DOI:
10.1039/D2SC05274A
(Edge Article)
Chem. Sci., 2022,
13, 13764-13773
Room temperature chemoselective hydrogenation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) C, CO and CN bonds by using a well-defined mixed donor Mn(I) pincer catalyst†
C, CO and CN bonds by using a well-defined mixed donor Mn(I) pincer catalyst†
Received
21st September 2022
, Accepted 2nd November 2022
First published on 3rd November 2022
Abstract
Chemoselective hydrogenation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CO and CN bonds in α,β-unsaturated ketones, aldehydes and imines is accomplished at room temperature (27 °C) using a well-defined Mn(I) catalyst and 5.0 bar H2. Amongst the three mixed-donor Mn(I) complexes developed, κ3-(R2PN3NPyz)Mn(CO)2Br (R = Ph, iPr, tBu); the tBu-substituted complex (tBu2PN3NPyz)Mn(CO)2Br shows exceptional chemoselective catalytic reduction of unsaturated bonds. This hydrogenation protocol tolerates a range of highly susceptible functionalities, such as halides (–F, –Cl, –Br, and –I), alkoxy and hydroxy, including hydrogen-sensitive moieties like acetyl, nitrile, nitro, epoxide, and unconjugated alkenyl and alkynyl groups. Additionally, the disclosed method applies to indole, pyrrole, furan, thiophene, and pyridine-containing unsaturated ketones leading to the corresponding saturated ketones. The CC bond is chemoselectively hydrogenated in α,β-unsaturated ketones, while the aldehyde's CO bond and imine's CN bond are preferentially reduced over the CC bond. A detailed mechanistic study highlighted the non-innocent behavior of the ligand in the (tBu2PN3NPyz)Mn(I) complex and indicated a metal–ligand cooperative catalytic pathway. The molecular hydrogen (H2) acts as a hydride source, whereas MeOH provides a proton for hydrogenation. DFT energy calculations supported the facile progress of most catalytic steps, involving a crucial turnover-limiting H2 activation.
C, CO and CN bonds in α,β-unsaturated ketones, aldehydes and imines is accomplished at room temperature (27 °C) using a well-defined Mn(I) catalyst and 5.0 bar H2. Amongst the three mixed-donor Mn(I) complexes developed, κ3-(R2PN3NPyz)Mn(CO)2Br (R = Ph, iPr, tBu); the tBu-substituted complex (tBu2PN3NPyz)Mn(CO)2Br shows exceptional chemoselective catalytic reduction of unsaturated bonds. This hydrogenation protocol tolerates a range of highly susceptible functionalities, such as halides (–F, –Cl, –Br, and –I), alkoxy and hydroxy, including hydrogen-sensitive moieties like acetyl, nitrile, nitro, epoxide, and unconjugated alkenyl and alkynyl groups. Additionally, the disclosed method applies to indole, pyrrole, furan, thiophene, and pyridine-containing unsaturated ketones leading to the corresponding saturated ketones. The CC bond is chemoselectively hydrogenated in α,β-unsaturated ketones, while the aldehyde's CO bond and imine's CN bond are preferentially reduced over the CC bond. A detailed mechanistic study highlighted the non-innocent behavior of the ligand in the (tBu2PN3NPyz)Mn(I) complex and indicated a metal–ligand cooperative catalytic pathway. The molecular hydrogen (H2) acts as a hydride source, whereas MeOH provides a proton for hydrogenation. DFT energy calculations supported the facile progress of most catalytic steps, involving a crucial turnover-limiting H2 activation.
Introduction
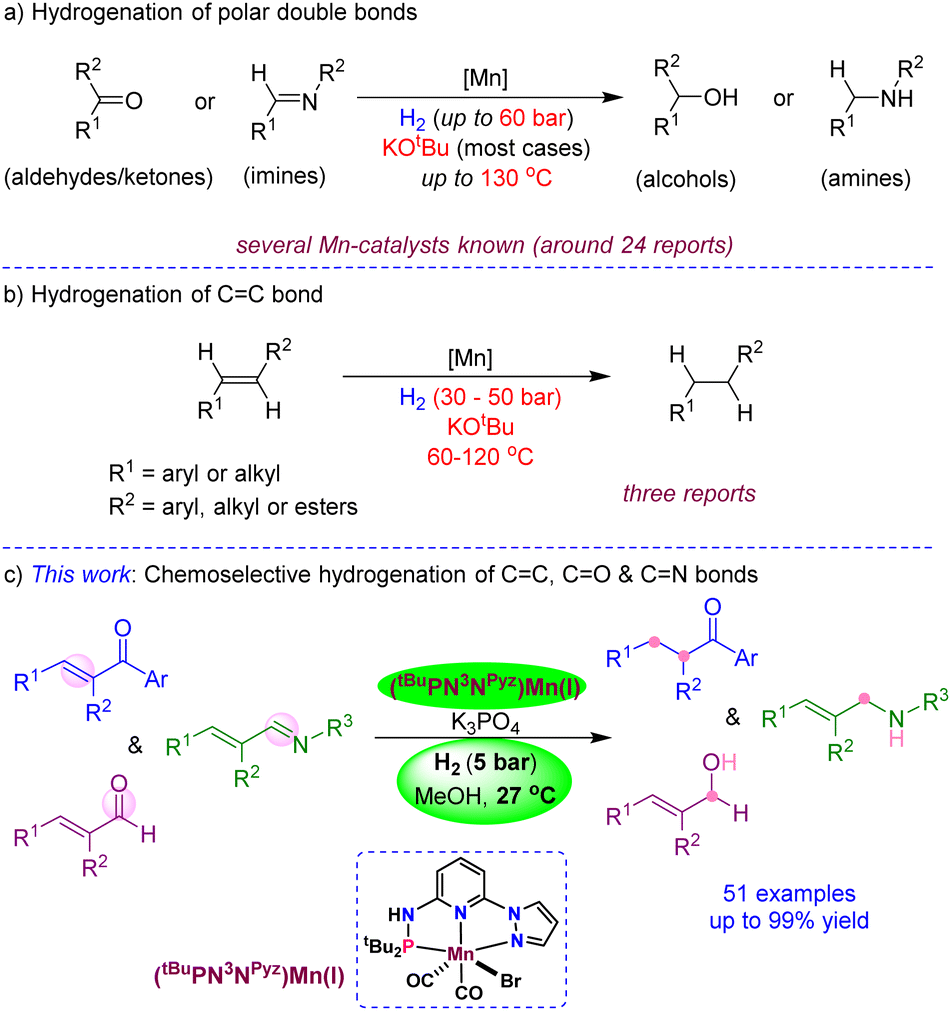
Chemoselective hydrogenation of unsaturated organic compounds is extremely important in academia and industry, as it plays a crucial role in the preparation of pharmaceutical intermediates, fragrances, fine chemicals and various bulk products. Particularly, the catalytic reductions using molecular hydrogen under ambient conditions represent one of the economical, atom-efficient and environmentally benign transformations.1 In this context, numerous heterogeneous catalysts are developed and demonstrated for the hydrogenation of various functional groups, which often require high reaction temperatures and/or pressures and result in poor selectivity.2 Over time, many well-defined, active and highly efficient homogeneous catalysts,3 derived from precious and non-precious transition metals, have been established for hydrogenation.4 In general, noble metal catalysis has dominated the field and accomplished very high catalytic turnovers and desired selectivity. However, the limited availability of noble metals in the earth's crust, and their expensiveness and underlying toxicity could limit their wide applications in hydrogenation in the future.
One of the fundamental research objectives in modern science is environmental benignity and sustainable development. Accordingly, the hydrogenations by catalysts based on low-toxic, earth-abundant and inexpensive non-precious metals have been given significant consideration.5 Particularly, the hydrogenations employing iron,6 cobalt7 and nickel6c are substantially explored and disclosed as active and highly efficient for reducing carbonyls, imines and nitriles.8 Being a less toxic, cheap and the third most abundant transition metal, manganese is elegantly demonstrated as a catalyst for the hydrogenation of aldehydes/ketones by Beller,9 Kempe,10 Sortais,11 Kirchner12 and others (Scheme 1a).13,14 Similarly, the hydrogenation of imines (or CN bond) using bidentate or pincer-ligated manganese catalysts was established.15 Meanwhile, the independent development of chiral pincer-manganese catalysts by Clarke,16 Beller,17 Han/Ding18 and others19 led to the asymmetric hydrogenation of ketones. In a significant advancement, Liu has shown the asymmetric hydrogenation of the CN bond in heteroaromatics using a chiral Mn-catalyst.20 Despite all developments on the Mn-catalyzed hydrogenations of multiple polar bonds, the chemoselective hydrogenation of CC bonds is highly challenging and extremely rare due to the associated high bond enthalpies. The group of Kirchner and Khusnutdinova independently used bidentate PP- and PN-ligated Mn(I) catalysts for the hydrogenation of alkenes using 50 bar and 30 bar H2 pressures, respectively, at elevated temperatures (Scheme 1b).21 Similarly, Topf has demonstrated CC hydrogenation in α,β-unsaturated carboxylic derivatives using 30–50 bar H2 at 100–120 °C; however, this protocol failed to provide chemoselective hydrogenation in α,β-unsaturated ketones.13c Though manganese-based catalysts promoted the hydrogenations of many CO and CN bonds and certain CC bonds,11b,13c,21 most of the reactions proceed at a high H2 pressure and at elevated temperatures, which is a significant drawback for practical applications. Additionally, most Mn-catalyzed hydrogenations require a large amount of a strong base (KOtBu) as an additive. As yet, a chemoselective hydrogenation protocol for reducing one unsaturated functional moiety in the presence of the other using a beneficial Mn-catalyst at ambient H2 pressure and temperature is unknown.22
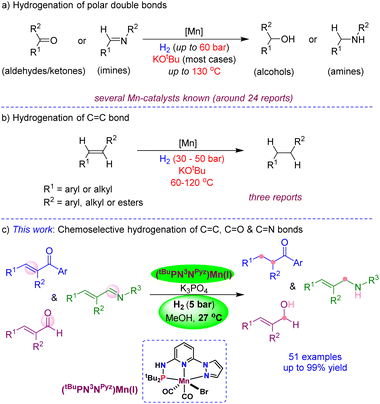 |
| | Scheme 1 Manganese-catalyzed hydrogenation of unsaturated bonds: (a) carbonyls and imines, (b) CC bond, and (c) chemoselective hydrogenation of CC, CO, and CN bonds. | |
To achieve more sustainability, efficiency and selectivity in Mn-catalysis, in this work, we developed the mixed-donor (PN3N)Mn(I) complexes and disclosed the chemoselective hydrogenation of CC, CO and CN bonds using 5 bar H2 and a mild base K3PO4 at room temperature (Scheme 1c). The notable features of the present protocol are (i) use of the 3rd most abundant transition metal as a catalyst, (ii) excellent chemoselectivity in the reduction of CC, CO and CN bonds, (iii) hydrogenation using 5 bar H2 and at room temperature (27 °C), (iv) use of a mild base and atom-efficient H2 source, and (v) broad substrates scope with excellent tolerance of hydrogen-sensitive functionalities.
