
Plasmon-enhanced photocatalytic overall water-splitting over Au nanoparticle-decorated CaNb2O6 electrospun nanofibers†
Jiaming
Zhang
,
Xiaoyi
Jiang
,
Jindou
Huang
,
Wei
Lu
and
Zhenyi
Zhang
 *
*
Key Laboratory of New Energy and Rare Earth Resource Utilization of State Ethnic Affairs Commission, Key Laboratory of Photosensitive Materials and Devices of Liaoning Province, School of Physics and Materials Engineering, Dalian Minzu University, 18 Liaohe West Road, Dalian, 116600, P. R. China. E-mail: zhangzy@dlnu.edu.cn
First published on 28th July 2022
Abstract
The design and construction of wide-bandgap semiconductor nanostructures is a promising way to achieve photocatalytic overall water-splitting, because their redox potentials easily simultaneously satisfy the thermodynamic requirements of water reduction and oxidation. However, the narrow light-harvesting range and the fast charge recombination of wide-bandgap semiconductors tremendously limit their photocatalytic activities. Herein, we developed a novel wide-bandgap semiconductor photocatalyst of CaNb2O6 nanofibers (NFs) fabricated by an electrospinning technique, followed by calcination. Upon UV-visible light irradiation, the as-electrospun CaNb2O6 NFs could split water into H2 and O2 without adding any cocatalyst and sacrificial agent. The H2-production rate of CaNb2O6 NFs was ∼7.7 times higher than that of CaNb2O6 nanoparticles (NPs). It is because the CaNb2O6 NFs with the NP-packed 1D nanostructure possess abundant homogeneous interfaces to facilitate inter-particle continuous charge migration, thereby prolonging the lifetimes of photoinduced electrons and holes toward both water reduction and oxidation. Notably, the introduction of Au NPs into the CaNb2O6 NFs could extend light absorption from the UV to the visible light range. The optimal sample of 1.0 at% Au NP-decorated CaNb2O6 NFs exhibited ∼13.1-fold enhancement of photocatalytic activity for overall water splitting as compared to CaNb2O6 NFs. This remarkable enhancement is attributed to an interesting plasmonic sensitization process, during which the visible-light-excited hot electrons in plasmonic Au NPs are able to transfer to the interband-excited CaNb2O6 across their hetero-interface for enhancing the photocatalytic water reduction. Meanwhile, the hot holes left in the Au NPs possess a longer lifetime for fulfilling the photocatalytic water oxidation.
 Zhenyi Zhang | Zhenyi Zhang is a professor in the School of Physics and Materials Engineering at Dalian Minzu University. He received his BS and PhD degrees from Northeast Normal University in 2007 and 2012, respectively. He worked as a postdoctoral research fellow in the Solar Fuels Laboratory at Nanyang Technological University in 2012–2013. His current research interests focus on the design and synthesis of low-dimensional semiconductor-based photoelectric materials for photocatalysis and sensing applications. |
1 Introduction
The development of sustainable green energy to replace environmentally hazardous fossil fuels is of great significance to alleviate the prominent global issues of energy shortage and atmospheric contamination.1–8 Photocatalytic water splitting over highly efficient semiconductor photocatalysts offers a promising approach to convert inexhaustible solar-energy into the non-polluting chemical fuel of H2.9–18 In theory, wide-bandgap semiconductors are more suitable to be used for photocatalytic overall water splitting as compared to narrow-bandgap semiconductors, because their conduction band (CB) and valence band (VB) positions are easier to straddle the redox potentials of water splitting (EH2/H2O = 0 V, EO2/H2O = +1.23 V, and pH = 0), thereby thermodynamically favouring the reduction and oxidation of water to H2 and O2 (Scheme 1A).19 To date, many wide-bandgap semiconductors, such as TiO2, SrTa2O6, and so on, have been investigated in the research area of photocatalytic water splitting for H2 production in the absence of sacrificial agents.20–23 Among them, columbite niobate compounds (ANb2O6: M = Ba, Sr, Ca, Mg, Ni, etc.) have been considered as one kind of up-and-coming photocatalysts for splitting pure water into H2 and O2 due to their suitable redox potentials and stable chemical structures.24–32 It has been reported that upon UV light irradiation, CaNb2O6 nanoparticles (NPs) are able to reduce pure water to produce H2 by virtue of their stronger redox potentials originating from the perovskite-like octahedral structure of [NbO6]7− groups (Scheme 1B).24,25,27 However, the achievement of broad-spectrum-driven overall pure-water splitting for producing both H2 and O2 is still a huge challenge for CaNb2O6 NPs, mainly owing to two drawbacks: (1) the fast recombination of photoinduced charge carriers in CaNb2O6 NPs tremendously limits the multi-hole-induced water-oxidation for the production of kinetically unfavourable O2;33 (2) the large bandgap energy of CaNb2O6 NPs impedes the absorption and utilization of visible light that comprises approximately 40% of solar irradiation.34 Thus, it is urgent to seek a rational tactic to synergistically enhance the photoinduced charge separation and visible light absorption of CaNb2O6 NPs.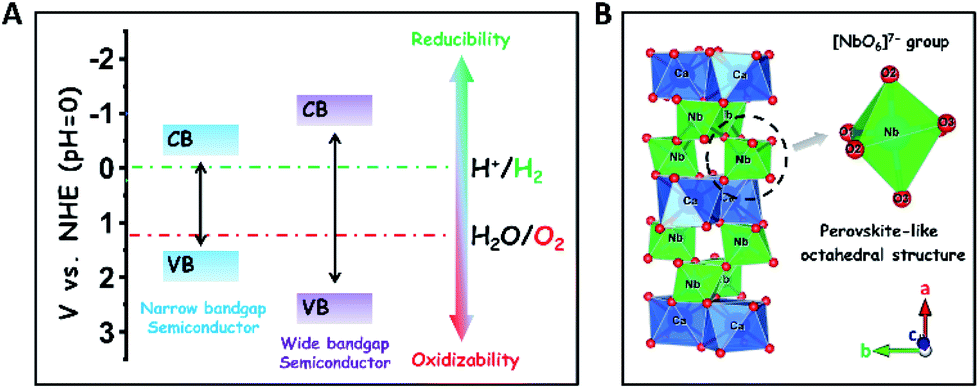 | ||
| Scheme 1 (A) Comparison of redox abilities between narrow- and wide-bandgap semiconductors for the photocatalytic reduction and oxidation of water to H2 and O2, respectively; (B) atomic arrangement structures of columbite CaNb2O6. | ||
In general, single-semiconductor NP-stacked nanostructures can provide abundant homogeneous interfaces to facilitate inter-particle continuous charge migration due to the low lattice mismatch between the NPs.35 This charge migration process would extend the lifetime of photoinduced charge carriers in semiconductors but without losing their intrinsic redox abilities. Notably, inorganic nanofibers (NFs) made by electrospinning possess a 1D ultra-long porous nanostructure composed of compactly packed NPs.36 In this context, the electrospinning fabrication of CaNb2O6 NFs seems to be an effective approach for hindering the charge-carrier recombination and achieving abundant active sites based on the unique 1D nanostructure properties. Importantly, various noble-metal nanostructures with different functional properties could be assembled uniformly in/on inorganic electrospun NFs for further manipulating their light harvesting and charge-carrier kinetic properties.37,38 For instance, the decoration of plasmonic NPs (Ag, Au, etc.) in/on wide-bandgap-semiconductor electrospun NFs could realize low-photon-energy-driven photocatalytic H2-production based on hot-electron transfer from Au to the contacted semiconductor due to the localized surface plasmon resonance (LSPR) of Au NPs.39–43 Thus, the introduction of highly stable plasmonic Au NPs into CaNb2O6 electrospun NFs is a prospective tactic to attain broad-spectrum-driven overall pure water splitting for stoichiometrically producing H2 and O2. Nevertheless, the relative exploration has been rarely reported up until now.
In this work, for the first time, we fabricated CaNb2O6 NFs through calcining electrospun precursor NFs of metal salts/polymer composites at 500, 600, and 700 °C, respectively. The structural characterization results indicate that the as-electrospun CaNb2O6 NFs with diameters of 60–100 nm were composed of tightly stacked NPs with grain sizes of 3–30 nm, thereby forming lots of homogeneous interfaces among the NPs. These homogeneous interfaces are conducive to prolonging the lifetime of photoinduced charge carriers through inter-particle charge migration, as evidenced by the steady-state and time-resolved photoluminescence (TRPL) spectra and photocurrent testing. Upon UV-visible light irradiation, the CaNb2O6 NFs could split pure water into H2 and O2 with the production rates of ∼9.95 and ∼4.96 μmol g−1 h−1, respectively, in the absence of a cocatalyst and sacrificial agent. However, the above stoichiometric water-splitting process could not be realized by using CaNb2O6 NPs as the photocatalyst (products: H2 and H2O2), because the fast recombination of photoinduced electron–hole pairs limits the four-hole-driven water-oxidation reaction.30 Furthermore, by combining 3D finite-difference-time-domain (FDTD) simulations with wavelength-controlled experiments, we demonstrated an interesting plasmon-enhanced photocatalytic overall water-splitting into H2 and O2 over the Au NP-decorated CaNb2O6 NFs. During the photocatalytic process, plasmonic Au NPs could absorb visible light to create energetic hot electrons and holes, which are able to enhance photocatalytic water reduction and oxidation only when the CaNb2O6 NFs were simultaneously excited to boost the hot electron transfer and reduce the reaction kinetic barrier of multi-hole-driven water oxidation.
