DOI:
10.1039/D2TA03978H
(Paper)
J. Mater. Chem. A, 2022,
10, 23384-23394
Carbon dots as an electron extractant for enhanced photocatalytic antibacterial activity of covalent organic frameworks†
Received
18th May 2022
, Accepted 3rd October 2022
First published on 26th October 2022
1 Introduction
Microbial infections have always been a huge threat to public health, and continuous efforts have been made to deal with microbial infections, such as the development of new drugs and antibiotics.1–5 Antibacterial photocatalytic therapy (APCT), an alternative sterilization technology, is one of the most effective sterilization methods, and has attracted extensive attention. The sterilization mechanism of APCT involves the generation of reactive oxygen species (ROS) through photoactivated photosensitizers and oxygen6,7 or the utilization of photocatalysts to oxidize cells by using photogenerated holes.8 Therefore, it is critical to select an appropriate photosensitizer for APCT. To date, many types of photosensitizers have been successfully explored and demonstrated to be effective in photodynamic therapy (PDT), such as silver nanoparticles,9 TiO2,10 copper,11 ZnO,12,13 g-C3N4,14 porphyrin derivatives,15 and phthalocyanines.16 However, their shortcomings limit their development in the field of photocatalysis: (1) a narrow absorption range of only UV absorption with low utilization of sunlight; (2) leakage of metal ions, which is a potential threat to human health and environmental safety; and (3) poor photothermal stability, resulting in the consumption of active ingredients during APCT.
Compared with other photosensitizers, covalent organic frameworks (COFs), a novel class of crystalline porous organic semiconductors linked by light elements through strong covalent bonds, have attracted widespread attention in photocatalytic therapy, owing to their nontoxicity, broad absorption range, high photothermal stability, tunable chemical structure, and electronic properties.17,18 Consequently, building crystalline COFs with suitable photoactive components and layered superposition sequences is an exciting direction to realize a new generation of highly active artificial photosystems. This ordered structure facilitates customized and regulated electronic structures and chemical functions of COFs at the molecular level. The strong photoelectric conversion ability of COFs makes them a promising material in the field of photocatalysis. However, there is still an inherent problem in COFs, the exciton binding energy of COFs is relatively high compared to that of inorganic semiconductors.19 This means that the separation and migration rates of photogenerated electron–hole pairs in pure COFs are limited only by changing the structure and tuning the bandgap.20,21 Therefore, the maximum extraction of carriers from photoexcited COFs is the most important concern in efficient photoredox catalysis. In contrast to the traditional methods for forming heterojunctions with metals and semiconductors, Long et al. engineered a metal–insulator–semiconductor (MIS) photosystem based on donor–acceptor (D–A) type COF photocatalysts with tunable photoelectronic conduction.22 In particular, with the inherent different electron affinities under light irradiation, such a D–A type COF link by an imine conjugated polymer skeleton, has intramolecular charge transfer (ICT) from D to A, and the electrons accumulate preferentially in receptors with efficient electron mobility through the delocalization of π-electrons.23 To achieve more efficient and faster carrier transfer, it is necessary to induce an internal driving force on the two-dimensional semiconductor plane to delocalize the optical carriers.
Based on the above, we constructed a binary system with a D–A type COF and selected carbon dots (CDs) as both the electron extractant and storage media. CDs are ideal candidate conductive media, owing to their high-water solubility and outstanding electron transfer and sinking efficiency, which can improve the dispersibility and photogenerated carrier separation efficiency of COFs.24,25 Considering the structural characteristics of COF semiconductors, the uniform distribution of CDs, with strong chemical affinity on the surface of COFs (C, N, and O), is conducive for π-electron emission from the skeleton of COFs and maximizing the π-electron traversing efficiency.
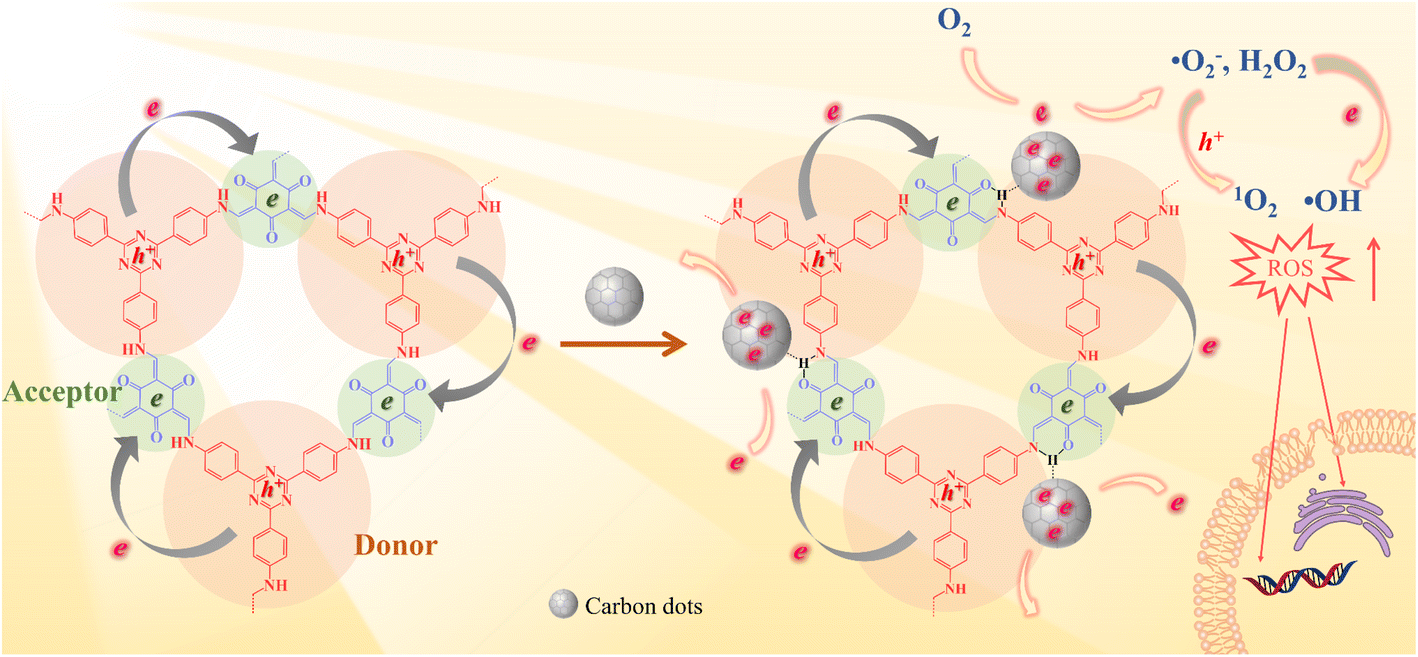
In this study, an intramolecular charge transfer system was constructed to extract π-electrons from photosensitive D–A COF semiconductors to CDs through hydrogen bonding, and a nanostructured photosystem was designed to demonstrate a high-efficiency bactericidal mechanism. The incorporation of CDs into a D–A type COF to construct antibacterial photocatalyst composites has not been reported to date. As illustrated in Scheme 1, under photoexcitation, photogenic electrons are circulated and transmitted from 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl) trianiline (TTA), an electron donor, to 1,3,5-triformylphloroglucinol (Tp), an electron acceptor. After Tp accepts the electrons, the remaining electrons flow back to the donor and rapidly recombine with the photogenic hole, with only a few electrons escaping. The amount of ROS produced is restricted. In contrast, with the loading of CDs, a charge transfer channel made of = C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O–H–N = hydrogen bonds is formed between the COF and CDs. The photogenerated electrons trapped within the porous structure of the COFs are extracted by CDs, causing electrons to accumulate in the CDs for more ROS evolution. The generated electrons (e−) reduce dissolved oxygen (O2) to a superoxide radical (˙O2−) and hydrogen peroxide (H2O2), the electrons further reduce H2O2 to hydroxyl radicals (˙OH), and ˙O2− can further be oxidized by holes (h+) to 1O2. Our study indicated that the prepared COF@CDs as APCT had better catalytic efficiency against Escherichia coli (E. coli) than a pure COF, and the in vitro disinfection efficiency was >95% upon activation with visible light for 60 min.
O–H–N = hydrogen bonds is formed between the COF and CDs. The photogenerated electrons trapped within the porous structure of the COFs are extracted by CDs, causing electrons to accumulate in the CDs for more ROS evolution. The generated electrons (e−) reduce dissolved oxygen (O2) to a superoxide radical (˙O2−) and hydrogen peroxide (H2O2), the electrons further reduce H2O2 to hydroxyl radicals (˙OH), and ˙O2− can further be oxidized by holes (h+) to 1O2. Our study indicated that the prepared COF@CDs as APCT had better catalytic efficiency against Escherichia coli (E. coli) than a pure COF, and the in vitro disinfection efficiency was >95% upon activation with visible light for 60 min.
