
Salt–solvent synchro-constructed robust electrolyte–electrode interphase for high-voltage lithium metal batteries†
Mingming
Fang‡
abc,
Juner
Chen‡
bc,
Boyang
Chen
abc and
Jianhui
Wang
 *abc
*abc
aSchool of Materials Science & Engineering, Zhejiang University, Hangzhou 310024, China
bKey Laboratory of 3D Micro/Nano Fabrication and Characterization of Zhejiang Province, School of Engineering, Westlake University, Hangzhou 310024, China. E-mail: wangjianhui@westlake.edu.cn
cInstitute of Advanced Technology, Westlake Institute for Advanced Study, Hangzhou 310024, China
First published on 16th May 2022
Abstract
Pairing a high-voltage nickel-rich cathode with a high-capacity lithium metal anode potentially realizes a battery energy density beyond 500 W h kg−1. However, there is lack of suitable electrolytes that are compatible with both the lithium metal anode and high-voltage cathode. Herein, we report an electrolyte design for high-voltage lithium metal batteries (LMBs) by using lithium difluoro(oxalato)borate salt (LiDFOB) and 1,2-ethylene sulfite solvent (ES) whose oxidation and reduction potentials are close to each other, respectively. This special salt–solvent combination induces a synergistic effect to synchro-construct a thin yet robust inorganic–organic interlocking protective film on both the cathode and anode, which is remarkably different from the conventional protective film that usually shows an inner inorganic–outer organic configuration. With a simple formula of “single salt single solvent” and a dilute salt concentration of 1 M (mol L−1), this electrolyte effectively alleviates various challenges faced by high-voltage LMBs, such as lithium dendrite growth, self-discharge, gas evolution and transition metal dissolution, and achieves the stable operation of an NCM622‖Li full cell (2.5 mA h cm−2, N/P = 4) at a high voltage up to 4.6 V. The capacity retention reaches ∼90% in 200 cycles with an average coulombic efficiency of 99.5%. In addition, this electrolyte demonstrates high ionic conductivity and stability in a wide temperature range, enabling high-voltage LMBs to charge and discharge at temperatures from −30 to 60 °C.
 Jianhui Wang | Jianhui Wang received his B.E. degree from Zhejiang University in 2006. He took a joint PhD training among Zhejiang University, National University of Singapore and Dalian Institute of Chemical Physics, and obtained his PhD degree in 2011. Then after one year post doc at Kyushu University, he joined the University of Tokyo in 2013 and served successively as a Project Researcher, JSPS Fellow and Chief Researcher. Since September 2018, he has been the Principal Investigator of the Energy Storage & Conversion Laboratory at Westlake University, aiming at the development of next-generation batteries and fuel cells. |
1 Introduction
Lithium-ion batteries have achieved great commercial success over the past 30 years. However, their energy density is approaching the theoretical limit (∼300 W h kg−1), yet failing to meet the increasing demands of energy density in electric vehicles, robots, and various portable electronic devices. Therefore, it is highly desirable to develop high-energy-density batteries beyond the conventional lithium-ion batteries.1–3 The increase of the energy density of a battery requires the increase of its operating voltage and/or capacity. Take a Ni-rich cathode, like LiNi0.6Mn0.2Co0.2O2 (NCM622), as an example, when the charging cut-off potential is raised from 4.3 to 4.6 V (vs. Li+/Li), it increases not only the operating voltage but also the specific capacity (from 170 to ∼210 mA h g−1), leading to an increase of energy density by ∼25%.4–6 As for the anode side, the substitution of graphite (372 mA h g−1, 0.1 V (vs. Li+/Li)) with lithium metal (3862 mA h g−1, 0 V (vs. Li+/Li)) will also be beneficial for both the operating voltage and capacity, further enhancing the overall energy density of the battery.7–9 Eventually, the combination of the high-voltage NCM622 cathode and the high-capacity lithium metal anode could lead to a high-voltage lithium metal battery (LMB) with an energy density >500 W h kg−1.10,11However, this high-energy-density LMB encounters severe interphase issues on both the cathode and anode (see Fig. 1a). First, when the potential rises above >4.5 V (vs. Li+/Li), the conventional electrolyte becomes unstable: the LiPF6 salt is prone to producing more HF, which will corrode the cathode material and accelerate the dissolution of transition metals;12,13 the carbonate solvent also undergoes an undesirable oxidative decomposition, causing gas evolution, thickened electrode–electrolyte interphases as well as impedance growth.14–16 Second, lithium metal is confronted with intrinsic problems of infinite volume changes and dendrite growth during lithium deposition/stripping, which inevitably damages the solid electrolyte interface (SEI), causing continuous consumption of the electrolyte and active lithium.17–20 The above issues dramatically deteriorate the battery performance and even bring severe safety hazards. Obviously, all of the aforementioned problems are closely related to the electrolyte. Therefore, it is urgent to develop new electrolytes to improve the interfacial stability between the electrolyte and the high-voltage cathode and lithium metal anode.
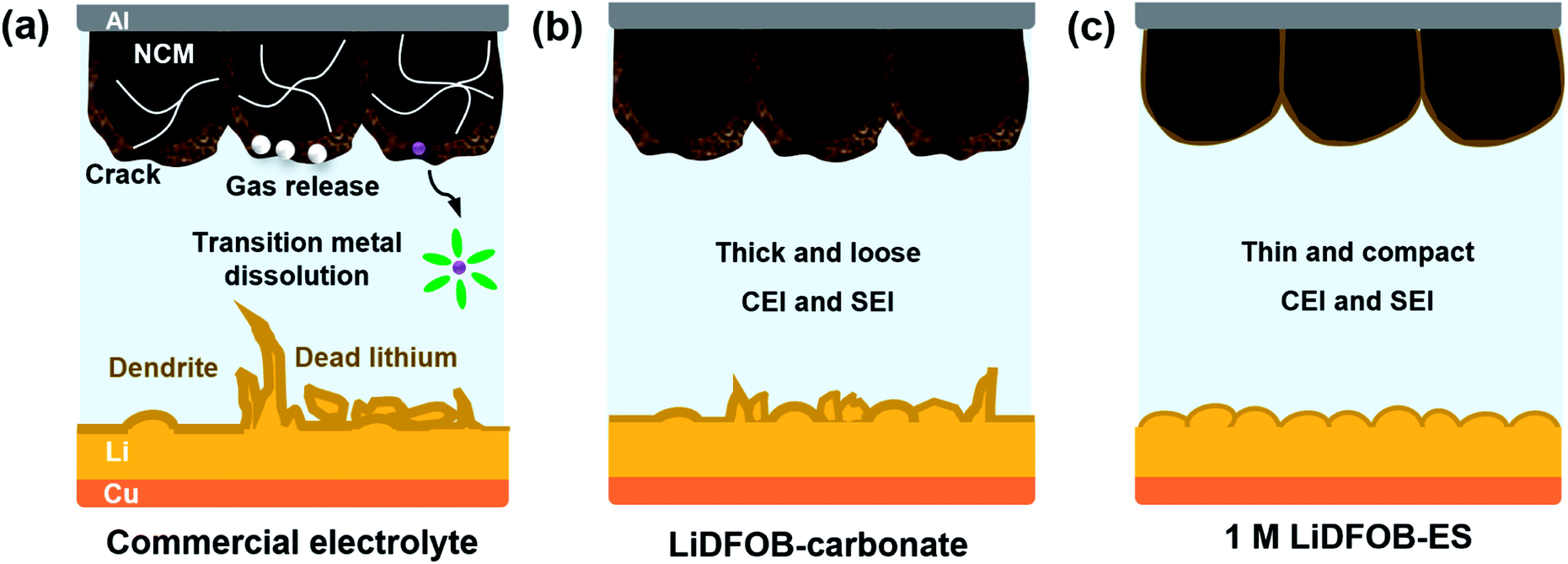 | ||
| Fig. 1 Electrolyte design strategy for high-voltage LMBs. (a) The commercial electrolytes of LiPF6–carbonate solvents usually suffer from various challenges with regard to both the anode and cathode: repeated rupture and formation of the SEI caused continuous consumption of the limited electrolyte and active lithium resources for the lithium metal anode, oxidative decomposition of the electrolyte which induced gas evolution, particle cracking, and transition metal dissolution for the high-voltage cathode. (b) The introduction of LiDFOB into the electrolyte can alleviate some issues to some degree, but the problem of continuous consumption of LiDFOB remains, which causes thickening of the SEI and CEI, and thus, a decline of battery performance. (c) We propose a new electrolyte design by selecting the salt and solvent with close redox potentials. This salt–solvent combination can induce a synergistic effect to synchro-construct an ultrathin and robust SEI/CEI, thus contributing to a stable operation of high-voltage LMBs. | ||
In recent years, salt-concentrated electrolytes,21–23 localized salt-concentrated electrolytes,24–26 all-fluorinated electrolytes,27,28 and sulfonamide-based electrolytes29 have been developed to remarkably improve the cycling performance of high voltage LMBs. Despite the improved performances, the salt-concentrated electrolytes suffer from high viscosity and cost, which impede their practical applications. Introducing an inert diluent such as hydrofluoroether to form localized salt-concentrated electrolytes can address the viscosity issue, but the hydrofluoroether solvents are more expensive than lithium salts, thus having no benefit of cost reduction.30 Most recently, the sulfonamide-based electrolyte was reported to enable the stable operation of an NCM811‖Li battery up to 4.7 V for 100 cycles.29 However, this electrolyte has a low ionic conductivity of 1.37 mS cm−2 at room temperature, posing significant difficulty for low-temperature application. Accordingly, there is still no electrolyte that meets all the requirements for the practical application of high-voltage LMBs—high stability, wide-temperature applicability, and low cost.