2 Results and discussion
Based on our proposed fabrication process (Fig. 1A), three CaNb2O6 samples named CaNb2O6-500 NFs, CaNb2O6-600 NFs, and CaNb2O6-700 NFs were fabricated through an electrospinning technique combined with the subsequent calcination at 500, 600, and 700 °C, respectively. Fig. 1B shows the X-ray diffraction (XRD) patterns of the as-fabricated samples. For the CaNb2O6-500 NFs, there is only one broad diffraction band (15–17°) on the XRD pattern, which could be originated from the residual carbon due to the incomplete decomposition of the polymer template at a relative low calcination temperature (Fig. S1†). Meanwhile, the lack of feature diffraction peaks of the CaNb2O6 crystal may be ascribed to its small grain size and poor crystallinity. Notably, when the calcination temperature was increased during the fabrication process, the feature diffraction peaks of orthorhombic fersmite CaNb2O6 (JCPDS, no. 11-619) appeared on the XRD patterns of both CaNb2O6-600 NFs and CaNb2O6-700 NFs, suggesting the formation of a CaNb2O6 phase-structure. Furthermore, according to the Debye–Scherrer formula (Fig. S2†), the average grain sizes of CaNb2O6-600 NFs and CaNb2O6-700 NFs were estimated to be ∼16 and ∼22 nm, respectively.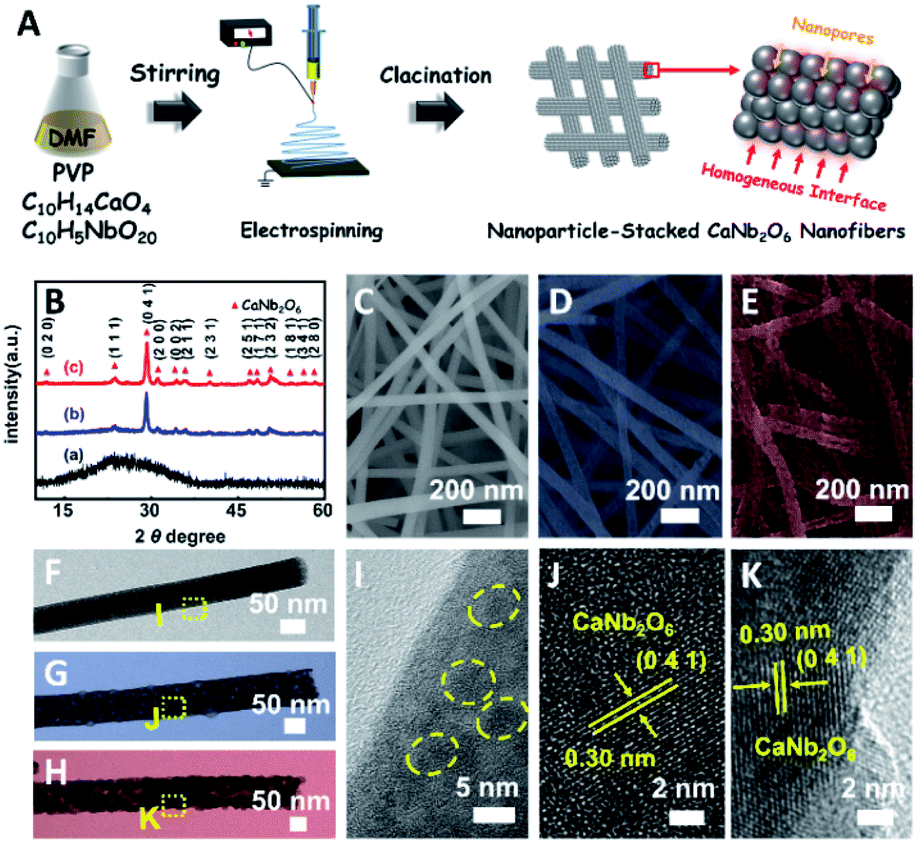 | ||
| Fig. 1 (A) Sketch map of the typical fabrication process of CaNb2O6 NFs; (B) XRD patterns of (a) CaNb2O6-500 NFs, (b) CaNb2O6-600 NFs, and (c) CaNb2O6-700 NFs; SEM images of (C) CaNb2O6-500 NFs, (D) CaNb2O6-600 NFs, and (E) CaNb2O6-700 NFs; TEM images of the individual (F) CaNb2O6-500 NF, (G) CaNb2O6-600 NF, and (H) CaNb2O6-700 NF; (I–K) HRTEM images from the corresponding yellow-colour squares labelled in images (F–H). | ||
The morphologies and microstructures of the above samples were investigated by using scanning electron microscopy (SEM) and transmission electron microscopy (TEM). As shown in Fig. 1C–E, all these samples have continuous nanofibrous structures with diameters of 60–100 nm and lengths of several micrometres. These NFs are aligned in random orientations and interweaved together. Furthermore, lots of nano-sized pores can be observed on the SEM images of CaNb2O6-600 NFs and CaNb2O6-700 NFs, while there is no distinguishable porous nanostructure on the SEM image of CaNb2O6-500 NFs. Further observations from TEM images (Fig. 1 F–H) indicate that all the as-fabricated CaNb2O6 NFs consist of compactly packed NPs with different mean grain sizes (∼3.5, ∼15.1 and ∼20.9 nm) depending on the calcination temperature during the fabrication process (Fig. S2†). During the fabrication process of CaNb2O6 NFs, the grain size of CaNb2O6 NPs in the NFs increased with the calcination temperature, thereby leading to larger nanopore sizes in the NFs produced at the higher calcination temperature (Fig. 1C–H and S3†). According to the TEM images in Fig. 1G and H, the mean pore sizes in the CaNb2O6-600 NFs and CaNb2O6-700 NFs are ∼8.9 and ∼14.3 nm, respectively (Fig. S2†). Meanwhile, an inter-particle homogeneous interface could be also found in the as-electrospun CaNb2O6-600 NFs and CaNb2O6-700 NFs (Fig. S3†). Fig. 1I–K show the high-resolution (HR) TEM images from the corresponding yellow-colour squares of Fig. 1F–H. As observed in Fig. 1I, the CaNb2O6-500 NFs contain lots of small-sized NPs (3–5 nm) with poor crystallinity. However, the selected area electron diffraction (SAED) pattern of the CaNb2O6-500 NFs displays the feature diffraction rings of orthorhombic fersmite CaNb2O6 (JCPDS, no. 11-619) (Fig. S4†), confirming the formation of polycrystalline CaNb2O6 NPs in the CaNb2O6-500 NFs. Furthermore, lattice fringes with interval distances of ∼0.30 nm could be observed on the HRTEM images of CaNb2O6-600 NFs and CaNb2O6-700 NFs, respectively, which agrees with the lattice spacing of the (041) plane of orthorhombic fersmite CaNb2O6. The above results prove the successful fabrication of CaNb2O6 NP-stacked 1D NFs by using the electrospinning and calcination methods.
The light absorption properties of the as-fabricated samples were investigated through UV-visible absorption spectra (Fig. 2A). For the CaNb2O6-600 NFs, the intrinsic absorption edge is located at ∼350 nm, which corresponds to the bandgap energy (Eg) of CaNb2O6 at ∼3.54 eV. In comparison with the CaNb2O6-600 NFs, the intrinsic absorption edge of CaNb2O6-700 NFs showed a red-shift of ∼25 nm, indicating a decreased bandgap (∼3.31 eV). This red-shift could be attributed to the larger grain-size of CaNb2O6 NPs in the CaNb2O6-700 NFs as compared to that in the CaNb2O6-600 NFs. Interestingly, the CaNb2O6-500 NFs present broad light absorption ranging from the UV to the visible light region. The visible light absorption is originated from the residual carbon-based impurities in the CaNb2O6-500 NFs due to the incomplete decomposition of the PVP template at a relatively low calcination temperature. To estimate the photocatalytic reduction and oxidation abilities of the as-fabricated samples, at first, we performed the half reactions of water splitting for producing H2 and O2 under UV-visible light irradiation in the presence of oxalic acid (H2C2O4) and silver nitrate (AgNO3) as the hole and electron sacrificial agents, respectively. As shown in Fig. 2B, all the as-fabricated samples can execute photocatalytic water-reduction for producing H2 with the rate order of CaNb2O6-600 NFs (∼182.50 μmol g−1 h−1) > CaNb2O6-700 NFs (∼147.50 μmol g−1 h−1) > CaNb2O6-500 NFs (∼50.00 μmol g−1 h−1). Among them, the lowest photocatalytic activity of CaNb2O6-500 NFs for H2 production is mainly due to their poor crystallization. Meanwhile, the photocatalytic activity of CaNb2O6-700 NFs is lower than that of the CaNb2O6-600 NFs, which may be attributed to the following reasons: (1) the narrower bandgap of CaNb2O6-700 NFs induces a weakened photocatalytic redox ability as compared to the CaNb2O6-600 NFs; (2) the smaller sizes of NPs in the CaNb2O6-600 NFs may provide more surface active sites for photocatalytic water splitting as compared to that of the CaNb2O6-700 NFs; (3) as compared to the CaNb2O6-700 NFs, the CaNb2O6-600 NFs possess smaller sizes of nanopores, which may cause the formation of more interfaces among the CaNb2O6-600 NPs for facilitating the separation of charge carriers. Interestingly, the photocatalytic O2-production rates of the as-fabricated samples also follow the sequence of CaNb2O6-600 NFs (∼22.50 μmol g−1 h−1) > CaNb2O6-700 NFs (∼21.00 μmol g−1 h−1) > CaNb2O6-500 NFs (∼18.00 μmol g−1 h−1), as shown in Fig. 2C. The above results suggest that all the as-fabricated samples are potentially useful for initiating photocatalytic overall water-splitting. And, the CaNb2O6-600 NFs possess the strongest photocatalytic redox ability among the as-fabricated three samples. Satisfactorily, when we directly introduced the as-fabricated samples into pure water, both H2 and O2 with a molar ratio of close to 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 released after UV-visible light irradiation. The gas production rates of CaNb2O6-600 NFs (H2: ∼9.95 μmol g−1 h−1; O2: ∼4.96 μmol g−1 h−1) were higher than those of both CaNb2O6-700 NFs (H2: ∼7.18 μmol g−1 h−1; O2: ∼3.58 μmol g−1 h−1) and CaNb2O6-500 NFs (H2: ∼4.34 μmol g−1 h−1; O2: ∼2.14 μmol g−1 h−1) (Fig. 2D).
