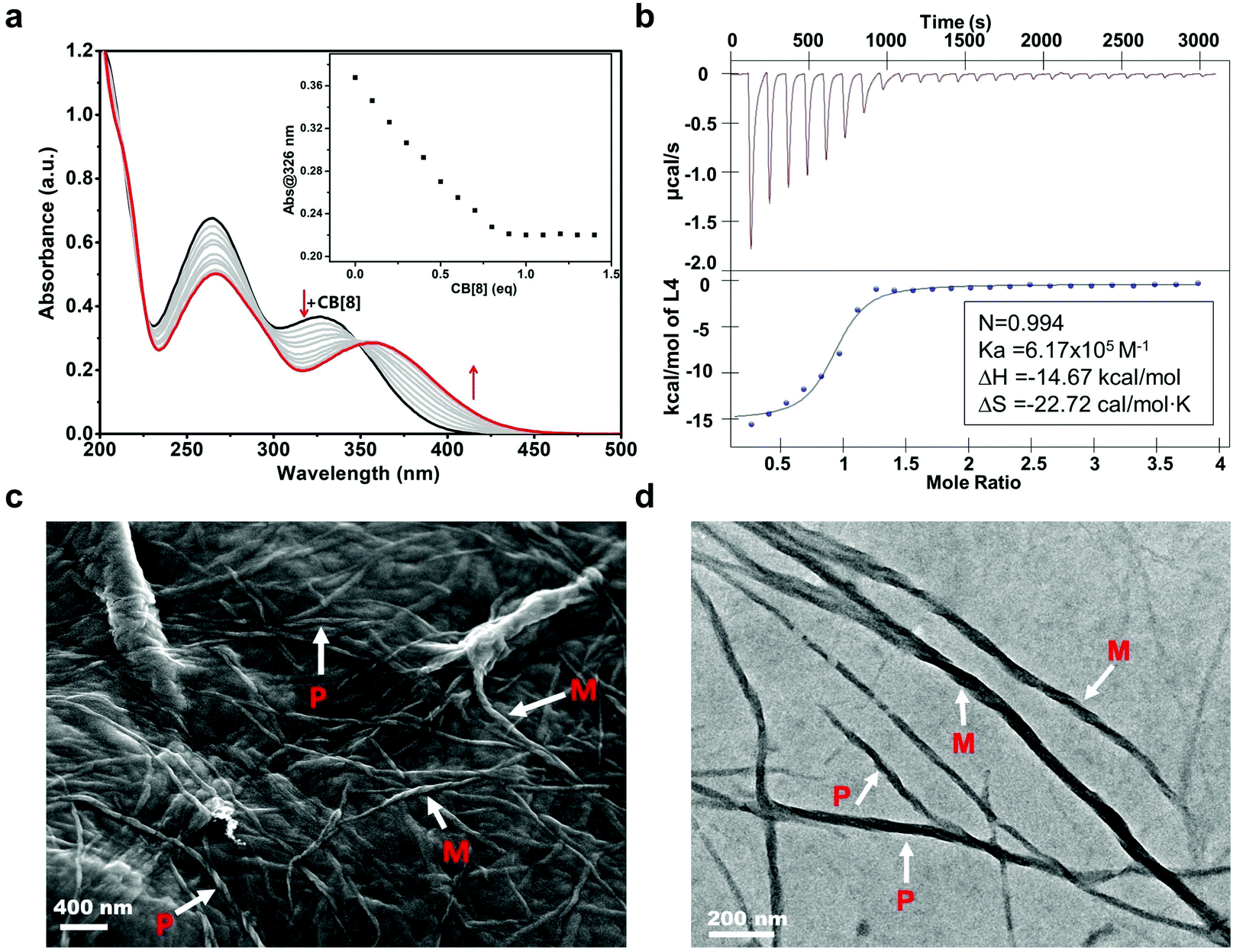 | ||
| Fig. 1 (a) UV-Vis absorption spectra of L4 (0.02 mM) titrated with CB[8] (0.2 mM); inset: absorbance at 326 nm varies with different equivalents of CB[8], (b) ITC data of L4 molecules coassembled with CB[8], showing a binding constant of k2 = 6.17 × 105 M−1; inset cell: [CB[8]] = 0.075 mM, syringe: [L4] = 1 mM, (c) the SEM image of the binary L4/CB[8] supramolecular fibers ([L4] = 0.05 mM), and (d) the TEM image of the L4/CB[8] supramolecular fibers ([L4] = 0.05 mM). | ||
A field emission scanning electron microscope (FE-SEM) and a transmission electron microscope (TEM) were used to investigate the morphology of the L4/CB[8] binary supramolecular structure. Assembled long helical nanofibers with an average diameter of around 20 nm and several hundreds of micrometers in length were observed by FE-SEM and TEM. These nanofibers showed both M-type (anticlockwise spiral structure) and P-type helical (clockwise spiral structure) morphologies (Fig. 1c and d). Although helical supramolecular structures were found, the signal was barely observed for the binary L4/CB[8] supramolecular system in circular dichroism (CD) spectroscopy (Fig. S3, ESI†). It is likely that the coassembly process of L4 and CB[8] was navigated towards the directions that were able to generate both clockwise and anticlockwise spiral nanofibers. As a result, the chirality of L4 molecules could not be presented effectively as M- and P-type nanofibers led to racemic properties.
Ionic surfactants have been reported to help chirality transfer from the molecular scale to the macroscopic scale due to their electrostatic and noncovalent interactions that could influence the supramolecular self-assembly.60,61 These surface species could also enhance the supramolecular chirality as their amphiphilic properties could guide the molecules to assemble towards specific directions. In order to generate sufficient chirality in L4/CB[8] supramolecules, we selected sodium dodecyl sulfate (SDS) as an anionic surfactant in this work. When dissolved in water, the negatively charged sulfate groups in SDS molecules could form electrostatic interactions with the positively charged viologen units in L4 molecules. UV-Vis absorption spectroscopy of L4 aqueous solution (0.01 mM) titrated with different amounts of SDS aqueous solution (0.01 M) was used to investigate the coassembly process of L4 and SDS molecules. As shown in Fig. 2a and Fig. S4 (ESI†), the L4/SDS binary system exhibited a nonlinear coassembly process due to the introduction of the SDS. When the amount of SDS was below 2.0 eq., the two absorption bands ranging from 240 to 400 nm decreased gradually, and the absorption band peaking at around 326 nm exhibited a slight red-shift to 336 nm. Such decline in absorbance was similar to the L4/CB[8] system as the L4/SDS supramolecular structure was gradually established through electrostatic interactions. However, different from CB[8], the SDS molecules were able to coassemble with L4 through the long alkyl chains via van der Waals interactions, and the coassembly process kept proceeding even when the molar amount of SDS was over 2.0 eq. The two absorption bands rebounded to ascend when more SDS was added to the solution. It is likely that the self-assembly process of the SDS molecules was able to influence the binary L4/SDS coassembly supramolecule. Both assembly modes could contribute to generating the L4/SDS supramolecular structure, leading to a redshift of the viologen peak from 336 nm to 353 nm. We ceased the titration when the amount of SDS reached 6.67 eq., where the absorption spectra barely changed, as shown in the inset of Fig. 2a. Besides, the Job's plot56 analysis based on UV-Vis absorption titrations, showing L4/(L4 + SDS) ≈ 0.33, indicates that the ratio of L4/SDS with both L4 and SDS was 1:2 (Fig. S5, ESI†).