In this work, we report a new electrolyte design that can overcome various challenges faced by LMBs under a high voltage and wide-temperature range while being cost-effective. We selected lithium difluoro(oxalato)borate (LiDFOB) as the salt because it can produce a protective film on both the cathode and anode surfaces; meanwhile, it passivates the aluminum collector and protects it from corrosion at high voltages without using high salt concentrations.31,32 And we selected 1,2-ethylene sulfite (ES) as the solvent owing to its reduction and oxidation potentials that are close to those of LiDFOB, respectively. This salt–solvent combination induces a synergistic effect to synchro-construct a thin yet robust protective film on both the cathode and anode, surmounting the long-lasting issue of continuous consumption of DFOB− in a conventional electrolyte such as LiDFOB–carbonate solvent (see Fig. 1b and c).33–36 Consequently, with the simple formula of “single salt single solvent” and a dilute concentration of 1 M, this electrolyte achieves the stable operation of NCM622‖Li full cells under a high cut-off voltage up to 4.6 V and a wide-temperature range from −30 to 60 °C.
2 Results and discussion
2.1 New electrolyte design for high-voltage LMBs
A robust electrode–electrolyte interphase is critical to realizing a high-voltage LMB. Owing to the good film-forming ability on both the cathode and anode, LiDFOB is widely used to passivate various electrodes, such as LiNixMnyCozO2, LiCoO2, and LiNi0.5Mn1.5O4 cathodes and graphite and lithium metal anodes.37–39 However, when applied in high-voltage LMBs with the voltage higher than 4.5 V, it still encounters difficulties of poor cycle life,36,40 low coulombic efficiency (CE),41,42 and unstable rate performance,43 which are generally ascribed to the continuous decomposition of the DFOB− anion that thickens the passivation layer and increases the impedance.33–36 From Cyclic Voltammetry (CV) curves of the Cu electrode in the electrolyte of LiDFOB–carbonate solvent (ethylene carbonate (EC) and propylene carbonate (PC)) (see Fig. 2a), a typical reduction decomposition at around 1.7 V (vs. Li+/Li) of DFOB− can be observed, which contributes to an SEI mainly composed of inorganic components such as LixBOy, LiF and Li2CO3.38,39 However, this film seems not very compact as a significant reduction current (∼0.02 mA cm−2) remained when the potential swept to lower potentials. At ∼0.8 V (vs. Li+/Li), the reduction of EC occurred, which contributes to an additional organic SEI layer on the LiDFOB-derived inorganic SEI. However, significant reduction currents of ∼0.03 mA cm−2 still existed at 0.4–0.8 V (vs. Li+/Li), indicating that this typical inner inorganic–outer organic SEI is still not compact or robust enough to inhibit the reduction decomposition of the electrolyte at low potentials.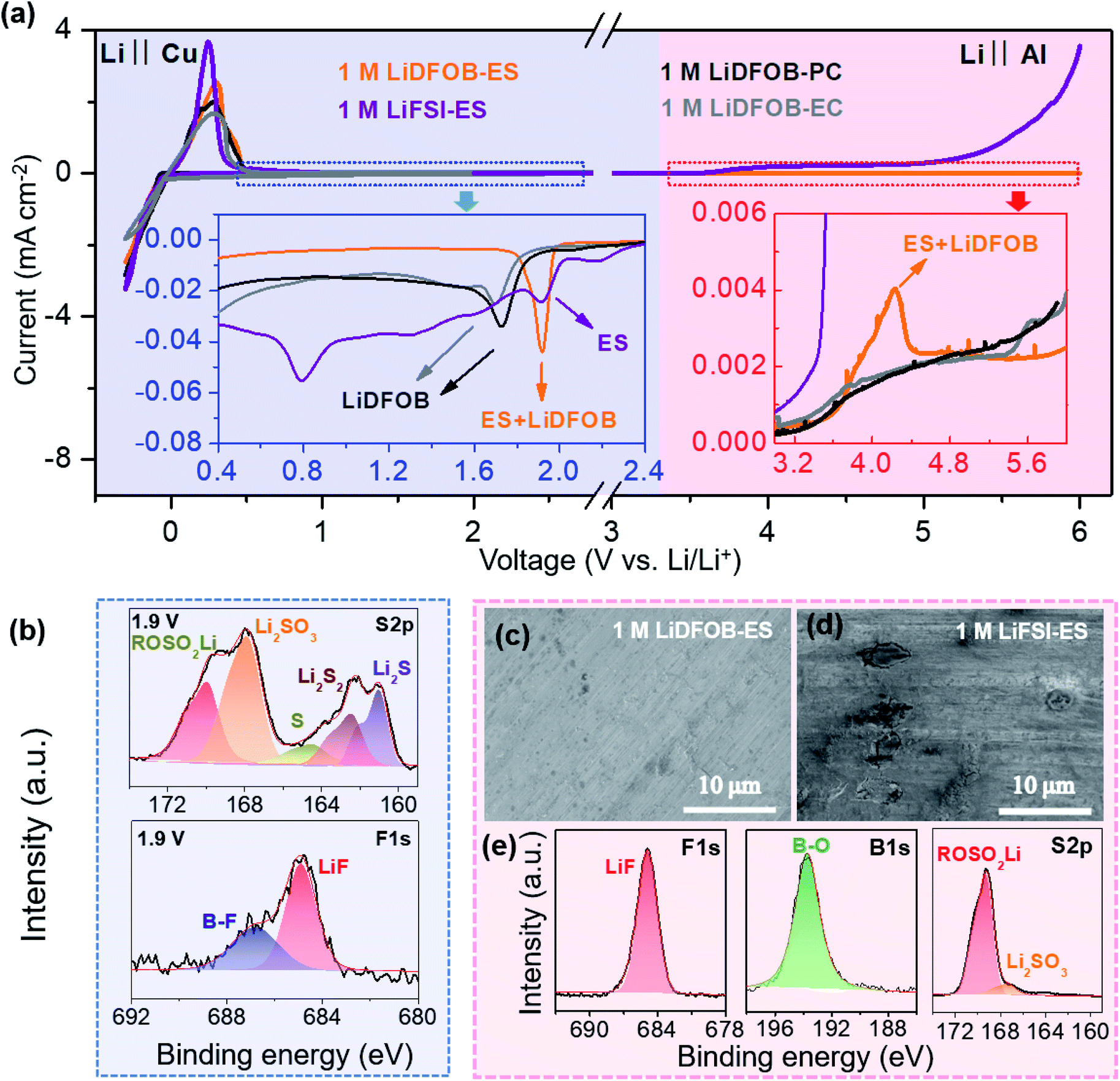 | ||
| Fig. 2 Salt–solvent synchro-constructed electrode–electrolyte interphase. (a) CV of a Cu electrode (light blue area) and LSV of an Al electrode (light red area) in various electrolytes. The scanning rate is 0.2 mV s−1. The insets show the enlarged view regions. (b) XPS spectra of the Cu electrode after a CP test at 1.9 V (vs. Li+/Li) in 1 M LiDFOB–ES electrolyte for 2 hours. (c, d) SEM images of the Al surface after the LSV test in 1 M LiDFOB–ES and 1 M LiFSI–ES electrolytes. (e) XPS spectra of the Al electrode after a CP test at 5 V (vs. Li+/Li) in 1 M LiDFOB–ES electrolyte for 6 hours. The close redox potentials of the LiDFOB salt and the ES solvent induce a peculiar salt–solvent synergistic effect to synchro-construct the SEI/CEI. | ||
To construct a better SEI, ES was selected as the solvent due to its reduction potential in the electrolyte (∼1.9 V vs. Li+/Li) being close to that of LiDFOB,44,45 which may alleviate the continuous reduction of the electrolyte at low potentials. As shown in Fig. 2a, the electrolyte 1 M LiDFOB–ES only exhibits a single sharp reduction peak at ∼1.9 V in the CV curve, showing some interaction between the ES and LiDFOB that induces the reduction of LiDFOB at a higher potential. We collected the Cu electrode after a chronoamperometry (CP) test at 1.9 V (vs. Li+/Li) for X-ray photoelectron spectroscopy (XPS) analysis. The results shown in Fig. 2b indicate that the reduction products contain both LiDFOB-derived components (LiF, LixBOy and Li2CO3) and ES-derived components (S, Li2Sx, LixSOy, ROSO2Li),46,47 revealing that both LiDFOB and ES have participated in the SEI formation. Notably, once the SEI formed, the reduction current of Cu in the 1 M LiDFOB–ES electrolyte sharply dropped to 7.1 μA, indicating that the SEI had effectively suppressed the reduction of the electrolyte at low potentials, considerably distinguishing its behavior from that in the LiDFOB–carbonate electrolytes. We also prepared other ES-based electrolytes using different salts, such as LiPF6, LiBF4 and LiFSI. We found that only the combination of LiDFOB and ES showed a single reduction peak and the smallest leakage current, revealing the importance of selecting an appropriate salt–solvent couple with a close reduction potential (see Fig. S1†).