:1 released after UV-visible light irradiation. The gas production rates of CaNb2O6-600 NFs (H2: ∼9.95 μmol g−1 h−1; O2: ∼4.96 μmol g−1 h−1) were higher than those of both CaNb2O6-700 NFs (H2: ∼7.18 μmol g−1 h−1; O2: ∼3.58 μmol g−1 h−1) and CaNb2O6-500 NFs (H2: ∼4.34 μmol g−1 h−1; O2: ∼2.14 μmol g−1 h−1) (Fig. 2D).
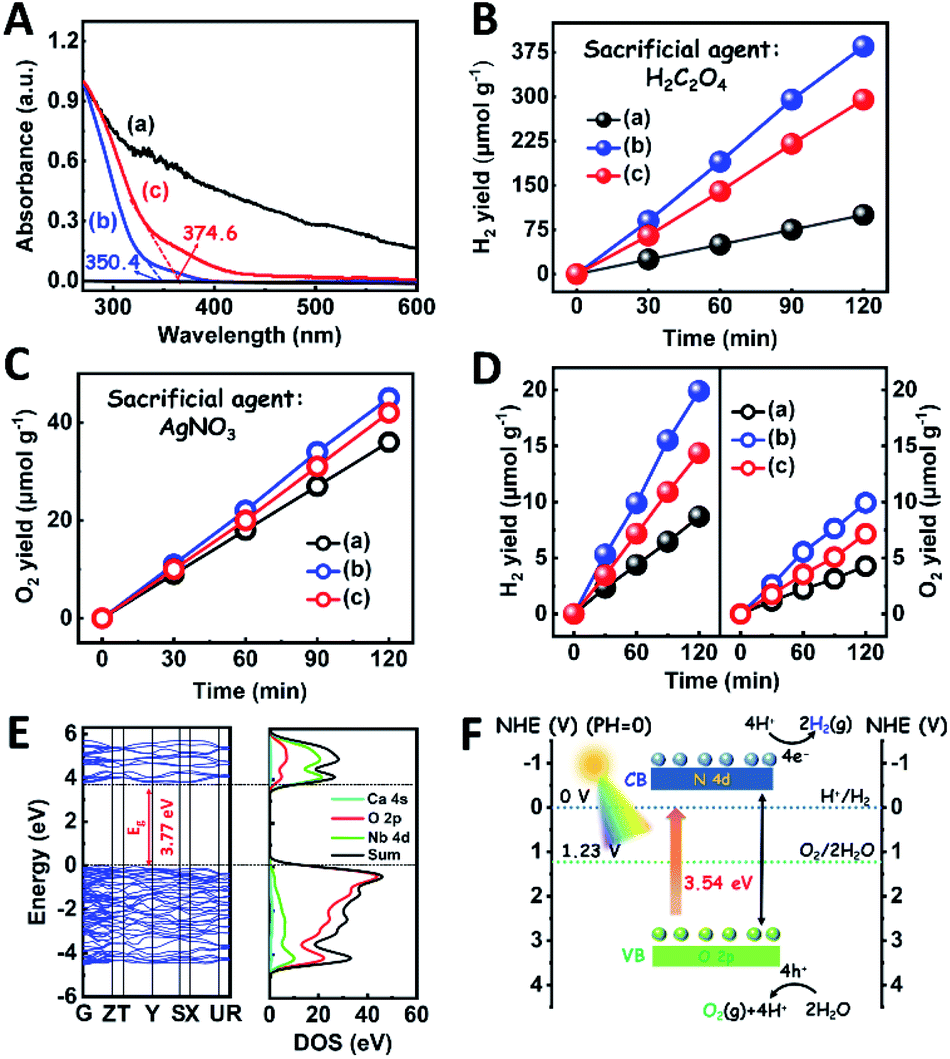 | ||
| Fig. 2 (A) UV-visible absorption spectra of the as-fabricated samples; plots of photocatalytic (B) H2 and (C) O2 yields versus UV-visible light irradiation time over different samples in the presence of H2C2O4 and AgNO3 as the hole and electron sacrificial agents, respectively; (D) plots of photocatalytic H2 and O2 yields versus UV-visible light irradiation time over different samples in pure water without adding any sacrificial agent: (a) CaNb2O6-500 NFs; (b) CaNb2O6-600 NFs; (c) CaNb2O6-700 NFs; (E) the calculated band structures and the DOS curve of CaNb2O6; (F) sketch map of the energy band structure and photocatalytic water-splitting process of CaNb2O6-600 NFs for H2 and O2 production. | ||
To further confirm the photocatalytic ability of CaNb2O6 crystals for water splitting, electronic band structures and density of states (DOS) simulations were carried out. As shown in Fig. 2E, the bandgap energy of CaNb2O6 crystals is simulated to be ∼3.77 eV, which is similar to the real value of CaNb2O6-600 NFs fabricated in our work. Furthermore, the valence band (VB) and conduction band (CB) of CaNb2O6 crystals are mainly contributed by the O 2p and Nb 4d orbitals, respectively.44 According to the results of UV-visible absorption spectroscopy and the Mott–Schottky plot (Fig. S5†), the VB and CB potentials of the CaNb2O6-600 NFs were estimated to be 3.1 V and −0.44 V versus the normal hydrogen electrode (NHE), respectively.45,46 It can be found that the CB potential of CaNb2O6-600 NFs is more negative than H+/H2 potential (0 V vs. NHE, pH = 0), while its VB potential is more positive than O2/H2O potential (1.23 V vs. NHE, pH = 0) (Fig. 2F). Thus, upon the interband excitation of CaNb2O6, the photoinduced electrons and holes on its CB and VB can enable the water reduction and oxidation reactions, respectively, for simultaneously producing H2 and O2. However, the CaNb2O6-600 NPs made by the sol–gel method with the calcination temperature at 600 °C (Fig. S6†) displayed a dramatic decrease of photocatalytic activity as compared to the CaNb2O6-600 NFs for pure water splitting. Upon UV-visible light irradiation, the H2 production rate was only ∼1.29 μmol g−1 h−1, which was ∼7.7 times lower than that of the CaNb2O6-600 NFs (Fig. 3A). However, the oxidation product of this photocatalytic water-splitting reaction was H2O2 rather than O2 that is a high-kinetically-required product (Fig. S7†). The lack of O2 products may be attributed to the fast recombination of photoinduced electron–hole pairs in the CaNb2O6-600 NFs, thereby hindering the four-hole-driven water-oxidation process. Interestingly, the Brunauer–Emmett–Teller (BET) analysis results indicate that the specific surface area of CaNb2O6-600 NFs (29.44 m2 g−1) was similar to that of the CaNb2O6-600 NPs (28.84 m2 g−1) (Fig. S8†). These results suggest that the improved kinetic process of photoinduced charge carriers in the CaNb2O6-600 NFs may play the leading role in increasing the photocatalytic activity for water splitting. To unravel the kinetic process of photoinduced charge carriers in the CaNb2O6-600 NPs and CaNb2O6-600 NFs, their steady-state and time-resolved photoluminescence (TRPL) spectra were recorded.47–49 As shown in Fig. 3B, both CaNb2O6-600 NPs and CaNb2O6-600 NFs show emission bands centred at ∼467 nm after 280 nm excitation. Meanwhile, the emission intensity of CaNb2O6-600 NFs is lower than that of the CaNb2O6-600 NPs, suggesting the suppressed recombination process of photoinduced charge carriers in the CaNb2O6-600 NFs. Fig. 3C displays the TRPL spectra of the above two samples, in which all the emission decay curves can be fitted by the biexponential kinetic function:
| I(t) = A1·exp (−t/τ1) + A2·exp(−t/τ2) + B | (1) |
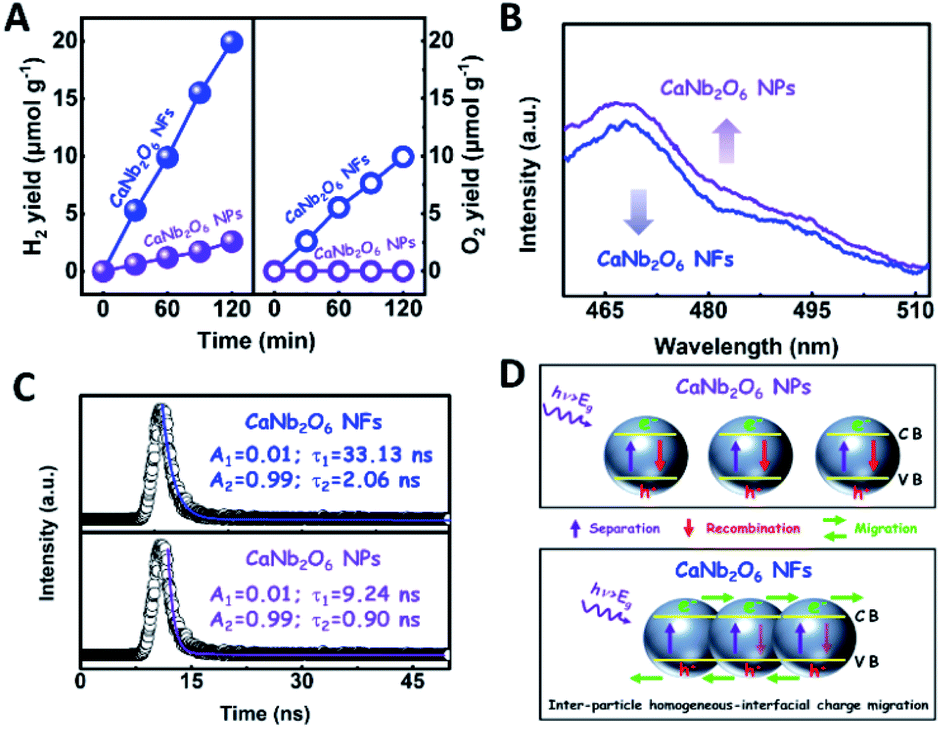 | ||