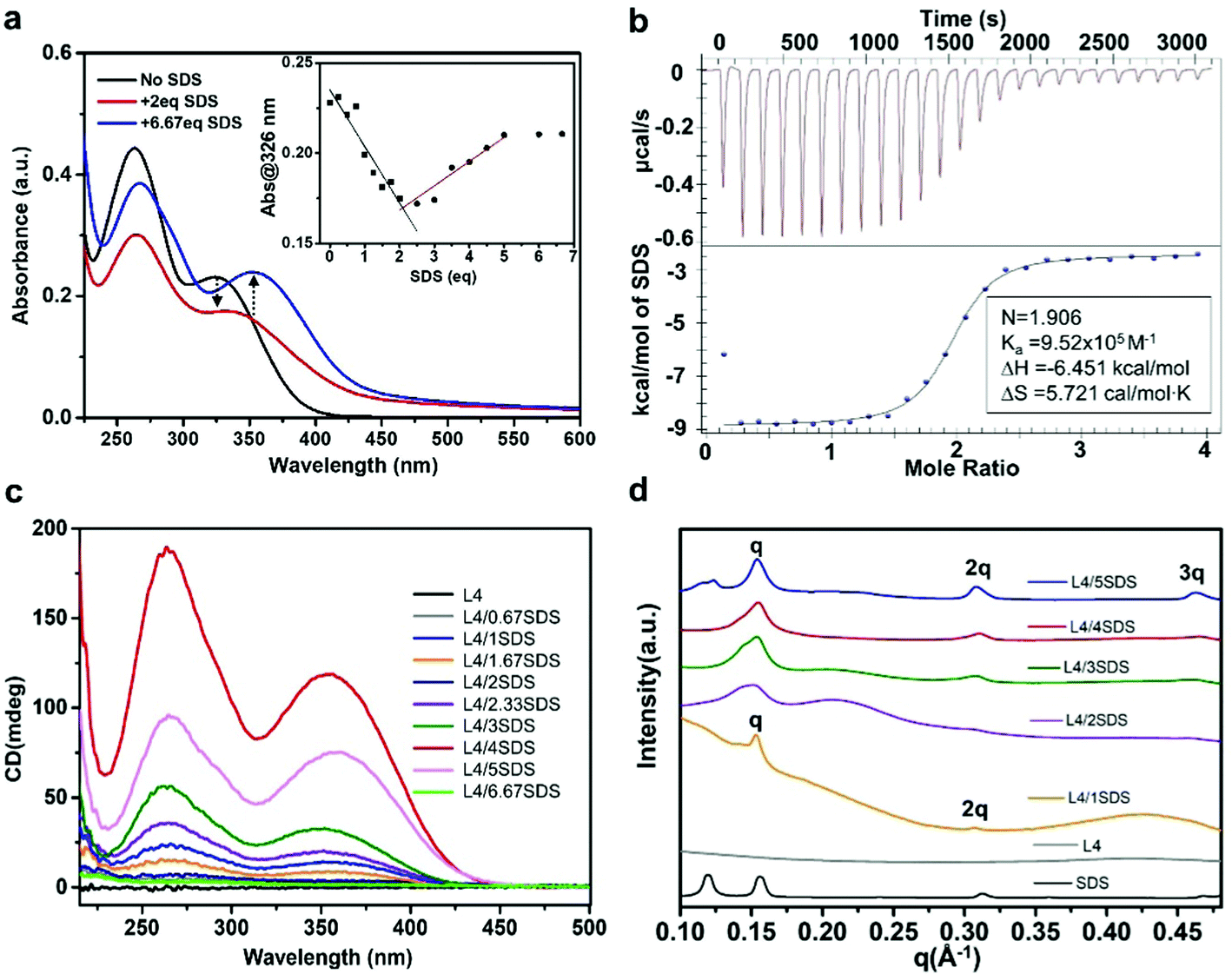 | ||
| Fig. 2 (a) The UV-Vis absorption spectra of aqueous L4 solution (0.01 mM) titrated with different amounts of SDS (0.01 M) (black trace: the absorption spectrum before adding SDS; red trace: the absorption spectrum after adding 2.0 eq. of SDS; blue trace: the absorption spectrum after adding 6.67 eq. of SDS; inset: absorbance at 326 nm varies with different equivalents of SDS). (b) ITC data of L4 molecules coassembled with SDS, showing a binding constant of k2 = 9.52 × 105 M−1; inset cell: [L4] = 0.08 mM, syringe: [SDS] = 1 mM, (c) CD spectra of the aqueous L4 solution (0.05 mM) titrated with SDS, and (d) the SAXS patterns of binary L4/SDS supramolecules with different molar ratios. | ||
The coassembly process of L4 and SDS was also investigated by ITC53,54 (Fig. 2b) and proton nuclear magnetic resonance (1H NMR) measurements (Fig. S6, ESI†). The L4/SDS binary system reached an equilibrium state when the molar ratio of L4 and SDS was 1:2 with a binding constant of k2 = 9.52 × 105 M−1. A binding constant (955831 M−1) of L4/SDS was obtained by fitting the 1:2 model in the supramolecular binding calculation website,56,57 and is almost consistent with the results of the ITC test. As this was about 1.5 times higher than that of the L4/CB[8] system, a relatively stronger electrostatic interaction was expected between the L4 and SDS molecules in the aqueous solution. According to the 1H NMR spectra, the proton peaks associated with the hydrogen atoms of the bipyridine units and the adjacent benzene rings of L4 tended to merge into smaller and broader peaks after adding the SDS to the aqueous solution (Fig. S6, ESI†). As a newly stable binary L4/SDS supramolecule was established via electrostatic interactions when the amount of SDS was 2.0 eq., the system experienced a totally different chemical environment that would lead to a sudden change in the NMR spectra even under low temperatures in D2O (Fig. S6b, ESI†). When the amount of SDS was over 4.0 eq., the self-assembly of excessive SDS molecules contributed to forming another different chemical environment. Besides, as the van der Waals interaction was much weaker than the electrostatic interaction, adding extra SDS to the L4 aqueous solution, after the L4 molar concentration reached 2.0 eq., would not affect the ITC signal. Several new peaks appeared at around 9.3 ppm, 8.7 ppm, 8.6 ppm, etc. in the 1H NMR spectra after the equivalent of SDS was over 2.0, indicating the appearance of the self-assembly of SDS molecules and a multistep coassembly of L4 and SDS.
It is possible that the binary L4/SDS supramolecule coassembled towards a specific pattern and generated chirality at the supramolecular level due to the strong electrostatic interaction. CD spectroscopy was therefore used to measure the chiral activity of this binary supramolecular system. As shown in Fig. 2c, we successfully observed a large positive Cotton effect of the aqueous L4/SDS solution with two broad absorption bands ranging from 225 nm to 435 nm, displaying a distinct positive upward (+) chirality of the binary L4/SDS supramolecular structure. In addition to the significant chirality transfer effect, the binary L4/SDS supramolecule also exhibited a chirality amplification as the CD signal increased gradually with the increasing amount of SDS. Interestingly, the CD signal continued to increase as the amount of SDS exceeded 2.0 eq., which was regarded as the equilibrium state of the binary system. When the amount of SDS reached 4.0 eq., the two absorption bands peaked at 264 nm and 364 nm with no further increase of the CD signal. These results revealed that after the initial coassembly of L4 and SDS (2.0 eq.), introducing an extra amount of SDS molecules could enhance the chiral activity of coassembly via the van der Waals interactions between the L4 and SDS molecules. Further addition of SDS molecules to reach a molar concentration of 6.67 eq. led to a decline of CD signals as the self-assembly with excessive SDS molecules would suppress the supramolecular chirality (Fig. 2c, pink and green traces). This is because excessive SDS would destroy the continuity of the further assembly of L4/SDS supramolecular units, resulting in the hindrance of chirality transferring and amplification at the supramolecular level.9
We then conducted SAXS measurements to further investigate the L4/SDS supramolecular packing structure. The molecular distance d of the supramolecule can be obtained through the equation d = 2π/q, where q is the scattering factor (vector). The L4 showed no diffraction peaks ascribed to a disordered assembled structure (Fig. 2d, gray trace). After coassembling with SDS, the scattering peaks of the samples were indexed as q1:q2 = 1:2 when the equivalent of SDS was low (≤1.0 eq.), and the associated d values were obtained as 4.08 nm and 2.03 nm. The d value 4.08 nm was close to the calculated length of the L4 molecule (estimated to be about 4.13 nm using Advanced Chemistry Development/ChemSketch molecular modeling software, Fig. S7, ESI†). As a result, a lamellar packing pattern was expected for the L4/SDS binary supramolecular structure. As the amount of SDS introduced was increased, this lamellar structure developed in a more sophisticated and stable fashion. When the equivalent of SDS was 5.0, there was a new peak q3 appearing in the SAXS pattern and the scattering peaks could be indexed q1:q2:q3= 1:2:3 with the corresponding d values of 4.08 nm, 2.03 nm, and 1.35 nm, respectively. The new peak q3 is likely to be associated with the self-assembly of the extra SDS molecules, which can also form the lamellar stacking mode (Fig. 2d, black trace). There is no doubt that SDS molecules facilitated chiral L4 molecules to construct such an ordered lamellar supramolecular structure that could show chirality at the supramolecular level.