On the other hand, in a high-voltage battery, the issues of anodic dissolution of Al collectors and oxidative decomposition of the electrolyte must also be overcome. We carried out linear sweep voltammetry (LSV) measurements of the Al electrode in different electrolytes (see Fig. 2a) and observed the polarized Al electrode using scanning electron microscopy (SEM) (see Fig. 2c and d). Obviously, all the LiDFOB-based electrolytes could effectively inhibit the corrosion dissolution of the Al collector at high voltages, benefiting from a passivation film on the Al surface resulting from the DFOB− at potentials of >4 V (vs. Li+/Li).31,32 Comparing the LSV curves of the LiFSI–ES electrolyte with the LiDFOB–carbonate solvent electrolytes, the former showed a stronger oxidation peak at >3.8 V (vs. Li+/Li), which should result from an extra contribution of ES oxidation. Indeed, from the XPS results of the polarized Al electrode after a CP test (Fig. S2† and 2e), both the LiDFOB-derived and ES-derived components can also be clearly observed, indicating that both LiDFOB and ES have synchro-constructed the protective film at high voltages as well. Moreover, among all the studied electrolytes, the oxidation current using 1 M LiDFOB–ES electrolyte was the smallest at >5.2 V (vs. Li+/Li), indicating that the LiDFOB–ES combination provided the most effective protection at high voltages. Accordingly, it is evident that the LiDFOB salt and ES solvent can synchro-construct robust electrolyte–electrode interphases on both the cathode and anode, facilitating the application of our 1 M LiDFOB–ES electrolyte in high-voltage LMBs.
2.2 Improved performance of the lithium metal anode
To examine the compatibility of the electrolyte with the lithium metal anode, we assembled Li‖Cu and Li‖Li cells to evaluate the CEs and cycling durability of lithium deposition/stripping. Obviously, 1 M LiDFOB–ES electrolyte reached a CE of 99.1%, which was significantly higher than that of commercial (1 M LiPF6/EC:DMC (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by vol)) and LiDFOB–carbonate electrolytes (see Fig. 3a). For the commercial electrolyte, large amounts of lithium dendrites were formed on the anode as revealed by SEM and Cryo-EM (see Fig. S3a and b†), which lowered the CE. This is because the carbonate solvents react with lithium metal to form a loose and porous organic-rich SEI, resulting in uneven lithium deposition. Using LiDFOB with a higher reduction potential as a solute, the LiDFOB-derived SEI suppressed the growth of lithium dendrites to a certain extent (see Fig. S3†). However, the performances of LiDFOB-based electrolytes are also profoundly influenced by solvents. Compared with carbonate solvents such as EC and PC, the LiDFOB–ES electrolyte showed the most even morphology of lithium deposition together with the lowest nucleation overpotential and the highest CE (see Fig. 3a), suggesting that the presence of ES contributes to a better SEI.
:1 by vol)) and LiDFOB–carbonate electrolytes (see Fig. 3a). For the commercial electrolyte, large amounts of lithium dendrites were formed on the anode as revealed by SEM and Cryo-EM (see Fig. S3a and b†), which lowered the CE. This is because the carbonate solvents react with lithium metal to form a loose and porous organic-rich SEI, resulting in uneven lithium deposition. Using LiDFOB with a higher reduction potential as a solute, the LiDFOB-derived SEI suppressed the growth of lithium dendrites to a certain extent (see Fig. S3†). However, the performances of LiDFOB-based electrolytes are also profoundly influenced by solvents. Compared with carbonate solvents such as EC and PC, the LiDFOB–ES electrolyte showed the most even morphology of lithium deposition together with the lowest nucleation overpotential and the highest CE (see Fig. 3a), suggesting that the presence of ES contributes to a better SEI.