Results and discussion
Synthesis of Mn(I) complexes
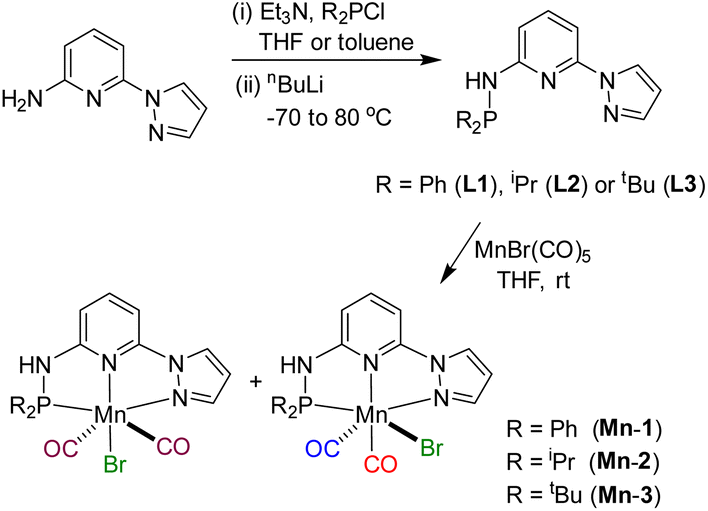
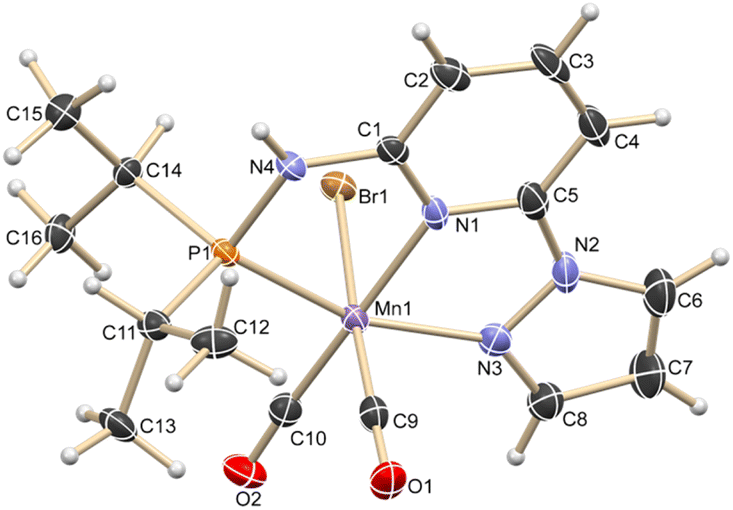
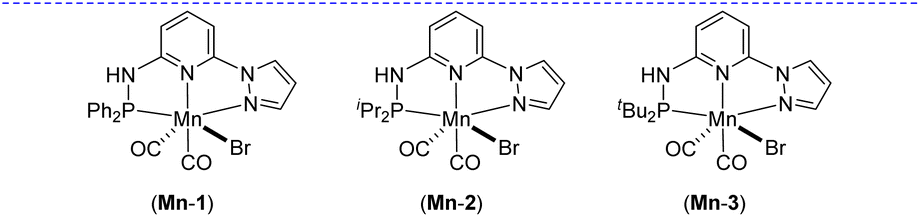
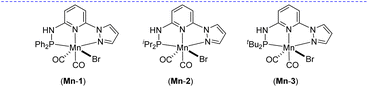
Based on our expertise in developing pincer-ligated complexes, we became interested in synthesizing mixed-donor PN3N ligands and pincer manganese complexes. We envisioned that a mixed-donor ligand with different electronic features of the donating center could suitably administer the electronic requirement at the metal center and can stabilize the active catalytic species. Moreover, the presence of NH on the side-arm could facilitate the non-innocent behavior of the ligand via an aromatization/dearomatization approach leading to the heterolytic activation of molecules. With this in mind, the isopropyl-tagged PN3N ligand L2 was synthesized in a few steps starting from 2,6-dibromopyridine, following a protocol similar to that for the synthesis of phenyl or tert-butyl PN3N ligands L1 and L3.23 Treatment of pincer ligands L1–L3 with Mn(CO)5Br in THF at room temperature afforded dicarbonyl Mn(I) complexes, (R2PN3NPyz)Mn(CO)2Br; Mn-1 (R = Ph), Mn-2 (R = iPr) and Mn-3 (R = tBu), respectively, in good yields (Scheme 2). These complexes were extensively characterized by multinuclear NMR spectroscopy, FT-IR, ESI-MS and elemental analysis. The 31P{1H}-NMR spectra of complexes Mn-1, Mn-2 and Mn-3 show peaks at 134.5, 136.9; 158.4, 159.8 and 175.3, 178.5 ppm, respectively. Two peaks for each complex at very similar chemical shift values indicate the formation of two geometrical isomers for each complex. Interestingly, all complexes displayed three IR peaks for carbonyls ranging between 1857 and 2053 cm−1. Similarly, a closer look at the 13C{1H}-NMR spectra of complexes indicates the three carbonyl signals. Moreover, the 1H and 13C{1H}-NMR spectra of all complexes display two sets of peaks. All these observations support the formation of two geometrical isomeric species in each complex Mn-1, Mn-2 and Mn-3. Considering the presence of three signals for carbonyls in each complex, we assume that one isomer of the Mn-complex would have two carbonyls that are trans to –Br and –Npy ligands accounting for two peaks (as indicated in the X-ray structure), whereas the other isomer would have two carbonyls trans to each other and display a single carbonyl peak (Scheme 2). Even though the assumed isomers are highly convincing, the probability of the mixture of a neutral (with –Br coordination) and a cationic (with Br as an anion) manganese species cannot be completely ruled out.11a,13b,16,17a All the complexes are further characterized by ESI-MS that show two prominent isotopic masses for 79Br and 81Br containing complexes. The molecular structure of Mn-2 was confirmed by a single crystal X-ray study (Fig. 1). As expected, the coordination geometry around the manganese is distorted octahedral with –Br and one –CO unit trans to each other, making the C9–Mn1–Br angle almost linear (175.54(15)°). The Mn1–C10 bond length (1.797(4) Å) is slightly longer than the Mn1–C9 bond length (1.773(4) Å), which might be due to the greater trans influence of –Npy than the –Br. Although each synthesized manganese complex contains two isomeric species, the generated active catalyst in the presence of a base is expected to be a single isomer (proved and discussed vide infra).
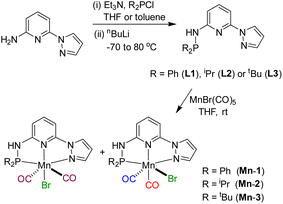 |
| | Scheme 2 Synthesis of (R2PN3NPyz) manganese pincer complexes. | |
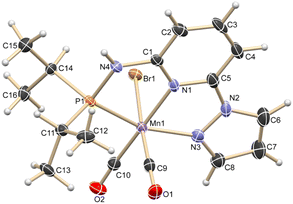 |
| | Fig. 1 ORTEP of compound Mn-2 showing the atom-numbering scheme. Displacement ellipsoids are drawn at the 50% probability level. Selected bond length (Å): Br1–Mn1, 2.5916(7); Mn1–C9, 1.773(4); Mn1–C10, 1.797(4); Mn1–N3, 2.011(3); Mn1–N1, 2.012(3); Mn1–P1, 2.2462(12). Selected bond angles (°): N3–Mn1–P1, 160.49(11); C10–Mn1–N1, 175.88(16); C9–Mn1–Br1, 175.54(14); C9–Mn1–C10, 87.40(19); C9–Mn1–N1, 96.23(16); N1–Mn1–Br1, 84.57(9). | |
Optimization for catalytic hydrogenation
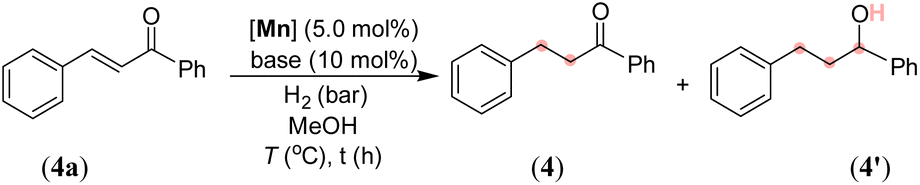
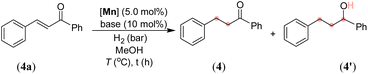
After synthesizing the sterically and electronically distinct mixed-donor (PN3N) manganese complexes, we have investigated their catalytic behavior for the chemoselective hydrogenation of α,β-unsaturated ketones using molecular hydrogen. We first screened the activity of (PN3N)Mn(I) complexes Mn-1, Mn-2, and Mn-3 for CC bond hydrogenation in (E)-3-phenyl-1-(phenyl)prop-2-en-1-one (4a) using a catalytic amount of KOtBu and 30 bar hydrogen pressure in methanol at 50 °C (see, Tables 1 and S1 in the ESI†). The use of Mn-1 as a catalyst led to 75% conversion of 4a, wherein chemoselective CC hydrogenated product 4 was obtained in 65% yield, and completely hydrogenated compound 4′ in 10% yield (entry 1). The electronically rich complex Mn-2 gave complete conversion of 4a; however, the product 4′ was obtained in 80% yield, and the remaining was allylic alcohol with only CO hydrogenation (entry 2). Interestingly, the bulky tBu-substituted complex Mn-3 gave a complete conversion of 4a with more chemoselectivity for 4 (entry 3). The formation of allylic alcohol in the presence of catalyst Mn-2 suggests the probability of a sequential CO hydrogenation-allylic alcohol isomerization leading to the uncontrolled hydrogenated compound 4′. The low steric effect in complex Mn-2 compared to Mn-3 might play a crucial role in chemoselective hydrogenation (1,2-hydrogenation of CO versus 1,4-hydrogenation of CC). Notably, the reaction at lower hydrogen pressure and room temperature (27 °C) significantly improved chemoselective CC bond hydrogenation without altering the overall conversion (entries 4 and 5). The use of other bases, such as NaOtBu, LiOtBu, and KOAc, led to low conversion, whereas the presence of mild bases K2CO3 or K3PO4 provided complete conversion with good chemoselectivity for 4 using 10 bar H2 and at room temperature (entries 6–10). The activity of a manganese catalyst using a catalytic amount of mild base K3PO4 is notable, as most of the Mn catalysis generally employs a strong base like KOtBu. The chemoselective hydrogenation of 4a also proceeded smoothly using 5 bar hydrogen pressure at room temperature (27 °C) and provided 4 in 96% isolated yield just in an hour (entry 11). Notably, the manganese catalysts Mn-1 and Mn-2 were less effective for hydrogenation under the optimized conditions (5 bar H2/27 °C/1 h), and provided <19% of product 4 (see the ESI†). Therefore, all chemoselective hydrogenations were conducted employing the Mn-3 catalyst and 5 bar H2 pressure at room temperature with the best-optimized reaction time. An in situ generated Mn(CO)5Br/L3 catalyst system is less effective, affording the hydrogenated product in 39% yield, highlighting the importance of a well-defined manganese catalyst (entry 12). However, a Mn(II) precursor with the L3 ligand (MnCl2/L3 or MnBr2/L3) did not provide hydrogenation (entries 13 and 14). Similarly, the Mn(CO)5Br precursor and bidentate N-donor or P-donor ligand systems were ineffective. Hydrogenation did not proceed in the absence of a catalyst, a base or H2, which suggests the importance of these components for the reaction.