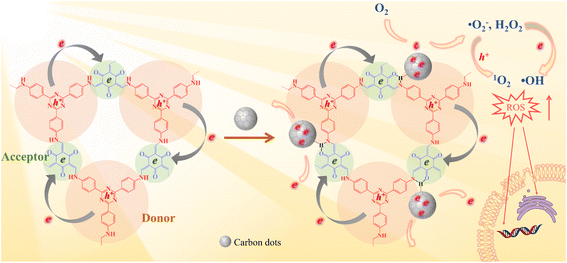 |
| | Scheme 1 Schematic illustration of a photocatalytic antibacterial system over the COF and COF@CD composite. | |
2 Results and discussion
2.1 Characterization of the as-synthesized materials
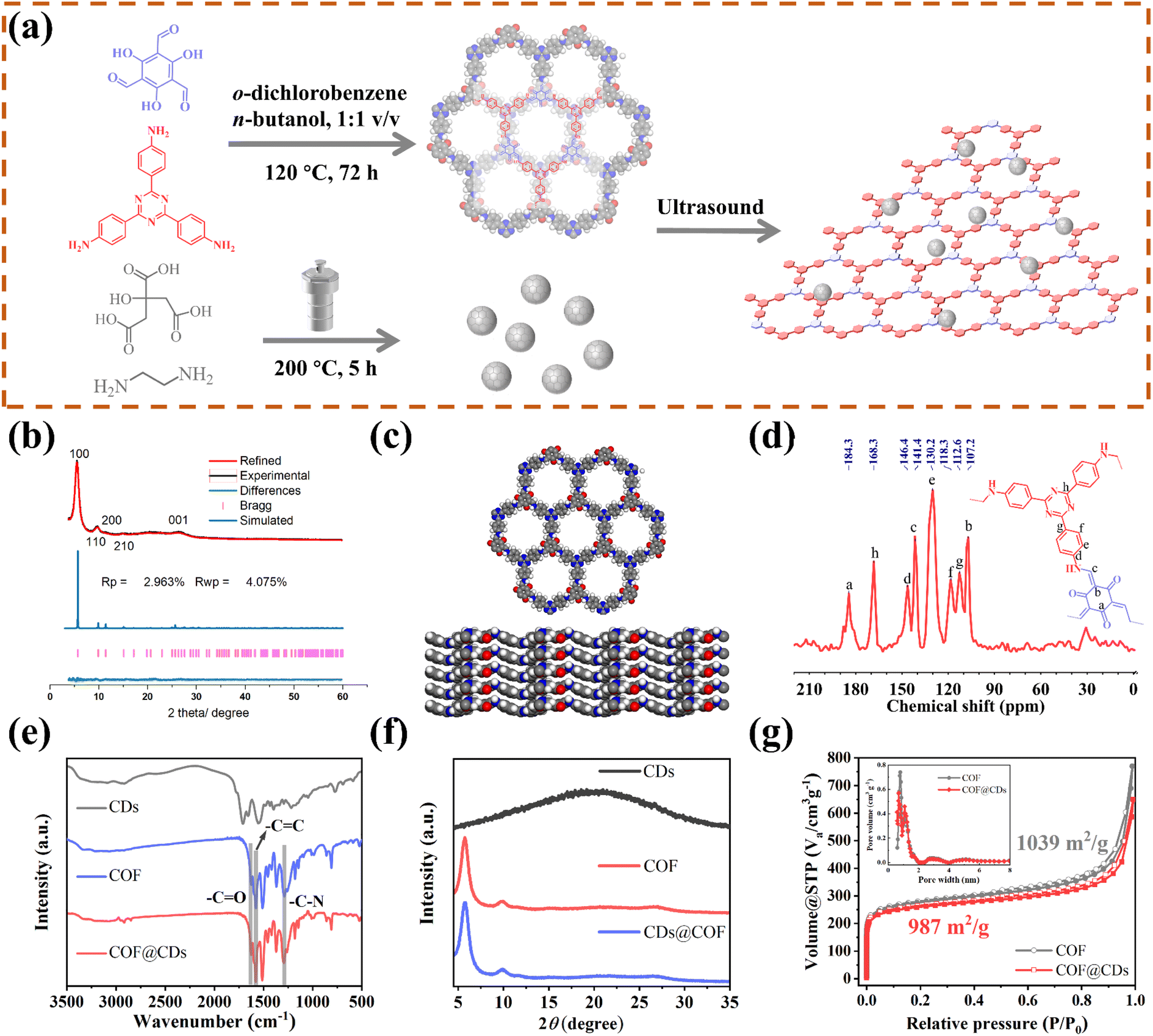
As schematically depicted in Fig. 1a, we synthesized a two-dimensional D-A type COF via Schiff base condensation between 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)trianiline (TTA) as the donor and 1,3,5-triformylphloroglucinol (Tp) as the acceptor in a mixed solution of o-dichlorobenzene/n-butanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, by vol.). The CDs were prepared by condensing citric acid and ethylenediamine through the hydrothermal method. The as-obtained TTATp-COF and CDs were sonicated in deionized water for 1 h. After evaporation to remove the solvent, the CDs were assembled on the surface of the COF nanosheet to form the COF@CD composite via hydrogen bonding. To evaluate the crystallinity of the TTATp-COF, powder X-ray diffraction (PXRD) was performed to compare the structural simulation results with the diffraction results (Fig. 1b). The PXRD patterns of the COF show several strong diffraction peaks indexed to the hexagonal crystalline phase in the region of 1.5–10°.26,27 The PXRD pattern of the TTATp-COF indicated that there were five diffraction peaks at 2θ = 5.7°, 9.8°, 11.5°, 15.3°, and 26.6°, which were assigned to the (100), (110), (200), (210), and (001) lattice planes of the polymer, respectively. The X-ray peak corresponding to (001) attributed to the topology of the amine monomer was broad and weak, and it was bent and not linear.28 We used the Material Studio software for Pawley refinement, which showed good consistency of experimental diffraction patterns, as proved by the low residual values (Rp = 2.963%, Rwp = 4.075%). As shown in Fig. 1c, the diffraction pattern was in good agreement with the simulated eclipsed AA-stacking model of TTATp-COF structures. The atomic-level formation of a COF was accessed by solid-state NMR spectra and FT-IR spectra. From 13C NMR spectra (Fig. 1d), the COF exhibited characteristic peaks for carbonyl carbons (CO) (marked as a) at 184.3 ppm, amine carbon (C–N) (marked as c) at 141.4 ppm, and exocyclic carbons (CC) (marked as b) at 107.2 ppm, which directly shows the structure of enol to keto tautomerism. The FT-IR spectra of the COF shown in Fig. 1e further confirmed the formation of a β-ketonamine framework structure. The characteristic stretching bands at 1628 cm−1, 1579 cm−1 and 1291 cm−1 can be assigned to CO, CC and C–N bonds, respectively. The XPS survey spectra (Fig. S1†) also reveal the structural components of COF for CO, CC, CN, and C–N bonds. COF@CDs have almost the same spectra with a slight displacement of binding energy compared to that of the COF. The PXRD, 13C NMR and FT-IR spectra demonstrated the successful synthesis of a crystalline covalent organic framework. The surface groups of CDs were investigated by XPS analysis (Fig. S2†). High-resolution XPS spectra revealed that they were mainly composed of carbon, nitrogen and oxygen. After loading CDs, we compared the FT-IR spectra and XRD patterns of CDS, the COF and COF@CDs. Owing to the low loading content (4%) and weak infrared stretching vibration, it is difficult to observe the vibration band from CDs in the COF@CD composite (Fig. 1e). The COF@CD composite exhibits similar XRD patterns to COF, suggesting that the crystalline structure of the COF is retained after incorporating CDs (Fig. 1f). These results indicated that the loading of CDs does not change the crystal structure of the COF. The porosity of the COF was determined by nitrogen adsorption–desorption measurement at 77 K (Fig. 1g). The COF featured a type II sorption isotherm, indicative of a typical micropore filling character. The Brunauer–Emmett–Teller (BET) surface area for the COF and COF@CDs was calculated to be 1039 and 987 m2 g−1, respectively. After incorporation of CDs, both the BET surface area and the number of micropores below 1 nm decreased slightly. It is difficult for the majority of CDs to enter the nanopores of the COF, thereby being positioned accordingly at the periphery.
:1, by vol.). The CDs were prepared by condensing citric acid and ethylenediamine through the hydrothermal method. The as-obtained TTATp-COF and CDs were sonicated in deionized water for 1 h. After evaporation to remove the solvent, the CDs were assembled on the surface of the COF nanosheet to form the COF@CD composite via hydrogen bonding. To evaluate the crystallinity of the TTATp-COF, powder X-ray diffraction (PXRD) was performed to compare the structural simulation results with the diffraction results (Fig. 1b). The PXRD patterns of the COF show several strong diffraction peaks indexed to the hexagonal crystalline phase in the region of 1.5–10°.26,27 The PXRD pattern of the TTATp-COF indicated that there were five diffraction peaks at 2θ = 5.7°, 9.8°, 11.5°, 15.3°, and 26.6°, which were assigned to the (100), (110), (200), (210), and (001) lattice planes of the polymer, respectively. The X-ray peak corresponding to (001) attributed to the topology of the amine monomer was broad and weak, and it was bent and not linear.28 We used the Material Studio software for Pawley refinement, which showed good consistency of experimental diffraction patterns, as proved by the low residual values (Rp = 2.963%, Rwp = 4.075%). As shown in Fig. 1c, the diffraction pattern was in good agreement with the simulated eclipsed AA-stacking model of TTATp-COF structures. The atomic-level formation of a COF was accessed by solid-state NMR spectra and FT-IR spectra. From 13C NMR spectra (Fig. 1d), the COF exhibited characteristic peaks for carbonyl carbons (CO) (marked as a) at 184.3 ppm, amine carbon (C–N) (marked as c) at 141.4 ppm, and exocyclic carbons (CC) (marked as b) at 107.2 ppm, which directly shows the structure of enol to keto tautomerism. The FT-IR spectra of the COF shown in Fig. 1e further confirmed the formation of a β-ketonamine framework structure. The characteristic stretching bands at 1628 cm−1, 1579 cm−1 and 1291 cm−1 can be assigned to CO, CC and C–N bonds, respectively. The XPS survey spectra (Fig. S1†) also reveal the structural components of COF for CO, CC, CN, and C–N bonds. COF@CDs have almost the same spectra with a slight displacement of binding energy compared to that of the COF. The PXRD, 13C NMR and FT-IR spectra demonstrated the successful synthesis of a crystalline covalent organic framework. The surface groups of CDs were investigated by XPS analysis (Fig. S2†). High-resolution XPS spectra revealed that they were mainly composed of carbon, nitrogen and oxygen. After loading CDs, we compared the FT-IR spectra and XRD patterns of CDS, the COF and COF@CDs. Owing to the low loading content (4%) and weak infrared stretching vibration, it is difficult to observe the vibration band from CDs in the COF@CD composite (Fig. 1e). The COF@CD composite exhibits similar XRD patterns to COF, suggesting that the crystalline structure of the COF is retained after incorporating CDs (Fig. 1f). These results indicated that the loading of CDs does not change the crystal structure of the COF. The porosity of the COF was determined by nitrogen adsorption–desorption measurement at 77 K (Fig. 1g). The COF featured a type II sorption isotherm, indicative of a typical micropore filling character. The Brunauer–Emmett–Teller (BET) surface area for the COF and COF@CDs was calculated to be 1039 and 987 m2 g−1, respectively. After incorporation of CDs, both the BET surface area and the number of micropores below 1 nm decreased slightly. It is difficult for the majority of CDs to enter the nanopores of the COF, thereby being positioned accordingly at the periphery.