| Fig. 3 (A) Plots of photocatalytic H2 and O2 yields versus UV-visible light irradiation time over the CaNb2O6 NPs and CaNb2O6 electrospun NFs in pure water; (B) steady-state PL spectra and (C) time-resolved transient PL decay of CaNb2O6 NPs and CaNb2O6 NFs; (D) sketch maps of the kinetic processes of photoinduced charge carriers in the CaNb2O6 NPs and CaNb2O6 NFs, respectively. | ||
In order to further extend the light absorption range and improve the charge-carrier kinetic process of the optimal CaNb2O6-600 NFs, the different contents of plasmonic Au NPs (atomic ratio of Au to Ca: 0.25, 0.5, 0.75, 1.0, and 1.25 at%) were introduced into the CaNb2O6-600 NFs by using the electrospinning and calcination methods. It is expected that the plasmonic Au NPs absorb and utilize visible light to further enhance the photocatalytic water-splitting activity (Fig. 4A). As shown in Fig. 4B, the diffraction peaks of all the as-fabricated Aux/CaNb2O6 NFs (x = 0.25, 0.5, 0.75, 1.0, and 1.25) can be perfectly indexed to orthorhombic fersmite CaNb2O6 (JCPDS, no. 11-619), indicating the unchanged phase structure of CaNb2O6 NFs after the introduction of Au NPs. However, the characteristic diffraction peaks of Au NPs were not observed on the XRD patterns of the as-fabricated Aux/CaNb2O6 NFs. This phenomenon may be attributed to the low content and small sizes of Au NPs in the CaNb2O6 nanofibrous matrix, thereby leading to weak crystal-diffraction signals beyond the XRD probing limit. Furthermore, all the Aux/CaNb2O6 NFs maintain the 1D porous nanofiber structures. Their structural parameters (several micrometres in lengths and 60–100 nm in diameters) are similar to those of the pristine CaNb2O6 NFs (Fig. 4C, D and S10†). Fig. 4D shows the dark-field scanning TEM (STEM) image and the corresponding element-mapping images of the Au1/CaNb2O6 NFs. The element-mapping images show that the Ca, Nb, Au, and O elements co-exist in the Au1/CaNb2O6 NFs. However, the Au and Nb elements in the NFs cannot be distinguished by the TEM-element mapping due to their similar Energy-Dispersive X-ray Spectroscopy (EDS) spectra (Fig. S11A†). Nevertheless, the bright Au NPs can be still found on the dark-field STEM of Au1/CaNb2O6 NFs (Fig. S11B†). Further investigation by using the TEM image indicates that the Au NPs with a mean size of ∼10.03 nm were dispersed randomly on/in the CaNb2O6 electrospun NFs (Fig. S12†). Fig. 4E shows the HRTEM image of the spherical NP selected from the yellow colour square of Fig. 4D. The interplanar distance of ∼0.234 nm corresponds well to the lattice spacing of the Au (111) plane. The formation of metallic Au can be also confirmed by X-ray photoelectron spectroscopy (XPS). As observed in Fig. 4F, the Au 4f7/2 and Au 4f5/2 peaks of Au1/CaNb2O6 NFs are positioned at 83.4 and 87.1 eV, respectively, which are in accordance with the corresponding values of the metallic Au0 state.50 Meanwhile, the valence states of Ca and Nb ions in both the CaNb2O6 and Au1/CaNb2O6 NFs are +2 and +5, respectively (Fig. S13†).
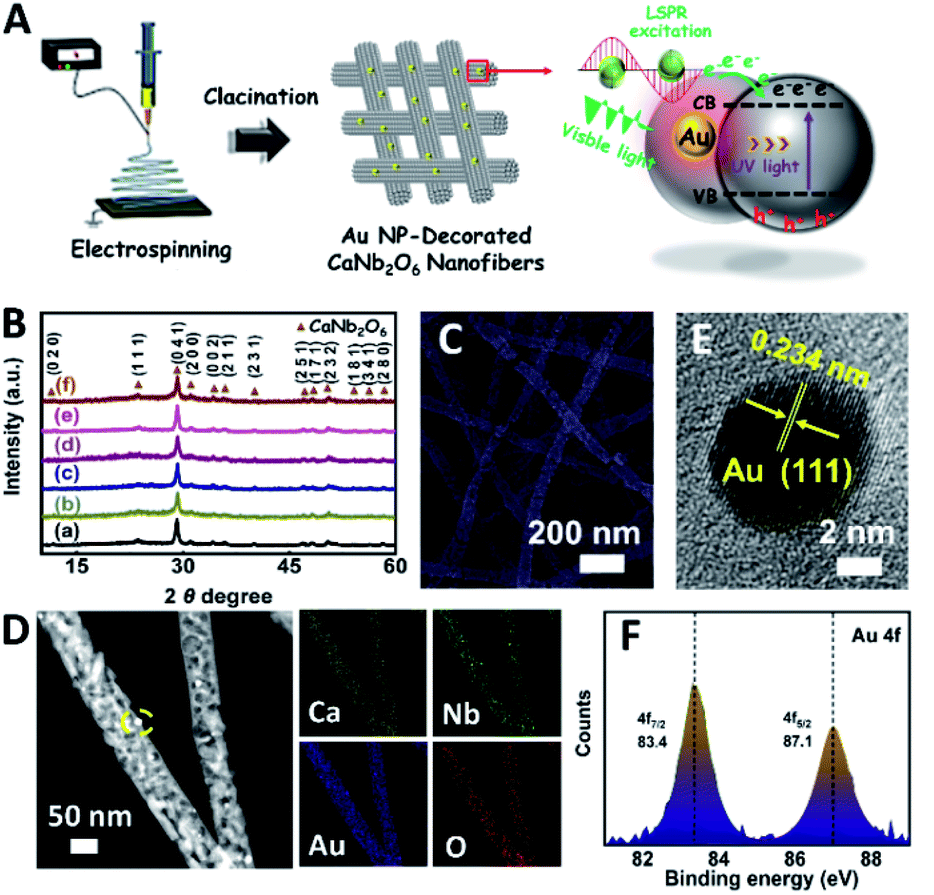 | ||
| Fig. 4 (A) Sketch map showing the typical fabrication process of the Au NP-decorated CaNb2O6 NFs; (B) XRD patterns of (a) CaNb2O6-600 NFs, (b) Au0.25/CaNb2O6 NFs, (c) Au0.5/CaNb2O6 NFs, (d) Au0.75/CaNb2O6 NFs, (e) Au1/CaNb2O6 NFs and (f) Au1.25/CaNb2O6 NFs; (C) SEM image of Au1/CaNb2O6 NFs; (D) dark-field STEM and the corresponding elemental mapping images of Au1/CaNb2O6 NFs; (E) HRTEM image from the yellow-coloured square of Fig. D; (F) XPS spectrum: Au 4f core level for the Au1/CaNb2O6 NFs. | ||
The light absorption properties of the as-fabricated Aux/CaNb2O6 NFs were investigated in comparison with those of the CaNb2O6-600 NFs through UV-visible absorption spectra. As shown in Fig. 5A, the intense UV-light absorption of all the as-fabricated samples can be assigned to the intrinsic absorption of CaNb2O6. Compared to the CaNb2O6-600 NFs, all the Aux/CaNb2O6 NFs exhibited obvious visible-light absorption bands at ∼550 nm (the inset of Fig. 5A). These absorption bands are attributed to the LSPR of Au NPs in the CaNb2O6 NFs.51 The similar position (∼550 nm) of plasmonic absorption bands suggests that the sizes of Au NPs were almost unchanged in the as-fabricated Aux/CaNb2O6 NFs.