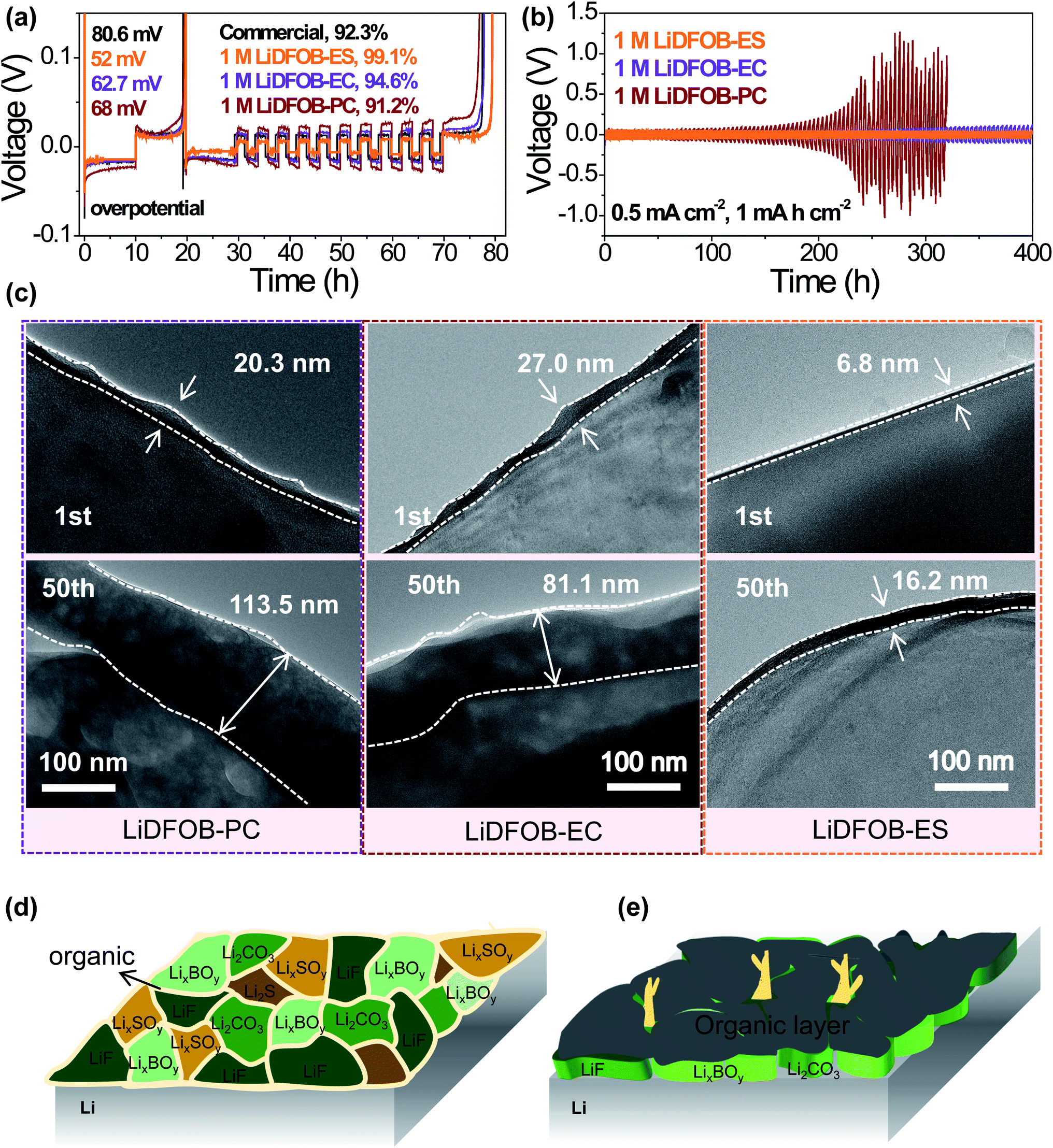 | ||
| Fig. 3 Electrochemical performance and SEI characterization of the Li-metal anode. (a) Li coulombic efficiency in a Li‖Cu cell using different electrolytes. (b) The cycling performance of a Li‖Li symmetric cell using different electrolytes. (c) Cryo-EM images of SEI after 1st and 50th Li plating in different electrolytes. (d, e) Schematic diagram of the SEI structure derived from 1 M LiDFOB–ES and 1 M LiDFOB–carbonate electrolytes. In 1 M LiDFOB–ES electrolyte, the salt–solvent synchro-constructed an ultrathin and robust inorganic–organic interlocking SEI, which is remarkably different from the conventional inner inorganic–outer organic SEI. The commercial electrolyte is 1 M LiPF6/EC:DMC (1:1 by vol). | ||
The long-term stability of the Li anode in various electrolytes can be found in Fig. 3b. During the initial 100 hours, all three electrolytes showed good cycle performance. However, the polarization voltage of cells using 1 M LiDFOB-PC sharply increased after 200 hours' operation, indicating an unstable SEI. As reported in the previous literature,48,49 PC makes little contribution to the formation of a good SEI. Thus, the SEI in the LiDFOB–PC electrolyte should be mainly from LiDFOB. However, this SEI is not stable as we could observe, via cryo-EM, a dramatic increase of the SEI thickness from 20.3 to 113.5 nm in 50 cycles (see Fig. 3c and S4†), which subsequently also caused the increase of interface impedance (see Fig. S5†). When using EC as a solvent, a much more smooth voltage curve of the Li‖Li cell was obtained, probably due to the contribution of EC to the SEI. However, the polarization voltage became larger after long cycling, which was consistent with the increased thickness of the SEI from 27.0 to 81.1 nm (see Fig. 3c). In sharp contrast, the cell using 1 M LiDFOB–ES electrolyte showed the smallest polarization in the long cycling even at a large current density of 5 mA cm−2 (see Fig. S6†), implying the formation of a robust and highly Li+-conductive SEI. Indeed, from Cryo-EM observation, this SEI was very thin (∼6.8 nm) and remained stable during the cycles (see Fig. 3b and c), thus benefiting a uniform lithium deposition (see Fig. S3 and S7†).
The chemical composition of the SEI formed on Li metal using 1 M LiDFOB–ES electrolyte was investigated by XPS (see Fig. S8†). The LiDFOB-derived inorganic components, such as LixBOy, LiF and Li2CO3, and the ES-derived inorganic/organic components, such as S, Li2Sx, LixSOy, ROSO2Li, and some polymers, can be clearly identified, indicating that both LiDFOB and ES simultaneously participate in the SEI formation, consistent with the results shown in Fig. 2b. The presence of both inorganic and organic components in the SEI together with the ultra-thin characteristic reflects a new inorganic–organic interlocking configuration of the SEI (see Fig. 3d), which is remarkably different from the conventional one with an inner inorganic–outer organic structure (see Fig. 3e). Therefore, it is the close reduction potentials of LiDFOB and ES that induce a synergistic effect to synchro-construct the SEI, which effectively suppresses the excessive reduction of LiDFOB and produces a thin yet robust SEI, thus contributing to an improved electrochemical performance of the Li metal anode.
2.3 Improved performance of the NCM622 cathode at 4.6 V
Besides stabilizing the lithium metal anode, an ideal electrolyte must have good compatibility with a high-voltage cathode to realize a high energy density LMB. For the NCM622 cathode, increasing the upper cut-off voltage theoretically increases the reversible capacity, but it is difficult to realize because the commercial electrolyte undergoes oxidative decomposition at high voltages, causing various issues, such as unstable CEI, gas evolution, transition metal dissolution and self-discharge. As shown in Fig. 4a and S9,† upon increasing the charging cut-off voltage from 4.3 to 4.7 V, the NCM622‖Li half-cell using the commercial electrolyte showed an increase of capacity by ∼16% (176–204 mA h g−1), but it also caused a dramatic increase of the cell impedance by one order, which suggests that the CEI is unstable at high voltages. By contrast, the capacity of the cell using 1 M LiDFOB–ES electrolyte increased by ∼24% (175–218 mA h g−1) with just a small increase of the cell impedance, indicating much higher stability at high voltages. Considering that the average output voltage of the battery also increased from 3.81 to 3.95 V, the energy density successfully increased by ∼33%.To confirm the enhanced stability of our designed electrolyte as compared to the commercial electrolyte, we systematically evaluated the cell at high voltages with regard to self-discharge, leakage current and gas evolution. Obviously, the 1 M LiDFOB–ES electrolyte showed remarkable improvements in all these aspects as can be found in Fig. 4b–d. For the charged cell using 1 M LiDFOB–ES electrolyte, more than 83% capacity was retained after 153 hours′ storage, whereas no capacity was left in 55 hours′ storage for that using the commercial electrolyte (see Fig. 4b). Under a harsher condition of constant voltage of 4.6 V, the leakage current for the cell using 1 M LiDFOB–ES electrolyte remained unchanged (11 μA) during the 36 hours test, which is only one eighth of that using the commercial electrolyte (see Fig. 4c). In addition, the oxidative gas product CO2 generally observed in the commercial electrolyte at high voltages was absent in the LiDFOB–ES electrolyte (see Fig. 4d).