The supramolecular morphology of the binary L4/SDS structure can be characterized by TEM. As shown in Fig. 3a, the L4/SDS initially coassembled to small nanofibers (∼20 nm in diameter and ∼250 nm in length on average) when the amount of SDS was relatively low (0.5 eq.). As these nanofibers were too small, the CD signal was barely observed in CD measurements accordingly (Fig. 2c). The nanofibers continued to grow into larger fibers with the increasing SDS (Fig. 3b). A few L4/SDS long fibers were found to exhibit M-type helical morphologies and P-type helical fibers were hardly found, which implied that the coassembly of L4 and SDS was guided towards a direction in which the M-type fibers were mainly generated (Fig. 3c and d). The major M-type morphology of the L4/SDS supramolecule resulted in large CD signals and the continuous introduction of SDS would enhance the CD signals (Fig. 2c). When the amount of SDS was over 4.0 eq., these microfibers stopped growing, and a few thin films were thus observed (Fig. 3e and f) due to the self-assembly of the excess SDS molecules that did not participate in the coassembly process (Fig. S8, ESI†). The magnification of chirality halted as these self-assembled thin films might have wrapped up M-type helical fibers and impeded supramolecular chirality. These results also were consistent well with the measurements of CD spectroscopy as shown in Fig. 2c.
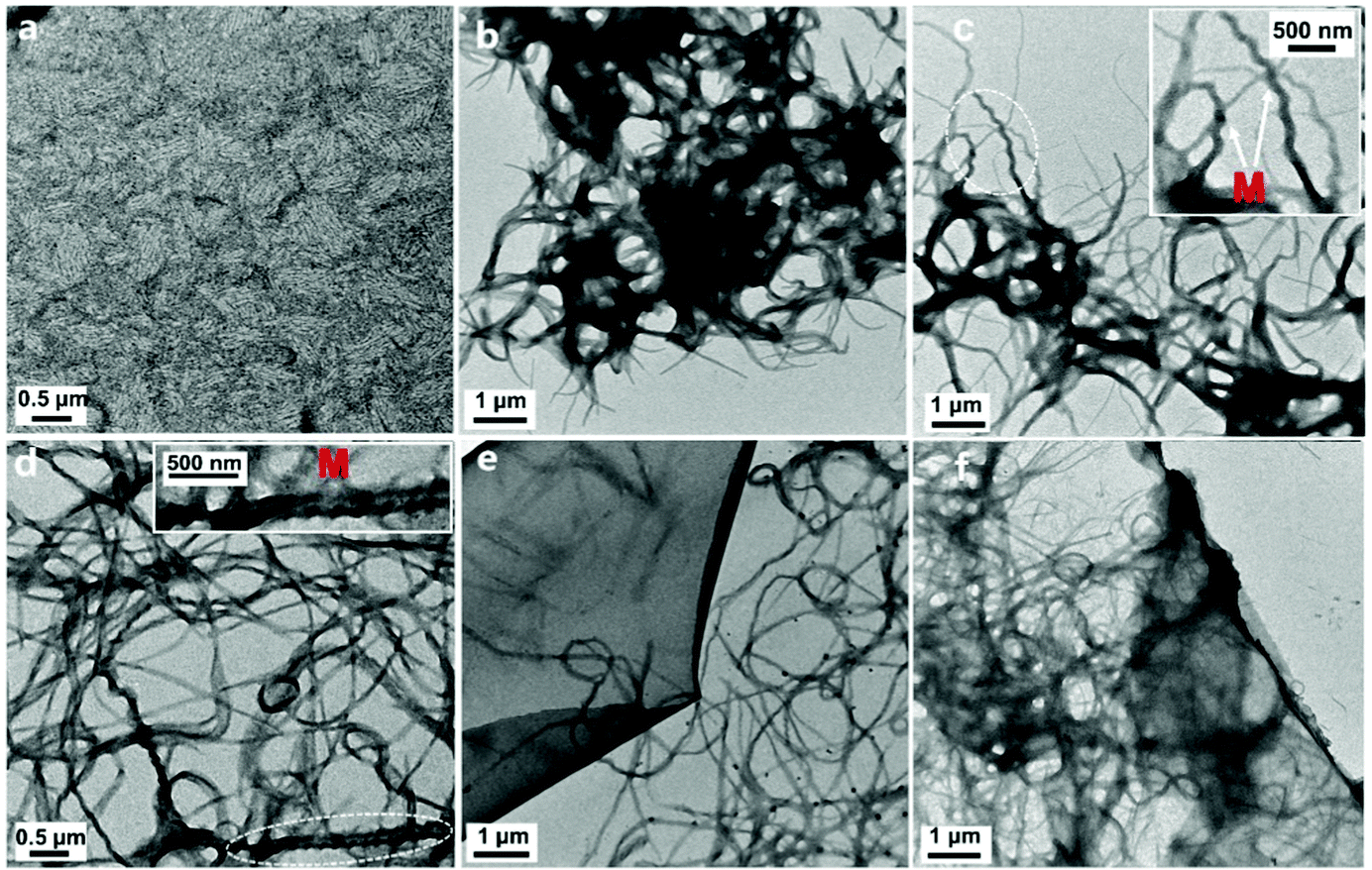 | ||
| Fig. 3 The TEM images of L4/SDS supramolecules with different equivalents of SDS in the solutions: (a) 0.5 eq., (b) 1.0 eq., (c) 2.0 eq., (d) 4.0 eq., (e) 5.0 eq., and (f) 6.67 eq. ([L4] = 0.05 mM). | ||
Since the chirality of L4 molecules can be effectively delivered and amplified to the supramolecular structure by coassembling with SDS, it is promising for the L4/CB[8] host–guest supramolecule to present chirality by taking advantage of this approach. As the binding constant k2 (9.52 × 105 M−1) of SDS and L4 was larger than the binding constant k1 (6.17 × 105 M−1) of CB[8] and L4, directly adding SDS to the L4/CB[8] system would result in the dissociation of the L4/CB[8] supramolecular structure (Fig. S9, ESI†) and the silence of the CD signal (Fig. S10a and b, ESI†). Herein, we mainly focused on adding CB[8] to the L4/SDS chiral supramolecular system.
We preliminarily investigated the chirality in the ternary coassembled system of L4/aSDS/bCB[8] (a and b represent molar equivalents of SDS and CB[8], respectively) with various amounts of SDS and CB[8] using CD spectroscopy measurements. According to the ITC measurements discussed above, one L4 molecule could either coassemble with two SDS molecules or one CB[8] molecule to reach a steady equilibrium state with associated ratios of 1:2 and 1:1, respectively (Fig. 1b and 2b). Therefore, the parameters a and b should follow the equation a/2 + b ≈ 1 in order to obtain a steady ternary coassemble supramolecule. As shown in Fig. 4a, the chirality of the ternary structure varied at different ratios of a and b. When the amount of SDS was relatively high and the amount of CB[8] was low, where 1 ≤ a ≤ 2 and 0 ≤ b < 0.5, the CD signals were positive. The CD signals started to decline dramatically after the introduction of CB[8] to the binary L4/SDS system. When the amount of CB[8] reached 0.5 eq. (a = 1.0, b = 0.5), the CD spectrum displayed a pattern similar to a flat trace that was close to 0, as there was almost no CD signal (Fig. 4a, gray dashed trace). It seemed that the supramolecular chirality was deeply suppressed when CB[8] started participating in the coassembly with L4/SDS supramolecules. When the amount of SDS decreased and the amount of CB[8] continued to increase, where 0 ≤ a < 1 and 0.5 < b < 1, chirality got inversed surprisingly as the CD signals turned from positive upward (+) to negative downward (−). When the molar ratio of L4:SDS:CB[8] was 1:0.67:0.67 (i.e. ∼3:2:2), the chirality inversion reached the farthest as the CD signal displayed a mirror symmetry to the L4/SDS (1:0.67) sample along the axis of the wavelength. The large change of chirality, especially the CD inversion, suggested that the coassembled structure in the ternary L4/SDS/CB[8] system might be different from the binary L4/SDS supramolecule.