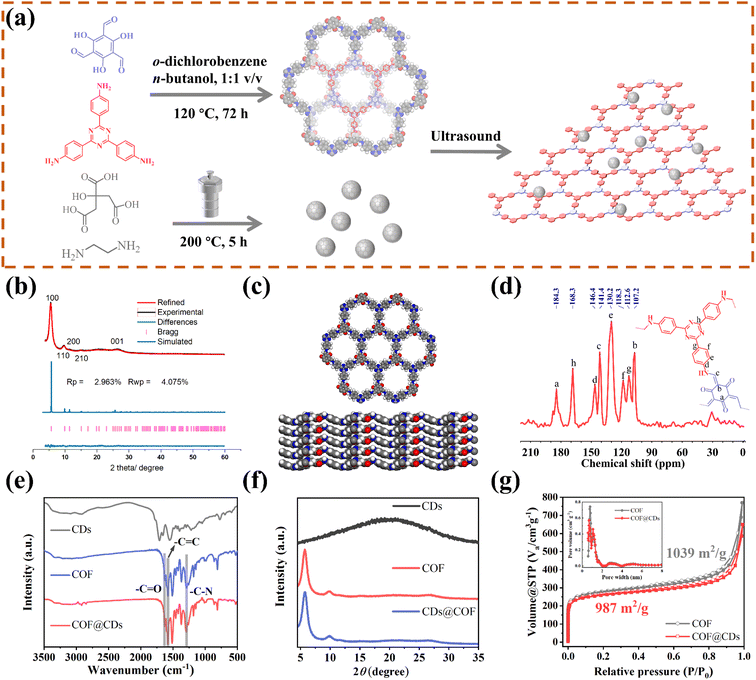 |
| | Fig. 1 (a) Scheme of the synthesis of the COF, CDs, and their composites. (b) PXRD patterns of the experimentally observed (black) and Pawley refined (red) COF with their refinement differences (green), and the simulated patterns for the eclipsed AA stacking modes (blue). (c) Lattice structure of the COF in top and side views (gray: carbon; white: hydrogen; blue: nitrogen; red: oxygen). (d) 13C MAS solid-state NMR spectrum of the COF. (e) FT-IR spectrum and (f) PXRD patterns of CDs, the COF and COF@CDs. (g) Nitrogen adsorption and desorption isotherms for the COF and COF@CDs. Inset: profile of the calculated pore size distribution for the COF and COF@CDs. | |
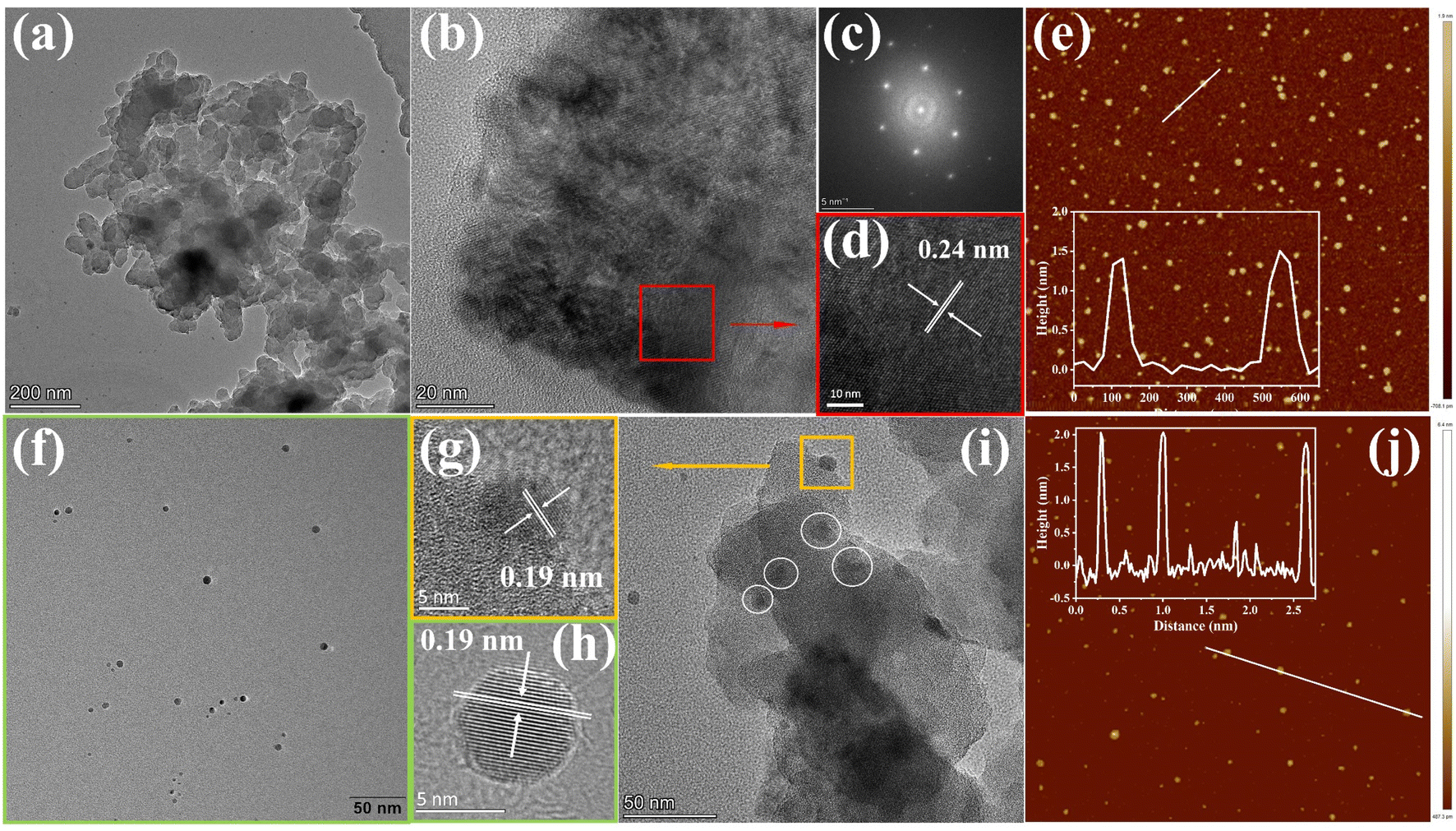
To gain insight into the surface morphology and microstructure, scanning electron microscopy (SEM), transmission electron microscopy (TEM), and atomic force microscopy (AFM) were exploited. The SEM images of the COF and COF@CDs were shown in Fig. S3(a and b)†, the overall morphology shows a two-dimensional sheet-like COF stacked layer by layer. The COF exhibited microstructures with ultrathin 2D layered π-interaction structures, as shown in Fig. 2a. High resolution electron transmission microscopy imaging (HR-TEM) (Fig. 2b), local magnified imaging (Fig. 2d) and fast Fourier transform (FFT) images (Fig. 2c) show the highly ordered and periodic structure of the COF with a lattice spacing of 0.24 nm, corresponding to the excellent crystallinity of XRD. As shown in Fig. 2f, the CDs have a regular spherical structure, with uniform dispersion without aggregation and a lattice spacing of 0.19 nm as clearly shown in Fig. 2h. Significantly, from the TEM imaging of COF@CDs in Fig. 2i, the individual crystallites highlighted by dot circles are uniformly distributed on the surface of the COF nanosheet, displayed an interplanar spacing of 0.19 nm as shown in Fig. 2g, which is ascribed to CDs. This intimate interface facilitates charge conduction for carrier separation in photocatalysis. The sizes of the COF and CDs were measured by AFM (Fig. 2e–j). The COF exhibited an ultrathin nanosheet structure and its average height and width were approximately 1.5 and 100 nm. The CDs were uniformly spherical with an average height of 2 nm. These morphological results prove that 0D CDs can be easily deposited onto a two dimensional COF nanosheet through strong hydrogen bonds.