Table 1 Optimization of reaction conditionsa
|
|
| Entry |
[Mn] |
Base |
T (°C)/t (h) |
Conv.b (%) |
4
(%) |
4′
(%) |
|
Reaction conditions: 4a (0.042 g, 0.20 mmol), base (0.02 mmol), [Mn] catalyst (0.01 mmol, 5 mol%), solvent (1.0 mL). Entries 1–4: 30 bar H2; entries 5–10: 10 bar H2; entries 11–14: 5 bar H2.
GC conversion, isolated yields are given in parentheses.
20% allylic alcohol was observed. All the catalysts Mn-1, Mn-2 and Mn-3 contain a mixture of two geometrical isomers.
|
| 1 |
Mn-1
|
KOtBu |
50/20 |
75 |
65 |
10 |
| 2c |
Mn-2
|
KOtBu |
50/20 |
100 |
— |
80 |
| 3 |
Mn-3
|
KOtBu |
50/20 |
100 |
63 (60) |
37 |
| 4 |
Mn-3
|
KOtBu |
27/20 |
100 |
81 (79) |
19 |
| 5 |
Mn-3
|
KOtBu |
27/20 |
100 |
91 (88) |
9 |
| 6 |
Mn-3
|
NaOtBu |
27/20 |
40 |
39 |
Trace |
| 7 |
Mn-3
|
LiOtBu |
27/20 |
74 |
73 |
Trace |
| 8 |
Mn-3
|
K2CO3 |
27/20 |
100 |
74 |
26 |
| 9 |
Mn-3
|
KOAc |
27/20 |
20 |
19 |
Trace |
| 10 |
Mn-3
|
K3PO4 |
27/20 |
100 |
88 |
12 |
|
11
|
Mn-3
|
K
3
PO
4
|
27/1 |
100
|
98 (96)
|
2
|
| 12 |
Mn(CO)5Br/L3 |
K3PO4 |
27/1 |
39 |
39 |
— |
| 13 |
MnBr2/L3 |
K3PO4 |
27/1 |
— |
— |
— |
| 14 |
MnCl2/L3 |
K3PO4 |
27/1 |
— |
— |
— |
|
Substrate scope of hydrogenation
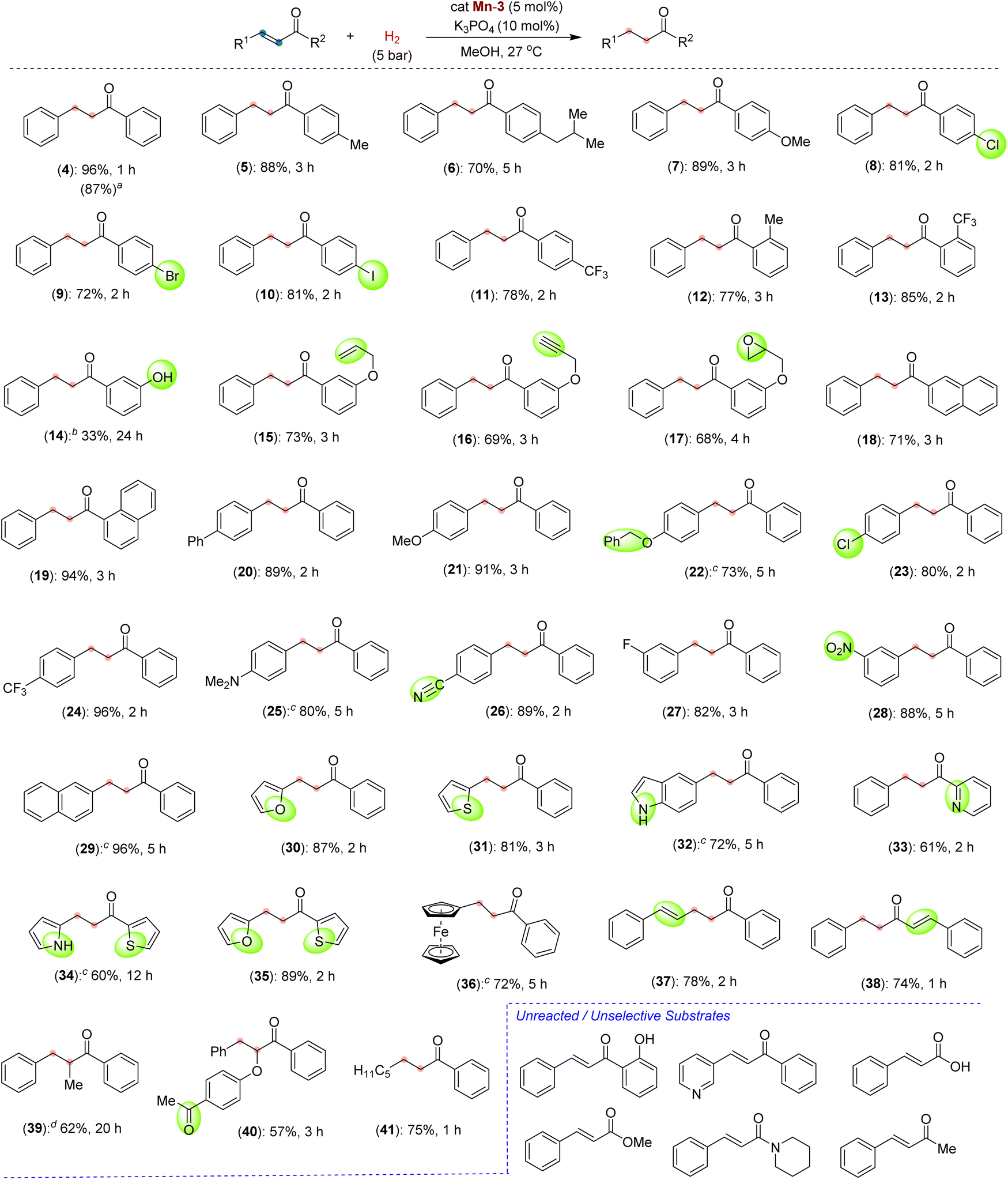
After successfully optimizing the reaction parameters for chemoselective CC bond hydrogenation in chalcone, we have explored the reaction scope using catalyst Mn-3, catalytic K3PO4 and 5 bar H2 at room temperature (Scheme 3). Depending upon the substrates, the hydrogenations were performed for different time intervals, and the best yields were reported. First, we checked the hydrogenation of α,β-unsaturated ketones with different substitutions on the benzoyl ring. Thus, the chalcones containing electron-donating alkyl and alkoxy substitutions at the para position of benzoyl reacted smoothly to give a good to an excellent yield of saturated ketones 5–7. The halogen substitutions, –Cl, –Br, –I, and –CF3, were well tolerated at the para position of the benzoyl ring affording the desired saturated ketones (8–11) in good yields. The tolerance of such functionalities is noteworthy as they can be employed for late-stage diversification. In addition to the para-substituted chalcones, the electron-rich and electron-deficient ortho-substituted compounds smoothly participated in hydrogenation (12 and 13). Chalcone having a phenolic –OH at the meta position of benzoyl, reacted slowly and afforded the product 14 in 33% yield. Interestingly, unsaturated ketones with sensitive and reducible functionalities, such as terminal alkene, alkyne, and epoxide, reacted chemoselectively to give compounds 15–17. The unsaturated ketones with the naphthyl moiety also reacted with good yields (18 and 19). A higher-scale hydrogenation of compound 4a (0.5 g, 2.4 mmol) provided the product 4 in 87% isolated yield (Scheme 3, in parenthesis), highlighting the potential practical application.