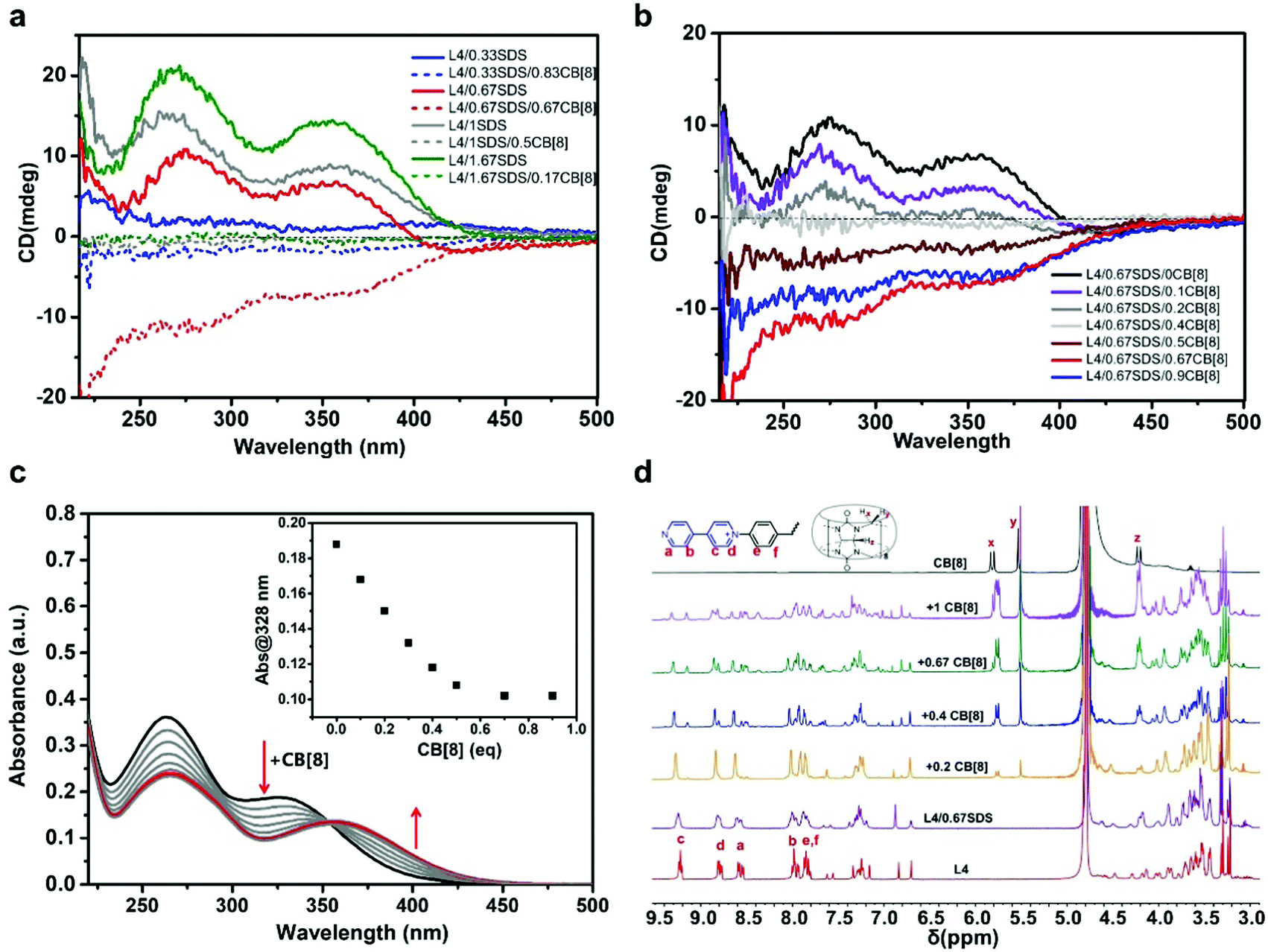 | ||
| Fig. 4 (a) CD spectra of L4/SDS and L4/SDS/CB[8] with different molar ratios, (b) CD spectra of aqueous L4/0.67SDS solution ([L4] = 0.05 mM) titrated by CB[8], (c) UV-Vis absorption spectra of aqueous L4/0.67SDS solution ([L4] = 0.01 mM) titrated with CB[8] (0.1 mM); inset: absorbance at 328 nm varies with different equivalents of CB[8], and (d) 1H NMR spectra of the L4/0.67SDS in D2O solution with different amounts of CB[8]. The concentration of L4 was regulated to 1 mM in order to get enough 1H NMR signal. | ||
It is also noticeable that the chirality changes highly depended on the molar ratio of each component. On one hand, when the equivalent of SDS was less than 1.0 (0 ≤ a < 1), L4 molecules were abundant before CB[8] was added, and each L4 molecule preferred to interact with one SDS molecule instead of two SDS molecules due to the steric effect. Therefore, there were some free L4 molecules and the L4/SDS coassembled units in the aqueous solution. In this case, most individual L4/SDS coassembled units contained one SDS molecule and one free viologen chain, leading to a preliminary layered supramolecular structure that could exhibit supramolecular chirality. When CB[8] was added, the CB[8] molecules directly interacted with the free viologen units of L4 molecules via host–guest interactions. These free viologen units might either belong to free L4 molecules or L4 molecules that had already coassembled with SDS molecules. As the CD signals started to decrease, CB[8] seemed to largely inhibit the chirality. However, instead of remaining constant, the CD signals reverted to the negative side and climbed to the lowest position when the ternary system reached equilibrium, leading to an obvious chirality inversion. It is possible that the incorporation of CB[8] molecules helped to disrupt the initial lamellar packing structure and establish a new ternary coassembled structure of L4, SDS, and CB[8]. The formed L4/SDS/CB[8] structure also showed a strong chirality but with negative CD signals. On the other hand, when the equivalent of SDS was over 1.0, there were no free L4 molecules in the solution. Even though the free viologen units could contribute to fabricating L4/SDS/CB[8] supramolecules, the newly formed structure was less stable and immature. Therefore, the CD signal intensity initially decreased as the negative CD signals counteracted the positive CD signals. However, the CD signals were not able to revert as the L4/SDS supramolecules that possessed positive CD signals still dominated in the solution. In both cases, some CB[8] molecules might also encapsulate the SDS and viologen chains by host–guest interactions. The encapsulation effect could impede CD spectra as it might destroy the original supramolecular structure and prohibit chirality expression. However, this encapsulation could not revert supramolecular chirality as no CD inversion was observed when saturated stable binary L4/SDS systems (1:2 and 1:4) were titrated with CB[8] (Fig. S11, ESI†), where the decrease of CD signals was mainly caused by host–guest interactions.