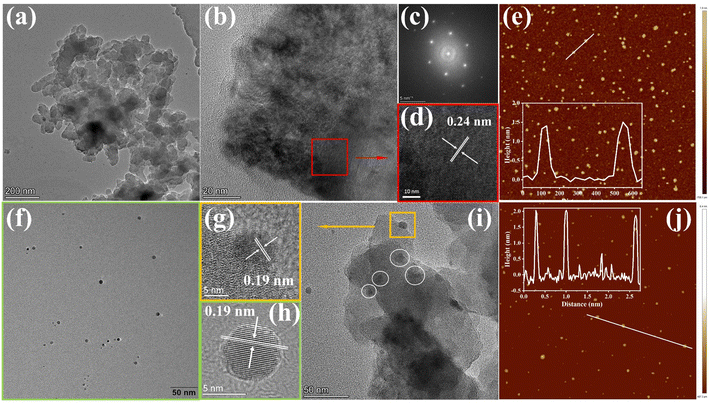 |
| | Fig. 2 (a) TEM, (b) and (d) HRTEM images of the COF and the corresponding (c) fast Fourier transform images. (f) TEM and (h) HRTEM images of CDs. (i) TEM and (g) HRTEM images of COF@CDs. AFM images of (e) the COF and (j) CDs. | |
2.2 Photocatalytic antibacterial activity
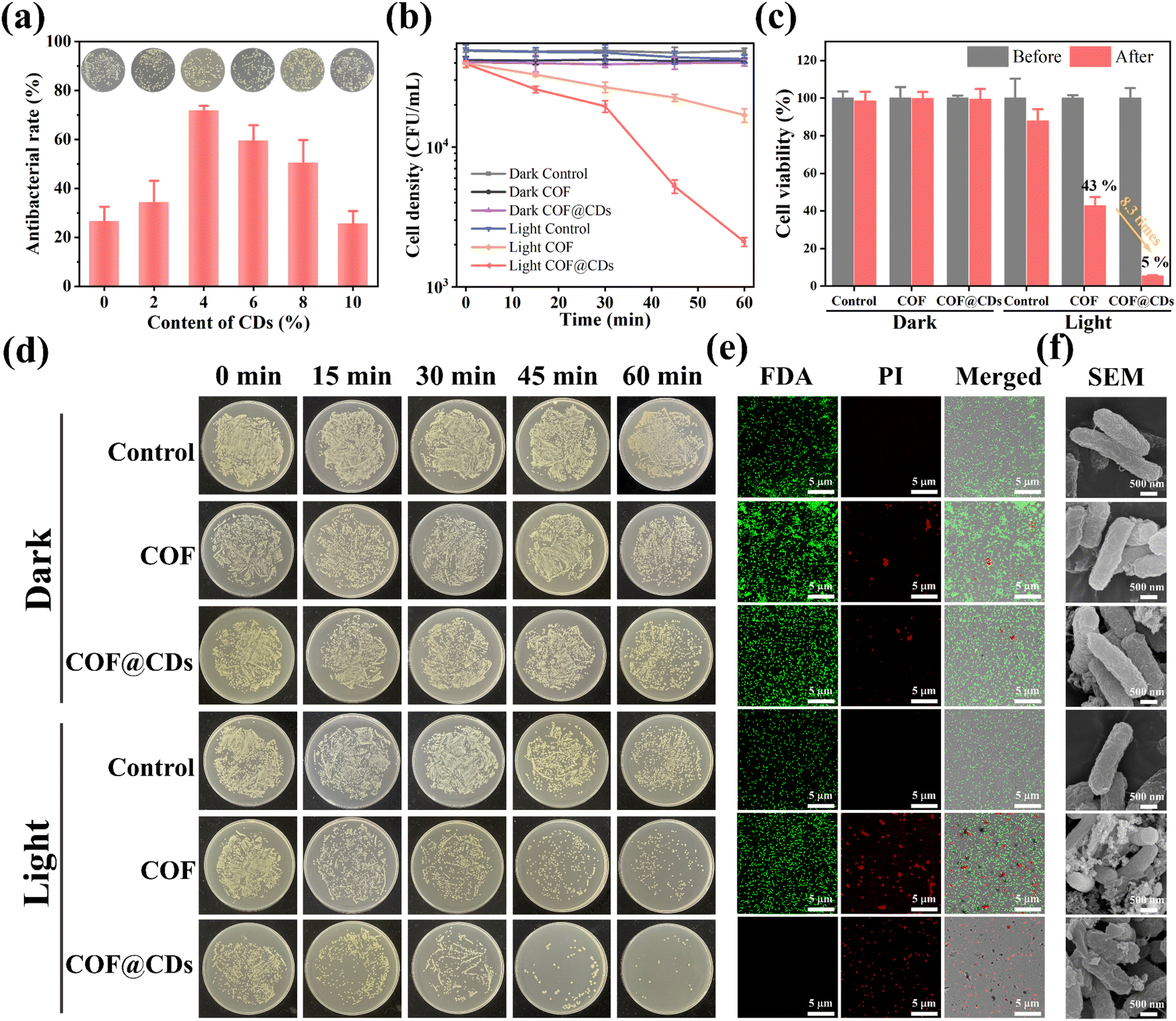
The successful fabrication of the TTATp-COF and CDs encouraged us to further investigate the photocatalytic antibacterial properties of COF@CDs. E. coli was selected as a model bacterium to evaluate the photocatalytic antibacterial activity of the as-prepared materials. We first evaluated the photocatalytic antibacterial effects of the composites with different loading contents of CDs. As shown in Fig. 3a, the antibacterial rate gradually increased from 0 to 4%. When the loading content was 4%, the sterilization rate of the composites reached the highest value, which was 2.7 times higher than that of the pure COF, but decreased gradually with a further increase in the loading capacity, and the sterilization rate of composites with a loading content of 10% was almost the same as that of the pure COF. The nanocomposites in experiments were all with a loading content of 4 wt% CDs. To evaluate the relationship between the photoexcitation time and bacterial inactivation rate more intuitively, we examined the bacterial counts quantified on LB agar plates and constructed a corresponding time-bacterial survival rate curve. An obvious bacterial colony was observed in the LB plate experiment, as shown in Fig. 3d. The statistical data from Fig. 3d are depicted in Fig. 3b, and the bacterial density of E. coli treated with a photocatalyst for 60 min in the dark and under light-only conditions without a photocatalyst was similar to that of the dark control group. However, the COF and COF@CD hybrid materials exhibited remarkable antibacterial activities under light conditions. After the addition of the COF, the bacterial density decreased with increasing irradiation time. The sterilization effect of COF@CDs was significantly stronger than that of the pure COF, with the density curve dropping more sharply and almost no colonies being formed on LB agar plates. We calculated the survival rate of E. coli after 60 min of each treatment, as shown in Fig. 3c. After 60 min of light irradiation, the cell viability of COF@CDs was only 5%, and the survival rate of bacteria decreased by 8.3 times compared to that of the COF. These results suggest that the combination of the COF and CDs greatly enhanced the antibacterial performance, and that the antibacterial activity was positively correlated with the irradiation time, which may be due to the production of more ROS.
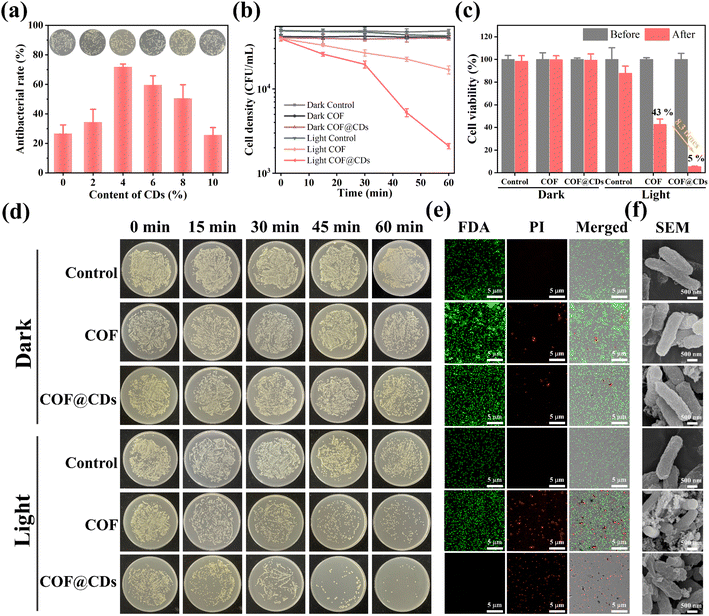 |
| | Fig. 3 (a) The antibacterial rate of different doping contents of CDs for the COF@CD composite. Inset: the corresponding E. coli colony growth. (b) The cell density curves of E. coli with the treatment of PBS, COF and COF@CDs under light and dark conditions at different time points. (c) The cell viability of E. coli treated with PBS, COF, and COF@CDs before and after light irradiation. (d) E. coli colony growth over time, corresponding to different treatment modes. (e) Confocal laser scanning microscopy and (f) SEM images of E. coli treated with PBS, COF, and COF@CDs under light and dark conditions after 1 h. | |
Additionally, antibacterial activity was investigated using the fluorescent live/dead staining method with confocal laser scanning microscopy (CLSM). Live and dead bacteria were stained with fluorescein diacetate (FDA) for green fluorescence and propidium iodide (PI) for red fluorescence. As shown in Fig. 3e, both E. coli cultures, except for the COF and COF@CDs treated with light groups showed bright green emission and no red fluorescence, indicating that the bacteria remained unaffected by the experimental treatments. As expected, a large amount of green fluorescence and scattered red fluorescence suggested partial membrane damage in the COF-treated bacteria under light exposure. The emission of red fluorescence increased significantly, and no green fluorescence was observed, indicating that the number of dead cells increased dramatically, demonstrating the severe membrane disruption capacity of COF@CDs under visible light. This result further highlights the synergistically enhanced antibacterial effect of the CD-integrated COF. The change in surface morphology of bacteria, as shown in the SEM images (Fig. 3f), reflects the influence of the samples on bacteria at the microscopic level. The smooth and intact cell walls of rod-shaped E. coli were damaged and roughened seriously by high concentrations of the COF and COF@CDs under light treatment. The cell structure of E. coli in the control group with light irradiation and the treatment group in the dark was intact without obvious shrinkage or damage, consistent with the results of the fluorescent live/dead staining method. Therefore, both the COF and COF@CD nanocomposites have antibacterial effects under light, but the composites have a higher sterilization efficiency than the pure COF. Photocatalytic antibacterial stability for COF@CDs was evaluated and is shown in Fig. S4.† There was no obvious decay of antibacterial activity of COF@CDs for two cycles under light irradiation. After irradiation and continuous agitation for 120 min, the antibacterial activity of COF@CDs began to decrease and the cell viability at 180 min was close to 40%. However, although the antibacterial efficiency for the fourth cycle was not as good as that for the first cycle, the sterilization rate maintains a similar level as the third cycle at 240 min, demonstrating the good photocatalytic antibacterial stability of COF@CDs.47,48
2.3 Detection of ROS
After the evaluation and confirmation of the antibacterial activity of the samples, we then explored the antibacterial mechanism to further understand why COF@CDs have a higher bactericidal efficiency than the pure COF. In APCT, ROS is the most beneficial sterilization tool, and the as-photogenerated ROS can inactivate proteins, damage nucleic acids, and cause peroxidation of lipids to disinfect bacteria.29–31 To evaluate the ability of the COF, CDs, and COF@CDs for generating ROS under light irradiation, 2,7-dichlorodihydro-diacetate fluorescein (DCFH-DA) was used as an indicator molecule, which can be rapidly oxidized to dichlorofluorescein (DCF), a fluorescent species, in the presence of ROS. As shown in Fig. 4a, after the absorption of light for 25 s, negligible fluorescence was observed in the control groups or the samples containing pure CDs. Obvious fluorescence was detected with the addition of the COF and COF@CDs, and the intensities of the composites were higher than those of the COF.