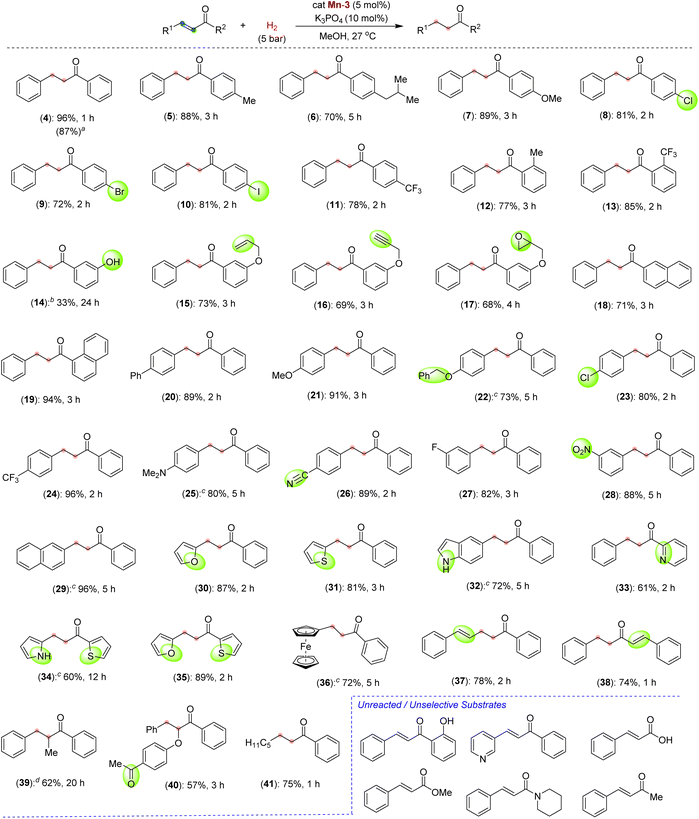 |
| | Scheme 3 Scope of Mn-catalyzed CC bond hydrogenation of α,β-unsaturated ketones. Reaction conditions: substrate, α,β-unsaturated ketone (0.20 mmol), Mn-3 (0.005 g, 0.01 mmol, 5 mol%), K3PO4 (0.0043 g, 0.02 mmol), MeOH (1.0 mL), H2 (5 bar). Yields are of isolated compounds. aReaction in 0.5 g scale. bReaction at 50 °C. cMixture of MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DCM (4:1) used. dReaction at 10 bar H2 and 50 °C. :DCM (4:1) used. dReaction at 10 bar H2 and 50 °C. | |
After this, we moved to check the effect and tolerability of different substitutions on the alkenyl–arenes towards chemoselective hydrogenation. Substrates with electron-rich substituents such as phenyl, methoxy and benzyloxy reacted efficiently, producing excellent yields of products 20–22. The survival of benzyl protection of phenolic –OH is notable, as such substrates are prone to hydrogenolysis under hydrogenation conditions. Similarly, the –Cl and –CF3 groups remained unaffected and delivered the saturated halo-ketones 23 and 24 in 80% and 96% yields, respectively. An amine functionality that could poison catalysis by binding to the metal is also sustained under optimized conditions (25). To our surprise, the highly desirous and hydrogenation-sensitive functionalities, –CN and –NO2 groups, could be tolerated to afford the products 26 and 28 in around 88% yields. A range of α,β-unsaturated ketones derived from heteroarenes, such as furanyl, thiophenyl, indolyl, pyrrolyl and pyridinyl, were successfully hydrogenated to afford the saturated heteroaryl ketones (30–35). The chemoselective hydrogenation of these heteroaryl-containing compounds is remarkable, as the heteroaryl rings often interfere with the reaction due to their coordination ability to metal. The hydrogenation of ketones having unprotected NH indolyl and pyrrolyl opens up a new avenue as they can further be diversified. An unsaturated ketone containing a ferrocene backbone provided selective hydrogenation to 36 in 72% yield. Interestingly, hydrogenation of a ketone containing extended conjugation provided selectively semi-hydrogenated product 37 in 78% yield. Similarly, in the substrate where the carbonyl group is in conjugation with two alkenes, one CC bond was selectively hydrogenated and provided a good yield of product 38. This hydrogenation protocol is also suitable for α,β-unsaturated ketones having a trisubstituted alkene to provide saturated ketones, albeit in moderate yields (39 and 40). A β-alkyl-α,β-unsaturated ketone, (E)-1-phenyloct-2-en-1-one could be hydrogenated to give compound 41 in 75% yield. Interestingly, in all these cases, an excellent chemoselective CC bond hydrogenation was observed in the presence of other H2-sensitive functionalities. Such chemoselective hydrogenation employing a Mn-catalyst is extremely rare.11b,12c,13c Unfortunately, the α,β-unsaturated ketones with free –OH at the ortho position, and carboxylate, ester and amide derivatives failed to participate in the reaction under the optimized conditions. However, the substrate (E)-4-phenylbut-3-en-2-one gave a mixture of highly unselective hydrogenated products.
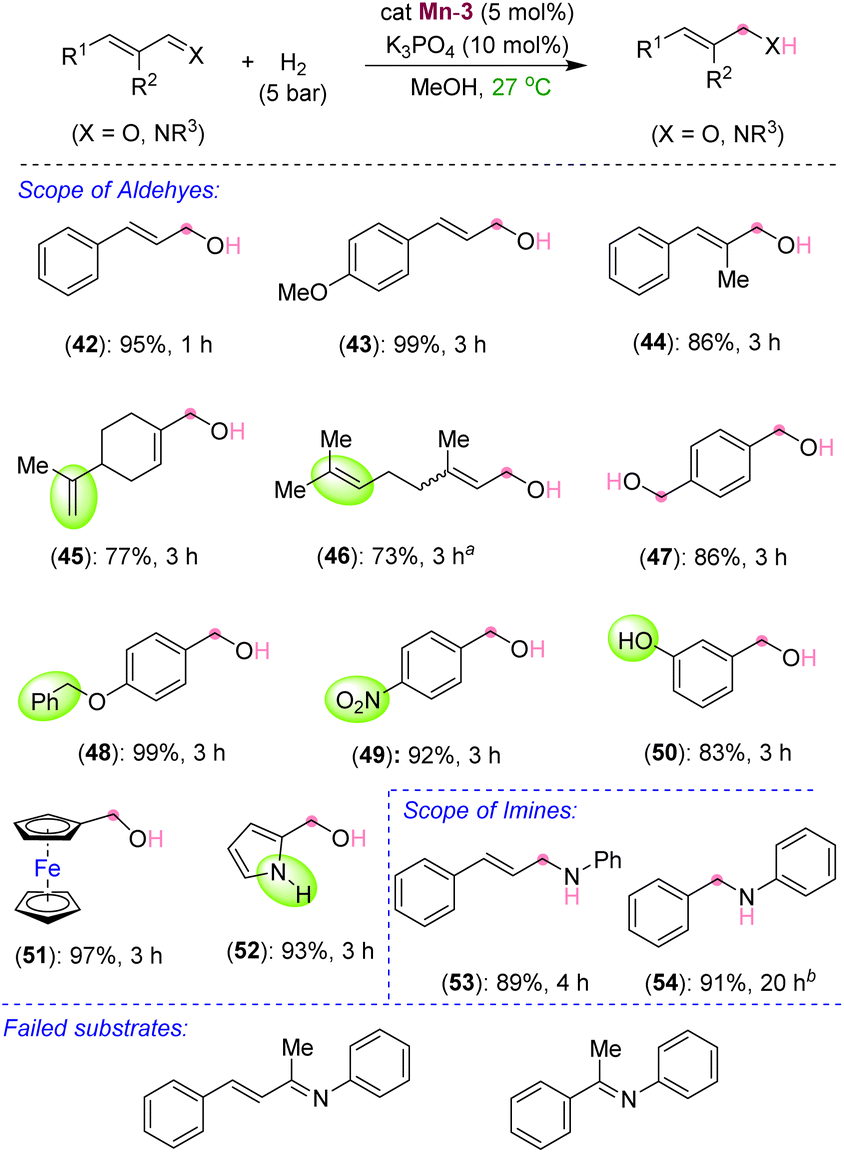
After exploring the scope and limitations of selective CC bond hydrogenation in unsaturated ketones, we were eager to know the reactivity of the synthesized manganese complex on the hydrogenation of α,β-unsaturated aldehydes and imines (Scheme 4). Surprisingly, under the standard reaction conditions, selective CO bond hydrogenation of cinnamaldehyde derivatives was observed, leading to the 3-phenylprop-2-en-1-ols (42–44) in excellent yields. Aliphatic and acyclic conjugated aldehydes also participated in the selective hydrogenation to unsaturated alcohols without harming the alkenyl groups, thus leading to the products 45 and 46 in 77% and 73% yields, respectively. The aromatic aldehydes containing benzyloxy, nitro, –OH, and pyrrolyl groups were also smoothly hydrogenated to the corresponding alcohols at room temperature (48–52). The tolerance of free –OH and –NH groups is highly impressive. Similarly, an α,β-unsaturated imine was chemoselectively hydrogenated to unsaturated amine (53) in good yield. Even a simple unconjugated N-aryl imine could be hydrogenated to amine in high yield (54). However, the attempted hydrogenations of ketone-derived imine analogues, such as (2E,3E)-N,4-diphenylbut-3-en-2-imine and (E)-N,1-diphenylethan-1-imine, failed under the optimized conditions and the unreacted starting compounds were quantitatively recovered (Scheme 4). We assume that the steric hindrance around the keto-derived imine inhibited its approach towards the manganese center, leading to an unsuccessful reaction. The chemoselective hydrogenations of the CC bond over the ketone carbonyl and that of the aldehyde's CO and imine's CN over the CC by using the newly developed catalyst are notable. Particularly, mild reaction conditions and the use of the catalytic K3PO4 base are significant. This catalyst can further be applied to novel catalytic approaches considering its advantage over other similar catalysts. Though, an excellent chemoselectivity was observed in the hydrogenation of α,β-unsaturated ketones, a trace formation of both CC and CO reductions was unavoidable in some cases.