As a result, free L4 molecules and viologen units were the keys to achieve negative CD signals for the ternary L4/SDS/CB[8] supramolecular structure. The chirality inversion did not result from the non-covalent interactions between two binary supramolecular systems (i.e. the interactions between the L4/SDS and L4/CB[8] supramolecules). As pure binary L4/CB[8] could not generate any CD signal, merely adding L4/CB[8] supramolecules to the L4/SDS supramolecular system would not cause such a large change in chirality. This was further confirmed by mixing stable L4/CB[8] supramolecules (molar ratio 2:2) with stable L4/SDS (molar ratio 1:2) supramolecular systems to achieve a molar ratio of 3:2:2, which was very similar to the molar ratio in the stable ternary L4/SDS/CB[8] coassembled supramolecules (molar ratio 1:0.67:0.67). Although CD signals were found to decrease due to the dilution of L4/SDS concentration by mixing, there was no chirality inversion (Fig. S12, ESI†). We also mixed L4, SDS, and CB[8] together with a molar ratio of 3:2:2 and negative CD signals were observed in the solution (Fig. S13, ESI†). Therefore, such chiral change and inversion would be due to the coassembly of L4, SDS, and CB[8], which generated a new ternary supramolecular structure.
We then mainly focused on investigating the coassembly mode of the L4/0.67SDS/bCB[8] system with various amounts of CB[8], because the L4/0.67SDS/0.67CB[8] structure showed the largest chirality inversion. CD spectroscopy was conducted once CB[8] was added to the L4/0.67SDS binary system (Fig. 4b). The two broad absorption bands around 275 nm and 356 nm of the aqueous L4/0.67SDS solution caused by the positive Cotton effect declined constantly with the increased amount of CB[8]. When the equivalent of CB[8] reached 0.4 eq., the CD signal was almost imperceptible as the spectrum trace profiled nearly a flat line. The CD signal reverted to negative as we continued to add CB[8], which indicated that the ternary L4/SDS/CB[8] supramolecule started to dominate. The negative CD signal arrived at the maximum as the quantity of CB[8] = 0.67 eq., where a stable ternary L4/SDS/CB[8] supramolecular system was fully developed. As expected, more CB[8] after 0.67 eq. would suppress the CD signal due to the CB[8] encapsulation effect by the host–guest interaction (Fig. 4b, blue trace). The artifacts derived from linear dichroism and linear birefringence were also excluded by recording the CD spectra of the isomeric molecule D4, which shares the same molecular structure with the L4 molecule but possesses an opposite S configuration (Fig. S14, ESI†). Moreover, the obtained ternary L4/SDS/CB[8] supramolecule exhibited a certain degree of thermal stability as there were almost no changes in the CD spectra of L4/SDS/CB[8] (1:0.67:0.67) when the sample was put in an environment under an elevated temperature from 25 °C to 80 °C for more than 2 h (Fig. S15, ESI†).
Both UV-Vis absorption spectroscopy and 1H NMR measurements were used to investigate the formation of the L4, SDS, and CB[8] coassembled system (the molar ratio of L4:SDS was set as 1:0.67 and the equivalent of CB[8] varied). As shown in Fig. 4c, the two broad absorption bands from 240 to 400 nm gradually declined after adding CB[8] to the L4/SDS aqueous solution. The absorbance of the small shoulder at around 328 nm gradually declined and showed a steady redshift as the ternary L4/SDS/CB[8] was gradually established. When the amount of CB[8] was 0.67 eq., this absorption band stopped changing and the peak stayed at around 355 nm, suggesting that the ternary supramolecule reached a steady state. Similar results were also observed in the UV-Vis absorption spectra of L4/0.33SDS and L4/1.5SDS aqueous solutions titrated with CB[8] (Fig. S16a and b, ESI†). Interestingly, distinct from the CD signal changes, excessive CB[8] would not cause significant changes in UV-Vis absorption spectra when the amount of SDS was largely over 2.0 eq., such as in the L4/SDS (1:4) aqueous solution (Fig. S17, ESI†). This might be due to the strong electrostatic interactions in the stable saturated L4/SDS system, which could not be disrupted by CB[8]. The 1H NMR measurements confirmed the formation of the ternary L4/SDS/CB[8] supramolecular system. As shown in Fig. 4d, when CB[8] was gradually added to the L4/0.67SDS binary aqueous solution, the proton peaks of the bipyridine unit and the adjacent benzene rings of the L4 molecule at around 8.01, 8.59, 8.81, and 9.25 ppm were observed to deform and new peaks were observed. Particularly, there were new peaks appearing at around 8.35, 8.50, 8.79, and 9.15 ppm, which indicated that the viologen units were experiencing a very different chemical environment in the L4/SDS/CB[8] supramolecular system. When CB[8] was excessive, where the amount was 1.0 eq., the intensity of the new proton peak on the viologen units further increased and the proton peaks corresponding to the viologen units split. This showed that the excessive CB[8] molecules might have wrapped the SDS and the viologen units via host–guest interactions and led to a supramolecular chirality prohibition. These results were also consistent with the results of Fig. S9–S11 (ESI†).
TEM was used to characterize the morphology changes of the L4/SDS/CB[8] supramolecules. When there is no CB[8] in the solution, the binary L4/0.67SDS supramolecules would form nanofibers (about 3.5 μm in length, Fig. 5a), which showed an agreement with those fibers in Fig. 3. It was also observed by SEM that these nanofibers were helical fibers with an M-type helical morphology that showed a positive CD signal in the CD spectroscopy (Fig. S18, ESI†). With the increasing amount of CB[8], these nanofibers began to disintegrate into shorter nanowires with different lengths (Fig. 5b). As the amount of CB[8] reached 0.4 eq., some microribbons and short nanowires were observed simultaneously (Fig. 5c). This demonstrated that CB[8] molecules would cause an instant change of the supramolecular morphology during the coassembly process. On one hand, no P-type helical fibers were found during the whole period. It was likely that these nanowires were too small, and the helical structure could not be observed directly in the assembled fibers. As for the two-dimensional structure of the microribbons, it is difficult to form three-dimensional helical structures like microfibers in the binary L4/SDS system. On the other hand, as the M-type fibers continued to dissociate with the introduction of CB[8], the newly formed nanowires and microribbons due to the formation of L4/SDS/CB[8] were expected to possess a different coassembly mode that could produce a negative CD signal. Finally, when the amount of CB[8] approached 0.67 eq., the microribbons of the L4/SDS/CB[8] supramolecular structure dominated (Fig. 5d).