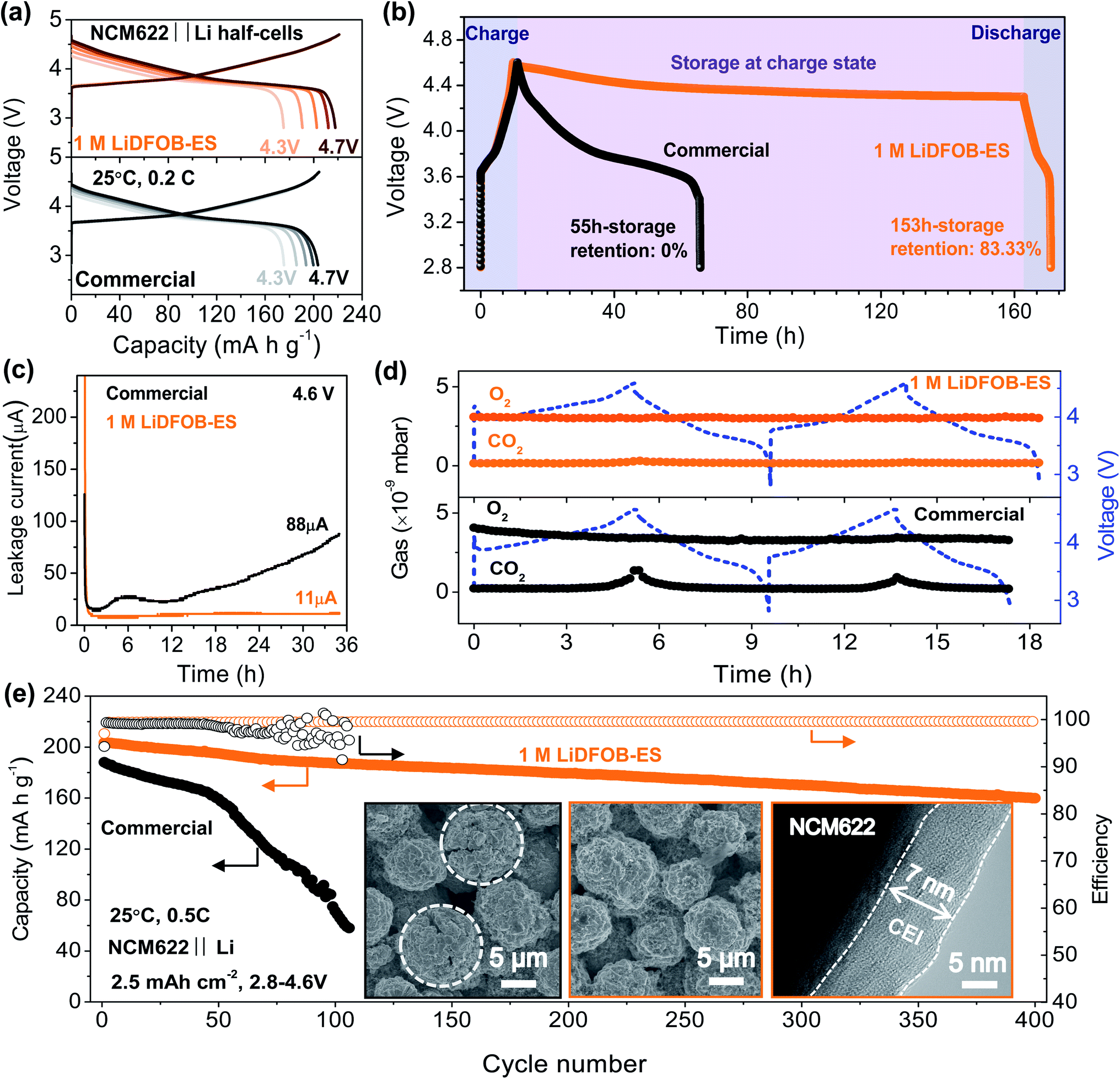 | ||
| Fig. 4 Electrochemical performance and CEI characterization of the NCM622 cathode at high voltages. (a) Charge–discharge curves of the NCM622‖Li half-cell at cut-off voltages from 4.3 to 4.7 V in the commercial and 1 M LiDFOB–ES electrolytes. (b) The self-discharge measurement after charging to 4.6 V. (c) Leakage currents dependent on time at a constant voltage of 4.6 V. (d) In situ DEMS analysis of the gas evolution during the first two charge–discharge cycles. (e) Cycling performance of the NCM622‖Li half-cell that had been pre-activated from 4.3 to 4.6 V as shown in (a). The insets show SEM and TEM images of the NCM622 particles after 400 cycles in the commercial (left) and 1 M LiDFOB–ES (middle and right) electrolytes. Charge and discharge were conducted at the same rate. The cutoff voltage for all cycles was 2.8–4.6 V. A 1C rate corresponds to 210 mA g−1 on the weight basis of the NCM622 active material. | ||
We then investigated the cycling performance of the cells. The cells using 1 M LiDFOB-PC, 1 M LiDFOB-EC and commercial electrolytes all displayed a rapid capacity decline after 50 cycles (see Fig. 4e and S10†). In contrast, the cell using 1 M LiDFOB–ES electrolyte delivered a high initial discharge capacity of 203.5 mA h g−1 at 0.5C and achieved a capacity retention of ∼80% over 400 cycles with a high average CE of 99.5% (Fig. 4e). Further we studied the structural change after the long-term cycling test. As shown in Fig. 4e, the NCM622 particles in the commercial electrolyte showed extensive cracking (Fig. 4e, left inset), whereas no cracks can be found on those in the 1 M LiDFOB–ES electrolyte (Fig. 4e, middle inset). The particle crack increases the electrochemical surface area between the electrolyte and electrode, which inevitably aggravates various side reactions, such as transition metal dissolution (see Fig. S11†). When observed by high-resolution transmission electron microscopy (HR-TEM), a thin and uniform CEI with a thickness of ∼7 nm was found on the NCM622 particles cycled in the LiDFOB–ES electrolyte (Fig. 4e, right inset), which is considerably thinner than the CEIs generated in the other electrolytes (>40 nm, see Fig. S12 and S13†). XPS results reveal that the CEI is mainly composed of LiF, LixBOy, LixSOy, and ROSO2Li (Fig. S14†), indicating that both LiDFOB and ES contribute to the formation of the CEI, consistent with the LSV results (see Fig. 2e). Based on the above results, we can conclude that the improved electrochemical performances of NCM622 in the 1 M LiDFOB–ES electrolyte can be ascribed to the formation of the thin and robust CEI, which prevents the electrolyte from continuous oxidation decomposition at high voltages and suppresses various subsequent deleterious reactions on the cathode, such as gas evolution, self-discharge, transition metal dissolution, and CEI thickening.