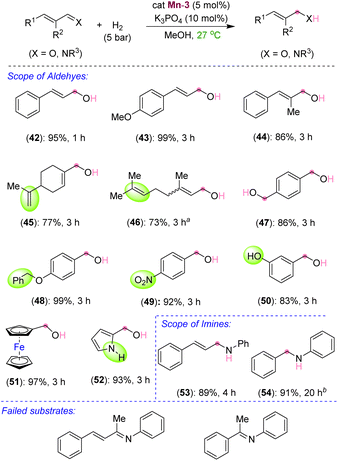 |
| | Scheme 4 Scope of Mn-catalyzed hydrogenation of aldehydes and imines. aReaction conditions: substrate (0.20 mmol), Mn-3 (0.005 g, 0.01 mmol, 5 mol%), K3PO4 (0.0043 g, 0.02 mmol), H2 (5 bar), MeOH (1.0 mL). Yields are of isolated compounds. aObtained as a mixture of E and Z isomers as the starting compound was also a mixture of both. bReaction performed at 50 °C. | |
Mechanistic aspects
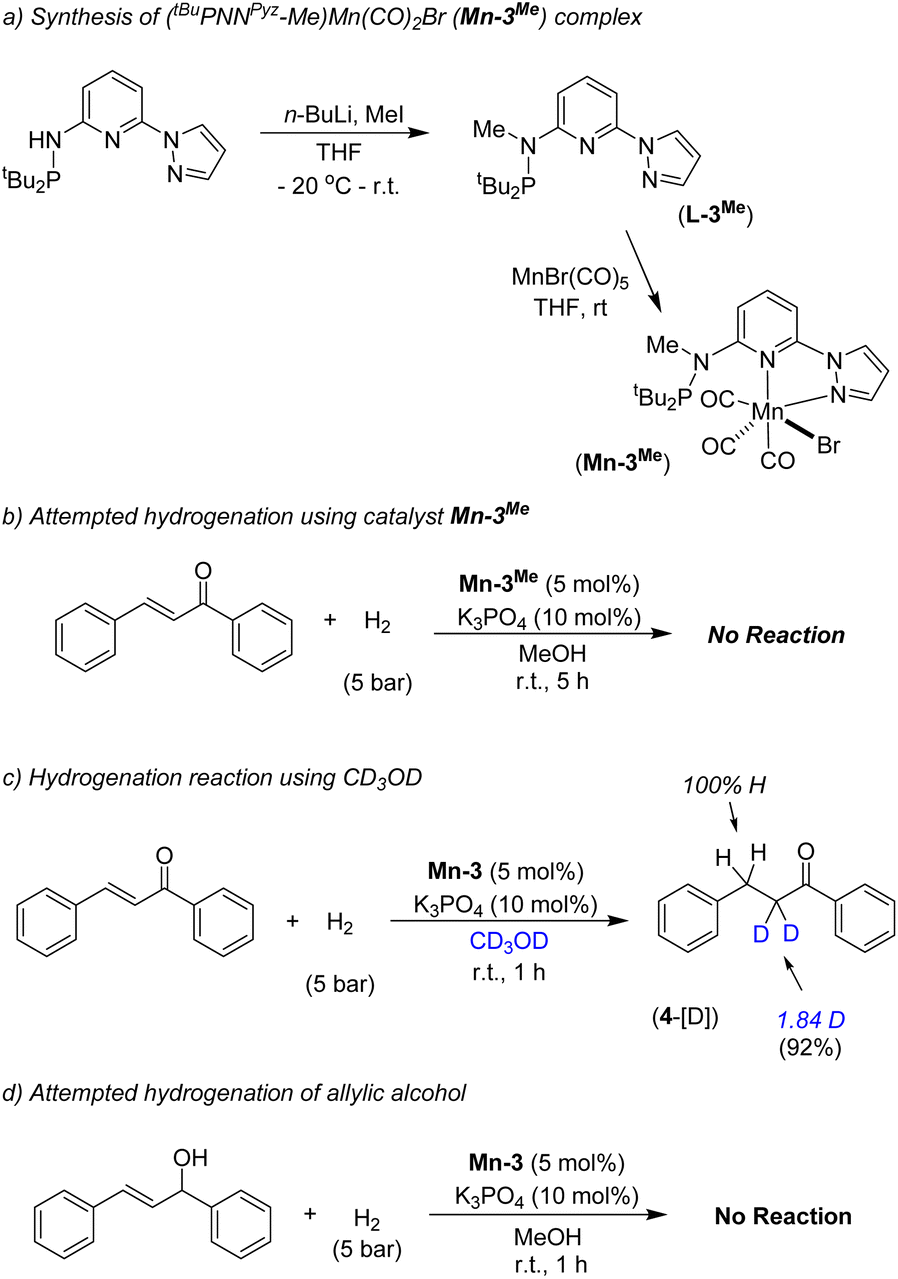
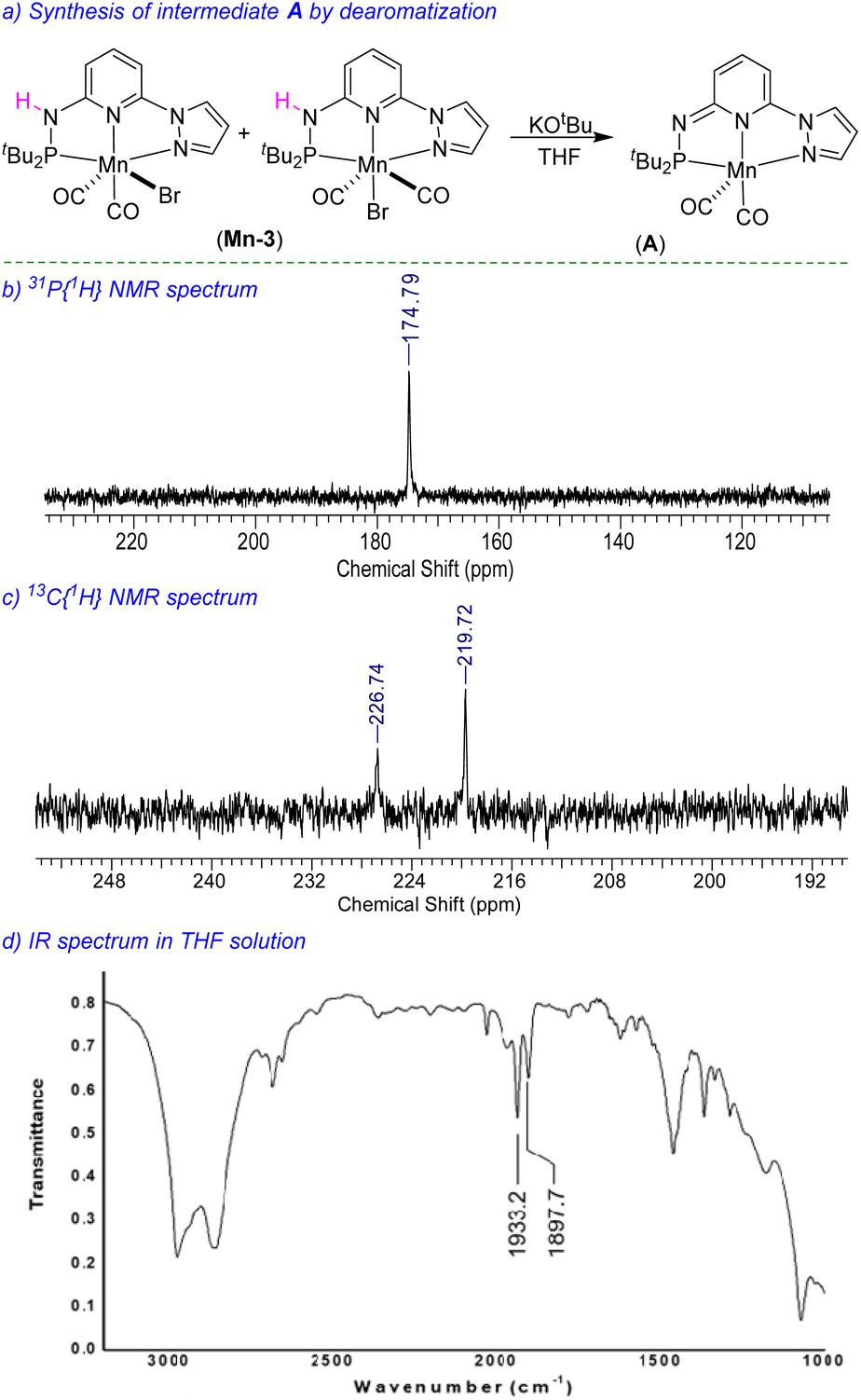
We have performed a few controlled experiments to understand the operating mode of the manganese catalyst. First, the N-methyl substituted ligand (L3-Me) and corresponding manganese complex, (tBuPN(Me)NPyz)Mn(CO)3Br (Mn-3Me) were synthesized and fully characterized by various analytical techniques (Scheme 5a). An attempted hydrogenation of 4a employing complex Mn-3Me as the catalyst under the standard hydrogenation conditions did not provide the hydrogenated product (Scheme 5b), and the starting compound (4a) was quantitatively recovered. This finding supports the crucial role of the N–H proton in the complex (tBuPN3(H)NPyz)Mn(CO)2Br (Mn-3) during hydrogenation. Furthermore, the treatment of Mn-3 with stoichiometric KOtBu produced dearomatized active intermediate A (Fig. 2). The 31P{1H} NMR spectrum of A showed a single peak (against two isomeric peaks for Mn-3), and two CO signals were observed in IR as well as 13C{1H} NMR spectra of A (against three peaks for CO in Mn-3). Interestingly, the employment of species A as a catalyst in hydrogenation without the use of an additional base provided a quantitative yield of 4. All these results suggest that the species A acts as an active catalyst and the catalytic reaction proceeds through the metal–ligand cooperation (dearomatization/aromatization) pathway.
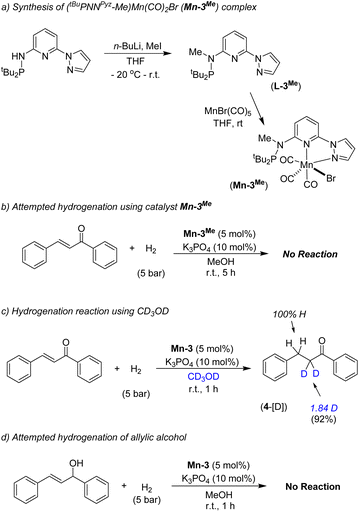 |
| | Scheme 5 Synthesis of Mn-3Me and controlled mechanistic experiments. | |
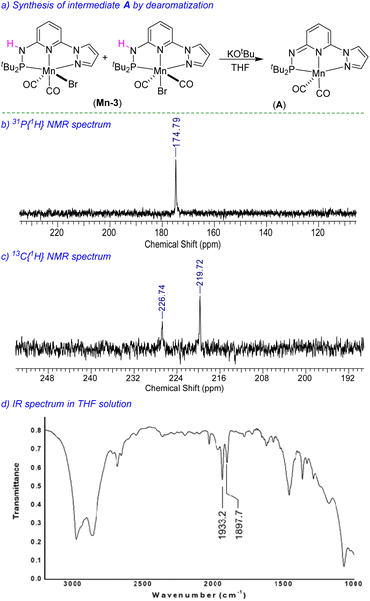 |
| | Fig. 2 Synthesis of active intermediate A and characterization spectra. | |
A hydrogenation reaction was performed using CD3OD as a solvent to thoroughly understand the hydrogenation process (Scheme 5c). The isolated hydrogenated product 4-[D] shows 92% deuterium incorporation at the alpha-methylene position (see 1H, 13C and deuterium NMR spectra in the ESI†). Moreover, the hydrogenation reaction did not occur in the presence of aprotic solvent (THF, dioxane or CH2Cl2). These observations indicate the necessity of a protic solvent as a proton source and tentatively support a Mn-enolate intermediate. All these findings are consistent with the low energy barrier observed for the protonation step (discussed vide infra). An attempted hydrogenation of allylic alcohol under the standard reaction conditions failed to give a hydrogenated product (Scheme 5d). This finding highlights that the hydrogenation of (E)-chalcone (4a) does not proceed via 1,2-hydrogen addition, instead a 1,4-addition of hydrogen occurs. The observed chemoselectivity and controlled studies allowed us to propose the following: (i) the Mn-3 catalyst prefers 1,2-hydrogen addition to CO or CN when steric hindrance on carbonyl's/imine's carbon is low (aldehyde's CO and imine's CN preferred over the CC), whereas (ii) the Mn-3 catalyst allowed 1,4-hydrogen addition when carbonyl's carbon is doubly substituted (i.e. in α,β-unsaturated ketones) due to more steric constraint. Mn–H might fail to approach the carbonyl's carbon in α,β-unsaturated ketones due to steric hindrance; instead it can access the β-carbon via 1,4-hydrogen addition. Notably, the Mn-2 catalyst, which is less bulky than the Mn-3 catalyst, allowed 1,2-hydrogen addition even in α,β-unsaturated ketone to form allylic alcohol. These findings are noteworthy in the consideration of catalyst developments for chemoselective functionalization.