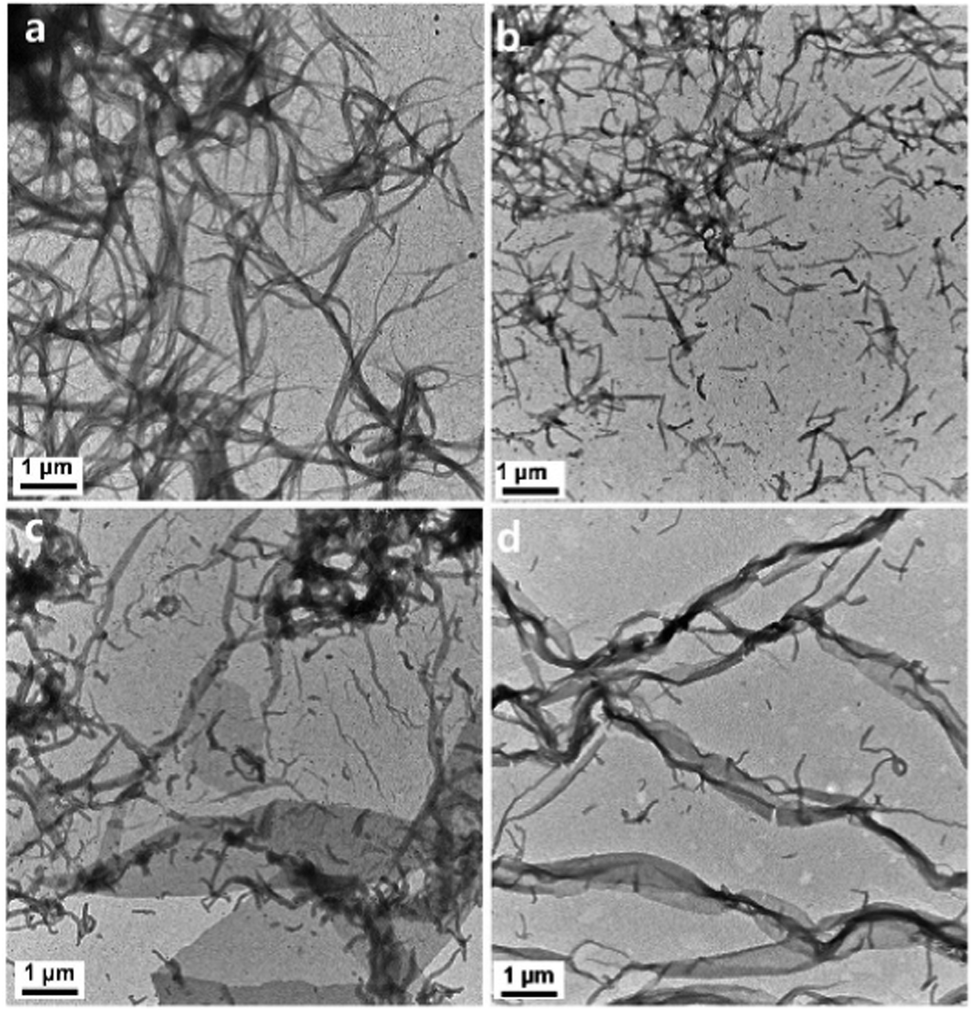 | ||
| Fig. 5 The TEM images of L4/0.67SDS/bCB[8] ternary coassembled with different equivalents of CB[8] (b values): (a) 0 eq., (b) 0.2 eq., (c) 0.4 eq., and (d) 0.67 eq. ([L4] = 0.05 mM). | ||
Lastly, in order to further understand the stacking pattern of the L4/SDS/CB[8] supramolecules, we also performed SAXS measurements in different L4/SDS/CB[8] systems. The supramolecular structure of L4/SDS (1:0.67) showed a typical lamellar stacking pattern with q1 = 0.154 Å−1 and q2 = 0.312 Å−1 (q1:q2 = 1:2) which was the same as the other L4/SDS binary systems as shown in Fig. 2d (Fig. 6a, green trace). However, the layered feature peaks started to fall and diminish when CB[8] was incorporated. At the beginning (L4:SDS:CB[8] = 1:0.67:0.2), there were barely any significant peaks associated with q1 and q2 and the ones related were much weaker and broader (Fig. 6a, yellow trace). As CB[8] continued to disrupt the old binary supramolecular arrangement, a new peak at q3 = 0.224 Å−1 was observed with a ratio of L4:SDS:CB[8] = 1:0.67:0.4, suggesting that a different supramolecular packing pattern was generated (Fig. 6a, black trace). More peaks were observed when the molar ratio became 1:0.67:0.67 with q4 = 0.340 Å−1, q5 = 0.365 Å−1, and q6 = 0.444 Å−1, respectively (Fig. 6a, red trace), as the newly ternary L4/SDS/CB[8] supramolecule structure dominated. The corresponding d values were calculated to be d3 = 2.80 nm, d4 = 1.85 nm, d5 = 1.72 nm, and d6 = 1.41 nm, respectively, and the ratio of 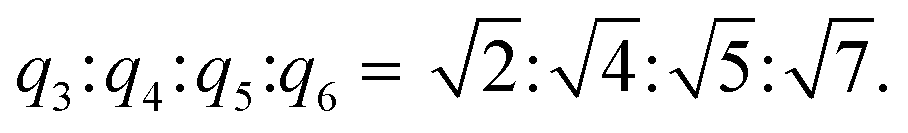 These d values and q ratios suggested a rectangular packing structure in the ternary L4/SDS/CB[8] supramolecule.62 More CB[8] would lead to the generation of a hexagonal packing structure,63 as another peak q7 = 0.293 Å−1 appeared in the SAXS pattern, and the ratio of q7:q4 was
These d values and q ratios suggested a rectangular packing structure in the ternary L4/SDS/CB[8] supramolecule.62 More CB[8] would lead to the generation of a hexagonal packing structure,63 as another peak q7 = 0.293 Å−1 appeared in the SAXS pattern, and the ratio of q7:q4 was 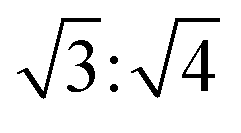 . The hexagonal packing mode was also found in the L4/CB[8] coassembled supramolecule with a q ratio of
. The hexagonal packing mode was also found in the L4/CB[8] coassembled supramolecule with a q ratio of 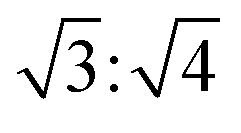 (Fig. S19, ESI†). This further demonstrated that spare CB[8] molecules were able to directly enclose the SDS and viologen units, which was consistent with the results obtained in Fig. 4d.
(Fig. S19, ESI†). This further demonstrated that spare CB[8] molecules were able to directly enclose the SDS and viologen units, which was consistent with the results obtained in Fig. 4d.
 | ||
| Fig. 6 (a) The SAXS patterns of L4/SDS/CB[8] with different molar ratios. (b) A graphic presentation of M-type helical fibers of L4/SDS and the chiral inversion after adding CB[8]. | ||
In order to understand the different molecular arrangements in binary and ternary assemblies, 1H NMR DOSY measurements of binary co-assembly L4/0.67SDS and co-assembly ternary L4/0.67SDS/0.67CB[8] were conducted. As shown in Fig. S20 (ESI†), the diffusion coefficient of L4/0.67SDS was slightly larger than that of the L4/0.67SDS/0.67CB[8] mixture (DL4/0.67SDS = 2.00 × 10−10 m2 s−1, DL4/0.67SDS/0.67CB[8] = 1.62 × 10−10 m2 s−1), indicating that the introduction of CB[8] interacting with the remaining free viologen in L4/0.67SDS changed the state of the original assembly and led to the formation of larger assemblies.64 In addition, we investigated the time-dependent morphological changes of binary and ternary supramolecules by TEM characterization. It was found that the change morphological trajectories of L4/0.67SDS (Fig. S21, ESI†) and L4/0.67SDS/0.67CB[8] (Fig. S22, ESI†) supramolecules over time were completely different, showing that their assembly ways and processes were completely irrelevant. Therefore, two completely opposite supramolecular chiral coassemblies were produced (Fig. 4b, black trace and red trace), which is consistent with the results of the SAXS test in Fig. 6.
Surfactants were used to regulate the chiral activities in supramolecular systems. Nevertheless, they are either responsive surfactants that could induce the supramolecular morphology changes and influence the chirality upon external stimuli, or surfactant molecules themselves containing chiral chains. Moreover, most of these cases usually involved one specific chirality manipulation.28,65–68 To the best of our knowledge, we found no previous examples of using non-chiral surfactant molecules to achieve the manipulation of multiple chiral activities in a multicomponent supramolecular system. First, the ionic surfactant molecule (SDS) formed a binary coassembly structure with the L4 molecule mainly through electrostatic interactions. As L4 molecules themselves could not generate a chiral supramolecular structure, SDS was expected to guide L4 molecules to assemble towards a specific direction (here is anticlockwise M-type) and to generate a lamellar stacking supramolecular structure. In this way, the chirality at the molecular level of L4 was effectively transferred to the supramolecular scale by the coassembly process. Second, by coassembling SDS, L4 and CB[8] molecules, a stable ternary L4/SDS/CB[8] supramolecular structure was prepared. In our case, the SDS molecules first helped to form the chiral supramolecule of L4/SDS that could express significant positive CD signals by guiding L4 assembly towards a specific direction. The introduction of CB[8] to the system might cause the dissociation of the binary supramolecule of L4/SDS as the perturbation of rigid CB[8] rings could destroy the lamellar packing motif. However, as the binding constant of k2 (SDS and L4) is larger than the k1 (CB[8] and L4), instead of replacing SDS, CB[8] incorporated a ternary coassembly process to establish a new stable ternary supramolecular system L4/SDS/CB[8]. The chirality was retained while causing negative CD signals as the packing mode changed to a rectangular pattern. As the ternary supramolecular structure highly depended on the molar quantity of each component, it offered a way to regulate supramolecular chirality in a precise manner. Here, the key to manipulate chirality inversion is to obtain both free viologen units and L4 molecules in the solution. On one hand, when the molar ratio of L4:SDS is more than 1, i.e., there were both free viologen units and L4 molecules in the solution that did not participate in the coassembly yet, CB[8] could be easily incorporated into the ternary coassembly process. It was likely that the original lamellar packing structure was not stable enough and disrupted by CB[8], resulting in a rectangular packing ternary L4/SDS/CB[8] supramolecular mode (Fig. 6b). This process also led to a significant chirality inversion as the supramolecular structure was changed. On the other hand, when the molar ratio of L4:SDS was not more than 1 (≤1), i.e. there were some free viologen units but almost no free L4 molecules, the chirality inversion could not be realized as there were not enough free L4 molecules to build a stable rectangular packing supramolecule. Even though in the ideal case where the L4:SDS = 1, the CD signals could not be managed to obvert negative by adjusting the amount of CB[8]. When the ratio became no more than 0.5 (L4:SDS = 1:2), where all viologen units had interacted with SDS molecules, CB[8] could only suppress supramolecular chirality by directly encapsulating the binary L4/SDS supramolecules, which led to a decrease of CD signals. Moreover, the special triangle molecular geometry of the L4 molecule itself might also facilitate the transformation of the supramolecular packing mode by incorporating the rigid structure of CB[8]. Although the binary L4/SDS supramolecule had lamellar stacking, the ternary supramolecule of L4/SDS/CB[8] could be built up into a rectangular stacking mode at a specific molar ratio (such as 3:2:2 of L4:SDS:CB[8]).