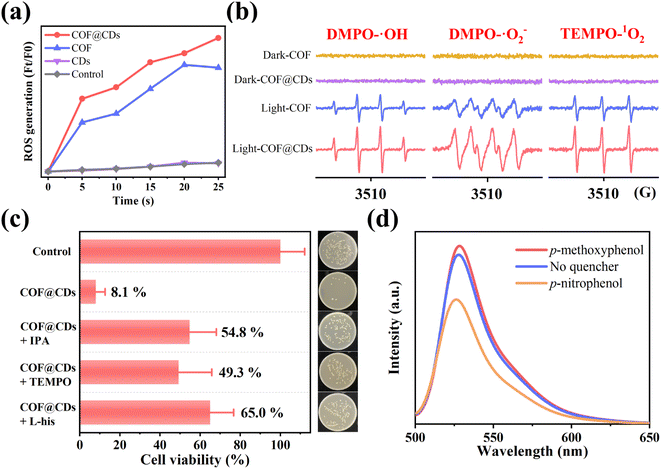 |
| | Fig. 4 (a) ROS generation efficiency of CDs, COF and COF@CDs in 25 s. (b) DMPO-˙OH, DMPO-˙O2− and TEMP-1O2 spin-trapping EPR spectra of the COF and COF@CDs under light and dark conditions. (c) Survival of E. coli after treatment with COF@CDs for 1 h in the absence and presence of isopropanol, TEMPO, and L-histidine. Inset: growth of the corresponding E. coli colony. (d) ROS fluorescent intensity in the absence and presence of p-nitrophenol and p-methoxyphenol after 25 s. | |
We further used electron paramagnetic resonance spectroscopy (EPR) to detect the types and intensity of ROS with 5,5-dimethyl-1-pyrroline N-oxide (DMPO) and 2,2,6, 6-tetramethyl-1-piperidine (TEMP) as spin probes. As shown in Fig. 4b, regardless of the type of ROSs (DMPO-˙OH, DMPO-˙O2−, and TEMP-1O2), both the COF and COF@CDs could generate ROS under illumination, and the characteristic peak intensity of COF@CDs was higher than that of the COF. There were no signals of ROS to be detected for samples in the dark, revealing that both ˙OH, ˙O2−, and 1O2 were excited by visible light. This measurement further confirmed that both the COF and COF@CDs have a high ROS production efficiency and that COF@CDs have a better production efficiency.
To explore which reactive oxygen species plays a major role in the photocatalytic disinfection of COF@CDs, ROS quenching experiments were performed. ˙OH, ˙O2−, and 1O2 were quenched with isopropanol (IPA), TEMPO and L-histidine (L-his), respectively. As shown in Fig. 4c, compared to the control group, the cell viability significantly increased by 41–57% for COF@CDs with the introduction of ROS scavengers, indicating that ROS were the dominant antibacterial factors and multiple reactive oxygen species cooperated in the photocatalytic disinfection system. The inset intuitively displays the E. coli colony growth. We then used p-nitrophenol, an electron scavenger, and p-methoxyphenol, a hole scavenger, in ROS-quenching experiments, to explore the role of electrons and holes generated by COF@CDs in ROS production. As shown in Fig. 4d, the fluorescence intensity of DCF was greatly reduced in the presence of p-nitrophenol, while there was a weak enhancement in the presence of p-methoxyphenol. This result reveals that electrons play a leading role in ROS production.17
2.4 Optical and electronic properties for the photocatalytic antibacterial mechanism
The photocatalytic bactericidal system involves three major steps: (1) absorption of light to generate electron–hole pairs by the COF, (2) separation and transfer of electron–hole pairs by CDs, and (3) surface redox reaction for ROS on CDs. To gain insight into the improved photocatalytic bactericidal performance of the COF@CD photocatalyst, the electronic properties of the samples were systematically investigated using photophysical and electrochemical measurements.
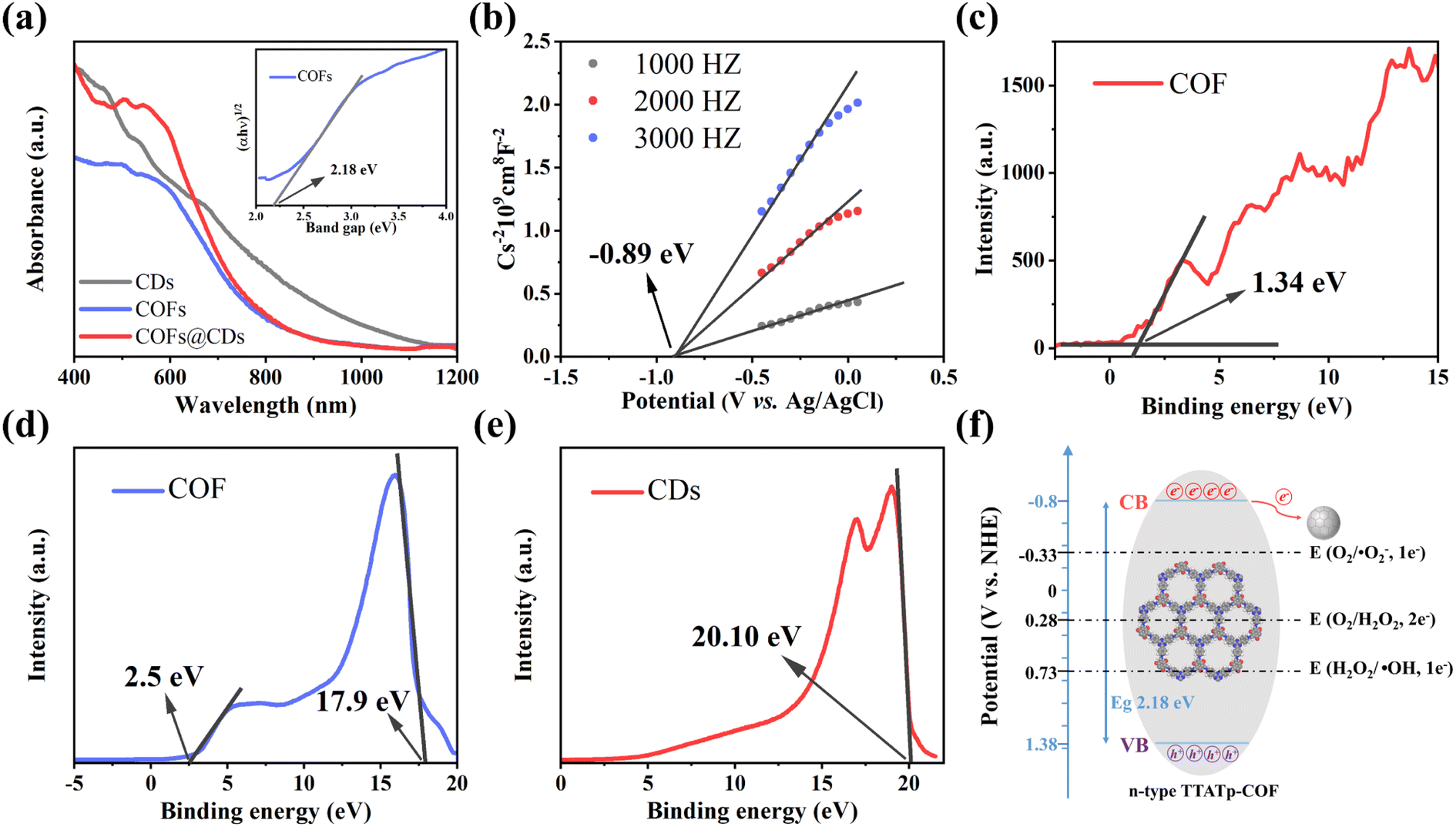
The UV-vis diffuse reflectance spectrum (DRS) measurements in Fig. 5a show the light absorption properties of the as-prepared samples. The COF exhibits broad optical absorbance, even covering the entire UV-vis region, resulting from the great enhancement of the delocalization of π-electrons within the 2D skeleton by the highly planar π-conjugated structure. CDs have a wider absorbance range, with strong absorbance extending into the near-infrared region. Compared to the pure COF, the absorption edge of the COF@CD composite did not shift noticeably, suggesting that the CDs were not incorporated into the COF lattice but were loaded on the COF surface, which is consistent with the XRD results. The corresponding optical band gap of the COF was calculated using the Kubelka–Munk equation: α(hν) = A(hν − Eg)1/2 (where α, ν, h, A, and Eg represent the absorption coefficient, light frequency, Planck's constant, constant value, and band gap energy, respectively).32 As shown in the inset of Fig. 5a, the optical band gap of the COF calculated from the Tauc plot was approximately 2.18 eV. According to the Mott–Schottky plots (Fig. 5b) measured at the frequencies of 1000, 2000, and 3000 Hz, the positive slope of the linear plots indicate that the TTATp-COF is an n-type semiconductor, and the derived flat band (FB) potential of the COF is approximately −0.89 eV vs. Ag/AgCl.33 For an n-type semiconductor, the bottom of the conduction band (CB) potential is generally 0.2 eV, which is more negative than the FB potential, and hence, the CB potential of COF nanosheets is −1.09 eV vs. Ag/AgCl, that is −0.89 eV vs. NHE, according to the equation, ENHE = EAg/AgCl + EAg/AgClθ; EAg/AgClθ = 0.1976 eV. Considering the Eg value of 2.18 eV, estimated from the UV-vis DRS, the valence band (VB) should be 1.38 eV. By calculating the results of VB-XPS in Fig. 5c, it can be directly obtained that the VB position of the TTATP-COF is located at 1.34 eV, which is almost the same as the above results.