DFT based calculations
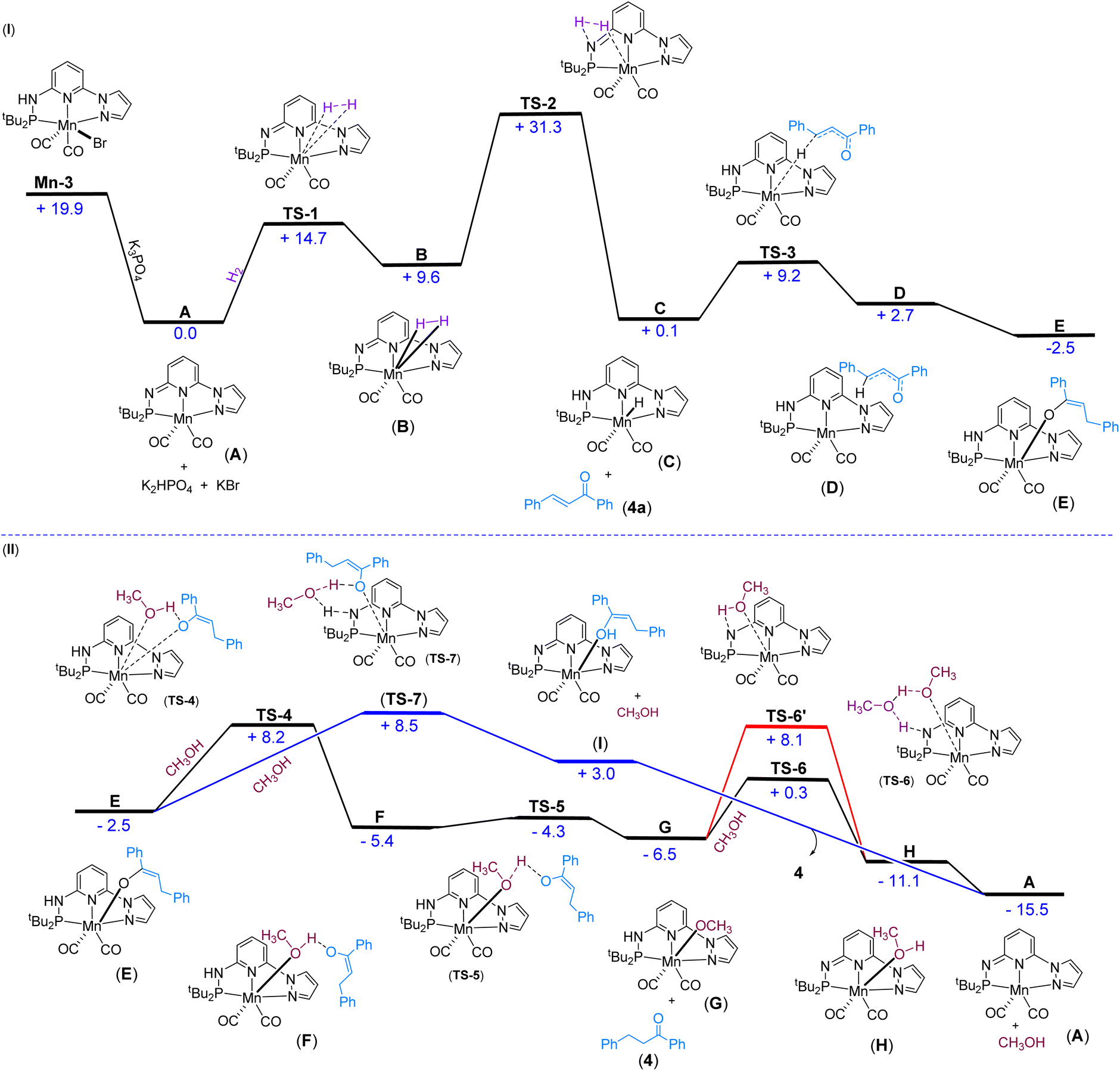
We have investigated the reaction mechanism of Mn-3 catalyzed hydrogenation of α,β-unsaturated carbonyl compound 4a using density functional theory (DFT) calculations (Fig. 3). Initially, the generation of the active catalyst A would occur when the pre-catalyst Mn-3 reacts with K3PO4 (Fig. 3(I)). In this step, the Mn–Br and N–H bonds break, forming the catalyst A, K2HPO4 and KBr. This step is thermodynamically favorable (ΔG = −19.9 kcal mol−1). Next, the H2 molecule adds to catalyst A, forming the intermediate Bvia a barrier of 14.7 kcal mol−1 (TS-1). Then, the reaction proceeds through TS-2 with a barrier of 21.7 kcal mol−1, in which the H–H bond breaks, and the Mn–H and N–H bonds form, generating the intermediate C. The overall barrier for the H2 activation (31.3 kcal mol−1) seems reasonable, considering that a minimum of 5 bar H2 pressure is essential for the reaction. In the next step, the reaction crosses the barrier of 9.1 kcal mol−1 (TS-3), wherein 4a reacts with C to form the intermediate D by hydride (H−) migration from Mn–H to 4a. In the next step, the intermediate D rearranges and forms a new intermediate E. This step is found to be thermodynamically favorable (ΔG = −5.2 kcal mol−1).
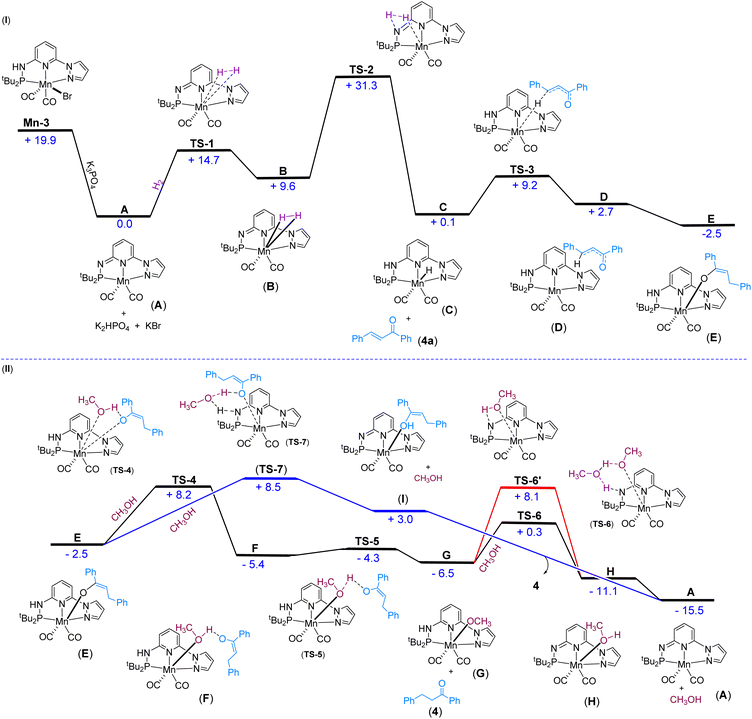 |
| | Fig. 3 Free energy profile for (tBuPNNPyz) Mn(I)-catalyzed hydrogenation: (I) Mn-3 to intermediate E, (II) intermediate E to regeneration of active catalyst A. The free energy values are given in kcal mol−1. | |
Starting with the intermediate E, two approaches are considered for protonation (Fig. 3(II)). In the first possibility, the methanol protonates the substrate step-wise, as shown in Fig. 3(II). Thus, MeOH can provide a proton to the substrate and coordinates with Mn concurrently. In this process, the Mn–O bond breaks and a different Mn–O bond forms via the transition state TS-4 with a barrier of 10.7 kcal mol−1, leading to the formation of the intermediate F. The reaction crosses a barrier of 1.1 kcal mol−1 (TS-5) in the following step, leading to product 4 and intermediate G. Then, the reaction could proceed in two different ways: with the assistance of solvent (MeOH) TS-6 and without the assistance of solvent TS-6′, which has barriers of 6.8 kcal mol−1 and 14.6 kcal mol−1, respectively. The energy values indicate that the reaction will proceed through the transition state TS-6 with the assistance of a solvent (MeOH), leading to the formation of intermediate H. In the next step, intermediate H is converted into A after releasing methanol. This step is thermodynamically favorable (ΔG = −4.4 kcal mol−1). The overall low barrier for the protonation process tentatively supports the experimental observation, wherein a reversible protonation was assumed.