Conclusions
In summary, we have fabricated a new chiral supramolecular system with multicomponents that involve positively charged chiral L4 molecules, macrocyclic structures CB[8], and SDS by a direct supramolecular coassembly method. The chirality of the L4 molecule could be transferred and magnified at the supramolecular scale. Moreover, we were able to regulate the chirality precisely and to achieve an instant large chirality inversion by adjusting the quantity of each component. SDS surfactant molecules could guide the L4 molecule to assemble towards a defined direction, which generated M-type helical fibers and facilitated the supramolecular chirality expression effectively. In some specific cases, such as the 1L4/0.67SDS/0.67CB[8] (3:2:2) supramolecular system, the layered packing pattern of the formed L4/SDS supramolecule could be destroyed by CB[8] molecules and result in a new rectangular packing mode during the coassembly process, causing a supramolecular chiral reversion. Our results demonstrate a feasible way to achieve chirality regulation in a supramolecule that contains multiple components. It may also afford a promising opportunity to design and fabricate controllable advanced chiral materials with high complexity and fidelity in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grants 22025503, 21790361, 21871084, and 21672060), the Shanghai Municipal Science and Technology Major Project (grant 2018SHZDZX03), the Fundamental Research Funds for the Central Universities, the Program of Introducing Talents of Discipline to Universities (grant B16017), the Program of Shanghai Academic/Technology Research Leader (19XD1421100), and the Shanghai Science and Technology Committee (grant 17520750100). We thank the NSFC/China for financial support. F. T. also thanks the Shanghai Sailing Program (21YF1409200) and the Fundamental Research Funds for the Central Universities. We thank the Research Center of Analysis and Test of East China University of Science and Technology for help on the characterization.Notes and references
- T. Zhao, J. Han, P. Duan and M. Liu, Acc. Chem. Res., 2020, 53, 1279–1292 CrossRef CAS PubMed.
- D. Xia, P. Wang, X. Ji, N. M. Khashab, J. L. Sessler and F. Huang, Chem. Rev., 2020, 120, 6070–6123 CrossRef CAS.
- G. A. Hembury, V. V. Borovkov and Y. Inoue, Chem. Rev., 2008, 108, 1–73 CrossRef CAS PubMed.
- J. Ryssy, A. K. Natarajan, J. Wang, A. J. Lehtonen, M. K. Nguyen, R. Klajn and A. Kuzyk, Angew. Chem., Int. Ed., 2021, 60, 5859–5863 CrossRef CAS.
- K. C. Peters, S. Mekala, R. A. Gross and K. D. Singer, J. Mater. Chem. C, 2020, 8, 4675–4679 RSC.
- W.-G. Qiao, J.-B. Xiong, Y.-X. Yuan, H.-C. Zhang, D. Yang, M. Liu and Y.-S. Zheng, J. Mater. Chem. C, 2018, 6, 3427–3434 RSC.
- Z. Zong, P. Zhang, H. Qiao, A. Hao and P. Xing, J. Mater. Chem. C, 2020, 8, 16224–16233 RSC.
- D. Yang, P. Duan, L. Zhang and M. Liu, Nat. Commun., 2017, 8, 15727 CrossRef CAS PubMed.
- B. Yue, L. Yin, W. Zhao, X. Jia, M. Zhu, B. Wu, S. Wu and L. Zhu, ACS Nano, 2019, 13, 12438–12444 CrossRef CAS.
- H. Wang, K. Wang, Y. Xu, W. Wang, S. Chen, M. Hart, L. Wojtas, L. P. Zhou, L. Gan, X. Yan, Y. Li, J. Lee, X. S. Ke, X. Q. Wang, C. W. Zhang, S. Zhou, T. Zhai, H. B. Yang, M. Wang, J. He, Q. F. Sun, B. Xu, Y. Jiao, P. J. Stang, J. L. Sessler and X. Li, J. Am. Chem. Soc., 2021, 143, 5826–5835 CrossRef CAS PubMed.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS.
- Y.-J. Choi, W.-J. Yoon, M. Park, D.-G. Kang, G. Bang, J. Koo, S.-I. Lim, S. Park and K.-U. Jeong, J. Mater. Chem. C, 2019, 7, 3231–3237 RSC.
- J. Hu, T. Zhu, C. He, Y. Zhang, Q. Zhang and G. Zou, J. Mater. Chem. C, 2017, 5, 5135–5142 RSC.
- P. Xing and Y. Zhao, Acc. Chem. Res., 2018, 51, 2324–2334 CrossRef CAS PubMed.
- L. Perez-Garcia and D. B. Amabilino, Chem. Soc. Rev., 2002, 31, 342–356 RSC.
- T. Kakuta, T. A. Yamagishi and T. Ogoshi, Acc. Chem. Res., 2018, 51, 1656–1666 CrossRef CAS.
- I. Neira, A. Blanco-Gomez, J. M. Quintela, M. D. Garcia and C. Peinador, Acc. Chem. Res., 2020, 53, 2336–2346 CrossRef CAS PubMed.
- C. Wang, D. Zhang and D. Zhu, Langmuir, 2007, 23, 1478–1482 CrossRef CAS.
- S. Bahring, L. Martin-Gomis, G. Olsen, K. A. Nielsen, D. S. Kim, T. Duedal, A. Sastre-Santos, J. O. Jeppesen and J. L. Sessler, Chem. – Eur. J., 2016, 22, 1958–1967 CrossRef.
- S. Bera, B. Xue, P. Rehak, G. Jacoby, W. Ji, L. J. W. Shimon, R. Beck, P. Kral, Y. Cao and E. Gazit, ACS Nano, 2020, 14, 1694–1706 CrossRef CAS.
- M. A. Mateos-Timoneda, M. Crego-Calama and D. N. Reinhoudt, Chem. Soc. Rev., 2004, 33, 363–372 RSC.
- G. Liu, J. Sheng, H. Wu, C. Yang, G. Yang, Y. Li, R. Ganguly, L. Zhu and Y. Zhao, J. Am. Chem. Soc., 2018, 140, 6467–6473 CrossRef CAS PubMed.
- L. Ji, Y. Sang, G. Ouyang, D. Yang, P. Duan, Y. Jiang and M. Liu, Angew. Chem., Int. Ed., 2019, 58, 844–848 CrossRef CAS PubMed.
- C. Wang, L. Y. Zhu, J. F. Xiang, Y. X. Yu, D. Q. Zhang, Z. G. Shuai and D. B. Zhu, J. Org. Chem., 2007, 72, 4306–4312 CrossRef CAS.