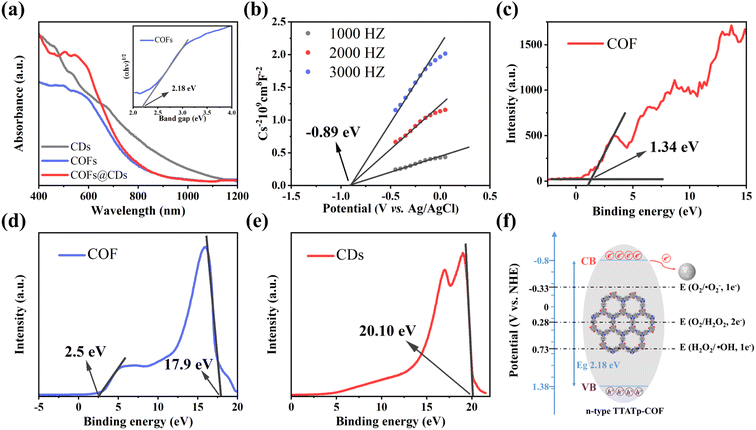 |
| | Fig. 5 (a) UV-vis diffuse-reflectance spectrum of CDs, COF, and COF@CDs. Inset: the Tauc-plot of the COF. (b) Mott–Schottky plot of the COF. (c) XPS valence band spectra of the COF. UPS spectra of (d) the COF and (e) CDs. (f) Band structure diagram of the COF. | |
To better understand the improved interface charge transfer efficiency, the band alignment of the COF and CDs was analyzed using ultraviolet photoelectron spectroscopy (UPS) measurements. As shown in Fig. 5d and e, the UPS results show that the secondary electron cutoff values of the COF and CDs are 17.9 and 20.10 eV; by subtracting the He I excitation energy (21.2 eV), the work functions are calculated to be 3.3 and 1.1 eV, respectively. Because of the smaller work function of the COF, the photogenerated electrons on the COF nanosheet are spontaneously transferred to CDs, driven by thermodynamics. An electron transfer pathway from the COF to CDs was built.34
Based on the above measurement results, we calculated the band distribution alignment of COF@CDs and proposed a possible production process of reactive oxygen species in APCT, as shown in Fig. 5f. When excited with photon energies exceeding the bandgap, the photogenerated electrons on the surface of the COF make transitions and are captured by CDs, leading to effective separation of electron–hole pairs. Since the CB potential of the COF (−0.8 eV vs. NHE) was more negative than the redox potential of O2/˙O2− (−0.33 eV vs. NHE), the electrons (e−) of the CB could reduce O2 adsorbed on the surface of COF@CDs to ˙O2−, which could be further oxidized by holes (h+) leaving the VB of the COF to 1O2.35 Furthermore, the COF has sufficient reduction ability to reduce O2 to H2O2 under irradiation (O2/H2O2 = +0.28 eV vs. NHE).36,37 In addition, the VB potential of the COF (+1.38 eV vs. NHE) was positive compared to that of the oxidation potential of OH−/˙OH (+1.99 eV vs. NHE), and it was thermodynamically forbidden for h+ to oxidize H2O to ˙OH.38 However, ROS detection measurements have demonstrated that the COF and COF@CDs can produce ˙OH under visible light irradiation, which has been attributed to the decomposition of H2O2 into ˙OH (+0.73 eV vs. NHE). The photoinduced electrons on the CB of the COF are transferred to CDs, which serves as an electron conduction and storage medium, efficiently decelerating the recombination of electron–hole pairs and prolonging the lifetime of charge carriers, resulting in increased ROS production, as shown in the following formulae eqn (1)–(5).
| | | COF@CDs + hv → e− + h+ | (1) |
| | | ˙O2− + e− + 2H+ → H2O2 | (4) |
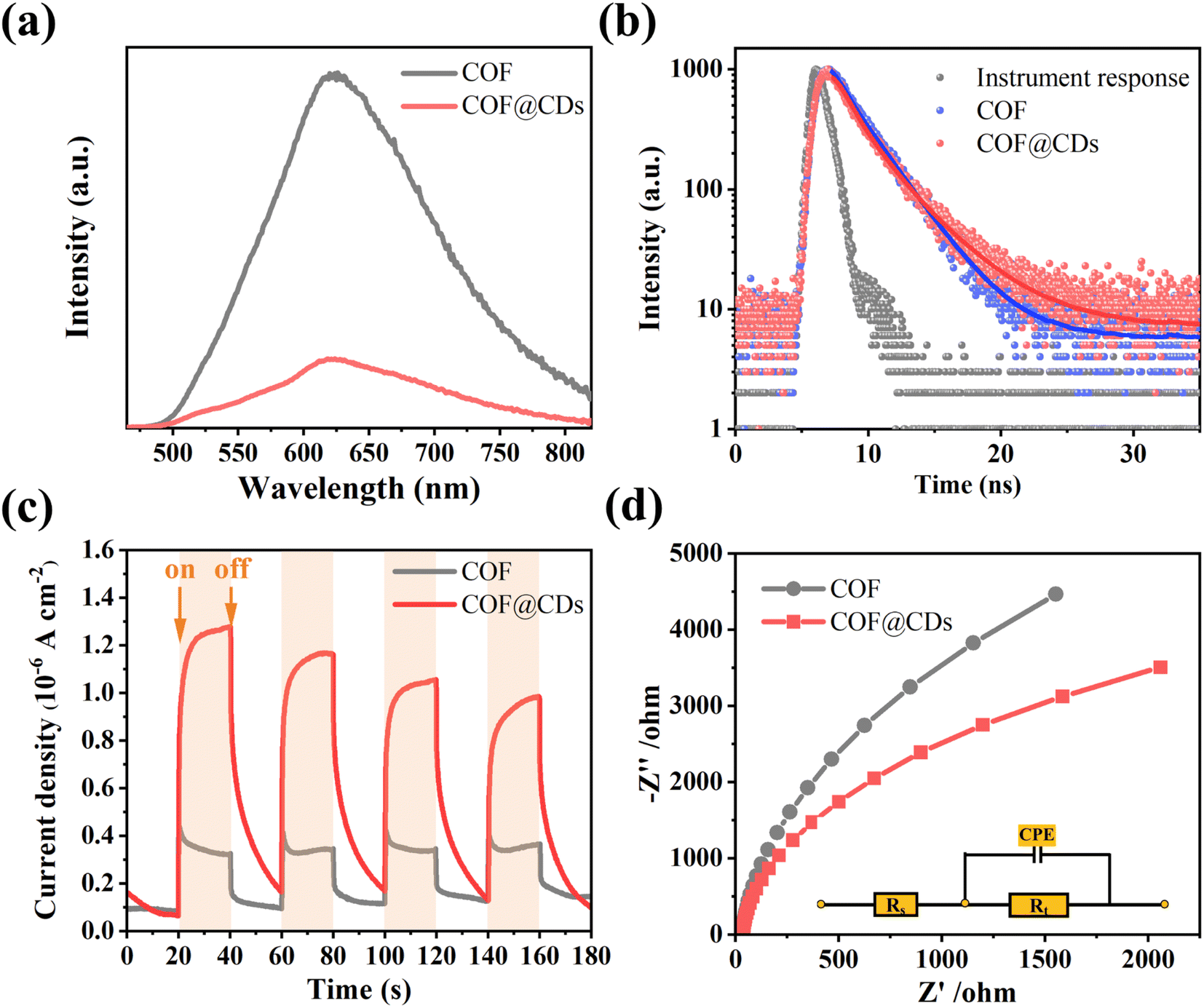
The charge carrier dynamics are also reflected by steady-state photoluminescence (PL) spectra and time-correlated single-photon counting (TCSPC) measurements. As shown in Fig. 6a, the COF exhibited characteristic emission peaks at 623 nm. When CDs were immobilized on the COF, significant quenching of the emission was observed. There is a charge transfer channel between the COF and CDs, which can facilitate photoinduced electron transfer from the excited state of the COF to CDs, and the charge density is redistributed, leading to effective suppression of the photoexcited carrier recombination of the COF. We further analyzed the inactivation of the stimulated COF by monitoring the emission decay. As shown in Fig. 6b, COF@CDs exhibited longer excited-state lifetimes (2.57 ns) than the COF (2.34 ns). CDs can efficiently accept electrons from the COF and retard carrier recombination.39 It is worth noting that the energetic π-electrons trapped in the COF skeleton can smoothly traverse to CDs, consequently achieving more efficient electron–hole pair migration and a larger photoexcitation rate. To further confirm that the CDs had excellent electron transfer capability, transient photocurrent response and electrochemical impedance spectroscopy (EIS) of different samples were investigated. As shown in Fig. 6c, the strength of the transient photocurrent response intensity for several on–off cycles of COF@CDs was much higher than that of the pure COF, which was beneficial to the redox reaction for ROS production.40 Similarly, from the EIS in Fig. 6d, higher electronic conductivity can be seen based on the smaller Nyquist plot radius for COF@CDs than for the COF during charge transfer processes, implying a decrease in charge transfer resistance.41 In Fig. S5,† the polarization curve shows the maximum overpotential of the pure COF. After loading CDs, the overpotential decreased, indicating that the CDs can accelerate the separation of photogenerated carriers.