In the second possibility of protonation, the solvent MeOH directly shuttles the protons between Mn–O and N–H through a transition state TS-7 with a barrier of 11.0 kcal mol−1, leading to the formation of intermediate I (Fig. 3(II)). In the next step, intermediate I is converted into the active species A and product 4. This step is thermodynamically favorable (ΔG = −18.5 kcal mol−1). A perusal of the two pathways based on turnover frequency (TOF) analysis indicates that the TOF would be the same for both approaches (Fig. 3(II)), because the main intermediate (TDI) (A) along the pathways and the main transition state (TDTS) (TS-2) are present in the early part of the cycles, which are common to both the proposed protonation pathways (for more information on the TOF analysis, see the ESI†). The calculated energy barrier values indicated a maximum barrier for H2 activation, which would be feasible at room temperature considering the reaction's 5.0 bar H2 pressure requirement. Therefore, H2 activation could be proposed as a probable turnover-limiting step. Notably, the protonation of the final compound by MeOH is very facile, which corroborates the experimental findings.
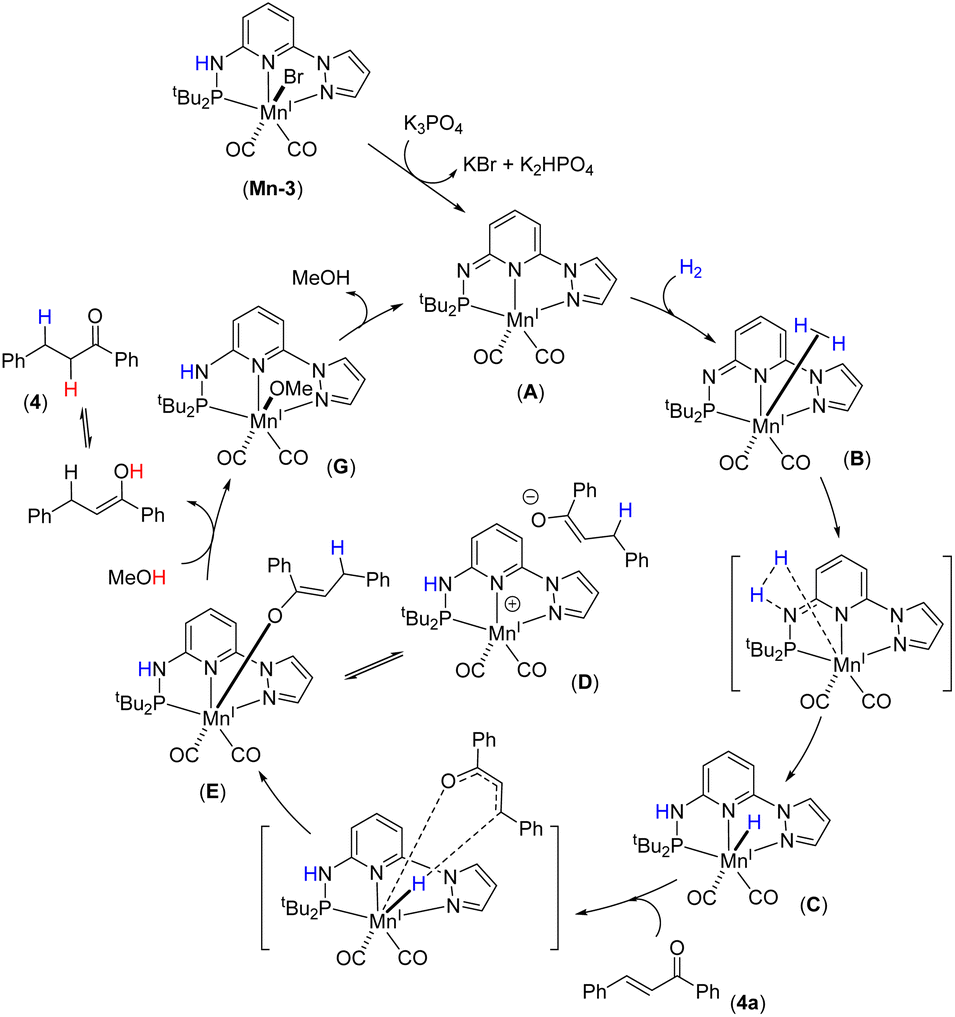
Catalytic cycle
Based on the experimental findings, DFT calculations and literature precedents,12a,15c,21b we proposed a tentative catalytic cycle for the Mn-catalyzed chemoselective hydrogenation of α,β-unsaturated ketones (Fig. 4). Initially, the complex Mn-3 would transform into the active catalyst A by dearomatization in the presence of K3PO4. We have identified the species A by NMR and IR analyses. The similar conversion of a metal complex into dearomatized species is well documented.12a,21b,24 Moreover, the species A as an active catalyst was verified. The H2 molecule will coordinate to Mn(I) species, followed by metal–ligand (M–L) cooperative activation of H2 leading to species C through transition state TS-2. Next, the reaction of substrate 4a with C leads to 1,4-hydride migration resulting in intermediate D (or E). The protonation of the semi-hydrogenated species in intermediate D (or E) by methanol provides the hydrogenated product 4. Finally, the resulting intermediate G would lead to the regeneration of active catalyst A. The DFT energy calculations supported all these elementary catalytic steps. H2 activation by the Mn(I) species has a high barrier and is a pivotal step in the hydrogenation process. Therefore, H2 activation by Mn(I)–ligand cooperation can be assumed to be the turnover-limiting step.
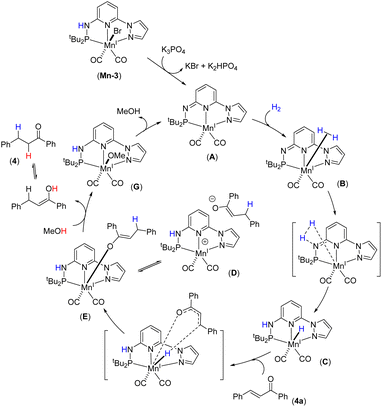 |
| | Fig. 4 Plausible catalytic cycle. | |
Conclusions
In summary, we have developed an efficient protocol for the chemoselective hydrogenation of CC, CO and CN bonds in α,β-unsaturated ketones, aldehydes and imines catalyzed by the well-defined pincer-ligated Mn(I) complex. The employment of a mild base, moderate H2 pressure and room temperature (27 °C) in Mn-catalyzed hydrogenation are highly advantageous to the commonly employed KOtBu base and extreme reaction conditions. The mixed donor Mn(I) complexes, κ3-(R2PN3NPyz)Mn(CO)2Br (R = Ph, iPr, tBu) were synthesized and meticulously characterized by various techniques. Though both the iPr and tBu-substituted Mn(I) complexes are efficient for the hydrogenation of α,β-unsaturated ketones, the (tBu2PNNPyz)Mn(CO)2Br complex as a catalyst provided exceptional chemoselectivity for the reduction of the CC bond (1,4-hydrogen addition). Thus, using the beneficial molecular hydrogen and a mild K3PO4 base, the (tBu2PNNPyz)Mn(I) catalyst could hydrogenate diverse unsaturated ketones to saturated ketones at room temperature with the compatibility of sensitive functionalities, such as halides (–F, –Cl, –Br, and –I), alkoxy, hydroxy, epoxide, acetyl, nitrile, nitro, and unconjugated alkenyl and alkynyl groups. The CO bond in aldehydes and the CN bond in imines were preferentially hydrogenated (1,2-hydrogen addition) over the CC bond using the Mn-3 catalyst. A comprehensive mechanistic investigation by controlled studies endorsed the non-innocent behavior of the ligand in the Mn catalyst and supported a metal–ligand cooperation pathway. The DFT energy calculations highlighted a probable turnover-limiting H2 activation with the facile progress of other elementary steps. Particularly, H2 provided a hydride source, and solvent MeOH acts as a proton source for hydrogenation. The DFT energy calculations unanimously supported the proposed mechanistic cycle.
Data availability
Crystallographic data for compound Mn-2 has been deposited at The Cambridge Crystallographic Data Centre (CCDC) under deposition number 2194541.†
Author contributions
A. B. S. and D. M. S. designed and performed the catalyst synthesis. A. B. S. carried out all catalytic and mechanistic experiments. A. B. S. and B. P. conceived the project, interpreted the results and co-wrote the original draft. R. G. G. performed XRD and analysed the data. P. B. performed DFT calculations under the supervision of K. V. All authors contributed to the analysis of the results and preparation of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by SERB, New Delhi, India (CRG/2020/000554). A. B. S. and D. M. S. thank CSIR-New Delhi for research fellowships. We acknowledge the National Supercomputing Mission (NSM) for providing computing resources of ‘PARAM Brahma’ at IISER Pune, which is implemented by C-DAC and supported by the Ministry of Electronics and Information Technology (MeitY) and Department of Science and Technology (DST), Government of India.
Notes and references
-
(a)
P. N. Rylander, Catalytic Hydrogenation in Organic Synthesis, in The Catalytic Hydrogenation in Organic Syntheses, Academic Press, New York, 1979, pp. 1–12 Search PubMed;
(b)
P. G. Andersson and I. J. Munslow, Modern Reduction Methods, Wiley-VCH Verlag GmbH & Co., 2008 CrossRef.
-
(a) J. Pritchard, G. A. Filonenko, R. van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2015, 44, 3808–3833 RSC;
(b) M. Tamura, Y. Nakagawa and K. Tomishige, J. Jpn. Petroleum Inst., 2019, 62, 106–119 CrossRef CAS;
(c) Y. Wang, M. Wang, Y. Li and Q. Liu, Chem, 2021, 7, 1180–1223 CrossRef CAS.
-
J. G. de Vries and C. J. Elsevier, Handbook of Homogeneous Hydrogenation, Weinheim Wiley-VCH, 2007 Search PubMed.
-
(a) R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40–73 CrossRef CAS;
(b) J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, Chem. Rev., 2011, 111, 1713–1760 CrossRef CAS PubMed;
(c) M. L. Clarke, Catal. Sci. Technol., 2012, 2, 2418–2423 RSC;
(d) D.-S. Wang, Q.-A. Chen, S.-M. Lu and Y.-G. Zhou, Chem. Rev., 2012, 112, 2557–2590 CrossRef CAS PubMed;
(e) S. Werkmeister, K. Junge and M. Beller, Org. Pro. Res. Dev., 2014, 18, 289–302 CrossRef CAS;
(f) Y.-M. He, Y. Feng and Q.-H. Fan, Acc. Chem. Res., 2014, 47, 2894–2906 CrossRef CAS PubMed.
-
(a) G. A. Filonenko, R. van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2018, 47, 1459–1483 RSC;
(b) T. Zell and R. Langer, ChemCatChem, 2018, 10, 1930–1940 CrossRef CAS;
(c) L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119, 2681–2751 CrossRef CAS PubMed;
(d) T. Irrgang and R. Kempe, Chem. Rev., 2019, 119, 2524–2549 CrossRef CAS PubMed.