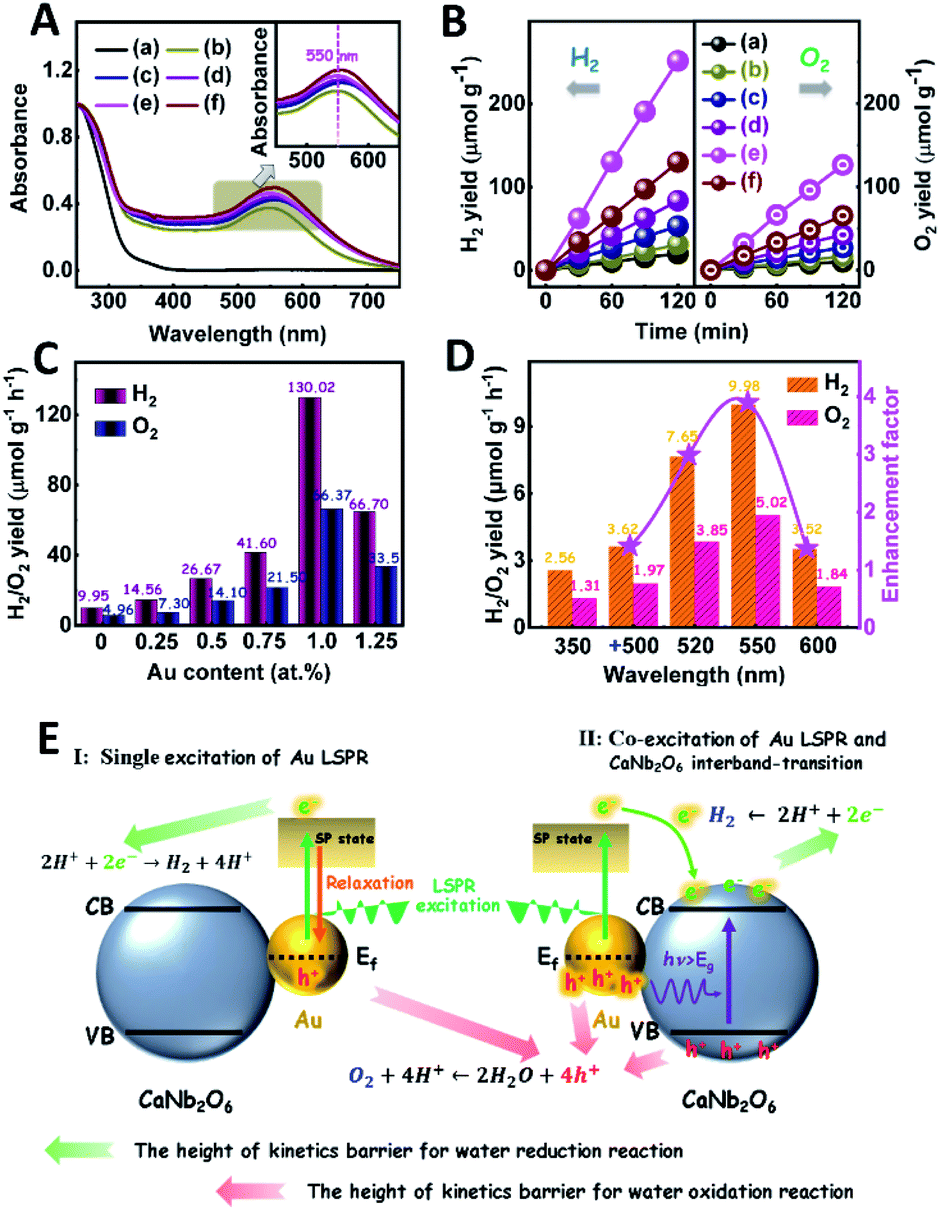 | ||
| Fig. 5 (A) UV-visible absorption spectra of the as-fabricated samples; (B) plots of photocatalytic H2 and O2 yields versus UV-visible light irradiation time over different samples in pure water without adding any sacrificial agent: (a) CaNb2O6-600 NFs, (b) Au0.25/CaNb2O6 NFs, (c) Au0.5/CaNb2O6 NFs, (d) Au0.75/CaNb2O6 NFs, (e) Au1/CaNb2O6 NFs and (f) Au1.25/CaNb2O6 NFs; (C) photocatalytic H2 and O2 production rates over Au/CaNb2O6 NFs with different Au contents under UV-visible light irradiation; (D) photocatalytic H2 and O2 yield rates over Au1/CaNb2O6 NFs under 350 nm irradiation in the absence or presence of secondary beam irradiation with the wavelength at 500 ± 15, 520 ± 15, 550 ± 15, and 600 ± 15 nm, respectively; (E) schematic diagram showing the plasmon-mediated photocatalytic overall water splitting over Au/CaNb2O6 NFs after the single excitation of Au LSPR or the co-excitation of Au LSPR and CaNb2O6 interband-transition. | ||
The photocatalytic activities of the as-fabricated Aux/CaNb2O6 NFs for overall water splitting were evaluated under UV-visible light irradiation in the absence of any sacrificial agent. The control experiment results indicate that no H2 and O2 were detected in the absence of either the as-fabricated NFs or the light irradiation. As shown in Fig. 5B, all the Aux/CaNb2O6 NFs exhibit enhanced photocatalytic activities for overall water splitting as compared to the pure CaNb2O6 NFs (H2: ∼9.95 μmol g−1 h−1; O2: ∼4.96 μmol g−1 h−1). The enhanced photocatalytic activity of Aux/CaNb2O6 NFs may be attributed to the LSPR of Au NPs that can induce hot-electron transfer from the Au to CaNb2O6 components. Please note that the LSPR absorption of Au NPs is unmatched with the light absorption of CaNb2O6 NFs, and therefore no plasmon resonance energy transfer process occurs in the Aux/CaNb2O6 NFs. Notably, the optimal sample of Au1/CaNb2O6 NFs possesses the H2 and O2 production rates of ∼130.02 and ∼66.37 μmol g−1 h−1, respectively (Fig. 5C). These gas-production rates are about 13.1 times higher than the corresponding rates of the CaNb2O6-600 NFs. However, when the content of Au NPs was higher than 1.0 at%, the photocatalytic activity of Au NP-decorated CaNb2O6 NFs was decreased (Fig. 5C). This can be ascribed to the “shield effect” of Au NPs to reduce the light absorption and active sites of the CaNb2O6 NFs.
To further recognize the contribution of Au LSPR to the photocatalytic activity of the Au1/CaNb2O6 NFs, we performed photocatalytic overall water-splitting upon dual-beam irradiation with different photon energies, in which one beam was for the CaNb2O6 interband excitation (350 nm), and the other was for Au LSPR excitation (500–600 nm). Upon single irradiation at 350 nm, the Au1/CaNb2O6 NFs are able to split pure water into H2 and O2 with the production rates of ∼2.56 and ∼1.31 μmol g−1 h−1, respectively (Fig. 5D). However, there was no detectable gas release over the Au1/CaNb2O6 NFs, when we adjusted the single irradiation wavelength at 500 ± 15, 520 ± 15, 550 ± 15 and 600 ± 15 nm, respectively, to only excite the Au LSPR. Please note that the above excitation wavelength (500–600 nm) is selected according to the UV-visible absorption spectrum of the Au1/CaNb2O6 NFs (Fig. 5A). These results suggest that the plasmon-induced hot electrons and holes over Au NPs cannot directly initiate the photocatalytic reaction of pure water splitting, because the ultra-fast relaxation of plasmonic hot-excitons in Au NPs cannot satisfy the reaction kinetic requirements of water oxidation and reduction for producing O2 and H2. Interestingly, when the above dual-beam irradiation was simultaneously used to execute the photocatalytic water splitting, the H2 and O2 production rates of the Au1/CaNb2O6 NFs could be obviously enhanced as compared to the corresponding rates obtained upon single irradiation at 350 ± 15 nm (Fig. 5D). Furthermore, the enhancement factors, obtaining from the ratio values of the H2-production rates of Au1/CaNb2O6 NFs under dual-beam irradiation to the H2-production rate of Au1/CaNb2O6 NFs under the single irradiation at 350 ± 15 nm, were basically correlated with the plasmonic absorption band of Au NPs in the Au1/CaNb2O6 NFs (Fig. S14†). These observations indicate that the LSPR-induced hot electrons and holes over Au NPs could enhance the photocatalytic activity of the Au1/CaNb2O6 NFs for water splitting only when the CaNb2O6 component was simultaneously excited to boost the hot electron transfer and unlock reaction kinetic barrier. The introduction of plasmonic Au NPs into the CaNb2O6 NFs can extend the light absorption and utilization range from the UV to the visible light region, thereby realizing the UV-visible-light-driven photocatalytic overall water splitting over the Au/CaNb2O6 NFs (Fig. S15†). According to the above results, the single excitation of Au-LSPR in the Au1/CaNb2O6 NFs could not directly initiate photocatalytic overall water splitting for producing H2 and O2 in the absence of a sacrificial agent. This may be due to the fast relaxation of plasmonic hot electrons and the high kinetic requirement of water oxidation to O2 (four-hole reaction) (Fig. 5E). Interestingly, semiconductors of such CaNb2O6 possess high conductivity after interband excitation,49 which could boost the hot electron transfer in the Au1/CaNb2O6 NFs. Meanwhile, the transfer process of hot electrons in the Au1/CaNb2O6 NFs could also prolong the lifetime of plasmon-induced hot holes in the Au NPs, thereby promoting the hot-hole transfer from Au to H2O for reducing the kinetic reaction barrier of water oxidation to O2 (Fig. 5E). Thus, the plasmon-induced hot electron transfer process in the Au1/CaNb2O6 NFs could extend the lifetimes of plasmonic hot excitons for enhancing the photocatalytic activity of overall water splitting.
The plasmon-induced electron transfer process in the Au1/CaNb2O6 NFs can be deduced based on the Bode phase plots through the following equation:52
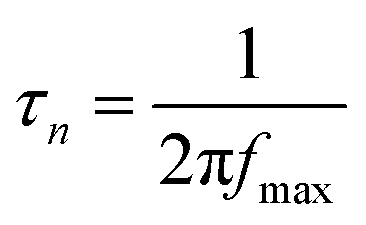 | (2) |
As observed in Fig. S16,† upon single irradiation at 350 nm, the fmax value of the Au1/CaNb2O6 NFs is ∼13.11 Hz. Interestingly, when we employed dual-beam irradiation with two wavelengths at 350 ± 15 and 550 ± 15 nm to simultaneously excite the CaNb2O6 interband transition and the Au LSPR, its fmax value could reduce to ∼5.81 Hz. Therefore, it can be calculated that the electron lifetime of the Au1/CaNb2O6 NFs (∼27.3 ms) obtained under dual-beam irradiation was about 2.2 times longer than that obtained under single beam irradiation (∼12.1 ms). The longer electron lifetime means that the co-excitation of Au LSPR and CaNb2O6 interband transition could effectively extend the electron transfer process due to the hot electron transfer form Au NPs to the contacted CaNb2O6 NFs. As a result, upon dual-beam irradiation (350 nm + 550 nm), more available electrons and holes could be accumulated on the CaNb2O6 and Au components for fulfilling the photocatalytic splitting of pure water into H2 and O2, respectively.