- A. Wu, Y. Guo, X. Li, H. Xue, J. Fei and J. Li, Angew. Chem., Int. Ed., 2021, 60, 2099–2103 CrossRef CAS PubMed.
- J. Liang, A. Hao, P. Xing and Y. Zhao, ACS Nano, 2021, 15, 5322–5332 CrossRef CAS.
- C. Kulkarni, A. K. Mondal, T. K. Das, G. Grinbom, F. Tassinari, M. F. J. Mabesoone, E. W. Meijer and R. Naaman, Adv. Mater., 2020, 32, e1904965 CrossRef.
- Z. Zhou, J. Zhou, L. Chen, Q. Zhao, C. Zhang and G. Ge, Chem. Commun., 2020, 56, 15345–15348 RSC.
- H. Jintoku, M. Dateki, M. Takafuji and H. Ihara, J. Mater. Chem. C, 2015, 3, 1480–1483 RSC.
- S. K. Nisha and S. K. Asha, J. Mater. Chem. C, 2014, 2, 2051–2060 RSC.
- M. Grzelczak, L. M. Liz-Marzan and R. Klajn, Chem. Soc. Rev., 2019, 48, 1342–1361 RSC.
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS.
- Z. Wang, Y. Li, A. Hao and P. Xing, Angew. Chem., Int. Ed., 2021, 60, 3138–3147 CrossRef CAS PubMed.
- Z. Wang, A. Hao and P. Xing, Angew. Chem., Int. Ed., 2020, 59, 11556–11565 CrossRef CAS PubMed.
- J. Zhao and P. Xing, ChemPlusChem, 2020, 85, 1511–1522 CrossRef CAS.
- D. H. Qu, Q. C. Wang, Q. W. Zhang, X. Ma and H. Tian, Chem. Rev., 2015, 115, 7543–7588 CrossRef CAS.
- H. Gholami, D. Chakraborty, J. Zhang and B. Borhan, Acc. Chem. Res., 2021, 54, 654–667 CrossRef CAS.
- T. Jiang, X. Wang, J. Wang, G. Hu and X. Ma, ACS Appl. Mater. Interfaces, 2019, 11, 14399–14407 CrossRef CAS.
- G. Yu, K. Jie and F. Huang, Chem. Rev., 2015, 115, 7240–7303 CrossRef CAS.
- M. Olesinska, G. Wu, S. Gomez-Coca, D. Anton-Garcia, I. Szabo, E. Rosta and O. A. Scherman, Chem. Sci., 2019, 10, 8806–8811 RSC.
- G. C. Yu, W. Yu, L. Shao, Z. H. Zhang, X. D. Chi, Z. W. Mao, C. Y. Gao and F. H. Huang, Adv. Funct. Mater., 2016, 26, 8999–9008 CrossRef CAS.
- C. Stoffelen, J. Voskuhl, P. Jonkheijm and J. Huskens, Angew. Chem., Int. Ed., 2014, 53, 3400–3404 CrossRef CAS.
- C. Stoffelen and J. Huskens, Nanoscale, 2015, 7, 7915–7919 RSC.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS.
- G. Das, S. K. Sharma, T. Prakasam, F. Gandara, R. Mathew, N. Alkhatib, N. Saleh, R. Pasricha, J. C. Olsen, M. Baias, S. Kirmizialtin, R. Jagannathan and A. Trabolsi, Chem. Commun., 2019, 2, 2399–3669 Search PubMed.
- X. Ma and H. Tian, Acc. Chem. Res., 2014, 47, 1971–1981 CrossRef CAS.
- E. Pazos, P. Novo, C. Peinador, A. E. Kaifer and M. D. Garcia, Angew. Chem., Int. Ed., 2019, 58, 403–416 CrossRef CAS PubMed.
- K. Moon, J. Grindstaff, D. Sobransingh and A. E. Kaifer, Angew. Chem., Int. Ed., 2004, 43, 5496–5499 CrossRef CAS PubMed.
- S. Kaabel, J. Adamson, F. Topic, A. Kiesila, E. Kalenius, M. Oeren, M. Reimund, E. Prigorchenko, A. Lookene, H. J. Reich, K. Rissanen and R. Aav, Chem. Sci., 2017, 8, 2184–2190 RSC.
- C. Hu, N. Ma, F. Li, Y. Fang, Y. Liu, L. Zhao, S. Qiao, X. Li, X. Jiang, T. Li, F. Shen, Y. Huang, Q. Luo and J. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 4603–4613 CrossRef CAS PubMed.
- Z. Yin, G. Song, Y. Jiao, P. Zheng and X. Zhang, CCS Chem., 2019, 1, 335–342 CrossRef CAS.
- Q. Zhang, D. H. Qu, Q. C. Wang and H. Tian, Angew. Chem., Int. Ed., 2015, 54, 15789–15793 CrossRef CAS.
- Z. Huang, X. Chen, G. Wu, P. Metrangolo, D. Whitaker, J. A. McCune and O. A. Scherman, J. Am. Chem. Soc., 2020, 142, 7356–7361 CrossRef CAS PubMed.
- Q. Wang, Q. Zhang, Q. W. Zhang, X. Li, C. X. Zhao, T. Y. Xu, D. H. Qu and H. Tian, Nat. Commun., 2020, 11, 158 CrossRef.
- S. Zhang, H. J. Sun, A. D. Hughes, R. O. Moussodia, A. Bertin, Y. Chen, D. J. Pochan, P. A. Heiney, M. L. Klein and V. Percec, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9058–9063 CrossRef CAS.
- Y. Liu, Q. Zhang, S. Crespi, S. Chen, X. K. Zhang, T. Y. Xu, C. S. Ma, S. W. Zhou, Z. T. Shi, H. Tian, B. L. Feringa and D. H. Qu, Angew. Chem., Int. Ed., 2021, 60, 16129–16138 CrossRef CAS.
- D. Brynn Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC.
- X. Xiao, J.-X. Liu, Z.-F. Fan, K. Chen, Q.-J. Zhu, S.-F. Xue and Z. Tao, Chem. Commun., 2010, 46, 3741–3743 RSC.
- F. Biedermann and W. M. Nau, Angew. Chem., Int. Ed., 2014, 53, 5694–5699 CrossRef CAS PubMed.
- J. Liang, P. Guo, X. Qin, X. Gao, K. Ma, X. Zhu, X. Jin, W. Xu, L. Jiang and P. Duan, ACS Nano, 2020, 14, 3190–3198 CrossRef CAS.
- X. Wang, W. Zhi, C. Ma, Z. Zhu, W. Qi, J. Huang and Y. Yan, JACS Au, 2021, 1, 156–163 CrossRef CAS.
- Z. Shen, Y. Jiang, T. Wang and M. Liu, J. Am. Chem. Soc., 2015, 137, 16109–16115 CrossRef CAS.
- M. Ichihara, A. Suzuki, K. Hatsusaka and K. Ohta, Liq. Cryst., 2007, 34, 555–567 CrossRef CAS.
- Y. C. Yang, H. Hu, Y. Y. Guo, A. D. Xia, J. F. Xu and X. Zhang, Macromol. Rapid Commun., 2020, 41, 2000080 CrossRef CAS.
- L. Zhou, J. Yue, Y. Fan and Y. Wang, Langmuir, 2018, 34, 12924–12933 CrossRef CAS.
- E. J. Creatto, F. Ceccacci, G. Mancini and E. Sabadini, Langmuir, 2018, 34, 13288–13295 CrossRef CAS PubMed.
- R. A. Musgrave, P. Choi, R. L. Harniman, R. M. Richardson, C. Shen, G. R. Whittell, J. Crassous, H. Qiu and I. Manners, J. Am. Chem. Soc., 2018, 140, 7222–7231 CrossRef CAS.
- S. Sakamoto, S. Fujii, K. Yoshida and K. Sakurai, Langmuir, 2016, 32, 12434–12441 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc03975j |
| This journal is © The Royal Society of Chemistry 2022 |