-
(a) I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed;
(b) T. Zell and D. Milstein, Acc. Chem. Res., 2015, 48, 1979–1994 CrossRef CAS PubMed;
(c) S. Chakraborty, P. Bhattacharya, H. Dai and H. Guan, Acc. Chem. Res., 2015, 48, 1995–2003 CrossRef CAS PubMed;
(d) A. Fürstner, ACS Cent. Sci., 2016, 2, 778–789 CrossRef PubMed.
-
(a) A. Mukherjee and D. Milstein, ACS Catal., 2018, 8, 11435–11469 CrossRef CAS;
(b) W. Liu, B. Sahoo, K. Junge and M. Beller, Acc. Chem. Res., 2018, 51, 1858–1869 CrossRef CAS PubMed;
(c) W. Ai, R. Zhong, X. Liu and Q. Liu, Chem. Rev., 2019, 119, 2876–2953 CrossRef CAS PubMed.
-
(a) R. M. Bullock, Science, 2013, 342, 1054–1055 CrossRef CAS PubMed;
(b) W. Zuo, A. J. Lough, Y. F. Li and R. H. Morris, Science, 2013, 342, 1080–1083 CrossRef CAS PubMed;
(c) M. R. Friedfeld, M. Shevlin, J. M. Hoyt, S. W. Krska, M. T. Tudge and P. J. Chirik, Science, 2013, 342, 1076–1080 CrossRef CAS PubMed;
(d) M. R. Friedfeld, H. Zhong, R. T. Ruck, M. Shevlin and P. J. Chirik, Science, 2018, 360, 888–893 CrossRef CAS PubMed.
- S. Elangovan, C. Topf, S. Fischer, H. Jiao, A. Spannenberg, W. Baumann, R. Ludwig, K. Junge and M. Beller, J. Am. Chem. Soc., 2016, 138, 8809–8814 CrossRef CAS PubMed.
- F. Kallmeier, T. Irrgang, T. Dietel and R. Kempe, Angew. Chem., Int. Ed., 2016, 55, 11806–11809 CrossRef CAS PubMed.
-
(a) A. Bruneau-Voisine, D. Wang, T. Roisnel, C. Darcel and J.-B. Sortais, Catal. Commun., 2017, 92, 1–4 CrossRef CAS;
(b) D. Wei, A. Bruneau-Voisine, T. o. Chauvin, V. Dorcet, T. Roisnel, D. A. Valyaev, N. l. Lugan and J.-B. Sortais, Adv. Synth. Catal., 2018, 360, 676–681 CrossRef CAS;
(c) R. Buhaibeh, O. A. Filippov, A. Bruneau-Voisine, J. Willot, C. Duhayon, D. A. Valyaev, N. Lugan, Y. Canac and J.-B. Sortais, Angew. Chem., Int. Ed., 2019, 58, 6727–6731 CrossRef CAS PubMed;
(d) R. Buhaibeh, C. Duhayon, D. A. Valyaev, J.-B. Sortais and Y. Canac, Organometallics, 2021, 40, 231–241 CrossRef CAS.
-
(a) M. Glatz, B. Stöger, D. Himmelbauer, L. F. Veiros and K. Kirchner, ACS Catal., 2018, 8, 4009–4016 CrossRef CAS PubMed;
(b) S. Weber, B. Stöger and K. Kirchner, Org. Lett., 2018, 20, 7212–7215 CrossRef CAS PubMed;
(c) S. Weber, J. Brünig, L. F. Veiros and K. Kirchner, Organometallics, 2021, 40, 1388–1394 CrossRef CAS PubMed.
-
(a) H.-J. Pan and X. Hu, Angew. Chem., Int. Ed., 2020, 59, 4942–4946 CrossRef CAS PubMed;
(b) W. Yang, I. Y. Chernyshov, R. K. A. van Schendel, M. Weber, C. Müller, G. A. Filonenko and E. A. Pidko, Nat. Commun., 2021, 12, 12 CrossRef CAS PubMed;
(c) T. Vielhaber and C. Topf, Appl. Catal. A: Gen., 2021, 623, 118280 CrossRef CAS.
- For reviews, see:
(a) B. Maji and M. K. Barman, Synthesis, 2017, 49, 3377–3393 CrossRef CAS;
(b) M. Garbe, K. Junge and M. Beller, Eur. J. Org. Chem., 2017, 2017, 4344–4362 CrossRef CAS;
(c) F. Kallmeier and R. Kempe, Angew. Chem., Int. Ed., 2018, 57, 46–60 CrossRef CAS PubMed;
(d) N. Gorgas and K. Kirchner, Acc. Chem. Res., 2018, 51, 1558–1569 CrossRef CAS PubMed.
-
(a) D. Wei, A. Bruneau-Voisine, D. A. Valyaev, N. Lugan and J.-B. Sortais, Chem. Commun., 2018, 54, 4302–4305 RSC;
(b) Y. Wang, L. Zhu, Z. Shao, G. Li, Y. Lan and Q. Liu, J. Am. Chem. Soc., 2019, 141, 17337–17349 CrossRef CAS PubMed;
(c) F. Freitag, T. Irrgang and R. Kempe, J. Am. Chem. Soc., 2019, 141, 11677–11685 CrossRef CAS PubMed;
(d) Z. Wang, L. Chen, G. Mao and C. Wang, Chin. Chem. Lett., 2020, 31, 1890–1894 CrossRef CAS;
(e) Y. Wang, S. Liu, H. Yang, H. Li, Y. Lan and Q. Liu, Nat. Chem., 2022, 14, 1233–1241 CrossRef CAS PubMed.
- M. B. Widegren, G. J. Harkness, A. M. Z. Slawin, D. B. Cordes and M. L. Clarke, Angew. Chem., Int. Ed., 2017, 56, 5825–5828 CrossRef CAS PubMed.
-
(a) M. Garbe, K. Junge, S. Walker, Z. Wei, H. Jiao, A. Spannenberg, S. Bachmann, M. Scalone and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 11237–11241 CrossRef CAS PubMed;
(b) M. Garbe, Z. Wei, B. Tannert, A. Spannenberg, H. Jiao, S. Bachmann, M. Scalone, K. Junge and M. Beller, Adv. Synth. Catal., 2019, 361, 1913–1920 CrossRef CAS.
-
(a) L. Zhang, Y. Tang, Z. Han and K. Ding, Angew. Chem., Int. Ed., 2019, 58, 4973–4977 CrossRef CAS PubMed;
(b) L. Zhang, Z. Wang, Z. Han and K. Ding, Angew. Chem., Int. Ed., 2020, 59, 15565–15569 CrossRef CAS PubMed.
-
(a) F. Ling, H. Hou, J. Chen, S. Nian, X. Yi, Z. Wang, D. Song and W. Zhong, Org. Lett., 2019, 21, 3937–3941 CrossRef CAS PubMed;
(b) L. Zeng, H. Yang, M. Zhao, J. Wen, J. H. R. Tucker and X. Zhang, ACS Catal., 2020, 10, 13794–13799 CrossRef CAS;
(c) F. Ling, J. Chen, S. Nian, H. Hou, X. Yi, F. Wu, M. Xu and W. Zhong, Synlett, 2020, 31, 285–289 CrossRef CAS;
(d) C. S. G. Seo, B. T. H. Tsui, M. V. Gradiski, S. A. M. Smith and R. H. Morris, Catal. Sci. Technol., 2021, 11, 3153–3163 RSC.
-
(a) C. Liu, M. Wang, S. Liu, Y. Wang, Y. Peng, Y. Lan and Q. Liu, Angew. Chem., Int. Ed., 2021, 60, 5108–5113 CrossRef CAS PubMed;
(b) C. Liu, M. Wang, Y. Xu, Y. Li and Q. Liu, Angew. Chem., Int. Ed., 2022, 61 DOI:10.1002/anie.202202814.
-
(a) S. Weber, B. Stöger, L. F. Veiros and K. Kirchner, ACS Catal., 2019, 9, 9715–9720 CrossRef CAS;
(b) S. M. W. Rahaman, D. K. Pandey, O. Rivada-Wheelaghan, A. Dubey, R. R. Fayzullin and J. R. Khusnutdinova, ChemCatChem, 2020, 12, 5912–5918 CrossRef CAS.
- For selected chemoselective hydrogenation using 4d metals, see:
(a) Q. Hu, Y. Hu, Y. Liu, Z. Zhang, Y. Liu and W. Zhang, Chem.–Eur. J., 2017, 23, 1040–1043 CrossRef CAS PubMed;
(b) W. Chang, X. Gong, S. Wang, L.-P. Xiao and G. Song, Org. Biomol. Chem., 2017, 15, 3466–3471 RSC;
(c) M. Soto, R. G. Soengas and H. Rodríguez-Solla, Adv. Synth. Catal., 2020, 362, 5422–5431 CrossRef CAS;
(d) Y. Gu, J. R. Norton, F. Salahi, V. G. Lisnyak, Z. Zhou and S. A. Snyder, J. Am. Chem. Soc., 2021, 143, 9657–9663 CrossRef CAS PubMed.
-
(a) D. Gong, W. Liu, T. Chen, Z.-R. Chen and K.-W. Huang, J. Mol. Catal. A Chem., 2014, 395, 100–107 CrossRef CAS;
(b) D. Gong, X. Zhang and K.-W. Huang, Dalton Trans., 2016, 45, 19399–19407 RSC;
(c) H. Chen, W. Pan, K.-W. Huang, X. Zhang and D. Gong, Polym. Chem., 2017, 8, 1805–1814 RSC.
-
(a) T. P. Gonçalves and K.-W. Huang, J. Am. Chem. Soc., 2017, 139, 13442–13449 CrossRef PubMed;
(b) T. Shimbayashi and K.-i. Fujita, Catalysts, 2020, 10, 635 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental procedures and characterization data, including 1H and 13C NMR of all compounds, CIF and DFT (.xyz) file]. CCDC 2194541 (for comp. Mn-2). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc05274a |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Dipesh M.
Sharma
ab,
Priyam
Bajpai
bc,
Rajesh G.
Gonnade
ab,
Dipesh M.
Sharma
ab,
Priyam
Bajpai
bc,
Rajesh G.
Gonnade