2.4 Performance of full cells over a wide temperature range
As the 1 M LiDFOB–ES electrolyte can stabilize both the high voltage cathode and lithium metal anode, we assembled a high-voltage full cell by pairing 50 μm Li foil with a high-loading NCM622 cathode (roughly 2.5 mA h cm−2, N/P = 4). At 25 °C, the NCM622‖Li full cell exhibited a capacity retention of 88.2% over 200 cycles with an average CE of 99.5% (see Fig. 5b and e). In contrast, the commercial electrolyte showed a rapid capacity decline after 80 cycles. Moreover, the battery using the 1 M LiDFOB–ES electrolyte also allows a fast charge–discharge rate (Fig. S15†), e.g., high capacities of 214.6 and 162.5 mA h g−1 at 0.1C and at 5C, respectively, substantiating the low impedance for Li+ transport in the battery.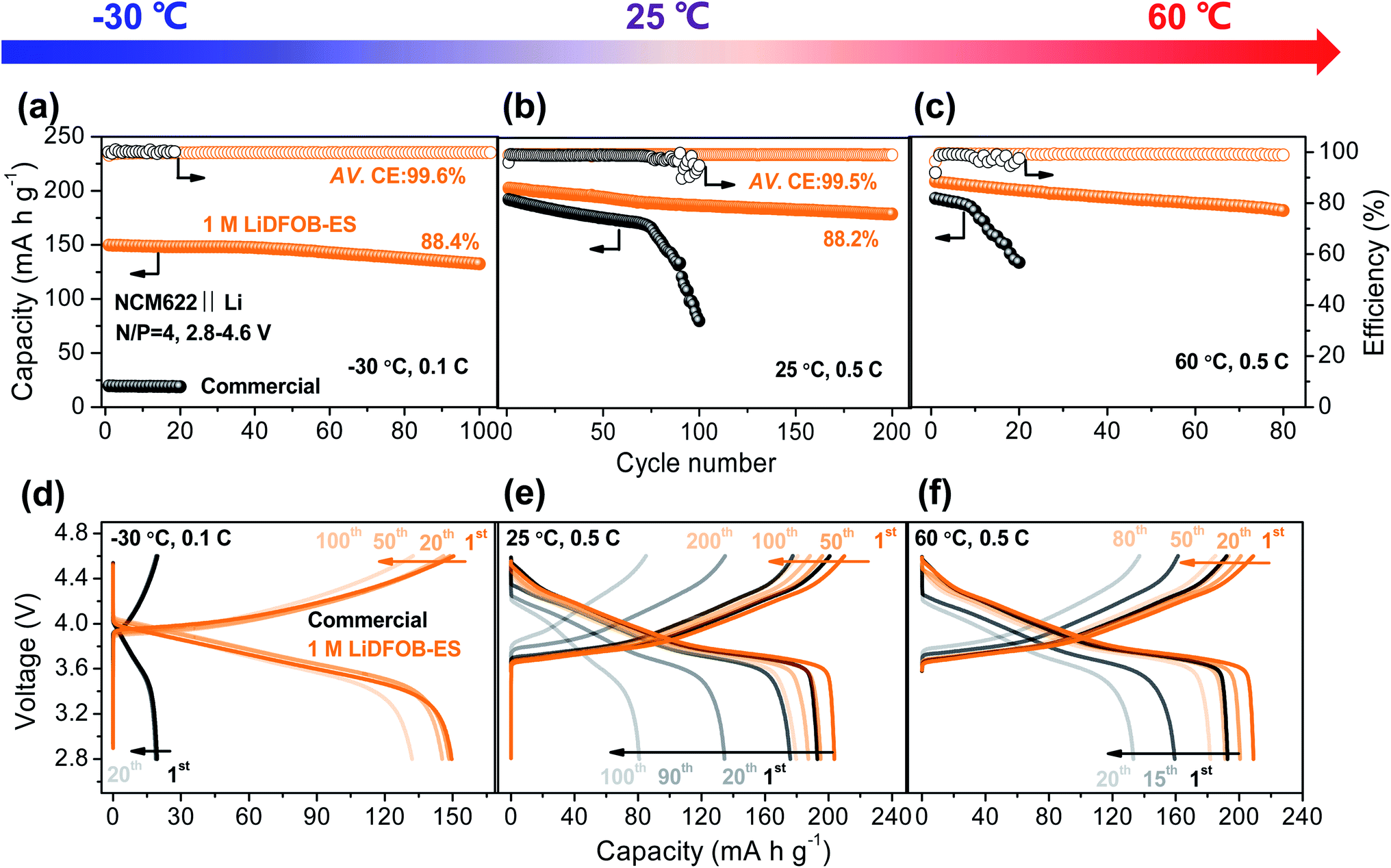 | ||
| Fig. 5 Electrochemical performances of the NMC622‖Li full-cells in a wide temperature range. (a–c) Cycling performance of the full cell in the commercial and 1 M LiDFOB–ES electrolytes at temperatures from −30 to 60 °C and their corresponding voltage profiles (d–f). Charge and discharge were conducted at the same rate. The cutoff voltage for all cycles was 2.8–4.6 V. A 1C rate corresponds to 210 mA g−1 on the weight basis of the NCM622 active material. The active material mass loading of NCM622 was approximately 11.8 mg cm−2. The negative-to-positive capacity ratio (N/P) is 4. | ||
It is worth noting that the 1 M LiDFOB–ES electrolyte has good ionic conductivity (0.44 mS cm−1 at −40 °C) and thermal stability (negligible evaporation at 100 °C) in a wide-temperature range, far superior to the commercial electrolyte (see Fig. S16 and Table S1†), showing the potential for a wide-temperature operation. As shown in Fig. 5a and d, even at a low temperature of −30 °C, the 1 M LiDFOB–ES electrolyte enabled the cell to deliver a high reversible capacity of 150 mA h g−1 with an ultrahigh CE of 99.6%, whereas only 20 mA h g−1 was obtained for the cell using the commercial electrolyte. Moreover, at a high temperature of 60 °C (see Fig. 5c and f), the cell using the 1 M LiDFOB–ES electrolyte also realized a capacity retention of 87.1% after 80 cycles with an average CE of 99.1%. In sharp contrast, the cell using the conventional commercial electrolyte failed in just a few cycles. Consequently, we confirm that our simple electrolyte formula of “single salt single solvent” with a dilute salt concentration—1 M LiDFOB–ES—enables a stable operation of LMBs under harsh conditions of high voltages (up to 4.6 V), fast rates (0.1–5C) and wide temperatures (−30 to 60 °C) (see Table S2† for the summary of previous work).
3 Conclusion
We have demonstrated that a simple electrolyte formula of 1 M LiDFOB–ES without any additive enables a stable charge–discharge operation of an NCM622|Li full cell at a high cut-off voltage up to 4.6 V, achieving a capacity retention of 88.2% over 200 cycles with an average CE of 99.5%. Increasing the cut-off voltage from 4.3 to 4.6 V successfully increases the specific capacity of NCM622 from 176 to 212 mA h g−1, leading to a significant increase of energy density by ∼27%. A series of examinations have been employed to show that the LiDFOB–ES electrolyte effectively suppresses aggressive side reactions, lithium dendrite growth, self-discharge, gas evolution, transition metal dissolution, and impedance growth under harsh conditions. This improved performance can be mainly attributed to a unique interphase chemistry—the LiDFOB salt and ES solvent with close reduction (oxidation) potentials induce a synergistic effect to synchro-construct thin and robust inorganic–organic interlocking protective films on both the cathode and anode—for alleviating the various aforementioned issues faced by a high-voltage LMB. Moreover, this 1 M LiDFOB–ES electrolyte also enables a stable cycling of high-voltage LMBs at −30–60 °C owing to its high ionic conductivity and stability in the wide-temperature range. In conclusion, our electrolyte design demonstrates a pragmatic approach to the development of high-performance high-voltage LMBs with low cost, fast rate and wide-temperature applicability.4 Experimental
4.1 Electrolyte and electrode preparation
The salt, lithium difluoro(oxalato)borate (LiDFOB, HeFei Prospect New Energy Technology Ltd), and the solvents, 1,2-ethylene sulfite (ES), ethylene carbonate (EC) and propylene carbonate (PC) (Adamass), were of battery grade and used without further purification. Electrolyte preparations were performed in an Ar-filled glove box. The electrolytes were prepared by dissolving LiDFOB in solvents with a molarity of 1 M (mol L−1). The commercial electrolyte 1 M LiPF6/EC:DMC (1:1 by volume) was purchased from DodoChem Co., Ltd and used as the reference. All electrolytes were dried using a molecular sieve before testing. The water content was less than 2 ppm, as detected using a coulometric Karl Fischer titrator. The cathode was prepared by mixing LiNi0.6Mn0.2Co0.2 (NCM622, HeFei Prospect New Energy Technology Ltd), acetylene black (AB, Li-400, Denka Company Limited) and polyvinylidene difluoride (PVDF) with a weight ratio of 94.5:3:2.5 in N-methylpyrrolidone (NMP). The slurry was coated on Al foil (20 μm, DodoChem) using an automatic coater (HF-Kejing, MSK-AFA-I). The obtained electrodes were dried at 120 °C under vacuum overnight. The active material mass loading was approximately 11.8 mg cm−2. Li foil (Φ 12 mm, Guangdong Canrd New Energy Technology Co. Ltd) with a thickness of 500 μm was used as the anode in the NCM622‖Li half-cells. An ultrathin Li foil (Φ 12 mm, Tianjin Medium-Energy Lithium Co. Ltd) with a thickness of 50 μm was used as the anode in the NCM622‖Li full cells.