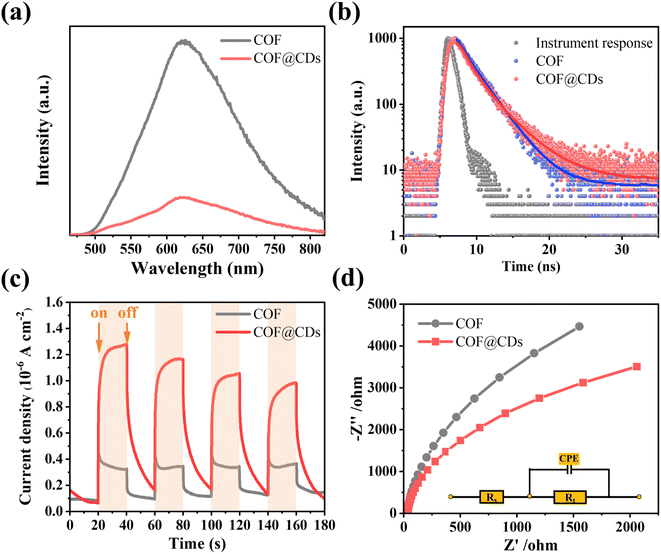 |
| | Fig. 6 (a) Photoluminescence spectra, (b) time-correlated single-photon counting measurements in DMF, (c) transient photocurrent response spectra, and (d) Nyquist plots from electrochemical impedance spectra of the COF and COF@CDs. | |
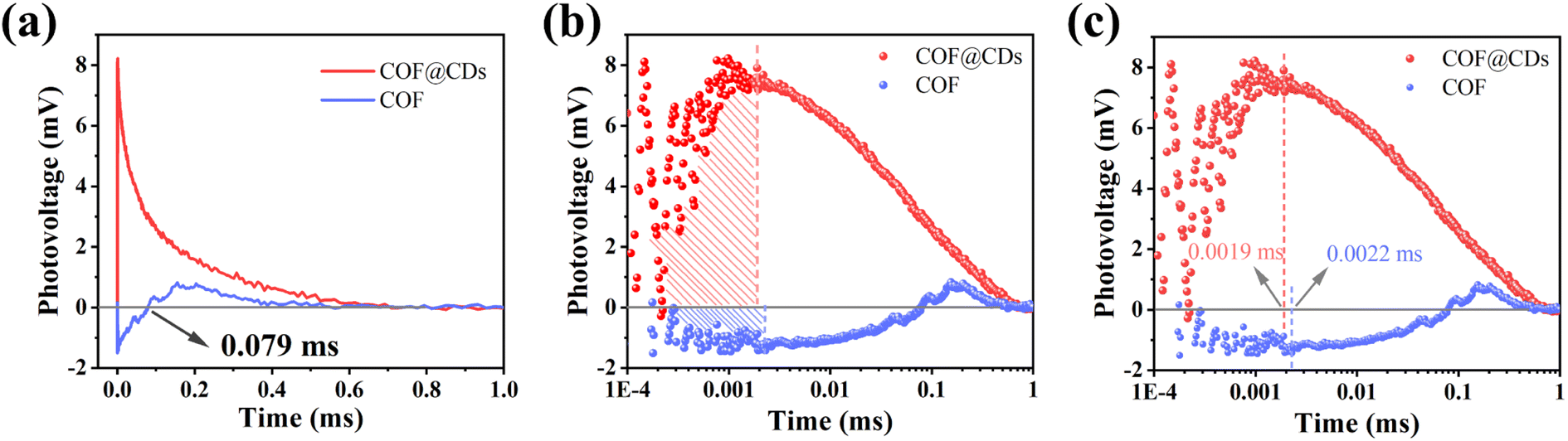
To study the dynamic information for delivery kinetics of photoinduced carriers, transient photovoltage (TPV) measurements were performed. As an important detection technique, TPV can directly reflect the photophysical process under working conditions, including the generation, separation, transport, accumulation, and recombination of photoinduced internal carriers. Fig. 7a shows the original TPV curves of the COF and COF@CDs. The transient photovoltage curve was positive for COF@CDs, but negative before 0.079 ms, and then became positive for the COF. A negative photovoltage response indicates electron accumulation on the surface, and a positive value suggests holes accumulation on the surface. It means that with irradiation, the electron–hole pairs were separated and then electrons escaped to the surface from the inside of the COF, showing a negative voltage. After 0.079 ms, the electrons were consumed by the reaction with oxygen, leaving the photogenerated holes that gradually accumulated at the interfaces of the COF, showing a positive voltage. The obvious difference for COF@CDs was that after loading CDs, a large number of electrons were rapidly extracted and stored by CDs, while the holes accumulated on the COF nanosheets, resulting in a positive signal on the TPV curve.42 As presented in Fig. 7b, the maximum photocarrier extraction efficiency is expressed by the shaded area. The value of COF@CDs is much larger than that of the COF, revealing that the CDs have an excellent charge storage capacity. The photocatalytic performance of charge extraction by CDs is superior to that of the COF.43–45 Here, we define an equation Tt/Tr = L to clarify the measurement of TPV,46 where, Tt is the time at which the photocarrier concentration reaches its maximum, Tr is the time required for complete recombination of the photoinduced carriers, and L is the ratio of 0 to 1. The smaller the L value, the faster the transfer and the longer the lifetime of the photogenerated carriers. The amplification diagram of Tt is shown in Fig. 7c, in which the Tt of COF@CDs is faster than that of the COF, illustrating the faster charge extraction rate of COF@CDs. The values of Tt, Tr and L for the COF and COF@CDs are listed in Table 1. The results indicated that COF@CDs had a smaller L value (0.0028) than the COF (0.004), suggesting a faster separation rate of electron–hole pairs and longer lifetime of charge carriers. CDs play an important role in efficiently extracting and storing electrons to accelerate electron transfer and prevent electron–hole recombination, thereby improving the process of APCT.
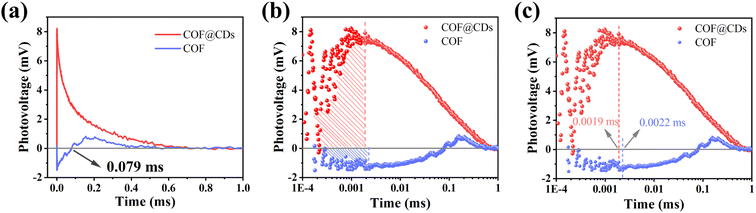 |
| | Fig. 7 (a) The transient photovoltage curves, the corresponding (b) amount of charge extraction and (c) maximum charge extraction time of the COF and COF@CDs. | |
Table 1 Parameters of materials measured by TPV
| Samples |
T
t (ms) |
T
r (ms) |
L
|
| COF |
0.0022 |
0.5495 |
0.004 |
| COF@CDs |
0.0019 |
0.6761 |
0.0028 |
2.5 Biosafety evaluation
To ensure the biosafety of COF@CD composites in biomedical applications, CCK-8 cytotoxicity tests were performed using human renal epithelial 293T cells as model cells. The CCK-8 results (Fig. S6†) showed that the survival rate of 293T cells increased with the addition of COF@CDs over a range of concentrations. When the concentration was increased to 200 μg mL−1, the survival rate of the cells decreased and the cell activity remained higher than that of the control group. COF@CDs did not elicit cytotoxicity toward 293T cells; in contrast, it has the effect of promoting cell proliferation. These results indicate that COF@CDs are non-toxic to humans and can be used in the treatment of bacterial infections, making them a promising antibacterial agent.
3 Conclusion
In summary, intramolecular charge transfer through CO–H–N hydrogen-bonding of the COF and CDs can greatly improve the separation and transfer of photogenerated charges confined to the organic π-orbitals of the COF. By elaborately regulating the loading content of CDs, the bactericidal rate against E. coli of the optimized COF@CD composite (4 wt% CDs) exhibited the highest value with 95% even at a low concentration of 50 μg mL−1 under visible light irradiation, the survival rate decreased by 8.3 times compared to that of the pure COF. Large amounts of ROS cause cell inactivation. Among them, ˙OH, ˙O2−, and 1O2 play a synergistic bactericidal role, while the photogenerated electrons are the key substances. TPV spectroscopy was performed to demonstrate that CDs, with excellent electron extraction and storage ability, accelerated the maximum electron storage time and greatly increased the electron extraction efficiency, which can significantly boost ROS production for antibacterial photocatalytic therapy of COF nanosheets. The COF@CD nanocomposite also exhibited high biosafety and low cytotoxicity against human renal epithelial 293T cells. This study provides a new strategy for the development of COFs and QD-based composites for efficient and safe photocatalytic disinfection.
Author contributions
Liang J designed and engineered the samples; Chen J and Huang X guided the characterization and optimization of the materials; Liang J and Li W performed the experiments; Shu W and Liu Y contributed to the theoretical analysis; Liang J wrote the paper with support from Zhang H, Lei B and Zhang X. All authors contributed to the general discussion.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The work was supported by the National Natural Science Foundations of China (No. 12274144), the Ministry of Science and Technology of China (No. G2021030022L), the Guangdong Provincial Special Fund for Modern Agriculture Industry Technology Innovation Teams (No. 2022KJ122 and 2023KJ122), the Independent Research and Development Projects of Maoming Laboratory (No. 2021ZZ004), the Key Realm R&D Program of Guangdong Province (No. 2021B0707010003), the Guangzhou Science & Technology Project (No. 202007020005 and 202103000059), the Guangdong Provincial Science and Technology Project (No. 2021A0505050006 and 2022A1515010229), and the Project of GDUPS (2018) for Prof. Bingfu LEI.