As we know, the LSPR-induced localized electric-field enhancement behavior is important for the generation of hot electrons in plasmonic nanostructures.53 In general, the large enhancement of the electric-field intensity would boost the generation of hot electrons in plasmonic nanostructures.54–56 Thus, to further reveal the LSPR-induced hot electron generation in the Au1/CaNb2O6 NFs, 3D finite-difference time domain (FDTD) simulations were carried out. According to the TEM images of Au1/CaNb2O6 NFs (bottom images of Fig. 6B–D), we simplified the simulation model as the individual Au/CaNb2O6 NF, in which Au NPs were assembled in the CaNb2O6 NF with three typical positions (Fig. 6A). The LSPR excitation of Au NPs at 550 nm can remarkably enhance the localized electric-field intensities (|E|2/|E0|2) in the near surface regions of Au NPs in the Au/CaNb2O6 NF (Fig. 6B–D top images). Moreover, the enhancement of the electric-field intensity depends on the position of Au NPs in/on the CaNb2O6 NF. When the Au NP was positioned on the outer surface of CaNb2O6 NF, the LSPR-enhanced electric-field intensity could reach ∼36× in the Au/CaNb2O6 interface region. However, the embedment of Au NPs in the middle and near-surface of the CaNb2O6 NF could induce ∼4-fold and ∼9-fold enhancements of the electric-field intensities, respectively. The above simulation results indicate that the Au/CaNb2O6 interface can strongly concentrate the incident visible light to generate lots of hot electrons at the surface plasmon (SP) states of Au NPs. These hot electrons further transfer from Au NPs to the excited CaNb2O6 for enhancing the photocatalytic water-reduction activity of the CaNb2O6 NFs, while the hot holes left in Au NPs possess extended lifetimes for initiating the photocatalytic water oxidation. The above results and analyses suggest that when the Au/CaNb2O6 NFs encounter UV-visible light irradiation, the LSPR-induced hot electrons would move to the CaNb2O6 surface for reducing water to H2, while the hot holes stay on the Au surface for oxidizing water to O2. It also means that in the Au/CaNb2O6 interface region, the active sites for water reduction and oxidation are mainly positioned at the surface of the CaNb2O6 and Au components, respectively.
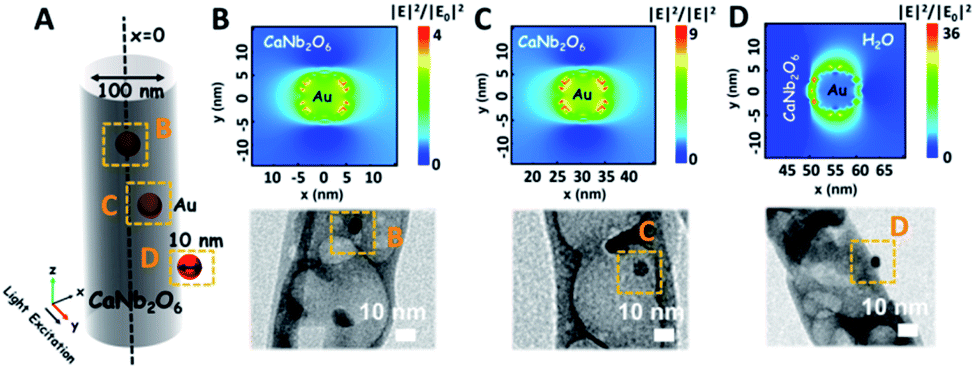 | ||
| Fig. 6 (A) Simplified structural model of the Au/CaNb2O6 NFs for the FDTD simulation; (B–D) simulated electric-field distributions and the corresponding TEM images of Au/CaNb2O6 NFs with the Au NP position at B, C, and D yellow squares of (A). | ||
To consolidate the above conclusion, the energy barrier to break the O–H bond of H2O on CaNb2O6 and Au/CaNb2O6 was calculated by nudged elastic band (NEB) calculations, respectively. As observed in Fig. 7A and B, the length of the O–H bond of the water molecule adsorbed on the CaNb2O6 surface is 1.107 Å, which is smaller than that at the Au/CaNb2O6 interface (1.357 Å), but is larger than that of the normal water molecule (0.99 Å). Meanwhile, the adsorption energy of H2O at the Au/CaNb2O6 interface is about 1.3 eV (Fig. S17A†), and the distance between the O atom of H2O and the adjacent Au atom is about 2.153 Å. It indicates that H2O can be a chemically bonded moiety on the Au surface. In comparison, the adsorption energy for H2O on the CaNb2O6 surface is about 0.69 eV (Fig. S17B†). This weaker interaction may decrease the activation effect on H2O on the CaNb2O6 surface. Thus, the hydrolytic energy barrier can be substantially reduced by the introduction of Au NPs into the CaNb2O6 NFs. Accordingly, the energy barrier for the reaction H2O* → OH* + H* occurring at the Au/CaNb2O6 interface (Ea = 0.14 eV) is much lower than the one for H2O* → OH* + H* on the CaNb2O6 surface (Ea = 0.47 eV) (Fig. 7A and B). The above results suggest that the Au/CaNb2O6 interface is beneficial to dissociate the water molecule due to the strong interactions between the O atom of H2O and the Au atom. Notably, in the final state (FS) of the H2O-adsorbed Au/CaNb2O6 interface (Fig. 7B), the O atom of H2O is bonded with Au, while the H atom of H2O is linked with CaNb2O6. This kind of chemical structure implies that for the Au/CaNb2O6 NFs the Au component serves as the active site for the photocatalytic O2 production, while the CaNb2O6 component acts as the active site for the H2 production. This result is consistent with our proposed mechanism of plasmon-enhanced photocatalytic overall water splitting.
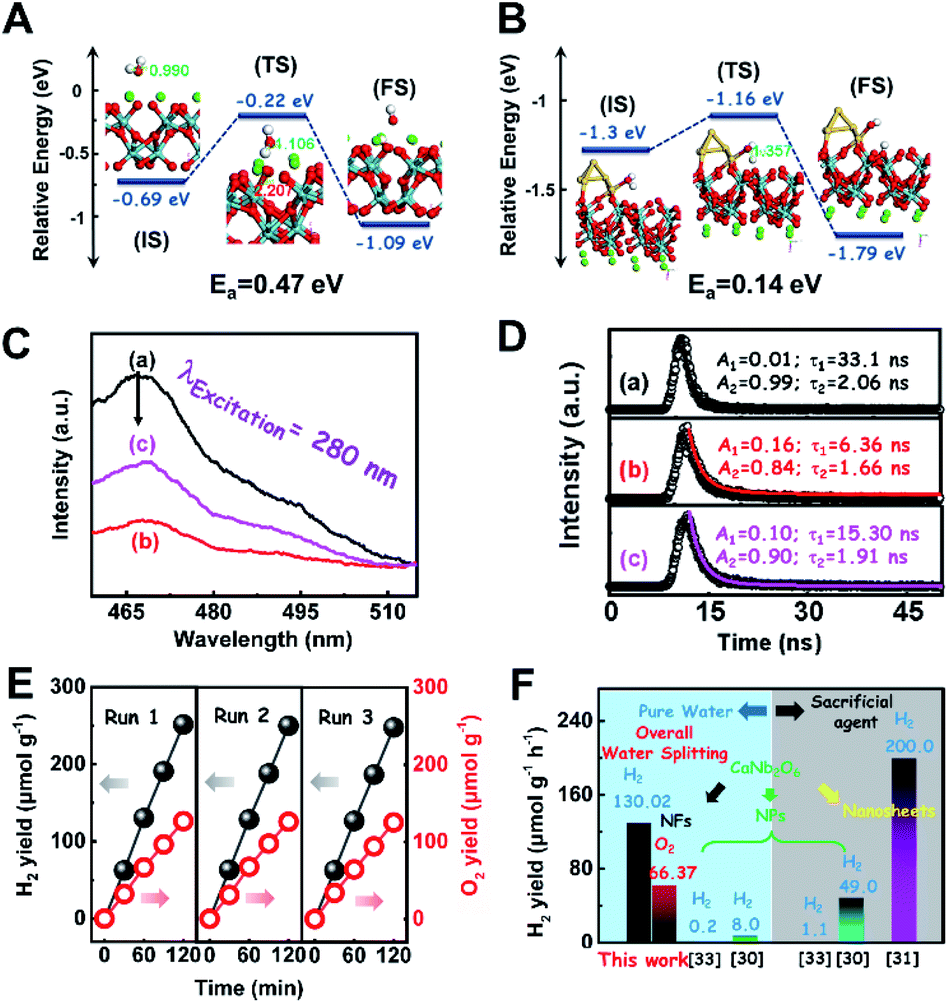 | ||
| Fig. 7 NEB calculations of the H2O decomposition processes on the (A) CaNb2O6 and (B) Au/CaNb2O6 surfaces (IS: initial state; TS: transition state; FS: final state); (C) steady-state PL spectra and (D) time-resolved transient PL decay of the as-fabricated samples: (a) CaNb2O6-600 NFs; (b) Pt1/CaNb2O6 NFs; (c) Au1/CaNb2O6 NFs; (E) cycling test of photocatalytic overall water splitting for the Au1/CaNb2O6 composite NFs; (F) comparison of the photocatalytic activity of the Au1/CaNb2O6 NFs for water splitting with that of CaNb2O6-based photocatalysts reported in the literature. | ||
To further assess the photocatalytic activity of the optimal Au1/CaNb2O6 NFs, 1.0 at% Pt NP-decorated CaNb2O6 NFs were fabricated as the control sample (Fig. S18 and S19†). It is well known that Pt is a commonly used cocatalyst and possesses the excellent “electron sink” effect and low overpotential for H2 production.51–53 However, to our surprise, the photocatalytic activity of Pt1/CaNb2O6 NFs for H2 and O2 production (∼37.50 and ∼19.00 μmol g−1 h−1) was much lower than that of the Au1/CaNb2O6 NFs (∼130.02 and ∼66.37 μmol g−1 h−1) upon UV-visible light irradiation. This result indicates that in comparison with the “electron sink” effect of Pt NPs, the LSPR of Au NPs is a more effective for enhancing the photocatalytic activity of CaNb2O6 NFs for water splitting.