4.2 Cell assembly and electrochemical measurements
Cu‖Li, NCM622‖Li half-cells, and NCM622‖Li full cells were assembled in standard 2032-type coin cells in an Ar-filled glove box for electrochemical performance measurements. Polyethylene was used as a separator for both half-cells and full cells. The amount of the electrolyte was 40 μL. In the full cells, the negative-to-positive capacity ratio (N/P) was controlled at 4. All cells were kept at the testing temperature for 2 h to reach temperature equilibrium before tests.Galvanostatic charge–discharge cycling and rate capability tests were performed using a Neware tester (CT-4008) within a voltage range of 2.8–4.6 V. For the cycling performance test, the cells were charged/discharged at 0.1C for −30 °C, and at 0.5C for 25 and 60 °C, respectively. For rate performance measurements, the cells were charged/discharged at the same rate (0.1–5C). A 1C rate corresponds to 210 mA g−1 on the weight basis of the NCM622 active material. Electrochemical impedance spectroscopy (EIS) was carried out on the NCM622‖Li half-cells at the discharge state with an amplitude of 10 mV over a frequency range of 0.1 mHz to 1 MHz using a Solartron 1470E electrochemical workstation. CV and LSV measurements were performed on a potentiostat (BioLogic, MPG-2) in a coin cell using Cu and Al foils as the working electrode, respectively, and lithium foil as the counter electrode. The scan rate was 0.2 mV s−1. The CE of Li metal at various temperatures was investigated on Cu‖Li coin cells using the method proposed by Aurbach et al. Before testing, 5 mA h cm−2 of Li was deposited onto the Cu foil at 0.5 mA cm−2 followed by a stripping process to 1 V for SEI formation. During testing, 5 mA h cm−2 (QR) of Li was first deposited followed by 10 cycles of 1 mA h cm−2 (QC) plating and stripping before finally stripping all Li to 1 V (QF). The accurate CE was obtained with the following equation:
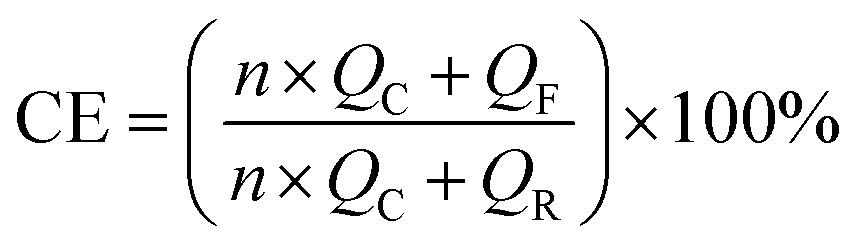
The electrochemical floating test was performed in coin cells with NMC622 and Li metal as the cathode and anode, respectively, in different electrolytes. The cells were first charged to 4.6 V at 0.1C and then maintained for 35 h. The leakage current was monitored using a Neware tester.
5 Characterization
5.1 Measurements of the physicochemical properties of electrolytes
Weight losses of the electrolytes upon heating were measured on a thermogravimetric analyzer (Mettler-Toledo, 3+/1600 HT). The samples were sealed in an Al pan with a pinhole for gas escape during the measurements. The ramping rate is 5 °C min−1 and the purge Ar flow is 50 ml min−1. The ionic conductivity was studied by AC impedance spectroscopy in a symmetrical Pt‖electrolyte‖Pt cell using a Solartron 1470E electrochemical workstation with a frequency range of 0.1 mHz to 1 MHz over a temperature range of −50 to 100 °C. A standard KCl solution was used to calibrate the cell constant. The viscosity and density of solutions were evaluated using a kinematic viscometer (Anton Paar, SVM 3001).5.2 Characterization of the material morphology and composition
The morphologies of deposited Li were characterized using a Field Emission Scanning Electron Microscope (Hitachi, Regulus 8230). A capacity of 2 mA h cm−2 of lithium was plated on the Cu working electrodes with a current density of 0.5 mA cm−2. Cycled Li and NMC622 electrodes were obtained by disassembling CR2032 coin cells in the glove box and washing the cycled electrodes with dimethyl carbonate (DMC). The morphology and microstructure were studied by high-resolution SEM (Hitachi, Regulus 8230) and Cryo-EM (Glacios). The surface analysis of the cycled electrodes was performed using an X-ray photoelectron spectrometer (XPS, ESCALAB Xi+) with Al-Kα radiation. A charge neutralizer was applied to compensate for the sample surface charge. The binding energy was calibrated using F 1s peak at 684.9 eV. The studied electrodes were subjected to a rinse in the DMC solvent followed by vacuum drying before XPS measurements. Thereafter, the samples were transferred from the Ar-filled glove box to the instrumental chambers using an instrumental accessory known as the “Sample Transfer Vessel” without exposure to air. The concentrations of metal ions in the electrolyte were detected by ICPMS (iCAP RQ, Thermo Fisher).5.3 In situ DEMS measurements
In situ DEMS measurements were used for detecting the gas evolution in the studied cells using different electrolytes during the galvanostatic charge–discharge process. The DEMS cells were assembled inside an argon-filled glovebox, comprising a lithium metal anode and a NCM622 cathode that is separated by a polypropylene separator (2500 Celgard). Before the DEMS measurement, the prepared cells were rested for 8 h and pure Ar gas was flushed for 3 h to remove the air in the cells. The generated gases were analyzed using a quadrupole mass spectrometer (QAS 100, Shanghai Linglu). The galvanostatic charge–discharge test was conducted at 0.1C using a Solartron 1470E electrochemical workstation.5.4 Cryo-EM transfer procedure
To prepare the Cryo-EM sample, the 200-mesh Cu grid was used as the working electrode in a Cu‖Li coin cell. Li metal with an areal capacity of 0.25 mA h cm−2 was deposited onto the Cu grid by applying a current of 0.5 mA cm−2 for 30 min. Then the cells were immediately disassembled in the glovebox and washed briefly with DMC to remove the residual electrolyte. The obtained Cu grid sample was placed in a grid box and transferred out of the glovebox. The sealed grid box was plunged directly into a bath of liquid nitrogen. The sample transfer and loading were carried out in liquid nitrogen without any exposure to air. The deposited Li was then observed by cryo-EM using an aberration-corrected Titan Glacios TEM operated at 200 kV. All cryo-EM images were acquired at low temperature (−178 °C) under the low dose condition (10 e−1 A−2 s−1 × 10 s) using a Falcon 3 camera.Author contributions
J. W., M. F., and J. C. designed the experiments. M. F. conducted the experiments. All authors contributed to the discussion. J. W. conceived and led the project. M. F. and J. C. contributed equally to this work.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the Westlake Education Foundation and National Natural Science Foundation of China (grant No. 21975207). The authors thank the Westlake Instrumentation and Service Centers for Physical Sciences and Molecular Sciences for characterization support.References
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 RSC.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef PubMed.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- S. Ahmed, S. E. Trask, D. W. Dees, P. A. Nelson, W. Lu, A. R. Dunlop, B. J. Polzin and A. N. Jansen, J. Power Sources, 2018, 403, 56–65 CrossRef CAS.
- S. Liu, J. Su, J. Zhao, X. Chen, C. Zhang, T. Huangb, J. Wu and A. Yua, J. Power Sources, 2018, 393, 92–98 CrossRef CAS.
- W. Li, B. Song and A. Manthiram, Chem. Soc. Rev., 2017, 46, 3006–3059 RSC.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 RSC.
- X. B. Cheng, R. Zhang, C. Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS PubMed.
- K. Yan, Z. Lu, H.-W. Lee, F. Xiong, P.-C. Hsu, Y. Li, J. Zhao, S. Chu and Y. Cui, Nat. Energy, 2016, 1, 16010 CrossRef CAS.
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS.
- J. Liu, Z. Bao, Y. Cui, E. J. Dufek, J. B. Goodenough, P. Khalifah, Q. Li, B. Y. Liaw, P. Liu, A. Manthiram, Y. S. Meng, V. R. Subramanian, M. F. Toney, V. V. Viswanathan, M. S. Whittingham, J. Xiao, W. Xu, J. Yang, X.-Q. Yang and J.-G. Zhang, Nat. Energy, 2019, 4, 180–186 CrossRef CAS.
- C. Zhan, J. Lu, A. Jeremy Kropf, T. Wu, A. N. Jansen, Y. K. Sun, X. Qiu and K. Amine, Nat. Commun., 2013, 4, 2437 CrossRef PubMed.
- N. P. W. Pieczonka, Z. Liu, P. Lu, K. L. Olson, J. Moote, B. R. Powell and J.-H. Kim, J. Phys. Chem. C, 2013, 117, 15947–15957 CrossRef CAS.