References
- K. E. Jones, N. G. Patel, M. A. Levy, A. Storeygard, D. Balk, J. L. Gittleman and P. Daszak, Nature, 2008, 451, 990–993 CrossRef CAS PubMed.
- C. Liu, D. Kong, P. Hsu, H. Yuan, H. Lee, Y. Liu, H. Wang, S. Wang, K. Yan and D. Lin, Nat. Nanotechnol., 2016, 11, 1098–1104 CrossRef CAS PubMed.
- J. Guo, Y. Qian, B. Sun, Z. Sun, Z. Chen, H. Mao, B. Wang and F. Yan, ACS Appl. Bio Mater., 2019, 2, 4418–4426 CrossRef CAS.
- J. Shi, M. Wang, Z. Sun, Y. Liu, J. Guo, H. Mao and F. Yan, Acta Biomater., 2019, 97, 247–259 CrossRef CAS PubMed.
- L. Wang, X. Zhang, X. Yu, F. Gao, Z. Shen, X. Zhang, S. Ge, J. Liu, Z. Gu and C. Chen, Adv. Mater., 2019, 31, 1901965 CrossRef PubMed.
- M. Lan, S. Zhao, X. Wei, K. Zhang, Z. Zhang, S. Wu, P. Wang and W. Zhang, Dyes Pigm., 2019, 170, 107574 CrossRef CAS.
- W. Wang, G. Li, D. Xia, T. An, H. Zhao and P. K. Wong, Environ. Sci.: Nano, 2017, 4, 782–799 RSC.
- Z. Zhang, L. Wang, W. Liu, Z. Yan, Y. Zhu, S. Zhou and S. Guan, Natl. Sci. Rev., 2021, 8, nwaa155 CrossRef CAS PubMed.
- Y. Wu, L. Zhang, Y. Zhou, Y. Li, Q. Liu, J. Hu and J. Yang, Chinese J. Catal., 2019, 40, 691–702 CrossRef CAS.
- X. Pan, W. Dong, J. Zhang, Z. Xie, W. Li, H. Zhang, X. Zhang, P. Chen, W. Zhou and B. Lei, ACS Appl. Mater. Interfaces, 2021, 13, 39446–39457 CrossRef CAS PubMed.
- A. Schlachter and P. D. Harvey, J. Mater. Chem. C, 2021, 9, 6648–6685 RSC.
- J. Podporska-Carroll, A. Myles, B. Quilty, D. E. McCormack, R. Fagan, S. J. Hinder, D. D. Dionysiou and S. C. Pillai, J. Hazard. Mater., 2017, 324, 39–47 CrossRef CAS.
- B. Leng, X. Zhang, S. Chen, J. Li, Z. Sun, Z. Ma, W. Yang, B. Zhang, K. Yang and S. Guo, J. Mater. Sci. Technol., 2021, 94, 67–76 CrossRef CAS.
- J. Guo, J. Zhou, Z. Sun, M. Wang, X. Zou, H. Mao and F. Yan, Acta Biomater., 2022, 146, 370–384 CrossRef CAS.
- P. Chanhom, N. Charoenlap, C. Manipuntee and N. Insin, J. Magn. Magn. Mater., 2019, 475, 602–610 CrossRef CAS.
- S. Xu, W. Lu, S. Chen, Z. Xu, T. Xu, V. K. Sharma and W. Chen, Chem. Eng. J., 2019, 375, 121949 CrossRef CAS.
- L. Zhang, S. Wang, Y. Zhou, C. Wang, X. Zhang and H. Deng, Angew. Chem., Int. Ed., 2019, 58, 14213–14218 CrossRef CAS PubMed.
- G. Lin, H. Ding, R. Chen, Z. Peng, B. Wang and C. Wang, J. Am. Chem. Soc., 2017, 139, 8705–8709 CrossRef CAS PubMed.
- T. Banerjee, F. Podjaski, J. Kröger, B. P. Biswal and B. V. Lotsch, Nat. Rev. Mater., 2021, 6, 168–190 CrossRef CAS.
- H. Dong, X. Meng, X. Zhang, H. Tang, J. Liu, J. Wang, J. Wei, F. Zhang, L. Bai and X. Sun, Chem. Eng. J., 2020, 379, 122342 CrossRef CAS.
- M. Gao, C. Li, H. Tang, X. Sun, H. Dong and F. Zhang, J. Mater. Chem. A, 2019, 7, 20193–20200 RSC.
- J. Ming, A. Liu, J. Zhao, P. Zhang, H. Huang, H. Lin, Z. Xu, X. Zhang, X. Wang and J. Hofkens, Angew. Chem., Int. Ed., 2019, 131, 18458–18462 CrossRef.
- P. B. Pati, G. Damas, L. Tian, D. L. Fernandes, L. Zhang, I. B. Pehlivan, T. Edvinsson, C. M. Araujo and H. Tian, Energy Environ. Sci., 2017, 10, 1372–1376 RSC.
- H. L. Wu, X. B. Li, C. H. Tung and L. Z. Wu, Adv. Mater., 2019, 31, 1900709 CrossRef.
- Q. Chen, Y. Liu, X. Gu, D. Li, D. Zhang, D. Zhang, H. Huang, B. Mao, Z. Kang and W. Shi, Appl. Catal., B, 2022, 301, 120755 CrossRef CAS.
- S. Kandambeth, A. Mallick, B. Lukose, M. V. Mane, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2012, 134, 19524–19527 CrossRef CAS PubMed.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed.
- G. Das, T. Prakasam, M. A. Addicoat, S. K. Sharma, F. Ravaux, R. Mathew, M. Baias, R. Jagannathan, M. A. Olson and A. Trabolsi, J. Am. Chem. Soc., 2019, 141, 19078–19087 CrossRef CAS.
- Q. Xin, H. Shah, A. Nawaz, W. Xie, M. Z. Akram, A. Batool, L. Tian, S. U. Jan, R. Boddula and B. Guo, Adv. Mater., 2019, 31, 1804838 CrossRef CAS PubMed.
- J. Liang, W. Li, J. Chen, X. Huang, Y. Liu, X. Zhang, W. Shu, B. Lei and H. Zhang, ACS Appl. Bio Mater., 2021, 4, 6937–6945 CrossRef CAS PubMed.
- J. Hynek, J. Zelenka, J. i. Rathouský, P. Kubát, T. s. Ruml, J. Demel and K. Lang, ACS Appl. Mater. Interfaces, 2018, 10, 8527–8535 CrossRef CAS PubMed.
- B. Ran, L. Ran, J. Hou and X. Peng, Small, 2022, 2107333 CrossRef CAS PubMed.
- Y. Zhang, X. Xu, Q. Liao, Q. Wang, Q. Han, P. Chen and K. Xi, J. Mater. Chem. B, 2022, 10, 3285–3292 RSC.
- J. Bai, R. Shen, K. Zhou, Z. Jiang, P. Zhang and X. Li, Chinese J. Catal., 2022, 43, 359–369 CrossRef CAS.
- H. Zhan, Q. Zhou, M. Li, R. Zhou, Y. Mao and P. Wang, Appl. Catal., B, 2022, 310, 121329 CrossRef CAS.
- J. Xiong, X. Li, J. Huang, X. Gao, Z. Chen, J. Liu, H. Li, B. Kang, W. Yao and Y. Zhu, Appl. Catal., B, 2020, 266, 118602 CrossRef CAS.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS PubMed.
- D. Xia, W. Wang, R. Yin, Z. Jiang, T. An, G. Li, H. Zhao and P. K. Wong, Appl. Catal., B, 2017, 214, 23–33 CrossRef CAS.
- X. Meng, J. Sheng, H. Tang, X. Sun, H. Dong and F. Zhang, Appl. Catal., B, 2019, 244, 340–346 CrossRef CAS.
- R. Miao, H. Liu, Q. Lei, L. Zhong, L. Zhang, J. He, Z. Ma and Y. Yao, Chem. Eng. J., 2022, 440, 135873 CrossRef CAS.
- J. Yang, H. Miao, J. Jing, Y. Zhu and W. Choi, Appl. Catal., B, 2021, 281, 119547 CrossRef CAS.
- Y. Han, J. Wu, Y. Li, X. Gu, T. He, Y. Zhao, H. Huang, Y. Liu and Z. Kang, Appl. Catal., B, 2022, 304, 120983 CrossRef CAS.
- H. Nie, Y. Liu, Y. Li, K. Wei, Z. Wu, H. Shi, H. Huang, Y. Liu, M. Shao and Z. Kang, Nano Res., 2022, 15, 1786–1795 CrossRef CAS.
- F. Li, Y. Liu, Q. Chen, X. Gu, W. Dong, D. Zhang, H. Huang, B. Mao, Z. Kang and W. Shi, Chem. Eng. J., 2022, 439, 135759 CrossRef CAS.
- Y. Liu, X. Wang, Y. Zhao, Q. Wu, H. Nie, H. Si, H. Huang, Y. Liu, M. Shao and Z. Kang, Nano Res., 2022, 1–8 Search PubMed.
- Y. Lei, L. Gu, W. He, Z. Jia, X. Yang, H. Jia and Z. Zheng, J. Mater. Chem. A, 2016, 4, 5474–5481 RSC.
- X. Ren, W. Liu, H. Zhou, J. Wei, C. Mu, Y. Wan, X. Yang, A. Nie, Z. Liu and X. Yang, Chem. Eng. J., 2022, 431, 134176 CrossRef CAS.
- L. Mao, B. Lu, J. Shi, Y. Zhang, X. Kang, Y. Chen, H. Jin and L. Guo, Catal. Today, 2022 DOI:10.1016/j.cattod.2022.03.035.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ab,
Xuejie
Zhang
ab,
Xuejie
Zhang