To clarify the “electron sink” effect of metal NPs (Au and Pt) on the enhancement of photocatalytic activity for water splitting, the steady-state PL spectroscopy spectra of the Au1/CaNb2O6 and Pt1/CaNb2O6 NFs were tested in comparison with that of the pure CaNb2O6 NFs. As observed in Fig. 7C, the CaNb2O6 NFs present an intense emission band with the peak at ∼467 nm after UV light irradiation at 280 nm. However, the introduction of noble-metal NPs into CaNb2O6 NFs could obviously decrease the PL intensity of CaNb2O6 NFs. Meanwhile, the PL intensity of the Au1/CaNb2O6 NFs is lower than that of CaNb2O6 NFs, but is higher than that of the Pt1/CaNb2O6 NFs. The reduced PL intensity reveals either a faster transfer process with a shorter lifetime or a slower recombination process with a longer lifetime of the photoinduced electrons.57 To further understand the reason for the PL quenching of the above noble-metal NP-decorated CaNb2O6 NFs, TRPL spectroscopy measurement was carried out, and the corresponding results are given in Fig. 7D. It can be seen that the PL decay curves of all the above samples can be fitted by a biexponential function (eqn (1)). According to the two emission lifetimes obtained from the fitting function (listed in the inset of Fig. 7B), the average emission lifetime could be calculated for comparing the overall emission decay behaviors of the above samples (eqn (3)).
| τA = (A1τ12 + A2τ22)/(A1τ1 + A2τ2) | (3) |
The results show that the noble-metal NP-decorated CaNb2O6 NFs possess shorter average lifetimes as compared to the CaNb2O6-600 NFs (∼5.94 ns). The quenched PL intensities and the shortened lifetimes of the noble-metal NP-decorated CaNb2O6 NFs indicate the existence of a non-radiative quenching channel due to the photoinduced interfacial electron transfer from CaNb2O6 NFs to the adhesive noble metal NPs. When only exciting the interband transition of CaNb2O6 in the noble-metal NP-decorated CaNb2O6 NFs, the noble-metal NPs can serve as electron sinks to accept the photoinduced electrons from the CaNb2O6 NFs, thereby suppressing the recombination process of photoinduced charge carriers in the CaNb2O6 NFs. Further investigations found that the average lifetime of the Pt1/CaNb2O6 NFs (∼3.61 ns) was shorter than that of the Au1/CaNb2O6 NFs (∼5.52 ns). The shorter average lifetime of the Pt1/CaNb2O6 NFs manifests that as for the role of the “electron sink” effect, Pt NPs are more effective for enhancing the photocatalytic activity than Au NPs. However, in our work, the actual photocatalytic activity of the Pt1/CaNb2O6 NFs was much lower than that of the Au1/CaNb2O6 NFs upon UV-visible light irradiation (please note that Au LSPR can be excited by visible light irradiation). This result suggests that the LSPR of Au NPs plays the leading role in enhancing the photocatalytic activity of the Au1/CaNb2O6 NFs. Finally, the stability of the as-fabricated Au1/CaNb2O6 NFs was investigated through a cycling test on photocatalytic overall water splitting upon UV-visible light irradiation. As shown in Fig. 7E and S20,† after three cycles, the Au1/CaNb2O6 NFs with the original phase structure still possess ∼94.9% of their initial photocatalytic activity in the first cycle, which reveals the good photocatalytic stability of the Au1/CaNb2O6 NFs. Importantly, our work is the first attempt to fabricate an electrospun CaNb2O6-based NF photocatalyst for realizing the stoichiometric splitting of pure water into H2 and O2 upon UV-visible irradiation. Furthermore, the photocatalytic H2-production activity of our produced Au1/CaNb2O6 NFs in the absence of a sacrificial agent is higher than that of all the reported CaNb2O6-based photocatalysts (Fig. 7F).
3 Conclusions
In summary, CaNb2O6 NFs with a NP-stacked 1D porous nanostructure have been fabricated by using an electrospinning technique combined with a high-temperature calcination process. The above unique nanostructure led to the formation of many continuous homogeneous interfaces in the CaNb2O6 NFs. These homogeneous interfaces could boost the inter-particle charge migration for generating long-lived charge carriers. Thus, the photocatalytic activity of CaNb2O6 NFs for pure water splitting was ∼7.7 times higher than that of the CaNb2O6 NPs under UV-visible light irradiation. More importantly, the introduction of plasmonic Au NPs into CaNb2O6 NFs could effectively extend the visible-light absorption range and further enhance the photoinduced charge transfer, thereby leading to ∼13.1-fold enhancement of the photocatalytic activities for both H2 and O2 production as compared to the pure CaNb2O6 NFs. We have also demonstrated that the co-excitation of Au LSPR and CaNb2O6 interband transition is the prerequisite for realizing this remarkable enhancement of photocatalytic water splitting. The excited CaNb2O6 could not only create highly active charge carriers to reduce the reaction kinetic barrier of water splitting, but also enhance its conductivity to boost the hot electron transfer for improving the hot exciton separation. Thus, the plasmon-excited hot electrons and holes over Au NPs can enhance the photocatalytic activity of Au/CaNb2O6 NFs for splitting pure water into H2 and O2. Our work reported a new wide-bandgap fersmite semiconductor photocatalyst of 1D CaNb2O6 NFs for photocatalytic splitting of pure water into H2 and O2. It also provides a new plasmon-dominant synergy strategy to simultaneously extend the light absorption range and improve the charge-carrier kinetics of 1D wide-bandgap semiconductor NFs for remarkably enhancing photocatalytic overall water splitting.4 Experimental
4.1 Fabrication of the samples
300000) was slowly added to the above mixture solution under vigorous stirring for 16 h. Afterward, this mixture solution of PVP/DMF/C10H14CaO4/C10H5NbO20 was filled into a plastic syringe for electrospinning. After the electrospinning treatment, the NFs of the PVP/C10H14CaO4/C10H5NbO20 composite could be obtained by applying an electric voltage of 10 kV with a distance of ∼15 cm between the needle tip and the tinfoil receiver. Finally, the as-electrospun composite NFs were heated to 500 °C, 600 °C and 700 °C, respectively, with a rising time of 4 h and kept for 2 h at the required temperature. The CaNb2O6 NFs fabricated through the calcination treatment at 500 °C, 600 °C and 700 °C were denoted as CaNb2O6-500 NFs, CaNb2O6-600 NFs, and CaNb2O6-700 NFs respectively.
4.2 Characterization
An X-ray diffractometer (XRD; XRD-6000, Shimadzu) with a Cu Kα line of 0.1541 nm was used to study the crystallization and the phase transition. The morphology and structure of the as-prepared samples were observed by using a scanning electron microscope (SEM; S-4800, Hitachi) and transmission electron microscope (TEM; JEM-2100, JEOL). UV-vis absorption spectra were recorded with a Lambda 750 UV/Vis/NIR spectrophotometer (PerkinElmer, USA). X-ray photoelectron spectroscopy (XPS) was performed on a VG-ESCALAB LKII instrument with a Mg Kα ADES (hν = 1253.6 eV) source at a residual gas pressure below 10−8 Pa. Photoluminescence (PL) spectra were recorded using a fluorescence spectrophotometer (F-4600, Hitachi) equipped with a 150 W Xe lamp as the excitation light source. The decay curves of the as-prepared samples were obtained on an FLS920 fluorescence lifetime spectrophotometer (Edinburgh Instruments, UK) under the excitation of a hydrogen flash lamp with the wavelength at 280 nm (nF900; Edinburgh Instruments). Thermogravimetry and differential scanning calorimetry (TG-DSC) were implemented by using a NETZSCH STA 449F3 simultaneous thermal analyser under an air atmosphere in the range of 30–800 °C. Fourier transform infrared spectroscopy (FT-IR) spectra were recorded by using a Nicolet iS10 FT-IR spectrometer in the range of 500–4000 cm−1. The specific surface areas of the samples was investigated through a Micromeritics ASPA-2020 instrument and analysed by the Brunauer–Emmett–Teller (BET) method.4.3 Computational method
4.4 Photocatalytic overall water splitting
5 mg of the as-fabricated samples and 10 mL of deionised water were added into a 40 mL quartz reactor, regularly. Then, the quartz reactor was blocked with a rubber plug, followed by the ventilation of argon gas for 15 minutes to drive off the residual air. Subsequently, the quartz reactor was exposed to UV-vis light (300 W Xe lamp without a filter, PLS-SXE300UV). The composition of the gas product from the upper space above the liquid in the quartz reactor was periodically analysed by using a gas chromatograph (GC) equipped with a thermal conductivity detector (TCD) (Analytical Instrument, SP-3420A, Beifen-Ruili). The amount of H2O2 produced during the photocatalytic oxidation of H2O over the CaNb2O6 NPs was tested through the following process: The photocatalytic reaction solution was centrifuged at 8000 rpm for 5 min to remove the residual photocatalyst. 1 mL of potassium hydrogen phthalate (C8H5KO4, 0.1 M) and 1 mL of KI solution (0.4 M) were added into the above solution for further analysis by UV-visible spectroscopy (PERSEE TU-1950) on the basis of the absorbance at 350 nm.4.5 Electrochemical measurement
The Bode phase plots were obtained by using an electrochemical workstation (Autolab PGSTAT302N). 5 mg of as-fabricated samples were coated on ITO glass with an effective area of 1 cm × 1 cm, which acted as working electrodes. Pt wire and Ag/AgCl electrodes acted as the counter and reference electrodes, respectively. And, 1 M Na2SO3 aqueous solution was used as the electrolyte. The photocurrent and Mott tests are similar to the above, and the electrolyte is 0.2 M Na2SO4.Author contributions
Jiaming Zhang: Data curation, Methodology, and Writing - Original Draft. Xiaoyi Jiang: Software, Formal analysis, and Validation. Jindou Huang: Software, Formal analysis, and Validation. Wei Lu: Validation and Writing - review & editing. Zhenyi Zhang: Conceptualization, Validation, Formal analysis, Resources, Writing - review & editing, and Supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant Nos. 12074055 and 51772041), Natural Science Foundation of Liaoning Province for Excellent Young Scholars (2022-YQ-13), Liaoning BaiQianWan Talents Program, and Dalian Science Foundation for Distinguished Young Scholars (No 2018RJ05).Notes and references
- S. Xu and E. A. Carter, Chem. Rev., 2019, 119, 6631–6669 CrossRef CAS PubMed.
- C. Li, Y. Xu, W. Tu, G. Chen and R. Xu, Green Chem., 2017, 19, 882–899 RSC.
- J. W. Wang, L. Jiang, H. H. Huang, Z. Han and G. Ouyang, Nat. Commun., 2021, 12, 4276 CrossRef CAS PubMed.
- H. Lei, Q. He, M. Wu, Y. Xu, P. Sun and X. Dong, J. Hazard. Mater., 2022, 421, 126696 CrossRef CAS PubMed.
- J. Li, X. Wu and S. Liu, Acta Phys. Sin. Ch. Ed., 2021, 37, 2009038 Search PubMed.
- Z. Jiang, Q. Chen, Q. Zheng, R. Shen, P. Zhang and X. Li, Acta Phys. Sin. Ch. Ed., 2021, 37, 2010059 Search PubMed.
- H. Chang, J. Liu, Z. Dong, D. Wang, Y. Xin, Z. Jiang and S. Tang, Acta Phys. Sin. Ch. Ed., 2021, 40, 1595–1603 CAS.
- M. Li, R. Guan, J. Li, Z. Zhao, J. Zhang, C. Dong, Y. Qi and H. Zhai, Acta Phys. Sin. Ch. Ed., 2020, 39, 1437–1443 CAS.
- A. Kumar, V. Navakoteswara Rao, A. Kumar, M. Venkatakrishnan Shankar and V. Krishnan, ChemPhotoChem, 2020, 4, 427–444 CrossRef CAS.
- G. Sun, S. Mao, D. Ma, Y. Zou, Y. Lv, Z. Li, C. He, Y. Cheng and J. Shi, J. Mater. Chem. A., 2019, 7, 15278 RSC.
- P. Niu, J. Dai, X. Zhi, Z. Xia, S. Wang and L. Li, InfoMat, 2021, 3, 931–961 CrossRef CAS.
- D. Ma, Z. Wang, J. Shi, Y. Zou, Y. Lv, X. Ji, Z. Li, Y. Cheng and L. Wang, J. Mater. Chem. A., 2020, 8, 11031 RSC.
- A. Kumar and V. Krishnan, Adv. Funct. Mater., 2021, 2009807 CrossRef CAS.
- G. Sun, B. Xiao, H. Zheng, J. Shi, S. Mao, C. He, Z. Li and Y. Cheng, J. Mater. Chem. A., 2021, 9, 9735 RSC.
- A. Kumar, V. Navakoteswara Rao, A. Kumar, A. Mushtaq, L. Sharma, A. Halder, S. K. Pal, M. V. Shankar and V. Krishnan, ACS Appl. Energy Mater., 2020, 3, 12134–12147 CrossRef CAS.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- N. Som, V. Mankad and P. Jha, Int. J. Hydrogen Energy, 2018, 47, 21634–21641 CrossRef.
- S. Kumar, A. Kumar, A. kumar and V. Krishnan, Catal. Rev.: Sci. Eng., 2019, 62, 1–60 Search PubMed.
- A. C. Malingowski, P. W. Stephens, A. Huq, Q. Huang, S. Khalid and P. G. Khalifah, Inorg. Chem., 2012, 51, 6096–6103 CrossRef CAS PubMed.
- C. Xu and P. lv, Chin. J. Struct. Chem., 2021, 40, 1223–1230 CAS.
- X. Ning and G. Lu, Nanoscale, 2020, 12, 1213–1223 RSC.
- S. Fang and Y. H. Hu, Int. J. Energy Res., 2019, 43, 1082–1098 CrossRef.
- N. C. Hildebrandt, J. Soldat and R. Marschall, Small, 2015, 11, 2051–2057 CrossRef CAS PubMed.
- Z. Wang, J. Hong, S.-F. Ng, W. Liu, J. Huang, P. Chen and W.-J. Ong, Acta Phys.-Chim. Sin., 2020, 2011033 Search PubMed.
- A. Kumar, A. Kumar and V. Krishnan, ACS Catal., 2020, 10, 10253–10315 CrossRef CAS.
- Y. Su, L. Peng, C. Du and X. Wang, J. Phys. Chem. C, 2011, 116, 15–21 CrossRef.
- A. Kumar, M. Kumar, V. Navakoteswara Rao, M. Venkatakrishnan Shankar, S. Bhattacharya and V. Krishnan, J. Mater. Chem. A, 2021, 9, 17006–17018 RSC.
- O. I. Velikokhatnyi and P. N. Kumta, J. Power Sources, 2012, 202, 190–199 CrossRef CAS.
- S. Liu, P. Li, N. Umezawa, W. Zhou, H. Abe and G. Wang, Catal. Sci. Technol., 2019, 9, 3619–3622 RSC.
- I.-S. Cho, S. Lee, J. H. Noh, D. W. Kim, D. K. Lee, H. S. Jung, D.-W. Kim and K. S. Hong, J. Mater. Chem., 2010, 20, 3979 RSC.
- I.-S. Cho, D. W. Kim, C. M. Cho, J.-S. An, H.-S. Roh and K. S. Hong, Solid State Sci., 2010, 12, 982–988 CrossRef CAS.
- Y. Zhang, C. Liu, G. Pang, S. Jiao, S. Zhu, D. Wang, D. Liang and S. Feng, Eur. J. Inorg. Chem., 2010, 2010, 1275–1282 CrossRef.
- I.-S. Cho, S. T. Bae, D. H. Kim and K. S. Hong, Int. J. Hydrogen Energy, 2010, 35, 12954–12960 CrossRef CAS.
- I.-S. Cho, S. T. Bae, D. K. Yim, D. W. Kim and K. S. Hong, J. Am. Ceram. Soc., 2009, 92, 506–510 CrossRef CAS.
- Y. Hong, S.-H. Jeon, H. Jeong and H. Ryu, Ceram. Int., 2021, 47, 7814–7823 CrossRef CAS.
- S. Choi, S. Kim, S. lim and H. Park, J. Phys. Chem. C, 2010, 114, 16475–16480 CrossRef CAS.
- Z. Zhang, Z. Wang, S. Cao and C. Xue, J. Phys. Chem. C, 2013, 117, 25939–25947 CrossRef CAS.
- Z. Zhang, X. Jiang, B. Liu, L. Guo, N. Lu, L. Wang, J. Huang, K. Liu and B. Dong, Adv. Mater., 2018, 30, 1705221 CrossRef PubMed.
- X. Fang, Q. Shang, Y. Wang, L. Jiao, T. Yao, Y. Li, Q. Zhang, Y. Luo and H. L. Jiang, Adv. Mater., 2018, 30, 1705112 CrossRef PubMed.
- H.-M. Chiu, T.-H. Yang, Y.-C. Hsueh, T.-P. Perng and J.-M. Wu, Appl. Catal., B, 2015, 163, 156–166 CrossRef CAS.
- H. Sun, S. Zeng, Q. He, P. She, K. Xu and Z. Liu, Dalton Trans., 2013, 1–3 Search PubMed.
- S. K. Cushing, J. Li, F. Meng, T. R. Senty, S. Suri, M. Zhi, M. Li, A. D. Bristow and N. Wu, J. Am. Chem. Soc., 2012, 134, 15033–15041 CrossRef CAS PubMed.
- Z. Liu, W. Hou, P. Pavaskar, M. Aykol and S. B. Cronin, Nano Lett., 2011, 11, 1111–1116 CrossRef CAS PubMed.
- I. S. Cho, D. W. Kim, T. H. Noh, S. Lee, D. K. Yim and K. S. Hong, J. Nanosci. Nanotechnol., 2010, 10, 1196–1202 CrossRef CAS PubMed.
- S. Cao, B. Shen, T. Tong, J. Fu and J. Yu, Adv. Funct. Mater., 2018, 28, 1800136 CrossRef.
- T. Tachikawa, T. Ochi and Y. Kobori, ACS Catal., 2016, 6, 2250–2256 CrossRef CAS.
- Y.-J. Hsiao, C.-W. Liu, B.-T. Dai and Y.-H. Chang, J. Alloys Compd., 2009, 475, 698–701 CrossRef CAS.
- E. V. Pean, S. Dimitrov, C. S. De Castro and M. L. Davies, Phys. Chem. Chem. Phys., 2020, 22, 28345–28358 RSC.
- P. Bera, K. Priolkar, A. Gayen, P. R. Sarode, M. S. Hegde, S. Emura, R. Kumashiro, V. Jayara and G. N. Subbanna⊥, Chem. Mater., 2013, 15, 2049–2060 CrossRef.
- X. Jiang, J. Huang, Z. Bi, W. Ni, G. Gurzadyan, Y. Zhu and Z. Zhang, Adv. Mater., 2022, e2109330 CrossRef PubMed.
- W. Guo, Q. Qin, L. Geng, D. Wang, Y. Guo and Y. Yang, J. Hazard. Mater., 2016, 308, 374–385 CrossRef CAS PubMed.
- X. Wang, C. Liow, A. Bisht, X. Liu, T. C. Sum, X. Chen and S. Li, Adv. Mater., 2015, 27, 2207–2214 CrossRef CAS PubMed.
- L. Zhang, N. Ding, L. Lou, K. Iwasaki, H. Wu, Y. Luo, D. Li, K. Nakata, A. Fujishima and Q. Meng, Adv. Funct. Mater., 2019, 29, 1806774 CrossRef.
- S. Cabrini, G. Lérondel, A. M. Schwartzberg, T. Mokari, J. Lee, S.-H. Lee, M. S. Kim, H. Shin and J. Kim, Proc. SPIE, 2014, 9161, 916109 CrossRef.
- G.-Y. Yao, Q.-L. Liu and Z.-Y. Zhao, Catalysts, 2018, 8, 236 CrossRef.
- H. Song, X. Meng, T. D. Dao, W. Zhou, H. Liu, L. Shi, H. Zhang, T. Nagao, T. Kako and J. Ye, ACS Appl. Mater. Interfaces, 2018, 10, 408–416 CrossRef CAS PubMed.
- M. Dozzi, A. Candeo, G. Marra, C. DAndrea, G. Valentini and E. Selli, J. Phys. Chem. C, 2018, 133, 14326–14335 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ta05332b |
| This journal is © The Royal Society of Chemistry 2022 |