- J. Xia, R. Petibon, D. Xiong, L. Ma and J. R. Dahn, J. Power Sources, 2016, 328, 124–135 CrossRef CAS.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- J. Wang, Q. Zheng, M. Fang, S. Ko, Y. Yamada and A. Yamada, Adv. Sci., 2021, 8, 2101646 CrossRef CAS PubMed.
- C. Fang, J. Li, M. Zhang, Y. Zhang, F. Yang, J. Z. Lee, M. H. Lee, J. Alvarado, M. A. Schroeder, Y. Yang, B. Lu, N. Williams, M. Ceja, L. Yang, M. Cai, J. Gu, K. Xu, X. Wang and Y. S. Meng, Nature, 2019, 572, 511–515 CrossRef CAS PubMed.
- Z. Zhang, Y. Li, R. Xu, W. Zhou, Y. Li, S. T. Oyakhire, Y. Wu, J. Xu, H. Wang, Z. Yu, D. T. Boyle, W. Huang, Y. Ye, H. Chen, J. Wan, Z. Bao, W. Chiu and Y. Cui, Science, 2022, 375, 66–70 CrossRef CAS PubMed.
- Z. Piao, P. Xiao, R. Luo, J. Ma, R. Gao, C. Li, J. Tan, K. Yu, G. Zhou and H.-M. Cheng, Adv. Mater., 2022, 34, 2108400 CrossRef CAS PubMed.
- Z. Ju, G. Lu, O. Sheng, H. Yuan, S. Zhou, T. Liu, Y. Liu, Y. Wang, J. Nai, W. Zhang and X. Tao, Nano Lett., 2022, 22, 1374–1381 CrossRef CAS PubMed.
- J. Wang, Y. Yamada, K. Sodeyama, C. H. Chiang, Y. Tateyama and A. Yamada, Nat. Commun., 2016, 7, 12032 CrossRef CAS PubMed.
- J. Wang, Y. Yamada, K. Sodeyama, E. Watanabe, K. Takada, Y. Tateyama and A. Yamada, Nat. Energy, 2018, 3, 22–29 CrossRef CAS.
- S. Ko, Y. Yamada and A. Yamada, Joule, 2021, 5, 998–1009 CrossRef CAS.
- S. Chen, J. Zheng, D. Mei, K. S. Han, M. H. Engelhard, W. Zhao, W. Xu, J. Liu and J. Zhang, Adv. Mater., 2018, 30, 1706102 CrossRef PubMed.
- X. Ren, S. Chen, H. Lee, D. Mei, M. H. Engelhard, S. D. Burton, W. Zhao, J. Zheng, Q. Li, M. S. Ding, M. Schroeder, J. Alvarado, K. Xu, Y. S. Meng, J. Liu, J.-G. Zhang and W. Xu, Chem, 2018, 4, 1877–1892 CAS.
- X. Cao, X. Ren, L. Zou, M. H. Engelhard, W. Huang, H. Wang, B. E. Matthews, H. Lee, C. Niu, B. W. Arey, Y. Cui, C. Wang, J. Xiao, J. Liu, W. Xu and J.-G. Zhang, Nat. Energy., 2019, 4, 796–805 CrossRef CAS.
- X. Fan, L. Chen, O. Borodin, X. Ji, J. Chen, S. Hou, T. Deng, J. Zheng, C. Yang, S. C. Liou, K. Amine, K. Xu and C. Wang, Nat. Nanotechnol., 2018, 13, 715–722 CrossRef CAS PubMed.
- L. Suo, W. Xue, M. Gobet, S. G. Greenbaum, C. Wang, Y. Chen, W. Yang, Y. Li and J. Li, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 1156–1161 CrossRef CAS PubMed.
- W. Xue, M. Huang, Y. Li, Y. G. Zhu, R. Gao, X. Xiao, W. Zhang, S. Li, G. Xu, Y. Yu, P. Li, J. Lopez, D. Yu, Y. Dong, W. Fan, Z. Shi, R. Xiong, C.-J. Sun, I. Hwang, W.-K. Lee, Y. Shao-Horn, J. A. Johnson and J. Li, Nat. Energy., 2021, 6, 495–505 CrossRef CAS.
- Y. Yamada, J. Wang, S. Ko, E. Watanabe and A. Yamada, Nat. Energy, 2018, 3, 22–29 CrossRef.
- K. Park, S. Yu, C. Lee and H. Lee, J. Power Sources, 2015, 296, 197–203 CrossRef CAS.
- I. A. Shkrob, Y. Zhu, T. W. Marin and D. P. Abraham, J. Phys. Chem. C, 2013, 117, 23750–23756 CrossRef CAS.
- S. Huang, S. Wang, G. Hu, L.-Z. Cheong and C. Shen, Appl. Surf. Sci., 2018, 441, 265–271 CrossRef CAS.
- F. Zhang, C. Wang, D. Zhao, L. Yang, P. Wang, W. Li, B. Wang and S. Li, Electrochim. Acta, 2020, 337, 135727 CrossRef CAS.
- Y. Liu, P. Yu, Q. Sun, Y. Wu, M. Xie, H. Yang, T. Cheng and W. A. Goddard, ACS Energy Lett., 2021, 6, 2320–2327 CrossRef CAS.
- J. Cha, J.-G. Han, J. Hwang, J. Cho and N.-S. Choi, J. Power Sources, 2017, 357, 97–106 CrossRef CAS.
- A. J. Louli, A. Eldesoky, R. Weber, M. Genovese, M. Coon, J. deGooyer, Z. Deng, R. T. White, J. Lee, T. Rodgers, R. Petibon, S. Hy, S. J. H. Cheng and J. R. Dahn, Nat. Energy., 2020, 5, 693–702 CrossRef CAS.
- S. H. Kang, D. P. Abraham, A. Xiao and B. L. Lucht, J. Power Sources, 2008, 175, 526–532 CrossRef CAS.
- S. Jurng, Z. L. Brown, J. Kim and B. L. Lucht, Energy Environ. Sci., 2018, 11, 2600–2608 RSC.
- B. Liao, H. Li, M. Xu, L. Xing, Y. Liao, X. Ren, W. Fan, L. Yu, K. Xu and W. Li, Adv. Energy Mater., 2018, 8, 1800802 CrossRef.
- Q. Dong, F. Guo, Z. Cheng, Y. Mao, R. Huang, F. Li, H. Dong, Q. Zhang, W. Li, H. Chen, Z. Luo, Y. Shen, X. Wu and L. Chen, ACS Appl. Energy Mater., 2019, 3, 695–704 CrossRef.
- S. Li, W. Zhao, X. Cui, Y. Zhao, B. Li, H. Zhang, Y. Li, G. Li, X. Ye and Y. Luo, Electrochim. Acta, 2013, 91, 282–292 CrossRef CAS.
- L. Dong, F. Liang, D. Wang, C. Zhu, J. Liu, D. Gui and C. Li, Electrochim. Acta, 2018, 270, 426–433 CrossRef CAS.
- D. Zhao, P. Wang, X. Cui, L. Mao, C. Li and S. Li, Electrochim. Acta, 2018, 260, 536–548 CrossRef CAS.
- F. Ren, W. Zuo, X. Yang, M. Lin, L. Xu, W. Zhao, S. Zheng and Y. Yang, J. Phys. Chem. C, 2019, 123, 5871–5880 CrossRef CAS.
- Z. Wu, S. Li, Y. Zheng, Z. Zhang, E. Umesh, B. Zheng, X. Zheng and Y. Yang, J. Electrochem. Soc., 2018, 165, A2792–A2800 CrossRef CAS.
- Y. Zhang, Y. Zhong, Z. Wu, B. Wang, S. Liang and H. Wang, Angew. Chem., Int. Ed., 2020, 59, 7797–7802 CrossRef CAS PubMed.
- L. Xing, X. Zheng, M. Schroeder, J. Alvarado, A. von Wald Cresce, K. Xu, Q. Li and W. Li, Acc. Chem. Res., 2018, 51, 282–289 CrossRef CAS PubMed.
- S.-K. Jeong, M. Inaba, Y. Iriyama, T. Abe and Z. Ogumi, Electrochem. Solid State Lett., 2003, 6, A13–A15 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ta02267b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |